

Abstract
The voltage-dependent potassium channel Kv1.3 participates in peripheral insulin sensitivity. Genetic ablation of Kv1.3 triggers resistance to diet-induced weight gain, thereby pointing to this protein as a pharmacological target for obesity and associated type II diabetes. However, this role is under intense debate because Kv1.3 expression in adipose tissue raises controversy. We demonstrated that Kv1.3 is expressed in white adipose tissue from humans and rodents. Moreover, other channels, such as Kv1.1, Kv1.2, Kv1.4 and especially Kv1.5, from the same Shaker family are also present. Although elevated insulin levels and adipogenesis remodel the Kv phenotype, which could lead to multiple heteromeric complexes, Kv1.3 markedly participates in the insulin-dependent regulation of glucose uptake in mature adipocytes. Adipocyte differentiation increased the expression of Kv1.3, which is targeted to caveolae by molecular interactions with caveolin 1. Using a caveolin 1-deficient 3T3-L1 adipocyte cell line, we demonstrated that the localization of Kv1.3 in caveolar raft structures is important for proper insulin signaling. Insulin-dependent phosphorylation of the channel occurs at the onset of insulin-mediated signaling. However, when Kv1.3 was spatially outside of these lipid microdomains, impaired phosphorylation was exhibited. Our data shed light on the putative role of Kv1.3 in weight gain and insulin-dependent responses contributing to knowledge about adipocyte physiology.
Electronic supplementary material
The online version of this article (10.1007/s00018-018-2851-7) contains supplementary material, which is available to authorized users.
Keywords: Adipose tissue, Potassium channels, Caveolae, Differentiation, Insulin
Introduction
Adipose tissue (AT) is crucial for buffering nutrient availability and demand by storing excess calories and preventing the toxic accumulation of surplus nutrients in non-adipose tissues. Because of the high incidence of obesity in industrialized countries, AT has attracted many investigations. White adipose tissue (WAT), even in a lean person, represents approximately 20% of body weight, and it can increase by more than 50% in morbidly obese patients. The primary function of WAT is to store lipids coming from the diet or synthetized from carbohydrate precursors and to release free fatty acids in response to various neural and hormonal stimuli. Moreover, WAT actively communicates with body organs, secreting lipid and protein factors that mediate local and systemic effects in nutrient intake, metabolism and energy expenditure at numerous levels, immunological responses and vascular physiology. In addition, WAT is regulated by sympathetic innervation and hormones such as insulin, catecholamine, thyroid and steroid hormones [1].
Insulin and catecholamines modulate the membrane potential in adipocytes, eliciting intracellular signaling [2]. Therefore, the role of ion channels in adipocytes is under investigation. Voltage-dependent K+ (Kv) currents in human and rat WAT present an activation threshold between − 20 and − 30 mV and are responsive to tetraethylammonium (TEA) [3, 4]. WAT and brown adipose tissue (BAT) share a Kv channel repertoire [3–5], with Kv1.3 being a putative candidate [6]. This is further supported by the obesity-resistant phenotype displayed by Kv1.3-null mice and the anti-obesity effect of the Kv1.3 blocker Shk-186 [7, 8]. In this context, Kv1.3 inhibition increases insulin sensitivity via GLUT4 translocation [9, 10]. However, the role of Kv1.3 in peripheral insulin responses has been highly questioned in skeletal muscle and AT [11]. Thus, the role of other Kv isoforms, mainly Kv1.5, in adipocyte glucose signaling is reported [12]. However, a main participation of Kv1.3 in insulin signaling is localized in the olfactory bulb, and strong evidence from BAT studies situates this channel as the most encouraging potential target for diabetes and obesity [13, 14].
Kv1.3 partially localizes in lipid raft microdomains whose lipid environment regulates channel activity [15–17]. Certain ion channels localize into lipid rafts, serving as targets for adjacent signaling molecules [18]. The adipocyte membrane is enriched in omega-shaped lipid raft structures named caveolae [19]. Caveolae, crucial for adipocytes physiology, serve as organizing centers, participating in signaling transduction, mechanosensing and mechanoprotection, lipotoxicity protection, endocytosis and lipid regulation [19–21]. Caveolin (Cav) is the major protein-mediating caveolae biogenesis and structure. Cav forms a family of three integral membrane proteins (Cav 1, Cav 2 and the muscular isoform Cav 3) [21]. Cav has a caveolin scaffolding domain (CSD) that can bind to other proteins via a caveolin-binding domain (CBD) [22]. We have recently identified a CBD at the N-terminal domain of Kv1.3 that is responsible for Cav association and lipid raft targeting of the channel [23]. This interaction is physiologically relevant in lymphocytes because, upon activation, Kv1.3 situates in the immunological synapse (IS), which is also enriched in rafts [24].
Taking all of this into account, we investigated the association of Kv1.3 with Cav 1 in adipocytes and whether this interaction affects localization and insulin signaling of the channel in adipocyte physiology. We found that although Kv1.3 is not the only Kv in WAT, the expression of the channel increases during adipogenesis, recruiting new channels into caveolae. In addition, our results demonstrated that mislocation of Kv1.3 in rafts impaired insulin-dependent phosphorylation of the channel. The role of ion channels in the AT physiology is an open debate and our data clearly situate Kv1.3 as a target in this complex scenario.
Methods
Cell culture, tissue handling and sample preparation
Rat adipocytes were isolated from epididymal WAT extracted immediately after euthanasia with CO2. Animals were moistened with 70% ethanol before the abdominal cavity was opened and WAT extracted. The tissue was immediately washed in dissociation media (in mM: 120 NaCl, 5 KCl, 1 CaCl2, 5 HEPES, 5 glucose, 3% albumin fraction V, pH 7.4) and digested in collagenase solution (0.5 mg/ml collagenase type II, 0.5 mg/ml DNase type I in dissociation buffer) for 30 min at 37 °C under agitation (60–70 rpm). Next, the suspension was filtered through a 100 µm Millipore filter and collected in a centrifuge tube. Dissociation buffer was added to fill the tube, and the cell suspension was centrifuged (400×g for 10 min). The resulting floating fad pat containing mature adipocytes was saved in a new tube. The pellet, containing the stromal vascular fraction (SVF), was subjected to erythrolysis for 5 min at 37 °C in 1 ml erythrolysis buffer (in mM: 155 NH4Cl, 5.7 K2HPO4, 0.1 EDTA). Afterwards, dissociation buffer was added and cells were centrifuged to stop erythrolysis (400×g for 5 min). Finally, the pellet was resuspended in adipocyte medium (DMEM supplemented with 10% FBS, 25 mM HEPES, 10,000 U/ml penicillin G and 10 mg/ml streptomycin), and 250,000 cells were plated in 35 mm diameter dishes containing poly-lysinated coverslips. Cells were cultured in adipocyte media supplemented with 500 μIU/ml of insulin. Three drops of the floating fat pad containing mature adipocytes were added. The medium was replaced after 2 days with 10 or 100 μIU/ml insulin supplemented with complete medium, as indicated. Differentiated preadipocytes were used 4–7 days after culture (fresh medium was added every 2 days). All products were from Sigma-Aldrich except FBS, which was purchased from Linus.
Subcutaneous human fat tissue was obtained from normal-weight subjects (BMI < 27 kg/m2) undergoing abdominal surgery or surgical mammary reduction. Fresh human adipose tissue was immediately processed as above to obtain differentiated adipocytes for immunocytochemistry. Human breast biopsies were obtained and treated as previously described for immunohistochemistry [25]. All human procedures were in accordance with current local guidelines and the Declaration of Helsinki. Experiments and surgical protocols were performed in accordance with the guidelines approved by the ethical committee of the Universitat de Barcelona following the European Community Council Directive 86/609 EEC.
3T3-L1 pre-adipocytes were cultured in DMEM containing 10% NCS at 37 °C in a 7% CO2 atmosphere. To induce differentiation, post-confluent 3T3-L1 pre-adipocytes were treated with 0.5 mM IBMX, 10 mg/ml insulin and 1 μM dexamethasone in DMEM containing 10% FBS for 2 days. Then, cells were transferred into DMEM with 10 mg/ml insulin and 10% FBS for 2 additional days. Afterwards, cells were maintained in DMEM containing 10% FBS, and fresh medium was added every other day until day 9, when the cells were fully differentiated into adipocytes.
HEK 293 cells were grown in DMEM containing 10% FBS and 100 U/ml penicillin/streptomycin (Gibco). Jurkat T-lymphocytes were cultured in RPMI containing 10% FBS and supplemented with 100 U/ml penicillin/streptomycin and 2 mM l-glutamine (Gibco).
RNA isolation, RT-PCR analysis and real-time PCR
Total RNA from cells and rat WAT and brain was isolated using a NucleoSpin RNA II (Macherey-Nagel, Germany). RNA was treated with DNase I, and PCR controls were performed in the absence of retrotranscriptase. cDNA synthesis was performed using transcriptor RT (Roche Applied Science) with random hexanucleotide and oligo dT primers, according to the manufacturer’s instructions. Once cDNA was synthesized, the conditions were set for further PCR using the Phire Hot Start II DNA polymerase (Finnzymes, Thermo Scientific), following the manufacturer’s instructions, for 35 cycles. The final PCR reactions (20 μl) were electrophoresed in a 1% agarose TBE gel (in mM: 40 Tris, 20 acetic acid, 1 EDTA, pH 8.0). Primers, accession numbers, annealing temperatures and the amplicon lengths for every gene are shown in Table 1.
Table 1.
Gene name, accession number, primer sequence (F: forward; R: reverse), annealing temperature and amplicon length (bp) of the Kv channels analyzed
| Gene | Accession number | Sequence | Annealing (°C) | Amplicon (bp) |
|---|---|---|---|---|
| Kcna1 | NM_173095 | F: 5′ GTCATGGTCATCCTCATCTCCAT 3′ | 60.9 | 703 |
| R: 5′ ACAATGACAGGTACGGGCAGGGCA 3′ | ||||
| Kcna2 | NM_012970 | F: 5′ TCCCTGGGCACCCACAGGAC 3′ | 62.5 | 794 |
| R: 5′ TGCTGGCCTTGCTGAGCGTC 3′ | ||||
| Kcna3 | NM_019270 | F: 5′ CTCATCTCCATTGTCATCTTCTGA 3′ | 61 | 718 |
| R: 5′ TTGAATTGGAAACAATCAC 3′ | ||||
| Kcna4 | NM_012971 | F: 5′ GACCTGATGCCCAGTGGCTC 3′ | 63 | 777 |
| R: 5′ TGTGCCCTGAGTTCTCCAGGTG 3′ | ||||
| Kcna5 | NM_012972 | F: 5′ GGATCACTCCATCACCAG 3′ | 61 | 334 |
| R: 5′ GGCTTCCTCCTCTTCCTTG 3′ |
Real-time PCR (RT-PCR) was performed using a 7500 real-time system machine with TaqMan Universal Maxter Mix II, no uracil N-glycosylase, according to the manufacturer’s instructions (Applied Biosystems). Validated rat TaqMan probes (Applied Biosystems) were used on cDNA retrotranscribed from 0.5 µg RNA, as described above.
Electrophysiology
Ionic currents were recorded using the whole-cell configuration of the patch-clamp technique as described previously [3, 4]. K+ currents were recorded using an EPC-10 (HEKA), and the appropriate software was used for data recording and analysis. Ionic currents were capacitance and leakage current compensated by a P/4 protocol sampled at 10 kHz (Digidata 1440; Molecular Devices) and filtered at 2.9 kHz. Patch electrodes of 2–4 MΩ were fabricated in a vertical puller (L/M 3P-A, List-Medical) from soda glass (Deltalab). The composition of the external control solution was (in mM): 140 NaCl, 2.7 KCl, 2.5 CaCl2, 1 MgCl2, 10 HEPES and 5 glucose; pH 7.4. When indicated, a high K+ external solution was used (in mM: 62.7 NaCl, 80 KCl, 2.5 CaCl2, 1 MgCl2, 10 HEPES and 5 glucose; pH 7.4). The standard solution used to fill the electrodes contained (in mM): 90 K+-glutamate, 55 KCl, 2 MgCl2, 10 HEPES and 5 EGTA; pH 7.4. To evoke voltage-gated currents, cells were stimulated with 250 ms square pulses ranging from − 60 either to + 60 or to + 80 mV in 10 mV steps from a holding potential of − 80 mV. The peak amplitude (pA) was normalized using the capacitance values (pF). The activation voltage dependence was analyzed using the same protocol in high K+ external solution and the inward peak data was measured. The activation curve was fitted with the Boltzmann equation:
where k represents the slope factor, V is the test potential and V1/2 is the potential at which the conductance was half maximal. To analyze the cumulative inactivation, currents were elicited by a train of seven depolarizing voltage steps of 250 ms from − 60 to + 60 mV once every 400 ms.
Steady-state inactivation was studied by applying a train of 500 ms pulses, ranging from − 120 to + 60 mV in 10 mV steps (prepulse), followed by a 500 ms pulse at + 60 mV in high K+ external solution. The pulse peak amplitude was collected to obtain the inactivation curve fitted with the Boltzmann equation as follows:
where Imax is the peak current measured after the most hyperpolarizing prepulse, and Ic is the smallest non-zero peak current obtained after a depolarizing prepulse.
Data analysis was performed using FitMaster (HEKA) and Sigma Plot 10.0 software (Systat Software). All recordings were performed at room temperature (RT). Toxins, TEA, Bupivacaine and Psora-4, diluted in H2O or DMSO, respectively, were applied and the inhibition of the peak current at + 60 mV calculated.
Glucose uptake
Adipocytes were prepared from rat epididymal adipose tissue. Tissue was minced and digested with collagenase in Krebs–Ringer–HEPES (KRH) buffer (in mM: 137 NaCl, 4.7 KCl, 1.15 MgSO4, 1.18 KH2PO4, 2.5 CaCl2, 20 HEPES, pH 7.4). Digested sample was filtered, and the infranatant was removed, and the floating layer of adipocytes was washed four times with fresh buffer. Adipocytes were incubated in KRH buffer in the presence or absence of 10 µM insulin for 30 min at 37 °C. When indicated, cells were treated with 100 nM Margatoxin 15 min before insulin treatment. Glucose uptake started by adding 5 mM 2-deoxyglucose and 0.1 mCi/ml 2-deoxy[H3]glucose for 15 min. Next, transport was stopped by adding cold STOP solution containing 50 mM d-glucose in PBS. Cells were lysed for 1 h in 0.1 N NaOH, 0.1% SDS and radioactivity was measured. Uptake is expressed per mg of protein, determined using the Bradford assay.
Immunocytochemistry, plasma membrane lawns (PML) and transmission electron microscopy
Adipocytes and 3T3-L1 cells were seeded on poly-d-lysine-treated coverslips. Cells were washed in phosphate-buffered saline without K+ (PBS-K+) and fixed with 4% paraformaldehyde for 10 min. Cells were permeabilized using 0.1% Triton X-100, Gly 20 mM in PBS-K+ for 10 min at room temperature (RT). After 60 min in blocking solution (1% BSA, 20 mM Gly, 0.05% Triton X-100, PBS-K+), cells were treated with either mouse anti-Kv1.3 (1/20, Neuromab) or rabbit anti-Kv1.3 (1/20, Alomone), rabbit anti-Kv1.5 (1/20, Alomone), rabbit anti-caveolin and rabbit anti-clathrin heavy chain (1/100, BD Biosciences) antibody in 1% BSA, 20 mM Gly, 0.05% Triton X-100 in PBS-K+ and again incubated for 90 min. After 3 washes, preparations were incubated for 60 min with Alexa-Fluor-488 or with Cy5 conjugated antibody (1:200; Molecular Probes), washed and mounted in Mowiol (Calbiochem). All procedures were performed at RT.
Plasma membrane lawns preparations from isolated adipocytes and 3T3-L1 adipocytes were obtained via osmotic shock, as previously described, with minor modifications [26]. Briefly, cells were washed twice in PBS-K+ and cooled on ice for 5 min. Next, cells were incubated for 5 min in 1/3 KHMgE (in mM: 70 KCl, 30 HEPES, 5 MgCl2, 3 EGTA, pH 7.5) and gently washed with non-diluted KHMgE to induce hypotonic shock. Burst cells were removed from the coverslip by pipetting up and down. After two washes with KHMgE buffer, only membrane sheets remained attached. PML were fixed with fresh 4% paraformaldehyde for 10 min at RT and mounted in Mowiol mounting media.
For transmission electron microscopy, PML were treated as performed for immunocytochemistry, but visualized with different secondary antibodies. Kv1.3 and caveolin were recognized by anti-Kv1.3 monoclonal (1/20, Neuromab) and rabbit anti-caveolin (1/100, BD Biosciences) antibodies. Goat anti-mouse and anti-rabbit secondary antibodies, conjugated to 10 nm and 15 nm gold particles, recognized Kv1.3 and Cav1, respectively. Briefly, samples were further fixed with 2.5% glutaraldehyde in PBS for 30 min at RT. Next, samples were subjected to freeze-drying, washed and cryoprotected with 10% methanol. Samples were then cryofixed using slam-freezing (BAF-060, Bal-Tec) for 90 min at − 90 °C and 10–7 mbar pressure. Replicas were obtained by rotationally (136 rpm) evaporating 1 nm platinum through an electron cannon (at an angle of 23°). This was reinforced by evaporating 10 nm carbon (at an angle of 75°). Replicas were separated from the sample using 30% hydrofluoric acid. Finally, samples were washed and mounted over Formvar coated grilles.
Double-labeling indirect immunofluorescence to measure fluorescence resonance energy transfer (FRET)
We performed indirect double-labeling immunofluorescence combined with conventional confocal laser scanning microscopy to measure FRET as previously described [27]. Briefly, adipocytes were immunolabelled as detailed above. Kv1.3 was tagged with the donor fluorophore (A488) and clathrin or caveolin with the acceptor (Cy3 or A546, respectively). Samples were imaged with a Leica SP2 confocal microscope. Images were acquired [donor: Ar laser at 7% and emission filter at (525–560 nm); acceptor: He/Ne laser at 5% and emission filter (640–670 nm)] before and after A488 bleach (3 frames with laser 514 nm at 100%). FRET efficiency was calculated using the equation:
where FDafter is the donor fluorescence (A488) after and FDbefore is the donor fluorescence before acceptor (Cy3 or A546) bleach. Analysis was performed using ImageJ.
Oil Red O staining
3T3-L1 cells, washed twice in PBS and fixed in 4% paraformaldehyde at RT for 10 min, were stained with Oil Red O (stock solution: 0.5 g/100 ml dissolved in isopropanol; working solution: 60% Oil Red O stock solution and 40% distilled water) at RT for 1 h. Next, cells were washed with water, and the stained fat droplets in the cells were visualized by light microscopy.
Protein extraction, co-immunoprecipitation and western blot analysis
Cells, washed in cold PBS, were lysed on ice with lysis buffer (1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 50 mM Tris–HCl, pH 7.5) supplemented with 1 μg/ml of aprotinin, 1 μg/ml of leupeptin, 1 μg/ml of pepstatin and 1 mM of phenyl-methylsulfonyluoride to inhibit proteases. Homogenates were centrifuged at 16,000g for 15 min at 4 °C, and the protein content was measured using Bradford assay.
For immunoprecipitation, samples were precleared with 50 μl of protein A or G-Sepharose beads for 2 h at 4 °C with gentle mixing as part of the co-immunoprecipitation procedure. The beads were then removed by centrifugation at 1000×g for 30 s at 4 °C. Samples were incubated overnight with Sepharose beads previously coated with anti-caveolin (BD transduction), anti-Kv1.3 (Neuromab), anti-Kv1.5 (Alomone) or anti-phosphotyrosine (Sigma) antibody (4 ng/μg protein) at 4 °C with gentle agitation. The beads were removed by centrifugation at 1000×g for 30 s at 4 °C, washed four times in wash buffer (0.1% Triton X-100, 10% Glycerol, 150 mM NaCl, 50 mM HEPES, pH 7.4), and resuspended in 100 μl of Laemmli SDS buffer.
Protein samples (50 μg), raft fractions (50 μl) and immunoprecipitates were boiled in Laemmli SDS loading buffer and separated by 10% SDS-PAGE. Next, samples were transferred to PVDF membranes (Immobilon-P, Millipore) and blocked with 5% dry milk supplemented with 0.05% Tween 20 in PBS. The filters were then immunoblotted with specific antibodies: anti-caveolin (1/200, BD Biosciences), anti-Kv1.3 (1/200, Neuromab), anti-Kv1.3 (1/200, Alomone), anti-clathrin (1/1000, BD Biosciences), anti-flotillin (1/1000, BD Biosciences), anti-βactin (1/50.000, Sigma), anti-Glut4 (1/500, OSCRX), anti-Insulin Receptor β (1/500, BD Transduction Laboratories). Finally, filters were washed with 0.05% Tween 20 PBS and incubated with horseradish peroxidase-conjugated secondary antibodies (BioRad).
Raft isolation
Low density, Triton-insoluble complexes were isolated, as previously described [15, 16]. Briefly, cells were homogenized in 1 ml of 1% Triton X-100 MBS (150 mM NaCl, 25 mM 2-morpholinoethanesulfonic acid 1-hydrate (MES), pH 6.5). After three washes in PBS, sucrose in MBS was added to a final concentration of 40%. A 5–30% linear sucrose gradient was layered on top and further centrifuged (39,000 rpm) for 20–22 h at 4 °C in a Beckman SW41 rotor. Gradient fractions (1 ml) were collected from the top and analyzed by Western blot.
Statistics
The results are expressed as the mean ± SE. Student’s t test, Paired t test or one-way ANOVA and Tukey’s post hoc test were used for statistical analysis (GraphPad PRISM v5.01). p < 0.05 was considered statistically significant.
Results
Adipocytes exhibit multiple voltage-dependent K+ current phenotypes
Potassium currents in adipocytes have been reported for more than 20 years [4]. However, the identification and role of the molecular candidates is still an open debate. In this context, rat adipocytes were differentiated from SVF and voltage-dependent K+ currents were elicited (Fig. 1). Following 250 ms pulses from − 60 to + 60 mV and based on the C-type inactivation of Kv currents, adipocytes exhibited three different phenotypes: (1) currents with almost no inactivation (< 10%), (2) intermediate inactivation (10–30%) and (3) a pronounced (> 30%) inactivation (Fig. 1a–c). All three groups showed similar current densities and activation thresholds were around − 40 mV (Fig. 1d). This rather negative threshold could be due to the high external potassium in which these experiments were conducted [28]. Steady-state activation and inactivation parameters were analyzed (Fig. 1d–g). Half-activation voltages steadily shifted to more depolarized potentials concomitantly with minor inactivating phenotypes (Table 2). V0.5 was − 18.6 ± 2.6, − 15.3 ± 1.6 and − 11.7 ± 0.8 mV for > 30, 10–30 and < 10%, respectively (Fig. 1d, e). No differences in the k slope were found. In addition, steady-state inactivation showed similar behavior (Fig. 1f, g). Thus, V0.5 were − 27.4 ± 1.0, − 24.9 + 0.5 and − 21.2 ± 0.6 for > 30, 10–30 and < 10%, respectively, whereas k slopes were again similar (Table 2).
Fig. 1.

Multiple voltage-dependent K+ current phenotypes in adipocytes. Adipocytes were isolated from rat epididymal SVF and Kv currents were elicited by applying 250 ms pulses from − 60 to + 60 mV in 10 mV steps. a Kv currents with poor inactivating characteristics (< 10%). b Kv currents with intermediate inactivation (10–30%). c Kv currents with pronounced inactivation (> 30%). d Steady-state activation, measured in high K+ external solution, of three phenotypes observed in adipocytes. e Half-maximal activation (V0.5) of Kv currents. f Steady-state inactivation of Kv currents observed in the three groups of adipocytes. g Half-maximal inactivation (V0.5) of Kv currents. Circles and columns: white, < 10%; gray, 10–30%; black, > 30%. Statistical analysis was performed by one-way ANOVA (n = 4–8 independent cells for each group). For detailed post hoc analysis, see Table 2
Table 2.
Steady-state activation and inactivation of the three differentiated voltage-dependent K+ current phenotypes
| Group | Activation | Inactivation | ||
|---|---|---|---|---|
| V0.5 (mM) | k | V0.5 (mM) | k | |
|
Poor inactivation < 10% |
− 11.7 ± 0.8 | 12.8 ± 0.8 | − 21.2 ± 0.6 | 6.8 ± 0.5 |
|
Moderate inactivation 10–30% |
− 15.3 ± 1.6 | 17.9 ± 1.5 | − 24.9 ± 0.5 | 6.5 ± 0.4 |
|
Pronounced inactivation > 30% |
− 18.6 ± 2.6 | 12.9 ± 2.3 | − 27.4 ± 1.0 | 6.8 ± 0.8 |
Adipocytes were clearly differentiated in poor, moderate and pronounced for < 10, 10–30 and > 30% of inactivation, respectively. Values are mean ± SE from 4 to 12 different cells. Statistics: p = 0.0218 and p < 0.0001 for half-activation and half-inactivation voltages, respectively (one way ANOVA). Post-hoc Tuckey test: p < 0.05, < 10 vs > 30% (activation); p < 0.001, < 10 vs 10–30 and < 10 vs > 30% (inactivation)
Adipocytes express different Kv1 channels
Although evidence indicates that not only Kv1.3 but also Kv1.5 could notably contribute to Kv currents in adipocytes, others claim no expression [10–12]. Unlike Kv1.5, Kv1.3 shows pronounced current inactivation and a greater negative half-activation voltage [29]. In addition, during microglia proliferation, macrophage activation and vascular muscle hyperplasia, shifts between Kv1.3 and Kv1.5 are documented [29–31]. However, other isoforms, forming homo- or heteromeric structures with Kv1.3 and Kv1.5, could contribute to the heterogeneous phenotype observed in Fig. 1 [32–34]. In this scenario, we characterized the molecular entities contributing to the Kv currents in adipocytes. A complex repertoire containing Kv1.1–Kv1.5 isoforms was confirmed by an PCR analysis (Fig. 2a). Because adipocytes from different anatomical depots are intrinsically different as a result of genetic or developmental events [35] we also analyzed whether different fat depots shared Kv genetic background. The mRNA expression of members of the Kv1 family, further extended to other channels documented in WAT (http://www.proteinatlas.org/humanproteome) such as Kv2.1, KCa1.1 [36, 37], are shown in Fig. 2b. While denoting some differences, adipocytes from several fat depots shared similar K+ channel expression profile.
Fig. 2.

Adipocytes express a repertoire of Kv1 channels. a Kv1.1–Kv1.5 mRNA expression in rWAT. Total RNA was isolated from epididymal WAT and PCR was performed and products were run in a 1% TBE agarose gel. Brain was used as a positive control. (+) Presence or (−) absence of retrotranscriptase (RT) reaction. MM molecular markers in base pairs (bp). b mRNA expression of Kv1.1–Kv1.5, Kv2.1 and KCa1.1 in different rat fat depots. RT-PCR was performed as described in methods and relative expression calculated by applying 2(−∆∆Ct). 18s ribosomal RNA was used as a loading control and epididymal data was taken as reference (value of 1). Color code is indicated at the inset. c Kv currents were elicited in adipocytes from SVF by applying 250 ms pulses from − 60 to + 80 mV and back to − 60 mV. Pharmacological profile of Kv currents of adipocytes. K+ currents were measured and 10–100 nM MgTx, 100 nM Psora-4, 100 nM α-DTX, 100 nM IbTx, 2 mM TEA, and 100 µM Bupivacaine (Bpv) were added to the bath in independent experiments. The percentage of inhibition was calculated as the current blocked respect to the initial current. Values are the mean ± SE, n = 6–8. *p < 0.05, **p < 0.01, ***p < 0.001 vs control (no additions) Paired t test. d–j Kv currents were elicited in adipocytes differentiated from epididymal SVF by applying the indicated protocol. Kv currents with pronounced inactivation (> 30%, d) and poor inactivation (< 10%, e–j) were analyzed. d Currents were elicited 250 ms pulses from − 60 to + 80 mV and back to − 40 mV. e–j Currents were obtained by 250 ms pulses from − 60 to + 60 mV and back to − 60 mV. Poorly inactivating currents were inhibited by e 2 mM TEA, f 100 nM Psora-4 and g, h 20 nM MgTx. i, j Cumulative inactivation of Kv currents was obtained by a train of seven depolarizing voltage steps of 250 ms from − 60 to + 60 mV once every 400 ms. i Representative current traces. j Intensity of the current vs the applied pulse
K+ currents were recorded in the presence of different toxins and chemicals (IC50 and KD shown in Supplementary Table 1). Differentiated adipocytes exhibited Kv currents that were partially sensitive to MgTx (10–100 nM), which blocks Kv1.1, Kv1.2 and Kv1.3 (Fig. 2c). The presence of 100 nM Psora-4, which also blocks Kv1.3, Kv1.2 and Kv1.5, also inhibited Kv currents (Fig. 2c). The pharmacological profile of Kv currents was further extended by additional chemicals and toxins. While 2 mM TEA inhibited the currents by 50%, 100 nM α-DTX did not. IbTx (100 nM) revealed a minimal KCa1.1 contribution in the presence of 5 mM EGTA. Although this profile could suggest a complex Kv scenario in adipocytes, higher TEA concentrations (3–60 mM) inhibited Kv currents from 50 to 80% supporting Kv1.3. MgTx and Psora also suggested that Kv1.3, and possibly Kv1.2, could be involved. The effect of 100 nM Psora-4 would indicate the presence of Kv1.5 and also Kv1.2. TEA and MgTx would also suggest Kv1.1 and Kv1.2, but the limited effect of α-DTX gives to these channels a relative minor role [38, Supplementary Table 1]. While TEA would also support a role for Kv2.1, in this scenario 100 µM Bupivacaine would reinforce the contribution of Kv1.5. Our results could suggest the presence of not only homotetrameric but also multiple heteromeric Kv1 forms with mixed pharmacology, as previously described [29, 34]. To further demonstrate the participation of Kv1.3, we analyzed specific biophysical and pharmacological properties. Kv blockers were applied in cells exhibiting pronounced and poor inactivating Kv currents. Inactivating Kv currents were partially sensitive to 20 nM MgTx and the additional presence of 100 nM Psora-4, which also blocks Kv1.5, further inhibited Kv currents (Fig. 2d). In addition, TEA, Psora-4 and MgTx partially blocked poorly inactivating (< 10%) Kv currents (Fig. 2e–h). Interestingly, the MgTX effect was more notable in cells exhibiting pronounced (> 30%) inactivation. Finally, following a train of depolarizing pulses a cumulative inactivation, a characteristic of Kv1.3, was apparent in cells with poor inactivating phenotype (Fig. 2i, j).
Insulin remodels Kv currents in adipocytes
Kv channels participate in insulin-dependent regulation of body weight. Insulin promotes adipogenesis as well as enhances K+ currents in adipocytes [39]. In addition, high plasma insulin levels are associated with obesity and type II diabetes and Kv1.3 has been implicated in these processes [8, 9]. Therefore, we further characterized whether insulin remodeled Kv currents in differentiated adipocytes. In this scenario, SVF-derived adipocytes were cultured in physiologically low (10 μIU/ml) and high (100 μIU/ml) insulin media, mimicking plasma concentrations from healthy and obese humans, respectively. As expected, high insulin increased Kv currents (Fig. 3a). At + 60 mV depolarizing pulses, high insulin rose currents by 83% (1.2 ± 0.1 vs 2.2 ± 0.1 nA for 10 and 100 μIU/ml, respectively). Concomitantly, an increase in cell capacitance, most likely due to cell size augmentation by lipid deposition under high insulin treatment, was associated with 100 μIU/ml insulin (Fig. 3b). In this context, Kv current density was still elevated under high insulin, despite the cell growth (34.5 ± 2.1 and 47.9 ± 1.9 pA/pF for 10 and 100 µIU/ml, respectively at + 60 mV) (Fig. 3c). Interestingly, while 100 µIU/ml insulin notably reduced the C-type inactivation of Kv currents by 30% (Fig. 3d) slightly increased the percentage of cells that exhibited currents with cumulative inactivation (Table 3). Analyzing further, this specific electrophysiological feature, we found that high insulin levels remodeled the Kv current phenotype (Fig. 3e). Thus, while 100 μIU/ml insulin almost doubled the frequency of poor inactivating cells (20.5 vs 38.2%), it markedly decreased the abundance of pronounced inactivating cells (38.6 vs 22.4% of > 30%), with no major changes in intermediate (40.9 vs 39.5% of 10–30%) inactivation. Furthermore, the percentage of cumulative inactivation of the Kv currents remained similar (Table 3). These data indicates a remodeling of Kv channels under high insulin stimulation in WAT cells. Concomitantly, the mRNA expression of Kv1.1–1.5, Kv2.1, KCa1.1 and Kvβ1–3 regulatory subunits, which affect the inactivation properties of Kv1 channels, was differentially regulated (Fig. 3f). While high insulin (100 µIU/ml) increased the mRNA of Kv1.3, Kv2.1 and KCa1.1, the expression of Kv1.5 decreased. In this context, the mRNA of different Kvβ regulatory subunits was differentially regulated upon high insulin treatment. Our results indicate that multiple K+ channel subunits are expressed in adipocytes and their expression was remodeled under high insulin condition. In addition, the formation of not only homotetrameric but also variable heteromeric structures with altered pharmacology should not be discarded, as previously described [29, 34].
Fig. 3.

High insulin concentrations remodeled Kv currents in adipocytes. SVF-derived adipocytes were incubated 4–7 days in low (10 µIU/ml) or high (100 µIU/ml) insulin concentrations following what described in the methods section. From a holding potential of − 60 mV, Kv currents were elicited by applying 250 ms pulses from − 60 to + 60 mV in 10 mV steps. a Current–voltage relationship in both conditions. b Cell capacitance in pF. c Current density (pA/pF) vs voltage (mV) plot of K+ currents in both conditions. d Percentage of inactivation at + 60 mV. Data are the mean ± SE of 44 (10 µIU/ml) and 76 (100 µIU/ml) cells. Circles and columns: white, 10 µIU/ml; black, 100 µIU/ml insulin. Statistical significance is denoted as *p < 0.05, **p < 0.01 and ***p < 0.001 vs 10 µIU/ml Student’s t test. e Distribution of cells according to their inactivation percentage at + 60 mV: poor (< 10%), intermediate (10–30%) or pronounced (> 30%) inactivation, at each insulin concentration. A high insulin concentration triggers a notable switch in the Kv phenotype. Black, < 10%; gray; 10–30%; white, > 30%. f RT-PCR was performed from mRNA purified from isolated epididymal adipocytes as described in methods and the relative expression was calculated by applying 2(−∆∆Ct). 18s ribosomal RNA was used as a loading control and 10 µIU/ml data was taken as reference (value of 1). Adipocytes were incubated in the presence of 10 and 100 µIU/ml for 3 days. Data are the mean ± SE of three independent rats each performed in triplicate
Table 3.
Cumulative inactivation of Kv currents in adipocytes
| Group of cells | Insulin (µIU/ml) | Percentage of cells with cumulative inactivation | Percentage of cumulative inactivation |
|---|---|---|---|
| <10% inactivation | 10 | 50 | 11.7 ± 0.4 (2) |
| 100 | 54.5 | 16.1 ± 3.1 (6) | |
| 10–30% inactivation | 10 | 60 | 11.5 ± 2.9 (6) |
| 100 | 70 | 14.2 ± 1.9 (14) | |
| >30% inactivation | 10 | 41.2 | 15.4 ± 5.2 (7) |
| 100 | 46.7 | 13.9 ± 3.5 (7) |
Adipocytes were differentiated in poor (< 10%), moderate (10–30%) and pronounced (> 30%) inactivation of Kv currents. SVF-derived adipocytes were incubated 4–7 days in low (10 µIU/ml) or high (100 µIU/ml) insulin concentrations following what described in the methods section. The cumulative inactivation was measured applying a train of seven depolarizing voltage steps of 250 ms from − 60 to + 60 mV once every 400 ms. The percentage of inactivation was calculated from the remaining current at the end of the last pulse vs the first. Values are mean ± SE. Number of cells indicated in brackets
Kv1.3 is present in WAT and participates in the glucose metabolism
Multiple Kv currents suggested a complex scenario in white adipocytes but the C-type and cumulative inactivation (Figs. 1, 2, 3), the pharmacological profile (Fig. 2) and relevant bibliography pointed to an important contribution of Kv1.3. Although the physiological role of Kv1.3 in AT is an issue of controversy, we confirmed the presence of Kv1.3 in several WAT sources from human and rodents (Fig. 4). RNA was obtained from rat epididymal WAT and the expression of Kv1.3 was detected, as well as in Jurkat T-lymphocytes that were used as a positive control (Fig. 4a). The protein expression of Kv1.3 in rWAT and rat-isolated adipocytes was confirmed (Fig. 4b). Opposite evidence claim either the presence or the absence of Kv1.3 in human adipose samples [8, 11, 12]. However, we unequivocally characterize the channel expression in fat tissue from different human depots. Therefore, we performed immunohistochemistry in human adipose tissue from the mammary gland (Fig. 4c) and immunocytochemistry in isolated adipocytes from rat epididymal and human subcutaneous adipose tissue biopsies (Fig. 4d, e). In this scenario, all our samples positively stained for Kv1.3.
Fig. 4.
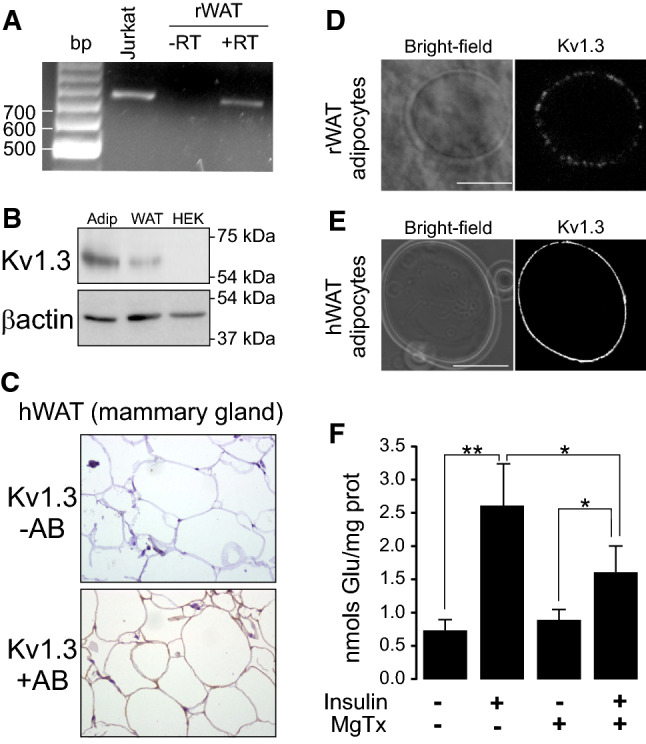
Human and rat WAT express Kv1.3 participating in insulin-activated glucose uptake in adipocytes. a RNA was extracted from rat WAT and RT-PCR was performed in the absence (−) or the presence (+) of retrotranscriptase (RT). Human Jurkat T-lymphocytes were used as a positive control. b Protein extracts from isolated rat adipocytes and WAT and human HEK 293 cells were analyzed for the expression of Kv1.3. β-actin was used as a loading and transfer control. c Human WAT from mammary gland biopsies was analyzed for the Kv1.3 staining in the absence (−) or the presence (+) of Kv1.3 antibody (AB). d Rat adipocytes from SVF were isolated and analyzed by immunohistochemistry for the presence of Kv1.3. e Adipocytes from subcutaneous tissue of human fat were stained for the expression of Kv1.3. Bars represent 50 µm. f Kv1.3 participates in insulin-dependent glucose uptake augmentation in adipocytes. Adipocytes were incubated with (+) or without (−) of 10 µM insulin for 30 min and glucose uptake was measured in the presence (+) or the absence (−) of 100 nM Margatoxin as described in the methods section. Values represent the mean ± SE, n = 4–6. *p < 0.05, **p < 0.01 Student’s t test
Furthermore, the role of Kv channels, and specifically Kv1.3, on insulin-dependent glucose uptake is controversial [8, 10–12, 14, 40]. Therefore, we next analyzed whether Kv1.3 participates in insulin signaling (Fig. 4f). MgTx was used because α-DTX discarded the participation of Kv1.1 and Kv1.2 (Fig. 2c, Supplementary Table 1). In adipocytes, 100 nM MgTx significantly reduced insulin-induced (10 µM insulin) glucose transport with no major effects on basal uptake, which points to the participation of Kv1.3 during glucose influx upon insulin signaling.
Caveolin interaction governs localization and insulin-dependent phosphorylation of Kv1.3
Kv1.3 participates in insulin signaling and channel subcellular localization is crucial in cell physiology [9, 10, 41, 42]. The insulin pathway inducing glucose uptake initiates in caveolae. The insulin receptor (IR) interacts with Cav and insulin-dependent GLUT4 translocation recruits the carrier to caveolae. Thus, an altered Cav 1/caveolae expression affects insulin signaling in adipose cells [43]. MgTx pointed to Kv1.3 participating in insulin-dependent glucose uptake. Moreover, Kv1.3 localizes in lipid rafts via interacting with Cav 1 [23]. Therefore, we wanted to study caveolar Kv1.3 targeting and whether an altered localization could affect the insulin signaling in AT. WAT express high levels of Cav 1 and caveolae is an important component of the adipocyte membrane [43]. Plasma membrane lawn preparations (PML) demonstrated that omega-shaped caveolae are abundant membrane structures of the adipocyte (Fig. 5a). Because insulin-mediated signaling initiates in caveolae and the turnover of the channel involves a clathrin-mediated endocytosis [44, 45], we analyzed the colocalization of Kv1.3 with both structures (Fig. 5b–g). PML indicated that Kv1.3 colocalized with Cav 1 almost threefold higher than with clathrin (Fig. 5h). As expected, Kv1.3 coimmunoprecipitated with Cav 1 (Fig. 5i) and clathrin (not shown). Moreover, Kv1.5, also expressed in WAT, coimmunoprecipitated with Kv1.3 and Cav 1. Although Kv1.5 does not interact with Cav 1, this result is not surprising because Kv1.5 may form heterotetrameric complexes with Kv1.3 [29]. To further demonstrate that Kv1.3 physically associated with Cav 1 in adipocytes, we performed FRET immunoassays (Fig. 5j–r). Kv1.3 physically interacted with Cav 1 but not with clathrin (Fig. 5s).
Fig. 5.

Kv1.3 targets to caveolae in adipocytes. Kv1.3 physically interacted with caveolin 1 in plasma membrane lawn preparations (PML) from adipocytes. a Electron micrograph of a representative PML from rat adipocytes. Multiple omega-shaped structures, recognized as caveolae, highlighted by white arrowheads, are widely distributed throughout the adipocyte plasma membrane. Bar represents 250 nm. b–h High colocalization of Kv1.3 with caveolin but not clathrin in PML. b–d Kv1.3 and caveolin; e–g Kv1.3 and clathrin. Color code: green, Kv1.3 (b, e); red, caveolin (c) and clathrin (f); Merge channel in yellow (d, g). Scale bars: 10 µm (b–d), 50 µm (e–g). h Kv1.3 colocalization with caveolin and clathrin in PML. A pixel by pixel analysis was performed and the percentage of colocalization was analyzed. Values are the mean ± SE of n > 20 cells. i Kv1.3 coimmunoprecipitated with caveolin 1 and Kv1.5 in isolated adipocytes. Total protein extracts were immunoprecipitated (IP) against Kv1.5, Kv1.3 and Cav. Next, samples were immunoblotted (IB) against Kv1.3 and Cav. SM, starting material; −, immunoprecipitated in the absence of antibody. j–s Kv1.3 associates with caveolin 1, but not clathrin, in adipocytes. j–l Immunofluorescent FRET analysis of the tetrameric Kv1.3 channels. Kv1.3 was immunodetected with anti-C-terminal polyclonal (j) and monoclonal (k) Kv1.3 antibodies and further immunostained with Alexa-Fluor-488 (donor) or Cy3-conjugated (acceptor) conjugated antibodies, respectively. l Normalized intensity prior to (pre-B) and after photobleaching (post-B). m–o Kv1.3 associated with caveolin. m Kv1.3 was immunolabelled with monoclonal anti-Kv1.3 and stained with A488. n Caveolin with polyclonal anti-Cav and Cy3. o Normalized intensity. p–r Kv1.3 did not associate with clathrin. p Kv1.3 was immunolabelled with polyclonal anti-Kv1.3 and stained with A488. q Clathrin with monoclonal anti-clathrin and A546. r Normalized intensity. s Efficiency of the indirect immunofluorescence FRET analysis. Bars represent 10 µm. ***p < 0.001 vs Kv1.3/Kv1.3 Student’s t test (n > 10 cells)
Insulin is an important trophic factor in adipocyte differentiation. To decipher whether Kv1.3 targeting to caveolae is important for adipogenesis, we used the pre-adipocyte 3T3-L1 cell line [43, 46]. In the presence of 10 mg/ml insulin, 3T3-L1 cells differentiated to adipocytes as demonstrated by morphological changes, which include the formation of fatty body droplets (Fig. 6a). Adipocyte differentiation was further documented by an increase in Cav 1 and GLUT4 expression. In this scenario, the expression of Kv1.3 augmented by almost threefold (Figs. 6b and see also 7a, b). The localization of Kv1.3 was also analyzed by immunocytochemistry (Fig. 6c–k). As expected, Kv1.3 targeted to the cell surface. Electron micrographs further demonstrated that the destination of Kv1.3 in 3T3-L1 adipocytes was mostly caveolae, because of its presence in characteristic omega-shaped structures that stained positively for Cav 1 (Fig. 6l–n).
Fig. 6.

Adipocyte differentiation increases Kv1.3 and targets the channel to caveolae. 3T3-L1 pre-adipocytes were differentiated to adipocytes. a Oil red O staining showed an increased adiposity of 3T3-L1 adipocytes. b 3T3-L1 adipogenesis elevated the expression of Cav1, Glut 4 as well as Kv1.3, although to a lesser extent. c–k Representative confocal images of Kv1.3/Cav 1 colocalization in 3T3-L1 pre-adipocytes (c–g) and adipocytes (h–k). c, h Bright field; d, i Kv1.3 in green; f–j Cav 1 in red; g–k merge in yellow. Bars represent 10 µm. l–n Electron micrographs of Kv1.3 located in caveolae stained with Cav 1 in 3T3-L1 adipocytes. Arrowhead points to Kv1.3 tagged with 15 nm gold particles. Cav 1 stained with 10 nm gold particles. Bars represent 200 nm
Fig. 7.

The depletion of Cav 1 in 3T3-L1 adipocytes mistargeted Kv1.3 lipid raft localization as well as impaired insulin-dependent phosphorylation of the channel. Lipid rafts were isolated from 3T3-L1 pre-adipocytes and adipocytes. A sucrose gradient, from low (1)- to high (12)-density fractions was applied, and the expression of Kv1.3, clathrin (non-raft marker) and caveolin (lipid raft marker) was analyzed. In addition, the 3T3-L1 Cav 1− cell line, with a silenced expression of Cav 1, was used. a Low expression of Kv1.3 in lipid rafts from 3T3-L1 wild type (WT) pre-adipocytes. b Increased expression of Kv1.3 targeting to rafts in 3T3-L1 WT adipocytes. Note the exacerbated augmentation of Cav 1 due to adipocyte differentiation. c Low expression of Kv1.3 in lipid rafts from 3T3-L1 Cav1− pre-adipocytes. d A minor increase of Kv1.3 is concomitant with reduced channel localization in rafts in 3T3-L1 Cav 1− adipocytes. Note the limited augmentation of Cav 1 due to adipocyte differentiation in this cell line. e Increase in Kv1.3 expression during adipocyte differentiation in 3T3-L1 WT and Cav 1− cells. *p < 0.05 vs pre-adipocytes (Student’s t test, n = 3-5). f Percentage of Kv1.3 in lipid rafts. Kv1.3 expression in caveolin-positive floating fractions was analyzed and relativized to the total amount of Kv1.3. *p < 0.05, ***p < 0.001 (Student’s t test, n = 3–5). White columns represent WT, black columns represent Cav 1− cells. g The reduction of Cav 1 in Cav 1− 3T3-L1 cell line impairs the insulin-dependent phosphorylation of Kv1.3. WT and Cav 1− cells were incubated with (+) or without (−) insulin, as described in methods. Total cell lysates were immunoprecipitated (IP) against phosphotyrosines (P-Tyr) and immunoblotted (IB) against the insulin receptor (InsR) and Kv1.3. Note an impairment in the phosphorylation of InsR and Kv1.3 in Cav 1− cells in the presence (+) of insulin
To further demonstrate that the newly synthetized Kv1.3 targeted to caveolar lipid rafts, these microdomains were isolated from pre-adipocytes and differentiated fatty 3T3-L1 cells. Adipogenesis notably increased the abundance of caveolar lipid rafts (Fig. 7a, b) with no apparent changes in clathrin. In this context, Kv1.3 augmented its localization into high-buoyancy fractions in adipocytes (Fig. 7f). The depletion of Cav 1 in 3T3-L1 adipocytes (3T3-L1 cav1−) reduced the maximal insulin response, diminishing the abundance of IR and GLUT4 at the cell surface [43]. Therefore, we used this Cav 1-deficient 3T3-L1 cell line to analyze whether Kv1.3 localization was affected (Fig. 7c, d). The expression of Kv1.3 was augmented in 3T3-L1 cav1− adipocytes, but to a lesser extent than in WT cells (Fig. 7e). Concomitantly, Kv1.3 targeting to lipid rafts was clearly impaired by 50% (Fig. 7f). Kv1.5 was not located in lipid rafts microdomains as previously described [15] and, as expected, no changes were observed in 3T3-L1 cav1− cells (Supplementary Fig. 1). Evidence demonstrates that insulin-dependent signaling initiates in caveolar rafts [43], Kv1.3 targets to raft microdomains [15] and Kv1.3 is phosphorylated by the IR upon insulin stimulation [47, 48]. Therefore, we finally analyzed whether the depletion of Cav 1, which impaired Kv1.3 raft localization, could affect insulin-dependent Kv1.3 phosphorylation in adipocytes. Thus, the phosphorylation of IR and Kv1.3 was clearly diminished in Cav 1− depleted cells (Fig. 7g). Our results clearly demonstrated that (1) Kv1.3 targets caveolae due to an association with caveolin, (2) adipocyte differentiation increases raft localization and channel expression and (3) this emplacement is important for proper insulin-dependent signaling, which in turn would have important consequences in WAT physiology.
Discussion
The function of Kv channels in the physiology of WAT is under investigation. Contradictory evidence is well documented [3, 4, 8, 10, 12, 13, 39, 40]. The main subject of the debate is the participation of Kv1.3 because Kv1.3 null mice exhibit a lean phenotype and present an altered insulin sensitivity, which points to this channel as a target in type II diabetes and obesity [6, 8–13, 40]. In this complex scenario, our work documents several important facts: (1) Kv1.3 is undoubtedly expressed in WAT from human and rodents; (2) although other isoforms, specially Kv1.5, are also present and although insulin remodeled the Kv phenotype of adipocytes, the pharmacological and biophysical properties pointed to Kv1.3 as an important contributor to Kv currents; and finally, (3) the abundance of Kv1.3 increased during adipogenesis in mouse 3T3-L1 cells, and this channel, via a physical interaction with Cav 1, was targeted to caveolar lipid rafts for proper insulin-dependent phosphorylation.
A decrease in Kv1.3 function facilitates a lean phenotype by enhancing insulin sensitivity, increasing GLUT4 translocation and glucose uptake via Ca2+ signaling in adipocytes [9, 10]. However, these results have been debated [8, 11, 12, 40]. In fact, from a metabolic point of view, any increase in glucose influx would enhance lipogenesis rather than generating a lean phenotype. Our data would support this because under high insulin, similar to type II diabetes and obesity, cell size was augmented, and adipogenesis increased Kv1.3 expression. In this complex scenario, while evidence demonstrates that Kv1.3 is expressed in WAT and that channel inhibition increases glucose transport [9], other findings support neither expression nor glucose uptake affectation in adipocytes [8, 11, 12]. The fact that Kv1.3−/− (KO) mice fed a high-fat diet gain less weight and are less obese than littermate controls is at the top of this debate [6]. Moreover, KO animals remain euglycemic with low blood insulin levels [9]. However, for the latter it was demonstrated that although meal regime was similar between Kv1.3 KO and wild type animals, basal metabolic rate was higher in the KO mice [7]. This was further investigated in a double gene-targeted deletion of Kv1.3 and melanocortin-4 receptor (MC4R) [7]. Thus, Kv1.3 abrogation in this genetic model of obesity triggers reductions in adiposity and body weight due to increased locomotor activity and energy expenditure [7, 14]. The last evidence provided by Chandy and coworkers described that the lean phenotype resulting from inhibiting Kv1.3 with ShK-186 is most likely due to an increase in BAT activity rather than WAT participation [8]. Along these lines, other channels such as TRPV2 or TASK-1 play a role in BAT thermogenesis and could also be a target for human obesity therapy [49, 50]. In fact, WT mice fed with a hypercaloric-fat diet reduced the number of mitochondria in mitral cells from the olfactory bulb, whereas this is not observed in Kv1.3 KO mice [51]. Note that sensory neurons from the olfactory bulb are a hot spot for the Kv1.3 function in the nervous system controlling food intake [14]. In this complex scenario, we found that selective inhibition of Kv1.3 diminished, rather increased, the insulin-dependent glucose uptake, which could be, in a sense, a reduction of fuel intake, which in turn would generate less fat deposition. This open controversy could have a surprising explanation. AT is a very complex endocrine tissue. In addition to the origin, metabolic status, age, sex and endocrine situation of the sample, the implication of beige adipose tissue may shed some light on this debate. BAT adipocytes appear upon thermogenic insults in WAT locations. Such brown cells, derived from precursors, are similar to classical white adipocytes. This BAT is often named inducible, beige, or bright. The thermogenic function of beige adipocytes, which differs from that of classical BAT, is not known, but some data suggest a major role in protection against obesity [52]. It is tempting to speculate that this heterogeneous composition would merge evidence. To generate a further complex and exciting scenario, our and other’s data suggest that the formation of heterotetrameric structures with other shaker channels, such as Kv1.5, as well as the contribution of other Kv1 isoforms that can be remodeled under several physiological situations, such as insulin resistance and obesity, should be contemplated [29, 33, 34]. We found that exposure to high insulin concentrations, similar to those observed in type II diabetes, triggered a switch in the K+ current phenotype in adipocytes. Our data would support that formation of hybrid complexes is highly possible. Similar remodeling has been documented in macrophages, microglia and smooth muscle [29–31]. Our work would incorporate adipocytes, under hyperinsulinemia, in this ever-growing list.
In addition to this intense debate that deserves further research, our and other’s glucose transport data strongly support that Kv1.3 is somehow involved in insulin sensitivity in the AT [9]. Indeed, a T-1645C polymorphism in KCNA3 (the human Kv1.3 gene) is associated with impaired insulin sensitivity and altered glucose tolerance [53]. This channel, which is crucial for the immunological response, concentrates in lipid raft structures upon IS formation and leukocyte activation [24]. We have recently described that this targeting is mostly mediated via an association with caveolin [23]. In adipocytes, insulin activates and recruits IR into caveolae [46]. IR phosphorylates Kv1.3 in tyrosines, which inhibits the channel activity [47, 48, 54]. Concomitantly, an insulin-dependent Kv1.3-mediated GLUT4 translocation to the membrane triggers an increase in glucose uptake [10]. All this endocrine regulation is spatially located within caveolae in AT [46]. We demonstrated that, similar to Cav 1 and GLUT4, Kv1.3 increases during adipogenesis, and these newly synthetized channels are properly located in caveolae. We found that Kv1.3 targeting to these raft domains in adipocytes is through physical interactions with Cav 1. An important part of our contribution is focused on the localization of Kv1.3 and the importance of this spatial regulation for insulin-dependent signaling. We demonstrated, using a Cav 1 deficient 3T3-L1 cell line, that Kv1.3 targeting away from lipid rafts and insulin-dependent phosphorylation of the channel, which is crucial for proper insulin-dependent signaling, is impaired in Cav 1− adipocytes.
Caveolae act as signaling platforms approximating effector molecules and their targets. The importance of Cav 1 in adipocyte physiology is well known because Cav depletion reduces GLUT4 and IR levels, decreasing their stability, and affecting glucose transport [46]. The effect of Cav depletion in the insulin-signaling cascade leading to reduced insulin-enhanced glucose transport could be aggravated by targeting Kv1.3 away from lipid raft structures. In this scenario, although Kv1.3 could be normally expressed, the channel would undergo less phosphorylation and thereby, as previously demonstrated for insulin and epidermal growth factor, the function would be mostly preserved [44, 54]. It is tempting to speculate that if Kv1.3 inhibition improves insulin sensitivity by abrogating the inflammatory state similar to what observed in leukocytes [55], an altered localization of the channel would maintain the function, thereby impairing the insulin-dependent response. Moreover, in a metabolic context, reduced channel inhibition would counteract elevated mitochondrial function, facilitating an obese phenotype, insulin resistance and the appearance of type II diabetes.
Obesity and related metabolic disorders are currently a global epidemic which requires novel and reliable pharmacological strategies. The medical and social implications derived from these diseases will represent a significant cost to healthcare systems. Kv1.3 participates in peripheral insulin sensitivity and is being considered as a new target for obesity treatment. Therefore, our results are of relevance because it clearly contributes to knowledge of adipocyte physiology.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1. Kv1.5 did not target to lipid rafts. Lipid rafts were isolated from 3T3-L1 Wt and 3T3-L1 Cav 1− adipocytes. A sucrose gradient, from low (1)- to high (12)-density fractions was applied, and the expression of Kv1.5, clathrin (non-raft marker) and caveolin (lipid raft marker) was analyzed. (A) Expression of Kv1.5 in sucrose fractions from (A) 3T3-L1 wild type (WT) and (B) 3T3-L1 Cav 1− adipocytes. Kv1.5 distribution, out rafts, was independent of the expression of Cav 1. Note the limited augmentation of Cav 1 due to adipocyte differentiation in this cell line (PDF 124 kb)
Acknowledgements
Supported by the Ministerio de Economia y Competitividad (MINECO, Spain) Grants (BFU2014-54928-R and BFU2017-87104-R) and Fondo Europeo de Desarrollo Regional (FEDER). MPV and JC contributed equally and hold fellowships from the MINECO and the Fundación Tatiana Pérez de Guzmán el Bueno, respectively. Authors thank Dr. C. López-Iglesias (CCiTUB, Universitat de Barcelona) for her help in electronic Microscopy and to Dr. J. Peinado-Onsurbe for the access to human samples. The English editorial assistance of the American Journal Experts is also acknowledged.
References
- 1.Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89:2548–2556. doi: 10.1210/jc.2004-0395. [DOI] [PubMed] [Google Scholar]
- 2.Cheng K, Groarke J, Osotimehin B, Haspel HC, Sonenberg M. Effects of insulin, catecholamines, and cyclic nucleotides on rat adipocyte membrane potential. J Biol Chem. 1981;256:649–655. [PubMed] [Google Scholar]
- 3.Ramirez-Ponce MP, Mateos JC, Bellido JA. Human adipose cells have voltage-dependent potassium currents. J Membr Biol. 2003;196(2):129–134. doi: 10.1007/s00232-003-0631-1. [DOI] [PubMed] [Google Scholar]
- 4.Ramírez-Ponce MP, Mateos JC, Carrión N, Bellido JA. Voltage-dependent potassium channels in white adipocytes. Biochem Biophys Res Commun. 1996;223(2):250–256. doi: 10.1006/bbrc.1996.0880. [DOI] [PubMed] [Google Scholar]
- 5.Wilson SM, Lee SC, Shook S, Pappone PA. ATP and beta-adrenergic stimulation enhance voltage-gated K current inactivation in brown adipocytes. Am J Physiol Cell Physiol. 2000;279:C1847–C1858. doi: 10.1152/ajpcell.2000.279.6.C1847. [DOI] [PubMed] [Google Scholar]
- 6.Xu J, Koni PA, Wang P, Li G, Kaczmarek L, Wu Y, Li Y, Flavell RA, Desir GV. The voltage-gated potassium channel Kv1.3 regulates energy homeostasis and body weight. Hum Mol Genet. 2003;12:551–559. doi: 10.1093/hmg/ddg049. [DOI] [PubMed] [Google Scholar]
- 7.Tucker K, Overton JM, Fadool DA. Kv1.3 gene-targeted deletion alters longevity and reduces adiposity by increasing locomotion and metabolism in melanocortin-4 receptor-null mice. Int J Obes (Lond) 2008;32:1222–1232. doi: 10.1038/ijo.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Upadhyay SK, Eckel-Mahan KL, Mirbolooki MR, Tjong I, Griffey SM, Schmunk G, Koehne A, Halbout B, Iadonato S, Pedersen B, Borrelli E, Wang PH, Mukherjee J, Sassone-Corsi P, Chandy KG. Selective Kv1.3 channel blocker as therapeutic for obesity and insulin resistance. Proc Natl Acad Sci USA. 2013;110:E2239–E2248. doi: 10.1073/pnas.1221206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu J, Wang P, Li Y, Li G, Kaczmarek LK, Wu Y, Koni PA, Flavell RA, Desir GV. The voltage-gated potassium channel Kv1.3 regulates peripheral insulin sensitivity. Proc Natl Acad Sci USA. 2004;101:3112–3117. doi: 10.1073/pnas.0308450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Wang P, Xu J, Desir GV. Voltage-gated potassium channel Kv1.3 regulates GLUT4 trafficking to the plasma membrane via a Ca2+-dependent mechanism. Am J Physiol Cell Physiol. 2006;290:C345–C351. doi: 10.1152/ajpcell.00091.2005. [DOI] [PubMed] [Google Scholar]
- 11.Straub SV, Perez SM, Tan B, Coughlan KA, Trebino CE, Cosgrove P, Buxton JM, Kreeger JM, Jackson VM. Pharmacological inhibition of Kv1.3 fails to modulate insulin sensitivity in diabetic mice or human insulin-sensitive tissues. Am J Physiol Endocrinol Metab. 2011;301:E380–E390. doi: 10.1152/ajpendo.00076.2011. [DOI] [PubMed] [Google Scholar]
- 12.Ngala RA, Zaibi MS, Langlands K, Stocker CJ, Arch JR, Cawthorne MA. Stimulation of glucose uptake in murine soleus muscle and adipocytes by 5-(4-phenoxybutoxy)psoralen (PAP-1) may be mediated by Kv1.5 rather than Kv1.3. PeerJ. 2014;2:e614. doi: 10.7717/peerj.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fadool DA, Tucker K, Pedarzani P. Mitral cells of the olfactory bulb perform metabolic sensing and are disrupted by obesity at the level of the Kv1.3 ion channel. PLoS One. 2011;6:e24921. doi: 10.1371/journal.pone.0024921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tucker K, Overton JM, Fadool DA. Diet-induced obesity resistance of Kv1.3−/− mice is olfactory bulb dependent. J Neuroendocrinol. 2012;24:1087–1095. doi: 10.1111/j.1365-2826.2012.02314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinez-Marmol R, Villalonga N, Sole L, Vicente R, Tamkun MM, Soler C, Felipe A. Multiple Kv1.5 targeting to membrane surface microdomains. J Cell Physiol. 2008;217:667–673. doi: 10.1002/jcp.21538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vicente R, Villalonga N, Calvo M, Escalada A, Solsona C, Soler C, Tamkun MM, Felipe A. Kv1.5 association modifies Kv1.3 traffic and membrane localization. J Biol Chem. 2008;283:8756–8764. doi: 10.1074/jbc.M708223200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bock J, Szabo I, Gamper N, Adams C, Gulbins E. Ceramide inhibits the potassium channel Kv1.3 by the formation of membrane platforms. Biochem Biophys Res Commun. 2003;305:890–897. doi: 10.1016/S0006-291X(03)00763-0. [DOI] [PubMed] [Google Scholar]
- 18.Martens JR, O’Connell K, Tamkun M. Targeting of ion channels to membrane microdomains: localization of Kv channels to lipid rafts. Trends Pharmacol Sci. 2004;25:16–21. doi: 10.1016/j.tips.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 19.Pilch PF, Souto RP, Liu L, Jedrychowski MP, Berg EA, Costello CE, Gygi SP. Cellular spelunking: exploring adipocyte caveolae. J Lipid Res. 2007;48:2103–2111. doi: 10.1194/jlr.R700009-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Sinha B, Koster D, Ruez R, Gonnord P, Bastiani M, Abankwa D, Stan RV, Butler-Browne G, Vedie B, Johannes L, Morone N, Parton RG, Raposo G, Sens P, Lamaze C, Nassoy P. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell. 2011;144:402–413. doi: 10.1016/j.cell.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hnasko R, Lisanti MP. The biology of caveolae: lessons from caveolin knockout mice and implications for human disease. Mol Interv. 2003;3:445–464. doi: 10.1124/mi.3.8.445. [DOI] [PubMed] [Google Scholar]
- 22.Couet J, Li S, Okamoto T, Ikezu T, Lisanti MP. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J Biol Chem. 1997;272:6525–6533. doi: 10.1074/jbc.272.10.6525. [DOI] [PubMed] [Google Scholar]
- 23.Perez-Verdaguer M, Capera J, Martinez-Marmol R, Camps M, Comes N, Tamkun MM, Felipe A. Caveolin interaction governs Kv1.3 lipid raft targeting. Sci Rep. 2016;6:22453. doi: 10.1038/srep22453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panyi G, Vamosi G, Bacso Z, Bagdany M, Bodnar A, Varga Z, Gaspar R, Matyus L, Damjanovich S. Kv1.3 potassium channels are localized in the immunological synapse formed between cytotoxic and target cells. Proc Natl Acad Sci USA. 2004;101:1285–1290. doi: 10.1073/pnas.0307421100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bielanska J, Hernandez-Losa J, Perez-Verdaguer M, Moline T, Somoza R, Ramon YCS, Condom E, Ferreres JC, Felipe A. Voltage-dependent potassium channels Kv1.3 and Kv1.5 in human cancer. Curr Cancer Drug Targets. 2009;9:904–914. doi: 10.2174/156800909790192400. [DOI] [PubMed] [Google Scholar]
- 26.Moreno C, Oliveras A, de la Cruz A, Bartolucci C, Munoz C, Salar E, Gimeno JR, Severi S, Comes N, Felipe A, Gonzalez T, Lambiase P, Valenzuela C. A new KCNQ1 mutation at the S5 segment that impairs its association with KCNE1 is responsible for short QT syndrome. Cardiovasc Res. 2015;107:613–623. doi: 10.1093/cvr/cvv196. [DOI] [PubMed] [Google Scholar]
- 27.König P, Krasteva G, Tag C, König IR, Arens C, Kummer W. FRET–CLSM and double-labeling indirect immunofluorescence to detect close association of proteins in tissue sections. Lab Investig. 2006;86:853–864. doi: 10.1038/labinvest.3700443. [DOI] [PubMed] [Google Scholar]
- 28.Safronov BV, Vogel W. Modulation of delayed rectifier K+ channel activity by external K+ ions in Xenopus axon. Pflug Arch. 1995;430:879–886. doi: 10.1007/BF01837400. [DOI] [PubMed] [Google Scholar]
- 29.Vicente R, Escalada A, Villalonga N, Texido L, Roura-Ferrer M, Martin-Satue M, Lopez-Iglesias C, Soler C, Solsona C, Tamkun MM, Felipe A. Association of Kv1.5 and Kv1.3 contributes to the major voltage-dependent K+ channel in macrophages. J Biol Chem. 2006;281:37675–37685. doi: 10.1074/jbc.M605617200. [DOI] [PubMed] [Google Scholar]
- 30.Kotecha SA, Schlichter LC. A Kv1.5 to Kv1.3 switch in endogenous hippocampal microglia and a role in proliferation. J Neurosci. 1999;19:10680–10693. doi: 10.1523/JNEUROSCI.19-24-10680.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cidad P, Miguel-Velado E, Ruiz-McDavitt C, Alonso E, Jimenez-Perez L, Asuaje A, Carmona Y, Garcia-Arribas D, Lopez J, Marroquin Y, Fernandez M, Roque M, Perez-Garcia MT, Lopez-Lopez JR. Kv1.3 channels modulate human vascular smooth muscle cells proliferation independently of mTOR signaling pathway. Pflug Arch. 2015;467:1711–1722. doi: 10.1007/s00424-014-1607-y. [DOI] [PubMed] [Google Scholar]
- 32.Baronas VA, Yang RY, Kurata HT. Extracellular redox sensitivity of Kv1.2 potassium channels. Sci Rep. 2017;7:9142. doi: 10.1038/s41598-017-08718-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takimoto K, Levitan ES. Altered K+ channel subunit composition following hormone induction of Kv1.5 gene expression. Biochemistry. 1996;35:14149–14156. doi: 10.1021/bi961290s. [DOI] [PubMed] [Google Scholar]
- 34.Coleman SK, Newcombe J, Pryke J, Dolly JO. Subunit composition of Kv1 channels in human CNS. J Neurochem. 1999;73:849–858. doi: 10.1046/j.1471-4159.1999.0730849.x. [DOI] [PubMed] [Google Scholar]
- 35.Lee MJ, Wu Y, Fried SK. Adipose tissue heterogeneity: implication of depot differences in adipose tissue for obesity complications. Mol Asp Med. 2013;34:1–11. doi: 10.1016/j.mam.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.You MH, Song MS, Lee SK, Ryu PD, Lee SY, Kim DY. Voltage-gated K+ channels in adipogenic differentiation of bone marrow-derived human mesenchymal stem cells. Acta Pharmacol Sin. 2013;34:129–136. doi: 10.1038/aps.2012.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishizuka M, Horinouchi W, Yamada E, Ochiai N, Osada S, Imagawa M. KCNMA1, a pore-forming α-subunit of BK channels, regulates insulin signalling in mature adipocytes. FEBS Lett. 2016;590:4372–4380. doi: 10.1002/1873-3468.12465. [DOI] [PubMed] [Google Scholar]
- 38.Alexander SP, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD, Pawson AJ, Sharman JL, Southan C, Davies JA. The concise guide to pharmacology 2017/18: voltage-gated ion channels. Br J Pharmacol. 2017;174(Suppl 1):S160–S194. doi: 10.1111/bph.13884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramirez-Ponce MP, Mateos JC, Bellido JA. Insulin increases the density of potassium channels in white adipocytes: possible role in adipogenesis. J Endocrinol. 2002;174:299–307. doi: 10.1677/joe.0.1740299. [DOI] [PubMed] [Google Scholar]
- 40.Jaimes-Hoy L, Gurrola GB, Cisneros M, Joseph-Bravo P, Possani LD, Charli JL. The Kv1.3 channel blocker Vm24 enhances muscle glucose transporter 4 mobilization but does not reduce body-weight gain in diet-induced obese male rats. Life Sci. 2017;181:23–30. doi: 10.1016/j.lfs.2017.05.027. [DOI] [PubMed] [Google Scholar]
- 41.Nicolaou SA, Szigligeti P, Neumeier L, Lee SM, Duncan HJ, Kant SK, Mongey AB, Filipovich AH, Conforti L. Altered dynamics of Kv1.3 channel compartmentalization in the immunological synapse in systemic lupus erythematosus. J Immunol. 2007;179:346–356. doi: 10.4049/jimmunol.179.1.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nicolaou SA, Neumeier L, Steckly A, Kucher V, Takimoto K, Conforti L. Localization of Kv1.3 channels in the immunological synapse modulates the calcium response to antigen stimulation in T lymphocytes. J Immunol. 2009;183:6296–6302. doi: 10.4049/jimmunol.0900613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gonzalez-Munoz E, Lopez-Iglesias C, Calvo M, Palacin M, Zorzano A, Camps M. Caveolin-1 loss of function accelerates glucose transporter 4 and insulin receptor degradation in 3T3-L1 adipocytes. Endocrinology. 2009;150:3493–3502. doi: 10.1210/en.2008-1520. [DOI] [PubMed] [Google Scholar]
- 44.Martinez-Marmol R, Comes N, Styrczewska K, Perez-Verdaguer M, Vicente R, Pujadas L, Soriano E, Sorkin A, Felipe A. Unconventional EGF-induced ERK1/2-mediated Kv1.3 endocytosis. Cell Mol Life Sci. 2016;73:1515–1528. doi: 10.1007/s00018-015-2082-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martinez-Marmol R, Styrczewska K, Perez-Verdaguer M, Vallejo-Gracia A, Comes N, Sorkin A, Felipe A. Ubiquitination mediates Kv1.3 endocytosis as a mechanism for protein kinase C-dependent modulation. Sci Rep. 2017;7:42395. doi: 10.1038/srep42395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ros-Baro A, Lopez-Iglesias C, Peiro S, Bellido D, Palacin M, Zorzano A, Camps M. Lipid rafts are required for GLUT4 internalization in adipose cells. Proc Natl Acad Sci USA. 2001;98:12050–12055. doi: 10.1073/pnas.211341698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bowlby MR, Fadool DA, Holmes TC, Levitan IB. Modulation of the Kv1.3 potassium channel by receptor tyrosine kinases. J Gen Physiol. 1997;110:601–610. doi: 10.1085/jgp.110.5.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fadool DA, Levitan IB. Modulation of olfactory bulb neuron potassium current by tyrosine phosphorylation. J Neurosci. 1998;18:6126–6137. doi: 10.1523/JNEUROSCI.18-16-06126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun W, Uchida K, Suzuki Y, Zhou Y, Kim M, Takayama Y, Takahashi N, Goto T, Wakabayashi S, Kawada T, Iwata Y, Tominaga M. Lack of TRPV2 impairs thermogenesis in mouse brown adipose tissue. EMBO Rep. 2016;17:383–399. doi: 10.15252/embr.201540819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Y, Zeng X, Huang X, Serag S, Woolf CJ, Spiegelman BM. Crosstalk between KCNK3-mediated ion current and adrenergic signaling regulates adipose thermogenesis and obesity. Cell. 2017;171(836–848):e813. doi: 10.1016/j.cell.2017.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kovach CP, Al Koborssy D, Huang Z, Chelette BM, Fadool JM, Fadool DA. Mitochondrial ultrastructure and glucose signaling pathways attributed to the Kv1.3 ion channel. Front Physiol. 2016;7:178. doi: 10.3389/fphys.2016.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giralt M, Villarroya F. White, brown, beige/brite: different adipose cells for different functions? Endocrinology. 2013;154:2992–3000. doi: 10.1210/en.2013-1403. [DOI] [PubMed] [Google Scholar]
- 53.Tschritter O, Machicao F, Stefan N, Schafer S, Weigert C, Staiger H, Spieth C, Haring HU, Fritsche A. A new variant in the human Kv1.3 gene is associated with low insulin sensitivity and impaired glucose tolerance. J Clin Endocrinol Metab. 2006;91:654–658. doi: 10.1210/jc.2005-0725. [DOI] [PubMed] [Google Scholar]
- 54.Fadool DA, Tucker K, Phillips JJ, Simmen JA. Brain insulin receptor causes activity-dependent current suppression in the olfactory bulb through multiple phosphorylation of Kv1.3. J Neurophysiol. 2000;83:2332–2348. doi: 10.1152/jn.2000.83.4.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Villalonga N, David M, Bielanska J, Vicente R, Comes N, Valenzuela C, Felipe A. Immunomodulation of voltage-dependent K+ channels in macrophages: molecular and biophysical consequences. J Gen Physiol. 2010;135:135–147. doi: 10.1085/jgp.200910334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. Kv1.5 did not target to lipid rafts. Lipid rafts were isolated from 3T3-L1 Wt and 3T3-L1 Cav 1− adipocytes. A sucrose gradient, from low (1)- to high (12)-density fractions was applied, and the expression of Kv1.5, clathrin (non-raft marker) and caveolin (lipid raft marker) was analyzed. (A) Expression of Kv1.5 in sucrose fractions from (A) 3T3-L1 wild type (WT) and (B) 3T3-L1 Cav 1− adipocytes. Kv1.5 distribution, out rafts, was independent of the expression of Cav 1. Note the limited augmentation of Cav 1 due to adipocyte differentiation in this cell line (PDF 124 kb)