

Abstract
Classical dendritic cells (cDCs) play a pivotal role in the early events that tip the immune response toward persistence or viral control. In vitro studies indicate that HIV infection induces the dysregulation of cDCs through binding of the LILRB2 inhibitory receptor to its MHC-I ligands and the strength of this interaction was proposed to drive disease progression. However, the dynamics of the LILRB2/MHC-I inhibitory axis in cDCs during early immune responses against HIV are yet unknown. Here, we show that early HIV-1 infection induces a strong and simultaneous increase of LILRB2 and MHC-I expression on the surface of blood cDCs. We further characterized the early dynamics of LILRB2 and MHC-I expression by showing that SIVmac251 infection of macaques promotes coordinated up-regulation of LILRB2 and MHC-I on cDCs and monocytes/macrophages, from blood and lymph nodes. Orientation towards the LILRB2/MHC-I inhibitory axis starts from the first days of infection and is transiently induced in the entire cDC population in acute phase. Analysis of the factors involved indicates that HIV-1 replication, TLR7/8 triggering, and treatment by IL-10 or type I IFNs increase LILRB2 expression. Finally, enhancement of the LILRB2/MHC-I inhibitory axis is specific to HIV-1 and SIVmac251 infections, as expression of LILRB2 on cDCs decreased in naturally controlled chikungunya virus infection of macaques. Altogether, our data reveal a unique up-regulation of LILRB2 and its MHC-I ligands on cDCs in the early phase of SIV/HIV infection, which may account for immune dysregulation at a critical stage of the anti-viral response.
Electronic supplementary material
The online version of this article (10.1007/s00018-017-2712-9) contains supplementary material, which is available to authorized users.
Keywords: ILT4, HLA class I, Immune checkpoint, ITIM, Innate immunity, LILR, SIRPa
Introduction
Classical dendritic cells (cDCs) include all dendritic cells other than plasmacytoid dendritic cells (pDCs) [1]. The regulation of cDC functions in the early phase of viral infection plays a critical role in the immune balance toward control or persistence of the virus. Indeed, cDCs are crucial components of the processes that bridge innate and adaptive immunity, due to their unique capacity to sense viruses and prime T cell responses [2]. Hence, dysregulation of cDCs represents an attractive way for viruses to subvert immune defenses at the onset of infection. cDCs from HIV-infected patients exhibit altered functions, including poor maturation capacity and inefficient antigen presentation to CD4+ T cells [3]. Furthermore, SIV infection of rhesus macaques induces dysregulation of early cDC immune responses that correlates with disease progression [4, 5]. However, the molecular basis of these dysfunctions is still poorly understood.
LILRB2 (also known as ILT4) is an inhibitory receptor specifically expressed on myeloid cells that plays an important role in the regulation of cDC functions and was proposed as an immune checkpoint molecule [6–9]. The main ligands of LILRB2 are MHC-I molecules that bind to LILRB2 through trans and cis interactions, resulting in the recruitment of SHP1/2 phosphatases which leads to the inhibition of on-going signaling pathways [10–13]. In vitro studies indicate that in HIV infection, strengthened interaction of LILRB2 with its MHC-I ligands induces the dysregulation of cDCs which is characterized by an altered capacity to stimulate CD4+ T cells and produce cytokines [14]. It was also reported that the strength of the LILRB2/MHC-I interaction is correlated with the level of cDC dysregulation and consequently the rate of disease progression in HIV-infected patients [15, 16]. The potency of the LILRB2/MHC-I inhibitory axis can be modulated by genetic polymorphisms of MHC-I haplotypes [15–17], as well as variations in the sequence of the HIV-Gag antigenic peptides presented by MHC-I [7]. The simultaneous enhancement of LILRB2 and MHC-I surface expression could also be a potent way for HIV to dysregulate cDC functions in early infection that is a critical stage for the installation of efficient immune responses against viruses. However, the characterization of LILRB2 and MHC-I expression on cDCs from blood and secondary lymphoid tissues at the onset of HIV infection is very difficult in humans. Indeed, longitudinal studies aiming to study immune responses in the first days of HIV infection are hampered by the late diagnosis of the disease and the difficulty of accessing lymphoid tissues in patients. In this context, the infection of macaques by SIV represents a unique model to longitudinally assess the dynamics of immune responses in vivo [18]. Moreover, macaques possess a LILRB2-like inhibitory receptor that is highly homologous with human LILRB2, making this model suitable for characterizing the modulation of LILRB2 expression during viral infections [19].
In this study we have investigated the dynamic of expression of LILRB2 and MHC-I on cDCs during the early immune response against HIV and SIV. For this purpose we first analyzed cDCs from primary HIV-1-infected patients and then used the SIV-infected macaque model to decipher the early dynamics of LILRB2 and MHC-I expression on cDCs in vivo. Our data reveal that the simultaneous expression of LILRB2 and its MHC-I ligands transiently increases at the surface of cDCs from blood and lymph nodes during the first days of infection. In contrast to SIV infection, LILRB2 levels decreased rapidly on cDCs after CHIKV infection in macaques which is characterized by efficient immune responses leading to rapid control of viral replication [20, 21], supporting our hypothesis that early dysregulation of cDC functions hinders the initiation of an efficient host immune response to control viral infection. Overall, these findings show coordinated enhancement of LILRB2 and MHC-I expression on cDCs during the acute phase of SIV/HIV infection. They suggest a major role of the LILRB2/MHC-I inhibitory axis in the early dysregulation of cDCs that could considerably attenuate the efficiency of immune responses and result in viral persistence.
Materials and methods
Ethics statement
All enrolled patients gave written informed consent to participate in the study. The C021 CODEX cohort and this substudy protocol were approved by the ethics review committee of Ile de France VII. The C06 ANRS PRIMO cohort and this substudy were approved by the Paris-Cochin ethics review committee.
For animal experimentation adult cynomolgus macaques (Macaca fascicularis) imported from Mauritius were housed in the facilities of the Infectious Disease Models and Innovative Therapies (IDMIT) Center, part of the “Commissariat à l’Energie Atomique et aux Energies Alternatives” (CEA, Fontenay-aux-Roses, France). Non-human primates (NHP) were used in accordance with French national regulations, under the supervision of national veterinary inspectors (CEA Permit Number A 92-032-02). The CEA complies with the Standards for Human Care and Use of Laboratory Animals, of the Office for Laboratory Animal Welfare (OLAW, USA) under OLAW Assurance number #A5826-01. The use of NHP at the CEA is in conformity with the recommendations of European Directive (2010/63, recommendation No. 9). The animals were used under the supervision of the veterinarians in charge of the animal facility. These studies were approved and accredited under statement numbers A 13-028, A14-067, A-16-035 by the ethics committee “Comité d’Ethique en Expérimentation Animale du CEA” registered under the number 44 by the French Ministry of Research. Animals were housed under controlled conditions of humidity, temperature, and light (12-h light/12-h dark cycles). Water was available ad libitum. Animals were monitored and fed commercial monkey chow and fruit 1–2 times daily by trained personnel. The macaques were provided with environmental enrichment, including toys, novel foodstuffs, and music, under the supervision of the CEA Animal Welfare Body.
Patient samples
Patients with primary HIV infection (PHI) were recruited from the multicenter ANRS CO6 PRIMO cohort. PHI was defined by a negative or indeterminate HIV ELISA associated with positive plasma HIV-RNA or p24 antigenaemia, an evolving western blot profile (no anti-Pol antibodies), or HIV seropositivity after a negative antibody test < 6 months before. All patients were antiretroviral-naive at enrolment in the cohort. HIV-controllers (HIC) patients were defined as patients infected by HIV-1 for ≥ 5 years who never received antiretroviral treatment and whose last five consecutive plasma HIV RNA values were < 400 copies/mL. They were recruited in the ANRS CO21 Codex cohort. All enrolled patients gave written informed consent to participate in this study. All recovered samples from HIV-infected patients were received as cryopreserved peripheral blood mononuclear cells (PBMCs) and stored at −180 °C. Biological samples from six HIV-1 seronegative donors (HIV-) were obtained from the Etablissement Français du Sang (EFS). PBMCs were isolated from fresh whole blood by density gradient in lymphocyte separation medium (Eurobio, Abcys). Red blood cells were lysed by hypotonic shock and cell counts performed using a Vi-CELL (Beckman-coulter). Cells were frozen in SVF/10% DMSO and stored at −180 °C for further analysis.
Animals, infections, and sample collection
Among the cynomolgus macaques included in our studies, males weighed ≥ 5 kg and females weighed ≥ 3.5 kg. Animals were intravenously administered 1000 animal infectious dose 50% (1000 AID50) of the isolate SIVmac251. NHPs enrolled in the CHIKV infection longitudinal study were infected with 103 PFU (100 AID50) of CHIKV isolate LR2006-OPY1 by inoculation into the saphenous vein. Blood and lymph node sampling was performed at various time points before and after infections and collected under general anesthesia by intra-muscular injection of 10 mg/kg ketamine (Rhone-Mérieux). Experimental procedures (animal handling, virus inoculations, and tissues sampling) were conducted after sedating the animals with ketamine chlorydrate (Rhône-Mérieux, Lyon, France, 10 mg/kg). Macaques were killed at defined time points by sedation with ketamine chlorhydrate (10 mg/kg) followed by intravenous injection of 180 mg/kg sodium pentobarbital (CEVA santé animale S.A., La ballastiere). Tissues not needed for this project were shared. Samples of spleen and peripheral and mesenteric lymph nodes, as well as blood, were harvested. Plasma was isolated from EDTA blood samples by centrifugation for 10 min at 950×g, and cryopreserved at −80 °C. Animal plasma was processed for viral RNA extraction using a Viral RNA and DNA isolation kit (Macherey–Nagel), followed by titration of viral RNA copy numbers by quantitative real-time RT-PCR. Tissue samples were collected in RPMI 1640 GlutaMAX™ Medium (Thermo Fischer) for further cell preparation.
SIV vRNA quantification in plasma
Blood plasma was isolated from EDTA-treated blood samples by centrifugation for 10 min at 1500×g, and stored frozen at − 80 °C. Viral RNA was prepared from 200 μL cell-free plasma using the High Pure viral RNA kit (Roche Diagnostics, Meylan), according to the manufacturer’s instructions. Absolute concentrations of plasma viral RNA (vRNA) were determined by RT-qPCR assay with the Superscript III Platinum one-step quantitative RT-PCR system (Invitrogen), using the following SIV gag primers and probe: Forward GCAGAGGAGGAAATTACCCAGTAC/Reverse: CAATTTTACCCAGGCATTTAATGTT/Probe: TGTCCACCTGCCATTAAGCCCGA. The quantification limit (QL) was estimated to be 111 copies/mL and the detection limit (DL) 37 copies/ml.
Cell preparations, antibodies, and FACS analysis
For longitudinal follow-up studies of SIV- and CHIKV-infected animals, 500 µL whole blood was stained with monoclonal antibodies (Table S1) for 15 min at room temperature. RBCs were lysed by hypotonic shock using diluted Lysing Solution 10X Concentrate (BD biosciences) for 15 min at room temperature, followed by washing in PBS and fixation using CellFIX (BD biosciences). Peripheral and mesenteric lymph node and spleen cells were obtained by mechanical dissociation using a GentleMACS dissociator (Miltenyi Biotec). Suspensions were passed through a 70-µm-pore size cell strainer, followed by a second mechanical disruption with a 3-mL needle-free syringe and passage through a second 70-µm-pore cell strainer, before washing with PBS. For spleen cells, we performed density gradient isolation using lymphocyte separation medium (Eurobio, Abcys) followed by the lysis of red blood cells by hypotonic shock. Cell counts were determined with a Vi-CELL (Beckman-coulter) and surface staining was performed on 1 × 107 isolated cells from tissues. Except for blood cells, staining of cells extracted from tissues was performed after saturation of Fc receptors with 5% healthy macaque serum at 4 °C for 20 min. To discriminate live from dead cells in our analysis, samples were incubated with the amine-reactive dye LIVE/DEAD® Fixable Blue Dead Cell Stain Kit and exposed to UV (Life technologies) for 15 min at room temperature. Cells were then labeled with monoclonal antibodies (Table S1) for 15 min at room temperature, washed in PBS, and fixed in CellFIX (BD biosciences). Monoclonal antibody 17E5.3E9 was generated by the Baylor Research Institute, using the amino acid sequence shown in Figure S1A which corresponds to the extracellular part of human LILRB2 protein, following the procedure reported in Banchereau et al. [22]. Screening by ELISA assays indicated no cross-reactivity of the antibody with LILRB1 and reactivity with soluble human LILRB4. The reactivity of the 17E5.3E9 antibody for LILRB2 was first evaluated by comparing the expression profile of blood immune cell subsets by flow cytometry in humans and cynomolgus macaque (Figure S1B). The specificity of the 17E5.3E9 antibody against LILRB2 in cynomolgus macaque was further confirmed by staining blood neutrophils before or after up-regulation of LILRB2 induced by granules exocytosis as previously reported [23]. As shown in Figure S1C, after stimulation for 30 min of cynomolgus macaque or human neutrophils with TNF-a (100 ng/ml), staining with 17E5.3E9 antibody revealed an up-regulation of LILRB2 expression on neutrophils of both species.
For the study of HIV-1-infected patients, including non-infected donors, cryopreserved PBMCs were rapidly thawed and washed with RPMI 1640 GlutaMAX™ Medium, (Thermo Fischer). For each sample, 5 million PBMCs were stained using the LIVE/DEAD® kit, to select live cells, followed by surface antibody staining (Table S1) for 15 min. Cells were washed and fixed using CellFIX (BD biosciences). Data for all samples were acquired on an LSR Fortessa (BD biosciences) equipped with five lasers (355, 405, 488, 561 and 640 nm) and analyzed using FlowJo v9.9 (FlowJo LLC, USA). For longitudinal follow-up studies, data acquisition was performed after calibration (using BD cytometer setup and tracking beads), using automated application settings. Isotype-specific antibodies for LILRB2, MHC-I, and CD86 were used as intra-assay controls. Absolute cell counts were calculated from leukocyte counts obtained by automated cell counting (Beckman Coulter) combined with flow cytometry data. Absolute cDC, lymphocyte, and monocyte counts were determined as the product of total leukocyte counts and the percentage of cells in the CD45+ gate.
Generation of monocyte-derived dendritic cells (MoDC)
PBMCs were isolated from fresh whole blood of healthy donors by density gradient in lymphocyte separation medium (Eurobio, Abcys). Red blood cells were lysed by hypotonic shock. Monocytes (CD14+) were purified from PBMCs by positive selection with anti-CD14 magnetic beads (Miltenyi Biotec). Sorted monocytes were differentiated into immature MoDCs by culture in RPMI 1640 Medium, GlutaMAX™ (Thermo Fischer) supplemented with 10% SVF and 1% antibiotic cocktail, along with GM-CSF (10 ng/ml) and IL-4 (25 ng/ml) (PeproTech) for 6 days.
HIV-1 strain
Infections were performed with HIV-1BaL (R5-monocytotropic). The HIV-1BaL viral strain was amplified for 10 days in IL-2/PHA-stimulated PBMCs, from three blood donors. Cell supernatants were collected 7 and 10 days post-infection and stored at − 20 °C. Viral replication was measured by quantifying reverse transcriptase (RT) activity in the cell-culture supernatants, using the Lenti RT Activity Kit (Cavidi), and TCID50 was calculated using Kärber’s formula. Virus was concentrated by centrifugation on Vivaspin 100,000 kDa columns (Sartorius) at 1500×g for 30 min.
Infection and stimulation of MoDCs
Immature MoDCs (5 × 106 cells) were pulsed with HIV-1 BaL (10-1MOI) by spinoculation at 1200×g for 60 min followed by incubation at 37 °C, in the presence of 5% CO2, for another hour. Pulsed cells were then washed three times and cultured for up to 8 days. Culture supernatants were discarded and replenished with fresh medium every 3 days. For in vitro stimulation of immature MoDC, cells were cultured in media alone or supplemented with various concentrations of the TLR7/8 Agonist, R848 (Invivogen) for 24 h or with the cytokines IFN-a, IL-10, or TNF-a (Miltenyi) individually for 48 h.
Analysis of LILRB2 and MHC-I expression in HIV-1 BaL-infected MoDCs
MoDCs were harvested and stained for surface expression analysis of LILRB2 and MHC-I by flow cytometry 5 and 8 days after pulsing with HIV-1 BaL. To discriminate dead from live cells during analysis, MoDCs were incubated with LIVE/DEAD® Fixable Blue Dead Cell Stain Kit (Life technologies) for 15 min, followed by incubation with anti-LILRB2 (PE, clone 42D1, eBioscience) and anti-MHC-I (APC/Cy7, clone W6/32, Biolegend) antibodies. MoDCs were then fixed, permeabilized (Cytofix/Cytoperm Buffer, BD), and stained with anti-HIV p24 (FITC, clone KC57, Beckman Coulter) antibody. Cells were washed and fixed using CellFIX (BD biosciences) and data for each sample were acquired on an LSR Fortessa (BD Biosciences).
Statistical analysis
To assess surface expression of LILRB2, MHC-Class I and CD86-specific isotype control antibodies for each marker were used to calculate the Mean Fluorescent Intensity as previously described [24]. The nonparametric Mann–Whitney U test was used to compare data between healthy donors and HIV-infected patients and the paired nonparametric Wilcoxon signed-rank test was used to compare data of PHI individuals before and after cART. We performed the Wilcoxon matched-pairs signed rank test to statically analyze LILRB2 and MHC-I levels at various time points during the longitudinal study of NHP infected by SIV. The non-parametric Mann–Whitney U test was used to analyze data between noninfected and HIV-infected MoDC for the in vitro MoDC assays, and the paired nonparametric Wilcoxon signed-rank test was used to compare data from the same cells at various times after HIV infection or various TLR7/8 agonist or cytokine concentrations. All statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software).
Results
The expression of LILRB2 and MHC-I are simultaneously increased on the surface of cDCs during early HIV infection
Since cDCs play a key role in the installation of the host’s anti-viral immune response and that their functions are tightly regulated by LILRB2-MHC-I interactions, we first assessed the expression level of both LILRB2 and its MHC-I ligands in cDCs during primary HIV infection. To this end, we analyzed blood samples from HIV-1-infected patients collected less than 30 days after infection (primary HIV+) and after 1 year of combined antiretroviral treatment (cART). Primary infected HIV+ patients exhibited high viral loads at inclusion, whereas viral loads were almost undetectable in these patients after 1 year of cART (Table 1). We also included HIV-infected elite controllers samples collected 5 years following infection at least and who naturally control HIV replication, and samples from non HIV-infected healthy donors.
Table 1.
Clinical and demographic characteristics of HIV-1-infected patients and control subjects
| Study groups | Treatment history | Gender | Age (median, range) | CD4 T-cell count, cell/µL (median, range) | Plasma viral load (log RNA copies/mL) (median, range) | Time since HIV-1 infection |
|---|---|---|---|---|---|---|
| Primary infected HIV-1+ patients (n = 7) | Naive | M | 37 (24–46) | 470 (258–669) | 8.9 × 106 (0.3–18.2) | < 30 Days |
| Primary treated HIV-1+ patients (n = 7) | cART | M | 843 (570–1247) | Undetectable < 50 | > 12 Months | |
| HIV-1+ Controllers (n = 6) | Naive | M | 38 (25–49) | 827 (652–1180) | Undetectable < 50 | > 5 Years |
| HIV-1 negative (n = 6) | Naive | M | 35 (25–45) | 856 (634–1412) | N/A | N/A |
cART combination anti-retroviral therapy, M male, N/A not applicable
We established and applied a flow cytometry gating strategy to discriminate cDCs (CD1c+) among the PBMC samples (Figure S2). Both LILRB2 and MHC-I levels were highly elevated on cDCs from HIV+ patients in acute phase of viral infection (Fig. 1). In contrast, the cDCs from these patients exhibited much lower levels of LILRB2 and MHC-I after 1 year of cART (Fig. 1a, b). Similarly, LILRB2 and MHC-I levels were lower on cDCs from elite controllers than those from primary HIV-infected patients. LILRB2 levels were slightly higher on cDCs from elite controllers than those from healthy donors, in agreement with a previous study [25]. We also evaluated the expression of the CD86 co-stimulatory molecule and the level of HLA-DR maturation marker. Prior to cART initiation, untreated HIV primary-infected patients show an increased expression of CD86 (Fig. 1c), whereas HLA-DR expression is decreased on cDCs (Figure S3). These expression profiles highlight the partial activation/maturation of cDCs during HIV-1 infection, as previously reported [26]. Finally, since cDCs interact with CD4+ T cells during early infection to shape adaptive immune responses, we also compared MHC-I levels on CD4+ T cells in samples from primary HIV-infected patients before and after 1 year of cART. MHC-I expression on CD4+ T cells was higher during primary HIV infection, before treatment, than after 1 year of cART, but the difference was not statistically significant (Fig. 1d).
Fig. 1. Characterization of LILRB2 and MHC-I expression level on cDCs from patients during the early phase of HIV-1 infection.

a–c Analysis of LILRB2, MHC-I, and CD86 surface expression on cDCs from blood samples of early HIV-1 infected patients before (primary HIV+) or after 1 year of cART (HIV+ cART+), HIV-1 infected elite controller patients (HIC), and HIV-1 non-infected controls (HIV-). d Analysis of CD4+ T cells in these samples to evaluate surface expression of MHC-I. Data are represented as mean fluorescence intensity with statistical analysis performed using the Mann–Whitney U test between HIV- (n = 6), HIC (n = 6), and primary HIV+ patients (n = 7) and the Wilcoxon signed-rank test for HIV+ primary patients before and after cART (p < 0.05 is considered significant)
These results demonstrate that HIV infection promotes the simultaneous increase of LILRB2 and MHC-I expression on cDCs from blood during the early immune response against HIV in humans.
LILRB2 is similarly distributed among human and cynomolgus macaque immune cell subsets
We further characterized the dynamics of LILRB2 expression on cDCs during HIV infection using the model of experimental infection of cynomolgus macaques with pathogenic SIVmac251. We first confirmed that LILRB2 is similarly distributed among immune cell subsets of humans and macaques as the expression profile of LILRB2 on immune cell subsets from non-human primates was still unknown. Flow cytometry analyses showed that the proportion of blood inflammatory monocytes (CD14midCD16high) was higher in cynomolgus macaques than in humans (Fig. 2), in agreement with a previous study [27]. We used both CD11c and CD1c markers since CD11c alone is not sufficient to accurately distinguish cDCs in macaques [27, 28]. LILRB2 was expressed on all cDCs, pDCs, and monocyte subsets in both humans and cynomolgus macaques, but at different levels, depending on the cell population (Fig. 2b and d). Similarly to humans, the level of LILRB2 was significantly higher on cDCs than other myeloid cell subsets in cynomolgus macaques (Fig. 2e). In contrast, we detected no surface expression of LILRB2 on lymphoid cell subsets, including T cells and B cells of either species. Expression levels of immunoregulatory receptors may vary according to tissue localization. Thus, we also investigated the expression level of LILRB2 on immune cell subsets from secondary lymphoid organs where cDCs shape adaptive immune responses against viruses. As shown in Figure S4, cDCs isolated from peripheral and mesenteric lymph nodes or spleen of cynomolgus macaques showed higher levels of LILRB2 than cDCs from peripheral blood. Moreover, analysis of T and B cells did not reveal expression of LILRB2 on these cell populations in secondary lymphoid organs.
Fig. 2. Comparison of the LILRB2 expression pattern between human and cynomolgus macaque immune cell subsets.
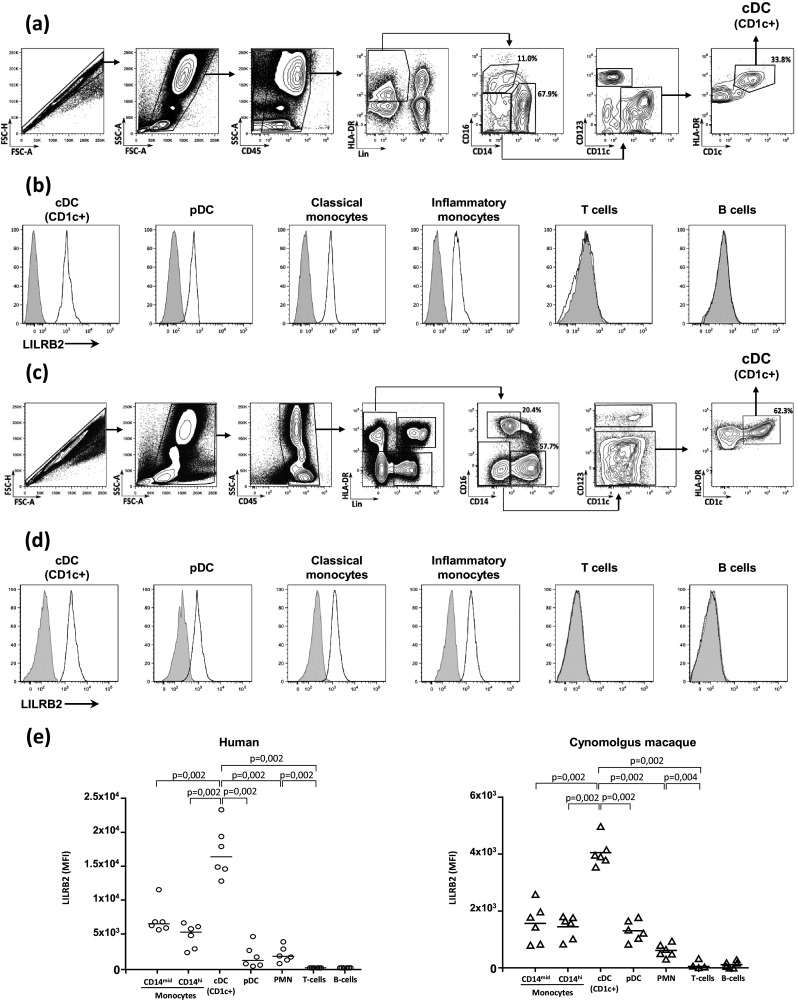
Flow cytometry gating strategy to characterize blood immune cell subsets in humans (a) and cynomolgus macaques (c). Surface expression of LILRB2 was assessed using monoclonal antibodies against human (clone 42D1) or cynomolgus macaque (clone 17E5.3E9) LILRB2. The distribution of LILRB2 expression is shown for various immune cell subsets, including cDCs, pDCs, classical (CD14high CD16−) and inflammatory (CD14mid CD16high) monocytes, T and B cells in healthy (b) human donors and (d) cynomolgus macaques. e Comparison of LILRB2 surface expression on whole blood immune cell subsets of healthy human donors (n = 6) and cynomolgus macaques (n = 6). Results shown are representative of at least three independent experiments. Statistical analyses were carried-out using the Mann–Whitney U test for both human and cynomolgus macaque samples (p < 0.05 is considered significant)
Overall, these results show that LILRB2 is similarly distributed among immune cell subsets in humans and cynomolgus macaques. Our data also show that LILRB2 levels are higher in cDCs from lymph nodes and spleen than those from peripheral blood, suggesting tight regulation of cDCs by LILRB2 in secondary lymphoid tissues.
Expression of LILRB2 and MHC-I are coordinately increased on cDCs during early SIV infection of cynomolgus macaques
We next investigated the dynamics of LILRB2 expression in blood and secondary lymphoid tissues during early SIV infection in vivo. Indeed, this model does not only allow the study of the dynamics of immune events before and during the first days following viral infection, but also permits access to secondary lymphoid organs where immune responses are shaped. Hence, we first infected three female cynomolgus macaques with the SIVmac251 strain and followed the surface expression of LILRB2 on cDCs from blood and lymph nodes over time by flow cytometry. Viral load was already detectable in peripheral blood 2 days after SIVmac251 infection and reached a peak 10 days following infection (mean = 6.2 ± SD = 0.15 log viral RNA copies) (Fig. 3a). As previously reported, a transient decrease of cDC counts in peripheral blood is induced between 9 and 10 days post-infection by SIVmac251 [29, 30], concomitant with the typical transient reduction of CD4+ T cell counts during acute infection.
Fig. 3. Analysis of the dynamics of LILRB2 and MHC-I expression on cDCs during infection of cynomlogus macaques by SIV.
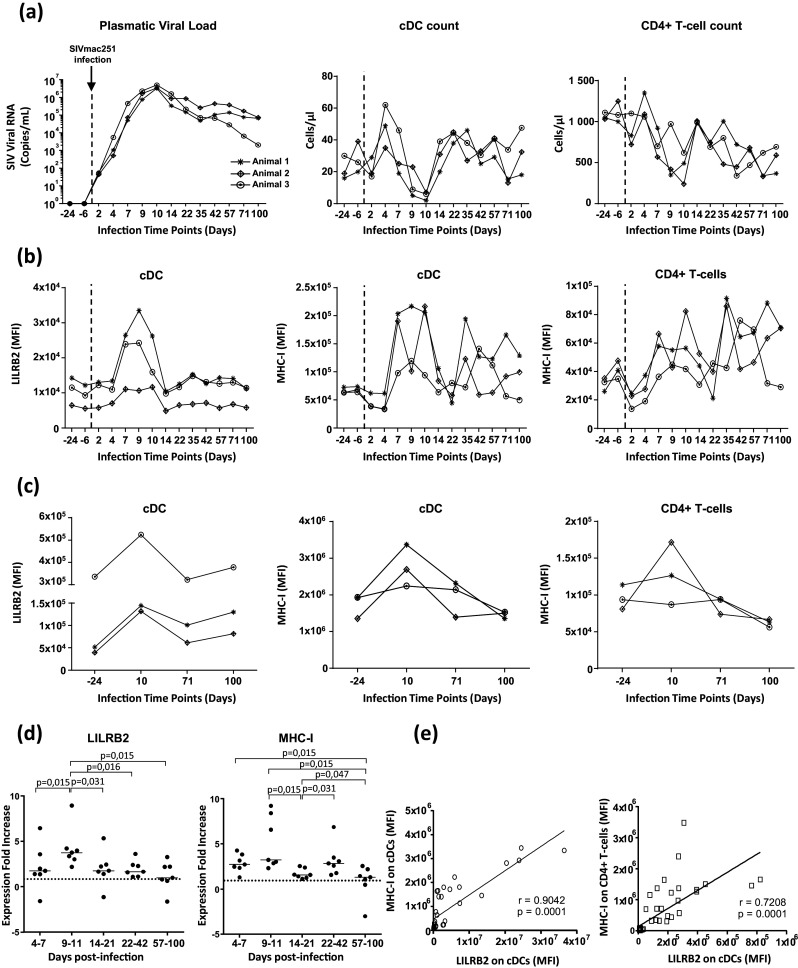
a Evolution of the viral load in the plasma of three female cynomolgus macaques infected by SIVmac251 at 1000 AID50/ml. The cell counts of both cDCs (CD1c+) and CD4+ T cells in blood measured by flow cytometry during SIV infection is also represented. b Flow cytometry analysis of LILRB2 and MHC-I expression on blood cDCs and MHC-I expression on blood CD4+ T cells during SIV infection. c Longitudinal follow-up of LILRB2 and MHC-I surface expression on cDCs from peripheral lymph nodes of cynomolgus macaques during SIV infection. The characterization of MHC-I surface expression on CD4+ T cells is also shown. d Modulation of LILRB2 and MHC-I levels on blood cDCs from seven cynomolgus macaques followed at various time points during early (days 4–7, days 9–11, days 14–21, days 22–44) and advanced (days 57–100) phases of SIV infection. Data are shown as the fold change of surface expression relative to baseline (represented with dashed line). Statistical analyses were carried-out using the Wilcoxon matched-pairs signed rank test (p < 0.05 is considered significant). e Positive correlation between LILRB2 levels on cDCs and MHC-I levels on cDCs (left) or CD4+ T cells (right). Symbols (circles or squares) represent data from ten cynomolgus macaques sampled at various time points after SIVmac251 infection. The Pearson R correlation coefficient and p values are shown on each plot (p < 0.05 is considered significant)
LILRB2 expression strongly increased on cDCs during the initial phase of SIVmac251 infection (Fig. 3b). Indeed, LILRB2 levels were already higher 4 days following SIVmac251 infection and reached a peak between 9 and 10 days post-infection, before returning to initial levels 14 days post-infection. MHC-I levels also increased on cDCs during the early phase of SIV infection with kinetics close to those of LILRB2 (Fig. 3b). We also assessed the level of MHC-I ligands on CD4+ T cells. MHC-I levels on CD4+ T cells rapidly decreased and then returned to normal between 2 and 7 days post-infection (Fig. 3b). Moreover, MHC-I levels on CD4+ T cells 9–10 days post-infection were higher than those measured before SIV infection.
We also assessed the expression of LILRB2 and MHC-I ligands on cDCs from peripheral lymph nodes of these animals. As shown in Fig. 3c, expression of LILRB2 and MHC-I is increased on cDCs from peripheral lymph nodes at day 10 post-infection but decreased during the later phase of SIV infection (Fig. 3c). Moreover, we investigated the dynamics of LILRB2 expression on monocytes and macrophages, as the function of these myeloid cell subsets is also altered after SIV or HIV infection. There was a transient decrease of monocyte counts at day 10 post-infection (Figure S5). Both expression of LILRB2 and MHC-I were transiently up-regulated on blood monocytes during the early phase of SIVmac251 infection between days 4 and 14 (Figure S5B and D) as observed for cDCs. In contrast to LILRB2, the expression of MHC-I also showed an increase between days 22 and 42 post-infection on blood cDCs and monocytes (Figs. 3b and S5B). This enhancement of MHC-I expression could result from the activation of CD8 T cell response and IFN-g production that are induced in this window of time as previously reported [31, 32]. Moreover, analysis of macrophages from peripheral lymph nodes revealed a large increase of LILRB2 and MHC-I levels, at day 10 post-infection that decreased during the later phase (Figure S5C).
The modulation of innate immune responses against HIV can differ according to sex [33]. Thus, we analyzed the dynamics of LILRB2 expression on cDCs in a group of male cynomolgus macaques infected by the same stock of SIVmac251. LILRB2 and MHC-I levels also increased on cDCs and monocytes from peripheral blood (Figure S6B and C), as well as cDCs and macrophages from peripheral lymph nodes (Figure S6D) during the early phase of SIV infection. These data show that the increase of LILRB2 expression on cDCs is independent of sex and confirm that the LILRB2/MHC-I inhibitory axis is enhanced during the early immune response against SIV.
Having shown that expression of LILRB2 is strongly enhanced on cDCs in early phase of SIV infection we next studied modulation of the SIRPa inhibitory receptor (also known as CD172a), which shares structural similarities with LILRB2 and regulates cDC functions [34]. SIRPa levels on cDCs from blood or lymph nodes remained stable during SIV infection, suggesting that the expression of LILRB2 specifically increases during early immune responses against SIV (Figure S7).
Finally, we extended our study to additional cynomologus macaques (n = 7) to achieve statistical significance and thus demonstrate that both LILRB2 and MHC-I levels significantly increase on cDCs and monocytes in blood during the early phase of SIV infection (Figs. 3d and S5D). Further analysis showed that LILRB2 levels on cDCs strongly correlated with those of its MHC-I ligands found on cDCs or CD4+ T cells during SIV infection (Fig. 3e).
These data demonstrate that SIVmac251 infection promotes a large and specific increase in the expression of LILRB2 and its MHC-I ligands on cDCs, as well as other myeloid cells, in blood and lymph nodes during early immune responses. These results also show that these mechanisms occur independently of the sex of the animals.
HIV infection and TLR7/8 stimulation enhance the expression of LILRB2 on human monocyte-derived dendritic cells (MoDCs)
We next investigated the factors that could account for increased expression of LILRB2 on cDCs during the early immune responses against HIV and SIV. Indeed, although it is well established that LILRB2 plays an important role in the regulation of cDC functions, the events involved in the regulation of LILRB2 expression are still poorly characterized. Hence, we first assessed the direct impact of HIV infection on LILRB2 expression by co-incubating monocyte-derived dendritic cells (MoDCs) with HIV-1. A time-course analysis of MoDC infection by HIV-1 (R5 strain) showed that nearly 30% of MoDCs were productively infected after 8 days of co-incubation (Fig. 4a, b).
Fig. 4. Analysis of LILRB2 and MHC-I expression on MoDCs after infection with HIV-1BAL or stimulation of TLR7/8 with R848.
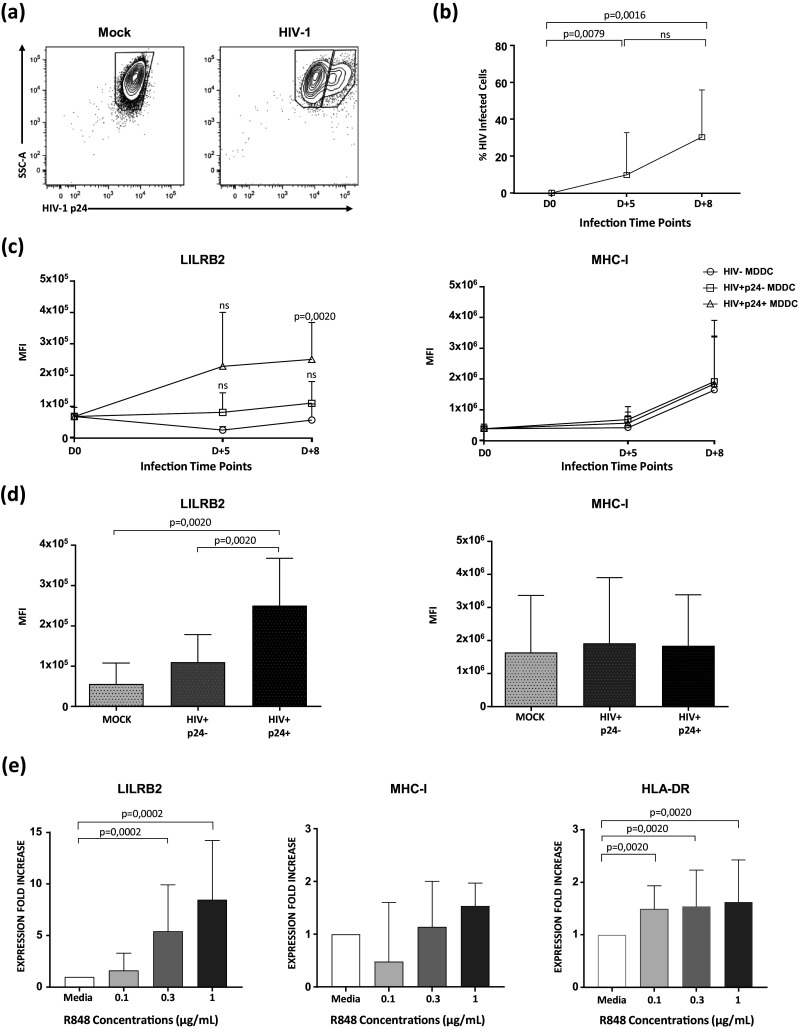
a Representative flow cytometry dot-plots from non-infected (Mock) and HIV-1 infected (HIV-1BAL) MoDCs. Intracellular staining of MoDCs for p24 was carried-out 8 days post-infection. b Kinetics of HIV-1BAL replication in MoDCs either five (D + 5) or eight (D + 8) days post-infection. The percentage of HIV-infected cells was calculated by measuring the proportion of MoDCs positive for p24 antigen expression (n = 10 ± SD = 18.2, Mann–Whitney U test for p value, with p < 0.05 considered significant). c LILRB2 and MHC-I expression on either MoDCs alone, MoDCs incubated with HIV-1, but not expressing p24, or MoDCs incubated with HIV-1 and expressing p24 (n = 10 ± SD = 1.3 × 105, Wilcoxon Paired test for p value, with p < 0.05 considered significant). d Quantification of LILRB2 and MHC-I expression on non-infected and HIV-1 infected MoDCs with or without productive p24 Ag 8 days after HIV-1 infection (n = 10 ± SD = 2.7 × 105, Wilcoxon Paired test for p value, with p < 0.05 considered significant). Data are representative of seven independent experiments. e Modulation of LILRB2, MHC-I, and HLA-DR expression on unstimulated (media alone) MoDCs or MoDCs stimulated with various doses of R848. Values represent the fold change of mean fluorescence intensity relative to that of unstimulated cells (n = 16 ± SD = 3.6, Wilcoxon Paired test for p value, with p < 0.05 considered significant)
Further analysis showed that LILRB2 expression was up-regulated on MoDCs exposed to HIV (Fig. 4c, d). Remarkably, LILRB2 expression was significantly higher in MoDCs showing intracellular p24 staining confirming productive HIV replication. However, HIV infection had no significant impact on the modulation of MHC-I expression in MoDCs.
TLR7/8 could also be involved in the regulation of LILRB2 expression, as they are expressed by cDCs and recognize viral RNA. Therefore, we next stimulated MoDCs with various doses of the TLR7/8 agonist R848. Stimulation of TLR7/8 with R848 induced a large increase in LILRB2 levels on MoDCs in a dose dependent manner (Fig. 4e). MHC-I levels were modulated to a much lower extent by R848 stimulation than LILRB2 levels, in agreement with data of MoDC infected by HIV. Finally, HLA-DR expression, used as positive control, was also enhanced on MoDCs but to a lower extent than LILRB2.
Altogether, these data show that HIV-1 can directly promote the expression of LILRB2 in MoDCs, through infection associated with viral replication, and suggest that HIV sensing by TLR7/8 could also enhance LILRB2 expression.
Regulation of LILRB2 expression in MoDCs is differentially affected by type-I IFN, IL-10, and TNF-a
A cytokine cascade is rapidly induced by HIV-1 infection and cytokines are important regulators of cDC functions. Moreover, previous studies indicate that type-I IFNs, TNF-a, and IL-10 are among the first cytokines highly induced during early HIV infection [32]. Thus, we also investigated the impact of these cytokines on the modulation of LILRB2 expression by flow cytometry. Stimulation of MoDCs with type-I IFNs significantly enhanced the surface expression of LILRB2 in a dose dependent manner (Fig. 5a). The surface expression of MHC-I and HLA-DR was also significantly enhanced under these experimental conditions. In contrast, stimulation of MoDCs with type-I IFNs did not alter the surface expression of SIRPa. In addition, treatment of MoDCs with IL-10 significantly increased LILRB2 and MHC-I expression in a dose dependent manner and, to a lesser extent, the expression of HLA-DR or SIRPa (Fig. 5b). Finally, stimulation of MoDCs with TNF-a revealed no increase of LILRB2 expression, but increased that of MHC-I, HLA-DR, and SIRPa (Fig. 5c).
Fig. 5. Impact of cytokine stimulation on the modulation of LILRB2 and MHC-I expression on MoDCs.
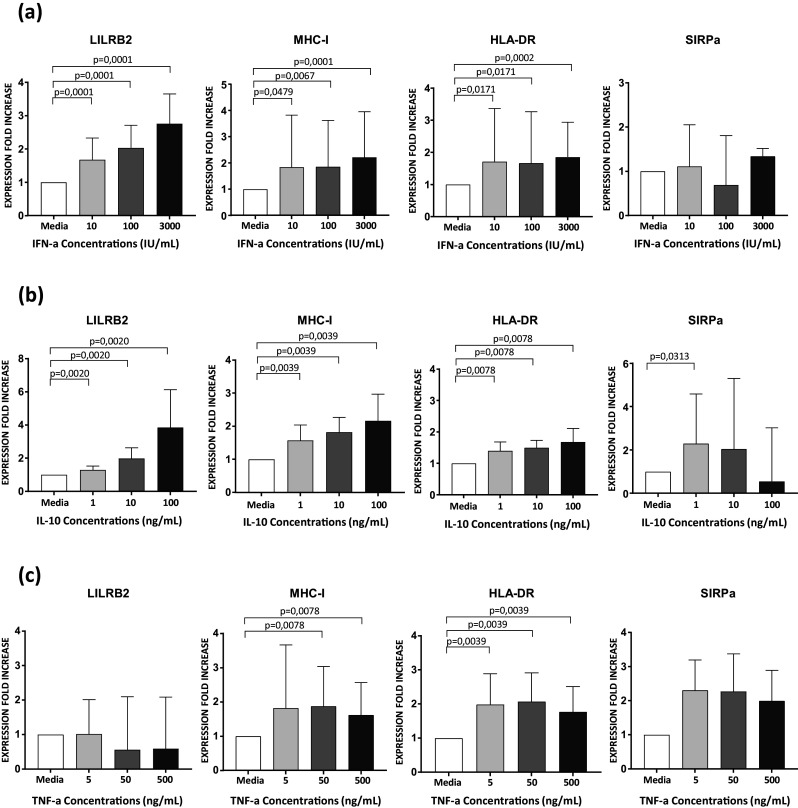
a Characterization of LILRB2, MHC-I, HLA-DR, and SIRPa surface expression on unstimulated (media alone) or stimulated MoDCs with various doses of type-I IFNs for 48 h (n = 16 ± SD = 3.3, Wilcoxon Paired test for p value, with p < 0.05 considered significant). b Analysis of MoDCs after stimulation with IL-10 for 48 h and quantification of the surface expression of LILRB2, MHC-I, HLA-DR, and SIRPa (n = 16 ± SD = 5.1, Wilcoxon Paired test for p value, with p < 0.05 considered significant). c Unstimulated MoDCs and MoDCs stimulated for 48 h with various doses of TNF-a were cultured to assess the modulation of LILRB2, MHC-I, HLA-DR and SIRPa surface expression (n = 16 ± SD = 1.8, Wilcoxon Paired test for p value, with p < 0.05 considered significant). Values represent the fold change of mean fluorescence intensity relative to that of unstimulated. Results shown are representative of 6 independent experiments
These data show that cytokines produced during the early phase of HIV-1 infection have a variable impact on the regulation of LILRB2 surface expression and suggest that the modulation of LILRB2 expression can occur through various mechanisms in cDCs.
LILRB2 expression on cDCs is down-modulated during early immune responses of cynomolgus macaques infected by chikungunya virus
Finally, we characterized LILRB2 surface expression in the context of a naturally controlled primary infection, such as chikungunya virus (CHIKV) in cynomolgus macaques [20], to determine whether the increase of LILRB2 levels on cDCs induced during early immune responses is specific to SIV infection. CHIKV infection of cynomolgus macaques induced a high viral load, peaking 2 days after virus inoculation (mean = 9.1 ± SD = 0.4 log viral RNA copies) (Fig. 6a). CD14 surface expression on monocytes rapidly decreased 4 days post-infection (Fig. 6b), probably through a shedding mechanism [35]. CHIKV infection was also accompanied by a modification of blood leukocyte counts, characterized by a transient decrease of circulating cDCs and lymphocytes 2 days post-infection (Figure S8A and B). Moreover, the number of granulocytes transiently peaked 2 days post-infection, whereas the number of monocytes in the blood peaked 7 days post-infection (Figure S8B).
Fig. 6. Longitudinal analysis of LILRB2 expression on cDCs during infection of cynomolgus macaques by CHIKV.
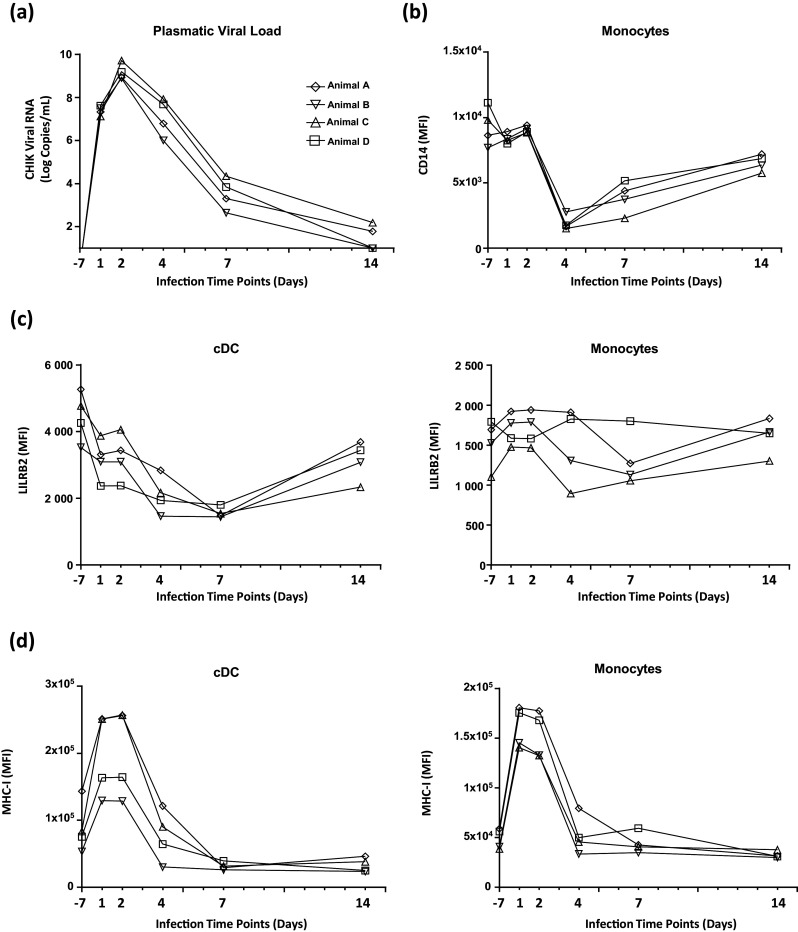
a Evolution of plasma viral load during infection of four cynomolgus macaques infected with the CHIKV-LR2006-OPY1 strain at 100 AID50/mL. b Flow cytometry analysis of cell surface CD14 expression on monocytes during the acute phase of CHIKV infection. c Time-course analysis of LILRB2 expression level on cDCs and monocytes from peripheral blood during the infection of cynomolgus macaques by CHIKV. d Analysis of MHC-I expression level on blood cDCs and monocytes during the infection of cynomolgus macaques by CHIKV
The analysis of the dynamics of LILRB2 surface expression during CHIKV infection showed that LILRB2 levels on cDCs decreased between baseline and 7 days post-infection, whereas they remained stable on monocytes (Fig. 6c). However, MHC-I levels on cDCs and monocytes increased during early immune responses against CHIKV (Fig. 6d).
These results show that, in contrast to SIV infection, LILRB2 expression is down-modulated on cDCs during the installation of an efficient immune response against CHIKV and suggest that up-regulation of LILRB2 on cDCs is associated with the dysregulation of innate responses and viral persistence mechanisms.
Discussion
In vitro studies have suggested that the strength of the interaction between LILRB2 and MHC-I plays a critical role in the dysregulation of cDCs induced by HIV [16, 36]. This was proposed to drive disease progression [15]. However, the dynamics of the LILRB2/MHC-I inhibitory axis that tip the balance during cDC-mediated early immune responses towards viral persistence or control remains unknown.
In this study we first investigated the modulation of LILRB2 and MHC-I expression on cDCs during the acute phase of HIV-1 infection. Our data show that cDCs express significantly higher levels of LILRB2 and MHC-I before cART initiation. These results are concordant with previous transversal studies carried-out on blood mDCs (CD11c+) of HIV-1 infected patients aiming to measure the expression of either LILRB2 or MHC-I, separately [36–38]. However, the simultaneous expression of LILRB2 and its MHC-I ligands during early HIV-1 infection has not been assessed. Studies on primary HIV-infected patients are limited to one blood sample, due to immediate cART administration. We therefore used the cynomolgus macaque model to track the dynamics of LILRB2 and MHC-I expression during infection with SIVmac251.
The analysis of LILRB2 distribution revealed very similar expression patterns in human and macaque immune cell subsets. These results indicate that cynomolgus macaques are more suitable for the study of the LILR family in vivo than mice in which PIR-B, the proposed counterpart of LILRB2, is highly expressed on B cells and contains six extracellular Ig-like domains, instead of the four contained in LILRB2 of humans and macaques [39, 40]. In this experimental species we demonstrate that pathogenic SIVmac251 infection induces transient, high expression of LILRB2 and its MHC-I ligands on cDCs from blood and peripheral lymph nodes during the early immune response (Fig. 3 and S6). Altogether, our results strongly suggest that the coordinated increase of LILRB2 and MHC-I expression on the same cell could enhance cis interactions, providing inhibitory signals and leading to the dysregulation of cDCs. In this regard, it was previously reported that the cis interaction of LILRB2 with MHC-I on the surface of the same cell is a potent way to generate inhibitory signals that impair the production of cytokines by mastocytes [12, 13]. We also showed increased MHC-I expression on CD4+ T cells during early SIV infection, which may favor the dysregulation of cDCs through binding to LILRB2. Indeed, after virus up-take, cDCs mainly interact with CD4+ T cells to shape the adaptive immune response during the early phase of the infection.
We could also observe that a pool of peripheral cDCs was rapidly depleted from blood following SIV infection, concomitant with the enhancement of LILRB2 expression (Fig. 3). Such a reduction of cDC levels has already been reported during the acute phase of SIV and HIV infection and is partially explained by the migration of cDCs to lymph nodes [29]. The increase of LILRB2 surface expression and the migration of blood cDCs toward lymph nodes occur in parallel. We thus speculate that cDCs arriving to lymph nodes are highly prone to dysregulation through the LILRB2/MHC-I inhibitory axis. In support of our hypothesis, several studies have shown that cDCs that have migrated to peripheral lymph nodes during the acute phase of SIV infection have an immature phenotype and dysfunctional characteristics [4, 5, 41].
The induced increase of LILRB2 expression during SIV infection peaked at day 9 and return to basal level, around 14 days post infection. LILRB2 expression remained significantly higher in primary HIV-infected patients that had been infected for more than 2 weeks. These discrepancies could result from differences between natural HIV infection and the standardized experimental procedure used in SIV model. Indeed, cynomolgus macaques are infected intravenously by high dose (1000 AID50/mL) from the same bulk of viral strain (SIVmac251). Moreover, the average of the viral load levels detected in primary HIV-infected patients (8,9E + 06 log HIV RNA copies/mL) is higher than that of the viral load peak of SIV-infected cynomolgus macaques (3,93E + 06 log SIV RNA copies/mL). Therefore, it is also possible that increase of LILRB2 expression is still detected in patients because of elevated viral load and cytokines environment resulting from high HIV production.
We also sought to determine whether the increase of LILRB2 expression on cDCs induced during the acute phase of SIV infection was specific to LILRB2. For this purpose we tested antibodies directed against human LILRB1 or LILRB4 on cynomolgus macaque blood samples. However, these antibodies did not reveal any staining on the immune cell subsets of macaque cynomolgus. Therefore, we could not compare the dynamics of LILRB2 surface expression to that of other inhibitory receptors belonging to the LILR family in cynomolgus macaques. However, we could follow the dynamics of the SIRPa inhibitory receptor. SIRPa shares structural similarities with LILRB2, including extracellular Ig-like and intracellular ITIM domains, and regulates cDC functions through the recruitment of SHP1/2 phosphatases [42]. Moreover, SIRPa is a critical marker for the identification of cDCs across tissues and species, including humans, macaques, and mice [43]. Our data show that SIV infection has no impact on the expression of SIRPa in cDCs from blood and lymph nodes during early immune responses and strongly suggest that the up-regulation of LILRB2 on cDCs is specifically targeted by SIV (Figure S7).
Our study shows that SIV infection also enhances the expression of LILRB2 on monocytes and macrophages in vivo during early immune responses (Figure S5 and S6). The increase of LILRB2 activity on monocytes could negatively affect their function, in particular, their differentiation into cDCs, contributing to the loss of cDCs induced by apoptosis following SIV and HIV infection [44]. The induced up-regulation of LILRB2 on macrophages is in accordance with recent studies demonstrating the early dysregulation of macrophages from peripheral lymph nodes in SIV-infected rhesus macaques [5]. The early dysfunctions of macrophages caused by LILRB2 could also disturb immune responses in secondary lymphoid tissues, since macrophages play an important role in the innate immune responses against viruses through the production of soluble factors and antigen-presentation.
In this study we also investigated the dynamics of LILRB2 expression on cDCs during CHIKV infection in vivo. Surface expression of LILRB2 on cDCs decreased during the early immune response and CHIKV replication was rapidly controlled (Fig. 6). Moreover, peripheral cDC levels decreased during the acute phase of CHIKV infection, suggesting the migration of cDCs with less LILRB2 on their surface toward peripheral lymph nodes (Figure S8). Expression of MHC-I on cDCs and monocytes rapidly and transiently increased between 2 and 4 days after CHIKV infection. In the presence of low amounts of LILRB2, the increased MHC-I expression could favor the availability of MHC-I ligands for CD8+ T-cell receptors. Indeed, previous reports indicate that CD8+ T cells are rapidly activated during the early phase of CHIKV infection in macaques and humans [45, 46].
Early HIV and SIV infection are characterized by the production of a high viral load, associated with concomitant induction of inflammatory and immuno-regulatory processes, including the production of cytokines. Given the complexity of the mechanisms induced in the early phase of the immune response against HIV and SIV, we set up an in vitro assay to characterize the factors that could be involved in the enhancement of LILRB2 expression. Our data show that the increase in LILRB2 expression requires productive infection of MoDCs, suggesting that sustained intracellular replication of HIV-1 is involved in this mechanism (Fig. 4). The potential direct role of HIV-1 in the regulation of LILRB2 expression is strengthened by the fact that stimulation of TLR7/8 induces the up-regulation of LILRB2, suggesting a relationship between these signaling pathways. CHIKV contains a single stranded RNA and might be expected to enhance LILRB2 expression in cDCs. However, Schilte et al. have reported that CHIKV is unable to trigger TLR7/8 pathways in cDCs and other hematopoietic cells [47]. Moreover, CHIKV is not able to infect cDCs or MoDCs, in contrast to HIV [20]. Since TLR3 could also sense HIV RNA, future investigations will be helpful to determine the impact of this pathway on LILRB2 expression.
Our in vitro data also indicate that the immunomodulatory cytokines IL-10 and type-I IFNs, which are produced during the early immune response against HIV, enhance the surface expression of LILRB2 (Fig. 5). In contrast, TNF-a had no effect on the regulation of LILRB2. Further studies will be required to determine whether type-I IFNs act on LILRB2 expression through a canonical pathway to initiate the resolution of inflammatory processes, which is necessary for control of the virus, and whether the simultaneous presence of HIV and cytokines act synergistically on the expression of LILRB2 on cDCs.
Previous in vitro studies have reported that the strength of the interaction between LILRB2 and MHC-I drives the dysregulation of cDCs of HIV-infected patients. These studies indicate that the strength of the LILRB2/MHC-I interaction is regulated by HIV-derived peptides or genetic variation of MHC-I haplotypes [7, 15, 16]. However, we have previously demonstrated that a slight increase of LILRB2 expression on neutrophils, resulting from granule exocytosis, directly affects neutrophil functions [23]. Indeed, neutrophils with higher levels of LILRB2 on their surface are more prone to inhibition by MHC-I ligands, resulting in the attenuation of phagocytosis and the production of reactive oxygen species [23, 39]. The upregulation of LILRB2 expression on cDCs during HIV/SIV infection that we report here is much higher than that on neutrophils, as it results from sustained protein production. Moreover, it is accompanied by increased MHC-I ligand expression. We propose that the simultaneous increase of LILRB2 expression and that of its MHC-I ligands, induced during the early phase of the immune response against HIV, considerably alter cDC functions. This hypothesis is strengthened by previous studies demonstrating that high expression of LILRB2 on cDCs drives potent immune-regulatory mechanisms, leading to the attenuation of CD4+ and CD8+ T cell responses [6, 22].
In summary, our data show specific enhancement of the LILRB2/MHC-I inhibitory axis in cDCs during the early immune response that may strongly influence mechanisms that tip the balance toward SIV or HIV persistence (Figure S9). However, further investigations will be necessary to determine the impact of LILRB2/MHC-I interaction on the efficiency of immune responses against SIV and HIV.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1. Generation and reactivity of anti-LILRB2 antibody (clone 17E5.3E9). (A) Amino acid sequence of human LILRB2 extracellular domain used to generate antibody 17E5.3E9. (B) Analysis of anti-LILRB2 antibody (clone 17E5.3E9) staining on cDCs, monocytes, neutrophils, eosinophils, CD4+ and CD8+ T cells, B cells and NK cells of healthy human and cynomolgus macaque blood samples. (C) Upregulation of LILRB2 is detected by 17E5.3E9 antibody on healthy human and cynomolgus macaque neutrophils stimulated with TNF-a. Flow cytometry analysis of LILRB2 at the surface of unstimulated (full line) or stimulated (dotted line) neutrophils for 30 min with TNF-a (100 ng/mL). Gray filled curve shows cells stained with isotype-matched control mAb. Data are representative of three independent experiments. (EPS 3132 kb)
Figure S2. Flow cytometry gating strategy used to identify cell subsets of PBMCs from HIV-1+ patients. cDCs (CD1c+) were identified as Lin−/HLA-DR+/CD16−/CD14−/CD123−/CD11c+/CD1c+. Results shown are representative between individuals. (EPS 1685 kb)
Figure S3. Characterization of HLA-DR expression level on cDCs during the early phase of HIV-1 infection. Analysis of HLA-DR surface expression on cDCs from blood samples of early HIV-1-infected patients before (primary HIV+) or after 1 year of cART (HIV+ cART+), HIV-1-infected elite controller patients (HIC), and HIV-1-non-infected controls (HIV-). Data are represented as mean fluorescence intensity with statistical analysis performed using the Mann–Whitney U test between HIC (n = 6), and primary HIV+ patients (n = 7) (p < 0,05 is considered significant). (EPS 1133 kb)
Figure S4. Characterization of LILRB2 expression on immune cell subsets from secondary lymphoid organs of cynomolgus macaques. (A) Flow cytometry gating strategy used to identify cDCs and T and B cells of peripheral lymph nodes and spleen of cynomolgus macaques. cDCs were identified as CD45+/Lin−(CD3/CD8/CD20)/HLA-DR+/CD163−/CD123−/CD1c+. (B) Flow cytometry analysis of LILRB2 expression on cDCs and on T and B cells from both peripheral and mesenteric lymph nodes, spleen, and peripheral blood (n = 4). (EPS 2019 kb)
Figure S5. Follow-up of LILRB2 and MHC-I expression on monocytes and macrophages during infection of cynomolgus macaques by SIVmac251. (A) Monocyte cell counts during SIVmac251 infection, at 1000 AID50/mL, of 3 three female cynomolgus macaques. LILRB2 and MHC-I surface expression profiles of (B) blood monocytes and (C) lymph node macrophages. (D) Modulation of LILRB2 and MHC-I surface expression on blood monocytes from seven cynomolgus macaques followed at various time points during early (days 4 to 7, days 9 to 11, days 14 to 21, days 22 to 42) and advanced (days 57 to 100) phases of SIVmac251 infection. Data are shown as the fold change of surface expression relative to baseline (represented with dashed line). Statistical analyses were carried-out using the Wilcoxon matched-pairs signed rank test (p < 0.05 is considered significant). (EPS 1393 kb)
Figure S6. Analysis of LILRB2 and MHC-I expression during SIV infection of three male cynomolgus macaques. (A) Plasma viral load of SIVmac251 infection at 1000 AID50/mL. (B) Modulation of LILRB2 (left panel) and MHC-I (right panel) expression on blood cDCs and (C) monocytes (CD14+/CD16). (D) Follow-up of LILRB2 surface expression on cDCs and macrophages isolated from peripheral lymph nodes of cynomolgus macaques during early (D + 14) and late (D + 85) phases of SIVmac251 infection. The surface expression of LILRB2 and MHC-I was determined by the mean fluorescence Intensity of each marker by flow cytometry. (EPS 1284 kb)
Figure S7. Follow-up of SIRPa inhibitory receptor expression on cDCs during SIV infection of cynomolgus macaques. (A) Modulation of SIRPa expression on blood cDCs of three female cynomolgus macaques infected with 1000 AID50/mL of SIVmac251. (B) SIRPa expression on cDCs from secondary lymphoid tissue was evaluated from sampled peripheral lymph nodes at various time points after SIV infection. Data are represented as the mean Fluorescence Intensity. (EPS 1119 kb)
Figure S8. Evolution of blood leucocyte counts during CHIKV infection of cynomolgus macaques. The evolution of cell counts of four cynomolgus macaques infected with the CHIKV-LR2006-OPY1 strain at a dose of 100 AID50/mL. (A) Follow-up of cDC counts analyzed by flow cytometry using Trucount tubes (BD biosciences). (B) Analysis of absolute blood cell numbers to follow the evolution of lymphocyte, monocyte, and granulocyte numbers using automated cell counts (HmX AL System, Beckman Coulter). (EPS 1111 kb)
Figure S9. Schematic of LILRB2/MHC-I inhibitory axis regulation in cDCs of lymph nodes during HIV/SIV infection. (1) IL-10 and IFN-I stimulation along with HIV-1 sensing by TLR7/8 induce (2) the upregulation of LILRB2 expression allowing higher (3) Cis or Trans interactions with MHC-I ligands leading to (4) cDC dysfunctions. (EPS 4300 kb)
Table S1. Monoclonal antibodies used for flow cytometry analysis of human and cynomolgus macaque samples. (EPS 1180 kb)
Acknowledgements
We thank the IDMIT infrastructure staff and Florian Meurisse for technical assistance. We also thank the ANRS cohorts CO6 PRIMO and CO21 CODEX for helpful collaboration and Anne Sophie Beignon for critical reading of the manuscript and helpful discussions. These works were supported by grants from the “Agence Nationale de Recherches sur le SIDA et les hépatites virales” under statement numbers 14-067, 16–035, University of Paris-Sud (Attractivité), European Union FP7 project (grant agreement no. 261202 (ICRES)), French government “Programme d’Investissements d’Avenir” (PIA) under Grant ANR-11-INBS-0008 funding the Infectious Disease Models and Innovative Therapies (IDMIT, Fontenay-aux-Roses, France) infrastructure and PIA grant ANR-10-EQPX-02-01 funding the FlowCyTech facility. G. Palomino was supported by a post-doctoral fellowship from Brazil government (Programa Ciência sem Fronteiras).
References
- 1.Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 3.Miller E, Bhardwaj N. Dendritic cell dysregulation during HIV-1 infection. Immunol Rev. 2013;254:170–189. doi: 10.1111/imr.12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wijewardana V, Soloff AC, Liu X, Brown KN, Barratt-Boyes SM. Early myeloid dendritic cell dysregulation is predictive of disease progression in simian immunodeficiency virus infection. PLoS Pathog. 2010;6:e1001235. doi: 10.1371/journal.ppat.1001235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wonderlich ER, Wu WC, Normolle DP, Barratt-Boyes SM. Macrophages and myeloid dendritic cells lose T cell-stimulating function in simian immunodeficiency virus infection associated with diminished IL-12 and IFN-alpha production. J Immunol. 2015;195:3284–3292. doi: 10.4049/jimmunol.1500683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang CC, Ciubotariu R, Manavalan JS, Yuan J, Colovai AI, Piazza F, Lederman S, Colonna M, Cortesini R, Dalla-Favera R, Suciu-Foca N. Tolerization of dendritic cells by T(S) cells: the crucial role of inhibitory receptors ILT3 and ILT4. Nat Immunol. 2002;3:237–243. doi: 10.1038/ni760. [DOI] [PubMed] [Google Scholar]
- 7.Lichterfeld M, Kavanagh DG, Williams KL, Moza B, Mui SK, Miura T, Sivamurthy R, Allgaier R, Pereyra F, Trocha A, Feeney M, Gandhi RT, Rosenberg ES, Altfeld M, Allen TM, Allen R, Walker BD, Sundberg EJ, Yu XG. A viral CTL escape mutation leading to immunoglobulin-like transcript 4-mediated functional inhibition of myelomonocytic cells. J Exp Med. 2007;204:2813–2824. doi: 10.1084/jem.20061865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ristich V, Liang S, Zhang W, Wu J, Horuzsko A. Tolerization of dendritic cells by HLA-G. Eur J Immunol. 2005;35:1133–1142. doi: 10.1002/eji.200425741. [DOI] [PubMed] [Google Scholar]
- 9.Kang X, Kim J, Deng M, John S, Chen H, Wu G, Phan H, Zhang CC. Inhibitory leukocyte immunoglobulin-like receptors: immune checkpoint proteins and tumor sustaining factors. Cell Cycle. 2016;15:25–40. doi: 10.1080/15384101.2015.1121324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colonna M, Samaridis J, Cella M, Angman L, Allen RL, O’Callaghan CA, Dunbar R, Ogg GS, Cerundolo V, Rolink A. Human myelomonocytic cells express an inhibitory receptor for classical and nonclassical MHC class I molecules. J Immunol. 1998;160:3096–3100. [PubMed] [Google Scholar]
- 11.Fanger NA, Cosman D, Peterson L, Braddy SC, Maliszewski CR, Borges L. The MHC class I binding proteins LIR-1 and LIR-2 inhibit Fc receptor-mediated signaling in monocytes. Eur J Immunol. 1998;28:3423–3434. doi: 10.1002/(SICI)1521-4141(199811)28:11<3423::AID-IMMU3423>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 12.Masuda A, Nakamura A, Maeda T, Sakamoto Y, Takai T. Cis binding between inhibitory receptors and MHC class I can regulate mast cell activation. J Exp Med. 2007;204:907–920. doi: 10.1084/jem.20060631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mori Y, Tsuji S, Inui M, Sakamoto Y, Endo S, Ito Y, Fujimura S, Koga T, Nakamura A, Takayanagi H, Itoi E, Takai T. Inhibitory immunoglobulin-like receptors LILRB and PIR-B negatively regulate osteoclast development. J Immunol. 2008;181:4742–4751. doi: 10.4049/jimmunol.181.7.4742. [DOI] [PubMed] [Google Scholar]
- 14.Lichterfeld M, Yu XG. The emerging role of leukocyte immunoglobulin-like receptors (LILRs) in HIV-1 infection. J Leukoc Biol. 2012;91:27–33. doi: 10.1189/jlb.0811442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bashirova AA, Martin-Gayo E, Jones DC, Qi Y, Apps R, Gao X, Burke PS, Taylor CJ, Rogich J, Wolinsky S, Bream JH, Duggal P, Hussain S, Martinson J, Weintrob A, Kirk GD, Fellay J, Buchbinder SP, Goedert JJ, Deeks SG, Pereyra F, Trowsdale J, Lichterfeld M, Telenti A, Walker BD, Allen RL, Carrington M, Yu XG. LILRB2 interaction with HLA class I correlates with control of HIV-1 infection. PLoS Genet. 2014;10:e1004196. doi: 10.1371/journal.pgen.1004196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang J, Goedert JJ, Sundberg EJ, Cung TD, Burke PS, Martin MP, Preiss L, Lifson J, Lichterfeld M, Carrington M, Yu XG. HLA-B*35-Px-mediated acceleration of HIV-1 infection by increased inhibitory immunoregulatory impulses. J Exp Med. 2009;206:2959–2966. doi: 10.1084/jem.20091386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones DC, Kosmoliaptsis V, Apps R, Lapaque N, Smith I, Kono A, Chang C, Boyle LH, Taylor CJ, Trowsdale J, Allen RL. HLA class I allelic sequence and conformation regulate leukocyte Ig-like receptor binding. J Immunol. 2011;186:2990–2997. doi: 10.4049/jimmunol.1003078. [DOI] [PubMed] [Google Scholar]
- 18.Sodora DL, Allan JS, Apetrei C, Brenchley JM, Douek DC, Else JG, Estes JD, Hahn BH, Hirsch VM, Kaur A, Kirchhoff F, Muller-Trutwin M, Pandrea I, Schmitz JE, Silvestri G. Toward an AIDS vaccine: lessons from natural simian immunodeficiency virus infections of African nonhuman primate hosts. Nat Med. 2009;15:861–865. doi: 10.1038/nm.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slukvin RL, II, Grendell DS, Rao AL, Hughes, Golos TG. “Cloning of rhesus monkey LILRs”. Tissue Antigens. 2006;67:331–337. doi: 10.1111/j.1399-0039.2006.00579.x. [DOI] [PubMed] [Google Scholar]
- 20.Labadie K, Larcher T, Joubert C, Mannioui A, Delache B, Brochard P, Guigand L, Dubreil L, Lebon P, Verrier B, de Lamballerie X, Suhrbier A, Cherel Y, Le Grand R, Roques P. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J Clin Invest. 2010;120:894–906. doi: 10.1172/JCI40104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rougeron V, Sam IC, Caron M, Nkoghe D, Leroy E, Roques P. Chikungunya, a paradigm of neglected tropical disease that emerged to be a new health global risk. J Clin Virol. 2015;64:144–152. doi: 10.1016/j.jcv.2014.08.032. [DOI] [PubMed] [Google Scholar]
- 22.Banchereau J, Zurawski S, Thompson-Snipes L, Blanck JP, Clayton S, Munk A, Cao Y, Wang Z, Khandelwal S, Hu J, McCoy WHt, Palucka KA, Reiter Y, Fremont DH, Zurawski G, Colonna M, Shaw AS, Klechevsky E. “Immunoglobulin-like transcript receptors on human dermal CD14+ dendritic cells act as a CD8-antagonist to control cytotoxic T cell priming”. Proc Natl Acad Sci USA. 2012;109:18885–18890. doi: 10.1073/pnas.1205785109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baudhuin J, Migraine J, Faivre V, Loumagne L, Lukaszewicz AC, Payen D, Favier B. Exocytosis acts as a modulator of the ILT4-mediated inhibition of neutrophil functions. Proc Natl Acad Sci USA. 2013;110:17957–17962. doi: 10.1073/pnas.1221535110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maecker HT, Frey T, Nomura LE, Trotter J. Selecting fluorochrome conjugates for maximum sensitivity. Cytometry A. 2004;62:169–173. doi: 10.1002/cyto.a.20092. [DOI] [PubMed] [Google Scholar]
- 25.Huang J, Burke PS, Cung TD, Pereyra F, Toth I, Walker BD, Borges L, Lichterfeld M, Yu XG. Leukocyte immunoglobulin-like receptors maintain unique antigen-presenting properties of circulating myeloid dendritic cells in HIV-1-infected elite controllers. J Virol. 2010;84:9463–9471. doi: 10.1128/JVI.01009-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dillon SM, Robertson KB, Pan SC, Mawhinney S, Meditz AL, Folkvord JM, Connick E, McCarter MD, Wilson CC. Plasmacytoid and myeloid dendritic cells with a partial activation phenotype accumulate in lymphoid tissue during asymptomatic chronic HIV-1 infection. J Acquir Immune Defic Syndr. 2008;48:1–12. doi: 10.1097/QAI.0b013e3181664b60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugimoto C, Hasegawa A, Saito Y, Fukuyo Y, Chiu KB, Cai Y, Breed MW, Mori K, Roy CJ, Lackner AA, Kim WK, Didier ES, Kuroda MJ. Differentiation kinetics of blood monocytes and dendritic cells in macaques: insights to understanding human myeloid cell development. J Immunol. 2015;195:1774–1781. doi: 10.4049/jimmunol.1500522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dutertre CA, Jourdain JP, Rancez M, Amraoui S, Fossum E, Bogen B, Sanchez C, Couedel-Courteille A, Richard Y, Dalod M, Feuillet V, Cheynier R, Hosmalin A. TLR3-responsive, XCR1+, CD141(BDCA-3)+/CD8alpha+-equivalent dendritic cells uncovered in healthy and simian immunodeficiency virus-infected rhesus macaques. J Immunol. 2014;192:4697–4708. doi: 10.4049/jimmunol.1302448. [DOI] [PubMed] [Google Scholar]
- 29.Malleret B, Karlsson I, Maneglier B, Brochard P, Delache B, Andrieu T, Muller-Trutwin M, Beaumont T, McCune JM, Banchereau J, Le Grand R, Vaslin B. Effect of SIVmac infection on plasmacytoid and CD1c+ myeloid dendritic cells in cynomolgus macaques. Immunology. 2008;124:223–233. doi: 10.1111/j.1365-2567.2007.02758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wijewardana V, Kristoff J, Xu C, Ma D, Haret-Richter G, Stock JL, Policicchio BB, Mobley AD, Nusbaum R, Aamer H, Trichel A, Ribeiro RM, Apetrei C, Pandrea I. Kinetics of myeloid dendritic cell trafficking and activation: impact on progressive, nonprogressive and controlled SIV infections. PLoS Pathog. 2013;9:e1003600. doi: 10.1371/journal.ppat.1003600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karlsson I, Malleret B, Brochard P, Delache B, Calvo J, Le Grand R, Vaslin B. Dynamics of T-cell responses and memory T cells during primary simian immunodeficiency virus infection in cynomolgus macaques. J Virol. 2007;81:13456–13468. doi: 10.1128/JVI.01619-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, Lebedeva M, DeCamp A, Li D, Grove D, Self SG, Borrow P. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. 2009;83:3719–3733. doi: 10.1128/JVI.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Addo MM, Altfeld M. Sex-based differences in HIV type 1 pathogenesis. J Infect Dis. 2014;209(Suppl 3):S86–S92. doi: 10.1093/infdis/jiu175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saito Y, Iwamura H, Kaneko T, Ohnishi H, Murata Y, Okazawa H, Kanazawa Y, Sato-Hashimoto M, Kobayashi H, Oldenborg PA, Naito M, Kaneko Y, Nojima Y, Matozaki T. Regulation by SIRPalpha of dendritic cell homeostasis in lymphoid tissues. Blood. 2010;116:3517–3525. doi: 10.1182/blood-2010-03-277244. [DOI] [PubMed] [Google Scholar]
- 35.Le-Barillec K, Si-Tahar M, Balloy V, Chignard M. Proteolysis of monocyte CD14 by human leukocyte elastase inhibits lipopolysaccharide-mediated cell activation. J Clin Invest. 1999;103:1039–1046. doi: 10.1172/JCI5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang J, Al-Mozaini M, Rogich J, Carrington MF, Seiss K, Pereyra F, Lichterfeld M, Yu XG. Systemic inhibition of myeloid dendritic cells by circulating HLA class I molecules in HIV-1 infection. Retrovirology. 2012;9:11. doi: 10.1186/1742-4690-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benlahrech A, Yasmin A, Westrop SJ, Coleman A, Herasimtschuk A, Page E, Kelleher P, Gotch F, Imami N, Patterson S. Dysregulated immunophenotypic attributes of plasmacytoid but not myeloid dendritic cells in HIV-1 infected individuals in the absence of highly active anti-retroviral therapy. Clin Exp Immunol. 2012;170:212–221. doi: 10.1111/j.1365-2249.2012.04647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang J, Yang Y, Al-Mozaini M, Burke PS, Beamon J, Carrington MF, Seiss K, Rychert J, Rosenberg ES, Lichterfeld M, Yu XG. Dendritic cell dysfunction during primary HIV-1 infection. J Infect Dis. 2011;204:1557–1562. doi: 10.1093/infdis/jir616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Favier B. Regulation of neutrophil functions through inhibitory receptors: an emerging paradigm in health and disease. Immunol Rev. 2016;273:140–155. doi: 10.1111/imr.12457. [DOI] [PubMed] [Google Scholar]
- 40.Takai T. A novel recognition system for MHC class I molecules constituted by PIR. Adv Immunol. 2005;88:161–192. doi: 10.1016/S0065-2776(05)88005-8. [DOI] [PubMed] [Google Scholar]
- 41.Wijewardana V, Bouwer AL, Brown KN, Liu X, Barratt-Boyes SM. Accumulation of functionally immature myeloid dendritic cells in lymph nodes of rhesus macaques with acute pathogenic simian immunodeficiency virus infection. Immunology. 2014;143:146–154. doi: 10.1111/imm.12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. 2014;32:25–50. doi: 10.1146/annurev-immunol-032713-120142. [DOI] [PubMed] [Google Scholar]
- 43.Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S, Van Gassen S, Chen J, Poidinger M, De Prijck S, Tavernier SJ, Low I, Irac SE, Mattar CN, Sumatoh HR, Low GH, Chung TJ, Chan DK, Tan KK, Hon TL, Fossum E, Bogen B, Choolani M, Chan JK, Larbi A, Luche H, Henri S, Saeys Y, Newell EW, Lambrecht BN, Malissen B, Ginhoux F. Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity. 2016;45:669–684. doi: 10.1016/j.immuni.2016.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laforge M, Campillo-Gimenez L, Monceaux V, Cumont MC, Hurtrel B, Corbeil J, Zaunders J, Elbim C, Estaquier J. HIV/SIV infection primes monocytes and dendritic cells for apoptosis. PLoS Pathog. 2011;7:e1002087. doi: 10.1371/journal.ppat.1002087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roques P, Ljungberg K, Kummerer BM, Gosse L, Dereuddre-Bosquet N, Tchitchek N, Hallengard D, Garcia-Arriaza J, Meinke A, Esteban M, Merits A, Le Grand R, Liljestrom P. Attenuated and vectored vaccines protect nonhuman primates against Chikungunya virus. JCI Insight. 2017;2:e83527. doi: 10.1172/jci.insight.83527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wauquier N, Becquart P, Nkoghe D, Padilla C, Ndjoyi-Mbiguino A, Leroy EM. The acute phase of Chikungunya virus infection in humans is associated with strong innate immunity and T CD8 cell activation. J Infect Dis. 2011;204:115–123. doi: 10.1093/infdis/jiq006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schilte C, Couderc T, Chretien F, Sourisseau M, Gangneux N, Guivel-Benhassine F, Kraxner A, Tschopp J, Higgs S, Michault A, Arenzana-Seisdedos F, Colonna M, Peduto L, Schwartz O, Lecuit M, Albert ML. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J Exp Med. 2010;207:429–442. doi: 10.1084/jem.20090851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Generation and reactivity of anti-LILRB2 antibody (clone 17E5.3E9). (A) Amino acid sequence of human LILRB2 extracellular domain used to generate antibody 17E5.3E9. (B) Analysis of anti-LILRB2 antibody (clone 17E5.3E9) staining on cDCs, monocytes, neutrophils, eosinophils, CD4+ and CD8+ T cells, B cells and NK cells of healthy human and cynomolgus macaque blood samples. (C) Upregulation of LILRB2 is detected by 17E5.3E9 antibody on healthy human and cynomolgus macaque neutrophils stimulated with TNF-a. Flow cytometry analysis of LILRB2 at the surface of unstimulated (full line) or stimulated (dotted line) neutrophils for 30 min with TNF-a (100 ng/mL). Gray filled curve shows cells stained with isotype-matched control mAb. Data are representative of three independent experiments. (EPS 3132 kb)
Figure S2. Flow cytometry gating strategy used to identify cell subsets of PBMCs from HIV-1+ patients. cDCs (CD1c+) were identified as Lin−/HLA-DR+/CD16−/CD14−/CD123−/CD11c+/CD1c+. Results shown are representative between individuals. (EPS 1685 kb)
Figure S3. Characterization of HLA-DR expression level on cDCs during the early phase of HIV-1 infection. Analysis of HLA-DR surface expression on cDCs from blood samples of early HIV-1-infected patients before (primary HIV+) or after 1 year of cART (HIV+ cART+), HIV-1-infected elite controller patients (HIC), and HIV-1-non-infected controls (HIV-). Data are represented as mean fluorescence intensity with statistical analysis performed using the Mann–Whitney U test between HIC (n = 6), and primary HIV+ patients (n = 7) (p < 0,05 is considered significant). (EPS 1133 kb)
Figure S4. Characterization of LILRB2 expression on immune cell subsets from secondary lymphoid organs of cynomolgus macaques. (A) Flow cytometry gating strategy used to identify cDCs and T and B cells of peripheral lymph nodes and spleen of cynomolgus macaques. cDCs were identified as CD45+/Lin−(CD3/CD8/CD20)/HLA-DR+/CD163−/CD123−/CD1c+. (B) Flow cytometry analysis of LILRB2 expression on cDCs and on T and B cells from both peripheral and mesenteric lymph nodes, spleen, and peripheral blood (n = 4). (EPS 2019 kb)
Figure S5. Follow-up of LILRB2 and MHC-I expression on monocytes and macrophages during infection of cynomolgus macaques by SIVmac251. (A) Monocyte cell counts during SIVmac251 infection, at 1000 AID50/mL, of 3 three female cynomolgus macaques. LILRB2 and MHC-I surface expression profiles of (B) blood monocytes and (C) lymph node macrophages. (D) Modulation of LILRB2 and MHC-I surface expression on blood monocytes from seven cynomolgus macaques followed at various time points during early (days 4 to 7, days 9 to 11, days 14 to 21, days 22 to 42) and advanced (days 57 to 100) phases of SIVmac251 infection. Data are shown as the fold change of surface expression relative to baseline (represented with dashed line). Statistical analyses were carried-out using the Wilcoxon matched-pairs signed rank test (p < 0.05 is considered significant). (EPS 1393 kb)
Figure S6. Analysis of LILRB2 and MHC-I expression during SIV infection of three male cynomolgus macaques. (A) Plasma viral load of SIVmac251 infection at 1000 AID50/mL. (B) Modulation of LILRB2 (left panel) and MHC-I (right panel) expression on blood cDCs and (C) monocytes (CD14+/CD16). (D) Follow-up of LILRB2 surface expression on cDCs and macrophages isolated from peripheral lymph nodes of cynomolgus macaques during early (D + 14) and late (D + 85) phases of SIVmac251 infection. The surface expression of LILRB2 and MHC-I was determined by the mean fluorescence Intensity of each marker by flow cytometry. (EPS 1284 kb)
Figure S7. Follow-up of SIRPa inhibitory receptor expression on cDCs during SIV infection of cynomolgus macaques. (A) Modulation of SIRPa expression on blood cDCs of three female cynomolgus macaques infected with 1000 AID50/mL of SIVmac251. (B) SIRPa expression on cDCs from secondary lymphoid tissue was evaluated from sampled peripheral lymph nodes at various time points after SIV infection. Data are represented as the mean Fluorescence Intensity. (EPS 1119 kb)
Figure S8. Evolution of blood leucocyte counts during CHIKV infection of cynomolgus macaques. The evolution of cell counts of four cynomolgus macaques infected with the CHIKV-LR2006-OPY1 strain at a dose of 100 AID50/mL. (A) Follow-up of cDC counts analyzed by flow cytometry using Trucount tubes (BD biosciences). (B) Analysis of absolute blood cell numbers to follow the evolution of lymphocyte, monocyte, and granulocyte numbers using automated cell counts (HmX AL System, Beckman Coulter). (EPS 1111 kb)
Figure S9. Schematic of LILRB2/MHC-I inhibitory axis regulation in cDCs of lymph nodes during HIV/SIV infection. (1) IL-10 and IFN-I stimulation along with HIV-1 sensing by TLR7/8 induce (2) the upregulation of LILRB2 expression allowing higher (3) Cis or Trans interactions with MHC-I ligands leading to (4) cDC dysfunctions. (EPS 4300 kb)
Table S1. Monoclonal antibodies used for flow cytometry analysis of human and cynomolgus macaque samples. (EPS 1180 kb)