

Abstract
Age-related macular degeneration (AMD) is the predominant cause of visual loss in old people in the developed world, whose incidence is increasing. This disease is caused by the decrease in macular function, due to the degeneration of retinal pigment epithelium (RPE) cells. The aged retina is characterised by increased levels of reactive oxygen species (ROS), impaired autophagy, and DNA damage that are linked to AMD pathogenesis. Mitophagy, a mitochondria-specific type of autophagy, is an essential part of mitochondrial quality control, the collective mechanism responsible for this organelle’s homeostasis. The abundance of ROS, DNA damage, and the excessive energy consumption in the ageing retina all contribute to the degeneration of RPE cells and their mitochondria. We discuss the role of mitophagy in the cell and argue that its impairment may play a role in AMD pathogenesis. Thus, mitophagy as a potential therapeutic target in AMD and other degenerative diseases is as well explored.
Keywords: Cell senescence, Mitochondrial DNA damage, Nrf2/PGC-1α axis, Oxidative damage, Ubiquitin
Introduction: AMD and autophagy
Age-related macular degeneration (AMD) is an eye disease characterised by a progressive decrease in macular function. It is the leading cause of visual impairment in the Western world in the elderly. There are approximately 50 million people suffering from AMD worldwide and the number is expected to increase threefold over the next 20 years. Thus, AMD is becoming a major global public health issue [1]. AMD is a complex disease with multifactorial etiology including ageing, family history, smoking, high blood pressure, obesity, hypercholesterolemia, and arteriosclerosis [2, 3].
The disease affects photoreceptors, choriocapillaris, and the retinal pigment epithelium (RPE) in the sensitive macula, which is responsible for colour and sharp central vision (Fig. 1a) [4, 5]. AMD can be divided into two distinct forms: dry (atrophic) and wet (exudative). A total of 80% of patients suffer from the dry subtype, for which no efficient treatment exists. Wet AMD development is strongly associated with upregulation of the vascular endothelial growth factor (VEGF), which is the principal therapy target to inhibit detrimental neovascularisation process caused by this growth factor [6].
Fig. 1.
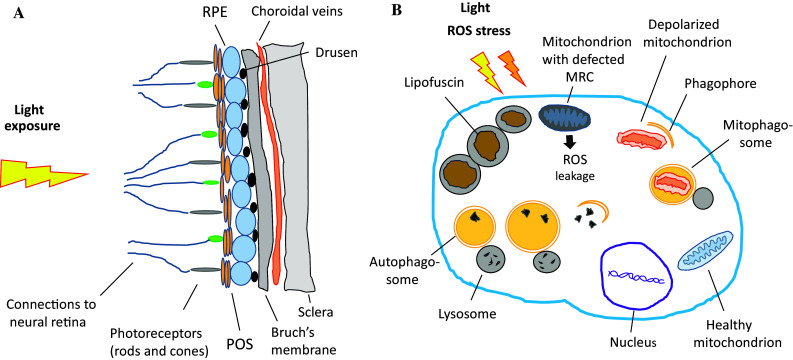
a The simplified partial structure of the aged retina showing the position of retinal pigment epithelium (RPE). The retina is exposed to high levels of light. Extracellular drusen formations between RPE and the Bruch’s membrane is a sign of ageing and the progression of age-related macular degeneration (AMD). POS, photoreceptor outer segment. b The aged RPE cell. Lipofuscin deposits, located in lysosomes, are their hallmarks, and enough accumulated, they contribute to the development of AMD. Light and reactive oxygen species (ROS) exposure produce continuous stress. The increase in defective mitochondria load fosters ROS exposure internally. The cell uses autophagy and mitophagy for degeneration of the damaged material. Their activities are weakened in aged cells, thus augmenting the accumulation of dense indissoluble lipofuscin waste. MRC mitochondrial respiratory chain
Age-related macular degeneration-related loss of vision is primarily associated with a progressive degeneration and cell death of the RPE, which secondarily evokes adverse effects on the rod and cone cells. The RPE cells are pre-disposed to chronic oxidative stress due to their high levels of oxygen consumption, exposure to lipid peroxidation products derived from photoreceptor outer segment (POS) and continuous light stimuli. Oxidatively damaged molecules, including carboxyethylpyrrole, malondialdehyde, 4-hydroxynonenal, and glycation end products, accumulate in the macular area, and are the source of chronic oxidative stress as well [7–9]. Golestaneh and associates [10, 11] have recently found that RPE cells from AMD patients produce more ROS than those derived from control normal donors. These cells from AMD sufferers were unable to increase the expression of superoxide dismutase (SOD) during oxidative stress [12]. In senescent RPE cells, the ability to respond to oxidative stress is weakened, resulting in an accumulation of auto-oxidative lipofuscin in the lysosomes of RPE cells and extracellular drusen formation between RPE and Bruch’s membrane [13, 14] (Fig. 2b; fundus of a healthy eye presented in Fig. 2a). In addition to the oxidative stress and formation of molecular aggregates, immunologic processes are involved in AMD pathogenesis. Generation of inflammatory-related molecules in the Bruch’s membrane and recruitment of macrophages, dendritic cells, complement activation, and microglial activation in the macular area have been documented [5, 15, 16]. In addition, ROS-induced DNA damage in RPE cells can contribute to the development of AMD [17–19].
Fig. 2.

a Fundus photograph of a healthy eye. b Fundus photograph of a dry AMD eye. The arrow indicates a yellowish zone of drusen accumulation in the macular area. Drusen are located between retinal pigment epithelium and Bruch’s membrane. The photographs are from Imaging Data Source data bank, Department of Ophthalmology, Kuopio University Hospital, Kuopio
A number of retinal pathologies, including AMD, are connected with mitochondrial dysfunction [20]. Decreased numbers of mitochondria, loss of mitochondria cristae [the folds in the inner mitochondrial membrane (IMM)], reduction of the mitochondrial matrix density as well as mutations in mitochondrial DNA (mtDNA) (“Mitochondrial DNA damage”) have been reported in studies examining the RPE of AMD donors [18, 21, 22]. Therefore, mitochondria appear to be an important target for study trying to survey of AMD pathology, and possibly provide promising for therapeutic targets.
Autophagy is a collective term for the complex lysosomal clearance processes utilised by cells to eliminate large unwanted structures such as cytoplasmic material (ubiquitinated macromolecules), organelles [as exemplified by damaged mitochondria, endoplasmic reticulum (ER), and peroxisomes], and pathogens. Moreover, it is an important means for survival in the starvation state when the cell literally “eats itself” [23–25]. Autophagy can be further subdivided into microautophagy, chaperone-mediated autophagy, and macroautophagy. The last of these is the best studied and is referred hereafter as autophagy. It is initiated upon induction (as defined by nutrition shortage) by the formation of isolation membranes, which originate from endoplasmic reticulum or other cellular membrane (Figs. 1b, 3). Then, they elongate as phagophore membranes and engulf a portion of the cytosol to form double membrane autophagosomes, which invaginate material for degradation. Furthermore, autophagosomes undergo multiple steps, including the attachment of the small GTPase Rab7, which signals their maturation, and is likewise needed for the transport of autophagosomes along microtubuli [26]. Finally, they fuse with primary lysosomes forming autolysosomes, and their contents are then degraded by lysosomal enzymes and recycled. This whole route is regulated by at least 41 autophagy-related genes (Atgs) identified so far [27, 28].
Fig. 3.
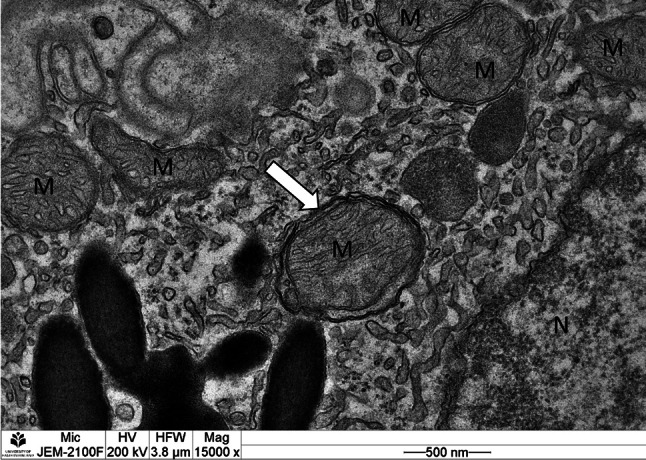
An early phase of mitophagy (the white arrow) is seen as the formation of double membrane, wrapped around a mitochondrion, in a mouse primary retinal pigment epithelial (RPE) cell. Picture is captured by transmission electron microscopy (×15,000 magnification). The photograph is from Ophthalmology Research Unit, University of Eastern Finland, Kuopio. M damaged mitochondrion, N nucleus
Autophagy is stimulated in response to many stress stimuli that are constantly present in the RPE such as hypoxia, oxidative stress, inflammation, or unfolded proteins [14, 29, 30]. Furthermore, autophagic proteins are intensely expressed in the eye [31, 32]. Autophagy, besides of the phagolytic degradation, is important in the daily turnover of POS too [33]. This all indicates that autophagy is essential in the fitness of both the neuroretina and the RPE. Failure of autophagy in aged cells, including RPE cells, results in the accumulation of protein aggregates, cellular degeneration and eventually cell death [28, 34]. Vice versa, functional autophagy reduces the toxicity of the protein aggregates and may prevent age-related modifications of RPE [30]. To date, there is strong evidence that weakened autophagy flux increases lipofuscinogenesis and RPE damage in AMD [11, 31, 35–39]. When autophagic capacity declines simultaneously with increased ROS production and protein aggregation as in RPE degeneration and AMD, it may activate inflammasomes, which provoke a low-grade inflammation in retinal cells and accelerate ageing process [40, 41]. Autophagy was for long considered as a non-selective means of bulk degradation, especially during starvation, where it provides nutrition to the cell. However, more recently, several selective types of autophagy have been revealed. These include the degradation of cytoplasmic aggregates (aggrephagy), lipid droplets (lipophagy), exogenous pathogens (xenophagy), and of organelles (for instance nucleophagy and ribophagy) [42–44]. The last group includes mitophagy. Although mitophagy is closely connected to general autophagy, it functions in several distinct pathways. In fact, mitophagy could be regarded not only just a type of autophagy, but also an essential and distinct part of the mechanisms that govern mitochondrial quality control. Figure 3 represents the action of mitophagy, revealed by transmission electron microscopy.
Impaired mitochondria stimulate non-selective autophagy
Increased generation of ROS or decreased ATP production has been shown to induce general autophagy. Mitochondria may play a key role in the regulation of autophagy by ROS signaling in mammalian cells [45–47]. It is especially important in RPE, where oxygen consumption and thus ROS production are excessive. Under starvation conditions, H2O2 is accumulated in mitochondria, leading to the inhibition of Atg4 activity. This results in an increase in the lipidation of Atg8 (a yeast gene) mammalian orthologues MAPK1LC3 and GATE-16, which are both involved in canonical autophagy [47]. In agreement with this, it has been shown that autophagosome marker proteins Atg5 and MAPK1LC3 localise in mitochondria during nutrient deprivation [48]. In addition, regions of MAPK1LC3 overlap with markers of the outer mitochondrial membrane (OMM), but not with those of the IMM, and matrix markers, probably excluding their involvement of mitophagy [49]. Autophagy may be regulated by the proteins located on mitochondrial surface, including beclin-1, which localises to the mitochondria and ER [50]. Beclin-1 is regulated by several proteins, including activating molecule in beclin-1-regulated autophagy (AMBRA1) and the anti-apoptotic factor Bcl-2 [51, 52]. In normal conditions, Bcl-2 interacts with AMBRA1 at mitochondrial surface and beclin-1 in ER to inhibit autophagy. However, in starvation, AMBRA1 dissociates from Bcl-2 and associates with beclin-1 at the reticulum and mitochondria surface to promote autophagy [52, 53]. The role of AMBRA1 and Bcl-2 in the selective autophagy of mitochondria, mitophagy, is discussed in “Ubiquitin-mediated mitophagy: PINK1 and Parkin”. A growing body of evidence suggests the involvement of non-selective autophagy in the management of mitochondrial fitness. On the other hand, mitochondria may have a role of signaling platform to regulate autophagy.
Mitophagy
Mitophagy, an autophagic pathway for mitochondrial quality control
Mitophagy is a principal mechanism of mitochondrial quality control eliminating aged, and damaged mitochondria [54–58]. Therefore, mitophagy is important for cellular homeostasis and cell survival, because the maintenance of a balance between normal and damaged mitochondria is essential for efficient energy production in the cell. Moreover, unnecessary mitochondria may be the sources of ROS, cytochrome c and other apoptosis-related factors, which may induce cell damage and eventually death. Oxidative damage to membrane proteins and anomalous protein aggregation might eventually form aqueous pores and to induce the mitochondrial permeability transition (MPT). This can further promote mitophagy. The loss of mitochondrial membrane potential (MMP) is a marker of mitophagy, but is generally not considered as a primary mitophagy-inducing factor [55, 59]. However, in the study by Kim and Lemasters [60], directly, photoirradiated mitochondria lost their MMP and were subsequently labelled with MAPK1LC3, thus indicating that this loss might directly promote mitophagy, at least in extreme conditions. In addition, mitophagy occurs in developmental regulation. Mitophagy participates in the differentiation of retinal ganglion cells (RGCs), which are projecting neurons of the retina [61]. It is also necessary for the reticulocyte maturation [62].
Some mitochondria-associated ER membrane (MAM) proteins are directly involved in autophagy/mitophagy. They act to bring mitochondria into close proximity of ER and serve as a communication platform [63]. Hamasaki and others [64] have shown that ER–mitochondria contact sites are themselves the origins of the autophagosome membrane. In addition, they revealed at least two MAM-related autophagic proteins, Atg14L and Atg5. The former is an early autophagy marker and an essential pro-autophagic protein, which is relocalised to MAMs, inducing autophagosome formation under starvation. Atg5 is another important MAM-resident protein required for the formation of autophagosomes, which are directly associated with MAMs. In general, MAM proteins and the ER–mitochondria junctions can have an impact in many diseases, including cancers and AMD [65].
Mitophagy and other types of selective autophagy are tightly regulated and need to react to various stimuli. Multiple levels and several mechanisms seem to be involved in this regulation. The upstream control for the initiation of mitophagy requires at least the mitogen-activated protein kinase (MAPK) pathway regulation [66, 67]. It is not involved in induction of non-selective, general autophagy, and so far, its exact mechanism and function in mitophagy is unclear. It has been suggested that the upstream control is involved in the phosphorylation phase, such that in the initial activation of mitophagy receptors and mediators launch the process.
Evidently, mitophagy represents an extreme attempt of a cell to maintain homeostasis disrupted by malfunctioning mitochondria. Mitophagy and mitochondrial biogenesis are balanced acts controlled by an intricate regulatory network [68]. Proper coordination of them is essential for stress resistance and longevity. Age-related decline of mitophagy increases the amount of redundant and non-functional cellular elements and this leads to impaired functioning of the cell. Factors that contribute to mitochondrial homeostasis play a role in the pathogenesis of many age-related disorders, such as neurodegenerative and cardiovascular disorders and cancers. Therefore, mitophagy presents an emerging potential as a target for therapy in these diseases.
Ubiquitin-mediated mitophagy: PINK1 and Parkin
Around the turn of the millennium, two genes, PINK1 (phosphatase and tensin homologue-induced putative kinase 1) and Park2 [RING-IBR-RING (RBR) E3 ubiquitin ligase, Parkin] were found to be associated with Parkinson’s disease in certain Japanese and Italian families, respectively. Follow-up functional studies revealed that both of them cooperate in the degradation of damaged mitochondria via mitophagy [69–73]. An overview of PINK1/Parkin-derived mitophagy is presented in Fig. 4.
Fig. 4.
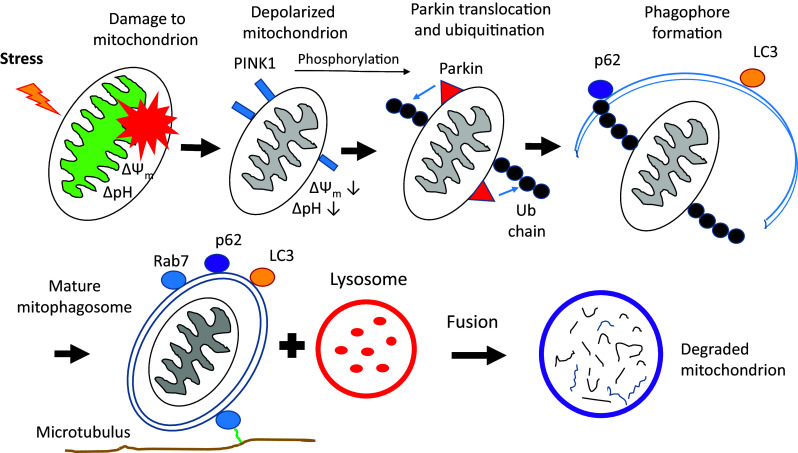
The PINK1-Parkin-derived pathway of mitophagy. In this presentation functional mitochondrion with regular pH and membrane potential encounters damage due to external or internal stress (e.g., reactive oxygen species excess). PINK1 is recruited to the outer mitochondrial membrane. Thereafter Parkin is phosphorylated and translocated. Activated Parkin ubiquitinates the outer membrane. Ubiquitination initiates the phagophore (isolation membrane formation) around the damaged mitochondrion by the general autophagy apparatus. LC3 and p62 are participating in this process directing the enveloped material to autophagic degradation. After maturation steps, including Rab7 attachment, and transport along microtubuli, thus formed mature “mitophagosome” fuses with the lysosome. ΔΨm mitochondrial membrane potential, LC3 MAPK1LC3, p62 Sequestosome1/p62, Rab7 small GTPase Rab7, Ub ubiquitin
Phosphatase and tensin homologue-induced putative kinase is a mitochondrial serine/threonine kinase, occurring in three isoforms, which may be targeted to the OMM with its kinase domain facing the cytoplasm, facilitating its physical interaction with Parkin, which resides in that cellular compartment [74, 75]. Parkin can subsequently catalyze a variety of ubiquitination reactions, ranging from mono- to poly-ubiquitination. All three isoforms of PINK1 may participate in the PINK1-Parkin interaction [76]. PINK1 is proposed to direct the mitochondrial localisation of Parkin through phosphorylation [77]. In steady-state PINK1 and Parkin control mitochondrial fission and fusion processes, as PINK1 is an upstream regulator of Parkin. PINK1 is kept at low levels in functional mitochondria and Parkin is evenly distributed throughout the cytoplasm.
In mitochondria with normal MMP, PINK1 is imported to IMM by the action of translocases of the outer (TOM) and inner membrane complexes. Furthermore, it is cleaved by the rhomboid protease PARL, and then degraded [78]. When the MMP is lost, the import of PINK1 into the IMM is prevented, and it remains on the OMM. Then, PINK1 is bound to the TOM complex as a dimer with the Tom7 subunit being the most important factor for this [79, 80].
Recently, it was found that heat-shock protein 70 (Hsp70) stabilised PINK1, thus mediating mitophagy [81]. The Hsp system is another quality-control mechanism and protein turnover regulator in the cell besides autophagy, and this discovery suggests that it can augment mitophagy. Moreover, Hsp system can protect RPE cells against oxidative stress and in this way can be linked to AMD pathogenesis [14, 82].
Parkin is an important element of the ubiquitination-mediated mitophagy pathway [77, 83]. In basal conditions, Parkin is in an auto-inhibited conformation, and its ubiquitin ligase activity remains dormant. The activation of Parkin is regulated by Bcl-2 and occurs when it is translocated to mitochondria. After this translocation, PINK1, located at OMM of damaged mitochondrion, phosphorylates Ser-65 of Parkin [84, 85]. Subsequently, Parkin acts to ubiquitinate mitochondrial surface proteins, including Mitofusin 1/2 (Mfn1/2) and voltage-dependent anion channel 1 (VDAC1), probably to prevent refusion and mitochondrial clustering. This ubiquitination results in the recruitment of adaptor proteins, including histone deacetylase 6 (HDAC6) and sequestosome-like protein receptors (p62/SQSTM1, NBR1, NDP52, TOLLIP, TAX1BP1, and optineurin) that link ubiquitinated substrates to the autophagic complex. MAPK1LC3 and autophagosomal membranes are then co-recruited when damaged mitochondria are aggregated and transported. This results in the engulfment of mitochondria by the autophagosome membrane, forming a “mitophagosome”, its maturation and transport, and final fusion with lysosomes in an HDAC6-dependent manner [26, 86]. Furthermore, SQSTM/p62 expression is blocked when PINK1 is knocked down, indicating the SQSTM1/p62 expression as a consequence of mitophagy induction [87]. PINK1 can drive low-level mitophagy as well. It recruits autophagy receptors NDP52 and optineurin independently of Parkin, suggesting the participation of at least one other Ub E3 ligase in mitophagy [88].
Activating molecule in beclin-1 regulated autophagy is involved in the PINK/Parkin2 mitophagy pathway, besides its participation in non-selective autophagy (“Impaired mitochondria stimulate non-selective autophagy”). AMBRA1 binds Parkin and then MAPK1LC3 with its MAPK1LC3 interacting region (LIR) domain [52]. Likewise, beclin 1 interacts with Parkin and regulates its localisation to the mitochondria [89]. Moreover, AMBRA1 has been shown to act in non-selective autophagy of mitochondria, independently of the PINK/Parkin2-regulated pathway, which has been discussed above [52]. As AMBRA1 is involved in both non-selective autophagy and PINK/Parkin2 regulated mitophagy, this highlights its role as an important regulator of the mitochondrial turnover.
SMAD-specific E3 ubiquitin protein ligase 1 (Smurf1), another E3 ubiquitin ligase, may interact in PINK1/Parkin-mediated mitophagy [90]. Mice deficient in the Smurf1 gene accumulate damaged mitochondria in various organs. In addition, Smurf1 interacts with SQSTM1/p62, suggesting its involvement in mitophagy/autophagy.
Recently, Lemasters [56] has proposed that autophagic degradation of the whole mitochondria occurs in fact according to two types. Type 1, which resembles the general autophagy, would represent the non-selective form of mitophagy. Mitochondrial damage, produced by photoirradiation, promotes the type 2, in which the autophagic phagophore formation does not occur and it is not associated with mitochondrial fission. In this type, the depolarised mitochondria would merge directly with MAPK1LC3-containing structures, after the associations of mitochondria with PINK1 and Parkin. Furthermore, inhibition of the phosphatidylinositol-3-kinase (PI3K) (class III) pathway, the established upstream signaling pathway for the nutrient deficiency-stimulated autophagy, does not block the type 2 mitophagy, implying that it is independent of beclin-1. Further research is still needed for the elucidation of these processes.
Receptor-mediated mitophagy
Receptor-mediated mitophagy is independent of ubiquitination. The Nix protein (known also as Bnip3L) is an integral OMM protein receptor, which regulates the selective removal of damaged mitochondria without protein ubiquitination in mammals [91, 92]. Nix gene-deficient mice develop anemia due to weakened receptor-mediated mitophagy [93, 94]. Nix has a ZXXY structure [Z aromatic (Trp/Phe/Tyr), Y large hydrophobic (Leu/Ile/Val), and X any amino acid residue] that facilitates its interaction with Atg8 family proteins MAPK1LC3 and GABARAP via an LIR domain [91, 95]. Therefore, Nix is a typical autophagy receptor, which brings mitochondria to autophagosomes via direct interaction with the Atg8 proteins [91].
Recent findings indicate that the phosphorylation of Ser-34 and 35 residues in the LIR domain of Nix increases its ability for interaction with MAPK1LC3. In addition, this contact is stronger, compared to that observed with the non-phosphorylated protein. This is due to additional hydrogen bonds between phosphorylated serines and nitrogen-containing basic amino acids in MAPK1LC3 [96].
The ubiquitination of mitochondrial proteins by the E3 ligase Parkin is assisted by Nix [97, 98]. However, the localisation of Nix in the mitochondrial outer membrane predisposes it to deliver mitochondria to autophagosomes without any ubiquitination, which may be useful for distinguishing between ubiquitination- and receptor-mediated mitophagy subtypes.
The Bnip3 protein, a Nix homologue, is another candidate for being a mitophagy receptor [99, 100]. This protein may initiate mitophagy by disturbing the interaction between beclin 1 and Bcl-2 proteins [101]. Bnip3 has an LIR domain to facilitate its binding to the MAPK1LC3/GABARAP proteins. Therefore, Nix and Bnip3 may act together to initiate mitophagy through the same mechanism underlined by MAPK1LC3/GABARAP binding. They may have overlapping functions as mitophagy receptors in normal conditions when damaged mitochondria should be removed to prevent cell death [100].
Another mitophagy receptor is FUNDC1, an integral mitochondrial outer membrane protein [102, 103]. Its role as a receptor has been described in hypoxia-induced mitophagy. In basal conditions, FUNDC1 is phosphorylated and blocked by casein kinase 2 and kinase Scr. In hypoxia as well as mitochondrial depolarisation conditions, FUNDC1 dephosphorylation by a mitophagic Ser/Thr phosphatase PGAM5 is triggered and mitophagy thus activated [104]. FUNDC1 interacts with MAPK1LC3 through its LIR motif. Another novel mitophagy receptor, FKBP8 (also known as FKBP38) belongs to FK506-binding protein (FKBP) family and is similarly anchored to OMM like Nix, and has an LIR domain as well. It recruits lipidated MAPK1LC3 to the damaged mitochondria and increases Parkin-independent mitophagy. Surprisingly, FKBP8 escapes from mitochondria before degradation, even though it acts as the receptor for the degradation process [105].
Some recently found PINK1/Parkin-independent Ub-ligases act on mitophagy signaling pathways as well. Glycoprotein 78 (Gp78), an E3 ubiquitin ligase, is involved in Parkin-independent mitophagy [106]. Gp78 regulates mitochondrial dynamics (fusion/fission) and mitophagy by regulating Mfn1. Mitochondrial E3 ubiquitin ligase (Mul1), a multifunctional ubiquitinase and sumoylase, ubiquitinates Mfn1 and thus regulates mitochondrial dynamics [107]. Mul1 interacts with Unc-like kinase 1 (ULK1) and the E2 conjugating enzyme Ube2E3, which both act in autophagy/mitophagy. Mul1 has an LIR domain in its RING finger domain and its binding with GABARAP requires the presence of Ube2E3, suggesting that an important role that Mul1 performs in the mediation of mitophagy [108].
Mitochondrial quality control and ageing
Mitochondrial DNA damage
The relation between the impairment of mitochondrial function and ageing and age-related diseases has been established in a wide range of organisms (e.g., [109–111]. The common consensus for ageing asserts that the process is started initially by molecular damage, which in turn leads to cellular and tissue degradation, and finally to the loss of an organ’s or tissue’s functionality [112]. The free radical theory proposes that ROS are as the main source of agents, which damage DNA, proteins, and lipids [113]. Damage to mtDNA is considered to be a major type of injury, in regard to degenerative diseases.
Mutations in this organelle’s DNA lead to compromised mitochondrial redox functions. They induce a decline in the activity of enzymes in the citric acid cycle as well as a decrease in the capacity of the mitochondrial respiratory chain (MRC). This diminishes cellular energy production, but increases ROS formation by impaired MRC, which may damage further the mtDNA. This can lead to the accumulation of lesions resulting in mitochondrial dysfunctions, which have been linked to several human diseases, including neurodegenerative disorders and cancer [114, 115]. The decrease of mitochondrial dynamics due to mtDNA mutations is well documented. Mitochondria can thus influence or regulate many important key features of ageing [20, 112]. Mitophagy might be an important means to regulate as well as manage the removal of the mitochondria with mutated mtDNA, and these might have implications for AMD pathology [17].
As the extent of mtDNA damage increases with ageing, this could be causative or just associative with senescence [116]. There might be genetic variation between individuals in greater susceptibility to mtDNA damage, associated with AMD [117]. mtDNA damage in RPE cells can have impact on mitochondria function and connect these lesions to the development of AMD [17–19]. Eight times more mtDNA damage, compared to nuclear DNA, has been reported in RPEs of AMD donors. This has been alleged to be much higher than in normal ageing when mtDNA lesions are usually maintained at low levels and located in the common deletion region of mtDNA. However, mtDNA damages in AMD donors have been observed to the mitochondria genome-wide [18]. Furthermore, less efficient DNA damage response, compared to that in the nucleus, suggests that mitochondria are the weak link in the RPE functionality faced with relentless pressure that ROS generation bares on that tissue [20].
A possible mechanism of AMD is associated with the dynamics of Alu transposons that represent commonly dispersed short-interspersed repeat elements in the human genome [118]. Normally, the level of Alu transcription is low, but it increases in stress conditions [119, 120]. The amount of Alu transcript is regulated by RNase III DICER1, which is crucial for miRNA processing. Dysfunctional or deficient of DICER1 disturbs the functioning of the miRNA network and results in the accumulation of toxic Alu transcripts [121]. To search for the mechanism underlying toxicity of non-degraded Alu retrotransposons, Tarallo and associated showed that deficient DICER1 or excess of Alu RNA activated the NLRP3 inflammasome and TLR-independent MyD88 (myeloid differentiation primary response 88) signaling [122]. No physical interactions between Alu RNA and NLRP3 were observed, while the Alu retrotransposone-induced mitochondria ROS production prior to the priming of inflammasome. Recently, Kerur and others showed a connection with mitochondrial damage, as an accumulation of Alu RNA-induced cytosolic escape of mtDNA with the involvement of cyclic GMP–AMP synthase (cGAS) [123]. It was shown that Alu RNA-induced opening of the mitochondrial permeability transition pole in RPE cells resulting in mitochondria swelling, rupture, and release of mtDNA into the cytosol. It seems that mtDNA is an important factor for the toxic action of Alu RNA in RPE cells and in the pathophysiology of AMD.
Mitochondria, mitophagy, and the cell senescence
It has been assumed that the mechanisms that drive to senescence, a state of irreversible cell cycle arrest, might as well promote age-related diseases [124, 125]. Cell senescence, induced by stress associated with ageing, is linked with a deficit in health and fitness. The phenotype of senescent cells includes the inability to replicate, adoption of immunogenic activity consisting of the pro-inflammatory, senescent-associated secretory phenotype (SASP), which includes increased expression of various cytokines, chemokines, growth factors as well as persistent DNA damage signaling [125–127].
It has been established that ageing and damaging of mitochondria are interconnected. Mitochondrial dysfunction is a significant cause of ageing, and senescent cells contribute to the senescence-associated mitochondrial dysfunction (SAMD) [125, 128, 129]. Damaged mitochondria produce ROS, which increase the likelihood of mtDNA damage (“Mitochondrial DNA damage”) and this leads to senescent growth arrest [130]. In addition, dysfunctional mitochondria release potentially detrimental molecular patterns, consisting of ROS and mtDNA fragments, which can trigger inflammasome activity or SASP [41, 127]. Thus, the failure of mitophagy can lead a reduced capacity to “clean” damaged mitochondria, resulting in inflammation. This can be related to AMD pathogenesis [131]. It should be noted that SASP includes the increased expression of VEGF, a factor that is directly related to the wet form of AMD (“Introduction: AMD and autophagy”) [132].
Autophagy is a cellular mechanism that provides nutrients in starvation and quality control, as it removes non- and dysfunctional cellular components. In regard of the senescence, autophagy has been found to have contradictory roles. In addition, autophagy can act as a pro-ageing mechanism, as it can promote oncogene-induced senescence. Overexpression of unc-like kinase 3 (ULK3), an autophagy gene, promotes autophagy and induces the Ras oncogene. Knockdown of autophagy genes Atg5 and Atg7 can bypass senescence [133]. On the other hand, autophagy is known as an anti-senescence process [134]. Mitochondrial damage increases the expression of autophagy genes MAPK1LC3, Atg5, and Atg8 as well as enhancing mitophagy, and thus postpones senescence. Inhibition of Mammalian target of rapamycin complex 1 (mTORC1), a suppressor of autophagy, leads to the restraint of aged phenotypes. This “duality” is explained by the complex nature of the autophagic/mitophagic processes. Moreover, the timing of the autophagic inhibition is essential. In young cells, autophagy inhibition can lead to senescence, whereas in old cells, autophagy upregulation can be important for the establishment of the aged phenotype [125].
Mitophagy seems to be independent of the general autophagy, in the regard of senescence. Mitophagic activity is found to be reduced in aged cells [135, 136]. mTORC1 activity is elevated in aged cells and lysosomal overload can disrupt the terminal events of autophagy/mitophagy. This leads to the accumulation of lipofuscin within the lysosomes, a hallmark of AMD (“Introduction: AMD and autophagy”). Mitophagy can be inhibited by p53, which interacts with Parkin and prevents its localisation on damaged mitochondria [137]. The suppression of PINK1 in aged cells leads to a lowered mitophagy [138]. In addition, PINK1 deficiency suppresses mitochondrial fission, which is important for the elimination of dysfunctional parts of the mitochondrial “network”, thus further promoting the senescent phenotype [135].
The PGC-1α-Nrf2 axis
Transcription factor Nrf2 [nuclear factor (erythroid-derived 2)-related factor 2, known also as NFE2L2] has a key role in the maintaining of cellular homeostasis in stress. It is a powerful activator of genes that contain electrophile-response elements in their promoter sequences and many of them code for antioxidant proteins. In these genes, electrophile-response elements are also called antioxidant response elements (AREs) [139]. Moreover, Nrf2 regulates mitochondrial biogenesis and ROS metabolism as well as the expression of many nuclear-encoded genes, like mitochondrial transcription factor A (Tfam), which in turn regulates mtDNA transcription, translation, and repair. Keap1 (Kelch-like ECH-associated protein 1) regulates Nrf2 by its targeting for ubiquitination. However, in conditions where there is increased oxidative stress, Keap1 is modified, Nrf2 is released and thus becomes activated. Furthermore, Keap-1-Nrf2 and autophagy are cross-regulated. For example, SQSTM1/p62 is a target for Nrf2. SQSTM1/p62 can further sequester Keap1 and this positive feedback coordinates Nrf2 and autophagy activities [140]. In RPE cells, a knock-out of Nrf2 gene resulted in the downregulation of SQSTM/p62 expression, but in an Nrf2-gene knock-out animal model, its expression was not changed in these cells. Therefore, the pathways of Nrf2 in the regulation of autophagy/mitophagy might be more complicated and remains to be characterised closer [141]. An increase in Nrf2 gene activity and the subsequent upregulating of ROS-induced damage-preventing proteins has been found in the RPE of AMD donor eyes. This suggests that it is one compensatory mechanism due to increased ROS [8].
Nuclear factor (erythroid-derived 2)-related factor 2 is, moreover, a downstream transcription factor for PGC-1α (peroxisome proliferator-activated receptor gamma coactivator-1 alpha) gene. The Nrf2 and PGC-1α proteins interact with each other as well (the latter as co-factor) activating mitochondria-related genes, mentioned earlier in this section [142, 143].
It has been shown that in the RPE maturation, the expression of the PGC-1α gene increases in mitochondria. In addition, it is a strong activator of mitochondrial function and antioxidant capacity in RPE cells [144]. Furthermore, PGC-1α is positively regulated by energy sensor NAD-dependent histone deacetylase Sirtuin 1 (Sirt1) and AMP-activated protein kinase (AMPK) [145, 146]. These both link it to autophagy. Recently, PGC-1α gene, along with Sirt1 protein, has been found to be repressed in RPE cells from AMD patients in which disintegrated mitochondria were found [10].
Gene knock-out animal models of PGC-1α and Nrf2 have been employed in the study of the RPE [141, 147]. When either of these genes is disrupted, signs of degeneration and the loss of function are evident. In RPE cells, Nrf2 gene knock-out degenerative signs (drusen, lipofuscin, and expression of inflammatory proteins) and development of autophagy-related vacuoles has been found. PGC-1α absence leads to dysregulation of all major pathways engaged in the retinal damage and apoptosis, restoring, and rejuvenation. Recently, a double-knock-out animal model of PGC-1α and Nrf2 genes has been established. Unpublished preliminary results from our laboratory show increased autolysosomes, increased expression of autophagy markers, damaged mitochondria, RPE degeneration, and finally age-related visual loss in double-knock-out mice. These findings link mitochondrial damage to PGC-1α and Nrf2 gene deficiencies, and these may have an impact in autophagic and mitophagic clearance in the RPE, and thus be involved in AMD pathogenesis. This justifies the suitability of this double-knock-out animal model for AMD studies.
Therapeutic potential of mitophagy in AMD
Lemasters [55], who introduced the term mitophagy, considered it as targeted defense against oxidative stress, malfunctioning mitochondria and ageing. With regard to RPE ageing and AMD, the mitochondrial damage has been proposed as a possible source of oxidative stress, due to high oxygen consumption and intensive light exposure in the retina. This increased stress causes mtDNA damage, oxidation of other molecules, and the decrease of cellular antioxidant capacity [148]. Mitophagy could be considered as an important protective mechanism of RPE cells against oxidative stress, which prevents retinal degeneration.
Weakening of mitochondrial activity and decrease in mitochondrial quality have been linked with ageing and the progress of age-related diseases, such as cardiovascular and neurodegenerative disorders, and cancers in the elderly [68, 149]. The RPE of AMD eyes manifest the decrease of mitochondria number, defective fission/fusion processes, disorganised cristae, and lower levels of MRC proteins. In addition, decreased levels of heat-shock proteins involved in the import of nuclear-coded proteins, suggests that severe defects in mitochondrial biogenesis due to shortage of cytosolic proteins, is an important factor [19, 21]. Weakened antioxidant protection combined with the shortage of mitochondrial capacity may strongly reduce RPE function and contribute to the onset of AMD. While senescent RPE cells might have sufficient energy at the resting state, they cannot efficiently display stress response, which demands supplementary ATP and thus reduces preventative capacity [150].
Advanced age results in mitochondrial decay, bioenergetics deficit (reduced MMP), shortage of antioxidant defense, and increased sensitivity to oxidative stress in RPE cells [151]. In addition, it has been found that while the number of mitochondria decreases and their length increases with age in the RPE from both aged human donors and rhesus monkeys [151, 152]. The elongation of mitochondria is associated with metabolic/oxidative stress. Furthermore, elongated mitochondria form parallel clusters and unexpectedly, these are oriented orthogonally (perpendicularly) to the basal membrane of the cell. The increased flavoprotein fluorescence (FPF), correlated with elevated mitochondrial dysfunction, has been observed in nonexudative (dry) AMD. The heterogeneity of FPF indicates that there is increased variability in the severity of the damages in the eyes observed in advanced AMD [153]. These findings further suggest that the age-related mitochondrial dysfunctions might be involved in the pathogenesis of AMD.
Some genetic variants of the MRC complex I are as well associated with an increased risk of AMD [154]. Inhibition of complex I function leads to ROS increase and subsequently reduces cell viability, by causing mitotic catastrophe. Mitophagy is later activated, since PINK1 and Parkin are localised to depolarised mitochondria [155]. In RPE cells, mitophagy can prevent the cell death, but when it is further blocked, it causes non-apoptotic cell death [156].
Terluk and associates [19] have found that mtDNA damages occur already in the early phases of AMD and they are limited to RPE, in both the macular and peripheral regions. These can be detected before the occurrence of the disease symptoms. Early intervention to prevent DNA damages could be useful in the attenuation of this disease’s progression. This could be done via modulation of mitophagic activity when mitochondria with damaged DNA and disturbed expression of important proteins involved in MRC could be removed.
Excess iron has been reported too in AMD retinas. This contributes to the excess ROS formation catalyzed by Fe3+ ions [157]. The accumulation of iron is accompanied by ROS production, as well as increased expression of the Divalent metal transporter 1 (DMT 1) and the decrease of Ferroportin 1 (FPN 1) gene expressions. These changes have been detected in dysfunctional mitochondria and linked to several neurodegenerative diseases. As this happens in the secluded space of mitochondria, causing the decrease of MRC complex I activity [158], it is likely to be important in the development of AMD via dysfunctional mitochondria.
Mitochondrial ferritin (FtMt), an iron-storage protein, controls antioxidant capacity via regulation of Fe storage. It suppresses the Fenton reaction, in which Fe2+ reacts with H2O2 resulting in the production of hydroxyl radicals [159]. The expression of FtMt increases in ageing and in neurodegenerative maladies [160]. A mutation in the FtMt gene has been associated with AMD occurrence [161]. Furthermore, the increase of FtMt triggers mitophagy via hypoxia-inducible factor-1α (HIF-1α) regulated pathway in RPE cells. However, FtMt may cause the induction of in wet AMD via an increase in VEGF secretion [162]. FtMt may be important in the protection of RPE cells by inducing mitophagy, reducing age-related stress, and exerting an antioxidant effect in mitochondria.
The alteration of protein phosphorylation induced by oxidative stress has been recently studied in RPE cells in interactome network scale [163]. Phosphorylation is a survival mechanism for proteins in chronic oxidative stress. However, this has a positive correlation with AMD progression. Moreover, general mitochondrial protein phosphorylation leads to alterations in mitochondrial activity, which could lead to increased mitophagic clearance of non-functional organs.
As no effective treatment for AMD exists so far, except for the moderate success of anti-VEGF treatment for the wet AMD [6], new prospects promising any alleviation from this disease or its progression would be worth examining. The enhancement of mitophagy, for more efficient removal of malfunctional mitochondria, lies within this area. This is likewise in connection with the augmenting of the energy metabolism of the mitochondria in aged cells, where the function of RPE cells decreases. These could be of potential use in the future AMD therapy. In the AMPK–mTOR energy-sensor route [164], AMPK, which increases autophagy/mitophagy, can be induced by metformin, a diabetes type 2 drug in use, and AICAR (5-aminoimidazole-4-carboxamide-1-β-ribofuranoside). The autophagy inhibitor mTOR is further suppressed by AMPK and its activity can be reduced by rapamycin [68, 165]. In addition, metformin induces MRC complex I function [166], blocks pro-inflammatory NF-kappa B signaling [167], and inhibits the secretory-associated senescence phenotype (“Mitochondria, mitophagy, and the cell senescence”) [168]. It can be thus used as a senostatic drug, which inhibits the senescent phenotype. Moreover, metformin induces mitophagy by reducing the abundance of cytosolic tumour suppressor factor p53, which has been found to inhibit mitophagy via interaction with Parkin [169]. This is an interesting example of “two-way feat” of p53 for autophagic action, as it can directly activate mitophagy as well [170].
The PGC-1α-Nrf2 axis (“The PGC-1α-Nrf2 axis”) could be targeted in mitochondria-related AMD therapy. Mitochondrial biogenesis can be enhanced by inducing PGC-1α via Sirt1, which can induce autophagy/mitophagy. Resveratrol, a polyphenolic natural compound, stimulates Sirt1 indirectly by elevating NAD+ levels and by activating AMPK [68, 171, 172]. Several other polyphenolic compounds (e.g., quercetin, berberine, catechin, ferulic acid, and tyrosol) are Sirt1 agonists as well, they sustain the stimulating action of resveratrol, and as such have a potential in therapy of many degenerative diseases [173]. Keap1-Nrf2 interaction inhibitors could be useful as inducers of mitophagy for the treatment of pathological conditions characterised by impaired mitochondrial quality control [174]. For example, electrophilic inducers carbonyl cyanide p-trifluoromethoxyphenylhydrazone, dimethylfumarate, curcumin, and tert-butylhydroquinone attach to Keap1 and thus prevent the degradation of Nrf2, leading to activation of mitochondrial biogenesis and autophagy/mitophagy.
In addition, the augmentation of Hsp70 might be useful in the mitophagy stimulation, since it regulates the stability of PINK1 [81] (“Ubiquitin-mediated mitophagy: PINK1 and Parkin”). Delivery of exogenous (recombinant) Hsp70 might be a strategy against RPE degeneration [82], not only as the augmentation of the chaperone activity itself and the protection against oxidative stress, but in the maintenance of mitophagy.
Spermidine, a ubiquitous polyamine compound has been found to be beneficial for slowing down ageing. This might be due to its capacity to upregulate autophagy. Spermidine increases autophagic flux and promotes mitophagy. It acts as acetylase inhibitor, though independently of Sirt1 [175]. Urolithin A, a metabolite of natural bioactive polyphenolic ellagitannins, has been found to induce mitophagy in mammals [176]. Remarkable, it reduces the number of mitochondria, but maintains the respiratory capacity in short-term dispensing. The long-term exposure promotes the biogenesis of mitochondria and their number. Other effective compounds include antibiotics and plant-derived metabolites [177]. For example, naturally occurring antibiotic actinonin induces mitophagy, possibly via depletion of mitophagic ribosomes and RNA decay [178, 179]. Recently, activation of mitophagy via Parkin and PINK1 modulation has been studied by a screen of chemical compounds, and by the use of artificial ligands, also called neo-substrates, such as ATP analogue kinetin triphosphate [180, 181]. The natural-based and synthetic compounds could be of a promising use in the treatment of mitophagy-dependent disorders, and generally in the anti-ageing therapies.
Senolytic therapy targets senescent cells to be destroyed or inhibit their secretory phenotype to eliminate their influence on other cells. In a recent, unpublished chemical screening, many compounds have been found, which can modify autophagy and/or mitophagy [125]. Many of them could act as senolytics via modification of the interactions between mitophagy, SAMD, and SASP (“Mitochondria, mitophagy, and the cell senescence”), for example, by the release of apoptosis-stimulating factors monitoring lowering of MMP [124, 125]. Therefore, targeting senescent cells by mitophagy induction is a promising perspective strategy to fight AMD, especially since senescence and not apoptosis can be responsible for death of retinal cells in AMD [182, 183].
Conclusions
Non-selective autophagy, mitophagy, and other selective types of autophagy act as a central node in a network controlling cellular development, lifespan, and death. Notably, non-selective autophagy is crucial as a pro-survival mechanism and it is upregulated in critical situations as well as ageing when other quality-control pathways (such as proteasomes) could fail to function. On the other hand, mitophagy is crucial for the mitochondrial quality control and it is more tightly controlled than non-selective autophagy. It is, however, induced in specific stress situations to prevent the accumulation of damaged mitochondria, although there seems so far to be no direct evidence of mitophagy activation solely by ageing [184].
Disturbances in autophagic clearance have been alleged to be associated with many degenerative diseases including AMD. Mitochondria may stimulate general autophagy, and ROS-dependent destructive mechanisms seem to underline this effect. In addition, mitochondria are targeted by mitophagy, the mitochondria-selective type of autophagy especially when they are affected by damaging agents and become dysfunctional. The efficacy of mitophagy may be regulated both positively and negatively by the dynamics of mitochondria. Mitophagy may contribute to the maintenance of mitochondria and this fact may be relevant to control important biological processes. Therefore, the interplay between mitophagy and mitochondria may belong to fundamental mechanisms of energy and metabolism homeostasis in eukaryotic cells, and in the development of age-related maladies, which could be related to the reduced of mitochondrial activity.
Despite the important role of mitophagy, several questions concerning fundamental aspects of its regulation are still open. First of all, what is the difference between mitophagy pathways activated by different factors? Second, what is the range in which Parkin may activate mitophagy by ubiquitinating/degrading mitochondrial surface proteins? Finally, what are the precise mechanisms of the mitophagy regulation by the interactions between PINK1, Parkin, and their substrates on the mitochondrial surface? These and other questions should be addressed in the future research on mitophagy.
In summary, the recuperation of mitochondrial quality and activity might be useful in anti-ageing therapies. Deeper understanding of the role of cellular autophagic clearance mechanisms, including mitophagy against ROS, is essential for searching for effective therapeutic tools against age-related degenerative diseases, such as AMD.
Acknowledgements
The authors thank Mr. Thomas Dunlop for checking the language of the manuscript.
Abbreviations
- AMBRA1
Activating molecule in beclin-1-regulated autophagy
- AMD
Age-related macular degeneration
- AMPK
AMP-activated protein kinase
- ARE
Antioxidant-binding element
- Atg
Autophagy-related gene
- DMT1
Divalent metal transporter 1
- ER
Endoplasmic reticulum
- FPN1
Ferroportin 1
- FPF
Flavoprotein fluorescence
- FtMt
Mitochondrial ferritin
- Gp78
Glycoprotein 78
- HDAC6
Histone deacetylase 6
- HIF-1α
Hypoxia-inducible factor-1α
- Hsp
Heat-shock protein
- IMM
Inner mitochondrial membrane
- Keap1
Kelch-like ECH-associated protein 1
- LIR
MAPK1LC3-interacting region
- MAM
Mitochondria-associated endoplasmic reticulum membrane
- MAPK
Mitogen-activated protein kinase
- MAPK1LC3
Microtubule-associated protein 1 light-chain 3
- Mfn
Mitofusin
- MMP
Mitochondrial membrane potential
- MPT
Mitochondrial permeability transition
- MRC
Mitochondrial respiratory chain
- mtDNA
Mitochondrial DNA
- mTORC1
Mammalian target of rapamycin complex 1
- Mul1
Mitochondrial E3 ubiquitin ligase
- Nrf2
Nuclear factor (erythroid-derived 2)-related factor 2
- OMM
Outer mitochondrial membrane
- PGC-1α
Peroxisome proliferator-activated receptor gamma coactivator-1α
- PI3K
Phosphatidylinosotole-3-kinase
- PINK1
Phosphatase and tensin homologue-induced putative kinase
- POS
Photoreceptor outer segment
- ROS
Reactive oxygen species
- RGC
Retinal ganglion cell
- RPE
Retinal pigment epithelium
- SAMD
Senescence-associated mitochondrial dysfunction
- SASP
Senescent-associated secretory phenotype
- Sirt1
NAD-dependent deacetylase Sirtuin 1
- Smurf1
SMAD-specific E3 ubiquitin protein ligase 1
- SOD
Superoxide dismutase
- SQSTM1/p62
Sequestosome1/p62
- Tfam
Mitochondrial transcription factor A
- TIM
Translocase of the inner mitochondrial membrane
- TOM
Translocase of the outer mitochondrial membrane
- Ub
Ubiquitin
- VDAC
Voltage-dependent anion channel
- VEGF
Vascular endothelial growth factor
References
- 1.Wong WL, Su X, Li X, Cheung CMG, Klein R, Cheng C-Y, Wong TY. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2(2):e106–e116. doi: 10.1016/S2214-109X(13)70145-1. [DOI] [PubMed] [Google Scholar]
- 2.Black JRM, Clark SJ. Age-related macular degeneration: genome-wide association studies to translation. Genet Med. 2015;18(4):283–289. doi: 10.1038/gim.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaarniranta K, Sinha D, Blasiak J, Kauppinen A, Veréb Z, Salminen A, Boulton ME, Petrovski G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy. 2013;9(7):973–984. doi: 10.4161/auto.24546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhutto I, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol Aspects Med. 2012;33(4):295–317. doi: 10.1016/j.mam.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinnunen K, Petrovski G, Moe MC, Berta A, Kaarniranta K. Molecular mechanisms of retinal pigment epithelium damage and development of age-related macular degeneration. Acta Ophthalmol. 2012;90(4):299–309. doi: 10.1111/j.1755-3768.2011.02179.x. [DOI] [PubMed] [Google Scholar]
- 6.Klettner A, Roider J. Treating age-related macular degeneration—interaction of VEGF-antagonists with their target. Mini Rev Med Chem. 2009;9(9):1127–1135. doi: 10.2174/138955709788922665. [DOI] [PubMed] [Google Scholar]
- 7.Blasiak J, Szaflik JP. DNA damage and repair in age-related macular degeneration. Front Biosci (Landmark Ed) 2011;16(4):1291–1301. doi: 10.2741/3789. [DOI] [PubMed] [Google Scholar]
- 8.Handa JT. How does the macula protect itself from oxidative stress? Mol Aspects Med. 2012;33(4):418–435. doi: 10.1016/j.mam.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klettner A. Oxidative stress induced cellular signaling in RPE cells. Front Biosci (Schol Ed) 2012;4(2):392–411. doi: 10.2741/S275. [DOI] [PubMed] [Google Scholar]
- 10.Golestaneh N, Chu Y, Cheng SK, Cao H, Poliakov E, Berinstein DM. Repressed SIRT1/PGC-1α pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J Transl Med. 2016;14(1):344. doi: 10.1186/s12967-016-1101-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Golestaneh N, Chu Y, Xiao YY, Stoleru GL, Theos AC. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017;8(1):e2537. doi: 10.1038/cddis.2016.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCord JM, Edeas MA. SOD, oxidative stress and human pathologies: a brief history and a future vision. Biomed Pharmacother. 2005;59(4):139–142. doi: 10.1016/j.biopha.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Jarrett SG, Boulton ME. Consequences of oxidative stress in age-related macular degeneration. Mol Aspects Med. 2012;33(4):399–417. doi: 10.1016/j.mam.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaarniranta K, Salminen A, Eskelinen EL, Kopitz J. Heat shock proteins as gatekeepers of proteolytic pathways-Implications for age-related macular degeneration (AMD) Ageing Res Rev. 2009;8(2):128–139. doi: 10.1016/j.arr.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Mettu PS, Wielgus AR, Ong SS, Cousins SW. Retinal pigment epithelium response to oxidant injury in the pathogenesis of early age-related macular degeneration. Mol Aspects Med. 2012;33(4):376–398. doi: 10.1016/j.mam.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 16.Tuo J, Grob S, Zhang K, Chan CC. Genetics of immunological and inflammatory components in age-related macular degeneration. Ocul Immunol Inflamm. 2012;20(1):27–36. doi: 10.3109/09273948.2011.628432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hyttinen JMT, Błasiak J, Niittykoski M, Kinnunen K, Kauppinen A, Salminen A, Kaarniranta K. DNA damage response and autophagy in the degeneration of retinal pigment epithelial cells—implications for age-related macular degeneration (AMD) Ageing Res Rev. 2017;36:64–77. doi: 10.1016/j.arr.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Karunadharma PP, Nordgaard CL, Olsen TW, Ferrington DA. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Investig Ophthalmol Vis Sci. 2010;51(11):5470–5479. doi: 10.1167/iovs.10-5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terluk MR, Kapphahn RJ, Soukup LM, Gong H, Gallardo C, Montezuma SR, Ferrington DA. Investigating mitochondria as a target for treating age-related macular degeneration. J Neurosci. 2015;35(18):7304–7311. doi: 10.1523/jneurosci.0190-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barot M, Gokulgandhi MR, Mitra AK. Mitochondrial dysfunction in retinal diseases. Curr Eye Res. 2011;36(12):1069–1077. doi: 10.3109/02713683.2011.607536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feher J, Kovacs I, Artico M, Cavallotti C, Papale A, Balacco Gabrieli C. Mitochondrial alterations of retinal pigment epithelium in age-related maculrt degeneration. Neurobiol Aging. 2006;27(7):596–607. doi: 10.1016/j.neurobiolaging.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 22.Jarrett SG, Lin H, Godley BF, Boulton ME. Mitochondrial DNA damage and its potential in the retina degeneration. Prog Retin Eye Res. 2008;27(6):596–607. doi: 10.1016/j.preteyeres.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 23.Shang F, Taylor A. Roles for the ubiquitin-proteasome pathway in protein quality control and signaling in the retina: implications in the pathogenesis of age-related macular degeneration. Mol Aspects Med. 2012;33(4):446–466. doi: 10.1016/j.mam.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reggiori F. Autophagy: new questions from recent answers. ISRN Mol Biol. 2012;2012:738718. doi: 10.5402/2012/738718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12(9):814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hyttinen JMT, Niittykoski M, Salminen A, Kaarniranta K. Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim Biophys Acta. 2013;1833(3):503–510. doi: 10.1016/j.bbamcr.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 27.Harnett MM, Pineda MA, Latré de Laté P, Eason RJ, Besteiro S, Harnett W, Langsley G. From Christian de Duve to Yoshinori Ohsumi: more to autophagy than just dining at home. Biomed J. 2017;40(1):9–22. doi: 10.1016/j.bj.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signalling regulation. Curr Opin Cell Biol. 2010;22(2):124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arjamaa O, Nikinmaa M, Salminen A, Kaarniranta K. Regulatory role of HIF-1alpha in the pathogenesis of age-related macular degeneration (AMD) Ageing Res Rev. 2009;8(4):349–358. doi: 10.1016/j.arr.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 30.Salminen A, Kaarniranta K. Regulation of the aging process by autophagy. Trends Mol Med. 2009;15(5):217–224. doi: 10.1016/j.molmed.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 31.Krohne TU, Stratmann NK, Kopitz J, Holz FG. Effects of lipid peroxidation products on lipofuscinogenesis and autophagy in human retinal pigment epithelial cells. Exp Eye Res. 2010;90(3):465–471. doi: 10.1016/j.exer.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 32.Wang AL, Boulton ME, Dunn WA, Jr, Rao HV, Cai J, Lukas TJ, Neufeld AH. Using LC3 to monitor autophagy flux in the retinal pigment epithelium. Autophagy. 2009;5(8):1190–1193. doi: 10.4161/auto.5.8.10087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JY, Zhao H, Martinez J, Doggett TA, Kolesnikov AV, Tang PH, Ablonczy Z, Chan CC, Zhou Z, Green DR, Ferguson TA. Noncanonical autophagy promotes the visual cycle. Cell. 2013;154(2):365–376. doi: 10.1016/j.cell.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 35.Kaarniranta K. Autophagy-hot topic in AMD. Acta Ophthalmol. 2010;88(4):387–388. doi: 10.1111/j.1755-3768.2009.01840.x. [DOI] [PubMed] [Google Scholar]
- 36.Mitter SK, Rao HV, Qi X, Cai J, Sugrue A, Dunn WA, Jr, Grant MB, Boulton ME. Autophagy in the retina: a potential role in age-related macular degeneration. Adv Exp Med Biol. 2012;723:83–90. doi: 10.1007/978-1-4614-0631-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryhänen T, Hyttinen JMT, Kopitz J, Rilla K, Kuusisto E, Mannermaa E, Viiri J, Holmberg CI, Immonen I, Meri S, Parkkinen J, Eskelinen EL, Uusitalo H, Salminen A, Kaarniranta K. Crosstalk between Hsp70 molecular chaperone, lysosomes and proteasomes in autophagy-mediated proteolysis in human retinal pigment epithelial cells. J Cell Mol Med. 2009;13(9B):3616–3631. doi: 10.1111/j.1582-4934.2008.00577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Viiri J, Amadio M, Marchesi N, Hyttinen JMT, Kivinen N, Sironen R, Rilla K, Akhtar S, Provenzani A, D’Agostino VG, Govoni S, Pascale A, Agostini H, Petrovski G, Salminen A, Kaarniranta K. Autophagy activation clears ELAV1/HuR-mediated accumulation of SQSTM1/p62 during proteasomal inhibition in human retinal pigment epithelial cells. PLoS One. 2013;8(7):e69563. doi: 10.1371/journal.pone.0069563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang AL, Lukas TJ, Yuan M, Du N, Tso MO, Neufeld AH. Autophagy and exosomes in the aged retinal pigment epithelium: possible relevance to drusen formation and age-related macular degeneration. PLoS One. 2009;4(1):e4160. doi: 10.1371/journal.pone.0004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kauppinen A, Niskanen H, Suuronen T, Kinnunen K, Salminen A, Kaarniranta K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells-Implications for age-related macular degeneration (AMD) Immunol Lett. 2012;147(1–2):29–33. doi: 10.1016/j.imlet.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 41.Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging. 2012;4(3):166–175. doi: 10.18632/aging.100444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boya P, Esteban-Martínez L, Serrano-Puebla A, Gómez-Sintes R, Vilarejo-Zori B. Autophagy in the eye: development, degeneration, and aging. Prog Retin Eye Res. 2016;55:206–245. doi: 10.1016/j.preteyeres.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 43.Hyttinen JMT, Amadio M, Viiri J, Pascale A, Salminen A, Kaarniranta K. Clearance of misfolded and aggregated proteins by aggrephagy and implication for aggregation diseases. Ageing Res Rev. 2014;18:16–28. doi: 10.1016/j.arr.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 44.Novak I. Mitophagy: a complex mechanism of mitochondrial removal. Antiox Redox Signal. 2012;17(5):794–802. doi: 10.1089/ars.2011.4407. [DOI] [PubMed] [Google Scholar]
- 45.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141(4):656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okamoto K, Kondo-Okamoto N. Mitochondria and autophagy: critical interplay between the two homeostats. Biochim Biophys Acta. 2012;1820(5):595–600. doi: 10.1016/j.bbagen.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 50.Funderburk SF, Wang QJ, Yue Z. The beclin 1–VPS34 complex—at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20(6):355–362. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cecconi F, Levine B. The role of autophagy in mammalian development: cell makeover rather than cell death. Dev Cell. 2008;15(3):344–357. doi: 10.1016/j.devcel.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strappazzon F, Nazio F, Corrado M, Cianfanelli V, Romagnoli A, Fimia GM, Campello S, Nardacci R, Piacentini M, Campanella M, Cecconi F. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2015;22(3):419–432. doi: 10.1038/cdd.2014.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Strappazzon F, Vietri-Rudan M, Campello S, Nazio F, Florenzano F, Fimia GM, Piacentini M, Levine B, Cecconi F. Mitochondrial BCL-2 inhibits AMBRA1–induced autophagy. EMBO J. 2011;30(7):1195–1208. doi: 10.1038/emboj.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kissová L, Deffieu M, Manon S, Camougrand N. Uth1p is involved in the autophagic degradation of mitochondria. J Biol Chem. 2004;279(37):39068–39074. doi: 10.1074/jbc.M406960200. [DOI] [PubMed] [Google Scholar]
- 55.Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005;8(1):3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- 56.Lemasters JJ. Variants of mitochondrial autophagy: types 1 and 2 mitophagy and micromitophagy (type 3) Redox Biol. 2014;2:749–754. doi: 10.1016/j.redox.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Michel S, Wanet A, De Pauw A, Rommelaere G, Arnould T, Renard P. Crosstalk between mitochondrial (dys)function and mitochondrial abundance. J Cell Physiol. 2012;227(6):2297–2310. doi: 10.1002/jcp.23021. [DOI] [PubMed] [Google Scholar]
- 58.Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Landi F, Bernabei R, Marzetti E. Fueling inflamm-aging through mitochondrial dysfunction: mechanisms and molecular targets. Int J Mol Sci. 2017;18(5):933. doi: 10.3390/ijms18050933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15(12):2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 60.Kim I, Lemasters JJ. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antiox Redox Signal. 2011;14(10):1919–1928. doi: 10.1089/ars.2010.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Esteban-Martínez L, Sierra-Filardi E, McGreal RS, Salazar-Roa M, Mariño G, Seco E, Durand S, Enot D, Graña O, Malumbres M, Cvekl A, Cuervo AM, Kroemer G, Boya P. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J. 2017;36(12):1688–1706. doi: 10.15252/embj.201695916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dengjel J, Abeliovich H. Roles of mitophagy in cellular physiology and development. Cell Tissue Res. 2017;367(1):95–109. doi: 10.1007/s00441-016-2472-0. [DOI] [PubMed] [Google Scholar]
- 63.Szymański J, Janikiewicz J, Michalska B, Patalas-Krawczyk P, Perrone M, Ziółkowski W, Duszyński J, Pinton P, Dobrzyń A, Więckowski MR. Interaction of mitochondria with the endoplasmic reticulum and plasma membrane in calcium homeostasis, lipid trafficking and mitochondrial structure. Int J Mol Sci. 2017;18(7):1576. doi: 10.3390/ijms18071576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495(7441):389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 65.Carreras-Sureda A, Pihán P, Hetz C. The unfolded protein response: at the intersection between endoplasmic reticulum function and mitochondrial bioenergetics. Front Oncol. 2017;7:55. doi: 10.3389/fonc.2017.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Farré JC, Subramani S. Mechanistic insights into selective autophagy pathways: lessons from yeast. Nat Rev Mol Cell Biol. 2016;17(9):537–552. doi: 10.1038/nrm.2016.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mao K, Wang K, Zhao M, Xu T, Klionsky DJ. Two MAPK-signaling pathways are required for mitophagy in Saccharomyces cerevisiae. J Cell Biol. 2011;193(4):755–767. doi: 10.1083/jcb.201102092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Palikaras K, Daskalaki I, Markaki M, Tavernarakis N. Mitophagy and age-related pathologies: development of new therapeutics by targeting mitochondrial turnover. Pharmacol Ther. 2017;178:157–174. doi: 10.1016/j.pharmthera.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 69.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin–mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 70.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189(2):211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1):1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85(2):257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrané J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA. 2010;107(1):378–383. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moore DJ. Parkin: a multifaced ubiquitin ligase. Biochem Soc Trans. 2006;34(Pt 5):749–753. doi: 10.1042/BST0340749. [DOI] [PubMed] [Google Scholar]
- 75.Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci USA. 2008;105(33):12022–12027. doi: 10.1073/pnas.0802814105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Geisler S, Holmström KM, Treis A, Skujat D, Weber SS, Fiesel FC, Kahle PJ, Springer W. The PINK1/Parkin–mediated mitophagy is compromised by PD-associated mutations. Autophagy. 2010;6(7):871–878. doi: 10.4161/auto.6.7.13286. [DOI] [PubMed] [Google Scholar]
- 77.Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, Kitada T, Kim JM, Chung J. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun. 2008;377(3):975–980. doi: 10.1016/j.bbrc.2008.10.104. [DOI] [PubMed] [Google Scholar]
- 78.Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy. 2013;9(11):1758–1769. doi: 10.4161/auto.24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lazarou M. Keeping the immune system in check: a role for mitophagy. Immunol Cell Biol. 2015;93(1):3–10. doi: 10.1038/icb.2014.75. [DOI] [PubMed] [Google Scholar]
- 80.Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012;22(2):320–333. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zheng Q, Huang C, Guo J, Tan J, Wang C, Tang B, Zhang H. Hsp70 participates in PINK1-mediated mitophagy by regulating the stability of PINK1. Neurosci Lett. 2017;662:264–270. doi: 10.1016/j.neulet.2017.10.051. [DOI] [PubMed] [Google Scholar]
- 82.Subrizi A, Toropainen E, Ramsay E, Airaksinen AJ, Kaarniranta K, Urtti A. Oxidative stress protection by exogenous delivery of rhHsp70 chaperone to the retinal pigment epithelium (RPE), a possible therapeutic strategy against RPE degeneration. Pharm Res. 2015;32(1):211–221. doi: 10.1007/s11095-014-1456-6. [DOI] [PubMed] [Google Scholar]
- 83.Rüb C, Wilkening A, Voos W. Mitochondnrial quality control by the Pink1/Parkin system. Cell Tissue Res. 2017;367(1):111–123. doi: 10.1007/s00441-016-2485-8. [DOI] [PubMed] [Google Scholar]
- 84.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol. 2014;205(2):143–150. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kazlauskaite A, Martínez-Torres RJ, Wilkie S, Kumar A, Peltier J, Gonzalez A, Johnson C, Zhang J, Hope AG, Peggie M, Trost M, van Aalten DM, Alessi DR, Prescott AR, Knebel A, Walden H, Muqit MM. Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK1-dependent phosphorylation and activation. EMBO Rep. 2015;16(8):939–954. doi: 10.15252/embr.201540352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Springer W, Kahle PJ. Regulation of PINK1-Parkin-mediated mitophagy. Autophagy. 2011;7(3):266–278. doi: 10.4161/auto.7.3.14348. [DOI] [PubMed] [Google Scholar]
- 87.Ivankovic D, Chau KY, Schapira AH, Gegg ME. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J Neurochem. 2016;136(2):388–402. doi: 10.1111/jnc.13412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Choubey V, Cagalinec M, Liiv J, Safiulina D, Hickey MA, Kuum M, Liiv M, Anwar T, Eskelinen EL, Kaasik A. BECN1 is involved in the initiation of mitophagy: it facilitates PARK2 translocation to mitochondria. Autophagy. 2014;10(6):1105–1119. doi: 10.4161/auto.28615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Orvedahl A, Sumpter R, Jr, Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, Forst CV, Wrana JL, Zhang YE, Luby-Phelps K, Xavier RJ, Xie Y, Levine B. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480(7375):113–117. doi: 10.1038/nature10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dötsch V, Ney PA, Dikic I. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11(1):45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946. doi: 10.1038/cdd.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454(7201):232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA. 2007;104(49):19500–19505. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rozenknop A, Rogov VV, Rogova NY, Löhr F, Güntert P, Dikic I, Dötsch V. Characterization of the interaction of GABARAPL-1 with the LIR motif of NBR1. J Mol Biol. 2011;410(3):477–487. doi: 10.1016/j.jmb.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 96.Rogov VV, Suzuki H, Marinković M, Lang V, Kato R, Kawasaki M, Buljubašić M, Šprung M, Rogova N, Wakatsuki S, Hamacher-Brady A, Dötsch V, Dikic I, Brady NR, Novak I. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep. 2017;7(1):1131. doi: 10.1038/s41598-017-01258-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW, 2nd, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem. 2010;285(36):27879–27890. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183(5):795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Azad MB, Chen Y, Henson ES, Cizeau J, McMillan–Ward E, Israels SJ, Gibson SB. Hypoxia induces autophagic cell death in apoptosis–competent cells through a mechanism involving BNIP3. Autophagy. 2008;4(2):195–204. doi: 10.4161/auto.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Novak I, Dikic I. Autophagy receptors in developmental clearance of mitochondria. Autophagy. 2011;7(3):301–303. doi: 10.4161/auto.7.3.14509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 102.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q. Mitochondrial outer–membrane protein FUNDC1 mediates hypoxia–induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14(2):177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 103.Wu W, Li W, Chen H, Jiang L, Zhu R, Feng D. FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy. 2016;12(9):1675–1676. doi: 10.1080/15548627.2016.1193656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C, Duan L, Wang X, Liu L, Liu X, Shen Y, Zhu Y, Chen Q. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. 2014;54(3):362–377. doi: 10.1016/j.molcel.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 105.Bhujabal Z, Birgisdottir ÅB, Sjøttem E, Brenne HB, Øvervatn A, Habisov S, Kirkin V, Lamark T, Johansen T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017;18(6):947–961. doi: 10.15252/embr.201643147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fu M, St-Pierre P, Shankar J, Wang PT, Joshi B, Nabi IR. Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol Biol Cell. 2013;24(8):1153–1162. doi: 10.1091/mbc.E12-08-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Peng J, Ren KD, Yang J, Luo XJ. Mitochondrial E3 ubiquitin ligase 1: a key enzyme in regulation of mitochondrial dynamics and functions. Mitochondrion. 2016;28:49–53. doi: 10.1016/j.mito.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 108.Ambivero CT, Cilenti L, Main S, Zervos AS. Mulan E3 ubiquitin ligase interacts with multiple E2 conjugating enzymes and participates in mitophagy by recruiting GABARAP. Cell Signal. 2014;26(12):2921–2929. doi: 10.1016/j.cellsig.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 109.Bernhardt D, Hamann A, Osiewacz HD. The role of mitochondria in fungal aging. Curr Opin Microbiol. 2014;22:1–7. doi: 10.1016/j.mib.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 110.Szklarczyk R, Nooteboom M, Osiewacz HD. Control of mitochondrial integrity in ageing and disease. Philos Trans R Soc Lond B Biol Sci. 2014;369(1646):20130439. doi: 10.1098/rstb.2013.0439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Payne BA, Chinnery PF. Mitochondrial dysfunction in aging: much progress but many unresolved questions. Biochim Biophys Acta. 2015;1847(11):1347–1353. doi: 10.1016/j.bbabio.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Diot A, Morten K, Poulton J. Mitophagy plays a central role in mitochondrial ageing. Mamm Genome. 2016;27(7–8):381–395. doi: 10.1007/s00335-016-9651-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ben-Meir A, Yahalomi S, Moshe B, Shufaro Y, Reubinoff B, Saada A. Coenzyme Q-dependent mitochondrial respiratory chain activity in granulosa cells is reduced with aging. Fertil Steril. 2015;104(3):724–727. doi: 10.1016/j.fertnstert.2015.05.023. [DOI] [PubMed] [Google Scholar]
- 115.Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, Janssen EM. Loss of phagocytic and antigen cross-presenting capacity in aging dendritic cells is associated with mitochondrial dysfunction. J Immunol. 2015;195(6):2624–2632. doi: 10.4049/jimmunol.1501006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kauppila TE, Kauppila JH, Larsson NG. Mammalian mitochondria and aging: an update. Cell Metab. 2017;25(1):57–71. doi: 10.1016/j.cmet.2016.09.017. [DOI] [PubMed] [Google Scholar]
- 117.Ferrington DA, Kapphahn RJ, Leary MM, Atilano SR, Terluk MR, Karunadharma P, Chen GK, Ratnapriya R, Swaroop A, Montezuma SR, Kenney MC. Increased retinal mtDNA damage in the CFH variant associated with age-related macular degeneration. Exp Eye Res. 2016;145:269–277. doi: 10.1016/j.exer.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Deininger P. Alu elements: know the SINEs. Genome Biol. 2011;12(12):236. doi: 10.1186/gb-2011-12-12-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pandey R, Bhattacharya A, Bhardwaj V, Jha V, Mandal AK, Mukerji M. Alu-miRNA interactions modulate transcript isoform diversity in stress response and reveal signatures of positive selection. Sci Rep. 2016;6:32348. doi: 10.1038/srep32348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang W, Wang WH, Azadzoi KM, Dai P, Wang Q, Sun JB, Zhang WT, Shu Y, Yang JH, Yan Z. Alu RNA accumulation in hyperglycemia augments oxidative stress and impairs eNOS and SOD2 expression in endothelial cells. Mol Cell Endocrinol. 2016;426:91–100. doi: 10.1016/j.mce.2016.02.008. [DOI] [PubMed] [Google Scholar]
- 121.Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, Kleinman ME, Ponicsan SL, Hauswirth WW, Chiodo VA, Kariko K, Yoo JW, Lee DK, Hadziahmetovic M, Song Y, Misra S, Chaudhuri G, Buaas FW, Braun RE, Hinton DR, Zhang Q, Grossniklaus HE, Provis JM, Madigan MC, Milam AH, Justice NL, Albuquerque RJ, Blandford AD, Bogdanovich S, Hirano Y, Witta J, Fuchs E, Littman DR, Ambati BK, Rudin CM, Chong MM, Provost P, Kugel JF, Goodrich JA, Dunaief JL, Baffi JZ, Ambati J. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011;471(7338):325–330. doi: 10.1038/nature09830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, Cho WG, Kaneko H, Fowler BJ, Bogdanovich S, Albuquerque RJ, Hauswirth WW, Chiodo VA, Kugel JF, Goodrich JA, Ponicsan SL, Chaudhuri G, Murphy MP, Dunaief JL, Ambati BK, Ogura Y, Yoo JW, Lee DK, Provost P, Hinton DR, Núñez G, Baffi JZ, Kleinman ME, Ambati J. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. 2012;149(4):847–859. doi: 10.1016/j.cell.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kerur N, Fukuda S, Banerjee D, Kim Y, Fu D, Apicella I, Varshney A, Yasuma R, Fowler BJ, Baghdasaryan E, Marion KM, Huang X, Yasuma T, Hirano Y, Serbulea V, Ambati M, Ambati VL, Kajiwara Y, Ambati K, Hirahara S, Bastos-Carvalho A, Ogura Y, Terasaki H, Oshika T, Kim KB, Hinton DR, Leitinger N, Cambier JC, Buxbaum JD, Kenney MC, Jazwinski SM, Nagai H, Hara I, West AP, Fitzgerald KA, Sadda SR, Gelfand BD, Ambati J. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat Med. 2018;24(1):50–61. doi: 10.1038/nm.4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Childs BG, Gluscevic M, Baker DJ, Laberge RM, Marquess D, Dananberg J, van Deursen JM. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. 2017;16(10):718–735. doi: 10.1038/nrd.2017.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Korolchuk VI, Miwa S, Carroll B, von Zglinicki T. Mitochondria in cell senescence: is mitophagy the weak link? EBioMedicine. 2017;21:7–13. doi: 10.1016/j.ebiom.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21(12):1424–1435. doi: 10.1038/nm.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K, Birch-Machin MA, Kirkwood TB, von Zglinicki T. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5:e110. doi: 10.1371/journal.pbio.0050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, Gerencser AA, Verdin E, Campisi J. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23(2):303–314. doi: 10.1016/j.cmet.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, Saretzki G, Rudolph KL, Kirkwood TB, von Zglinicki T. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010;6:347. doi: 10.1038/msb.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ferrington DA, Sinha D, Kaarniranta K. Defects in retinal pigment epithelial cell proteolysis and the pathology associated with age-related macular degeneration. Prog Retin Eye Res. 2016;51:69–89. doi: 10.1016/j.preteyeres.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Marazita MC, Dugour A, Marquioni-Ramella MD, Figueroa JM, Suburo AM. Oxidative stress-induced premature senescence dysregulates VEGF and CFH expression in retinal pigment epithelial cells: implications for age-related macular degeneration. Redox Biol. 2016;7:78–87. doi: 10.1016/j.redox.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavaré S, Arakawa S, Shimizu S, Watt FM, Narita M. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23(7):798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 135.Dalle Pezze P, Nelson G, Otten EG, Korolchuk VI, Kirkwood TB, von Zglinicki T, Shanley DP. Dynamic modelling of pathways to cellular senescence reveals strategies for targeted interventions. PLoS Comput Biol. 2014;10(8):e1003728. doi: 10.1371/journal.pcbi.1003728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, Sandri M, Muñoz-Cánoves P. Autophagy maintains stemness by preventing senescence. Nature. 2016;529(5784):37–42. doi: 10.1038/nature16187. [DOI] [PubMed] [Google Scholar]
- 137.Ahmad T, Sundar IK, Lerner CA, Gerloff J, Tormos AM, Yao H, Rahman I. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease. FASEB J. 2015;29(7):2912–2929. doi: 10.1096/fj.14-268276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS, Duncan SR, Rojas M, Shiva S, Chu CT, Mora AL. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Investig. 2015;125(3):521–538. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ohtsuji M, Katsuoka F, Kobayashi A, Aburatani H, Hayes JD, Yamamoto M. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J Biol Chem. 2008;283(48):33554–33562. doi: 10.1074/jbc.M804597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dodson M, Redmann M, Rajasekaran NS, Darley-Usmar V, Zhang J. KEAP1-NRF2 signalling and autophagy in protection against oxidative and reductive proteotoxicity. Biochem J. 2015;469(3):347–355. doi: 10.1042/BJ20150568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhao Z, Chen Y, Wang J, Sternberg P, Freeman ML, Grossniklaus HE, Cai J. Age-related retinopathy in NRF-2-deficient mice. PLoS One. 2011;6(4):e19456. doi: 10.1371/journal.pone.0019456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Phillipson OT. Management of the aging risk factor for Parkinson’s disease. Neurobiol Aging. 2014;35(4):847–857. doi: 10.1016/j.neurobiolaging.2013.10.07. [DOI] [PubMed] [Google Scholar]
- 143.Ping Z, Zhang LF, Cui YJ, Chang YM, Jiang CW, Meng ZZ, Xu P, Liu HY, Wang DY, Cao XB. The protective effects of salidroside from exhaustive exercise-induced heart injury by enhancing the PGC-1 α -NRF1/NRF2 pathway and mitochondrial respiratory function in rats. Oxid Med Cell Longev. 2015;2015:876825. doi: 10.1155/2015/876825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Iacovelli J, Rowe GC, Khadka A, Diaz-Aguilar D, Spencer C, Arany Z, Saint-Geniez M. PGC-1α induces human RPE oxidative metabolism and antioxidant capacity. Investig Ophthalmol Vis Sci. 2016;57(3):1038–1051. doi: 10.1167/iovs.15-17758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA. 2007;104(29):12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Salminen A, Kaarniranta K, Kauppinen A. Crosstalk between oxidative stress and SIRT1: impact on the aging process. Int J Mol Sci. 2013;14(2):3834–3859. doi: 10.3390/ijms14023834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Egger A, Samardzija M, Sothilingam V, Tanimoto N, Lange C, Salatino S, Fang L, Garcia-Garrido M, Beck S, Okoniewski MJ, Neutzner A, Seeliger MW, Grimm C, Handschin C. PGC-1α determines light damage susceptibility of the murine retina. PLoS One. 2012;7(2):e31272. doi: 10.1371/journal.pone.0031272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Liang FQ, Godley BF. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res. 2003;76(4):397–403. doi: 10.1016/S0014-4835(03)00023-X. [DOI] [PubMed] [Google Scholar]
- 149.Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. 2016;61(5):654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rohrer B, Bandyopadhyay M, Beeson C. Reduced metabolic capacity in aged primary retinal pigment epithelium (RPE) is correlated with increased susceptibility to oxidative stress. Adv Exp Med Biol. 2016;854:793–798. doi: 10.1007/978-3-319-17121-0_106. [DOI] [PubMed] [Google Scholar]
- 151.He Y, Ge J, Burke JM, Myers RL, Dong ZZ, Tombran-Tink J. Mitochondria impairment correlates with increased sensitivity of aging RPE cells to oxidative stress. J Ocul Biol Dis Inform. 2010;3(3):92–108. doi: 10.1007/s12177-011-9061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gouras P, Ivert L, Neuringer M, Nagasaki T. Mitochondrial elongation in the macular RPE of aging monkeys, evidence of metabolic stress. Graefes Arch Clin Exp Ophthalmol. 2016;254(6):1221–1227. doi: 10.1007/s00417-016-3342-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Field MG, Comer GM, Kawaji T, Petty HR, Elner VM. Noninvasive imaging of mitochondrial dysfunction in dry age-related macular degeneration. Ophthalmic Surg Lasers Imaging. 2012;43(5):358–365. doi: 10.3928/15428877-20120712-02. [DOI] [PubMed] [Google Scholar]
- 154.SanGiovanni JP, Arking DE, Iyengar SK, Elashoff M, Clemons TE, Reed GF, Henning AK, Sivakumaran TA, Xu X, DeWan A, Agrón E, Rochtchina E, Sue CM, Wang JJ, Mitchell P, Hoh J, Francis PJ, Klein ML, Chew EY, Chakravarti A. Mitochondrial DNA variants of respiratory complex I that uniquely characterize haplogroup T2 are associated with increased risk of age-related macular degeneration. PLoS One. 2009;4(5):e5508. doi: 10.1371/journal.pone.0005508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278(10):8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- 156.Lee SY, Oh JS, Rho JH, Jeong NY, Kwon YH, Jeong WJ, Ryu WY, Ahn HB, Park WC, Rho SH, Yoon YG, Jeong SY, Choi YH, Kim HY, Yoo YH. Retinal pigment epithelial cells undergoing mitotic catastrophe are vulnerable to autophagy inhibition. Cell Death Dis. 2014;5:e1303. doi: 10.1038/cddis.2014.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Blasiak J, Szaflik J, Szaflik JP. Implications of altered iron homeostasis for age-related macular degeneration. Front Biosci (Landmark Ed) 2011;16:1551–1559. doi: 10.2741/3804. [DOI] [PubMed] [Google Scholar]
- 158.Mena NP, Urrutia PJ, Lourido F, Carrasco CM, Núñez MT. Mitochondrial iron homeostasis and its dysfunctions in neurodegenerative disorders. Mitochondrion. 2015;21:92–105. doi: 10.1016/j.mito.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 159.Drysdale J, Arosio P, Invernizzi R, Cazzola M, Volz A, Corsi B, Biasiotto G, Levi S. Mitochondrial ferritin: a new player in iron metabolism. Blood Cells Mol Dis. 2002;29(3):376–383. doi: 10.1006/bcmd.2002.0577. [DOI] [PubMed] [Google Scholar]
- 160.Wang L, Yang H, Zhao S, Sato H, Konishi Y, Beach TG, Abdelalim EM, Bisem NJ, Tooyama I. Expression and localization of mitochondrial ferritin mRNA in Alzheimer’s disease cerebral cortex. PLoS One. 2011;6(7):e22325. doi: 10.1371/journal.pone.0022325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Stenirri S, Santambrogio P, Setaccioli M, Erba BG, Pia Manitto M, Rovida E, Ferrari M, Levi S, Cremonesi L. Study of FTMT and ABCA4 genes in a patient affected by age-related macular degeneration: identification and analysis of new mutations. Clin Chem Lab Med. 2011;50(6):1021–1029. doi: 10.1515/cclm-2011-0854. [DOI] [PubMed] [Google Scholar]
- 162.Wang X, Yang H, Yanagisawa D, Bellier JP, Morino K, Zhao S, Liu P, Vigers P, Tooyama I. Mitochondrial ferritin affects mitochondria by stabilizing HIF-1α in retinal pigment epithelium: implications for the pathophysiology of age-related macular degeneration. Neurobiol Aging. 2016;47:168–179. doi: 10.1016/j.neurobiolaging.2016.07.025. [DOI] [PubMed] [Google Scholar]
- 163.Sripathi SR, He W, Prigge CL, Sylvester O, Um JY, Powell FL, Neksumi M, Bernstein PS, Choo DW, Bartoli M, Gutsaeva DR, Jahng WJ. Interactome mapping guided by tissue-specific phosphorylation in age-related macular degeneration. Int J Sci Eng Res. 2017;8(2):680–699. doi: 10.14299/ijser.2017.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hyttinen JMT, Petrovski G, Salminen A, Kaarniranta K. 5′-Adenosine monophosphate-activated protein kinase-mammalian target of rapamycin axis as therapeutic target for age-related macular degeneration. Rejuvenation Res. 2011;14(6):651–660. doi: 10.1089/rej.2011.1220. [DOI] [PubMed] [Google Scholar]
- 165.Bové J, Martínez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011;12(8):437–452. doi: 10.1038/nrn3068. [DOI] [PubMed] [Google Scholar]
- 166.Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab. 2014;20(6):953–966. doi: 10.1016/j.cmet.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 167.Saisho Y. Metformin and inflammation: its potential beyond glucose-lowering effect. Endocr Metab Immune Disord Drug Targets. 2015;15(3):196–205. doi: 10.18632/aging.100444. [DOI] [PubMed] [Google Scholar]
- 168.Moiseeva O, Deschênes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, Bourdeau V, Pollak MN, Ferbeyre G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell. 2013;12(3):489–498. doi: 10.1111/acel.12075. [DOI] [PubMed] [Google Scholar]
- 169.Song YM, Lee WK, Lee YH, Kang ES, Cha BS, Lee BW. Metformin restores Parkin-mediated mitophagy, suppressed by cytosolic p53. Int J Mol Sci. 2016;17(1):122. doi: 10.3390/ijms17010122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9(10):691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 171.Cantó C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20(2):98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, Criollo A, Galluzzi L, Malik SA, Vitale I, Michaud M, Madeo F, Tavernarakis N, Kroemer G. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010;1:e10. doi: 10.1038/cddis.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Giovannini L, Bianchi S. Role of nutraceutical SIRT1 modulators in AMPK and mTOR pathway: evidence of a synergistic effect. Nutrition. 2017;34:82–96. doi: 10.1016/j.nut.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 174.Georgakopoulos ND, Frison M, Alvarez MS, Bertrand H, Wells G, Campanella M. Reversible Keap1 inhibitors are preferential pharmacological tools to modulate cellular mitophagy. Sci Rep. 2017;7(1):10303. doi: 10.1038/s41598-017-07679-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Morselli E, Mariño G, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, Cabrera S, Bénit P, Rustin P, Criollo A, Kepp O, Galluzzi L, Shen S, Malik SA, Maiuri MC, Horio Y, López-Otín C, Andersen JS, Tavernarakis N, Madeo F, Kroemer G. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol. 2011;192(4):615–629. doi: 10.1083/jcb.201008167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet-Dit-Félix AA, Williams EG, Jha P, Lo Sasso G, Huzard D, Aebischer P, Sandi C, Rinsch C, Auwerx J. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med. 2016;22(8):879–888. doi: 10.1038/nm.4132. [DOI] [PubMed] [Google Scholar]
- 177.Fivenson EM, Lautrup S, Sun N, Scheibye-Knudsen M, Stevnsner T, Nilsen H, Bohr VA, Fang EF. Mitophagy in neurodegeneration and aging. Neurochem Int. 2017;109:202–209. doi: 10.1016/j.neuint.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Richter U, Lahtinen T, Marttinen P, Myöhänen M, Greco D, Cannino G, Jacobs HT, Lietzén N, Nyman TA, Battersby BJ. A mitochondrial ribosomal and RNA decay pathway blocks cell proliferation. Curr Biol. 2013;23(6):535–541. doi: 10.1016/j.cub.2013.02.019. [DOI] [PubMed] [Google Scholar]
- 179.Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, Holmström KM, Fergusson MM, Yoo YH, Combs CA, Finkel T. Measuring in vivo mitophagy. Mol Cell. 2015;60(4):685–696. doi: 10.1016/j.molcel.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hasson SA, Fogel AI, Wang C, MacArthur R, Guha R, Heman-Ackah S, Martin S, Youle RJ, Inglese J. Chemogenomic profiling of endogenous PARK2 expression using a genome-edited coincidence reporter. ACS Chem Biol. 2015;10(5):1188–1197. doi: 10.1021/cb5010417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hertz NT, Berthet A, Sos ML, Thorn KS, Burlingame AL, Nakamura K, Shokat KM. A neo-substrate that amplifies catalytic activity of Parkinson’s-disease-related kinase PINK1. Cell. 2013;154(4):737–747. doi: 10.1016/j.cell.2013.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Blasiak J, Piechota M, Pawlowska E, Szatkowska M, Sikora E, Kaarniranta K. Cellular senescence in age-related macular degeneration: can autophagy and DNA damage response play a role? Oxid Med Cell Longev. 2017;2017:5293258. doi: 10.1155/2017/5293258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kozlowski MR. RPE cell senescence: a key contributor to age-related macular degeneration. Med Hypotheses. 2012;78(4):505–510. doi: 10.1016/j.mehy.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 184.Knuppertz L, Osiewacz HD. Orchestrating the network of molecular pathways affecting ageing: role of non-selective autophagy and mitophagy. Mech Ageing Dev. 2016;153:30–40. doi: 10.1016/j.mad.2016.01.003. [DOI] [PubMed] [Google Scholar]