

Abstract
The human Far Upstream Element (FUSE) Binding Protein 1 (FUBP1) is a multifunctional DNA- and RNA-binding protein involved in diverse cellular processes. FUBP1 is a master regulator of transcription, translation, and RNA splicing. FUBP1 has been identified as a potent pro-proliferative and anti-apoptotic factor by modulation of complex networks. FUBP1 is also described either as an oncoprotein or a tumor suppressor. Especially, FUBP1 overexpression is observed in a growing number of cancer and leads to a deregulation of targets that includes the fine-tuned MYC oncogene. Moreover, recent loss-of-function analyses of FUBP1 establish its essential functions in hematopoietic stem cell maintenance and survival. Therefore, FUBP1 appears as an emerging suspect in hematologic disorders in addition to solid tumors. The scope of the present review is to describe the advances in our understanding of the molecular basis of FUBP1 functions in normal cells and carcinogenesis. We also delineate the recent progresses in the understanding of the master role of FUBP1 in normal and pathological hematopoiesis. We conclude that FUBP1 is not only worth studying biologically but is also of clinical relevance through its pivotal role in regulating multiple cellular processes and its involvement in oncogenesis.
Keywords: P21, P53, FIR, KH domain, Leukemia, c-KIT
Introduction
The human Far Upstream Element Binding Protein 1 (initially FUSE Binding Protein, FBP, and recently renamed FUBP1) is a protein-coding gene located on the reverse strand of chromosome 1 (1p31.1). FUBP1 is a master regulator of transcription, splicing, and translation through its bindings to single-stranded DNA (ssDNA) and RNA. Moreover, FUBP1 is an ATP-dependent DNA helicase V 3′–5′ that has an activity on both DNA-DNA and RNA-RNA duplexes. Multiple functions of FUBP1 can be associated with its helicase properties or can be mediated by interactions with other protein partners like the FUBP Interacting Repressor (FIR).
FUBP1 functions as an important activator or repressor of transcription and translation of its target genes [1]. FUBP1 promotes cell proliferation, inhibits apoptosis, and enhances cell migration by modulating complex networks. For example, FUBP1 is a well-known activator of MYC expression [2–8]. Importantly, FUBP1 endorses oncogenic or tumor suppressor roles [6, 8–15]. Overexpression of FUBP1 can lead to alterations in the expression of its target genes that often include deregulation of the oncogene MYC. Conversely, tumor suppressor functions of FUBP1 are associated with FUBP1 loss-of-function, although the exact involvement of FUBP1 in these tumors remains to be elucidated.
An emerging area of research unravels additional activities of FUBP1 in hematopoiesis. Indeed, FUBP1 has been recently described as a master player for expansion and self-renewal of hematopoietic stem cells (HSCs) [2, 16, 17]. Hematopoiesis is a continuous and precisely controlled process in which pluripotent HSCs produce all mature blood cells upon differentiation including erythrocytes, megakaryocytes, myeloid cells (monocyte, basophil, neutrophil, and eosinophil), and lymphoid cells (B- and T-lymphocytes and natural killer). Therefore, transcription regulators like FUBP1 are required to control the maintenance and the differentiation of HSCs. These functions of FUBP1 in hematopoiesis contribute to increase our knowledge on the multifunctional FUBP1 protein and raise questions about its role in hematological malignancies, a field that remains open for exploration.
The present review provides an overview of the structure, characteristics, and function of FUBP1. Its surprising dual role in DNA and RNA binding will be summarized. FUBP1 regulatory mechanisms in cancer will then be depicted. We will next discuss the binding sequence specificity of FUBP1. Finally, we will delineate the recent advances in FUBP1 functions in normal and pathological hematopoiesis. Altogether, this review presents an overview of current understanding about the master regulator FUBP1 and highlights that FUBP1 represents an attractive target for future molecular cancer therapies.
FUBP1 is a DNA- and RNA-binding protein
Molecular and structural properties of FUBP1
The far upstream element binding protein family
FUBP1 belongs to an evolutionary ancient and conserved family of single-stranded (ss) DNA-binding regulators including FUBP1, FUBP2 (also known as KH-type splicing regulatory, KHSRP), and FUBP3 [18]. Although the three members share highly conserved sequences and structures [19, 20], their functions and expression pattern diverge. The FUBPs are reported to participate in a wide variety of biological processes including mRNA synthesis or degradation, splicing, RNA transport, or translational regulation [1, 20, 21]. While FUBP1 and FUBP3 bind ssDNA and RNA, FUBP2 is more often described as an exclusive RNA-binding protein [21]. FUBP2 is involved in several post-transcriptional aspects of RNA metabolism including alternative splicing of pre-mRNA, mRNA decay, mRNA localization, and miRNA biogenesis [21, 22]. FUBP3 is implicated in transcriptional and post-transcriptional regulation, although its biological role remains largely unexplored [23, 24]. FUBP1 and FUBP2 share the most functional similarity in contrast to FUBP3 that is only weakly localized in the nucleus and does not bind the FUBP-Interacting Repressor (FIR) [20]. The three FUBP members may cooperate to fine-tune the regulation of common target genes. However, FUBP1 is the sole FUBP family member that has been involved in hematopoiesis, so far.
Expression of FUBP1
FUBP1 is highly conserved among species. Human FUBP1 shares more than 90% similarity with mammalian phylogenies, 73% homology with the less evolved vertebrate zebrafish (Danio rerio), and orthologs have been identified in invertebrate species such as Caenorhabditis elegans and Drosophila melanogaster (where FUBP1 ortholog is called Psi) [14, 19, 25]. Since FUBP1 is expressed in a wide variety of species including in vertebrates and invertebrates, it seems that FUBP1 was present in the common ancestor of Animals [25]. Furthermore, human FUBP1 gene is widely expressed among tissues, although it exhibits different spatial–temporal patterns of expression [26]. For example, FUBP1 is less expressed in mature cells compared to progenitor cells, in the myeloid lineage [2, 19].
Functional characteristics of FUBP1 transcript and protein
The human FUBP1 gene spans 31,187 bp and displays two transcript variants (variant 1, NM_001303433 and variant 2, NM_003902) [25]. The variant 1 is composed of 20 protein-coding exons and encodes a 655 amino acid protein, called the isoform 1. The variant 2 lacks an alternate in-frame exon in the 5′-coding region compared to variant 1, making it shorter. The variant 2, which is the representative form of FUBP1 mRNA, is also composed of 20 exons, but encodes a 644 amino acid protein (isoform 2). FUBP1, FUBP2, and FUBP3 proteins share the same architecture with three distinct domains composed of an N-terminal repression domain, a central domain allowing ssDNA- and RNA-binding, and a C-terminal transactivation domain (Fig. 1) [3, 18, 19]. A flexible glycine/proline-rich linker (amino acids 448–510) connects the central domain to the C-terminal domain (Fig. 1). The central domain contains four K-homology (KH) motifs, each followed by an amphipathic helix. KH motifs are important for protein binding to ssDNA or RNA [27]. In its inactive state, FUBP1 protein is folded into an enclosed conformation allowing its N-terminal domain to repress the C-terminal transactivation domain [18, 19]. Upon activation, FUBP1 undergoes a conformational change to release the C-terminal domain, but the exact mechanism behind FUBP1 activation has not been studied in depth yet. Its C-terminal domain displays an unusual repeated sequence consisting of three tyrosine-rich motifs which are required for transcriptional activation of FUBP1, as replacement of tyrosines by non-phosphorylatable residues greatly impairs activation [3, 18, 19]. The C-terminal domain is also the binding site of the Transcriptional Factor IIH (TFIIH) p89 subunit, a complex involved in transcriptional initiation and promoter escape [3, 18].
Fig. 1.

Schematic representation of FUBP1 protein domains with its associated mutations in leukemia identified so far. FUBP1 encompasses three domains, one inhibitory domain in N-terminal, one central domain containing four KH subdomains for DNA and RNA bindings and one transactivation domain in C-terminal. Few somatic mutations of FUBP1 have been described in leukemia [147–149] and are represented here. All mutations produce truncated proteins that are not functional, except for the two SNV identified in B-cell precursors ALL. NLS: nuclear localization signal, ALL: acute lymphoblastic leukemia
Nuclear localization of FUBP1 protein is driven by three nuclear localization signals (NLS), one in the N-terminal domain, one in the central domain, and one in the C-terminal domain. However, under various stress stimuli such as viral infection and apoptosis, FUBP1 can translocate from nucleus to cytoplasm [28, 29]. When cells suffer from these stimuli, FUBP1 is cleaved by caspase-3 and -7 at the caspase consensus site (DQPD, amino acids 71–74) located within the N-terminal NLS, leading to a decrease in its overall nuclear level [30]. The role of the FUBP1-truncated proteins currently remains unclear. FUBP1 protein may also be modulated by post-translational modifications such as phosphorylation, acetylation, mono-methylation, di-methylation, sumoylation, and ubiquitination [31–33]. Besides caspases 3-7, few FUBP1 regulators are described such as P38, USP22 proteins, and miR-16 [5, 32, 34, 35].
FUBP1 is a multifunctional DNA-binding protein
Characterization of the FUBP1 DNA-binding mechanism
FUBP1 interacts with ssDNA. Single-stranded DNA is produced during all aspects of DNA metabolism: during replication, DNA repair, recombination, and in response to torsional stress upon initiation of the transcriptional machinery [7, 36].
Proteins with KH domains like FUBP1 have the property to form stable complexes with both DNA and RNA. Although FUBP1 binding to single-stranded nucleic acids is facilitated by its four KH domains, only the two subdomains KH3 and KH4 are required for DNA or RNA bindings in vitro [3]. The amphipathic helix adjacent to each KH domain also supports FUBP1 binding by promoting the intramolecular folding required to form a functional DNA-binding domain [3]. KH4 and KH3 FUBP1-subdomains are connected by a glycine-rich (GXXG) loop that allows flexibility between the bindings of the two subdomains. The first KH protein identified was the heterogeneous nuclear ribonucleoprotein K (hnRNP-K). Interestingly, both FUBP1 and hnRNP-K are known to bind MYC promoter. The hnRNP-K binds to the CT element located at − 100 to 150 bp, while FUBP1 binds to the FUSE element on the non-coding strand of MYC, located − 1500 bp upstream of the P1 promoter (or − 1700 bp upstream of the P2 promoter) [3, 37–39]. The FUSE element on MYC is an AT-rich sequence, 29 nucleotide-long 5′-TATATTCCCTCGGGATTTTTTATTTTGTG-3′ [3, 40]. On this FUBP1-DNA-binding sequence, the KH4 and KH3 subdomains recognize, respectively, 5′-TATTCC-3′ and 5′-ATTTTT-3′ short sequences, separated by six nucleotides forming a DNA bridge, where FUBP1 might not bind [41]. KH1 and KH2 subdomain-binding properties are less documented, but they could bind MYC in 3′ of the KH3-binding motif [42].
Besides the FUSE sequence on MYC gene, FUBP1 interacts with a large spectrum of sequences across a wide range of affinities [43]. Structural modelling of KH3 and KH4 subdomains has led to the identification of the creation of a groove, where amino acids interact with single-stranded nucleic acids. The KH4 forms a narrow groove (approx. 10 Å). The center of the groove forms a hydrophobic pocket that captures negatively charged ssDNA. At the same time, the negatively charged sugar-phosphate backbone of nucleotides is attracted to the groove and binds the hydrophilic and positively charged edges of the FUBP1 groove through H-bonds or salt bridges [41]. The narrowness of the groove formed by KH4 favors pyrimidines over purines due to the presence of only one carbon nitrogen ring. On the contrary, the KH3 subdomain forms a wider groove that allows accommodating purine-rich sequences without severe structural distortion [44]. A Systematic Evolution of Ligands by EXponential enrichment (SELEX) experiment described the optimal sequence for full-length FUBP1 binding sequence. According to SELEX, FUBP1 KH2-3-4 subdomains favor the interaction with a similar set of tetramers T(T/C)GT, whereas the optimal binding sequence for KH1 is (T/G)TG(T/C) [43]. This KH1-bound sequence specificity may be important for FUBP1 and FIR functions, because KH1 contact bases seem to differ in footprint assays upon FUBP1/FIR/FUSE or FUBP1/FUSE bindings. Each subdomain is optimally separated by 4–5 nucleotides on DNA [43]. Nevertheless, the conformation of the flexible linker may accept up to 7 nucleotides between KH3 and KH4 subdomains (35Å) [41]. Taking this optimal sequence in consideration, the FUSE sequence from MYC deviates from the perfect consensus, showing that FUBP1 accepts binding sites that do not correspond to the optimal sequence. Indeed, the 29 nucleotide-long FUSE sequence is not retrieved anywhere else in the human genome, although several studies have identified other FUBP1-bound sequences called “FUSE-like”. A description of different FUBP1-bound sequences is provided in Table 1. These variable sequences definitely distinguish FUBP1 from traditional transcription factors. Although FUBP1 does not display strict binding sequence dependence, the scrutiny of the few FUSE-like sequences described reveals a preference for AT- or GT-rich stretches, as mentioned previously. However, the specificity by which FUBP1 binds DNA to regulate its target genes is still not well understood.
Table 1.
Summary of the current literature on DNA and RNA-FUBP1 binding regions and related functions
| Gene | Localization, role of FUBP1 and DNA or RNA sequence | References | Motif | |
|---|---|---|---|---|
| DNA bindings | MYC |
− 1.7 kb upstream the P2 promoter (strand −) Transcriptional activation |
[3, 34, 41, 43, 55] | A/T-rich |
| GTATATTCCCTCGGGATTTTTTATTTTGTGTTATTCCACGGCATGAAAAA | ||||
| USP29 |
− 2.5 kb upstream TSS (strand −) Transcriptional activation |
[5] | T- and GT- rich | |
| TTTGCATTACTTTTTTTTTTGTTTGTTTTTGAGATGGAGTTTTGCTCTTGTTGCCC | ||||
| c-KIT |
+ 30 kb upstream TSS (strand +) Transcriptional activation |
[44] | A/T-rich | |
| AGTTTATTCCTATGGGGATATAAAAGTGTGTCAG | ||||
| CDKN1A |
− 2.7 kb upstream TSS (strand −) Transcriptional repression |
[8, 11, 32] | T- and GT- rich | |
| CTGGCTTTTTGTTTTCATTTTGTTTTTTTGTTTTGTTTTGTTTTTTGAGACAA | ||||
| BIK |
− 1.2 kb upstream TSS (strand −) Transcriptional repression |
[11] | T- and GT- rich | |
| CTTTTGTGGGGTTTTTTTGTTTGTTTTTGTTTTTGTTTTTTTGA | ||||
| RNA bindings | PKD2 |
3′UTR Translational repression |
[80] |
AU- and AUUUA-rich |
| 691nt…CCUAAAGAUUUAUCUGCAUAUUCUUUUUCCCAUGUGGCUCUACUCAUUUGCAACUGAAUUUAAUGUU…1044nt | ||||
| GAP-43 |
3′UTR mRNA degradation |
[81] | CU-rich | |
| GUCCACUUUCCUCUCUCUCUCUCUCU | ||||
| COX2 |
3′UTR Unknown |
[73] |
AU- and AUUUA-rich |
|
| AAGUCUAAUGAUCAUAUUUAUUUAUUUAUAUGAACCAUGUCUAUUAAUUUAAUUAUUUAAUAAUAUUUAUAUUAAA | ||||
| P27 |
5′UTR: 32–40 nucleotides upstream the start codon Translational activation |
[85] | ||
| GCGAAGAG | ||||
| Triadin |
Exon 10 on an exonic splicing silencer Suppression of the exon 10 splicing |
[94] | AU-rich | |
| GUCGAUAUAUGAUUGA | ||||
| MDM2 |
Intron 11 on an intronic splicing enhancer Activation of the exon 11 splicing inclusion |
[78] | U- and GU-rich | |
| AAUUCCCUUUCUUGUGUGUAUGGU | ||||
| DMD |
Intron 38 on an intronic splicing enhancer Activation of the exon exon 39 splicing inclusion |
[79] | U- and GU-rich | |
| GGAUUUUUGUGUGUGUUUAAAUAACAUGUCUUAUUA | ||||
| Hepatitis C virus |
3′UTR Enhances replication. |
[74] | U- and UC-rich | |
| Poly(U/UC) region | ||||
| EV71 |
5′UTR, in the IRES Enhances viral protein synthesis. |
[86, 88] | U- and AU- rich | |
| 636nt…CAAUUGUUUACCUAUUUAUUGGUUUUGUACCAUUAUCACUGAAGUCUGUGAUCACUCUCAAAUUCAUUUUGACCC…745nt | ||||
The specific nucleotides (nt) required for FUBP1 binding are underlined
Transcriptional activation of target genes
Myc
c-MYC protein, encoded by MYC gene, plays critical roles in cell cycle regulation, metabolism, apoptosis, differentiation, cell adhesion, and tumorigenesis [45]. As a global transcription factor, MYC regulates 10–15% of genes in the human genome [46]. In 1990, FUBP1 was described for the first time to bind the FUSE element in MYC promoter [2]. Since then, series of experiments have explored how FUBP1 positively regulates the transcription of the proto-oncogene MYC [1]. Interestingly, FUBP1 alone is not sufficient to activate MYC expression, but it is required for its maximal activation [47].
The regulation of MYC by FUBP1 involves other factors like FIR and TFIIH. TFIIH is a multifunctional RNA polymerase II transcription factor playing a dual role in both transcription and DNA excision repair [48, 49]. TFIIH is a ten subunit protein complex composed of the core complex formed by 6 subunits (p8, p34, p44, p52, p62, and p89), the CAK [cyclin-dependent kinase (CDK)-activating kinase] subcomplex (formed by CDK7, MAT1, and cyclin H), and the p80 linking the core and the CAK. The p89 subunit (also called ERCC3 or XPB) has a 3′–5′ helicase activity and the p80 subunit (or ERCC2, XPD) has a 5′–3′ ATP-dependent helicase activity [50]. TFIIH generates an open complex to initiate the transcription. TFIIH is required on relaxed DNA or on supercoiled DNA like MYC promoter [51, 52].
For MYC activation, the torsion stress induced by the transcription allows the non-coding strand of the FUSE sequence to recruit FUBP1. At the same time, TFIIH interacts physically with FUBP1 on the C-terminal transactivation domain of FUBP1 via the p62, p80, and p89 subunits of TFIIH, forming a DNA-loop [47]. Subsequently, FUBP1 stimulates the helicase activity of p89 that allows the transcription of MYC. However, the expression of MYC needs to be rigorously controlled, since even a transient excess of unscheduled MYC activity increases tumorigenicity [53]. To maintain a correct level of MYC expression, a rapid negative feedback mechanism occurs, mediated by the repressor FIR. FIR is an alternatively spliced variant of PUF60 (also called SIAHBP1 or RoBP1) that lacks 17 amino acids in the N-terminus due to truncation of exon 5. FIR is a DNA- and RNA-binding protein, encompassing a repression domain that binds TFIIH at the N-terminal, two RNA recognition motifs in the central domain, and an U2AF homology motif at the C-terminus [42].
For MYC repression, FIR protein interacts with the central DNA-binding domain of FUBP1 via its RNA recognition motifs. Then, FIR binds the FUSE sequence as a homodimer through its RNA recognition motifs forming a tripartite FUSE/FUBP1/FIR complex [42, 54, 55]. Next, this interaction brings FIR close to TFIIH which still interacts with FUBP1, allowing the N-terminus of FIR to interact physically with TFIIH and thereby to suppress its p89 helicase activity [54, 56]. The repression of TFIIH generates a torsional stress that changes the conformation of the FUSE element. Finally, the FUSE renatures, FUBP1 can no longer bind and is ejected from the FUSE. Subsequently, FIR homodimerizes on the FUSE instead of FUBP1, providing a stabilization of the FIR/FUSE interaction that brings MYC expression back to basal level until a new transcriptional cycle begins [57].
To summarize, FUBP1 binds on MYC promoter and activates its transcription through TFIIH helicase activity and FIR counteracts the whole system, suppressing the activation effect of FUBP1. Therefore, the FUSE/FUBP1/FIR complex represents a sensitive molecular tool for the fine-tuned regulation of MYC [4, 42, 54, 58].
However, some studies have reported that the mechanism of regulation of MYC by FUBP1 might not be ubiquitous, since MYC mRNA and protein levels are not affected by FUBP1 depletion in different cell types such as the hepatocellular carcinoma (HCC) cells Hep3B, normal human fibroblast, and Hela cells [6, 8, 32].
Usp29
An elegant study underscores that FUBP1 activates the transcription of the ubiquitin-specific peptidase 29 (USP29) [5]. Using electrophoretic mobility-shift assay (EMSA) and chromatin immunoprecipitation (ChIP)-PCR, the authors identified a FUSE-like sequence located 2.5 kb upstream of the transcription start site (TSS) of the human USP29 gene. Interestingly, the 54 bp USP29 FUSE-like sequence has a better matching score with the optimal FUBP1-binding site proposed by SELEX than the FUSE sequence found on MYC (Table 1) [43]. Parts of this sequence (up to 44 nucleotide-long) are retrieved several times in the human genome. In addition, in response to physiological stresses, FUBP1 cooperates with P38 (also called JTV1 or AIMP2) on USP29 promoter to upregulate its transcription [5].
Stathmins
Stathmin family includes 4 members: stathmin and stathmin like 2-3-4, encoded, respectively, by STMN1 (or OP18), STMN2 (SCG10), STMN3 (SCLIP), and STMN4 (RB3). These proteins are ubiquitous microtubule-destabilizing proteins, required for many cellular processes, such as cytoplasmic organization, cell motility, cell division, and cell adhesion [59, 60]. Consequently, stathmins overexpression are involved in tumor progression [61]. FUBP1 has been described to upregulate STMN1, STMN2, and STMN3 mRNA levels in non-small cell lung cancer (NSCLC) cells and at least STMN1 in HCC cells [6, 12, 62]. FUBP1 may participate in tumor metastasis through the regulation of stathmin family members. Interestingly, no FUSE-like sequence was found yet in the promoter (up to 2,000 bp) of human STMN1 and STMN3. Although several segments within the STMN1 promoter can be related with the optimal binding sequence for FUBP1, further studies are required to decipher their relevance.
Cyclins
The Cyclin D2 (CCND2) and in a lesser extent Cyclin D1 (CCND1) mRNA were both down-regulated in the absence of FUBP1 either in Hep3B HCC cell line or in adult long term-HSCs [8, 16]. Since Cyclin D1 and D2 are required for cell cycle G1/S transition, FUBP1 endorses a pro-proliferative function, consistently with MYC regulation.
Hematopoietic factors
FUBP1 also upregulates Hoxa10 mRNA in adult murine long-term HSCs [16]. HOXA10 is a member of the HOX gene family of transcription factors essential to the development of the embryo [63], and is implicated in hematopoietic differentiation and proliferation [64]. Moreover, FUBP1 cooperates with the major hematopoietic regulator RUNX1 (also named AML1) to activate the oncogene c-KIT by binding two regulatory regions, at + 700 bp and + 30 kb within the first intron of c-KIT [44]. c-KIT is implicated in cell growth control and stimulation of proliferation of HSCs and early committed hematopoietic lineage cells [65, 66]. In the human pre-B cell line Nalm6, the overexpression of FUBP1 and RUNX1 upregulates c-KIT mRNA and protein levels and amplifies its signaling pathway, contributing to cell proliferation and resistance to the c-KIT inhibitor imatinib mesylate. Using molecular biology and structural modelling, a FUSE-like sequence on the + 30 kb enhancer of c-KIT has been identified (Table 1) [44].
Transcriptional repression of target genes
Cyclin-dependent kinase inhibitors
FUBP1 binds to P21 (CDKN1A) gene to repress its transcription [8, 16, 32]. P21 is a well-known inhibitor of cell cycle that can arrest cell cycle progression in G1/S and G2/M transitions by inhibiting CDK4-6/cyclin-D and CDK2/cyclin-E, respectively [67]. P21 is also implicated in DNA repair and apoptosis inhibition [68, 69]. Rabenhorst et al. found four potential FUBP1-binding sites within the human P21 promoter (2.7 kb upstream of its TSS) [8]. In particular, a GT-rich sequence (50 nucleotide-long) was identified as essential for FUBP1 binding using an EMSA in Hep3B cells (Table 1). The binding of FUBP1 on this sequence was confirmed later by ChIP-qPCR in HeLa and Nalm6 cells [32, 44]. The depletion of FUBP1 upregulates P21 mRNA level in various cell types, such as normal human fibroblasts or human Hep3B, HeLa, MCF7, and H1299 cell lines [32]. However, in clear cell renal cell carcinoma (CCRCC) 786-O and caki-1 cells, FUBP1 mRNA was positively correlated with P21 mRNA and tumor growth [10]. In this case, the authors demonstrated that the upregulation of P21 mRNA level by FUBP1 resulted in apoptosis inhibition. This result is in line with proliferative and anti-apoptotic functions of FUBP1.
Two other cyclin-dependent kinase inhibitor proteins are regulated by FUBP1. The cell-cycle inhibitors P15 (CDKN2B) and P27 (CDKN1B) were both upregulated in the absence of FUBP1, respectively, in HCC cells and adult murine long-term HSCs [8, 16].
Proapoptotic factors
FUBP1 represses mRNA level of proapoptotic proteins such as NOXA, BIK (BCL2 Interacting Killer), TNFA (Tumor Necrosis Factor-Alpha), and TRAIL (Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand) in Hep3B cells [7]. The depletion of FUBP1 upregulates consistently Noxa mRNA in adult mouse long-term HSCs. A FUSE-like binding region was identified located at − 1.2 kb upstream the TSS in BIK promoter and concentration-dependent binding of FUBP1 to this site was validated [11]. This sequence encompasses GT-rich stretch consistently with P21 FUSE-like sequence and with the optimal sequence uncovered by SELEX (Table 1). By repressing these death-promoting proteins, FUBP1 supports cell survival.
Altogether, the normal transcriptional function of FUBP1 is characterized by the stimulation of cell proliferation, survival, and migration.
Regulation of viral replication
Indirect transcriptional repression: example with p53
During stress condition, FUBP1 binds directly to the DNA-binding domain of the tumor suppressor p53 to inhibit its recruitment to target promoters [70–72]. The protein p53 is involved in multiple central cellular processes, including DNA repair, genomic stability, senescence, cell cycle control, and apoptosis. Following hepatitis C virus (HCV) infection, FUBP1 inhibits p53 transactivation activity and regulates BCCIP and TCTP mRNA and protein, which are positive and negative regulators of p53, respectively [71]. Given that p53 is involved in cellular host cell defense during viral infection, by inhibiting p53, FUBP1 promotes viral expansion. Following human adenovirus infection, FUBP1-p53 complex is stabilized by the Early 1A (E1A) protein, inhibiting the binding of p53-regulated promoters. During adenovirus infection, Frost et al. showed that the depletion of FUBP1 leads to enhanced p53 promoter occupancy on several target genes, such as P21, GADD45A, and PIG3, followed by enhanced promoter histone acetylation and enhanced gene expression compared to control cells [72]. Indeed, FUBP1 interacts directly with E1A protein. E1A is the first gene transcribed after infection of human adenovirus and the protein encoded plays a pivotal role in the viral replication process of adenovirus. E1A interacts with large variety of protein, such as FUBP1, to reprogram host cell gene expression and induce S-phase. Consequently, FUBP1 indirectly prevents the p53 stress response pathway that would block viral replication.
FUBP1 is a multifunctional RNA-binding protein
Characterization of the FUBP1 RNA-binding mechanism
FUBP1 is implicated in different processes in RNA regulation such as post-transcriptional regulation, splicing, or viral replication. Even if FUBP1 is almost exclusively in the nucleus, a portion is present in the cytosol, enabling regulation of cytoplasmic RNA [47, 73, 74].
FUBP1 was described to bind AU-rich elements (AREs) via its KH subdomains. AREs are RNA cis-regulatory elements, 50–150 nucleotide-long, and usually include many copies of the AUUUA pentamer or UUAUUUAUU nonamer [75, 76]. AREs are present in 3′UTRs of many short-lived mRNA species. ARE-binding proteins such as FUBP1 stabilize or destabilize mRNA depending on the cellular context. Like FUBP1, FUBP2 is also known to interact with AREs to facilitate mRNA degradation [77]. However, other studies indicate that FUBP1 could bind GU-rich sequences, consistently with the GT-rich optimal sequence identified by SELEX [43, 78, 79]. Current knowledge about the FUBP1 RNA-binding sequences is described in Table 1. As it was reported for ssDNA-binding sequences, AU- and GU-rich stretches are retrieved, strengthening the sequence specificity of FUBP1 binding.
Translational regulation
Regulation of mRNA stability is a crucial step in post-transcriptional regulation of gene expression. It seems that FUBP1 exhibits a dual role as it both favors mRNA decay by binding in 3′UTR and promotes mRNA stabilization by binding to the 5′UTR. Furthermore, FUBP1 binds directly to the eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) strengthening its link to the translation initiation complex machinery [80].
3′UTR binding
FUBP1 interacts specifically with the 3′UTR of nucleophosmin (NPM), polycystic kidney disease 2 (PKD2), and growth-associated protein 43 (GAP43) mRNA to repress their translation [80–82].
NPM is an oncoprotein or a tumor suppressor implicated in different pathways such as ribosome biogenesis, chromatin remodelling, mRNA transport, DNA repair, embryogenesis, apoptosis, and cellular growth [83]. Through the direct repression of NPM translation, FUBP1 decreases cell proliferation [82]. The specific FUBP1-RNA-binding motif on NPM 3′UTR is not described yet.
PKD2 is a receptor forming a complex with PKD1 involved in calcium transport and calcium signaling in renal epithelial cells. FUBP1 has no effect on the PKD2 mRNA level, but suppresses its translation by binding to the 3′UTR (nucleotides 691–1044). This highly conserved region contains 68% of AU and two AREs (AUUUA) motifs in the human PKD2 gene (NM_000297.3) [80]. In zebrafish studies, fubp1 seems to regulate pkd2-associated disease phenotypes (tail curling and pronephric cyst) through the regulation of pkd2 protein level [80].
GAP43 encodes a membrane phosphoprotein crucial for axonal growth and formation of neural connections. FUBP1 induces GAP43 mRNA degradation via its binding on a 26 nucleotide-long pyrimidine-rich (CU-rich) sequence downstream of the end of the coding region (Table 1) [81]. Thus, FUBP1 may participate in neural development through GAP43 mRNA degradation. This result is consistent with another study, demonstrating that the transactivation domain of FUBP1 interacts with the survival motor neuron (SMN) protein in neurons [84]. Since FUBP1 and SMN both are highly expressed in developing neuronal tissue, the interaction between both factors might be critical for motor neuron development.
Moreover, FUBP1 was described to bind on the 3′UTR of COX2 mRNA, on a 76 nucleotide-long mRNA sequence that contains 6 AREs elements (Table 1). Even if FUBP1 specifically binds on this sequence, its binding was unaffected by inactivating mutations [73]. To ascertain the relevance of FUBP1 binding on the 3′UTR of COX2, further studies will be necessary.
5′UTR binding
FUBP1 binds the internal ribosome entry site (IRES) on the 5′UTR of P27 and activates its translation in human breast cancer MCF7 cells [85]. FUBP1 binds the 8 nucleotides 5′-GCGAAGAG-3′ upstream the codon start of P27 through its central domain. Moreover, the N-terminal domain of FUBP1 is important for P27 translational activation. Interestingly, this short sequence is neither AU, CU nor GU-rich. The authors proposed that it is more likely to be a structural element or a spacer between two KH contact sites, but the exact binding sites of the KH FUBP1-subdomains remain unknown [85].
Regulation of viral replication
FUBP1 activates the effective viral replication of HCV and enterovirus 71 (EV71), two positive single-stranded RNA viruses belonging to the Flaviviridae and Picornaviridae families, respectively [74, 86–88].
In an HCC cell line following HCV infection, FUBP1 binds a poly(U/UC) region within the 3′UTR of the HCV genomic RNA, a regulatory region required for HCV replication and infectivity [74]. The binding on this pyrimidine-rich region was consistent with the literature [41]. FUBP1 also interacts with HCV nonstructural viral proteins NS5A and NS5B, facilitating the HCV replication [71, 74]. Moreover, FUBP1 is essential for HCV replication in Huh7.5 cells interacting with p53, leading to the inhibition of the p53-mediated apoptosis, as mentioned in section I-2D [71].
FUBP1 binds on the IRES within the 5′ UTR of EV71 and enhances viral translation and viral growth in various cell lysates [74, 87, 88]. FUBP1 binds an AU-rich linker region (nucleotides 686 to 714), identified by EMSA (Table 1) [86, 88]. Following EV71 infection, FUBP1 is recruited to the 5′UTR IRES, and enhanced viral translation. As translation increases, the viral proteinase 2A cleaves FUBP1 to generate FUBP11−371 and FUBP1372−644 products [88]. The FUBP11−371 is a functional product that lacks the KH4 subdomain and the C-terminal domain. The authors have demonstrated that FUBP11−371 on its own is also able to promote EV71 translation by binding to the 5′UTR linker region located just upstream FUBP1 full-length binding site (nucleotides 656–674) [88]. In addition, FUBP2 and FUBP3 were also described to bind the IRES element of the EV71 5′UTR. Surprisingly, FUBP2 acts as negative regulator of EV71 viral translation, whereas FUBP3 serves as a positive regulator of EV71 replication-like FUBP1 [87–89]. Because all FUBP members are implicated in the viral recruitment and modulation of EV71, it is reasonable to assume that there is a common role between the FUBP members during viral infection.
On the other hand, FUBP1 is described to act as a negative regulator of the Japanese encephalitis virus (JEV), a single-stranded positive-sense RNA, and a member of Flaviviridae family and that is the leading cause of viral encephalitis in Asia. FUBP1 appeared to suppress JEV protein translation through its interaction with both the 5′ and the 3′ UTRs in neuroblastoma N18, NT2, and HeLa infected cells [28]. Thus, FUBP1 may participate in host antiviral defense response.
Therefore, FUBP1 may function as a negative or positive host factor for different members of the Flaviviridae family, such as JEV and HCV. It would be interesting to further investigate whether FUBP1 regulates viral functions of other RNA viruses.
Splicing regulation
Although FUBP2 and the drosophila FUBP1-ortholog Psi have been previously linked to splicing regulation for many years [90, 91] and the three FUBP family members have been identified by mass spectrometric analysis as spliceosome-associated factors [92], the emerging role of FUBP1 in splicing regulation has been only recently proven. Alternative splicing is a major source of genetic diversity in eukaryotes and implies 95% of human multi-exon genes [93]. FUBP1 can undergo exonic inclusion or exclusion based on its binding position on cis-acting splicing regulatory elements.
FUBP1 has been described to bind an exonic splicing silencer on exon 10 of the cardiac protein triadin to induce exon skipping [94]. Splicing process consists of two consecutive reactions before the release of the intron. FUBP1 induced a splicing repression during the second-step reaction, where normally, an –OH group at the 3′ end of the exon attacks the phosphodiester bond at the 3′ splice site yielding ligated exons [95]. FUBP1 blocks protein factors required for the second step of splicing and prevents exon ligation by binding an AU-rich sequence located on the exon 10 of triadin (Table 1) [94]. In this study, the splicing patterns of 51 transcripts regulated by other AU-rich binding proteins or related to cancer were examined under depletion or overexpression of FUBP1. Among them, FUBP1 decreases inclusion of exon 14 of the pre-mRNA encoding ACLY and exons 4–7 of the caspase 9 pre-mRNA, while FUBP1 facilitates inclusion of PTBP2 exon 10 and ENAH/MENA exon 11 [94]. Moreover, FUBP1, together with FUBP2, are splicing activators of SMN2, and lead to a shorter and nonfunctional SMN protein [96].
FUBP1 is also a positive splicing regulatory factor of MDM2 [78]. MDM2 is an oncogene that ubiquitinates p53 for degradation [97]. FUBP1 regulates MDM2 in minigene experiments containing exons 3-11-12 through the direct binding to two intronic regions, one upstream and the other one downstream of the exon 11 of MDM2. The upstream region does not reveal any specific FUBP1 consensus-binding motif, while the downstream-binding site contains a 120 nucleotide-long sequence enriched in AU residues [78]. The scrutiny of the 16 nucleotide-long specifically bound by FUBP1 revealed a GU-rich motif (Table 1). Therefore, FUBP1 may bind simultaneously the two identified intronic regions across exon 11 to facilitate better recruitment of the spliceosome or positive regulatory factors while masking or competing with negative regulatory factors [78]. The FUBP1-bound motif enriched in GU stretch is also retrieved in the splicing regulation of DMD gene by FUBP1 (Table 1) [79]. It has been shown that FUBP1 promotes the normal inclusion of exon 39 during splicing by binding an intronic splicing enhancer element in the intron 38, located about 80 bp upstream of DMD exon 39 [79]. Deregulation of the DMD gene, encoding the dystrophin, causes Duchenne or Becker muscular dystrophies, or cardiomyopathy [98]. Deregulation of FUBP1 could disrupt the proper alternative splicing of DMD and thus may modify the clinical phenotype of DMD-related pathologies. Recently, FUBP1 has been identified as a cofactor of the splicing factor U2AF2 (or U2AF65), a core component of the spliceosome [99]. On specific pre-mRNA targets, FUBP1 stabilizes U2AF2 bindings to control alternative splicing. For example, FUBP1 promotes exon 10 inclusion of PTBP2, by binding at least a distal intronic site, enhancing the binding of U2AF2 at this site [99]. FUBP1 favors also exon 8a inclusion of neuron-specific LSD1 promoting terminal neuronal differentiation [100].
Given the multiple molecular functions endorsed by FUBP1 (Fig. 2), and their impact on cell proliferation, migration, and survival, the role of FUBP1 in cancer must be discussed.
Fig. 2.
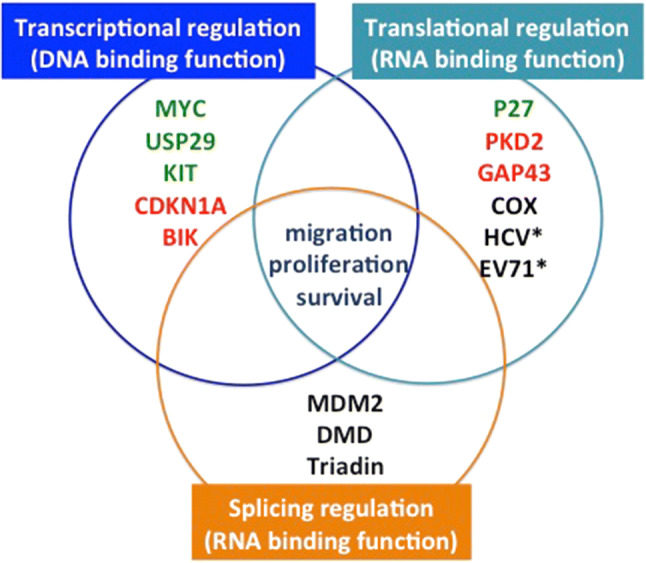
Scheme of the multiple functions and target genes of FUBP1 categorized between its role in the regulation of transcription, translation, and splicing. The list of genes is indicative and not exhaustive. Red: transcriptional repression, RNA degradation, translation repression; Green: transcriptional activation, translational activation. * are viruses
FUBP1 is implicated in cancer development.
FUBP1 as a tumor suppressor
FUBP1 loss-of-function
Several studies have highlighted in recent years new somatic mutations in FUBP1 gene in some subgroups of lower grade glioma (grades II and III). Gliomas are the most frequent cancers of the central nervous system. Lower grade gliomas originate either from the oligodendrocytes (oligodendrogliomas), astrocytes (astrocytoma), or a mix of these cell types (oligoastrocytomas) [101]. Twenty percent of brain tumors in adults are oligodendrogliomas. This subtype carries, in 70% of the cases, a chromosome translocation t(1;19) generating separated or combined allelic losses on 1p and 19q [102]. The combined deletion of chromosomes 1p and 19q in oligodendrogliomas results in monoallelic loss of FUBP1 gene on chromosome 1p (Table 2) and CIC gene on 19q, another important factor in oligodendrogliomas [13, 103]. In addition, FUBP1 mutations have been detected in 15–20% of oligodendrogliomas and 8% of oligoastrocytomas subtypes, especially in those with CIC mutation (23%) [13, 103, 104]. Although FUBP1 mutation is frequently observed with CIC mutation, the possible relationship between FUBP1 and CIC remains to be elucidated. Importantly, all FUBP1 mutations in lower grade gliomas inactivate their encoded proteins, as they alter splicing sites, produce stop codons, or generate out-of-frame insertions or deletions [13, 103–105]. No “hot-spot” mutation has been identified. Moreover, FUBP1 mutations in oligodendrogliomas are significantly and exclusively associated with two subtypes, the IDH1 mutation and the 1p/19q codeletion [13, 104, 105]. Therefore, in the 1p/19q codeletion, the deletion of FUBP1 is often associated with an inactivating mutation of the other allele, resulting in the complete loss of FUBP1 expression. Lower grade gliomas harbor intratumoral heterogeneity. Using whole-exome sequencing and targeted deep sequencing of multiple regional and temporal sampling, Suzuki et al. demonstrated the evidence of a clonal architecture of the mutations acquired in these tumors. They have highlighted ten major driving alterations (including IDH1 mutations and 1p/19q codeletion), and additional mutations such as FUBP1 and CIC mutations do not appear to be essential for establishment of oligodendrogliomas [106]. Moreover, mutations in FUBP1 are maintained from the primary tumor to recurrent oligodendroglioma or are newly acquired at recurrence, suggesting that these FUBP1 mutations confer a selective growth advantage during tumor clonal evolution [107]. Accordingly, FUBP1 mutations in these tumors do not seem to represent a founder event, but are more likely associated with maintenance and expansion of these cancer cells. Furthermore, FUBP1 loss-of-function was linked with unfavorable progression-free survival and overall survival especially in tumors carrying 1p/19q codeletion [104]. Altogether these data, in addition to the recent comprehensive analysis of oncogenic driver genes and mutations in > 9000 tumors across 33 cancer types by The Cancer Genome Atlas (TCGA), propose FUBP1 as a tumor suppressor in lower grade gliomas [108].
Table 2.
Summary of the current literature on FUBP1 deregulation in solid cancers
| Cancer | Deregulation | Downstream targets | Phenotypic effect | References |
|---|---|---|---|---|
| Oligodendrogliomas | Inactivating mutations of FUBP1 | NA | [9, 13, 103–106, 110, 159–162] | |
| Gliomas (other subtypes) | FUBP1 ↑ | MYC ↑ | G1/S cell cycle transition ↑ | [15] |
| Hepatocellular carcinoma | FUBP1 ↑ | MYC ↑ | Proliferation ↓ | [126] |
| Stathmin 1/3 ↑ | Migration ↑ | [6] | ||
| P21 ↓, P15 ↓, TNF-α ↓, TRAIL ↓, BIK ↓, NOXA ↓, CCND2 ↑ | Proliferation ↑, apoptosis ↓ | [8] | ||
| PE2 upregulates FUBP1 | Proliferation ↑, apoptosis ↓ | [163] | ||
|
P53 ↓, P21 ↓, BCCIP ↓, TCTP ↑ |
Cell sensitivity to irradiation ↓ | [70] | ||
| FIR upregulates FUBP1 | [7] | |||
| P21 ↓, BIK ↓, CCND2 ↑, TCTP ↑ | [11] | |||
| PI3K/AKT/mTOR upregulate FUBP1 | [164] | |||
| Gastric cancer | FUBP1 ↑ | NA | [35, 165] | |
| FUBP1 ↑ | MYC ↑ | Tumor growth ↑ | [34] | |
| Colorectal carcinoma | FIR ↑ and FIR splice variant: FIR del/exon2 | Absence of MYC repression | Resistance to apoptosis ↑, tumor growth ↑ | [56, 134] |
| FUBP1 ↑ | NA | [166] | ||
| Breast cancer | FUBP1 ↑ | MYC ↑ | Proliferation ↑, tumor growth ↑ | [34] |
| Clear cell renal cancer | FUBP1 ↑ | MYC ↑ | [24] | |
| MYC ↑, P21 ↑ | Proliferation ↑, apoptosis ↓ | [10] | ||
| Bladder cancer | FUBP1 ↑ | NA (not MYC) | [24] | |
| Ovarian cancer cells | FUBP1 ↑ | NA | [167] | |
| NA | Resistance ↑ | [117, 118] | ||
| Prostate cancer | FUBP1 ↑ | NA (not MYC) | [24] | |
| Nasopharyngeal carcinoma | FUBP1 ↑ | MYC ↑ | Proliferation ↑, colony formation ↑ and resistance ↑ | [116] |
| MYC | Resistance ↑ | [168] | ||
| Esophageal squamous cell carcinoma | FUBP1 ↑ | MYC ↑ | Proliferation ↑, G1/S cell cycle transition ↑ | [14] |
| Non-small cell lung cancer | FUBP1 ↑ | Stathmin 1/3 ↑ | Proliferation ↑, migration ↑ and matrix invasion ↑ | [12] |
| FIR ↑ FUBP1 | [130] | |||
| Sacral chordomas | FUBP1 ↑ | MYC ↑ | Proliferation ↑, invasion ↑ | [127] |
NA not available
Another aspect that remains largely unexplored is the potential function of FUBP1 splice variants. Yip et al. uncovered two FUBP1 infrequent somatic splice variant sites that could play a role in oligodendroglioma development [109]. Moreover, FUBP1 seems to be a tumor suppressor in human neuroblastoma, as higher expression correlates with better survival in all stages [110]. No recurrent mutations in FUBP1 have been described in neuroblastoma tumors so far. However, FUBP1 alternative splice variants are associated with patient survival [110]. First, a specific NAGNAG splice site was identified within FUBP1 exon 5 [111]. This splicing event can incorporate a serine at position 97 in the full-length wild-type FUBP1 isoform or can produce an isoform lacking this serine at position 97 (FUBP197−) [110]. Interestingly, FUBP197− increases MYC protein level much more than FUBP1 does, and a higher FUBP197− expression correlates with a worst event-free survival in MYCN-non-amplified neuroblastomas. Conversely, neuroblastoma patient survival is better when FUBP1 is higher expressed than FUBP197−. These data suggest that FUBP1 mutations observed in oligodendrogliomas are maybe not restricted to loss-of-function, but may compete with FUBP197− in a dominant-negative way [110].
Does splicing regulation by FUBP1 trigger tumor suppressor function?
First evidence for the role of splicing regulation by FUBP1 in cancer development came from the study of MDM2 pre-mRNA splicing. Indeed, splice patterns of MDM2 triggered by FUBP1 may be associated with the pathological p53 pathway [97]. Alternative splicing of MDM2 is a hallmark of several cancer types [112]. Interestingly, the majority of these alternative variants lack the p53-interaction domain, notably the MDM2-B (ALT1) isoform that is the most frequently expressed in human cancers. Knockdown of FUBP1 in HeLa cells induced the formation of this alternative isoform MDM2-B (skipping of internal exons 4 through 11), a stress-induced splice variant of MDM2, even under normal conditions [78]. Since MDM2-B triggers cell growth and tumorigenesis [113, 114], it is reasonable to assume that FUBP1 acts as a tumor suppressor by triggering the proper splicing of MDM2. More recently, analysis of whole-exome sequencing data across 33 tumor types from TCGA database identified 119 splicing factor genes, including FUBP1, with significant non-silent mutation patterns. This study demonstrated that FUBP1 loss-of-function mutation is associated with cassette exon events in low-grade gliomas [115]. Still, in low-grade gliomas, Hwang et al. demonstrated the role of FUBP1 in LSD1 + 8a isoform, establishing a direct role for FUBP1 in neuronal differentiation and explaining its tumor-suppressor function in the nervous system [100].
FUBP1 as an oncoprotein
Deregulation of FUBP1 is documented in a large diversity of neoplastic diseases. FUBP1 is an important oncoprotein overexpressed in a variety of malignancies, including HCC, NSCLC, breast cancer, CCRCC, colorectal carcinoma, glioma, bladder cancer, prostate cancer, gastric cancer, colorectal carcinoma, osteosarcoma, esophageal squamous cell carcinoma, ovarian cancer, nasopharyngeal carcinoma, and leukemia (Table 2 and Fig. 3).
Fig. 3.
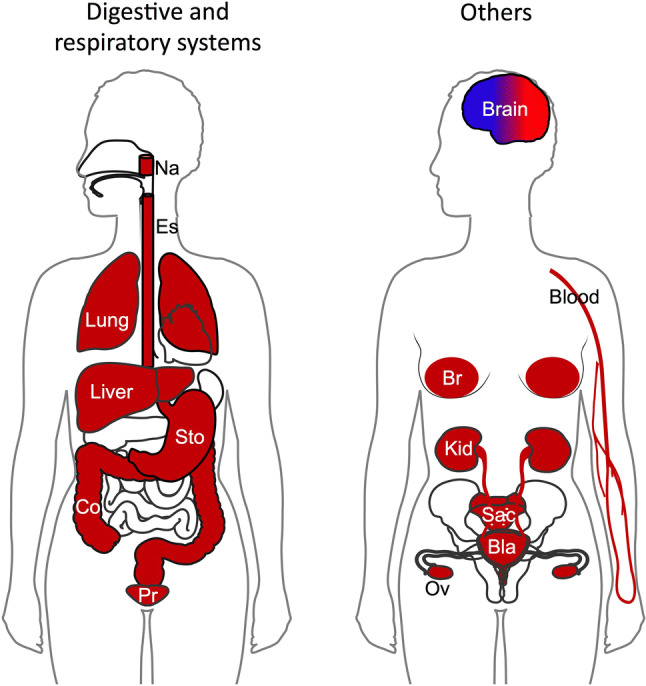
Illustration of the deregulation of FUBP1 in human cancers. Tissues in red represent the cancer-related studies where FUBP1 have been identified upregulated and act as an oncoprotein. Tissue in blue represents the cancer-related studies where FUBP1 is described as a tumor suppressor. Bla bladder, Br breast, Co colon, Es esophage, Kid kidney, Na nasopharynx, Ov ovary, Pr prostate, Sac sacrum, Sto stomach
FUBP1 may be considered as a cancer biomarker because its overexpression is often correlated with poor overall survival or lower event-free survival patient prognosis, such as in HCC, glioma, gastric cancer, esophageal squamous cell carcinoma, ovarian cancer, and nasopharyngeal carcinoma [1, 7, 14, 15, 34, 116, 117]. Moreover, FUBP1 overexpression correlates with an increase in chemotherapy and/or radiotherapy resistances in nasopharyngeal carcinoma, ovarian cancer and in the Hu7 HCC cells [70, 116–118]. FUBP1 expression can also be correlated with tumor size and stage in ovarian and CCRCC [10, 118] in contrast to gastric cancers [34]. Several mechanisms linking FUBP1 to tumorigenesis have been described, such as inappropriate expression of its target genes, deregulation of the FIR–FUBP1 system, or viral induction. Hence, the pharmacological inhibition of FUBP1 might represent a promising therapeutic strategy for patients with elevated FUBP1 expression. An overview of the FUBP1 inhibitors is described in Table 3.
Table 3.
Description of FUBP1-inhibitors (top of the list) and activator (last in the list) identified so far
| Compound | Chemical structure | Mechanism | Effect on FUBP1 target genes | IC50 |
|---|---|---|---|---|
| Benzoyl anthranilic Acid [150] |
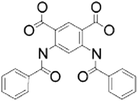
|
Interacts with the hydrophobic region of the KH domain of FUBP1, leading to an impaired DNA-binding | ↓ Binding on MYC | 350 μM |
| Pyrazolo[1,5a] pyrimidine [11] |
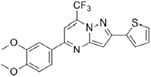
|
Inhibits or prevents the binding between FUBP1 and the FUSE |
↓ Binding on P21 and ↑ P21 mRNA ↓ Binding on BIK and ↑ BIK mRNA ↓ CCND mRNA in Hep3B cells |
11–24 μM depending on assays |
| GSK343 [128] |
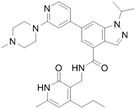
|
Competitive inhibitor | ↓ MYC protein level in Saos-2 cells | Not available (4 nM for EZH2) |
| Camptothecin (CPT) [156] |
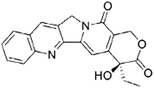
|
Interferes with the transcriptional control of FUBP1 |
↑ P21 mRNA ↑ BIK mRNA ↓ CCND2 mRNA ↓ TCTP mRNA in Hep3B and HepG2 cells. |
3.2 ± 0.6 mM |
| 7-ethyl-10-hydroxycamptothecin (SN-38) [156] |
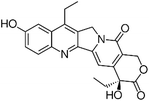
|
Interferes with the transcriptional control of FUBP1 |
↑ P21 mRNA ↑ BIK mRNA ↓ CCND2 mRNA ↓ TCTP mRNA in Hep3B and HepG2 cells. |
0.78 ± 0.2 mM - 1.9 ± 0.7 mM depending on assays |
| ACTIVATOR: pyrido-pyrimidinone SMN-C2 and SMN-C3 [144] |
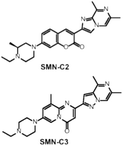
|
Binds to the AGGAAG motif on exon 7 of the SMN2 pre-mRNA Creates a new functional binding surface with FUBP1 |
↑ SMN2 splicing | EC50: 100 nM |
Inappropriate expression of FUBP1 target genes
FUBP1 overexpression is involved in tumor initiation and progression through regulation of key players in apoptosis, cell cycle, or cell motility. The role of FUBP1 in cancer development has been studies in HCC. HCC represents the most common primary liver cancer and the third leading cause of cancer mortality [119, 120]. Importantly, FUBP1 is overexpressed in more than 80% of human HCC compared to the normal liver and correlates with poor patient prognosis [6, 8]. MYC is a major oncogene in HCC, since MYC overexpression in hepatic cells leads to development of 30-60% of HCC [121–123]. One mechanism explaining MYC activation is its locus amplification in the chromosomal amplification of 8q24 [124, 125]. In addition, transcriptional activation by FUBP1 overexpression could be another mechanism supporting MYC up-regulation in HCC [126]. However, some studies did not detect significant correlation between FUBP1 and MYC expression, as FUBP1 knockdown does not influence MYC mRNA and protein levels in Hep3B cells [6, 8], but this could be explained by various MYC expression depending on HCC subtypes.
Other studies showed that FUBP1 overexpression was correlated with elevated MYC expression in breast cancer, CCRCC, gastric cancer, gliomas, esophageal squamous cell carcinoma, sacral chordomas, and some nasopharyngeal carcinoma, suggesting a powerful oncogenic role of FUBP1 in these cancers through MYC upregulation (Table 2) [10, 14, 34, 58, 126, 127]. On the contrary, FUBP1 overexpression was not associated with MYC level in bladder and prostate cancers also, even if MYC plays an important role in tumor progression in these cancers [24]. Elevated level of MYC in these cases might rather be due to MYC copy number amplification than a consequence of FUBP1 overexpression. Hence, the participation of FUBP1 in prostate and bladder cancers is still unknown, but suggests that FUBP1 regulates other target genes responsible for its oncogenic function.
The oncogenic effect of FUBP1 is, therefore, not always related to MYC deregulation. FUBP1 also promotes cancer cell proliferation by the transcriptional repression of cell cycle inhibitors P21, P15 and the activation of the positive cell cycle regulator Cyclin D2 in HCC (Table 2) [8]. FUBP1 is also implicated in the inhibition of p53 tumor suppressive activity during stress condition [70–72]. FUBP1 prevents the DNA-binding activity of p53, interfering with the regulation of its target genes including P21. Thus, overexpression of FUBP1 may be involved in tumor progression by inhibiting p53 activity. In addition, FUBP1 promotes cancer cell survival by the inhibition of TNF-α, TRAIL, BIK, and NOXA expressions in HCC cells [8]. Moreover, overexpression of FUBP1 enhances cancer cell migration through activation of stathmin family members in HCC cell lines and human NSCLC cells [6, 12]. These regulations can be investigated in the other cancers where FUBP1 is overexpressed. For example, independent overexpressions of stathmins and FUBP1 have been reported in breast cancer and osteosarcoma and are associated with an increase of tumor cell proliferation, migration, and matrix invasion [34, 128, 129]. However, a direct correlation between these two oncogenes remains uncertain in those pathologies.
Deregulation of the FIR–FUBP1 system
An alteration in FIR–FUBP1 regulatory mechanism can lead to inappropriate MYC expression. FIR regulates negatively FUBP1 to restore the basal transcription state of FUBP1 target genes [54]. An overexpression of FIR should lead to a decrease of MYC level, and conversely, an overexpression of FUBP1 should predict a low level of FIR. However, in cancers like HCC and NSCLC, an overexpression of both FIR, FUBP1, and MYC were observed compared to normal tissues [7, 12, 126, 130]. It seems worthy to investigate the oncogenic role of FUBP1 through the analysis of the all FIR/FUSE/FUBP1 system. First, a reason that could explain the upregulation of FIR is the frequent high copy number amplification of chromosome 8q22.1–24.3 in human cancers including breast, prostate, bladder, colon, lung, ovaries, pancreas, and brain cancers. Importantly, it is one of the most prominent genomic gains either in HCC (47%) or in NSCLC (41–44%) [131–133]. Interestingly, this frequent genomic gain includes both FIR locus 8q24.3 and MYC locus 8q24.21. Surprisingly, in most primary human HCC and NSCLC cells, FIR overexpression does not inhibit FUBP1 activity, but positively regulates its transcription through the induction of the transcription factor DP-1 (TFDP1), supporting cell proliferation and migration. Therefore, FIR can lose its suppressing activity on FUBP1 in tumor cells and gain tumorigenic properties by activating FUBP1 and subsequent MYC levels [7, 130]. One hypothesis that could explain this abnormal regulation is the presence of FIR alternative variants, which may promote tumor development by disabling full-length FIR to repress MYC [56]. Indeed, expression of different FIR splice variants, especially one lacking exon 2 (FIRΔexon2), is expressed in most human colorectal cancer, HCC and NSCLC cells in contrast to adjacent normal tissues [7, 56, 130, 134].
Viral induction
As another suggested mode of oncogenic action, FUBP1 may contribute to oncogenesis by promoting viral replication. For instance, FUBP1 enhances the replication of HCV [70, 74], a virus which constitutes a major cause of chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma worldwide [135, 136].
FUBP1 as a master regulator of hematopoiesis
Phenotypic effects of FUBP1 depletion in hematopoiesis
The phenotype of mice lacking Fubp1 expression was recently investigated. Murine Fubp1 gene was inactivated by gene-trapping [16] or gene-targeting by cre/loxP system (Fubp1 knockout, KO) [17] technologies. Heterozygous Fubp1+/− mice appeared to be normal, with no apparent anatomic or microscopic features [17]. However, the homozygous Fubp1−/− embryos die in utero at around E15.5 (ranging from E10.5 to birth) with severe anemia, suggesting an essential role for normal development of embryos [16, 17]. Fubp1−/− KO embryos are pale, puny, and thinner (20% reduction of the weight) compared to normal embryos at the same age, and have multiple morphological dysfunctions concerning different organs including hypoplastic thymus, spleen, and lungs, a hypertrophy of the cardiac ventricular wall, abnormalities in the central nervous system, and a poorly developed placenta, which is a main hematopoietic organ in early mouse development [17]. Nevertheless, the morphologic variation among the KO embryos was surprisingly broad, ranging from subtle to fulminant phenotypes, but correlated with the survival rate discrepancy. The anemic phenotype and the hypoplastic lymphoid and placental tissues clearly indicate a dysfunction in hematopoiesis. With the Fubp1−/− gene trap mice, a severe reduction in fetal and adult long-term HSCs was observed [16] that was not identified with the Fubp1 KO model [17]. Moreover, in a competitive transplantation experiment, the Fubp1-deficient long-term HSCs were not able to repopulate bone marrow and peripheral blood of irradiated mice compared to control cells. However, the multilineage differentiation was not affected by the absence of Fubp1 [16], although the functionality of the mature blood cells lacking Fubp1 was not investigated. In the KO Fubp1 experiments, a normal or even increased HSC numbers were observed, but HSCs were unable to produce the appropriate level of progenitors, suggesting that HSCs are well produced but not functional [17]. Altogether, these data showed that FUBP1 is a major regulator of hematopoiesis, in HSCs maintenance, expansion, and long-term blood reconstitution.
Molecular effects of FUBP1 depletion in hematopoiesis
To go further, the role of FUBP1 in hematopoiesis was investigated more deeply in FACS-isolated adult long-term HSCs depleted for Fubp1. In cells down-regulated for FUBP1, an increase in p21, Noxa, p27, Mds1, and Rac2 expression and a reduction in Fir, cyclin D2, Myc, and Hoxa10 were observed [16]. Functional inactivation of Fubp1 in long-term HSCs promotes cell cycle arrest and apoptosis, resulting in HSC ablation. The depletion of Fubp1 was also associated with a decrease of cells in S phase of the cell cycle and a prolonged generation time [16]. Moreover, because Fubp1 KO cells expressed a broader range of Myc level, FUBP1 may help the stabilization of Myc mRNA [17]. By the positive regulation of proliferative factors such as Cyclin D2 and Myc together with the repression of the proapoptotic protein Noxa and the cell cycle repressor p21, FUBP1 promotes long-term HSC proliferation and survival. Therefore, FUBP1 is essential for long-term HSC self-renewal and expansion. In the human pre-B cell line, Nalm6, FUBP1 depletion decreases the expression of c-KIT mRNA, with an opposite profile upon FUBP1 overexpression. By regulating the oncogene c-KIT in pre-B lymphoblasts, FUBP1 promotes cell proliferation in vitro and in vivo [44].
In addition, the role of FUBP1 in murine embryonic stem cells was also investigated using the CRISPR/Cas9 technology. Surprisingly, even if HSCs came from pluripotent embryonic stem cells, the absence of Fubp1 did not affect embryonic stem cell self-renewal or survival unlike to HSCs [137]. However, Fubp1 knockout significantly reduced the mRNA expression of mesoderm markers such as the Brachyury, Flk-1, SnaiI, FGFR1, and Bmp4 that are indispensable for mesodermal formation during early embryonic development. Ectoderm and endoderm marker expression was not altered in the absence of Fubp1 [137]. It was particularly interesting as all hematologic lineages came from mesoderm cells. The authors hypothesize that the impaired HSC self-renewal seen in Fubp1−/− gene trap mice [16] may be linked to this mesoderm differentiation defect. Moreover, the delayed mesoderm differentiation in Fubp1−/− embryonic bodies presents a reduced erythroid differentiation capacity. Altogether, these results further strengthen the implication of FUBP1 in HSC self-renewal and in erythroid differentiation (Table 4).
Table 4.
Summary of the current literature on FUBP1 in hematopoiesis and leukemia
| Cell types | Deregulation | Downstream targets | Phenotypic effect | References |
|---|---|---|---|---|
| Embryonic stem cells | Brachyury ↑, Flk-1 ↑, SnaiI ↑, FGFR1 ↑, Bmp4 ↑ | Mesodermal formation during early embryonic development | [137] | |
| HSCs | p21 ↓, Noxa ↓, p27 ↓, Mds1 ↓, Rac2 ↓, Fir ↑, cyclin D2 ↑, Myc ↑, Hoxa10 ↑ | Long-term HSC proliferation and survival (essential for HSC self-renewal) | [16, 17] | |
| Pre-B lymphoblasts | c-KIT ↑ | Cell proliferation | [44] | |
| Acute myeloid leukemia (AML) | FUBP1↑ | [143, 157] | ||
| Acute lymphoblastic leukemia (ALL) | FUBP1↑ | [157] | ||
| B-cell precursors ALL | FUBP1 SNV mutations | [148] | ||
| Chronic lymphocytic leukemia (CLL) | FUBP1 truncated protein | [147] | ||
| Mantle cell lymphoma following a CLL relapse | FUBP1 truncated protein | MYC ↑ | [149] |
FUBP1 expression and role in all hematopoietic lineages
Although FUBP1 is expressed in a wide variety of hematopoietic cell lines including lymphoid (such as Raji, BJAB, DHL4, DHLG, L428, Nalm6, REH and Jurkat) and myeloid cell lines (HL-60, U937, U266, and K562), the exact expression of FUBP1 across all lineages has not been fully elucidated so far. Recently, the presence of FUBP1 protein has been demonstrated in human CD34 + HSCs and hematopoietic progenitor cells such as multipotent progenitor, granulocyte macrophage progenitor, and multipotent lymphoid progenitor cells [44]. It appears that FUBP1 is less expressed in differentiated cells than progenitor cells, at least in the myeloid lineage [2, 19]. FUBP1 level is reduced in the human myeloblastic leukemia cell line HL-60 and in the human monoblastic cell line U-937 following induction of differentiation along monocytic or granulocytic pathways [2, 19]. Moreover, in those differentiated cells, the reduction of FUBP1 binding to MYC was well associated with a decrease in MYC expression [2]. This finding was important as the repression of MYC is required for the switch from HSC self-renewal to differentiation [138–140]. Subsequently, high MYC expression has been demonstrated to block the terminal differentiation into myeloid lineage [141, 142]. Since FUBP1 activates MYC transcription, the reduction of FUBP1 expression in differentiated cells may provide a hypothesis to explain how MYC is repressed during differentiation. Hence, by the fine-tuning of MYC, FUBP1 could be an important regulator that controls the switch between HSC self-renewal and differentiation especially in myeloid lineage. Otherwise, FUBP1 seems expressed in mature lymphocytes [19], suggesting that a different mechanism is involved in lymphoid differentiation.
Does FUBP1 play an oncogenic or tumor suppressor role in leukemia?
Due to its major role in hematopoiesis, the involvement of FUBP1 in blood cancers is an emerging area of research. Strong evidence that FUBP1 could have an oncogenic role in leukemia came from recent large-scale bioinformatic analyses of FUBP1 expression across hematologic cancers compared to various solid cancers. RNA-Seq-based transcriptome open-source data available via the Pediatric Cancer Data (St. Jude Children’s Research Hospital), TCGA Research Network, and the Cancer Cell Line Encyclopedia (CCLE) uncovered that FUBP1 mRNA is highly expressed in hematologic malignancies, including acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL), compared to other brain or solid cancers (Fig. 4). Moreover, FUBP1 mRNA levels were shown to be highly elevated in leukemia stem cell-enriched cell populations of AML patients compared to HSCs (Table 4) [143]. Hence, FUBP1 was identified as a potential leukemia stem cells-related gene profile signature (FRD = 0.065).
Fig. 4.

Overview of FUBP1 mRNA expression in different cancers. FUBP1 RNA-Seq-based transcriptome open-source data based upon data generated by a Pediatric Cancer Data (St. Jude Children’s Research Hospital) (https://pecan.stjude.cloud/home) [157]. b Cancer genome atlas (TCGA) research network: (http://cancergenome.nih.gov/) or c cancer cell line encyclopedia (CCLE) (https://portals.broadinstitute.org/ccle) [158]. These large-scale analyses reveal that FUBP1 is more overexpressed in AML and ALL compared to brain and solid cancers. FPKM fragments per kb per million reads. Numbers of patients are indicated within parentheses
Like in other cancers overexpressing FUBP1, a deregulation of its target genes can be predictable. This is supported by the fact that oncogenic functions of MYC in hematological malignancies are well characterized, although no related MYC deregulations are associated to FUBP1 upregulation so far. MYC is a major player in hematopoiesis, an overexpression is recurrently found in ALL and AML and is often associated with disease progression [144]. It is reasonable to hypothesize that the enhanced FUBP1 expression could be responsible for MYC mRNA overexpression in leukemia not related to chromosomal translocation or amplification of MYC. Another FUBP1 target well described in hematologic malignancies is c-KIT. c-KIT gene is frequently overexpressed in AML and more rarely in ALL [145], and constitutive c-KIT activity triggers B-ALL in mouse model [146]. Given that FUBP1 and RUNX1 overexpression in pre-B lymphoblasts triggers c-KIT oncogenic pathway activation and subsequent cell proliferation [44], it is very likely that FUBP1 deregulations could be implicated in the onset or the maintenance of some leukemic disorders.
In addition, a number of reports have identified mutations in FUBP1 that could have a functional impact on leukemogenesis (Fig. 1 and Table 4) [147–149]. DNA samples from 538 patients with chronic lymphocytic leukemia (CLL) were analyzed by whole-exome sequencing and revealed that FUBP1 might be a cancer driver gene in this leukemia subtype [147]. A gene is considered as a cancer candidate gene if it possesses a high non-synonymous mutation rate. Here, somatic mutations in FUBP1 gene were identified in 9 patients over the 538 (1.7%), enabling the discovery of 9 different mutations. The FUBP1 mutations observed in CLL are highly biased towards truncating events as the large majority are frameshift-inducing insertions and deletions and nonsenses mutations. Moreover, two deletion mutations in FUBP1 gene were enriched in the aggressive lymphoma (mantle cell lymphoma) following a CLL relapse. These two deletions located in the N-terminal domain cause frameshift, produce truncated proteins, and thus may have an impact on MYC regulation [149]. In B-cell precursors ALL, computational tools used for driver gene prediction also uncovered FUBP1 [148]. The mutations described in B-ALL are single-nucleotide variations (SNV) that still produce functional proteins (Fig. 1). Despite the low frequency of these FUBP1 mutations (3 patients over 182–1.6%), one SNV located in the C-terminal domain was predicted to be damaging. Thus, potentially cancer-driving mutations were newly identified in FUBP1, even if no hot spot appeared to emerge. However, the phenotypic effects of all these mutations remain to be elucidated.
Discussion and future perspectives
FUBP1 endorses several activities that play important roles in normal or pathological situations. How FUBP1 protein does manage all these diverse functions is an important question. First, FUBP1 can activate some genes and repress others. FUBP1 activation and repression domains might operate separately and independently. This dual role in gene regulation could be governed by cellular physiological conditions or molecular contexts that include chromatin organization or interacting partners like RUNX1 or p53. Identifying interacting partners that govern functional activity of FUBP1 is, therefore, an interesting goal for further studies.
Then, as transcription and RNA-processing events occur in the nucleus, whereas translation, and RNA stability or decay occur in the cytoplasm, FUBP1 DNA- or RNA-binding functions should depend on its subcellular localization. In other words, even if FUBP1 is almost exclusively nuclear, it seems coherent to assume that FUBP1 regulates mRNA translation or viral replication in the cytosol. According to the previous reports, extracellular signaling such as apoptotic signal induced by stress condition or viral infection triggers translocation of nuclear FUBP1 into the cytoplasm [28, 29]. Consequently, the cellular distribution of FUBP1 should be altered, enabling regulation of cytoplasmic RNA. FUBP1 protein can be involved in different processes in different contexts, even if they are contradictory. Nuclear FUBP1 promotes cell proliferation for example by activating MYC and repressing P21 transcriptions via DNA binding to their promoters, while cytoplasmic FUBP1 represses cell proliferation by activating P27 and repressing NPM translations via RNA-binding to untranslated regions. It is even more surprising, given that p27 (encoded by CDKN1B) and p21 (CDKN1A) both induce cell-cycle arrest and share a lot of functional characteristics. Therefore, it will be meaningful to determine how the switch between FUBP1 transcriptional and translational functions is orchestrated exactly and how the balance between FUBP1 different functions is regulated.
Even if an optimal DNA-binding sequence for FUBP1 has been identified [43], no strict consensus-binding site has been proposed. It is particularly interesting to see that FUBP1 protein can bind several nucleotides sequences according to its KH3 and KH4 subdomains properties [40, 43]. FUBP1 optimal sequence for RNA-binding is still unelucidated, although AU-rich and GU-rich sequences seem privileged by FUBP1, strengthening its binding sequence specificity. Although FUBP1 subdomains display different conformations during its binding on ssDNA or RNA [40], FUBP1 occupancies between ssDNA and RNA are consistent (Table 1). FUBP1 displays sequence preference for pyrimidine-rich sequences with a high T/U repetition rate. The requirement of the helicase activity of FUBP1 in its functions in transcription, translocation, splicing, and viral replication is still not fully demonstrated. In addition, the affinity of FUBP1 for either DNA versus RNA remains to be determined.
Another point of consideration is the potential cooperation of FUBP2 and FUBP3 with FUBP1. Strong evidence concerning this cooperation came from the common structural characteristics shared by the FUBP family members. FUBP members can functionally coordinate to regulate many targets [20]. For example FUBP1 and FUBP3 promote EV71 replication [87, 88], while FUBP2 represses EV71 viral translation [89]. In addition, FUBP2 downregulation correlated with an upregulation of FUBP1 expression in HCC cells [6], strengthening the interplay between FUBP members. Thus, it could be interesting to investigate FUBP1 regulation in the light of a potential partnership or compensation effect with FUBP2 and/or FUBP3.
FUBP1 deregulation is often retrieved in cancers. FUBP1 loss-of-function is associated with tumor suppressor role, while FUBP1 overexpression is associated with an oncogenic role strengthening the essential role of FUBP1 in the normal regulation of key cellular processes (Table 2). FUBP1 loss-of-function is triggered by chromosomal deletions and/or inactivating mutations. For now, lower grade gliomas are the sole cancers describing such FUBP1 phenotype. Through the regulation of an increasing number of targets, FUBP1 appears to be crucial for normal brain development. However, in contrast to oligodendroglioma, somatic mutations in FUBP1 do not always seem to be associated with tumor suppressor role as in leukemia [147–149]. Whether and how such FUBP1 mutations perturb the regulation of its target remains elusive.
While most studies support the idea that enhanced FUBP1 expression is often correlated with an overexpression of MYC in malignancies, FUBP1 deregulation in cancers is not systematically related to the transcriptional activation of the oncogene MYC. Even if FUBP1 is required for MYC maximal transcription and acts as sensor of MYC promoter activity, FUBP1 seems to combine several transcriptional effects in carcinogenesis. FUBP1 overexpression in cancer involves the regulation of an interaction network composed of either cell cycle regulator such as MYC, c-KIT, P15, P21, P27, Cyclin D1, and Cyclin D2, apoptotic regulators like NOXA, BIK, TNFa, and TRAIL, and cell division and cell motility regulators like stathmin members. Further studies will be needed to elucidate the functional relevance of FUBP1 in carcinogenesis.
FUBP1 is a novel key transcriptional regulator that regulates long-term HSC self-renewal and differentiation. While numerous studies reported deregulation in FUBP1 expression in solid cancers, hematological cancers are poorly studied. Large-scale analyses uncovered an overexpression of FUBP1 mRNA in some leukemia subtype and revealed potential cancer-driving mutations. Hence, the understanding of FUBP1 role in hematological malignancies represents a challenge due to its oncogenic capacities.
Overexpression of FUBP1 constitutes a typical feature of most human cancers and is often correlated with the clinical characteristics and prognosis of patients. It seems that FUBP1 expression could be considered as an independent prognostic factor in cancer. Because of its potent oncogenic capacities and its cancer signature profile, FUBP1 might be an important target for therapy. Small compounds have been described to activate FUBP1 binding. In particular, analogs of the pyrido-pyrimidinone RG-7916, SMN-C2 and SMN-C3 correct exon 7 splicing of SMN2 pre-mRNA by increasing the binding of the splicing modulators, FUBP1 and its homolog, FUBP2 [96]. On the contrary, small compounds that inhibit FUBP1 activity have also been identified (Table 3). Benzoyl anthranilic acid has been described to inhibit the DNA binding of FUBP1 by targeting its hydrophobic ssDNA-binding pocket [150]. Later, the pyrazolo[1,5a] pyrimidine molecule has been described, even if it displays a more reasonable IC50 and a better solubility compared to the benzoyl anthranilic acid [11], clinical use of this molecule seems arduous because of the high IC50. The GSK343 was also described to inhibit FUBP1 expression [128], although off-targets are predictable, as it is a major inhibitor of the H3K27 methyltransferase EZH2. GSK343 could be an interesting inhibitor in specific cancers, where FUBP1 and EZH2 are upregulated such as liver cancer [6, 126, 151], osteosarcoma [152, 153], prostate cancer [24, 154], and breast cancer [34, 155]. Moreover, a novel screen in the FDA-approved drug library has allowed to identify two FUBP1 inhibitors: the camptothecin and its derivative 7-ethyl-10-hydroxycamptothecin (SN-38), the active irinotecan metabolite [156]. SN-38 interferes with the binding of FUBP1 to its target DNA sequence and induces deregulation of FUBP1 target genes in HCC cells. SN-38 presents the huge advantage to have a low IC50 and to be already used in clinic in combination with others chemotherapeutics to treat carcinoma. In that line, an important aspect of shFUBP1 studies is that FUBP1 downregulation sensitizes HCC cells for apoptosis-inducing chemotherapeutic drugs like the mitomycin C or doxorubicin [6, 8]. Therefore, SN-38 represents a promising molecule for clinical treatment of FUBP1-related cancers in combination with chemotherapeutic drugs.
To conclude, FUBP1 is a multifunctional protein that plays crucial roles in many biological processes. Cellular and physiopathological significances of the effects triggered by FUBP1 are not fully understood and remain open for exploration. Altogether, this review shows that FUBP1 can be a new cancer biomarker and represents an attractive target for future molecular cancer therapies. Overall, the master and multifunctional regulator FUBP1 is interesting from both a biological and clinical point of view.
Abbreviations
- ALL
Acute lymphoblastic leukemia
- AML
Acute myeloid leukemia
- ARE
AU-rich elements
- CCRCC
Clear cell renal cell carcinoma
- ChIP
Chromatin immunoprecipitation
- CLL
Chronic lymphocytic leukemia
- EMSA
Electrophoretic mobility shift assay
- EV71
Enterovirus 71
- FACS
Fluorescence activated cell sorting
- FDA
Food and drug administration
- HCC
Hepatocellular carcinoma
- HCV
Hepatitis C virus
- HSC
Hematopoietic stem cells
- IRES
Internal ribosome entry site
- KO
Knock-out
- NLS
Nuclear localization signal
- NSCLC
Non-small cell lung cancer
- SELEX
Systematic evolution of ligands by exponential enrichment
- SNV
Single nucleotide variation
- ssDNA
Single-stranded DNA
- TSS
Transcription start site
Funding
This work was supported by Ligue Régionale contre le cancer (comité 22, 35, 56, 79, 41) (MBT, LD), SFR Biosit UMS CNRS 3480—INSERM 018 (MBT), Région Bretagne (LD, MBT), The Société Française d’Hématologie (LD), Rennes Métropole (MBT), the société française de lutte contre les cancers et les leucémies de l’enfant et de l’adolescent and the Fédération Enfants et Santé (MBT), a private donator Mrs. M-Dominique Blanc-Bert (MBT), Cancéropole Grand Ouest (LD), and the CNRS, Université de Rennes 1 and the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme (FP7/2007-2013) under REA grant agreement no. 291851 (MBT).
Compliance with ethical standards
Conflict of interest
The authors declare no competing financial interests.
References
- 1.Zhang J, Chen QM. Far upstream element binding protein 1: a commander of transcription, translation and beyond. Oncogene. 2013;32:2907–2916. doi: 10.1038/onc.2012.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avigan MI, Strober B, Levens D. A far upstream element stimulates c-myc expression in undifferentiated leukemia cells. J Biol Chem. 1990;265:18538–18545. [PubMed] [Google Scholar]
- 3.Duncan R, Bazar L, Michelotti G, et al. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 1994;8:465–480. doi: 10.1101/gad.8.4.465. [DOI] [PubMed] [Google Scholar]
- 4.Levens D. How the c-myc promoter works and why it sometimes does not. JNCI Monogr. 2008;2008:41–43. doi: 10.1093/jncimonographs/lgn004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J, Chung H-J, Vogt M, et al. JTV1 co-activates FBP to induce USP29 transcription and stabilize p53 in response to oxidative stress. EMBO J. 2011;30:846–858. doi: 10.1038/emboj.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malz M, Weber A, Singer S, et al. Overexpression of far upstream element binding proteins: a mechanism regulating proliferation and migration in liver cancer cells. Hepatology. 2009;50:1130–1139. doi: 10.1002/hep.23051. [DOI] [PubMed] [Google Scholar]
- 7.Malz M, Bovet M, Samarin J, et al. Overexpression of far upstream element (FUSE) binding protein (FBP)-interacting repressor (FIR) supports growth of hepatocellular carcinoma. Hepatology. 2014;60:1241–1250. doi: 10.1002/hep.27218. [DOI] [PubMed] [Google Scholar]
- 8.Rabenhorst U, Beinoraviciute-Kellner R, Brezniceanu M-L, et al. Overexpression of the far upstream element binding protein 1 in hepatocellular carcinoma is required for tumor growth. Hepatology. 2009;50:1121–1129. doi: 10.1002/hep.23098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baumgarten P, Harter PN, Tönjes M, et al. Loss of FUBP1 expression in gliomas predicts FUBP1 mutation and is associated with oligodendroglial differentiation, IDH1 mutation and 1p/19q loss of heterozygosity: FUBP1 expression in human gliomas. Neuropathol Appl Neurobiol. 2014;40:205–216. doi: 10.1111/nan.12088. [DOI] [PubMed] [Google Scholar]
- 10.Duan J, Bao X, Ma X, et al. Upregulation of far upstream element-binding protein 1 (FUBP1) promotes tumor proliferation and tumorigenesis of clear cell renal cell carcinoma. PLoS One. 2017;12:e0169852. doi: 10.1371/journal.pone.0169852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauck S, Hiesinger K, Khageh Hosseini S, et al. Pyrazolo[1,5a]pyrimidines as a new class of FUSE binding protein 1 (FUBP1) inhibitors. Bioorg Med Chem. 2016;24:5717–5729. doi: 10.1016/j.bmc.2016.09.015. [DOI] [PubMed] [Google Scholar]
- 12.Singer S, Malz M, Herpel E, et al. Coordinated expression of stathmin family members by far upstream sequence element-binding protein-1 increases motility in non-small cell lung cancer. Cancer Res. 2009;69:2234–2243. doi: 10.1158/0008-5472.CAN-08-3338. [DOI] [PubMed] [Google Scholar]
- 13.Bettegowda C, Agrawal N, Jiao Y, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science. 2011;333:1453–1455. doi: 10.1126/science.1210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang L, Zhu J, Zhang J, et al. Far upstream element-binding protein 1 (FUBP1) is a potential c-Myc regulator in esophageal squamous cell carcinoma (ESCC) and its expression promotes ESCC progression. Tumor Biol. 2016;37:4115–4126. doi: 10.1007/s13277-015-4263-8. [DOI] [PubMed] [Google Scholar]
- 15.Ding Z, Liu X, Liu Y, et al. Expression of far upstream element (FUSE) binding protein 1 in human glioma is correlated with c-Myc and cell proliferation. Mol Carcinog. 2015;54:405–415. doi: 10.1002/mc.22114. [DOI] [PubMed] [Google Scholar]
- 16.Rabenhorst U, Thalheimer FB, Gerlach K, et al. Single-stranded DNA-binding transcriptional regulator FUBP1 is essential for fetal and adult hematopoietic stem cell self-renewal. Cell Rep. 2015;11:1847–1855. doi: 10.1016/j.celrep.2015.05.038. [DOI] [PubMed] [Google Scholar]
- 17.Zhou W, Chung Y-J, Castellar ER, et al. Far upstream element binding protein plays a crucial role in embryonic development, hematopoiesis, and stabilizing MYC expression levels. Am J Pathol. 2016 doi: 10.1016/j.ajpath.2015.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duncan R, Collins I, Tomonaga T, et al. A unique transactivation sequence motif is found in the carboxyl-terminal domain of the single-strand-binding protein FBP. Mol Cell Biol. 1996;16:2274–2282. doi: 10.1128/MCB.16.5.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis-Smyth T, Duncan RC, Zheng T, et al. The far upstream element-binding proteins comprise an ancient family of single-strand DNA-binding transactivators. J Biol Chem. 1996;271:31679–31687. doi: 10.1074/jbc.271.49.31679. [DOI] [PubMed] [Google Scholar]
- 20.Chung H-J, Liu J, Dundr M, et al. FBPs are calibrated molecular tools to adjust gene expression. Mol Cell Biol. 2006;26:6584–6597. doi: 10.1128/MCB.00754-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Briata P, Bordo D, Puppo M, et al. Diverse roles of the nucleic acid-binding protein KHSRP in cell differentiation and disease. Wiley Interdiscip Rev RNA. 2016;7:227–240. doi: 10.1002/wrna.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gherzi R, Lee K-Y, Briata P, et al. A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol Cell. 2004;14:571–583. doi: 10.1016/j.molcel.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Gau B-H, Chen T-M, Shih Y-HJ, Sun HS. FUBP3 interacts with FGF9 3′ microsatellite and positively regulates FGF9 translation. Nucleic Acids Res. 2011;39:3582–3593. doi: 10.1093/nar/gkq1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weber A, Kristiansen I, Johannsen M, et al. The FUSE binding proteins FBP1 and FBP3 are potential c-myc regulators in renal, but not in prostate and bladder cancer. BMC Cancer. 2008;8:369. doi: 10.1186/1471-2407-8-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zerbino DR, Achuthan P, Akanni W, et al. Ensembl 2018. Nucleic Acids Res. 2018;46:D754–D761. doi: 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D. GeneCards: integrating information about genes, proteins and diseases. Trends Genet TIG. 1997;13:163. doi: 10.1016/S0168-9525(97)01103-7. [DOI] [PubMed] [Google Scholar]
- 27.Valverde R, Edwards L, Regan L. Structure and function of KH domains: structure and function of KH domains. FEBS J. 2008;275:2712–2726. doi: 10.1111/j.1742-4658.2008.06411.x. [DOI] [PubMed] [Google Scholar]
- 28.Chien H-L, Liao C-L, Lin Y-L. FUSE binding protein 1 interacts with untranslated regions of japanese encephalitis virus RNA and negatively regulates viral replication. J Virol. 2011;85:4698–4706. doi: 10.1128/JVI.01950-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang Q, Xia W, Ji Q, et al. Role of far upstream element binding protein 1 in colonic epithelial disruption during dextran sulphate sodium-induced murine colitis. Int J Clin Exp Pathol. 2014;7:2019–2031. [PMC free article] [PubMed] [Google Scholar]
- 30.Jang M, Park BC, Kang S, et al. Far upstream element-binding protein-1, a novel caspase substrate, acts as a cross-talker between apoptosis and the c-myc oncogene. Oncogene. 2009;28:1529–1536. doi: 10.1038/onc.2009.11. [DOI] [PubMed] [Google Scholar]
- 31.Kim MJ, Park B-J, Kang Y-S, et al. Downregulation of FUSE-binding protein and c-myc by tRNA synthetase cofactor p38 is required for lung cell differentiation. Nat Genet. 2003;34:330–336. doi: 10.1038/ng1182. [DOI] [PubMed] [Google Scholar]
- 32.Atanassov BS, Dent SYR. USP22 regulates cell proliferation by deubiquitinating the transcriptional regulator FBP1. EMBO Rep. 2011;12:924–930. doi: 10.1038/embor.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hornbeck PV, Zhang B, Murray B, et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Venturutti L, Cordo Russo RI, Rivas MA, et al. MiR-16 mediates trastuzumab and lapatinib response in ErbB-2-positive breast and gastric cancer via its novel targets CCNJ and FUBP1. Oncogene. 2016;35:6189–6202. doi: 10.1038/onc.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao D, Zhang Y, Song L. MiR-16-1 targeted silences far upstream element binding protein 1 to advance the chemosensitivity to adriamycin in gastric cancer. Pathol Oncol Res POR. 2017 doi: 10.1007/s12253-017-0263-x. [DOI] [PubMed] [Google Scholar]
- 36.Marceau AH. Functions of single-strand DNA-binding proteins in DNA replication, recombination, and repair. Methods Mol Biol Clifton NJ. 2012;922:1–21. doi: 10.1007/978-1-62703-032-8_1. [DOI] [PubMed] [Google Scholar]
- 37.Michelotti GA, Michelotti EF, Pullner A, et al. Multiple single-stranded cis elements are associated with activated chromatin of the human c-myc gene in vivo. Mol Cell Biol. 1996;16:2656–2669. doi: 10.1128/MCB.16.6.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Michelotti EF, Michelotti GA, Aronsohn AI, Levens D. Heterogeneous nuclear ribonucleoprotein K is a transcription factor. Mol Cell Biol. 1996;16:2350–2360. doi: 10.1128/MCB.16.5.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tomonaga T, Levens D. Activating transcription from single stranded DNA. Proc Natl Acad Sci USA. 1996;93:5830–5835. doi: 10.1073/pnas.93.12.5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Braddock DT. Molecular basis of sequence-specific single-stranded DNA recognition by KH domains: solution structure of a complex between hnRNP K KH3 and single-stranded DNA. EMBO J. 2002;21:3476–3485. doi: 10.1093/emboj/cdf352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Braddock DT, Louis JM, Baber JL, et al. Structure and dynamics of KH domains from FBP bound to single-stranded DNA. Nature. 2002;415:1051–1056. doi: 10.1038/4151051a. [DOI] [PubMed] [Google Scholar]
- 42.Hsiao H, Nath A, Lin C-Y, et al. Quantitative characterization of the interactions among c-myc transcriptional regulators FUSE, FBP, and FIR. Biochemistry (Mosc) 2010;49:4620–4634. doi: 10.1021/bi9021445. [DOI] [PubMed] [Google Scholar]
- 43.Benjamin LR, Chung H-J, Sanford S, et al. Hierarchical mechanisms build the DNA-binding specificity of FUSE binding protein. Proc Natl Acad Sci. 2008;105:18296–18301. doi: 10.1073/pnas.0803279105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Debaize L, Jakobczyk H, Avner S, et al. Interplay between transcription regulators RUNX1 and FUBP1 activates an enhancer of the oncogene c-KIT and amplifies cell proliferation. Nucleic Acids Res. 2018 doi: 10.1093/nar/gky756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stine ZE, Walton ZE, Altman BJ, et al. MYC, metabolism, and cancer. Cancer Discov. 2015;5:1024–1039. doi: 10.1158/2159-8290.CD-15-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dang CV. MYC on the Path to Cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He L, Liu J, Collins I, et al. Loss of FBP function arrests cellular proliferation and extinguishes c-myc expression. EMBO J. 2000;19:1034–1044. doi: 10.1093/emboj/19.5.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feaver WJ, Svejstrup JQ, Bardwell L, et al. Dual roles of a multiprotein complex from S. cerevisiae in transcription and DNA repair. Cell. 1993;75:1379–1387. doi: 10.1016/0092-8674(93)90624-Y. [DOI] [PubMed] [Google Scholar]
- 49.Schaeffer L, Roy R, Humbert S, et al. DNA repair helicase: a component of BTF2 (TFIIH) basic transcription factor. Science. 1993;260:58–63. doi: 10.1126/science.8465201. [DOI] [PubMed] [Google Scholar]
- 50.Compe E, Egly J-M. TFIIH: when transcription met DNA repair. Nat Rev Mol Cell Biol. 2012;13:343. doi: 10.1038/nrm3350. [DOI] [PubMed] [Google Scholar]
- 51.Parvin JD, Sharp PA. DNA topology and a minimal set of basal factors for transcription by RNA polymerase II. Cell. 1993;73:533–540. doi: 10.1016/0092-8674(93)90140-L. [DOI] [PubMed] [Google Scholar]
- 52.Goodrich JA, Tjian R. Transcription factors IIE and IIH and ATP hydrolysis direct promoter clearance by RNA polymerase II. Cell. 1994;77:145–156. doi: 10.1016/0092-8674(94)90242-9. [DOI] [PubMed] [Google Scholar]
- 53.Felsher DW, Bishop JM. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci USA. 1999;96:3940–3944. doi: 10.1073/pnas.96.7.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu J, He L, Collins I, et al. The FBP interacting repressor targets TFIIH to inhibit activated transcription. Mol Cell. 2000;5:331–341. doi: 10.1016/S1097-2765(00)80428-1. [DOI] [PubMed] [Google Scholar]
- 55.Liu J, Kouzine F, Nie Z, et al. The FUSE/FBP/FIR/TFIIH system is a molecular machine programming a pulse of c-myc expression. EMBO J. 2006;25:2119–2130. doi: 10.1038/sj.emboj.7601101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsushita K, Tomonaga T, Shimada H, et al. An essential role of alternative splicing of c-myc suppressor FUSE-binding protein-interacting repressor in carcinogenesis. Cancer Res. 2006;66:1409–1417. doi: 10.1158/0008-5472.CAN-04-4459. [DOI] [PubMed] [Google Scholar]
- 57.Crichlow GV, Zhou H, Hsiao H, et al. Dimerization of FIR upon FUSE DNA binding suggests a mechanism of c-myc inhibition. EMBO J. 2008;27:277–289. doi: 10.1038/sj.emboj.7601936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weber A, Liu J, Collins I, Levens D. TFIIH operates through an expanded proximal promoter to fine-tune c-myc expression. Mol Cell Biol. 2005;25:147–161. doi: 10.1128/MCB.25.1.147-161.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rubin CI, Atweh GF. The role of stathmin in the regulation of the cell cycle. J Cell Biochem. 2004;93:242–250. doi: 10.1002/jcb.20187. [DOI] [PubMed] [Google Scholar]
- 60.Mistry SJ, Atweh GF. Role of stathmin in the regulation of the mitotic spindle: potential applications in cancer therapy. Mt Sinai J Med N Y. 2002;69:299–304. [PubMed] [Google Scholar]
- 61.Belletti B, Nicoloso MS, Schiappacassi M, et al. Stathmin activity influences sarcoma cell shape, motility, and metastatic potential. Mol Biol Cell. 2008;19:2003–2013. doi: 10.1091/mbc.E07-09-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuner R, Muley T, Meister M, et al. Global gene expression analysis reveals specific patterns of cell junctions in non-small cell lung cancer subtypes. Lung Cancer Amst Neth. 2009;63:32–38. doi: 10.1016/j.lungcan.2008.03.033. [DOI] [PubMed] [Google Scholar]
- 63.Krumlauf R. Hox genes in vertebrate development. Cell. 1994;78:191–201. doi: 10.1016/0092-8674(94)90290-9. [DOI] [PubMed] [Google Scholar]
- 64.Lawrence HJ, Sauvageau G, Humphries RK, Largman C. The role of HOX homeobox genes in normal and leukemic hematopoiesis. Stem Cells Dayt Ohio. 1996;14:281–291. doi: 10.1002/stem.140281. [DOI] [PubMed] [Google Scholar]
- 65.Broudy VC. Stem cell factor and hematopoiesis. Blood. 1997;90:1345–1364. [PubMed] [Google Scholar]
- 66.Kitamura Y, Hirotab S. Kit as a human oncogenic tyrosine kinase. Cell Mol Life Sci CMLS. 2004;61:2924–2931. doi: 10.1007/s00018-004-4273-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gartel AL, Serfas MS, Tyner AL. p21–negative regulator of the cell cycle. Proc Soc Exp Biol Med. 1996;213:138–149. doi: 10.3181/00379727-213-44046. [DOI] [PubMed] [Google Scholar]
- 68.Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639–649. [PubMed] [Google Scholar]
- 69.Soria G, Speroni J, Podhajcer OL, et al. p21 differentially regulates DNA replication and DNA-repair-associated processes after UV irradiation. J Cell Sci. 2008;121:3271–3282. doi: 10.1242/jcs.027730. [DOI] [PubMed] [Google Scholar]
- 70.Dixit U, Liu Z, Pandey AK, et al. Fuse binding protein antagonizes the transcription activity of tumor suppressor protein. BMC Cancer. 2014 doi: 10.1186/1471-2407-14-925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dixit U, Pandey AK, Liu Z, et al. FUSE binding protein 1 facilitates persistent hepatitis C virus replication in hepatoma cells by regulating tumor suppressor p53. J Virol. 2015;89:7905–7921. doi: 10.1128/JVI.00729-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Frost JR, Mendez M, Soriano AM. Adenovirus 5 E1A-mediated suppression of p53 via FUBP1. J Virol. 2018 doi: 10.1128/jvi.00439-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sully G, Dean JLE, Wait R, et al. Structural and functional dissection of a conserved destabilizing element of cyclo-oxygenase-2 mRNA: evidence against the involvement of AUF-1 [AU-rich element/poly(U)-binding/degradation factor-1], AUF-2, tristetraprolin, HuR (Hu antigen R) or FBP1 (far-upstream-sequence-element-binding protein 1) Biochem J. 2004;377:629–639. doi: 10.1042/BJ20031484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang Z, Harris D, Pandey VN. The FUSE binding protein is a cellular factor required for efficient replication of hepatitis C virus. J Virol. 2008;82:5761–5773. doi: 10.1128/JVI.00064-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Caput D, Beutler B, Hartog K, et al. Identification of a common nucleotide sequence in the 3′-untranslated region of mRNA molecules specifying inflammatory mediators. Proc Natl Acad Sci USA. 1986;83:1670–1674. doi: 10.1073/pnas.83.6.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang T, Kruys V, Huez G, Gueydan C. AU-rich element-mediated translational control: complexity and multiple activities of trans-activating factors. Biochem Soc Trans. 2002;30:952–958. doi: 10.1042/bst0300952. [DOI] [PubMed] [Google Scholar]
- 77.Roberto Gherzi, Ching-Yi Chen, Michele Trabucchi, et al. The role of KSRP in mRNA decay and microRNA precursor maturation. Wiley Interdiscip Rev RNA. 2010;1:230–239. doi: 10.1002/wrna.2. [DOI] [PubMed] [Google Scholar]
- 78.Jacob AG, Singh RK, Mohammad F, et al. The splicing factor FUBP1 is required for the efficient splicing of oncogene MDM2 Pre-mRNA. J Biol Chem. 2014;289:17350–17364. doi: 10.1074/jbc.M114.554717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miro J, Laaref AM, Rofidal V, et al. FUBP1: a new protagonist in splicing regulation of the DMD gene. Nucleic Acids Res. 2015;43:2378–2389. doi: 10.1093/nar/gkv086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zheng W, Shen F, Hu R, et al. Far upstream element-binding protein 1 binds the 3′ untranslated region of PKD2 and suppresses its translation. J Am Soc Nephrol. 2016 doi: 10.1681/asn.2015070836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Irwin N, Baekelandt V, Goritchenko L, Benowitz LI. Identification of two proteins that bind to a pyrimidine-rich sequence in the 3′-untranslated region of GAP-43 mRNA. Nucleic Acids Res. 1997;25:1281–1288. doi: 10.1093/nar/25.6.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Olanich ME, Moss BL, Piwnica-Worms D, et al. Identification of FUSE-binding protein 1 as a regulatory mRNA-binding protein that represses nucleophosmin translation. Oncogene. 2011;30:77–86. doi: 10.1038/onc.2010.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Box JK, Paquet N, Adams MN, et al. Nucleophosmin: from structure and function to disease development. BMC Mol Biol. 2016 doi: 10.1186/s12867-016-0073-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Williams BY, Hamilton SL, Sarkar HK. The survival motor neuron protein interacts with the transactivator FUSE binding protein from human fetal brain. FEBS Lett. 2000;470:207–210. doi: 10.1016/S0014-5793(00)01320-X. [DOI] [PubMed] [Google Scholar]
- 85.Zheng Y, Miskimins WK. Far upstream element binding protein 1 activates translation of p27Kip1 mRNA through its internal ribosomal entry site. Int J Biochem Cell Biol. 2011;43:1641–1648. doi: 10.1016/j.biocel.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang P-N, Lin J-Y, Locker N, et al. Far upstream element binding protein 1 binds the internal ribosomal entry site of enterovirus 71 and enhances viral translation and viral growth. Nucleic Acids Res. 2011;39:9633–9648. doi: 10.1093/nar/gkr682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsai F-J, Lin C-W, Lai C-C, et al. Kaempferol inhibits enterovirus 71 replication and internal ribosome entry site (IRES) activity through FUBP and HNRP proteins. Food Chem. 2011;128:312–322. doi: 10.1016/j.foodchem.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 88.Hung C-T, Kung Y-A, Li M-L, et al. Additive promotion of viral internal ribosome entry site-mediated translation by far upstream element-binding protein 1 and an enterovirus 71-induced cleavage product. PLoS Pathog. 2016 doi: 10.1371/journal.ppat.1005959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lin J-Y, Li M-L, Shih S-R. Far upstream element binding protein 2 interacts with enterovirus 71 internal ribosomal entry site and negatively regulates viral translation. Nucleic Acids Res. 2009;37:47–59. doi: 10.1093/nar/gkn901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Min H, Turck CW, Nikolic JM, Black DL. A new regulatory protein, KSRP, mediates exon inclusion through an intronic splicing enhancer. Genes Dev. 1997;11:1023–1036. doi: 10.1101/gad.11.8.1023. [DOI] [PubMed] [Google Scholar]
- 91.Labourier E, Adams MD, Rio DC. Modulation of P-Element Pre-mRNA Splicing by a Direct Interaction between PSI and U1 snRNP 70 K Protein. Mol Cell. 2001;8:363–373. doi: 10.1016/S1097-2765(01)00311-2. [DOI] [PubMed] [Google Scholar]
- 92.Rappsilber J, Ryder U, Lamond AI, Mann M. Large-Scale proteomic analysis of the human spliceosome. Genome Res. 2002;12:1231–1245. doi: 10.1101/gr.473902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pan Q, Shai O, Lee LJ, et al. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 94.Li H, Wang Z, Zhou X, et al. Far upstream element-binding protein 1 and RNA secondary structure both mediate second-step splicing repression. Proc Natl Acad Sci. 2013;110:E2687–E2695. doi: 10.1073/pnas.1310607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Will CL, Lührmann R. Spliceosome structure and function. Cold Spring Harb Perspect Biol. 2011 doi: 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang J, Schultz PG, Johnson KA. Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc Natl Acad Sci USA. 2018;115:E4604–E4612. doi: 10.1073/pnas.1800260115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Freedman DA, Wu L, Levine AJ. Functions of the MDM2 oncoprotein. Cell Mol Life Sci CMLS. 1999;55:96–107. doi: 10.1007/s000180050273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Emery AEH. The muscular dystrophies. Lancet Lond Engl. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 99.Sutandy FXR, Ebersberger S, Huang L, et al. In vitro iCLIP-based modeling uncovers how the splicing factor U2AF2 relies on regulation by cofactors. Genome Res. 2018;28:699–713. doi: 10.1101/gr.229757.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hwang I, Cao D, Na Y, et al. Far upstream element-binding protein 1 regulates LSD1 alternative splicing to promote terminal differentiation of neural progenitors. Stem Cell Rep. 2018 doi: 10.1016/j.stemcr.2018.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ostrom QT, Bauchet L, Davis FG, et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro-Oncol. 2014;16:896–913. doi: 10.1093/neuonc/nou087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Barbashina V, Salazar P, Holland EC, et al. Allelic losses at 1p36 and 19q13 in gliomas: correlation with histologic classification, definition of a 150-kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene. Clin Cancer Res Off J Am Assoc Cancer Res. 2005;11:1119–1128. [PubMed] [Google Scholar]
- 103.Sahm F, Koelsche C, Meyer J, et al. CIC and FUBP1 mutations in oligodendrogliomas, oligoastrocytomas and astrocytomas. Acta Neuropathol (Berl) 2012;123:853–860. doi: 10.1007/s00401-012-0993-5. [DOI] [PubMed] [Google Scholar]
- 104.Chan AK-Y, Pang JC-S, Chung NY-F, et al. Loss of CIC and FUBP1 expressions are potential markers of shorter time to recurrence in oligodendroglial tumors. Mod Pathol Off J US Can Acad Pathol Inc. 2014;27:332–342. doi: 10.1038/modpathol.2013.165. [DOI] [PubMed] [Google Scholar]
- 105.Brat DJ, Verhaak RGW, Cancer Genome Atlas Research Network et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372:2481–2498. doi: 10.1056/nejmoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Suzuki H, Aoki K, Chiba K, et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat Genet. 2015;47:458–468. doi: 10.1038/ng.3273. [DOI] [PubMed] [Google Scholar]
- 107.Aihara K, Nagae G. Genetic and epigenetic stability of oligodendrogliomas at recurrence. Acta Neuropathol Commun. 2017 doi: 10.1186/s40478-017-0422-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bailey MH, Tokheim C, Porta-Pardo E, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173:371–385. doi: 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yip S, Butterfield YS, Morozova O, et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J Pathol. 2012;226:7–16. doi: 10.1002/path.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chen J, Hackett CS, Zhang S, et al. The genetics of splicing in neuroblastoma. Cancer Discov. 2015;5:380–395. doi: 10.1158/2159-8290.CD-14-0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bradley RK, Merkin J, Lambert NJ, Burge CB. Alternative splicing of RNA triplets is often regulated and accelerates proteome evolution. PLoS Biol. 2012;10:e1001229. doi: 10.1371/journal.pbio.1001229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bartel F, Taubert H, Harris LC. Alternative and aberrant splicing of MDM2 mRNA in human cancer. Cancer Cell. 2002;2:9–15. doi: 10.1016/S1535-6108(02)00091-0. [DOI] [PubMed] [Google Scholar]
- 113.Singh RK, Tapia-Santos A, Bebee TW, Chandler DS. Conserved sequences in the final intron of MDM2 are essential for the regulation of alternative splicing of MDM2 in response to stress. Exp Cell Res. 2009;315:3419–3432. doi: 10.1016/j.yexcr.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 114.Steinman HA, Burstein E, Lengner C, et al. An alternative splice form of Mdm2 induces p53-independent cell growth and tumorigenesis. J Biol Chem. 2004;279:4877–4886. doi: 10.1074/jbc.M305966200. [DOI] [PubMed] [Google Scholar]
- 115.Seiler M, Peng S, Agrawal AA, et al. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep. 2018;23:282–296. doi: 10.1016/j.celrep.2018.01.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liu Z-H, Hu J-L, Liang J-Z, et al. Far upstream element-binding protein 1 is a prognostic biomarker and promotes nasopharyngeal carcinoma progression. Cell Death Dis. 2015;6:e1920. doi: 10.1038/cddis.2015.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pénzváltó Z, Lánczky A, Lénárt J, et al. MEK1 is associated with carboplatin resistance and is a prognostic biomarker in epithelial ovarian cancer. BMC Cancer. 2014;14:837. doi: 10.1186/1471-2407-14-837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang J, Xiong X, Hua X, et al. Knockdown of FUSE binding protein 1 enhances the sensitivity of epithelial ovarian cancer cells to carboplatin. Oncol Lett. 2017;14:5819–5824. doi: 10.3892/ol.2017.6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yu MC, Yuan J-M, Lu SC. Alcohol, cofactors and the genetics of hepatocellular carcinoma. J Gastroenterol Hepatol. 2008;23:S92–S97. doi: 10.1111/j.1440-1746.2007.05293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sanyal AJ, Yoon SK, Lencioni R. The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist. 2010;15:14–22. doi: 10.1634/theoncologist.2010-S4-14. [DOI] [PubMed] [Google Scholar]
- 121.Kawate S, Fukusato T, Ohwada S, et al. Amplification of c-myc in hepatocellular carcinoma: correlation with clinicopathologic features, proliferative activity and p53 overexpression. Oncology. 1999;57:157–163. doi: 10.1159/000012024. [DOI] [PubMed] [Google Scholar]
- 122.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 123.Murakami H, Sanderson ND, Nagy P, et al. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Res. 1993;53:1719–1723. [PubMed] [Google Scholar]
- 124.Zimonjic DB, Keck CL, Thorgeirsson SS, Popescu NC. Novel recurrent genetic imbalances in human hepatocellular carcinoma cell lines identified by comparative genomic hybridization. Hepatol Baltimore, MD. 1999;29:1208–1214. doi: 10.1002/hep.510290410. [DOI] [PubMed] [Google Scholar]
- 125.Schlaeger C, Longerich T, Schiller C, et al. Etiology-dependent molecular mechanisms in human hepatocarcinogenesis. Hepatol Baltimore, MD. 2008;47:511–520. doi: 10.1002/hep.22033. [DOI] [PubMed] [Google Scholar]
- 126.Zubaidah RM, Tan GS, Tan SBE, et al. 2-D DIGE profiling of hepatocellular carcinoma tissues identified isoforms of far upstream binding protein (FUBP) as novel candidates in liver carcinogenesis. Proteomics. 2008;8:5086–5096. doi: 10.1002/pmic.200800322. [DOI] [PubMed] [Google Scholar]
- 127.Wen H, Ma H, Li P, et al. Expression of far upstream element-binding protein 1 correlates with c-Myc expression in sacral chordomas and is associated with tumor progression and poor prognosis. Biochem Biophys Res Commun. 2017;491:1047–1054. doi: 10.1016/j.bbrc.2017.08.008. [DOI] [PubMed] [Google Scholar]
- 128.Xiong X, Zhang J, Liang W, et al. Fuse-binding protein 1 is a target of the EZH2 inhibitor GSK343, in osteosarcoma cells. Int J Oncol. 2016;49:623–628. doi: 10.3892/ijo.2016.3541. [DOI] [PubMed] [Google Scholar]
- 129.Bièche I, Lachkar S, Becette V, et al. Overexpression of the stathmin gene in a subset of human breast cancer. Br J Cancer. 1998;78:701–709. doi: 10.1038/bjc.1998.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Müller B, Bovet M, Yin Y, et al. Concomitant expression of far upstream element (FUSE) binding protein (FBP) interacting repressor (FIR) and its splice variants induce migration and invasion of non-small cell lung cancer (NSCLC) cells. J Pathol. 2015;237:390–401. doi: 10.1002/path.4588. [DOI] [PubMed] [Google Scholar]
- 131.Brisbin AG, Asmann YW, Song H, et al. Meta-analysis of 8q24 for seven cancers reveals a locus between NOV and ENPP2 associated with cancer development. BMC Med Genet. 2011;12:156. doi: 10.1186/1471-2350-12-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Boelens MC, Kok K, van der Vlies P, et al. Genomic aberrations in squamous cell lung carcinoma related to lymph node or distant metastasis. Lung Cancer Amsterdam Netherlands. 2009;66:372–378. doi: 10.1016/j.lungcan.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 133.Moinzadeh P, Breuhahn K, Stützer H, Schirmacher P. Chromosome alterations in human hepatocellular carcinomas correlate with aetiology and histological grade—results of an explorative CGH meta-analysis. Br J Cancer. 2005;92:935–941. doi: 10.1038/sj.bjc.6602448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Matsushita K, Tomonaga T, Kajiwara T, et al. c-myc suppressor FBP-interacting repressor for cancer diagnosis and therapy. Front Biosci Landmark Ed. 2009;14:3401–3408. doi: 10.2741/3461. [DOI] [PubMed] [Google Scholar]
- 135.Saito I, Miyamura T, Ohbayashi A, et al. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proc Natl Acad Sci. 1990;87:6547–6549. doi: 10.1073/pnas.87.17.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shi ST, Lai MM. Hepatitis C viral RNA: challenges and promises. Cell Mol Life Sci CMLS. 2001;58:1276–1295. doi: 10.1007/PL00000939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wesely J, Steiner M, Schntgen F, et al. Delayed mesoderm and erythroid differentiation of murine embryonic stem cells in the absence of the transcriptional regulator FUBP1. Stem Cells Int. 2017;2017:1–12. doi: 10.1155/2017/5762301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wilson A, Murphy MJ, Oskarsson T, et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004;18:2747–2763. doi: 10.1101/gad.313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Reavie L, Gatta GD, Crusio K, et al. Hematopoietic stem cell differentiation regulated by a single ubiquitin ligase: substrate complex. Nat Immunol. 2010;11:207–215. doi: 10.1038/ni.1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hoffman B, Amanullah A, Shafarenko M, Liebermann DA. The proto-oncogene c-myc in hematopoietic development and leukemogenesis. Oncogene. 2002;21:3414–3421. doi: 10.1038/sj.onc.1205400. [DOI] [PubMed] [Google Scholar]
- 141.Freytag SO. Enforced expression of the c-myc oncogene inhibits cell differentiation by precluding entry into a distinct predifferentiation state in G0/G1. Mol Cell Biol. 1988;8:1614–1624. doi: 10.1128/MCB.8.4.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hoffman B, Liebermann DA, Selvakumaran M, Nguyen HQ. Role of c-myc in myeloid differentiation, growth arrest and apoptosis. Curr Top Microbiol Immunol. 1996;211:17–27. doi: 10.1007/978-3-642-85232-9_3. [DOI] [PubMed] [Google Scholar]
- 143.Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17:1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 144.Delgado MD, León J. Myc roles in hematopoiesis and leukemia. Genes Cancer. 2010 doi: 10.1177/1947601910377495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lennartsson J, Jelacic T, Linnekin D, Shivakrupa R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells. 2005;23:16–43. doi: 10.1634/stemcells.2004-0117. [DOI] [PubMed] [Google Scholar]
- 146.Weidemann RR, Behrendt R, Schoedel KB, et al. Constitutive Kit activity triggers B-cell acute lymphoblastic leukemia-like disease in mice. Exp Hematol. 2017;45:45–55. doi: 10.1016/j.exphem.2016.09.005. [DOI] [PubMed] [Google Scholar]
- 147.Landau DA, Tausch E, Taylor-Weiner AN, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–530. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lindqvist CM, Lundmark A, Nordlund J, et al. Deep targeted sequencing in pediatric acute lymphoblastic leukemia unveils distinct mutational patterns between genetic subtypes and novel relapse-associated genes. Oncotarget. 2016;27:64071–64088. doi: 10.18632/oncotarget.11773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Klener P, Fronkova E, Berkova A, et al. Mantle cell lymphoma-variant Richter syndrome: detailed molecular-cytogenetic and backtracking analysis reveals slow evolution of a pre-MCL clone in parallel with CLL over several years: mantle cell lymphoma-variant Richter syndrome. Int J Cancer. 2016;139:2252–2260. doi: 10.1002/ijc.30263. [DOI] [PubMed] [Google Scholar]
- 150.Huth JR, Yu L, Collins I, et al. NMR-driven discovery of benzoylanthranilic acid inhibitors of far upstream element binding protein binding to the human oncogene c-myc promoter. J Med Chem. 2004;47:4851–4857. doi: 10.1021/jm0497803. [DOI] [PubMed] [Google Scholar]
- 151.Au SL-K, Wong CC-L, Lee JM-F, et al. EZH2-mediated H3K27me3 is involved in epigenetic repression of deleted in liver cancer 1 in human cancers. PLoS One. 2013;8:e68226. doi: 10.1371/journal.pone.0068226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lv Y-F, Yan G-N, Meng G. Enhancer of zeste homolog two silencing inhibits tumor growth and lung metastasis in osteosarcoma. Sci Rep. 2015 doi: 10.1038/srep12999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Niforou KM, Anagnostopoulos AK, Vougas K, et al. The proteome profile of the human osteosarcoma U2OS cell line. Cancer Genom Proteom. 2008;5:63–78. [PubMed] [Google Scholar]
- 154.Varambally S, Dhanasekaran SM, Zhou M, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 155.Kleer CG, Cao Q, Varambally S, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Khageh Hosseini S, Kolterer S, Steiner M, et al. Camptothecin and its analog SN-38, the active metabolite of irinotecan, inhibit binding of the transcriptional regulator and oncoprotein FUBP1 to its DNA target sequence FUSE. Biochem Pharmacol. 2017;146:53–62. doi: 10.1016/j.bcp.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 157.Zhou X, Edmonson MN, Wilkinson MR, et al. Exploring genomic alteration in pediatric cancer using protein paint. Nat Genet. 2016;48:4–6. doi: 10.1038/ng.3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Barretina J, Caponigro G, Stransky N, et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Idbaih A, Ducray F, Dehais C, et al. SNP array analysis reveals novel genomic abnormalities including copy neutral loss of heterozygosity in anaplastic oligodendrogliomas. PLoS One. 2012;7:e45950. doi: 10.1371/journal.pone.0045950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Cahill DP, Louis DN, Cairncross JG. Molecular background of oligodendroglioma: 1p/19q, IDH, TERT, CIC and FUBP1. CNS Oncol. 2015;4:287–294. doi: 10.2217/cns.15.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Erdem-Eraslan L, Heijsman D, de Wit M, et al. Tumor-specific mutations in low-frequency genes affect their functional properties. J Neurooncol. 2015;122:461–470. doi: 10.1007/s11060-015-1741-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang L, Liu Y, Wang M. EZH2-, CHD4-, and IDH-linked epigenetic perturbation and its association with survival in glioma patients. J Mol Cell Biol. 2017 doi: 10.1093/jmcb/mjx056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ma J, Chen M, Xia S-K, et al. Prostaglandin E2 promotes liver cancer cell growth by the upregulation of FUSE-binding protein 1 expression. Int J Oncol. 2013;42:1093–1104. doi: 10.3892/ijo.2013.1782. [DOI] [PubMed] [Google Scholar]
- 164.Samarin J, Laketa V, Malz M, et al. PI3K/AKT/mTOR-dependent stabilization of oncogenic far-upstream element binding proteins in hepatocellular carcinoma cells. Hepatology. 2016;63:813–826. doi: 10.1002/hep.28357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhang F, Tian Q, Wang Y. Far upstream element-binding protein 1 (FUBP1) is overexpressed in human gastric cancer tissue compared to non-cancerous tissue. Onkologie. 2013;36:650–655. doi: 10.1159/000355659. [DOI] [PubMed] [Google Scholar]
- 166.Jia MY, Wang YJ. Far upstream element-binding protein 1(FUBP1) expression differs between human colorectal cancer and non-cancerous tissue. Neoplasma. 2014;61:533–540. doi: 10.4149/neo_2014_065. [DOI] [PubMed] [Google Scholar]
- 167.Gagné J-P, Gagné P, Hunter JM, et al. Proteome profiling of human epithelial ovarian cancer cell line TOV-112D. Mol Cell Biochem. 2005;275:25–55. doi: 10.1007/s11010-005-7556-1. [DOI] [PubMed] [Google Scholar]
- 168.Zhong Q, Liu Z, Lin Z-R, et al. The RARS-MADlLl fusion gene induces cancer stem cell-like properties and therapeutic resistance in nasopharyngeal carcinoma. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.ccr-17-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]