

Abstract
RNA-binding proteins (RBPs) and microRNAs (miRNAs) are the most important regulators of mRNA stability and translation in eukaryotic cells; however, the complex interplay between these systems is only now coming to light. RBPs and miRNAs regulate a unique set of targets in either a positive or negative manner and their regulation is mainly opposed to each other on overlapping targets. In some cases, the levels of RBPs or miRNAs regulate the cellular levels of one another and decreased levels of either results in changes in translation of their targets. There is growing evidence that these regulatory circuits are crucial in the development and progression of cancer; however, the rules underlying synergism and antagonism between miRNAs and RNA-binding proteins remain unclear. Synthetic biology seeks to develop artificial systems to better understand their natural counterparts and to develop new, useful technologies for manipulation of gene expression at the RNA level. The recent development of artificial RNA-binding proteins promises to enable a much greater understanding of the importance of the functional interactions between RNA-binding proteins and miRNAs, as well as enabling their manipulation for therapeutic purposes.
Keywords: miRNA, Designer RNA-binding proteins, Pentatricopeptide repeat, PUF domain, Synthetic biology, RNA–protein interactions, RNA interference
Introduction
Post-transcriptional regulation dominates a wide range of gene regulatory processes that control stability and turnover as well as the translation of mRNAs. Among the most well-known post-translational regulators are RNA-binding proteins (RBPs) of which a wide variety exist, each with a unique function and scope of target mRNAs, such that RNAs from transcription to degradation are bound to numerous different proteins. Recently, an additional group of regulators were discovered known as microRNAs (miRNAs). Since the initial discovery of miRNAs, it was found that the expression of many miRNAs as well as their activity was altered in many diseases including cancer, Parkinson’s disease and Huntington’s disease [1, 2].
miRNAs were first discovered to be involved in developmental timing in C. elegans, where it was found that a 22 nucleotide (nt) long regulatory RNA named lin-4 regulated the production of the protein lin-14 [3, 4]. Moreover, a second miRNA known as let-7 was discovered shortly after and is conserved among a large number of organisms including humans. These small regulatory non-coding RNAs are now known to play an important role in the post-transcriptional regulation of the majority (~ 60%) of genes and thousands have since been discovered [5, 6]. A common feature that defines all miRNAs is the processing they undergo from a long primary transcript to a ~ 22-nt mature miRNA (Fig. 1). Primary miRNAs (pri-miRNAs) transcribed by RNA polymerase II are processed in two subsequent steps to a mature active miRNA [5, 7, 8]. Initial processing occurs in the nucleus by the RNAse III enzyme Drosha in complex with the RNA-binding protein DiGeorge syndrome critical region 8 (DGCR8, also known as Pasha), generating a ~ 70 nt precursor miRNA (pre-miRNA) with a characteristic 2-nt 3′ overhang [5, 9, 10]. The 2-nt 3′ overhang characteristic of cleavage by Drosha is recognized by the Ran-GTPase-dependent Exportin 5 that subsequently facilitates the export of pre-miRNAs into the cytosol [10, 11]. Cytosolic pre-miRNA is recognized by a second RNAse III enzyme called Dicer, which cleaves the loop region of pre-miRNA to release the ~ 22 nt mature miRNA.
Fig. 1.
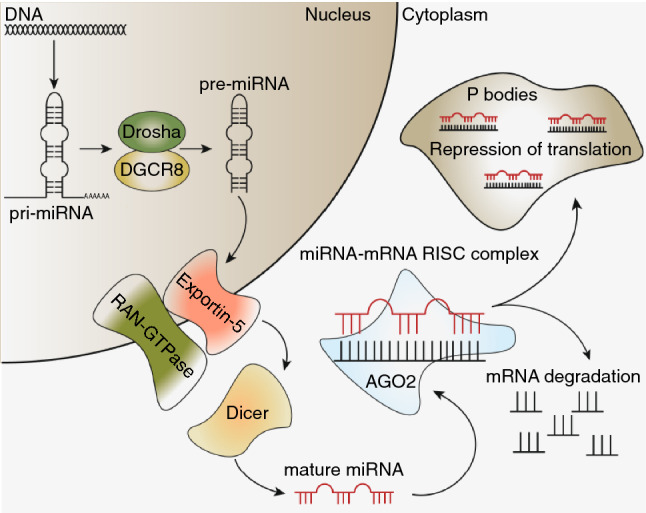
Schematic overview of the miRNA biogenesis pathway
Mature miRNA is loaded into the RNA-induced silencing complex (RISC) though binding to Argonaute 2 (Ago2) and subsequently binds mRNA with sequence complementarity in the so-called seed region (nucleotides 2–7), which is of key importance [5, 8, 12–14]. The majority of the miRNA recognition sites are located in the 3′-untranslated regions (3′-UTRs) of target mRNAs; however, studies have indicated that miRNAs also target sites within the coding regions as well as the 5′-UTRs of mRNAs [15–18]. While Ago2 is capable of cleaving most target mRNAs, there are a few miRNA targets in humans that lack the central sequence match (nucleotides 9–11) required for Ago2-dependent cleavage and degradation of target mRNAs [19, 20]. These mRNAs are bound to the miRNA-RISC and translocated to the cytoplasmic P bodies where translation is repressed through recruitment of GW182-containing complexes [5]. In addition, degradation of repressed mRNAs can potentially occur through recruitment of the CCR4-NOT deadenylase complex to the RISC, leading to the removal of the poly(A) tail and subsequent degradation [21]. While miRNAs are mostly known for repressing mRNA translation and their negative impact on mRNA stability, several studies have indicated that miRNAs are also capable of positively influencing translation as well as mRNA stability [22, 23].
RBPs, such as Drosha, Dicer and Ago2, play a very prominent role in the maturation and function of miRNA. RBPs are well known for their essential role in regulating protein expression, RNA metabolism, transport and localization of various RNA molecules. They play a critical role in regulating the balance between translation, repression and turnover of both mRNAs and ncRNAs to sustain cellular processes as well the ability to quickly adapt to any stress signal that may occur. In addition, they regulate maturation of mRNA and various small non-coding RNAs (ncRNAs) including miRNAs [24, 25]. While certain RBPs are particularly suitable for engineering purposes, others might be more challenging due to their essential functions in the cell such as Drosha and Dicer. RBPs are characterized by the presence of one or more RNA-binding domains (RBDs) and it is estimated that at least 800 structurally distinct domains exist. While some of these domains are widespread, others are quite rare and are often confined to certain organisms or specific functions. Among the most widespread of RNA-binding domains are: zinc fingers, RNA recognition motifs (RRMs), DEAD motifs and K homology (KH) domains. A distinct feature of RBPs is the presence of multiple RBDs as either single domains or as repeats. Each RBD on its own is often not particularly specific nor does it have high affinity for its target; however, RBDs often occur as repeats of a single or multiple RBDs. Multiple RBDs increase the affinity and specificity of the RBP through the combination of multiple weaker interactions. Furthermore, different combinations of RBDs and their structural arrangements allow for the specific recognition and regulation of a wide variety of RNAs [24, 25]. Often, other non-enzymatic or enzymatic domains are present which further widens the scope of targets which RBPs can regulate [24, 25].
RBPs can recognize their target mRNA in either a sequence-specific manner or in a non-sequence-specific manner where they recognize the secondary structure of RNAs, or a combination of the two. Sequence-specific RBPs are characterized by a primarily hydrophobic binding surface, which allows for maximal intermolecular contacts with the RNA [26]. The interactions between non-sequence-specific RBPs and RNA are achieved through binding of the positively charged side chains with the sugar–phosphate backbone of the target RNA. In this review, we give a brief overview of the widespread physical and regulatory interactions that occur between miRNAs and RBPs in regulating mRNA stability and translation and their role in the initiation and progression of cancer. We also give an overview of the recent engineering of artificial RBPs that may play an important role in understanding and manipulating these interactions and may give rise to new alternative therapies for cancer.
RNA-binding protein and miRNA-mediated post-transcriptional regulation
Roughly, 7.5% of all human protein coding genes encode for RBPs, reflecting their importance in everyday cellular processes as they govern many pathways in cells through transcriptional regulation of either mRNA or miRNA [24, 27, 28]. While many RBPs are implicated in binding to mRNAs to regulate their turnover and translation, a number of RBPs have been found to play an important role in intracellular localization of mRNA, such as insulin-like growth factor 2 mRNA-binding protein 1 (IMP1) [28–30]. Interestingly, the role of RBPs in transporting mRNAs is important in the formation of cell polarity and for its maintenance and is often altered in cancers [28, 29].
Recently, a large number of miRNA-interacting RBPs were found by Treiber et al., who identified proteins associating with 72 pre-miRNAs in 11 cell lines [31]. Using a proteomics-based pull-down approach, they identified 180 miRNA-interacting RBPs, some of which were previously identified and well characterized, such as Lin28A/B, TUT4 and TUT7. Further characterization of a subset of the potential miRNA-interacting RBPs, such as RBM10, DDX21 and MATR3, confirmed that they bind specifically to certain miRNA precursors. While these results are certainly exciting and could be of great interest to the field, for the purpose of this review we will focus on a number of well-characterized examples of cross-regulation between miRNA and RNA-binding proteins.
AU-rich elements (AREs) are cis-acting elements typically located in the 3′-UTR from where they exert control over mRNA turnover, transport and translation. One of the main RBP families that bind to these AU-rich elements is the mammalian RNA-binding protein family Hu, also known as embryonic lethal abnormal vision (Elav) in Drosophila, consisting of the ubiquitously expressed HuR (HuA) and the primarily neuronal proteins HuB, HuC and HuD [16, 32]. The RBP HuR is characterized by three separate RRM motifs that recognize ARE sequences, which are typically found in the 3′-UTR of mRNAs. Interestingly, HuR was found to bind sequences in the 5′-UTR as well as near splice sites in pre-mRNAs [32]. RBPs and miRNAs can regulate their target genes in a variety of different ways, such as lowering or increasing the expression of a target mRNA by competing for binding sites on the mRNA. An example of this type of regulation is that of RBP HuR and miR-331-3p and their target, the ERBB2 oncogene. Binding of HuR to mRNA typically relieves the repression of translation by miRNAs such as miR-331-3p, which represses translation of ERBB2 mRNA [16, 32]. Translation of ERBB2 is inhibited by miR-331-3p through binding to distinct sites within the 3′-UTR that can be inhibited through binding of HuR. When HuR outcompetes miR331-3p for binding to the 3′-UTR of ERBB2 it leads to an increase in expression of ERBB2 [15, 16, 32].
An additional way that RBPs can regulate their targets is to make the binding site of the miRNA more or less accessible, such as in the case with HuR and let-7 in their regulation of the oncogene MYC. While HuR typically increases translation of its target by competing for binding with the inhibitory miRNA, it can also aid in the miRNA-mediated repression of translation of certain genes such as the well-known oncogene MYC by facilitating binding of the miRNA let-7 [2, 33–35]. Repression of MYC translation occurs through HuR binding-mediated opening of the mRNA structure of MYC that facilitates the binding of let-7, leading to a reduction in translation [15, 36]. While HuR can certainly facilitate down-regulation of MYC through let-7, the effect is dependent on the expression levels of let-7, which has been found down-regulated in a number of cancers such as lung cancer [36]. Interestingly, MYC signalling can stimulate the overexpression of a family of oncogenic miRNAs known as the miR-17-92 cluster, which are overexpressed in a variety of haematopoietic malignancies [37]. Therefore this regulatory network can be quite complex, given that a large number of different miRNAs are regulated either positively or negatively by HuR, which is often found deregulated in a variety of different cancers [15, 16, 38].
Intriguingly, it has been shown that HuR can both positively or negatively influence translation of certain oncogenes such as COX2, ERBB2 and MYC. These genes have been associated with driving the formation of tumours and have also been linked to the progression of the disease and formation of metastases as well [16, 28, 32, 39]. This demonstrates the potential of HuR to act as both an oncogenic RBP and a tumour suppressor RBP depending on the miRNA and mRNA. With the discovery of miRNAs that exert a regulatory effect on the translation and/or decay of mRNAs, it was found that certain miRNAs act a tumour suppressors such as let-7, miR-7-5p and miR-331-3p, as well as oncomiRs (oncogenic microRNAs), as exemplifed by the previously mentioned miR17-92 cluster [33, 34, 40].
While miRNAs and RBPs can either regulate genes in concert or exhibit opposing regulatory effects, they are also capable of regulating the translation and the activity of each other. Such is the case for miR-519, which has been found to reduce HuR expression at the level of translation by binding to either the 3′-UTR or the coding region of HuR mRNA, which results in decreased proliferation [32, 41, 42]. Interestingly, expression of miR-519 is inversely correlated with the expression of HuR which was found in healthy tissue as well as in human tumours [41, 42]. Another example of such regulation is that of miR-16, which binds to HuR mRNA resulting in a reduction in the cellular levels of HuR, and binding of HuR to miRNA-16 rapidly reduces steady-state levels of miR-16 [42–44]. This regulatory effect HuR and miR-16 have on each other indirectly affects their regulatory targets such as COX2, TNFα, Bcl-2 and Mcl-1. In a number of cancers, the mRNA stability factor HuR is overexpressed, such as colorectal cancer (CRC), which increases the cytoplasmic levels of HuR, reduces miR-16 levels and promotes translation of its target genes [42–44].
Aberrations in the activity of either HuR, miRNAs or their target mRNAs can result in the abnormal expression of certain oncogenes as described previously [45]. In addition, in a number of different cancers RNA-binding proteins responsible for maturation of miRNAs were associated with carcinogenesis [42–44]. Aberrations in any of the RBPs involved in miRNA maturation, such as Dicer, Exportin 5 and Ago2, promote oncogenesis in a number of different cancers by lowering the levels of mature miRNAs [46–49]. However, the resulting decrease in the levels of mature miRNA is non-specific and affects all expressed miRNAs. This is in contrast to the targeted down-regulation of specific pri-miRNAs processing, which is modulated through aberrations in RBPs such as DEAD-box 5 (DDX5/p68) and DDX17 (p72), both of which are overexpressed in breast, prostate and colon tumours [46–48]. Interestingly, DDX5 can interact with p53 to increase the processing of specific miRNAs that suppress growth, which is abolished when p53 is lost [50]. On the other hand, DDX17 is a known target of YAP, which is the main target of the Hippo signalling pathway. In tumour cells this signalling pathway is lost, leading to the nuclear translocation of YAP where it binds DDX17 resulting in the inactivation of the microprocessor complex in miRNA biogenesis [51]. Therefore, the numerous regulatory interactions between RBPs and miRNAs provide a potentially fertile area for the development of new therapeutics.
Rationale for the design of engineered RNA-binding proteins for treatment of genetic diseases
In recent years, many researchers have been unravelling the complex molecular architecture underlying the various types of cancer to improve current therapies as well as design of new drugs. While the number of potential new drug targets has significantly increased, the number of drugs that are approved by the FDA for use in clinical practice is still very low [52]. Despite this, the efficacy of treatment will still be limited due to the heterogeneity of tumours and rise of new drug-resistant cell populations. This occurs with the non-specific classical chemotherapy drugs as well as specific targeted therapies. Increased specificity comes with the caveat of increased susceptibility to the occurrence of drug resistance through a single mutation of the drug target or downstream targets. Most of these novel drugs are small molecules aimed at inhibiting a specific function at the protein level by preventing either their activation, the binding of a ligand or both (for example, tyrosine kinase inhibitors of the EGFR, such as erlotinib) [52–54]. However, none aim at selectively inhibiting or promoting expression and/or translation of both oncogenes and tumour suppressor genes at the level of the DNA and RNA using designer DNA-binding proteins and RBPs. Translational regulation of mRNA by miRNAs and RBPs, as described earlier, is an indication of their potential for the design of new highly specific RBPs that can target any dysregulated mRNA or miRNA in cancer. It should be possible, through the use of engineered RBPs, to specifically modulate cellular levels of certain key oncogenic and tumour suppressor miRNAs or mRNA translation in cancer either directly or indirectly.
Design of novel RNA-binding protein for treatment of cancer
A recent article by Chen et al. [17] demonstrated the potential of using RBPs to suppress mature miR-21 levels in tumour cells. In this work, a redesigned RBP was used to significantly reduce cancer cell viability through a reduction in maturation of miR-21. The RNA recognition motif (RRM) of the human RNA-binding protein Rbfox1 was redesigned, called Fox-RRM*, by mutating four amino acids, namely R118D, E147R, N151S and E152T, to accommodate and stabilize a modified interaction with RNA. These mutations allowed for Fox-RRM* to specifically recognize the motif UGAAUC that is present in the terminal loop of pre-miR-21 (Fig. 2). The binding affinity of Fox-RRM* to pre-miR-21, as determined by using isothermal titration calorimetry (ITC), was dramatically increased compared to wild-type Rbfox-RRM. This was confirmed using electrophoretic mobility shift assays (EMSA), and FOX-RRM* was shown to bind specifically to the terminal loop of pre-miR-21. Processing of pri- and pre-miR-21 by Drosha and Dicer, respectively, was assessed using an in vitro processing assay comparing wild-type Rbfox-RRM with Fox-RRM*, demonstrating that levels of pri- and pre-miR-21 were progressively lost as the protein concentration increased. In addition, Dicer from Giardia intestinalis was engineered by replacing its PAZ domain, which recognizes the 2-nt overhang at the 3′-end of double-stranded RNA (dsRNA), with Fox-RRM*. The recombinant RRM*-Dicer was shown to be able to effectively degrade pre-miR-21 in a time-dependent in vitro cleavage assay. In addition, it was able to suppress mature miRNA-21 levels in a specific and efficient manner resulting in decreased cell viability in HeLa cells, suggesting that this could be an effective treatment for cancer.
Fig. 2.
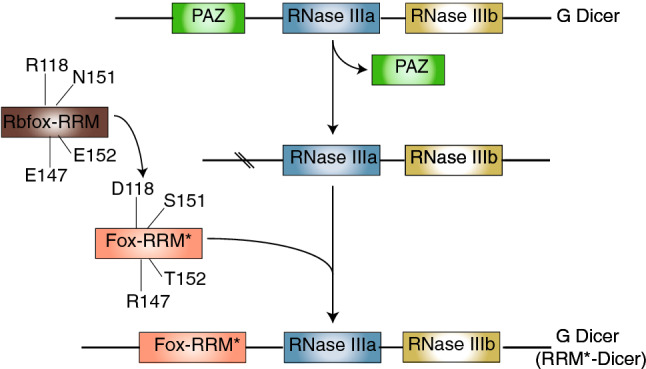
Schematic representation of the design of a novel miRNA binding protein [17]
Unfortunately, such an approach cannot simply be applied to other RRM domains or even other RBPs, as the typical affinity of a single RRM domain is only in the micromolar range and these have limited specificity on an individual basis. Rbfox-RRM and the human U1A-RRM are exceptions to this observation as they bind highly efficiently to a UGCAUG with nanomolar affinity or AUUGCAC with picomolar affinity in the secondary structures of their target RNAs, respectively [17, 55, 56]. High affinity in the nanomolar range or even lower is preferable as typically the protein concentration required to completely inhibit access to a target is higher than needed for effective binding. Such discrepancy between binding and inhibition has been demonstrated using Lin-28, which efficiently binds to the pre-miRNA Let-7 at 1 nM concentration; however, effective inhibition of Let-7 maturation occurs at a much higher concentration (~ µM) [57].
An alternative to the use of RRM domains for recognizing motifs in miR-21 or any other miRNA are the PUF proteins. PUF (Pumilio and FBF homology) proteins are a family of eukaryote-specific RBPs implicated in controlling development and differentiation through repression or activation of translation of their target RNAs [58–62]. PUF proteins bind to their target RNAs using a domain that typically consists of eight consecutive PUF repeats each approximately 36 amino acids in length that each can bind a single base (Fig. 3a). A set of specific residues within each PUF repeat facilitates RNA binding and, depending on the specific combination of amino acids in those positions, binds to a particular nucleotide with the exception of cytosine. Interestingly, no natural PUF proteins had been found that were capable of binding to cytosine [59]. Filipovska et al. showed that specific residues are required to bind cytosine that were identified through coupling of the evolution of PUF proteins to a life and death selection pressure using the yeast three-hybrid system in Saccharomyces cerevisiae [59]. The discovery of the amino acid code required for binding to cytosine has cleared a major hurdle that has deterred researchers so far from using PUF proteins for designing novel RBPs.
Fig. 3.
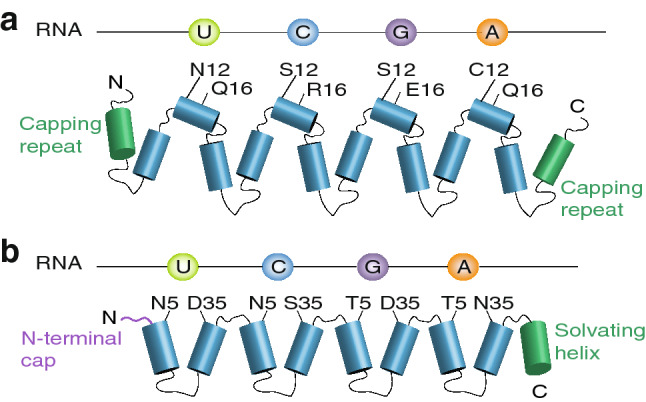
Schematic representations of the structures and binding codes of PUF (a) and PPR proteins (b)
The key advantage of having a simple one repeat:one nucleotide code for RNA recognition is the ability to target any RNA sequence of interest. As a result, a number of new applications for the use of PUF domains have been designed [63]. Ozawa and colleagues designed a split green fluorescent protein (GFP) system in which two fusion proteins are designed, each containing a PUF domain and either an N- or C-terminal fragment of GFP [64–68]. Upon binding to adjacent sites within the same RNA, the two GFP fragments come together and fluoresce. This method eliminates the background signal from GFP, as fluorescence is only observed when RNA-bound PUFs facilitate the folding of GFP fragments. An additional benefit of using this split GFP system is the increased specificity of the system for particular RNAs with the use of two PUF domains. A second application for PUFs involves their fusion to either a translational activator or translational repressor [69–73]. These fusion proteins were shown to be able to effectively reduce or increase translation of both luciferase reporters and endogenous mRNAs and could stimulate poly(A) tail addition or removal. The magnitude of translational control was proportional to the affinity of the PUF protein to its RNA target, providing additional flexibility to the approach. In another innovative example, Wang and colleagues attached a PUF protein to either a splicing activator or splicing repressor [74, 75]. They demonstrated that a fusion of a splicing repressor and a PUF domain designed to recognize a sequence in the exon extension region of Bcl could effectively increase the formation of the shorter pro-apoptotic Bcl-xS mRNA. In addition, several cell lines became increasingly more sensitive to cisplatin and paclitaxel when transfected with the previously mentioned fusion protein.
Another potential system for the sequence-specific targeting of RNAs is the pentatricopeptide repeat (PPR) RBPs, which contain short repeats of ~ 35 amino acids that are often found in tandem with between 2 and 33 individual PPR repeats within a single RBP (Fig. 3b) [76–79]. Each PPR repeat forms two anti-parallel α-helices that interact to establish a helix-turn-helix motif that through stacking of multiple repeats forms a super helix with an RNA-binding groove. Each PPR repeat is capable of recognizing a single nucleotide in a similar fashion to that of the PUFs, with the residues responsible for base recognition at positions 5 and 35 [76, 77, 79]. PPRs have been found to regulate many aspects of RNA metabolism, including RNA folding, stability, translation and modification [80–85], particularly in mitochondria and chloroplasts [86]. This nucleotide binding code has been used to design novel synthetic PPR proteins to bind diverse RNAs of interest. In addition, through adding regulatory domains to these repeat proteins or by harnessing their inherent ability to wrap around their ligands and block the activities of enzymes that act on them [87], these synthetic PPR proteins allow for the specific modulation of RNA metabolism and activity.
A key area for the development of potential RNA-targeting therapies is the issue of delivery. At present, the two main modes for delivering proteins into human cells use either viruses to deliver the genes encoding them, or direct protein delivery. The most common viral delivery approaches harness the adeno-associated virus (AAV) or lentiviruses. The latter results in the stable integration of the viral genome and is suited best for genetic engineering of actively dividing cells or stem cells [88, 89]. However, the former is best suited for short-term gene expression or when targeting terminally differentiated cells as the vectors remain in the cytoplasm. Protein delivery can be achieved using engineered virus-like particles (VLPs) or cell-penetrating peptides (CPPs). The former utilizes the viral protein Gag, also known as Gag-pol and is a polypeptide encoding for a number of different important viral proteins such as reverse transcriptase and integrase. Kaczmarczyk et al. designed a Gag-fusion protein based of an avian retrovirus, which cannot replicate in human nor does it contain a polymerase. Proteins of interest can be fused to the Gag protein, resulting in the formation of VLP particles containing up to 5000 copies of the Gag-fusion protein [90]. The latter are short synthetic or natural peptides (< 30 amino acids) that can either be fused to a protein of interest or co-administrated to enhance their uptake into the target cells [91].
Future strategies
Over the last few years, a number of highly specific RBPs have been identified and characterized such as the PUF and PPR proteins. With the modularity of these proteins and a known code to bind any nucleotide, it may be possible to engineer a RBD that can recognize any specific sequence of interest. In parallel, a large number of new examples of cross talk between RBPs and miRNA have been discovered. One such example of cross-regulation is miRNA synthesis, where a number of different RBPs interact and aid in the maturation of the miRNAs. A second case of cross-regulation occurs in the interactions between specific oncogenes and tumour suppressor genes, where translation is regulated either in concert or in opposition by specific miRNAs and RBPs. Interestingly, regulation of these genes has also been shown to occur through decreasing cellular levels of either their miRNA or RBP by preventing either their maturation (miRNAs) or inhibiting their translation (RBPs). RBDs with high specificity and affinity can be redesigned to recognize specific targets and subsequently exchanged with existing binding domains of proteins such as, but not limited to, nucleases, translational regulators and mRNA processing proteins. This will not only allow for the design of therapeutic strategies that enable strategic regulation of the transcriptome of tumour cells and aid in the treatment of cancer, but can also aid in our understanding of the biology underlying miRNA regulation as well as identification of their targets. The innovative example of a designed miRNA targeting artificial RBP and the potential of PUF and PPR proteins to create engineered mRNA and miRNA targeting RBPs has set the stage for an exciting new era in cancer research. These engineered RBPs could be a part of a multi-drug regimen to reinstate sensitivity to certain drugs to allow usage of lower doses of chemotherapeutics for a more efficient treatment while simultaneously minimizing the side effects.
Acknowledgements
Research in our groups has been supported by fellowships, scholarships and grants from the Australian Research Council (FT0991008, FT0991113, DP140104111 to A. F. and O. R.), the National Health and Medical Research Council (APP1058442, APP1045677 to A. F. and O. R.,APP1071081 and APP1084964 to P. L), and the Cancer Council Western Australia (to A. F. and O. R.).
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Erson AE, Petty EM. MicroRNAs in development and disease. Clin Genet. 2008;74:296–306. doi: 10.1111/j.1399-0004.2008.01076.x. [DOI] [PubMed] [Google Scholar]
- 2.Cowland JB, Hother C, Gronbaek K. MicroRNAs and cancer. APMIS. 2007;115:1090–1106. doi: 10.1111/j.1600-0463.2007.apm_775.xml.x. [DOI] [PubMed] [Google Scholar]
- 3.Lee RC. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-Y. [DOI] [PubMed] [Google Scholar]
- 4.Reinhart BJ, Slack FJ, Basson M, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 5.Hammond SM. An overview of microRNAs. Adv Drug Deliv Rev. 2015;87:3–14. doi: 10.1016/j.addr.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman RC, Farh KK-H, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee Y, Jeon K, Lee J-T, et al. MicroRNA maturation: stepwisee processing and subcellular localization. EMBO J. 2002;21:4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631–640. doi: 10.1016/j.cell.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 9.Denli AM, Tops BBJ, Plasterk RH, et al. Processing of primary microRNAs by the microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- 10.Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17:3011–3016. doi: 10.1101/gad.1158803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lund E, Güttinger S, Calado A, et al. Nuclear export of microRNA precursors. Science. 2004;80(303):95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi H, Tomari Y. RISC assembly: coordination between small RNAs and Argonaute proteins. Biochim Biophys Acta Gene Regul Mech. 2016;1859:71–81. doi: 10.1016/j.bbagrm.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Iwakawa HO, Tomari Y. The functions of MicroRNAs: mRNA decay and translational repression. Trends Cell Biol. 2015;25:651–665. doi: 10.1016/j.tcb.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 14.Meister G. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet. 2013;14:447–459. doi: 10.1038/nrg3462. [DOI] [PubMed] [Google Scholar]
- 15.Kim HH, Kuwano Y, Srikantan S, et al. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009;23:1743–1748. doi: 10.1101/gad.1812509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Epis MR, Barker A, Giles KM, et al. The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNA miR-331-3p in prostate cancer cells. J Biol Chem. 2011;286:41442–41454. doi: 10.1074/jbc.M111.301481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Yang F, Zubovic L, et al. Targeted inhibition of oncogenic miR-21 maturation with designed RNA-binding proteins. Nat Chem Biol. 2016;12:717–723. doi: 10.1038/nchembio.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kedde M, van Kouwenhove M, Zwart W, et al. A Pumilio-induced RNA structure switch in p27-3’ UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol. 2010;12:1014–1020. doi: 10.1038/ncb2105. [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Carmell MA, Rivas FV, et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305:1437–1441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- 20.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Boland A, Kuzuoǧlu-Öztürk D, et al. A DDX6-CNOT1 complex and W-binding pockets in CNOT9 reveal direct links between miRNA target recognition and silencing. Mol Cell. 2014;54:737–750. doi: 10.1016/j.molcel.2014.03.034. [DOI] [PubMed] [Google Scholar]
- 22.Ørom UA, Nielsen FC, Lund AH. MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell. 2008;30:460–471. doi: 10.1016/j.molcel.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 23.Vasudevan S, Tong Y, Steitz JA. Cell cycle control of microRNA-mediated translation regulation. Cell Cycle. 2008;7:1545–1549. doi: 10.4161/cc.7.11.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15:829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Auweter SD, Oberstrass FC, Allain FHT. Sequence-specific binding of single-stranded RNA: is there a code for recognition? Nucleic Acids Res. 2006;34:4943–4959. doi: 10.1093/nar/gkl620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neelamraju Y, Hashemikhabir S, Janga SC. The human RBPome: from genes and proteins to human disease. J Proteom. 2015;127:61–70. doi: 10.1016/j.jprot.2015.04.031. [DOI] [PubMed] [Google Scholar]
- 28.Pereira B, Billaud M, Almeida R. RNA-binding proteins in cancer: old players and new actors. Trends Cancer. 2017;3:506–528. doi: 10.1016/j.trecan.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Medioni C, Mowry K, Besse F. Principles and roles of mRNA localization in animal development. Development. 2012;139:3263–3276. doi: 10.1242/dev.078626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rackham O, Brown CM. Visualization of RNA-protein interactions in living cells: FMRP and IMP1 interact on mRNAs. EMBO J. 2004;23:3346–3355. doi: 10.1038/sj.emboj.7600341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Treiber T, Treiber N, Plessmann U, et al. A compendium of RNA-binding proteins that regulate MicroRNA biogenesis. Mol Cell. 2017;66:270–284. doi: 10.1016/j.molcel.2017.03.014. [DOI] [PubMed] [Google Scholar]
- 32.Srikantan S, Tominaga K, Gorospe M. Functional interplay between RNA-binding protein HuR and microRNAs. Curr Protein Pept Sci. 2012;13:372–379. doi: 10.2174/138920312801619394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302:1–12. doi: 10.1016/j.ydbio.2006.08.028. [DOI] [PubMed] [Google Scholar]
- 34.Cho WCS. OncomiRs: the discovery and progress of microRNAs in cancers. Mol Cancer. 2007;6:60. doi: 10.1186/1476-4598-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 36.Akao Y, Nakagawa Y, Naoe T. let-7 MicroRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull. 2006;29:903–906. doi: 10.1248/bpb.29.903. [DOI] [PubMed] [Google Scholar]
- 37.Babashah S, Soleimani M. The oncogenic and tumour suppressive roles of microRNAs in cancer and apoptosis. Eur J Cancer. 2011;47:1127–1137. doi: 10.1016/j.ejca.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 38.Meisner N-C, Filipowicz W. Properties of the regulatory RNA-binding protein HuR and its role in controlling miRNA repression. Adv Exp Med Biol. 2011;700:106–123. doi: 10.1007/978-1-4419-7823-3_10. [DOI] [PubMed] [Google Scholar]
- 39.Young LE, Moore AE, Sokol L, et al. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol Cancer Res. 2012;10:167–180. doi: 10.1158/1541-7786.MCR-11-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Webster RJ, Giles KM, Price KJ, et al. Regulation of epidermal growth factor receptor signaling in human cancer cells by MicroRNA-7. J Biol Chem. 2009;284:5731–5741. doi: 10.1074/jbc.M804280200. [DOI] [PubMed] [Google Scholar]
- 41.Abdelmohsen K, Kim MM, Srikantan S, et al. miR-519 suppresses tumor growth by reducing HuR levels. Cell Cycle. 2010;9:1354–1359. doi: 10.4161/cc.9.7.11164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abdelmohsen K, Srikantan S, Kuwano Y, Gorospe M. miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc Natl Acad Sci. 2008;105:20297–20302. doi: 10.1073/pnas.0809376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jing Q, Huang S, Guth S, et al. Involvement of microRNA in AU-rich element-mediated mRNA instability. Cell. 2005;120:623–634. doi: 10.1016/j.cell.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 45.Peng Y, Croce CM. The role of MicroRNAs in human cancer. Signal Transduct Target Ther. 2016;1:15004. doi: 10.1038/sigtrans.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Kouwenhove M, Kedde M, Agami R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat Rev Cancer. 2011;11:644–656. doi: 10.1038/nrc3107. [DOI] [PubMed] [Google Scholar]
- 47.Zhang L. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci USA. 2006;103:9136–9141. doi: 10.1073/pnas.0508889103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim MS, Oh JE, Kim YR, et al. Somatic mutations and losses of expression of microRNA regulation-related genes AGO2 and TNRC6A in gastric and colorectal cancers. J Pathol. 2010;221:139–146. doi: 10.1002/path.2683. [DOI] [PubMed] [Google Scholar]
- 49.Melo SA, Moutinho C, Ropero S, et al. A genetic defect in exportin-5 traps precursor MicroRNAs in the nucleus of cancer cells. Cancer Cell. 2010;18:303–315. doi: 10.1016/j.ccr.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki HI, Yamagata K, Sugimoto K, et al. Modulation of microRNA processing by p53. Nature. 2009;460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 51.Mori M, Triboulet R, Mohseni M, et al. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell. 2014;156:893–906. doi: 10.1016/j.cell.2013.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hebar A, Valent P, Selzer E. The impact of molecular targets in cancer drug development: major hurdles and future strategies. Expert Rev Clin Pharmacol. 2013;6:23–34. doi: 10.1586/ecp.12.71. [DOI] [PubMed] [Google Scholar]
- 53.Steins M, Thomas M, Geißler M. Recent results in cancer research. Cham: Springer; 2018. Erlotinib; pp. 1–17. [DOI] [PubMed] [Google Scholar]
- 54.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 55.Auweter SD, Fasan R, Reymond L, et al. Molecular basis of RNA recognition by the human alternative splicing factor Fox-1. EMBO J. 2006;25:163–173. doi: 10.1038/sj.emboj.7600918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oubridge C, Ito N, Evans PR, et al. Crystal structure at 1.92 A resolution of the RNA-binding domain of the U1A spliceosomal protein complexed with an RNA hairpin. Nature. 1994;372:432–438. doi: 10.1038/372432a0. [DOI] [PubMed] [Google Scholar]
- 57.Newman M, Thomson JM, Hammond SM. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA. 2008;14:1539–1549. doi: 10.1261/rna.1155108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wickens M, Bernstein DS, Kimble J, Parker R. A PUF family portrait: 3′UTR regulation as a way of life. Trends Genet. 2002;18:150–157. doi: 10.1016/S0168-9525(01)02616-6. [DOI] [PubMed] [Google Scholar]
- 59.Filipovska A, Razif MFM, Nygard KKA, Rackham O. A universal code for RNA recognition by PUF proteins. Nat Chem Biol. 2011;7:425–427. doi: 10.1038/nchembio.577. [DOI] [PubMed] [Google Scholar]
- 60.Filipovska A, Rackham O. Designer RNA-binding proteins: new tools for manipulating the transcriptome. RNA Biol. 2011;8:978–983. doi: 10.4161/rna.8.6.17907. [DOI] [PubMed] [Google Scholar]
- 61.Filipovska A, Rackham O. Modular recognition of nucleic acids by PUF, TALE and PPR proteins. Mol Biosyst. 2012;8:699. doi: 10.1039/c2mb05392f. [DOI] [PubMed] [Google Scholar]
- 62.Wang X, McLachlan J, Zamore PD, Hall TMT. Modular recognition of RNA by a human Pumilio-homology domain. Cell. 2002;110:501–512. doi: 10.1016/S0092-8674(02)00873-5. [DOI] [PubMed] [Google Scholar]
- 63.Wang Y, Wang Z, Tanaka Hall TM. Engineered proteins with Pumilio/fem-3 mRNA binding factor scaffold to manipulate RNA metabolism. FEBS J. 2013;280:3755–3767. doi: 10.1111/febs.12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ozawa T, Natori Y, Sato M, Umezawa Y. Imaging dynamics of endogenous mitochondrial RNA in single living cells. Nat Methods. 2007;4:413–419. doi: 10.1038/nmeth1030. [DOI] [PubMed] [Google Scholar]
- 65.Yoshimura H, Ozawa T. Real-time fluorescence imaging of single-molecule endogenous noncoding RNA in living cells. New York: Humana Press; 2018. pp. 337–347. [DOI] [PubMed] [Google Scholar]
- 66.Yoshimura H, Inaguma A, Yamada T, Ozawa T. Fluorescent probes for imaging endogenous β-actin mRNA in living cells using fluorescent protein-tagged pumilio. ACS Chem Biol. 2012;7:999–1005. doi: 10.1021/cb200474a. [DOI] [PubMed] [Google Scholar]
- 67.Yamada T, Yoshimura H, Inaguma A, Ozawa T. Visualization of nonengineered single mRNAs in living cells using genetically encoded fluorescent probes. Anal Chem. 2011;83:5708–5714. doi: 10.1021/ac2009405. [DOI] [PubMed] [Google Scholar]
- 68.Yoshimura H, Ozawa T. Methods in enzymology. Cambridge: Academic Press; 2016. Monitoring of RNA dynamics in living cells using PUM-HD and fluorescent protein reconstitution technique; pp. 65–85. [DOI] [PubMed] [Google Scholar]
- 69.Cooke A, Prigge A, Opperman L, Wickens M. Targeted translational regulation using the PUF protein family scaffold. TL-108. Proc Natl Acad Sci USA. 2011;108:15870–15875. doi: 10.1073/pnas.1105151108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cao J, Arha M, Sudrik C, et al. A universal strategy for regulating mRNA translation in prokaryotic and eukaryotic cells. Nucleic Acids Res. 2015;43:4353–4362. doi: 10.1093/nar/gkv290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Campbell ZT, Valley CT, Wickens M. A protein-RNA specificity code enables targeted activation of an endogenous human transcript. Nat Struct Mol Biol. 2014;21:732–738. doi: 10.1038/nsmb.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cao J, Arha M, Sudrik C, et al. Bidirectional regulation of mRNA translation in mammalian cells by using PUF domains. Angew Chem Int Ed Engl. 2014;53:4900–4904. doi: 10.1002/anie.201402095. [DOI] [PubMed] [Google Scholar]
- 73.Adamala KP, Martin-Alarcon DA, Boyden ES. Programmable RNA-binding protein composed of repeats of a single modular unit. Proc Natl Acad Sci USA. 2016;113:E2579–E2588. doi: 10.1073/pnas.1519368113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y, Cheong C-G, Hall TMT, Wang Z. Engineering splicing factors with designed specificities. Nat Methods. 2009;6:825–830. doi: 10.1038/nmeth.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang Y, Wang Z. Design of RNA-binding proteins: manipulate alternative splicing in human cells with artificial splicing factors. In: Clifton NJ, editor. Methods in molecular biology. New York: Humana Press; 2016. pp. 227–241. [DOI] [PubMed] [Google Scholar]
- 76.Coquille S, Filipovska A, Chia T, et al. An artificial PPR scaffold for programmable RNA recognition. Nat Commun. 2014;5:5729. doi: 10.1038/ncomms6729. [DOI] [PubMed] [Google Scholar]
- 77.Filipovska A, Rackham O. Pentatricopeptide repeats. RNA Biol. 2013;10:1426–1432. doi: 10.4161/rna.24769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schmitz-Linneweber C, Small I. Pentatricopeptide repeat proteins: a socket set for organelle gene expression. Trends Plant Sci. 2008;13:663–670. doi: 10.1016/j.tplants.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 79.Barkan A, Rojas M, Fujii S, et al. A combinatorial amino acid code for RNA recognition by pentatricopeptide repeat proteins. PLoS Genet. 2012;8:4–11. doi: 10.1371/journal.pgen.1002910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lopez Sanchez MIG, Mercer TR, Davies SMK, et al. RNA processing in human mitochondria. Cell Cycle. 2011;10:2904–2916. doi: 10.4161/cc.10.17.17060. [DOI] [PubMed] [Google Scholar]
- 81.Davies SMK, Lopez Sanchez MIG, Narsai R, et al. MRPS27 is a pentatricopeptide repeat domain protein required for the translation of mitochondrially encoded proteins. FEBS Lett. 2012;586:3555–3561. doi: 10.1016/j.febslet.2012.07.043. [DOI] [PubMed] [Google Scholar]
- 82.Rackham O, Filipovska A. The role of mammalian PPR domain proteins in the regulation of mitochondrial gene expression. Biochim Biophys Acta Gene Regul Mech. 2012;1819:1008–1016. doi: 10.1016/j.bbagrm.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 83.Liu G, Mercer TR, Shearwood AMJ, et al. Mapping of mitochondrial RNA-protein interactions by digital rnase footprinting. Cell Rep. 2013;5:839–848. doi: 10.1016/j.celrep.2013.09.036. [DOI] [PubMed] [Google Scholar]
- 84.Rackham O, Busch JD, Matic S, et al. Hierarchical RNA processing is required for mitochondrial ribosome assembly. Cell Rep. 2016;16:1874–1890. doi: 10.1016/j.celrep.2016.07.031. [DOI] [PubMed] [Google Scholar]
- 85.Siira SJ, Spåhr H, Shearwood AMJ, et al. LRPPRC-mediated folding of the mitochondrial transcriptome. Nat Commun. 2017;8:1532. doi: 10.1038/s41467-017-01221-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Small ID, Rackham O, Filipovska A. Organelle transcriptomes: products of a deconstructed genome. Curr Opin Microbiol. 2013;16:652–658. doi: 10.1016/j.mib.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 87.Spåhr H, Chia T, Lingford JP, et al. Modular ssDNA binding and inhibition of telomerase activity by designer PPR proteins. Nat Commun. 2018;9:2212. doi: 10.1038/s41467-018-04388-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cai Y, Mikkelsen JG. Lentiviral delivery of proteins for genome engineering. Curr Gene Ther. 2016;16:194–206. doi: 10.2174/1566523216666160527143702. [DOI] [PubMed] [Google Scholar]
- 89.Buchholz CJ, Friedel T, Büning H. Surface-engineered viral vectors for selective and cell type-specific gene delivery. Trends Biotechnol. 2015;33:777–790. doi: 10.1016/j.tibtech.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 90.Kaczmarczyk SJ, Sitaraman K, Young HA, et al. Protein delivery using engineered virus-like particles. Proc Natl Acad Sci. 2011;108:16998–17003. doi: 10.1073/pnas.1101874108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bolhassani A, Jafarzade BS, Mardani G. In vitro and in vivo delivery of therapeutic proteins using cell penetrating peptides. Peptides. 2017;87:50–63. doi: 10.1016/j.peptides.2016.11.011. [DOI] [PubMed] [Google Scholar]