

Abstract
Alzheimer’s disease (AD) is a multifactorial age-related brain disease. Numerous pathological events run forth in the brain leading to AD. There is an initial long, dormant phase before the clinical symptoms become evident. There is a need to diagnose the disease at the preclinical stage since therapeutic interventions are most likely to be effective if initiated early. Undoubtedly, the core cerebrospinal fluid (CSF) biomarkers have a good diagnostic accuracy and have been used in clinical trials as end point measures. However, looking into the multifactorial nature of AD and the overlapping pathology with other forms of dementia, it is important to integrate the core CSF biomarkers with a broader panel of other biomarkers reflecting different aspects of pathology. The review is focused upon a panel of biomarkers that relate to different aspects of AD pathology, as well as various studies that have evaluated their diagnostic potential. The panel includes markers of neurodegeneration: neurofilament light chain and visinin-like protein (VILIP-1); markers of amyloidogenesis and brain amyloidosis: apolipoproteins; markers of inflammation: YKL-40 and monocyte chemoattractant protein 1; marker of synaptic dysfunction: neurogranin. These markers can highlight on the state and stage-associated changes that occur in AD brain with disease progression. A combination of these biomarkers would not only aid in preclinical diagnosis, but would also help in identifying early brain changes during the onset of disease. Successful treatment strategies can be devised by understanding the contribution of these markers in different aspects of disease pathogenesis.
Keywords: Diagnosis, Neurofilament light, Neurodegeneration, Synaptic dysfunction, Neurogranin, Fatty acid-binding proteins, Neuroinflammation
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease whose pathology starts decades before the clinical symptoms appear [1]. The preclinical stage represents a dormant phase where neuropathological changes are accumulating but the person has normal cognition [2]. Numerous biochemical pathways have been described to explain the pathogenesis of AD. Starting with the identification of amyloid beta (Aβ) in 1985, as the main component of amyloid plaques [3], our understanding of Aβ and amyloid precursor protein (APP) metabolism, and tau pathology (neurofibrillary tangles and neuropil threads) has improved with time. Thorough research has been carried out to understand other aspects of AD pathogenesis and thereafter, numerous hypotheses have been put forth. AD may, therefore, be considered a result of a number of pathological changes in the brain, such as amyloidosis, neurodegeneration, inflammation, synaptic dysfunction, disruption of neuronal signaling and neuronal membranes, oxidative stress and mitochondrial dysfunction [4]. These changes direct the trajectory of preclinical AD to AD dementia and make AD a multifaceted disease.
Several AD drug trials have failed, probably because the treatments are initiated at an advanced stage where damage is too severe, and the drug is not able to demonstrate a clinical benefit because the brain is too compromised to benefit from a treatment [5–7]. It is imperative to initiate an early treatment and ensure that the correct patient population is included in the clinical trials. Therefore, there is an urgent need to diagnose AD and initiate treatment at the preclinical stage, so as to obtain a clinical benefit. The first step in devising successful treatment strategies is to identify biomarkers for accurate diagnosis of AD, and thereafter develop therapeutic strategies. It is essential to find an ideal biomarker that should also help in monitoring the mechanism of action and the biochemical effects of the treatment drug [8]. As per the regulatory bodies such as Food and Drug Administration (FDA) and European Medicine Agency (EMA), exploration and validation of a biomarker should be integrated with drug development to accelerate the journey towards development of an effective therapeutic intervention [9]. Clinical trials that aim at evaluating the effectiveness of therapeutic strategies can come up with reliable results when the therapeutic effect of these agents is monitored using markers that reflect over the molecular changes of the disease. As far as AD is concerned the promising markers in this context are the cerebrospinal fluid (CSF) markers [8]. The CSF biomarkers are the potential candidates to facilitate early diagnosis of AD because the AD pathological hallmarks start decades before the appearance of cognitive symptoms [10]. The core CSF biomarkers [CSF Aβ-42, total tau (T-tau) and phosphorylated tau (P-tau)] have been extensively studied and validated in relation to AD pathology, conversion and progression. There is a further need to explore and evaluate additional CSF biomarkers, which can aid in early and accurate diagnosis of AD, as well as in monitoring the downstream effects of a therapeutic intervention. As seen from the high failure rate of AD drug trials, it is extremely essential to explore additional CSF biomarkers which reflect on individual pathologies, meet the regulatory qualification and can help to enrich the clinical trial populations.
The CSF biomarkers as a part of AD diagnostic criteria
The biomarkers of AD have been divided into three main categories: the biomarkers of amyloid deposition (A), tau pathology (T) and neurodegeneration (N) (A/T/N) [11]. The biomarkers of amyloid accumulation include abnormal tracer retention on amyloid positron emission tomography (PET) imaging and CSF Aβ-42. The biomarkers of tau pathology include CSF P-tau or tau PET. The biomarkers of neurodegeneration include CSF T-tau and 18F-2-fluoro-2-deoxy-d-glucose positron emission tomography (FDG-PET) and brain atrophy via magnetic resonance imaging (MRI). The brain imaging techniques have been used as end points in clinical trials [12]. However, the limited accessibility, lack of molecular specificity, exposure to radioactivity and cost factor involved in neuroimaging markers particularly Aβ imaging, restricts their use in routine analysis [13]. Therefore, the CSF is being extensively studied worldwide, in AD biomarker research. The CSF is in direct contact with the brain and the biochemical changes occurring in the brain are reflected in it [14]. The CSF biomarkers have a causal relation to AD pathology and may provide an insight into the different aspects of AD pathogenesis. The core CSF biomarkers (decreased CSF Aβ-42 and elevated T-tau and P-tau) have shown a high specificity and sensitivity for AD diagnosis [15]. CSF Aβ-42 correlates well with Aβ pathology [16], whilst the correlation of the tau markers with pathology is less clear; recent data indicate that CSF T-tau and P-tau may be markers of a neuronal reaction to Aβ pathology, which with time will translate into full-blown pathology (neurodegeneration and tangle pathology) [17]. In any case, these markers are quite specific for identifying an individual with preclinical AD [18].
In the recent years, with the advances in our understanding of AD pathophysiology, it has become evident that the relation between clinical symptoms and disease pathology varies, and the cognitive impairment evolves gradually. As a result, in 2011 the National Institute on Aging (NIA) and the Alzheimer’s Association (AA) revised the diagnostic and research criteria for AD and included the CSF biomarkers in addition to the imaging markers [19]. In 2014, the International Working Group (IWG) reanalysed the pathological and topographical biomarkers of AD. Diagnostic changes were proposed for typical, atypical, mixed and preclinical AD. According to this, the pathological markers such as decreased CSF Aβ-42 and elevated T-tau and P-tau were considered as specific makers of disease pathology [18].
The need of additional CSF biomarkers
The research on core CSF biomarkers (CSF Aβ-42, T-tau and P-tau) began nearly 2 decades ago. Reduced CSF Aβ and elevated T-tau and P-tau were found in the CSF of AD patients. [20, 21]. This created a pathway for further research to look over into the diagnostic potential of these biomarkers, which reflect upon brain amyloidosis and neurodegeneration. Today, these biomarkers are extensively used in diagnosis and clinical trials. They have a high enough diagnostic accuracy and reflect upon the neuropathological hallmarks of AD: the neurofibrillary tangles and amyloid plaques [22]. Additional biomarkers are still needed to complement the core biomarkers for early diagnosis and prognosis of AD and get a better insight into the different pathogenic pathways associated with the AD.
The core CSF biomarkers are relatively stable in clinical AD and, therefore, do not serve as good markers in studying disease progression [23, 24]. The CSF Aβ-42 is sharply reduced in the preclinical phase of AD while the levels are found to be constant in the subsequent phases [25]. The altered levels of these core markers do not predict the rate of cognition decline as they do not correlate with the Mini Mental Status Examination (MMSE) [26]. In another multi-center longitudinal study, it was found that there was lack of association between the changes in CSF biomarkers and the rate of change or decline in cognition over a period of 4 years [27]. In addition, these markers do not perform well enough in differentiating AD from other forms of dementia due to partially coinciding pathologies [28]. The therapeutic strategies that aim at reducing amyloid load have failed to show a clinical benefit in spite of clearing Aβ [29]. Studies have shown that the reduced levels of CSF Aβ negatively correlate with the brain amyloid load [30]. However, this association does not match with the clinical diagnosis of AD. The discordance has been found mainly in the cognitively normal participants, which have reduced CSF Aβ but are amyloid negative as seen by PET. Therefore, CSF Aβ levels are altered in the preclinical stage [31–34]. This has led to the contamination of cohort groups due to the inclusion of CSF Aβ positives in the control group. This necessitates the need for exploration and evaluation of additional or novel biomarkers that aid in accurate diagnosis, correlate with cognitive function, but also help in better understanding the disease progression and different aspects of AD pathology.
AD is a multifaceted disease, and AD dementia is a result of a number of pathological changes in the brain (Fig. 1). Numerous proteins or other biomolecules play significant roles in these pathological pathways. A reduction or elevation of their levels in the CSF is associated with a pathological change, which can directly highlight upon the extent of damage, or can occur as a protective response against the damage. A detailed understanding of disease pathogenesis at molecular level through CSF biomarkers can help in designing new efficacious chemical entities for treatment. In addition, biomarkers can serve as targets for therapeutic agents aimed to combat the associated pathological change. In context of a clinical trial, a biomarker can serve as a surrogate end point. The time consuming end points associated with the ongoing trails in AD can be reduced with the application of additional makers [35, 36]. An early diagnosis aided through CSF biomarkers would ensure cohort uniformity through recruitment of correct patient population. This would help in improving clinical trial design and interpretation [37]. The clinical stages of AD are well defined and understood, but it is important to identify and understand the different pathophysiological stages of AD. CSF biomarkers would help in understanding and identifying these stages. To bring advancement in the field of AD biomarker and therapeutic research, it is of utmost important that new biomarkers in relation to AD pathogenesis be explored in the CSF and their potential to diagnose AD at preclinical stage be evaluated in well-established cohorts.
Fig. 1.
Alzheimer’s disease: a multifaceted disease
This review highlights upon the various CSF biomarkers that reflect upon different aspects of multifaceted AD, and also highlights upon the different studies conducted on these biomarkers in the past. Each biomarker helps to track different pathological events. An assessment of the levels of these markers in CSF might reveal an independent information or might unfold the association between individual pathologies. Altogether, the CSF measure of the biomarkers that relate to individual AD pathologies such as brain amyloidosis, neurodegeneration, synaptic dysfunction and neuroinflammation can help in better understanding the disease pathogenesis, accurate diagnosis and prognosis and thereby help in devising effective treatment strategies (Fig. 2).
Fig. 2.

CSF biomarkers for Alzheimer’s disease (AD) diagnosis and understanding different aspects of pathology
The biomarkers of amyloidogenesis and brain amyloidosis
Apolipoprotein E (ApoE)
Role in AD pathogenesis
ApoE is a glycoprotein, which is highly expressed in the liver and the Central Nervous System (CNS) [38]. In the CNS, it is mainly expressed by astrocytes and to some extent by the microglia [39, 40]. It is a constituent of lipoproteins, and in the CNS it is mainly confined to the HDL (high-density lipoproteins) [41]. In the brain, apoE plays a vital role in regulating cholesterol metabolism and transport [42]. ApoE plays a significant role in AD pathogenesis by affecting amyloid and tau pathology, and the isoforms have a differential role in pathogenesis (Fig. 3). Genome wide association studies (GWAS) have revealed that APOE locus, on chromosome 19, with ε4 variant as the major genetic risk for late onset Alzheimer’s disease (LOAD) [43, 44]. In response to neuronal injury, the expression of apoE is upregulated [45]. The three isoforms of apoE (E2, E3, E4) differentially affect cholesterol transport, metabolism and synaptic plasticity, repair and neurite growth. The E4 isoform is least effective in regulating cholesterol transport, efflux and synaptic plasticity [46, 47].
Fig. 3.

Role of apoE in the pathophysiology of AD (AD Alzheimer’s disease, ApoE apolipoprotein E, Aβ amyloid beta)
ApoE mediates clearance of Aβ in an isoform-dependent manner, through endocytosis of Aβ lipoprotein complexes, by affecting proteolytic degradation of Aβ and its transport across BBB. Lipidated apoE binds to Aβ to form Aβ lipoprotein complexes [48] and facilitates endocytosis of these complexes. ApoE binds with its receptors, low-density lipoprotein receptor (LDLR) and lipoprotein receptor-related protein (LRP1), and mediates the endocytosis of lipoproteins [49]. The three isoforms bind differentially with Aβ (Ε2 > Ε3 > Ε4), and differentially influence the lipidation of Aβ and hence the endocytosis [50]. ApoE also regulates proteolytic degradation of Aβ, and among the isoforms E4 isoform is the least efficient in promoting the degradation [51]. ApoE also influences Aβ clearance by regulating its transport across BBB, in an isoform-dependent manner. The E2 and E3 isoforms mediate faster clearance of Aβ through the BBB as compared to E4 [52]. This could be attributed to the effect of apoE on the integrity of tight junctions in BBB, which is impaired in the apoE4-BBB model and apoE4 knock-in mice [53]. ApoE also affects accumulation of Aβ by promoting formation of Aβ filaments [54]. The presence of apoE is essential for Aβ accumulation, which is isoform dependent. The E4 isoform promotes much higher accumulation than E2 and E3 [55, 56]. No amyloid deposits were found in APOE(−/−) transgenic mice (APPV717F+/−), that overexpresses the amyloid precursor protein, as compared to APOE(−/+) and APOE(+/+) [55]. Significant differences in Aβ deposition have been found in PDAPP mice (which develop age-dependent Aβ accumulation), according to the apoE isoform expressed. The amyloid load in hippocampus was two times higher in E4 mice compared to E3 and 4.6 times higher than E2 mice [56].
Neurodegeneration in AD is also influenced by apoE. ApoE affects neuroinflammation, and tau-mediated neurodegeneration. ApoE4 exacerbates neuronal death and modulates microglial activation [57], and overexpression of apoE4 results in tau hyperphosphorylation [58]. Higher tau levels have been found in P301S/E4 tau transgenic mice compared with P301S/E2 and P301S/E3 mice. The brain atrophy and neuroinflammation were much more in E4 mice as compared to E2 and E3 [57]. Recent data also suggest intriguing interactions between apoE isoforms and the activation state of disease-associated microglia, which may be part of the disease-promoting effect of apoE4 [59].
CSF biomarker studies pertaining to ApoE
ApoE is a major apolipoprotein found in the CSF [60]. Numerous studies have evaluated the levels of apoE in the CSF, so as to establish it as a potential marker (Table 1). To evaluate the CSF levels, researches have used methods such as enzyme-linked immuno sorbent assay (ELISA), mass spectrometry, multiplex assays and flow cytometry. Studies on CSF levels of apoE in AD show inconsistent results, with either decreased [61–64] or elevated [65–67] levels as compared to controls. As per some studies APOE genotype may influence CSF ApoE levels. Higher CSF levels of apoE have been reported in individuals having APOE ε4 alleles [68]. Strong positive correlations have been found between CSF apoE levels and CSF tau in AD patients as compared to controls [65]. The correlation between CSF apoE levels and CSF tau are also APOE genotype dependent [69]. The correlation between the two markers suggests that altered apoE levels in CSF could be attributed to the neurodegeneration in AD or vice versa. ApoE binds to protein tau in an isoform-dependent manner [70]. The association of apoE CSF levels with genotype, and genotype-dependent correlation between ApoE and CSF Tau, suggests that neurodegeneration is isoform influenced.
Table 1.
Studies conducted to evaluate the role of apoE as a potential CSF biomarker
| Study | Study groups | CSF levels in AD/study groups | Association with APOE genotype/core markers | Analysis method |
|---|---|---|---|---|
| Van Harten et al. [71] | Non-demented individuals (n = 430) classified into subjective cognitive decline (SCD, n = 207) or mild cognitive impairment (MCI, n = 213) | CSF levels were higher in MCI compared to SCD at baseline, on stratification of MCI and SCD into ε4 and non-ε4 carriers and significant difference was seen only in ε4 carriers | CSF levels were strongly associated with CSF T-tau and P-tau in APOE ε4 carriers | ELISA |
| Johansson et al. [72] | AD (n = 29), other dementias (n = 4), stable cognitive function (SMCI, n = 13) and healthy controls (n = 18) | CSF levels were significantly elevated in AD compared to other dementia and lower as compared to healthy controls | ApoE levels in CSF were not associated with APOE genotype | Luminex multiplex assay |
| Rezeli et al. [73] | AD (n = 13) and non-AD (n = 12) | No significant difference between study groups | ApoE levels in CSF were not associated with APOE genotype; CSF levels positively correlated with P-tau in ε4 non-carriers | Mass spectrometry |
| Richens et al. [74] | AD (n = 10) and controls (n = 18) | No significant difference between study groups | Luminex multiplex assay | |
| Toledo et al. [75] | Normal controls (n = 92), MCI (n = 149) and AD (n = 69) | CSF apoE levels were associated with cognitive decline and atrophy rate | Results were only significant in the group without the ε4 allele | Luminex multiplex assay |
| Perrin et al. [76] | Cognitively normal [clinical dementia rating (CDR 0, n = 24) and mild AD or probable AD (CDR 1, n = 24)] | Did not differ significantly between the two groups | ELISA | |
| Zhang et al. [61] | AD (n = 48), Parkinson’s disease (PD, n = 40), controls (n = 95) | Reduced in AD as compared to controls, but did not differ from PD | Multiplex assay | |
| Hesse et al. [68] | AD patients (n = 55) and normal controls (n = 21) | Reduced in AD as compared to the controls | Individuals having ε4 allele had higher apoE levels | ELISA |
| Fukuyama et al. [66] | AD patients, n = 25 [early onset Alzheimer’s disease (EOAD) and late onset Alzheimer’s disease (LOAD)] and normal controls (n = 36) | ApoE levels were reduced with age in normal controls and increased in AD (more distinctly in EOAD). The elevated levels were positively correlated with decline in cognition | ELISA | |
| Pirttilä et al. [64] | AD (n = 25); classified as APOE ε4 positive (n = 16) and negatives (n = 9), and as mild (n = 16) and moderate dementia (n = 9) | ApoE levels decreased with time in ε4 positives and in both mild and moderate dementia | ELISA | |
| Lindh et al. [65] | AD (n = 18), MCI (n = 9), other dementia disorders (ODD, n = 9) and age-matched healthy controls | Elevated in AD, MCI and ODD as compared to healthy controls and significantly increased in AD at follow-up | CSF levels significantly correlated with CSF tau in AD | ELISA |
| Merched et al. [67] | AD (n = 38), controls (n = 31) and those suffering from other neurological disorders (n = 47) | Significantly elevated in patients with LOAD as compared to controls and other neurological disorders | ELISA | |
| Landen et al. [63] | EOAD (n = 23), LOAD (n = 31), frontotemporal dementia (FTD, n = 16), vascular dementia (VaD, n = 25) and controls (n = 25) | Significantly reduced in all the groups compared to the controls, in EOAD compared to LOAD and FTD | ApoE levels in CSF were not influenced by APOE genotype | ELISA |
| Blennow et al. [62] | AD (n = 11), FTD (n = 10) and controls (n = 10) | Significantly reduced in AD as compared to the controls and FTD group. Also, significantly reduced in FTD as compared to controls | ELISA |
Thus, quantification of apoE levels can highlight upon state of amyloid and tau pathology in AD brains. Owing to the significant role of apoE in AD pathogenesis, further studies should be conducted in well-established cohorts to establish apoE as a potential CSF diagnostic and theragnostic biomarker. There have been inconsistencies with regard to CSF apoE levels. However, these inconsistencies could be attributed to a number of factors such as sample variability, variability in method or technique of analysis or unequal gender distribution in study groups.
Clusterin
Role in AD pathogenesis
Clusterin also called apolipoprotein J is a stress-induced chaperone glycoprotein which can stabilize stressed protein structures. It does so by binding to the hydrophobic surfaces of the partially unfolded proteins [77, 78]. In the brain, it is highly expressed by astrocytes [79]. It plays varied roles in AD pathology. Genome wide association studies (GWAS) have revealed that single nucleotide polymorphisms (SNP’s) associated with clusterin (CLU) gene are associated with AD [80]. Genetic variations have been located by resequencing of CLU-coding exons. These variations can lead to non-synonymous substitutions, insertions or deletion in β chain of clusterin affecting its further processing and functioning [81].
Clusterin affects amyloid pathology in multiple ways. It interacts with the Aβ peptides to form complexes. The antibodies specific to clusterin strongly stain the amyloid deposits in AD brain [82]. This interaction keeps Aβ solubilized and prevents its fibrillation, and also regulates its transport across the BBB [83–85]. The binding of clusterin with Aβ increases its clearance through BBB. A study on mice models has shown that Aβ clearance is increased by 83%, when bound to clusterin [85]. Another study conducted on Tg6799 mouse has found reduction in amyloid plaques and severity of cerebral amyloid angiopathy, upon intravenous administration of clusterin [86]. Clusterin levels are increased in response to Aβ accumulation. Higher intracellular clusterin levels were found upon exposure of Aβ in APP/PSEN1 mice and hippocampal neurons [87]. The levels are significantly increased in frontal cortex and in the hippocampus in AD [88]. The elevated levels are localized to the regions abundant in Aβ [89]. This could be attributed as a protective response to combat the excessive Aβ deposition within the brain tissue. It likely suppresses Aβ deposition in conjunction with ApoE. This is evident through the results obtained from a study conducted on PDAPP transgenic mice, to look at the influence of apoE and clusterin on Aβ accumulation. Aβ accumulation was higher and early in apoE(−/−) and clusterin(−/−) mice. In addition, the Aβ levels were elevated in CSF and brain interstitial fluid, in such mice [90].
It acts as a neuroprotectant by combating oxidative stress and apoptosis [91]. It prevents the mitochondrial transfer of activated Bcl-2-associated X (Bax) protein, a member of Bcl-2 protein family, which is known to accelerate apoptosis. Clusterin is also involved in double-stranded DNA break repair [92–94]. Clusterin also influences inflammation and immune response. The expression of clusterin by astrocytes is increased on treatment with Interleukin 2 [95]. It inhibits the membrane binding of membrane attack complex and regulates the nuclear factor kappa light chain enhancer of activated B cell (NF-κB) pathway [96, 97]. NF-κB is a transcription factor, whose activation causes reactivation of astrocytes, increases the expression of inflammatory mediators such as cytokines and free radicals [97]. Therefore, NF-κB is an inducer of neuroinflammation. Clusterin inhibits the NF-κB activity by stabilizing inhibitors of NF-κB (IκBs) [98].Therefore, clusterin plays varied roles in AD pathology and serves as neuroprotectant by combating apoptosis, regulating inflammation and immune response and preventing aggregation of Aβ (Fig. 4). It can certainly serve a potential stage and state AD biomarker.
Fig. 4.

Varied roles of clusterin in AD pathology (Bax protein Bcl-2-associated X protein, BBB blood brain barrier, DNA deoxyribonucleic acid, NF-κB nuclear factor kappa light chain enhancer of activated B cells, IκBs inhibitors of NF-κB). Yellow circle in the figure represents clusterin
CSF biomarker studies pertaining to clusterin
Numerous studies have evaluated the diagnostic potential of clusterin in CSF using different methods such as ELISA, mass spectrometry and multiplex assays. Most of the studies have reported that clusterin is significantly increased in CSF of AD patients (Table 2). The increased levels of clusterin could be attributed as a defence against neurodegeneration. CSF clusterin levels correlate well with the core CSF biomarkers (T-tau and P-tau, and Aβ-42), and are also significantly associated with CSF tau/Aβ ratio [99–101]. CSF clusterin levels were found to be associated with the entorhinal cortex atrophy rate among CSF Aβ-42-positive individuals. [102]. These correlations very likely suggest that CSF clusterin levels are elevated in relation to the pathological changes in the brain. Elevation in CSF levels of clusterin and the correlation with core biomarkers suggest that elevated levels of clusterin could be attributed as a protective response to the amyloidosis and increased neurodegeneration in the AD brain. Looking at the role of clusterin in AD pathogenesis, a further exploration of its role as an AD biomarker is needed in the CSF.
Table 2.
Studies conducted to evaluate the role of clusterin as a potential CSF biomarker
| Study | Study groups | CSF levels in AD/study groups | Association with APOE genotype/core markers | Analysis method |
|---|---|---|---|---|
| Deming et al. [99] | AD (n = 300) and controls (n = 373) | Clusterin CSF levels were significantly elevated in AD | CSF levels were not associated with APOE genotype | Multiplex immunoassay |
| Prikrylova Vranova et al. [103] | PD (n = 23), Parkinson’s disease dementia (PDD, n = 18), dementia with Lewy bodies (DLB, n = 15), AD (n = 18), progressive supranuclear palsy (PSP, n = 16), multiple system atrophy (MSA, n = 12) and control group (n = 21) | No significant difference between AD and other diagnostic groups | ELISA | |
| Jongbloed et al. [100] | Subjective memory complainers (SMC, n = 17) and volunteers (n = 14) | The clusterin CSF levels were reduced in SMC. The difference in the CSF levels between study groups was more clear with the CSF clusterin/plasma ratio | Significant positive correlation with CSF T-tau and P-tau | ELISA |
| Sihlbom et al. [104] | AD (n = 17) and controls (n = 16) | The clusterin CSF levels were elevated in AD | Mass spectrometry | |
| Nilselid et al. [101] | AD (n = 99) and controls (n = 39) | The clusterin CSF levels (native and deglycosylated) were elevated in AD | Both native and deglycosylated clusterin levels positively corrected with CSF tau and Aβ-42 | ELISA |
| Finehout et al. [105] | AD (n = 34) and controls (non-AD, n = 34) | The clusterin CSF levels were elevated in AD | Mass spectrometry | |
| Lidstrom et al. [106] | AD (n = 32), VaD (n = 20), PD (n = 17) and controls; longitudinal samples from patients with acute stroke | The clusterin CSF levels did not differ significantly in AD, VaD, PD or acute stroke, as compared to controls | CSF clusterin levels did not depend on the APOE ε4 status in AD | ELISA |
Aβ oligomers (AβOs)
Role in AD pathogenesis and biomarker studies
Neurodegeneration is a result of self-association of Aβ molecules and not just caused by the presence of Aβ. The oligomers of Aβ can be even more toxic than fibrillar Aβ aggregates [107]. They affect synapse composition, shape and density, thereby play a significant role in synaptic degeneration in AD [108]. Administration of cell-derived AβOs inhibit long-term potentiation of synaptic transmission, induced in rats [109]. The CSF levels of AβOs have been quantified in AD. Using a sensitive assay, it has been found that CSF levels of AβOs significantly increase in AD patients as compared to aged controls [110]. Lower levels of CSF AβOs have been reported in AD patients as compared to those with other forms of dementia [111]. In another study the ratio of AβOs/Aβ-42 was found to be significantly elevated in AD as compared to the non-AD group [112]. The diagnostic potential of AβOs in AD should be further explored using well-established cohorts.
Biomarkers of neuroinflammation
YKL-40/chitinase-3-like protein 1 (CHI3L1)
Role in AD pathogenesis
YKL-40 is a glycoprotein belonging to the family of 18 glycosyl hydrolases. It is also called human cartilage glycoprotein-39 (HC gp-39) or chitinase-3-like-1 protein (CHI3L1). It binds with chitin but does not have a chitinase activity [113]. It is secreted by the chondrocytes, synovial cells, vascular smooth muscle cells, macrophages and neutrophils [114, 115]. It is named based on the first three terminal amino acids: tyrosine (Y), lysine (K), and leucine (L) [115, 116]. YKL-40 plays a key role in inflammation, therefore, influences AD pathology. In response to neuroinflammation, the expression of YKL-40 is increased and is localized to astrocytes in the region of inflammation [117]. It is expressed by the microglia, and the expression of YKL-40 messenger ribonucleic acid (mRNA) is increased in AD [118]. Microglia and astrocytes are associated with senile plaques in AD and play a key role in immune response in the brain [119]. The microglia are activated in response to neurodegeneration. The plaque-associated activated microglia are large and mostly phagocytic [120]. They constantly scavenge the plaques, damaged neurons, infectious agents and promote inflammation in damaged tissue [121, 122]. Aβ, either alone or together with inflammatory mediators, sets up an activation cycle to activate the microglia and thereby generate an immune response in the brain [123]. Microglial activation thereby plays an important role in AD [124]. Therefore, microglial-expressed protein YKL-40 is a potential marker of neuroinflammation and plays a significant role in AD pathogenesis.
CSF biomarker studies pertaining to YKL-40
The CSF levels of YKL-40 are elevated in AD. Through numerous studies, it has been have found that increased levels of YKL-40 in CSF have prognostic and diagnostic utility as a biomarker for AD. YKL-40 aids in preclinical AD diagnosis and discriminating cognitively normal individuals from mild cognitive impairment (MCI) or AD patients (Table 3). The role of YKL-40 is also seen in differential diagnosis of dementia [125]. The levels of YKL-40 have been found to significantly correlate with MMSE scores [126]. Studies suggest YKL-40 is elevated early in the AD continuum and can serve as a valuable neuroinflammatory marker to detect early pathological changes and can even be used to study disease progression. The association of CSF YKL-40 with CSF T-tau and P-tau (Table 3) indicates that YKL-40 can help in tracking the neuroinflammation associated to neurodegeneration. Being a potential diagnostic and prognostic marker, it can serve as a target to combat AD-associated neuroinflammation. YKL-40 levels are consistently increased with age. This suggests that neuroinflammation occurs normally with aging. However, the still higher increase in ε4 carriers suggest that neuroinflammation is exacerbated with amyloidosis and neurodegeneration [127]. On the contrary, a recent study also indicates that inflammation could be driven by amyloidosis but, independent of the APOE ε4 status. In this study, the CSF levels were elevated in Aβ-positive individuals (low CSF Aβ), who were APOE ε4 non-carriers [128]. Therefore, YKL-40 can be used as a potential marker to stage the neuroinflammation associated with AD.
Table 3.
Studies conducted to evaluate the role of YKL-40 as a potential CSF biomarker
| Study | Study groups | CSF levels in AD/study groups | Association with core biomarkers/APOE genotype | Analysis method |
|---|---|---|---|---|
| Gispert et al. [129] | Controls (n = 49), preclinical AD (n = 19), MCI due to AD (n = 27), mild AD dementia (n = 15) | Elevated in preclinical AD, AD and MCI | Increased in APOE ε4 carriers within a study group; positively associated with P-tau | ELISA |
| Hoglund et al. [128] | Healthy older individuals, n = 129 (divided into high CSF Aβ, n = 86 and low CSF Aβ, n = 43) | No difference between two groups | Significantly elevated in APOE ε4 non-carriers who were Aβ positive | ELISA |
| Gispert et al. [130] | Normal controls (n = 53), preclinical AD (CSF Aβ < 500 pg/mL, n = 20), MCI due to AD (n = 28), mild AD dementia (n = 15) | Significantly increased in MCI due to AD compared to controls | No association with Aβ-42; positive linear association with P-tau | ELISA |
| Janelidze et al. [124] | Healthy controls (n = 53), stable mild cognitive impairment (sMCI, n = 62), MCI who later developed AD (MCI-AD, n = 35), AD dementia (n = 74), PDD/DLB (n = 47), vascular dementia (VaD, n = 34), and FTD (n = 33) | Significantly increased compared to healthy controls and sMCI. Elevated as compared with DLB, PDD but not with VaD or FTD. | Positively correlated with Aβ-42 in AD patients and with tau in all diagnostic groups | ELISA |
| Wennström et al. [125] | AD patients with mild to moderate dementia (n = 49), PD (n = 61), DLB (n = 36) and non-demented controls (n = 44) | Elevated compared to non-demented controls, DLB and PD patients | Not associated with CSF P-tau, T-tau or Aβ-42 | ELISA |
| Hellwig et al. [131] | AD dementia (n = 39), MCI due to AD (n = 13), MCI not due to AD (n = 29), non-AD dementia (n = 14) |
Significantly elevated in AD compared to MCI not due to AD or non-AD dementia. Also elevated in MCI-AD |
Significant correlation with T-tau and P-tau in the non-AD group; no association CSF with Aβ | ELISA |
| Kester et al. [132] | Cognitively normal (n = 37), MCI (n = 61), AD (n = 65) | Elevated in MCI and AD baseline levels in MCI-predicted progression to AD. Longitudinally increased in AD and MCI | ELISA | |
| Alcolea et al. [133] | Cognitively normal controls (n = 80) and amnestic MCI (n = 27) | Elevated significantly in amnesic MCI as compared to controls | Strong correlation with T-tau and P-tau; no correlation between YKL-40 and Aβ-42 | ELISA |
| Sutphen et al. [127] | Cognitively normal middle-aged individuals (CDR 0, n = 169), classified as early, mid and late | Significantly elevated in mid and late middle-aged individuals | Biomarker changes more evident in ε4 carriers | ELISA |
Monocyte chemoattractant protein 1 (MCP-1)
Role in AD pathogenesis
Chemokines are low-molecular weight cytokines. They are secondary inflammatory mediators induced by the primary mediators such as interleukin-1. These act as chemoattractants and direct leucocytes to the site of inflammation. They express their action through guanine nucleotide-associated protein (G protein)-coupled receptors. There are approximately 50 cytokines which are classified into four families; CC cytokines (have 2 adjacent cysteine residues at the N terminal), CXC cytokines (have two terminal cysteine residues separated by one amino acid), C cytokines (have 2 cysteine residues in total, one at N terminal and other at the downstream) and CX3C cytokines (have 2 cysteine residues separated by 3 amino acids at the N terminal) [134, 135]. Inflammation plays a significant role in AD pathogenesis. The cytokines and chemokines, being inflammatory mediators, are involved in AD pathogenesis. They are released by the astrocytes, which play a role in Aβ generation and degeneration [136]. The production of chemokines is increased in response to Aβ and plays an important role in migration of astrocytes. The treatment of neonatal astrocytes with Aβ significantly increased the production of MCP-1. In the same study, it was found that astrocytes from adult mice migrate in response to MCP-1, indicating the role of MCP-1 in astrogliosis and degradation of Aβ [137]. Deficiency of chemokine receptors in transgenic mice models has shown to promote early Aβ accumulation [138]. The astrocytes proliferate in response to neurodegeneration and increase the deposition of toxic Aβ [139]. Aβ itself increases the expression of chemokines and cytokines by astrocytes, by reactivating them. There is a continuous cycle of activation and reactivation of astrocytes leading to inflammation and neuronal injury. [140, 141]. Therefore, the chemokines being mediators of inflammation play a significant role in AD pathogenesis.
CSF biomarker studies pertaining to MCP-1
Many studies have demonstrated the role of CC chemokine, MCP-1 or CCL2 in AD diagnosis. Studies have reported elevated CSF levels of MCP-1 in AD (Table 4). MCP-1 levels in CSF are positively correlated with the decrease in MMSE scores and higher baseline levels predict a faster rate of cognitive decline in AD [142–144]. Therefore, MCP-1 could serve as a marker of cognitive decline along the AD continuum. MCP-1 plays an important role in AD-associated neuroinflammation and can serve a potential biomarker to track the same.
Table 4.
Studies conducted to evaluate the role of MCP-1 as a potential CSF biomarker
| Study | Study groups | CSF levels in AD/study groups | Association with core biomarkers/APOE genotype | Analysis method |
|---|---|---|---|---|
| Janelidze et al. [145] | Cognitively normal (n = 315), MCI (n = 449) and AD (n = 57) | Study participants were divided into Aβ positives and negatives. MCP-1 levels were not influenced by Aβ status | Electrochemiluminescence immunoassay [meso scale discovery, (MSD)] | |
| Wennström et al. [125] | AD patients with mild to moderate dementia (n = 49), PD (n = 61), DLB (n = 36) and non-demented controls (n = 44) | Elevated compared to non-demented controls; difference not detectable on age correction | Electrochemiluminescence immunoassay (MSD) | |
| Rosén et al. [146] | AD (n = 25) and controls (n = 25) | No significant difference between AD and controls | MSD assay | |
| Westin et al. [142] | Controls (n = 30) and MCI (n = 119) | Elevated in sMCI and even in MCI to AD converters; higher baseline values predict rate of future cognitive decline | Electrochemiluminescence immunoassay (MSD) | |
| Correa et al. [147] | AD (n = 22) and healthy controls (n = 27) | Significantly elevated in AD | Positively correlated with CSF P-tau and Aβ | ELISA |
| Choi et al. [148] | AD (n = 11), PD (n = 8), healthy controls (n = 13) | Elevated in AD and PD but not significantly as compared to controls | Multiplex immunoassay (Luminex xMAP technology) | |
| Galimberti et al. [143] | Amnestic MCI (n = 38), AD (n = 36), subjects with non-inflammatory affections of the nervous system (n = 41) | Significantly elevated in MCI and AD compared to controls | ELISA | |
| Blasko et al. [144] | AD (n = 23), FTD (n = 5), alcohol dementia (n = 10), major depression (n = 11) and healthy controls (n = 27) | Significantly increased in AD compared to controls (no difference in case of older controls | Positive correlation with T-tau but not with Aβ and P-tau | ELISA |
Biomarker of synaptic dysfunction
Neurogranin
Role in AD pathogenesis
Neurogranin is a calmodulin-binding, postsynaptic protein found in the dendrites [149]. It plays an important role in memory potentiation. It binds with calmodulin and releases the same when intracellular concentration of calcium increases. The released calmodulin binds with the calcium ions and activates a signal transduction pathway [150]. Synaptic dysfunction is linked to decline in cognition and occurs prior to neuronal degeneration [151, 152]. The brain levels of synaptic proteins including neurogranin are reduced in AD at an early stage. The synaptic dysfunction in terms of reduction in synapses is also seen in MCI which is higher in mild AD. Thus, synaptic dysfunction occurs early in AD and indicates disease progression [153–157]. Neurogranin regulates the calcium-dependent postsynaptic signaling triggered by calmodulin [158]. It has been found that neurogranin [Ng(+/+)] mice exhibit greater intracellular calcium concentration as compared to Ng(−/−) mice upon tetanic stimulation [159]. Expression of neurogranin reduces with aging [160]. Reduced brain levels of neurogranin can cause a dysregulation of post-synaptic signaling. Reduced neurogranin mRNA expression has been reported in hippocampal and retrosplenial regions of the brain in aged mice [160]. Therefore, a reduction of synaptic proteins such as neurogranin in the brain relates to synaptic dysfunction and the CSF levels of such proteins can be used for disease diagnosis and monitor the progression.
CSF biomarker studies pertaining to neurogranin
In the past few years, a number of researchers have evaluated the diagnostic and prognostic potential of the biomarker neurogranin. A number of assay methods have been developed to quantify neurogranin in the CSF and have reported elevated neurogranin levels in AD (Table 5). In a study conducted on various synaptic proteins including neurogranin in post-mortem brain samples, it was found that synaptic proteins discriminated dementia cases from controls with over 90% sensitivity and specificity [161]. The CSF neurogranin levels correlate with brain atrophy and amyloid load and also help in predicting decline in cognition. The CSF levels differ significantly between stable MCI (sMCI) and MCI to AD converters and between sMCI and AD [162–165]. Increased CSF levels of neurogranin are specific to AD and not seen in other neurodegenerative diseases [166, 167]. Therefore, it is a promising biomarker for early AD diagnosis, predicting progression and distinguishing AD from other forms of dementia. It can act as a theragnostic marker, which can help in monitoring biochemical effects of drugs used to improve synaptic function. Since, synaptic dysfunction is associated to cognitive decline, neurogranin can help in staging the rate of cognitive decline along the AD continuum. However, large longitudinal studies are needed to further validate the role of neurogranin in AD diagnosis and prognosis.
Table 5.
Studies conducted to evaluate the role of neurogranin as a potential CSF biomarker
| Study | Study groups | CSF levels in AD/study groups | Association with core biomarkers/APOE genotype | Analysis method |
|---|---|---|---|---|
| Hoglund et al. [128] | Healthy older individuals, n = 129 (divided into high CSF Aβ, n = 86 and low CSF Aβ, n = 43) | Significantly elevated in Aβ-positive group | Significantly elevated in APOE ε4 non-carriers who were Aβ positive | MSD assay |
| Tarawneh et al. [165] | Cognitively normal controls (CDR 0, n = 207) and AD (CDR: 0.5, 1, 2.; n = 95), non-AD dementias (n = 19) | Significantly elevated in participants with CDR ≥ 0.5 compared to CDR 0 and non-AD dementias | Positively correlated with CSF T-tau and P-tau | Two-site immunoassay (Erenna Singulex) |
| Mattsson et al. [168] | AD (n = 93), MCI (n = 187) and controls (n = 109) | Significantly predicted AD vs. controls; elevated in AD | CSF neurogranin levels were associated with Aβ positivity in all the groups; correlated strongly with T-tau | Electrochemiluminescence immunoassay (MSD) |
| Sanfilippo et al. [169] | Healthy controls (n = 44), MCI (n = 50), prodromal AD (n = 36), AD (n = 25), major depressive disorder (MDD, n = 6) | Significantly elevated in AD, prodromal AD vs. controls and MDD; significantly elevated in prodromal AD vs. MCI | Correlated positively with CSF T-tau and P-tau | ELISA |
| De Vos et al. [170] | Healthy controls (n = 29), MCI due to AD (n = 20), AD MCI due to AD (n = 20), AD, MCI due to AD and AD with high tau levels (n = 19) | Significantly elevated in MCI vs. controls but not in AD; no significant differences between MCI and AD | Correlated with T-tau in MCI and AD but not with Aβ-42 | ELISA |
| Kester et al. [164] | Cognitively normal (n = 37), MCI (n = 61) and AD (n = 65) | Significantly elevated in AD compared to controls; no difference between MCI and AD, MCI and cognitively normal | Strong positive correction with CSF T-tau and P-tau | Sandwich immunoassay (Erenna Singulex) |
| Kvartsberg et al. [163] | Three cohorts consisting of AD (n = 100), MCI (n = 40) and controls (n = 80) | Significantly elevated in AD vs. controls (using both methods); significantly elevated in MCI-AD vs. sMCI, AD vs. sMCI | Strong positive correction with CSF T-tau and P-tau in AD and controls | Mass spectrometry and ELISA |
| Hellwig et al. [131] | AD dementia (n = 39), MCI due to AD (n = 13), MCI not due to AD (n = 29), non-AD dementia (n = 14) | Significantly increased in AD, MCI due to AD compared to MCI not due to AD | Moderate correlation with CSF T-tau and P-tau but not with Aβ-42 | MSD assay |
| Portelius et al. [162] | Cognitively normal (n = 110), MCI (n = 173) and AD (n = 95) | Significantly elevated in progressive MCI, AD vs. controls | Negatively correlated with CSF Aβ-42 in sMCI, progressive MCI and AD; strong positive correlation with CSF T-tau and P-tau in all the study groups | Electrochemiluminescence immunoassay (MSD) |
| Thorsell et al. [171] | Study 1: AD (n = 11) and controls (n = 9); study 2: AD (n = 10), MCI (n = 10) and controls (n = 11) | Significantly increased in AD compared to controls, no significant difference between MCI vs. AD or controls | Positive correlation with CSF T-tau and P-tau in AD (study 1), all groups together (study 2); negative correlation with CSF Aβ-42 in MCI | Immunoblot analysis |
Biomarker of altered microglial activity
Soluble ectodomain of triggering receptor expressed on myeloid cells (sTREM2)
Role in AD pathogenesis
Ectodomain of triggering receptor expressed on myeloid cells (TREM2) is a transmembrane glycoprotein immune receptor expressed in a number of cells such as dendritic cells, osteoclasts, tissue macrophages and the microglia. It contains an ectodomain with three N-glycosylation residues, a transmembrane sequence and a short intracellular tail. Its functions are mediated via DNAX-activating protein of 12 kDa (DAP12) signaling [172, 173]. In the brain, it is expressed by the microglial cells and regulates microglial-mediated phagocytosis and clearance of apoptotic neurons [174, 175]. It plays an important role in regulating immune responses in the brain and the production of inflammatory cytokines [176, 177]. TREM2 is upregulated in mice with mutant APP and amyloid deposition [178]. The mutations associated with the TREM2 gene are associated with an increased risk for AD. GWAS, next generation sequencing, Sanger sequencing and genotyping have revealed that R47H TREM2 variant is a risk factor for AD, which can increase the risk of developing AD by two- to fourfold [179–182]. This can be associated to tau pathology, since carriers of the risk variant were found to possess higher levels of T-tau [183]. It has also been found that mutations in TREM2 reduce Aβ clearance [184]. TREM2 undergoes regulated membrane proteolytic processing by ADAM 10 (A disintegrin and metalloproteinase domain-containing protein 10) and γ secretase, and releases the soluble ectodomain sTREM2 into the extracellular space [185]. The sTREM2 is detectable in the CSF and the levels have been quantified in different neurological disorders such as AD, frontotemporal dementia (FTD) and multiple sclerosis [186, 187]. Since, TREM2 regulates microgliosis, the soluble fragment of the protein, sTREM2, could play a role in regulating TREM2-mediated microgliosis. The exact biological role of the soluble fragment is unclear. However, using in vitro and in vivo models, Zhong et al. have shown that sTREM2 promotes microglial survival and induces production of inflammatory cytokines [188]. In this study, it was found that administration of sTREM2-fc fusion protein increased the microglial viability, in both TREM2 knockout mice as well as wild type. Administration of sTREM2 reduced the microglial apoptosis induced by removal of granulocyte macrophage colony-stimulating factor (GM-CSF), in both knock out and wild mice. In addition, it was found that sTREM2 treatment activates the microglia by increased expression of inflammatory cytokines [188]. A significant reduction in microgliosis as well as microglial clustering around Aβ plaques has been found in Trem2−/−5XFAD mice as compared to the controls [189]. Therefore, sTREM2 likely plays a role in microgliosis, but further studies are needed to affirmatively elucidate the exact role of sTREM2.
CSF biomarker studies pertaining to sTREM2
Numerous studies have revealed that CSF levels of sTREM2 are altered in AD (Table 6). The levels are elevated in dominantly inherited AD cases years before the onset of symptoms [190], which highlights that microgliosis occurs prior to the onset of symptoms and later to brain amyloidosis. The Nasu–Hakola disease (NHD) TREM2 mutation carriers have lower CSF levels of sTREM2 [187]. This signifies that there is altered protein production in mutation carriers. Studied have found the CSF levels of TREM2 are increased in AD at early stage and correlate well with the markers of neurodegeneration and tau pathology. Therefore, microgliosis is most likely an early event that occurs along the AD and occurs in response to neurodegeneration. The CSF levels are lesser in AD as compared to MCI who later developed AD (MCI-AD). Thus, microgliosis increases from the preclinical AD to MCI-AD and there after reduces in AD, probably due to reduction in immune response [191]. Higher CSF levels in MCI patients are associated to increased gray matter volume. This reflects upon the protective response of microglia in response to neurodegeneration [192]. The role of TREM2 in regulating brain immune response, microgliosis and inflammation needs to be further explored. The CSF levels of sTREM2 can help in tracking the altered microgliosis along the disease trajectory and can serve as a potential stage biomarker for identifying early stages of AD and as theragnostic marker to monitor therapeutic effects of drugs administered at an early stage.
Table 6.
Studies conducted to evaluate the role of sTREM2 as a potential CSF biomarker
| Study | Study groups | CSF levels in AD/study groups | Association with core biomarkers/APOE genotype | Analysis method |
|---|---|---|---|---|
| Gispert et al. [129] | Controls (n = 49), preclinical AD (n = 19), MCI due to AD (n = 27), mild AD dementia (n = 15) | Elevated in AD and MCI (however, no significant difference upon age correction) | Positively associated with CSF P-tau, no significant difference between APOE ε4 carriers and non-carriers in any study group | ELISA |
| Heslegrave et al. [193] | AD (n = 37) and cognitively normal controls (n = 22) | Significantly elevated in AD | Significant positive correlation with CSF T-tau and P-tau but not with CSF Aβ-42 | Mass spectrometry |
| Suarez-Calvet et al. [194] | Controls (n = 150), preclinical AD (n = 63), MCI due to AD (n = 111) and AD dementia (n = 200) | Significantly elevated in MCI-AD compared to controls and AD dementia | Positively correlated with CSF T-tau and P-tau (stronger in preclinical AD, MCI due to AD and AD); not affected by APOE ε4 status | ELISA (MSD platform) |
| Henjum et al. [195] | Cohort 1: controls (n = 50), MCI (n = 21) and AD (n = 29); cohort 2: controls (n = 25) and AD (n = 25) | No statistical difference among the diagnostic groups | Positively correlated with CSF Aβ-42, T-tau and P-tau among controls | ELISA |
| Piccio et al. [187] | Cognitive normal (n = 107), AD (n = 73), FTD and TREM2 risk variant carriers (n = 40) | Significantly elevated in AD compared to controls (all non TREM2 risk variant carriers) | Highly correlated with CSF T-tau and P-tau levels but not with CSF Aβ-42 | ELISA |
| Gispert et al. [192] | Cognitively normal controls (n = 45), preclinical AD (n = 19), MCI due to AD (n = 27), and mild AD (n = 23) | Highest levels in MCI-AD; significantly higher than the controls and preclinical AD | Positively correlated with CSF T-tau and P-tau in all diagnostic groups | ELISA (MSD platform) |
Biomarkers reflecting neuronal membrane disruption (neurodegeneration)
Fatty acid-binding protein 3 (FABP3) or heart-type fatty acid-binding protein (HFABP)
Role in AD pathogenesis
The fatty acid-binding proteins (FABPs) are transport proteins for fatty acids and other lipophilic biomolecules. FABP3 is mainly expressed in the heart and skeletal muscles but has also been isolated from the brain [196]. In the brain, FABPs bind to long-chain polyunsaturated fatty acids (PUFA), such as docosahexaenoic acid (DHA) and arachidonic acid (ARA) and is involved in the transport of these fatty acids. These fatty acids are indispensable for maintaining neuronal membrane integrity, neurite growth and synapse formation. The DHA and ARA modulate neural membrane fluidity and permeability [197, 198]. The dietary supplementation of DHA has been found to improve spatial memory and reduce Aβ deposition in mice [199]. DHA also prevents Aβ-induced neuronal damage in vivo and in vitro [200]. Since HFABP or FABP3 regulates the transport of DHA and other fatty acids, it is likely to be associated with AD pathogenesis. The brain levels of FABP3 are reduced in such neurodegenerative diseases, which could be associated to altered signal transduction and membrane integrity [201]. FABPs are released following a cellular injury [202, 203]. Therefore, like other FABP’s, HFABP is likely to be associated with cellular dysfunction associated with AD. FABP3 is also associated with dopaminergic system and changes in dopaminergic system are likely to be associated with AD. It binds and regulates the dopaminergic D2 receptors, and overexpression of FABP3 promotes α-synuclein oligomerization [204–206]. Catalepsy behavior induced by haloperidol administration was found to be significantly increased in FABP3 knockout mice as compared to the wild type, indicating that FABP3 regulates D2 receptors [204]. In the same study, it was found that over expression of FABP3 increased D2 receptor sensitivity [204]. The association of FABP3 with dopaminergic system also signifies the role of FABP3 in AD pathogenesis.
CSF biomarker studies pertaining to FABP3
The CSF levels of FABP3 are elevated in AD and is a potential diagnostic marker for differential diagnosis of neurodegenerative diseases (Table 7). The elevated levels are significantly associated with brain atrophy in cases with low Aβ-42 and reflect on lipid dyshomeostasis in the CNS [207]. Therefore, elevated FABP3 levels in CSF might be associated to brain amyloidosis. The diagnostic accuracy of the core CSF biomarkers has been found to be increased in conjunction with FABP3. In addition, FABP3 and the ratio of FABP3/Aβ-42 are useful in predicting the progression of MCI subjects to AD. [208, 209]. In a recent study involving healthy aged individuals, the CSF levels of FABP3 were significantly elevated in Aβ-positive individuals (low CSF Aβ), compared to negative individuals (high CSF Aβ) [128]. Therefore, it is a good biomarker for predicting disease progression in early stages of disease and can help in identifying healthy aged individuals at risk of developing AD. The elevated CSF levels correlate with core markers of neuro degeneration (Table 7). The elevated levels in AD could likely be associated to the destruction of neurons.
Table 7.
Studies conducted to evaluate the role of FABP3 as a potential CSF biomarker
| Study | Study groups | CSF levels in AD/study groups | Association with core biomarkers/APOE genotype | Analysis method | |
|---|---|---|---|---|---|
| Hoglund et al. [128] | Healthy older individuals, n = 129 (divided into high CSF Aβ, n = 86 and low CSF Aβ, n = 43) | Significantly elevated in Aβ-positive group | Elevated significantly in APOE ε4 non-carriers who were Aβ positive | Electrochemiluminescence immunoassay (MSD) | |
| Bjerke et al. [210] | Non-demented women (n = 86) | Elevated in those who developed dementia and AD at follow-up; higher baseline levels associated to development of dementia | Strong correlation with CSF T-tau and P-tau at baseline | Electrochemiluminescence immunoassay (MSD) | |
| Biscetti et al. [211] | AD (n = 48), PD (n = 54), DLB (n = 40), PDD (n = 21), other neurological disorders (OND) as controls (n = 47) | Significantly elevated in AD, DLB compared to PD and OND | Significantly correlated with CSF T-tau levels CSF | ELISA | |
| Chiasserini et al. [212] | AD (n = 40), PD (n = 58), OND | Significantly elevated in AD compared to PD and OND | Positively correlated with CSF T-tau, P-tau but not with Aβ | Immunoassay | |
| Guo et al. [208] | Healthy controls (n = 92), MCI (n = 149), AD (n = 60) | Significantly elevated in AD compared to controls | Multiplex immunoassay (Luminex xMAP technology) | ||
| Desikan et al. [207] | Cognitively normal (n = 90), amnestic MCI (n = 139) and probable AD (n = 66) | Elevated in AD and MCI compared to controls | Significantly associated with CSF P-tau | Multiplex immunoassay (Luminex xMAP technology) | |
Biomarkers of neuronal structure and signaling disruption (markers of neurodegeneration)
Neurofilament light chain protein (NFL): marker of axonal degeneration
Role in AD pathogenesis
Neurofilaments are the proteins particularly found in neuronal axons. They are 10 nm in diameter and are essential for the axonal growth and the transmission of impulses along the axons [213]. These are heteropolymers composed of four subunits, namely neurofilament heavy, medium and light polypeptides and α-internexin [214]. Being elastic and fibrous, they maintain the shape of neurons and act as neuroskeletal supports [215]. NFL plays a role in protecting neurites from dystrophy and regulates pathways generating Aβ [216]. Significantly higher neocortical Aβ deposition was found in APP/PS1 NFL(−/−) mice as compared to APP/PS1 NFL(+/+) mice. The dystrophic neurites were also significantly higher in NFL(−/−) mice, in regions surrounding the plaques. In addition, higher microgliosis was found in such regions, in NFL(−/−) mice as compared to NFL(+/+) mice [216]. Neurofilaments are likely to be released from neuronal axons in response to neuronal damage in neurodegenerative diseases. NFL is mainly located in myelinated axons and white matter changes are associated with increased NFL levels in the CSF. Therefore, elevated levels of NFL in the CSF reflect on axonal degeneration [217] (Fig. 5). NFL is a specific biomarker of axonal degeneration, whose levels have been found to be elevated in a wide range of neurodegenerative diseases including AD. It is not a disease-specific biomarker but can aid in differential diagnosis of neurodegenerative disorders since its levels are elevated in FTD as compared to AD [218]. High CSF NFL levels predict high hippocampal atrophy rate in cognitively healthy older adults as well those at risk of AD [219]. In case of AD, it can help in tracking the different dynamic changes along the disease continuum.
Fig. 5.
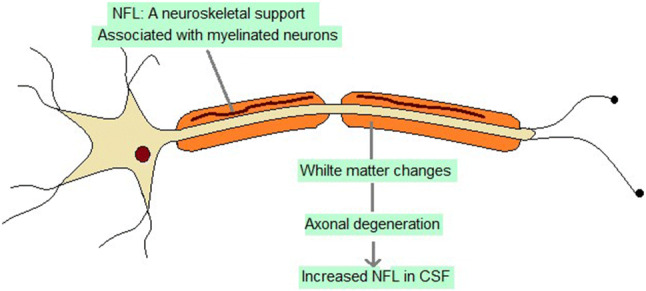
Role of NFL in AD pathogenesis (NFL neurofilament light chain, CSF cerebrospinal fluid)
CSF biomarker studies pertaining to NFL
The CSF levels of NFL are elevated in a wide range of neurodegenerative diseases including AD as compared to normal controls (Table 8). NFL levels are significantly elevated in AD compared to sMCI, and higher CSF levels in AD are associated with cognitive decline, white matter change, brain atrophy, and lower FDG-PET. The change in CSF levels and these associations are independent of Aβ positivity [168, 220]. Therefore, NFL reflects upon neuronal or axonal degeneration independent of Aβ pathology. Since the CSF levels of NFL are significantly elevated in AD compared to sMCI and associated to brain atrophy and cognitive decline, it can be used as potential biomarker to study disease progression and severity along the AD continuum. In addition, the diagnostic performance of core CSF biomarkers in differential diagnosis of early onset Alzheimer’s disease (EOAD) and FTD is improved in conjunction with the CSF levels of NFL [221]. Hence, it also has a potential to differentially diagnose a range of neurodegenerative diseases. But, the potential of NFL to identify individuals at risk of developing AD or its potential to identify preclinical AD needs to be further explored.
Table 8.
Studies conducted to evaluate the role of NFL as a potential CSF biomarker
| Study | Study groups | CSF levels in AD/study groups | Association with core biomarkers | Analysis method |
|---|---|---|---|---|
| Hoglund et al. [128] | Healthy older individuals, n = 129 (divided into high CSF Aβ, n = 86 and low CSF Aβ, n = 43) | No difference between the two groups | ELISA | |
| Zetterberg et al. [220] | AD (n = 95), MCI (n = 192) and cognitively normal individuals (n = 110) | Significantly elevated in AD, sMCI, MCI-AD as compared to controls; AD vs sMCI | Baselines levels correlated with low CSF Aβ, T-tau and P-tau | ELISA |
| Mattsson et al. [168] | AD (n = 93), MCI (n = 187) and controls (n = 109) | Elevated; Significantly predicted AD vs. controls | CSF NFL levels are not associated with Aβ positivity; correlated with T-tau | ELISA |
| Scherling et al. [222] | Normal controls (n = 47), AD (n = 50) various other neurodegenerative disorders such as FTD (n = 79), PD (n = 6) etc | Elevated significantly in AD as compared to normal controls; also in all FTD subgroups compared to normal controls and AD | ELISA | |
| Skillback et al. [218] | Healthy controls (n = 107), EOAD (n = 223), LOAD (n = 1194) FTD (n = 146), DLB (n = 114), PDD (n = 45), VaD (n = 465), mixed AD and VaD (n = 517), other dementias (n = 108), dementia not specified (n = 437) | Elevated in all groups as compared to controls; highest in FTD; significantly elevated in EOAD and LOAD compared to controls | ELISA | |
| Sjögren et al. [217] | Normal controls (n = 20), AD (n = 22), subcortical vascular dementia (SVD, n = 9) | Increased in AD and SVD compared with controls | ELISA | |
| Rosengren et al. [223] | Healthy controls (n = 39), AD (n = 37), FTD (n = 5) and VaD (n = 20) | Increased in AD, compared with controls; in FTD and VaD compared to AD and healthy controls | ELISA |
Visinin-like protein 1 (VILIP-1): marker of neuronal injury
Role in AD pathogenesis
VILIP-1 belongs to a large family of calcium-binding proteins called neuronal calcium sensors (NCSs) [224]. The VIPIL-1 protein is encoded by the visinin-like 1 (VSNL1) gene and contains 191 amino acids and weighs 22 kDa [225]. VILPI-1 is distributed in different regions of the brain [226]. The calcium ions (Ca2+) are involved in neuronal signaling and the NCSs mediate the action of these ions. In response to a high intracellular concentration of Ca2+, VILIP-1 gets reversibly translocated to the membrane components of the cell. This reversible interaction of VILIP-1 modulates signaling cascade in the neurons via activation of specific membrane-bound targets [227, 228]. Therefore, VILIP-1 plays an important role in neuronal signaling. The VILIP-1 regulates neuron ion channels, neuronal growth, survival, synaptic plasticity and activates cyclic adenosine monophosphate (cAMP) and cyclic guanine monophosphate (cGMP) signaling pathways [225]. Neurodegenerative disorders such as AD are associated with disturbed Ca2+ homeostasis in the neurons, which affect neuronal signaling by causing excessive activation of receptors, weakening the Ca2+ buffering capacity of neurons and deregulating the Ca2+ channels [229]. Aβ modulates this disturbed Ca2+ homeostasis by increasing the influx of Ca2+ by forming channels [230]. The NCSs such as VILIP-1 play a significant role in AD pathogenesis. The intracellular expression of VILIP-1 is reduced in AD brains as compared to controls. VILIP-1 has been found to be associated with extracellular plaques and NFTs in the brains of AD patients and its expression is associated with enhanced hyper phosphorylation of tau protein and cell death [231, 232]. In mild AD, there is a considerable loss of neurons in the entorhinal cortex [233, 234]. The levels of VILIP-1 are reduced in the entorhinal cortex of AD patients [235]. Therefore, it is a marker of neuronal injury. Figure 6 depicts the role of VILIP-1 in AD pathogenesis.
Fig. 6.

The role of VILIP-1 in AD pathogenesis (Aβ amyloid beta, AD Alzheimer’s disease)
This signifies that VILIP-1 is neurotoxic under a disturbed Ca2+ homeostasis. In AD, its intracellular expression is reduced. Increased expression promotes hyperphosphorylation and cell death which is reduced by calcium buffer protein. A disturbed Ca2+ balance causes the loss of vulnerable neurons and thereby the release of VILIP-1 extracellularly [225, 231, 232, 236].
CSF biomarker studies pertaining to VILIP-1
Numerous studies have been conducted to illustrate its role as a potential CSF diagnostic, prognostic and a differential biomarker. CSF levels of VILIP-1 aid in the early diagnosis of AD, distinguish AD from MCI, helps in identifying the patients with MCI likely to progress to AD, and in differentiating AD from other forms of dementia (Table 9). When used in combination with the core CSF markers, the diagnostic performance is improved [237]. VILIP-1 and VILIP-1/Aβ-42 ratio negatively correlates with MMSE [237, 238]. Baseline CSF levels of VILIP-1 are associated with rate of whole brain and regional brain atrophy in AD. VILIP-1 and the ratio of VILIP-1/Aβ-42 correlate significantly with the brain amyloid load. Therefore, VILIP-1 and the ratio of VILIP-1/Aβ-42 help in predicting the future cognitive decline. [239–243]. VILIP-1 can be used as a surrogate marker of neurodegeneration but, larger longitudinal studies are needed to validate the same. It can help in tracking the protective effects of neuroprotective therapeutic interventions.
Table 9.
Studies conducted to evaluate the role of VILIP-1 as a potential CSF biomarker
| Study | Study groups | CSF levels in AD/study groups | Association with core biomarkers | Analysis method |
|---|---|---|---|---|
| Hoglund et al. [128] | Healthy older individuals, n = 129 (divided into high CSF Aβ, n = 86 and low CSF Aβ, n = 43) | No difference between the two groups | Significantly elevated in APOE ε4 non-carriers who were Aβ positive | ELISA |
| Babic Leko et al. [238] | Healthy controls (n = 9), AD (n = 109), MCI (n = 45), VaD (n = 9), FTD (n = 18), DLB (n = 5), other dementias | Significantly elevated in AD vs. controls, MCI and DLB; no difference in AD vs. VaD and FTD | Positive correlation with CSF T-tau and P-tau in a mixed group of (healthy controls, AD, MCI) as well as in AD and MCI | ELISA |
| Mroczko et al. [240] | Cognitively normal controls (n = 18), AD (n = 33), MCI (n = 15) | Significantly elevated in AD vs. controls and MCI; also elevated in MCI vs. controls but not significantly | Significantly correlated with CSF P-tau in AD and controls; with Aβ-42 in controls | ELISA |
| Kester et al. [132] | Cognitively normal (n = 37), MCI (n = 61), AD (n = 65) | Elevated in AD and MCI vs. normal controls but not significantly; baseline levels in MCI predicted progression to AD. Longitudinally increased in AD and MCI | Microparticle based immunoassay (Erenna Singulex) | |
| Tarawneh et al. [242] | Normal controls (CDR 0, n = 64) and AD (n = 23) | Significantly elevated in AD vs. controls | Microparticle based immunoassay (Erenna Singulex) | |
| Sutphen et al. [127] | Cognitively normal middle-aged individuals (CDR 0, n = 169), classified as early, mid and late | Significantly elevated in late middle-aged individuals as compared to early and mid in non-ε4 carriers; | Biomarker changes more evident in ε4 carriers longitudinally | Microparticle based immunoassay (Erenna Singulex) |
| Luo et al. [239] | Normal controls (n = 40), AD (n = 61), DLB (n = 32) | Significantly elevated in AD vs. controls and DLB | Positively correlated with T-tau and P-tau | ELISA |
| Tarawneh et al. [243] | Normal controls (CDR 0, n = 211) and AD (CDR 0.5 and 1, n = 60) | Significantly elevated in AD vs. controls | Microparticle based immunoassay (Erenna Singulex) | |
| Tarawneh et al. [241] | Cognitively normal controls (n = 211), AD (n = 98), other dementias (n = 19) | Significantly elevated in AD vs. controls and other dementias | Correlated with T-tau and P-tau | Microparticle based immunoassay (Erenna Singulex) |
| Lee et al. [237] | Controls (n = 24) and AD (n = 33) | Significantly elevated in AD vs. controls | Strong positive correlation with CSF T-tau and P-tau; higher levels associated with APOE ε4 genotype | ELISA |
Conclusion
The multifaceted AD dementia is an amalgam of different pathological changes in the brain. The different pathological changes may represent a hierarchy of events that occur one after another or may follow their own trajectory, which ultimately leads to dementia due to AD. To get a deeper insight into different aspects of disease pathogenesis biomolecules/proteins involved in the associated biochemical pathways need to be explored and evaluated as disease biomarkers for disease diagnosis, prognosis and therapy. The CSF biomarkers would serve as reliable measures, to assess the time course of AD and the associated pathological changes along the continuum of the disease. A number of biomarkers in relation to different AD-associated pathological changes have been discussed in the current manuscript. They together or alone can aid in an accurate AD diagnosis starting from the preclinical phase and thereby can give a clear picture of the pathological changes that occur across the disease continuum. The use of multiple biomarkers can help in understanding the association of individual pathologies [244], and may provide an understanding about how one pathological change influences the other. Hence, these biomarkers in conjunction can improve the accuracy of diagnosis. It has been found that a biomarker model consisting of the biomarkers T-tau, NFL, neurogranin reflecting upon neurodegeneration, axonal damage and synaptic dysfunction, respectively, has a higher diagnostic accuracy (area under the receiver-operating curve (AUC) 85.5%) in classifying AD and controls [168]. The combination of CSF biomarkers, including YKL-40 could distinguish cognitively normal participants with clinical dementia rating (CDR) score of 0 from those with CDR > 0 with AUC 0.896 [76].
The CSF levels of these biomarkers change likely with the pathological change or event in the AD brain. The elevated CSF levels of clusterin can highlight upon the role of clusterin in binding with Aβ and preventing its fibrillization or its role in promoting the formation of soluble toxic Aβ oligomers. An elevated CSF levels of biomarkers YKL-40 and MCP-1 highlight upon neuroinflammation as a protective response to brain damage. These proteins are expressed by the astrocytes, which are activated in response to neurodegeneration and thereafter release inflammatory mediators. Elevated levels of sTREM2 highlight upon brain microgliosis as a response to phagocytise-accumulated Aβ. Therefore, these novel biomarkers can help in tracking inflammatory processes related to AD neurodegeneration. They can help in tracking stage and state-associated neuroinflammation in AD and combating the same with the therapeutic agents. Inflammation is associated with a number of psychiatric disorders [245]. These biomarkers can help in understanding the association of psychiatric disorders such as depression with AD. The dynamic changes in levels of VILIP-1, a biomarker of neuronal injury and NFL, a biomarker of axonal damage can alone or in conjunction provide an insight into the longitudinal cognitive changes associated with neurodegeneration. The cognitive decline associated with synaptic degeneration can be well accounted via CSF measure of neurogranin.
Hence, it can be concluded that the CSF biomarkers will certainly benefit in diagnosing AD at an early stage with much higher diagnostic accuracy either alone, together or in conjunction with the core CSF biomarkers. This would also aid in understanding the disease pathogenesis and progression. They can account for the lag between preclinical and clinical AD, and can act as indices of pathological change. They can serve as end point measures in clinical trials and accelerate the drug development process through the design of new drug molecules that can be targeted on the right individuals at the right stage. The complex nature of AD definitely directs us toward a strong rationale to use multiple biomarkers for understanding disease pathogenesis, and for a successful and accurate preclinical diagnosis, prognosis and treatment.
Acknowledgements
The authors thank the Australian Imaging, Biomarker and Lifestyle Study of Ageing (AIBL) Study Group (http://www.aibl.csiro.au) Edith Cowan University (ECU) and Deakin University. The AIBL study is a collaboration between Commonwealth Scientific and Industrial Research Organisation (CSIRO), ECU, The Florey Institute of Neuroscience and Mental Health (FINMH), National Ageing Research Institute (NARI), and Austin Health. It involves support from CogState Ltd., Hollywood Private Hospital, and Sir Charles Gairdner Hospital. The study receives funding from the National Health and Medical Research Council (NHMRC), Brightfocus Foundation USA, Dementia Australia Dementia Research Foundation (AADRF), the Dementia Collaborative Research Centres program (DCRC2), the Cooperative Research Centre (CRC) for Mental Health, the McCusker Alzheimer’s Research Foundation and Operational Infrastructure Support from the Government of Victoria. KD also thanks ECU HDR (Higher degree by research) Scholarship. KB is supported by the Torsten Söderberg foundation, Sweden. HZ is a Wallenberg Academy Fellow supported by grants from the Swedish Research Council, the European Research Council, the Olav Thon Foundation and the UK Dementia Research Institute at UCL.
Compliance with ethical standards
Conflict of interest
KB has served as a consultant or at advisory boards for Alzheon, BioArctic, Biogen, Eli Lilly, Fujirebio Europe, IBL International, Merck, Novartis, Pfizer, and Roche Diagnostics, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Ventures-based platform company at the University of Gothenburg. HZ has served at scientific advisory boards of Eli Lilly, Roche Diagnostics, Samumed, CogRx and Wave, has received travel support from Teva and is a co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Ventures-based platform company at the University of Gothenburg.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45(3):358–368. doi: 10.1002/1531-8249(199903)45:3<358::AID-ANA12>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 2.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Jr, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011;7(3):280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82(12):4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nat Rev Dis Prim. 2015;1:15056. doi: 10.1038/nrdp.2015.56. [DOI] [PubMed] [Google Scholar]
- 5.Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, Reichert M, Ketter N, Nejadnik B, Guenzler V, Miloslavsky M, Wang D, Lu Y, Lull J, Tudor IC, Liu E, Grundman M, Yuen E, Black R, Brashear HR. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):322–333. doi: 10.1056/NEJMoa1304839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris JC, Selkoe DJ. Recommendations for the incorporation of biomarkers into Alzheimer clinical trials: an overview. Neurobiol Aging. 2011;32(suppl 1):S1–S3. doi: 10.1016/j.neurobiolaging.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Sperling RA, Jack CR, Aisen PS (2011) Testing the right target and the right drug at the right stage. Sci Transl Med 3 (111):111–133. 10.1126/scitranslmed.3002609 [DOI] [PMC free article] [PubMed]
- 8.Blennow K. Biomarkers in Alzheimer’s disease drug development. Nat Med. 2010;16(11):1218–1222. doi: 10.1038/nm.2221. [DOI] [PubMed] [Google Scholar]
- 9.Arneric SP, Batrla-Utermann R, Beckett L, Bittner T, Blennow K, Carter L, Dean R, Engelborghs S, Genius J, Gordon MF, Hitchcock J, Kaplow J, Luthman J, Meibach R, Raunig D, Romero K, Samtani MN, Savage M, Shaw L, Stephenson D, Umek RM, Vanderstichele H, Willis B, Yule S. Cerebrospinal fluid biomarkers for Alzheimer’s disease: a view of the regulatory science qualification landscape from the coalition against major diseases CSF biomarker team. J Alzheimer’s Dis. 2016;55(1):19–35. doi: 10.3233/jad-160573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferreira D, Perestelo-Pérez L, Westman E, Wahlund L-O, Sarría A, Serrano-Aguilar P. Meta-review of CSF core biomarkers in Alzheimer’s disease: the state-of-the-art after the new revised diagnostic criteria. Front Aging Neurosci. 2014;6:47. doi: 10.3389/fnagi.2014.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jack CR, Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, Hampel H, Jagust WJ, Johnson KA, Knopman DS, Petersen RC, Scheltens P, Sperling RA, Dubois B. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 2016;87(5):539–547. doi: 10.1212/wnl.0000000000002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hampel H, Frank R, Broich K, Teipel SJ, Katz RG, Hardy J, Herholz K, Bokde ALW, Jessen F, Hoessler YC, Sanhai WR, Zetterberg H, Woodcock J, Blennow K. Biomarkers for Alzheimer’s disease: academic, industry and regulatory perspectives. Nat Rev Drug Discov. 2010;9(7):560–574. doi: 10.1038/nrd3115. [DOI] [PubMed] [Google Scholar]
- 13.Johnson KA, Fox NC, Sperling RA, Klunk WE. Brain imaging in Alzheimer disease. Cold Spring Harbor Perspect Med. 2012;2(4):a006213. doi: 10.1101/cshperspect.a006213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fishman RA (1992) Cerebrospinal fluid in diseases of the nervous system. WB Saunders Company, Philadelphia
- 15.Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L (2006) Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 10.1016/s1474-4422(06)70355-6 [DOI] [PubMed]
- 16.Ashton NJ, Scholl M, Heurling K, Gkanatsiou E, Portelius E, Hoglund K, Brinkmalm G, Hye A, Blennow K, Zetterberg H. Update on biomarkers for amyloid pathology in Alzheimer’s disease. Biomark Med. 2018;12(7):799–812. doi: 10.2217/bmm-2017-0433. [DOI] [PubMed] [Google Scholar]
- 17.Sato C, Barthelemy NR, Mawuenyega KG, Patterson BW, Gordon BA, Jockel-Balsarotti J, Sullivan M, Crisp MJ, Kasten T, Kirmess KM, Kanaan NM, Yarasheski KE, Baker-Nigh A, Benzinger TLS, Miller TM, Karch CM, Bateman RJ. Tau kinetics in neurons and the human central nervous system. Neuron. 2018;98(4):861–864. doi: 10.1016/j.neuron.2018.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K, DeKosky ST, Gauthier S, Selkoe D, Bateman R. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13(6):614–629. doi: 10.1016/S1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- 19.Jack CR, Albert M, Knopman DS, McKhann GM, Sperling RA, Carillo M, Thies W, Phelps CH. Introduction to revised criteria for the diagnosis of Alzheimer’s disease: National Institute on Aging and the Alzheimer Association Workgroups. Alzheimer’s Dement. 2011;7(3):257–262. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blennow K, Wallin A, Agren H, Spenger C, Siegfried J, Vanmechelen E. Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol. 1995;26(3):231–245. doi: 10.1007/bf02815140. [DOI] [PubMed] [Google Scholar]
- 21.Motter R, Vigo-Pelfrey C, Kholodenko D, Barbour R, Johnson-Wood K, Galasko D, Chang L, Miller B, Clark C, Green R, et al. Reduction of beta-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann Neurol. 1995;38(4):643–648. doi: 10.1002/ana.410380413. [DOI] [PubMed] [Google Scholar]
- 22.Blennow K, Hampel H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol. 2003;2(10):605–613. doi: 10.1016/S1474-4422(03)00530-1. [DOI] [PubMed] [Google Scholar]
- 23.Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature. 2009;461(7266):916–922. doi: 10.1038/nature08538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou B, Teramukai S, Yoshimura K, Fukushima M. Validity of cerebrospinal fluid biomarkers as endpoints in early-phase clinical trials for Alzheimer’s disease. J Alzheimer’s Dis. 2009;18(1):89–102. doi: 10.3233/jad-2009-1124. [DOI] [PubMed] [Google Scholar]
- 25.Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9(1):119–128. doi: 10.1016/s1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kester MI, van der Vlies AE, Blankenstein MA, Pijnenburg YA, van Elk EJ, Scheltens P, van der Flier WM. CSF biomarkers predict rate of cognitive decline in Alzheimer disease. Neurology. 2009;73(17):1353–1358. doi: 10.1212/WNL.0b013e3181bd8271. [DOI] [PubMed] [Google Scholar]
- 27.Toledo JB, Xie SX, Trojanowski JQ, Shaw LM. Longitudinal change in CSF Tau and Abeta biomarkers for up to 48 months in ADNI. Acta Neuropathol. 2013;126(5):659–670. doi: 10.1007/s00401-013-1151-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blennow K, Zetterberg H, Fagan AM. Fluid biomarkers in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(9):a006221. doi: 10.1101/cshperspect.a006221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet (London, England) 2008;372(9634):216–223. doi: 10.1016/s0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 30.Blennow K, Mattsson N, Schöll M, Hansson O, Zetterberg H. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol Sci. 2015;36(5):297–309. doi: 10.1016/j.tips.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 31.Mattsson N, Insel PS, Donohue M, Landau S, Jagust WJ, Shaw LM, Trojanowski JQ, Zetterberg H, Blennow K, Weiner MW. Independent information from cerebrospinal fluid amyloid-beta and florbetapir imaging in Alzheimer’s disease. Brain. 2015;138(Pt 3):772–783. doi: 10.1093/brain/awu367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palmqvist S, Mattsson N, Hansson O. Cerebrospinal fluid analysis detects cerebral amyloid-beta accumulation earlier than positron emission tomography. Brain. 2016;139(Pt 4):1226–1236. doi: 10.1093/brain/aww015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59(3):512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 34.Leuzy A, Carter SF, Chiotis K, Almkvist O, Wall A, Nordberg A. Concordance and diagnostic accuracy of [11C]PIB PET and cerebrospinal fluid biomarkers in a sample of patients with mild cognitive impairment and Alzheimer’s disease. J Alzheimer’s Dis. 2015;45(4):1077–1088. doi: 10.3233/jad-142952. [DOI] [PubMed] [Google Scholar]
- 35.Hsueh C-T, Liu D, Wang H. Novel biomarkers for diagnosis, prognosis, targeted therapy and clinical trials. Biomark Res. 2013;1(1):1. doi: 10.1186/2050-7771-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Counts SE, Ikonomovic MD, Mercado N, Vega IE, Mufson EJ (2016) Biomarkers for the early detection and progression of Alzheimer’s disease. Neurotherapeutics 1–19 [DOI] [PMC free article] [PubMed]
- 37.Fagan AM, Perrin RJ. Upcoming candidate cerebrospinal fluid biomarkers of Alzheimer’s disease. Biomark Med. 2012;6(4):455–476. doi: 10.2217/bmm.12.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science (New York, NY) 1988;240(4852):622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 39.Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW. Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochem Biophys Acta. 1987;917(1):148–161. doi: 10.1016/0005-2760(87)90295-5. [DOI] [PubMed] [Google Scholar]
- 40.Stone DJ, Rozovsky I, Morgan TE, Anderson CP, Hajian H, Finch CE. Astrocytes and microglia respond to estrogen with increased apoE mRNA in vivo and in vitro. Exp Neurol. 1997;143(2):313–318. doi: 10.1006/exnr.1996.6360. [DOI] [PubMed] [Google Scholar]
- 41.DeMattos RB, Brendza RP, Heuser JE, Kierson M, Cirrito JR, Fryer J, Sullivan PM, Fagan AM, Han X, Holtzman DM. Purification and characterization of astrocyte-secreted apolipoprotein E and J-containing lipoproteins from wild-type and human apoE transgenic mice. Neurochem Int. 2001;39(5–6):415–425. doi: 10.1016/S0197-0186(01)00049-3. [DOI] [PubMed] [Google Scholar]
- 42.Leduc V, Jasmin-Belanger S, Poirier J. APOE and cholesterol homeostasis in Alzheimer’s disease. Trends Mol Med. 2010;16(10):469–477. doi: 10.1016/j.molmed.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 43.Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, Schjeide BMM, Hooli B, DiVito J, Ionita I, Jiang H, Laird N, Moscarillo T, Ohlsen KL, Elliott K, Wang X, Hu-Lince D, Ryder M, Murphy A, Wagner SL, Blacker D, Becker KD, Tanzi RE. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am J Hum Genet. 2008;83(5):623–632. doi: 10.1016/j.ajhg.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Lince DH, Zismann VL, Beach TG, Leung D, Bryden L, Halperin RF, Marlowe L, Kaleem M, Walker DG, Ravid R, Heward CB, Rogers J, Papassotiropoulos A, Reiman EM, Hardy J, Stephan DA. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry. 2007;68(4):613–618. doi: 10.4088/JCP.v68n0419. [DOI] [PubMed] [Google Scholar]
- 45.Poirier J, Hess M, May PC, Finch CE. Astrocytic apolipoprotein E mRNA and GFAP mRNA in hippocampus after entorhinal cortex lesioning. Brain Res Mol Brain Res. 1991;11(2):97–106. doi: 10.1016/0169-328X(91)90111-A. [DOI] [PubMed] [Google Scholar]
- 46.Gong JS, Kobayashi M, Hayashi H, Zou K, Sawamura N, Fujita SC, Yanagisawa K, Michikawa M. Apolipoprotein E (ApoE) isoform-dependent lipid release from astrocytes prepared from human ApoE3 and ApoE4 knock-in mice. J Biol Chem. 2002;277(33):29919–29926. doi: 10.1074/jbc.M203934200. [DOI] [PubMed] [Google Scholar]
- 47.Michikawa M, Fan QW, Isobe I, Yanagisawa K. Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J Neurochem. 2000;74(3):1008–1016. doi: 10.1046/j.1471-4159.2000.0741008.x. [DOI] [PubMed] [Google Scholar]
- 48.Tokuda T, Calero M, Matsubara E, Vidal R, Kumar A, Permanne B, Zlokovic B, Smith JD, Ladu MJ, Rostagno A, Frangione B, Ghiso J. Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer’s amyloid beta peptides. Biochem J. 2000;348(Pt 2):359–365. [PMC free article] [PubMed] [Google Scholar]
- 49.Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10(5):333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harbor Perspect Med. 2012;2(3):a006312. doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang Q, Lee CD, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL. ApoE promotes the proteolytic degradation of Aβ. Neuron. 2008;58(5):681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV (2008) apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J Clin Invest 118(12):4002-4013 [DOI] [PMC free article] [PubMed]
- 53.Nishitsuji K, Hosono T, Nakamura T, Bu G, Michikawa M. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood–brain-barrier model. J Biol Chem. 2011;M111:225532. doi: 10.1074/jbc.M111.225532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma J, Yee A, Brewer HB, Jr, Das S, Potter H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature. 1994;372(6501):92–94. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- 55.Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, Ghetti B, Paul SM. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci. 1999;96(26):15233–15238. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM (2011) Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med 3(89):89ra57. 10.1126/scitranslmed.3002156 [DOI] [PMC free article] [PubMed]
- 57.Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, McCue L, Xiong C, Del-Aguila JL, Morris JC, Cruchaga C, Fagan AM, Miller BL, Boxer AL, Seeley WW, Butovsky O, Barres BA, Paul SM, Holtzman DM. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549(7673):523–527. doi: 10.1038/nature24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tesseur I, Van Dorpe J, Spittaels K, Van den Haute C, Moechars D, Van Leuven F. Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am J Pathol. 2000;156(3):951–964. doi: 10.1016/s0002-9440(10)64963-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi Y, Holtzman DM (2018) Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. 10.1038/s41577-018-0051-1 [DOI] [PMC free article] [PubMed]
- 60.Roheim PS, Carey M, Forte T, Vega GL. Apolipoproteins in human cerebrospinal fluid. Proc Natl Acad Sci. 1979;76(9):4646–4649. doi: 10.1073/pnas.76.9.4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang J, Sokal I, Peskind ER, Quinn JF, Jankovic J, Kenney C, Chung KA, Millard SP, Nutt JG, Montine TJ. CSF multianalyte profile distinguishes Alzheimer and Parkinson diseases. Am J Clin Pathol. 2008;129(4):526–529. doi: 10.1309/w01y0b808emeh12l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blennow K, Hesse C, Fredman P. Cerebrospinal fluid apolipoprotein E is reduced in Alzheimer’s disease. NeuroReport. 1994;5(18):2534–2536. doi: 10.1097/00001756-199412000-00032. [DOI] [PubMed] [Google Scholar]
- 63.Landen M, Hesse C, Fredman P, Regland B, Wallin A, Blennow K. Apolipoprotein E in cerebrospinal fluid from patients with Alzheimer’s disease and other forms of dementia is reduced but without any correlation to the apoE4 isoform. Dementia (Basel, Switzerland) 1996;7(5):273–278. doi: 10.1159/000106892. [DOI] [PubMed] [Google Scholar]
- 64.Pirttilä T, Koivisto K, Mehta PD, Reinikainen K, Kim KS, Kilkku O, Heinonen E, Soininen H, Riekkinen P, Sr, Wisniewski HM. Longitudinal study of cerebrospinal fluid amyloid proteins and apolipoprotein E in patients with probable Alzheimer’s disease. Neurosci Lett. 1998;249(1):21–24. doi: 10.1016/S0304-3940(98)00381-4. [DOI] [PubMed] [Google Scholar]
- 65.Lindh M, Blomberg M, Jensen M, Basun H, Lannfelt L, Engvall B, Scharnagel H, Marz W, Wahlund LO, Cowburn RF. Cerebrospinal fluid apolipoprotein E (apoE) levels in Alzheimer’s disease patients are increased at follow up and show a correlation with levels of tau protein. Neurosci Lett. 1997;229(2):85–88. doi: 10.1016/S0304-3940(97)00429-1. [DOI] [PubMed] [Google Scholar]
- 66.Fukuyama R, Mizuno T, Mori S, Yanagisawa K, Nakajima K, Fushiki S. Age-dependent decline in the apolipoprotein E level in cerebrospinal fluid from control subjects and its increase in cerebrospinal fluid from patients with Alzheimer’s disease. Eur Neurol. 2000;43(3):161–169. doi: 10.1159/000008157. [DOI] [PubMed] [Google Scholar]
- 67.Merched A, Blain H, Visvikis S, Herbeth B, Jeandel C, Siest G. Cerebrospinal fluid apolipoprotein E level is increased in late-onset Alzheimer’s disease. J Neurol Sci. 1997;145(1):33–39. doi: 10.1016/S0022-510X(96)00234-1. [DOI] [PubMed] [Google Scholar]
- 68.Hesse C, Larsson H, Fredman P, Minthon L, Andreasen N, Davidsson P, Blennow K. Measurement of apolipoprotein E (apoE) in cerebrospinal fluid. Neurochem Res. 2000;25(4):511–517. doi: 10.1023/A:1007516210548. [DOI] [PubMed] [Google Scholar]
- 69.Martinez-Morillo E, Hansson O, Atagi Y, Bu G, Minthon L, Diamandis EP, Nielsen HM. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathol. 2014;127(5):633–643. doi: 10.1007/s00401-014-1266-2. [DOI] [PubMed] [Google Scholar]
- 70.Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong L-M, Jakes R, Huang DY, Pericak-Vance M, Schmechel D, Roses AD. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci. 1994;91(23):11183–11186. doi: 10.1073/pnas.91.23.11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Harten AC, Jongbloed W, Teunissen CE, Scheltens P, Veerhuis R, van der Flier WM. CSF ApoE predicts clinical progression in nondemented APOEε4 carriers. Neurobiol Aging. 2017;57:186–194. doi: 10.1016/j.neurobiolaging.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 72.Johansson P, Almqvist EG, Bjerke M, Wallin A, Johansson JO, Andreasson U, Blennow K, Zetterberg H, Svensson J. Reduced cerebrospinal fluid concentration of apolipoprotein A-I in patients with Alzheimer’s disease. J Alzheimer’s Dis. 2017;59(3):1017–1026. doi: 10.3233/jad-170226. [DOI] [PubMed] [Google Scholar]
- 73.Rezeli M, Zetterberg H, Blennow K, Brinkmalm A, Laurell T, Hansson O, Marko-Varga G. Quantification of total apolipoprotein E and its specific isoforms in cerebrospinal fluid and blood in Alzheimer’s disease and other neurodegenerative diseases. EuPA Open Proteomics. 2015;8:137–143. doi: 10.1016/j.euprot.2015.07.012. [DOI] [Google Scholar]
- 74.Richens JL, Vere KA, Light RA, Soria D, Garibaldi J, Smith AD, Warden D, Wilcock G, Bajaj N, Morgan K, O’Shea P. Practical detection of a definitive biomarker panel for Alzheimer’s disease; comparisons between matched plasma and cerebrospinal fluid. Int J Mol Epidemiol Genet. 2014;5(2):53–70. [PMC free article] [PubMed] [Google Scholar]
- 75.Toledo JB, Da X, Weiner MW, Wolk DA, Xie SX, Arnold SE, Davatzikos C, Shaw LM, Trojanowski JQ. CSF Apo-E levels associate with cognitive decline and MRI changes. Acta Neuropathol. 2014;127(5):621–632. doi: 10.1007/s00401-013-1236-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perrin RJ, Craig-Schapiro R, Malone JP, Shah AR, Gilmore P, Davis AE, Roe CM, Peskind ER, Li G, Galasko DR, Clark CM, Quinn JF, Kaye JA, Morris JC, Holtzman DM, Townsend RR, Fagan AM. Identification and validation of novel cerebrospinal fluid biomarkers for staging early Alzheimer’s disease. PLoS ONE. 2011;6(1):e16032. doi: 10.1371/journal.pone.0016032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nuutinen T, Suuronen T, Kauppinen A, Salminen A. Clusterin: a forgotten player in Alzheimer’s disease. Brain Res Rev. 2009;61(2):89–104. doi: 10.1016/j.brainresrev.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 78.Humphreys DT, Carver JA, Easterbrook-Smith SB, Wilson MR. Clusterin has chaperone-like activity similar to that of small heat shock proteins. J Biol Chem. 1999;274(11):6875–6881. doi: 10.1074/jbc.274.11.6875. [DOI] [PubMed] [Google Scholar]
- 79.De Silva HV, Harmony JAK, Stuart WD, Gil CM, Robbins J. Apolipoprotein J: structure and tissue distribution. Biochemistry. 1990;29(22):5380–5389. doi: 10.1021/bi00474a025. [DOI] [PubMed] [Google Scholar]
- 80.Jun G, Naj AC, Beecham GW, et al. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with apoe genotypes. Arch Neurol. 2010;67(12):1473–1484. doi: 10.1001/archneurol.2010.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bettens K, Brouwers N, Engelborghs S, Lambert J-C, Rogaeva E, Vandenberghe R, Le Bastard N, Pasquier F, Vermeulen S, Van Dongen J. Both common variations and rare non-synonymous substitutions and small insertion/deletions in CLU are associated with increased Alzheimer risk. Mol Neurodegener. 2012;7(1):3. doi: 10.1186/1750-1326-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McGeer PL, Kawamata T, Walker DG. Distribution of clusterin in Alzheimer brain tissue. Brain Res. 1992;579(2):337–341. doi: 10.1016/0006-8993(92)90071-G. [DOI] [PubMed] [Google Scholar]
- 83.Matsubara E, Soto C, Governale S, Frangione B, Ghiso J. Apolipoprotein J and Alzheimer’s amyloid beta solubility. Biochem J. 1996;316(Pt 2):671–679. doi: 10.1042/bj3160671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Calero M, Rostagno A, Matsubara E, Zlokovic B, Frangione B, Ghiso J. Apolipoprotein J (clusterin) and Alzheimer’s disease. Microsc Res Tech. 2000;50(4):305–315. doi: 10.1002/1097-0029(20000815)50:4<305::aid-jemt10>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 85.Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, Zlokovic BV. Transport pathways for clearance of human Alzheimer’s amyloid β-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab. 2007;27(5):909–918. doi: 10.1038/sj.jcbfm.9600419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Qi X-M, Wang C, Chu X-K, Li G, Ma J-F. Intraventricular infusion of clusterin ameliorated cognition and pathology in Tg6799 model of Alzheimer’s disease. BMC Neurosci. 2018;19(1):2. doi: 10.1186/s12868-018-0402-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang P, Chen K, Gu Y, Guo Q, Hong Z, Zhao Q. β-Amyloid upregulates intracellular clusterin but not secretory clusterin in primary cultured neurons and APP mice. Curr Alzheimer Res. 2017;14(11):1207–1214. doi: 10.2174/1567205014666170531080948. [DOI] [PubMed] [Google Scholar]
- 88.Lidström AM, Bogdanovic N, Hesse C, Volkman I, Davidsson P, Blennow K. Clusterin (apolipoprotein J) protein levels are increased in hippocampus and in frontal cortex in Alzheimer’s disease. Exp Neurol. 1998;154(2):511–521. doi: 10.1006/exnr.1998.6892. [DOI] [PubMed] [Google Scholar]
- 89.Miners JS, Clarke P, Love S (2016) Clusterin levels are increased in Alzheimer’s disease and influence the regional distribution of Abeta. Brain Pathol (Zurich, Switzerland). 10.1111/bpa.12392 [DOI] [PMC free article] [PubMed]
- 90.DeMattos RB, Cirrito JR, Parsadanian M, May PC, O’Dell MA, Taylor JW, Harmony JAK, Aronow BJ, Bales KR, Paul SM, Holtzman DM. ApoE and clusterin cooperatively suppress Aβ levels and deposition: evidence that ApoE regulates extracellular Aβ metabolism in vivo. Neuron. 2004;41(2):193–202. doi: 10.1016/S0896-6273(03)00850-X. [DOI] [PubMed] [Google Scholar]
- 91.Viard I, Wehrli P, Jornot L, Bullani R, Vechietti J-L, French LE, Schifferli JA, Tschopp J. Clusterin gene expression mediates resistance to apoptotic cell death induced by heat shock and oxidative stress. J Investig Dermatol. 1999;112(3):290–296. doi: 10.1046/j.1523-1747.1999.00531.x. [DOI] [PubMed] [Google Scholar]
- 92.Wolter KG, Hsu Y-T, Smith CL, Nechushtan A, Xi X-G, Youle RJ. Movement of bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139(5):1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang H, Kim JK, Edwards CA, Xu Z, Taichman R, Wang C-Y (2005) Clusterin inhibits apoptosis by interacting with activated Bax. Nat Cell Biol 7:909. 10.1038/ncb1291. https://www.nature.com/articles/ncb1291#supplementary-information [DOI] [PubMed]
- 94.Shannan B, Seifert M, Boothman D, Tilgen W, Reichrath J. Clusterin and DNA repair: a new function in cancer for a key player in apoptosis and cell cycle control. J Mol Histol. 2006;37(5–7):183–188. doi: 10.1007/s10735-006-9052-7. [DOI] [PubMed] [Google Scholar]
- 95.Zwain IH, Grima J, Cheng CY. Regulation of clusterin secretion and mRNA expression in astrocytes by cytokines. Mol Cell Neurosci. 1994;5(3):229–237. doi: 10.1006/mcne.1994.1027. [DOI] [PubMed] [Google Scholar]
- 96.Kirszbaum L, Bozas S, Walker I. SP-40, 40, a protein involved in the control of the complement pathway, possesses a unique array of disulphide bridges. FEBS Lett. 1992;297(1–2):70–76. doi: 10.1016/0014-5793(92)80330-J. [DOI] [PubMed] [Google Scholar]
- 97.Wu Z-C, Yu J-T, Li Y, Tan L. Clusterin in Alzheimer’s disease. Adv Clin Chem. 2012;56:155. doi: 10.1016/B978-0-12-394317-0.00011-X. [DOI] [PubMed] [Google Scholar]
- 98.Santilli G, Aronow BJ, Sala A. Essential requirement of apolipoprotein J (clusterin) signaling for IκB expression and regulation of NF-κB activity. J Biol Chem. 2003;278(40):38214–38219. doi: 10.1074/jbc.C300252200. [DOI] [PubMed] [Google Scholar]
- 99.Deming Y, Xia J, Cai Y, Lord J, Holmans P, Bertelsen S, Holtzman D, Morris JC, Bales K, Pickering EH, Kauwe J, Goate A, Cruchaga C. A potential endophenotype for Alzheimer’s disease: cerebrospinal fluid clusterin. Neurobiol Aging. 2016;37:208.e201–208.e209. doi: 10.1016/j.neurobiolaging.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jongbloed W, Herrebout MA, Blankenstein MA, Veerhuis R. Quantification of clusterin in paired cerebrospinal fluid and plasma samples. Ann Clin Biochem. 2014;51(Pt 5):557–567. doi: 10.1177/0004563213503456. [DOI] [PubMed] [Google Scholar]
- 101.Nilselid AM, Davidsson P, Nagga K, Andreasen N, Fredman P, Blennow K. Clusterin in cerebrospinal fluid: analysis of carbohydrates and quantification of native and glycosylated forms. Neurochem Int. 2006;48(8):718–728. doi: 10.1016/j.neuint.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 102.Desikan RS, Thompson WK, Holland D, Hess CP, Brewer JB, Zetterberg H, Blennow K, Andreassen OA, McEvoy LK, Hyman BT, Dale AM. The role of clusterin in amyloid-beta-associated neurodegeneration. JAMA Neurol. 2014;71(2):180–187. doi: 10.1001/jamaneurol.2013.4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Prikrylova Vranova H, Henykova E, Mares J, Kaiserova M, Mensikova K, Vastik M, Hlustik P, Zapletalova J, Strnad M, Stejskal D, Kanovsky P. Clusterin CSF levels in differential diagnosis of neurodegenerative disorders. J Neurol Sci. 2016;361:117–121. doi: 10.1016/j.jns.2015.12.023. [DOI] [PubMed] [Google Scholar]
- 104.Sihlbom C, Davidsson P, Sjogren M, Wahlund LO, Nilsson CL. Structural and quantitative comparison of cerebrospinal fluid glycoproteins in Alzheimer’s disease patients and healthy individuals. Neurochem Res. 2008;33(7):1332–1340. doi: 10.1007/s11064-008-9588-x. [DOI] [PubMed] [Google Scholar]
- 105.Finehout EJ, Franck Z, Choe LH, Relkin N, Lee KH. Cerebrospinal fluid proteomic biomarkers for Alzheimer’s disease. Ann Neurol. 2007;61(2):120–129. doi: 10.1002/ana.21038. [DOI] [PubMed] [Google Scholar]
- 106.Lidstrom AM, Hesse C, Rosengren L, Fredman P, Davidsson P, Blennow K. Normal levels of clusterin in cerebrospinal fluid in Alzheimer’s disease, and no change after acute ischemic stroke. J Alzheimer’s Dis. 2001;3(5):435–442. doi: 10.3233/JAD-2001-3501. [DOI] [PubMed] [Google Scholar]
- 107.Sakono M, Zako T. Amyloid oligomers: formation and toxicity of Abeta oligomers. FEBS J. 2010;277(6):1348–1358. doi: 10.1111/j.1742-4658.2010.07568.x. [DOI] [PubMed] [Google Scholar]
- 108.Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27(4):796–807. doi: 10.1523/jneurosci.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 110.Savage MJ, Kalinina J, Wolfe A, Tugusheva K, Korn R, Cash-Mason T, Maxwell JW, Hatcher NG, Haugabook SJ, Wu G, Howell BJ, Renger JJ, Shughrue PJ, McCampbell A. A sensitive abeta oligomer assay discriminates Alzheimer’s and aged control cerebrospinal fluid. J Neurosci. 2014;34(8):2884–2897. doi: 10.1523/jneurosci.1675-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sancesario GM, Cencioni MT, Esposito Z, Borsellino G, Nuccetelli M, Martorana A, Battistini L, Sorge R, Spalletta G, Ferrazzoli D, Bernardi G, Bernardini S, Sancesario G. The load of amyloid-beta oligomers is decreased in the cerebrospinal fluid of Alzheimer’s disease patients. J Alzheimer’s Dis. 2012;31(4):865–878. doi: 10.3233/jad-2012-120211. [DOI] [PubMed] [Google Scholar]
- 112.Santos AN, Ewers M, Minthon L, Simm A, Silber RE, Blennow K, Prvulovic D, Hansson O, Hampel H. Amyloid-beta oligomers in cerebrospinal fluid are associated with cognitive decline in patients with Alzheimer’s disease. J Alzheimer’s Dis. 2012;29(1):171–176. doi: 10.3233/jad-2012-111361. [DOI] [PubMed] [Google Scholar]
- 113.Kazakova MH, Sarafian VS. YKL-40—a novel biomarker in clinical practice? Folia Med. 2009;51(1):5–14. [PubMed] [Google Scholar]
- 114.Volck B, Price PA, Johansen JS, Sorensen O, Benfield TL, Nielsen HJ, Calafat J, Borregaard N. YKL-40, a mammalian member of the chitinase family, is a matrix protein of specific granules in human neutrophils. Proc Assoc Am Phys. 1998;110(4):351–360. [PubMed] [Google Scholar]
- 115.Johansen JS. Studies on serum YKL-40 as a biomarker in diseases with inflammation, tissue remodelling, fibroses and cancer. Dan Med Bull. 2006;53(2):172–209. [PubMed] [Google Scholar]
- 116.Johansen JS, Williamson MK, Rice JS, Price PA. Identification of proteins secreted by human osteoblastic cells in culture. J Bone Miner Res. 1992;7(5):501–512. doi: 10.1002/jbmr.5650070506. [DOI] [PubMed] [Google Scholar]
- 117.Bonneh-Barkay D, Wang G, Starkey A, Hamilton RL, Wiley CA. In vivo CHI3L1 (YKL-40) expression in astrocytes in acute and chronic neurological diseases. J Neuroinflamm. 2010;7(1):1–8. doi: 10.1186/1742-2094-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflamm. 2006;3:27. doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines. Sci World J. 2012;2012:756357. doi: 10.1100/2012/756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sheng JG, Mrak RE, Griffin WS. Neuritic plaque evolution in Alzheimer’s disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathol. 1997;94(1):1–5. doi: 10.1007/s004010050664. [DOI] [PubMed] [Google Scholar]
- 121.Lee CY, Landreth GE (2010) The role of microglia in amyloid clearance from the AD brain. J Neural Transm (Vienna, Austria: 1996) 117(8):949–960. 10.1007/s00702-010-0433-4 [DOI] [PMC free article] [PubMed]
- 122.Neniskyte U, Neher JJ, Brown GC. Neuronal death induced by nanomolar amyloid beta is mediated by primary phagocytosis of neurons by microglia. J Biol Chem. 2011;286(46):39904–39913. doi: 10.1074/jbc.M111.267583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mrak RE, Griffin WS. Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005;26(3):349–354. doi: 10.1016/j.neurobiolaging.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 124.Janelidze S, Hertze J, Zetterberg H, Landqvist Waldo M, Santillo A, Blennow K, Hansson O. Cerebrospinal fluid neurogranin and YKL-40 as biomarkers of Alzheimer’s disease. Ann Clin Transl Neurol. 2016;3(1):12–20. doi: 10.1002/acn3.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wennström M, Surova Y, Hall S, Nilsson C, Minthon L, Hansson O, Nielsen HM. The inflammatory marker YKL-40 is elevated in cerebrospinal fluid from patients with Alzheimer’s but not Parkinson’s disease or dementia with Lewy bodies. PLoS ONE. 2015;10(8):e0135458. doi: 10.1371/journal.pone.0135458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Antonell A, Mansilla A, Rami L, Lladó A, Iranzo A, Olives J, Balasa M, Sanchez-Valle R, Molinuevo JL. Cerebrospinal fluid level of YKL-40 protein in preclinical and prodromal Alzheimer’s disease. J Alzheimer’s Dis. 2014;42(3):901–908. doi: 10.3233/JAD-140624. [DOI] [PubMed] [Google Scholar]
- 127.Sutphen CL, Jasielec MS, Shah AR, et al. Longitudinal cerebrospinal fluid biomarker changes in preclinical alzheimer disease during middle age. JAMA Neurol. 2015;72(9):1029–1042. doi: 10.1001/jamaneurol.2015.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hoglund K, Kern S, Zettergren A, Borjesson-Hansson A, Zetterberg H, Skoog I, Blennow K. Preclinical amyloid pathology biomarker positivity: effects on tau pathology and neurodegeneration. Transl Psychiatry. 2017;7(1):e995. doi: 10.1038/tp.2016.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gispert JD, Monté GC, Suárez-Calvet M, Falcon C, Tucholka A, Rojas S, Rami L, Sánchez-Valle R, Lladó A, Kleinberger G, Haass C, Molinuevo JL. The APOE ε4 genotype modulates CSF YKL-40 levels and their structural brain correlates in the continuum of Alzheimer’s disease but not those of sTREM2. Alzheimer’s Dement Diagn Assess Dis Monit. 2017;6:50–59. doi: 10.1016/j.dadm.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gispert JD, Monté GC, Falcon C, Tucholka A, Rojas S, Sánchez-Valle R, Antonell A, Lladó A, Rami L, Molinuevo JL. CSF YKL-40 and pTau181 are related to different cerebral morphometric patterns in early AD. Neurobiol Aging. 2016;38:47–55. doi: 10.1016/j.neurobiolaging.2015.10.022. [DOI] [PubMed] [Google Scholar]
- 131.Hellwig K, Kvartsberg H, Portelius E, Andreasson U, Oberstein TJ, Lewczuk P, Blennow K, Kornhuber J, Maler JM, Zetterberg H, Spitzer P. Neurogranin and YKL-40: independent markers of synaptic degeneration and neuroinflammation in Alzheimer’s disease. Alzheimer’s Res Ther. 2015;7:74. doi: 10.1186/s13195-015-0161-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kester MI, Teunissen CE, Sutphen C, Herries EM, Ladenson JH, Xiong C, Scheltens P, van der Flier WM, Morris JC, Holtzman DM, Fagan AM. Cerebrospinal fluid VILIP-1 and YKL-40, candidate biomarkers to diagnose, predict and monitor Alzheimer’s disease in a memory clinic cohort. Alzheimer’s Res Ther. 2015;7:59. doi: 10.1186/s13195-015-0142-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Alcolea D, Vilaplana E, Pegueroles J, Montal V, Sánchez-Juan P, González-Suárez A, Pozueta A, Rodríguez-Rodríguez E, Bartrés-Faz D, Vidal-Piñeiro D. Relationship between cortical thickness and cerebrospinal fluid YKL-40 in predementia stages of Alzheimer’s disease. Neurobiol Aging. 2015;36(6):2018–2023. doi: 10.1016/j.neurobiolaging.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 134.Graves DT, Jiang Y. Chemokines, a family of chemotactic cytokines. Crit Rev Oral Biol Med. 1995;6(2):109–118. doi: 10.1177/10454411950060020101. [DOI] [PubMed] [Google Scholar]
- 135.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354(6):610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 136.Kato S, Gondo T, Hoshii Y, Takahashi M, Yamada M, Ishihara T. Confocal observation of senile plaques in Alzheimer’s disease: senile plaque morphology and relationship between senile plaques and astrocytes. Pathol Int. 1998;48(5):332–340. doi: 10.1111/j.1440-1827.1998.tb03915.x. [DOI] [PubMed] [Google Scholar]
- 137.Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat Med. 2003;9:453. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- 138.El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13(4):432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 139.Blasko I, Stampfer-Kountchev M, Robatscher P, Veerhuis R, Eikelenboom P, Grubeck-Loebenstein B. How chronic inflammation can affect the brain and support the development of Alzheimer’s disease in old age: the role of microglia and astrocytes. Aging Cell. 2004;3(4):169–176. doi: 10.1111/j.1474-9728.2004.00101.x. [DOI] [PubMed] [Google Scholar]
- 140.Forloni G, Mangiarotti F, Angeretti N, Lucca E, De Simoni MG. Beta-amyloid fragment potentiates IL-6 and TNF-alpha secretion by LPS in astrocytes but not in microglia. Cytokine. 1997;9(10):759–762. doi: 10.1006/cyto.1997.0232. [DOI] [PubMed] [Google Scholar]
- 141.Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9(4):453–457. doi: 10.1038/nm838. [DOI] [PubMed] [Google Scholar]
- 142.Westin K, Buchhave P, Nielsen H, Minthon L, Janciauskiene S, Hansson O. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer’s disease. PLoS One. 2012;7(1):e30525. doi: 10.1371/journal.pone.0030525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Galimberti D, Schoonenboom N, Scheltens P, et al. Intrathecal chemokine synthesis in mild cognitive impairment and Alzheimer disease. Arch Neurol. 2006;63(4):538–543. doi: 10.1001/archneur.63.4.538. [DOI] [PubMed] [Google Scholar]
- 144.Blasko I, Lederer W, Oberbauer H, Walch T, Kemmler G, Hinterhuber H, Marksteiner J, Humpel C. Measurement of thirteen biological markers in CSF of patients with Alzheimer’s disease and other dementias. Dement Geriatr Cogn Disord. 2006;21(1):9–15. doi: 10.1159/000089137. [DOI] [PubMed] [Google Scholar]
- 145.Janelidze S, Mattsson N, Stomrud E, Lindberg O, Palmqvist S, Zetterberg H, Blennow K, Hansson O. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology. 2018;91(9):e867–e877. doi: 10.1212/wnl.0000000000006082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rosén C, Andersson CH, Andreasson U, Molinuevo JL, Bjerke M, Rami L, Lladó A, Blennow K, Zetterberg H. Increased levels of chitotriosidase and YKL-40 in cerebrospinal fluid from patients with Alzheimer’s disease. Dement Geriatr Cogn Disord Extra. 2014;4(2):297–304. doi: 10.1159/000362164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Correa JD, Starling D, Teixeira AL, Caramelli P, Silva TA. Chemokines in CSF of Alzheimer’s disease patients. Arq Neuropsiquiatr. 2011;69(3):455–459. doi: 10.1590/S0004-282X2011000400009. [DOI] [PubMed] [Google Scholar]
- 148.Choi C, Jeong JH, Jang JS, Choi K, Lee J, Kwon J, Choi KG, Lee JS, Kang SW. Multiplex analysis of cytokines in the serum and cerebrospinal fluid of patients with Alzheimer’s disease by color-coded bead technology. J Clin Neurol (Seoul, Korea) 2008;4(2):84–88. doi: 10.3988/jcn.2008.4.2.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gerendasy DD, Sutcliffe JG. RC3/neurogranin, a postsynaptic calpacitin for setting the response threshold to calcium influxes. Mol Neurobiol. 1997;15(2):131–163. doi: 10.1007/BF02740632. [DOI] [PubMed] [Google Scholar]
- 150.Hayashi Y. Long-term potentiation: two pathways meet at neurogranin. EMBO J. 2009;28(19):2859–2860. doi: 10.1038/emboj.2009.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27(5):457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 152.Bertoni-Freddari C, Fattoretti P, Casoli T, Caselli U, Meier-Ruge W. Deterioration threshold of synaptic morphology in aging and senile dementia of Alzheimer’s type. Anal Quant Cytol Histol (The International Academy of Cytology and American Society of Cytology) 1996;18(3):209–213. [PubMed] [Google Scholar]
- 153.Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68(18):1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- 154.Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R. Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci Lett. 1994;174(1):67–72. doi: 10.1016/0304-3940(94)90121-X. [DOI] [PubMed] [Google Scholar]
- 155.Fyfe I. Alzheimer disease: neurogranin in the CSF signals early Alzheimer disease and predicts disease progression. Nat Rev Neurol. 2015;11(11):609. doi: 10.1038/nrneurol.2015.178. [DOI] [PubMed] [Google Scholar]
- 156.Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr, Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology. 2001;56(1):127–129. doi: 10.1212/WNL.56.1.127. [DOI] [PubMed] [Google Scholar]
- 157.Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W, Jr, Kaye J, Manczak M. Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J Alzheimer’s Dis. 2005;7(2):103–117. doi: 10.3233/JAD-2005-7203. [DOI] [PubMed] [Google Scholar]
- 158.Kubota Y, Putkey JA, Waxham MN. Neurogranin controls the spatiotemporal pattern of postsynaptic Ca2+/CaM signaling. Biophys J. 2007;93(11):3848–3859. doi: 10.1529/biophysj.107.106849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Huang K-P, Huang FL, Jäger T, Li J, Reymann KG, Balschun D. Neurogranin/RC3 enhances long-term potentiation and learning by promoting calcium-mediated signaling. J Neurosci. 2004;24(47):10660–10669. doi: 10.1523/JNEUROSCI.2213-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mons N, Enderlin V, Jaffard R, Higueret P. Selective age-related changes in the PKC-sensitive, calmodulin-binding protein, neurogranin, in the mouse brain. J Neurochem. 2001;79(4):859–867. doi: 10.1046/j.1471-4159.2001.00646.x. [DOI] [PubMed] [Google Scholar]
- 161.Bereczki E, Francis PT, Howlett D, Pereira JB, Hoglund K, Bogstedt A, Cedazo-Minguez A, Baek JH, Hortobagyi T, Attems J, Ballard C, Aarsland D. Synaptic proteins predict cognitive decline in Alzheimer’s disease and Lewy body dementia. Alzheimer’s Dement. 2016;12(11):1149–1158. doi: 10.1016/j.jalz.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 162.Portelius E, Zetterberg H, Skillback T, Tornqvist U, Andreasson U, Trojanowski JQ, Weiner MW, Shaw LM, Mattsson N, Blennow K. Cerebrospinal fluid neurogranin: relation to cognition and neurodegeneration in Alzheimer’s disease. Brain. 2015;138(Pt 11):3373–3385. doi: 10.1093/brain/awv267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kvartsberg H, Duits FH, Ingelsson M, Andreasen N, Ohrfelt A, Andersson K, Brinkmalm G, Lannfelt L, Minthon L, Hansson O, Andreasson U, Teunissen CE, Scheltens P, Van der Flier WM, Zetterberg H, Portelius E, Blennow K. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer’s disease. Alzheimer’s Dement. 2015;11(10):1180–1190. doi: 10.1016/j.jalz.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 164.Kester MI, Teunissen CE, Crimmins DL, et al. Neurogranin as a cerebrospinal fluid biomarker for synaptic loss in symptomatic Alzheimer disease. JAMA Neurol. 2015;72(11):1275–1280. doi: 10.1001/jamaneurol.2015.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tarawneh R, D’Angelo G, Crimmins D, Herries E, Griest T, Fagan AM, Zipfel GJ, Ladenson JH, Morris JC, Holtzman DM. Diagnostic and prognostic utility of the synaptic marker neurogranin in Alzheimer disease. JAMA Neurol. 2016;73(5):561–571. doi: 10.1001/jamaneurol.2016.0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wellington H, Paterson RW, Portelius E, Tornqvist U, Magdalinou N, Fox NC, Blennow K, Schott JM, Zetterberg H. Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurology. 2016;86(9):829–835. doi: 10.1212/wnl.0000000000002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Portelius E, Olsson B, Hoglund K, Cullen NC, Kvartsberg H, Andreasson U, Zetterberg H, Sandelius A, Shaw LM, Lee VMY, Irwin DJ, Grossman M, Weintraub D, Chen-Plotkin A, Wolk DA, McCluskey L, Elman L, McBride J, Toledo JB, Trojanowski JQ, Blennow K. Cerebrospinal fluid neurogranin concentration in neurodegeneration: relation to clinical phenotypes and neuropathology. Acta Neuropathol. 2018;136(3):363–376. doi: 10.1007/s00401-018-1851-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mattsson N, Insel PS, Palmqvist S, Portelius E, Zetterberg H, Weiner M, Blennow K, Hansson O. Cerebrospinal fluid tau, neurogranin, and neurofilament light in Alzheimer’s disease. EMBO Mol Med. 2016;8(10):1184–1196. doi: 10.15252/emmm.201606540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sanfilippo C, Forlenza O, Zetterberg H, Blennow K (2016) Increased neurogranin concentrations in cerebrospinal fluid of Alzheimer’s disease and in mild cognitive impairment due to AD. J Neural Transm (Vienna, Austria: 1996) 123(12):1443–1447. 10.1007/s00702-016-1597-3 [DOI] [PubMed]
- 170.De Vos A, Struyfs H, Jacobs D, Fransen E, Klewansky T, De Roeck E, Robberecht C, Van Broeckhoven C, Duyckaerts C, Engelborghs S, Vanmechelen E. The cerebrospinal fluid neurogranin/BACE1 ratio is a potential correlate of cognitive decline in Alzheimer’s disease. J Alzheimer’s Dis. 2016;53(4):1523–1538. doi: 10.3233/jad-160227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Thorsell A, Bjerke M, Gobom J, Brunhage E, Vanmechelen E, Andreasen N, Hansson O, Minthon L, Zetterberg H, Blennow K. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer’s disease. Brain Res. 2010;1362:13–22. doi: 10.1016/j.brainres.2010.09.073. [DOI] [PubMed] [Google Scholar]
- 172.Bouchon A, Dietrich J, Colonna M (2000) Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol (Baltimore, MD: 1950) 164(10):4991–4995 [DOI] [PubMed]
- 173.Colonna M. TREMs in the immune system and beyond. Nat Rev Immunol. 2003;3(6):445–453. doi: 10.1038/nri1106. [DOI] [PubMed] [Google Scholar]
- 174.Schmid CD, Sautkulis LN, Danielson PE, Cooper J, Hasel KW, Hilbush BS, Sutcliffe JG, Carson MJ. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J Neurochem. 2002;83(6):1309–1320. doi: 10.1046/j.1471-4159.2002.01243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. 2009;109(4):1144–1156. doi: 10.1111/j.1471-4159.2009.06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Takahashi K, Rochford CDP, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201(4):647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL (2006) Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol (Baltimore, MD: 1950) 177(4):2051–2055 [DOI] [PubMed]
- 178.Frank S, Burbach GJ, Bonin M, Walter M, Streit W, Bechmann I, Deller T. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia. 2008;56(13):1438–1447. doi: 10.1002/glia.20710. [DOI] [PubMed] [Google Scholar]
- 179.Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368(2):107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jin SC, Benitez BA, Karch CM, Cooper B, Skorupa T, Carrell D, Norton JB, Hsu S, Harari O, Cai Y, Bertelsen S, Goate AM, Cruchaga C. Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum Mol Genet. 2014;23(21):5838–5846. doi: 10.1093/hmg/ddu277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JSK, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert J-C, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St. George-Hyslop P, Singleton A, Hardy J. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Slattery CF, Beck JA, Harper L, Adamson G, Abdi Z, Uphill J, Campbell T, Druyeh R, Mahoney CJ, Rohrer JD, Kenny J, Lowe J, Leung KK, Barnes J, Clegg SL, Blair M, Nicholas JM, Guerreiro RJ, Rowe JB, Ponto C, Zerr I, Kretzschmar H, Gambetti P, Crutch SJ, Warren JD, Rossor MN, Fox NC, Collinge J, Schott JM, Mead S. R47H TREM2 variant increases risk of typical early-onset Alzheimer’s disease but not of prion or frontotemporal dementia. Alzheimer’s Dement. 2014;10(6):602.e604–608.e604. doi: 10.1016/j.jalz.2014.05.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bertram L. The role of TREM2 R47H as a risk factor for Alzheimer’s disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson’s disease. Alzheimer’s Dement. 2015;1:10. doi: 10.1016/j.jalz.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleo A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sanchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 6(243):243ra286. 10.1126/scitranslmed.3009093 [DOI] [PubMed]
- 185.Colonna M, Wang Y. TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci. 2016;17(4):201–207. doi: 10.1038/nrn.2016.7. [DOI] [PubMed] [Google Scholar]
- 186.Piccio L, Buonsanti C, Cella M, Tassi I, Schmidt RE, Fenoglio C, Rinker J, 2nd, Naismith RT, Panina-Bordignon P, Passini N, Galimberti D, Scarpini E, Colonna M, Cross AH. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain. 2008;131(Pt 11):3081–3091. doi: 10.1093/brain/awn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Piccio L, Deming Y, Del-Aguila JL, Ghezzi L, Holtzman DM, Fagan AM, Fenoglio C, Galimberti D, Borroni B, Cruchaga C. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016;131(6):925–933. doi: 10.1007/s00401-016-1533-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhong L, Chen X-F, Wang T, Wang Z, Liao C, Wang Z, Huang R, Wang D, Li X, Wu L. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med. 2017;214(3):597–607. doi: 10.1084/jem.20160844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160(6):1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Suarez-Calvet M, Araque Caballero MA, Kleinberger G, Bateman RJ, Fagan AM, Morris JC, Levin J, Danek A, Ewers M, Haass C (2016) Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci Transl Med 8(369):369ra178. 10.1126/scitranslmed.aag1767 [DOI] [PMC free article] [PubMed]
- 191.Schindler SE, Holtzman DM. CSF sTREM2: marking the tipping point between preclinical AD and dementia? EMBO Mol Med. 2016;8(5):437–438. doi: 10.15252/emmm.201606245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Gispert JD, Suarez-Calvet M, Monte GC, Tucholka A, Falcon C, Rojas S, Rami L, Sanchez-Valle R, Llado A, Kleinberger G, Haass C, Molinuevo JL. Cerebrospinal fluid sTREM2 levels are associated with gray matter volume increases and reduced diffusivity in early Alzheimer’s disease. Alzheimer’s Dement. 2016;12(12):1259–1272. doi: 10.1016/j.jalz.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 193.Heslegrave A, Heywood W, Paterson R, Magdalinou N, Svensson J, Johansson P, Ohrfelt A, Blennow K, Hardy J, Schott J, Mills K, Zetterberg H. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol Neurodegener. 2016;11:3. doi: 10.1186/s13024-016-0071-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Suarez-Calvet M, Kleinberger G, Araque Caballero MA, Brendel M, Rominger A, Alcolea D, Fortea J, Lleo A, Blesa R, Gispert JD, Sanchez-Valle R, Antonell A, Rami L, Molinuevo JL, Brosseron F, Traschutz A, Heneka MT, Struyfs H, Engelborghs S, Sleegers K, Van Broeckhoven C, Zetterberg H, Nellgard B, Blennow K, Crispin A, Ewers M, Haass C. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol Med. 2016;8(5):466–476. doi: 10.15252/emmm.201506123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Henjum K, Almdahl IS, Arskog V, Minthon L, Hansson O, Fladby T, Nilsson LN. Cerebrospinal fluid soluble TREM2 in aging and Alzheimer’s disease. Alzheimer’s Res Ther. 2016;8(1):17. doi: 10.1186/s13195-016-0182-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yoshimoto K, Tanaka T, Somiya K, Tsuji R, Okamoto F, Kawamura K, Ohkaru Y, Asayama K, Ishii H. Human heart-type cytoplasmic fatty acid-binding protein as an indicator of acute myocardial infarction. Heart Vessels. 1995;10(6):304–309. doi: 10.1007/BF02911388. [DOI] [PubMed] [Google Scholar]
- 197.Janssen CI, Kiliaan AJ. Long-chain polyunsaturated fatty acids (LCPUFA) from genesis to senescence: the influence of LCPUFA on neural development, aging, and neurodegeneration. Prog Lipid Res. 2014;53:1–17. doi: 10.1016/j.plipres.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 198.Veerkamp JH, Zimmerman AW (2001) Fatty acid-binding proteins of nervous tissue. J Mol Neurosci 16(2–3):133–142. 10.1385/jmn:16:2-3:133(discussion 151–137) [DOI] [PubMed]
- 199.Hooijmans CR, Van der Zee CEEM, Dederen PJ, Brouwer KM, Reijmer YD, van Groen T, Broersen LM, Lütjohann D, Heerschap A, Kiliaan AJ. DHA and cholesterol containing diets influence Alzheimer-like pathology, cognition and cerebral vasculature in APPswe/PS1dE9 mice. Neurobiol Dis. 2009;33(3):482–498. doi: 10.1016/j.nbd.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 200.Tan Y, Ren H, Shi Z, Yao X, He C, Kang J-X, Wan J-B, Li P, Yuan T-F, Su H. Endogenous docosahexaenoic acid (DHA) prevents Aβ1–42 oligomer-induced neuronal injury. Mol Neurobiol. 2016;53(5):3146–3153. doi: 10.1007/s12035-015-9224-0. [DOI] [PubMed] [Google Scholar]
- 201.Cheon MS, Kim SH, Fountoulakis M, Lubec G. Heart type fatty acid binding protein (H-FABP) is decreased in brains of patients with Down syndrome and Alzheimer’s disease. J Neural Transm Suppl. 2003;67:225–234. doi: 10.1007/978-3-7091-6721-2_20. [DOI] [PubMed] [Google Scholar]
- 202.Pelsers MMAL, Hermens WT, Glatz JFC. Fatty acid-binding proteins as plasma markers of tissue injury. Clin Chim Acta. 2005;352(1):15–35. doi: 10.1016/j.cccn.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 203.Glatz JFC (1998) Fatty acid-binding protein as a plasma marker for the early detection of myocardial injury. In: Kaski JC, Holt DW (eds) Myocardial damage: early detection by novel biochemical markers. Springer Netherlands, Dordrecht, pp 73–84. 10.1007/978-94-017-2380-0_7
- 204.Shioda N, Yamamoto Y, Watanabe M, Binas B, Owada Y, Fukunaga K. Heart-type fatty acid binding protein regulates dopamine D2 receptor function in mouse brain. J Neurosci. 2010;30(8):3146–3155. doi: 10.1523/JNEUROSCI.4140-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Shioda N, Yabuki Y, Kobayashi Y, Onozato M, Owada Y, Fukunaga K. FABP3 protein promotes α-synuclein oligomerization associated with 1-methyl-1, 2, 3, 6-tetrahydropiridine-induced neurotoxicity. J Biol Chem. 2014;289(27):18957–18965. doi: 10.1074/jbc.M113.527341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Martorana A, Koch G (2014) Is dopamine involved in Alzheimer’s disease? Front Aging Neurosci. 10.3389/fnagi.2014.00252 [DOI] [PMC free article] [PubMed]
- 207.Desikan RS, Thompson WK, Holland D, Hess CP, Brewer JB, Zetterberg H, Blennow K, Andreassen OA, McEvoy LK, Hyman BT, Dale AM. Heart fatty acid binding protein and Abeta-associated Alzheimer’s neurodegeneration. Mol Neurodegener. 2013;8:39. doi: 10.1186/1750-1326-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Guo LH, Alexopoulos P, Perneczky R. Heart-type fatty acid binding protein and vascular endothelial growth factor: cerebrospinal fluid biomarker candidates for Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci. 2013;263(7):553–560. doi: 10.1007/s00406-013-0405-4. [DOI] [PubMed] [Google Scholar]
- 209.Chiasserini D, Parnetti L, Andreasson U, Zetterberg H, Giannandrea D, Calabresi P, Blennow K. CSF levels of heart fatty acid binding protein are altered during early phases of Alzheimer’s disease. J Alzheimer’s Dis. 2010;22(4):1281–1288. doi: 10.3233/jad-2010-101293. [DOI] [PubMed] [Google Scholar]
- 210.Bjerke M, Kern S, Blennow K, Zetterberg H, Waern M, Borjesson-Hanson A, Ostling S, Kern J, Skoog I. Cerebrospinal fluid fatty acid-binding protein 3 is related to dementia development in a population-based sample of older adult women followed for 8 years. J Alzheimer’s Dis. 2016;49(3):733–741. doi: 10.3233/jad-150525. [DOI] [PubMed] [Google Scholar]
- 211.Biscetti L, Eusebi P, Salvadori N, Frattini G, Simoni S, Mollenhauer B, Engelborghs S, Tambasco N, Calabresi P, Parnetti L, Chiasserini D (2016) Combination of cerebrospinal fluid h-fabp and core Alzheimer’s Disease biomarkers improves the differential diagnosis of neurodegenerative disorders. Alzheimer’s Dement 12(7 suppl):P201–P202. 10.1016/j.jalz.2016.06.353 [DOI]
- 212.Chiasserini D, Biscetti L, Eusebi P, Farotti L, Tambasco N, Calabresi P, Parnetti L. CSF levels of heart fatty acid binding protein in Parkinson’s and Alzheimer’s disease. Parkinsonism Relat Disord. 2016;22(Suppl 2):e33. doi: 10.1016/j.parkreldis.2015.10.037. [DOI] [Google Scholar]
- 213.Yuan A, Rao MV, Veeranna Nixon RA. Neurofilaments at a glance. J Cell Sci. 2012;125(14):3257–3263. doi: 10.1242/jcs.104729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Yan Y, Jensen K, Brown A. The polypeptide composition of moving and stationary neurofilaments in cultured sympathetic neurons. Cell Motil Cytoskelet. 2007;64(4):299. doi: 10.1002/cm.20184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wagner OI, Rammensee S, Korde N, Wen Q, Leterrier J-F, Janmey PA. Softness, strength and self-repair in intermediate filament networks. Exp Cell Res. 2007;313(10):2228–2235. doi: 10.1016/j.yexcr.2007.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Fernandez-Martos CM, King AE, Atkinson RAK, Woodhouse A, Vickers JC. Neurofilament light gene deletion exacerbates amyloid, dystrophic neurite, and synaptic pathology in the APP/PS1 transgenic model of Alzheimer’s disease. Neurobiol Aging. 2015;36(10):2757–2767. doi: 10.1016/j.neurobiolaging.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 217.Sjögren M, Blomberg M, Jonsson M, Wahlund LO, Edman Å, Lind K, Rosengren L, Blennow K, Wallin A. Neurofilament protein in cerebrospinal fluid: a marker of white matter changes. J Neurosci Res. 2001;66(3):510–516. doi: 10.1002/jnr.1242. [DOI] [PubMed] [Google Scholar]
- 218.Skillback T, Farahmand B, Bartlett JW, Rosen C, Mattsson N, Nagga K, Kilander L, Religa D, Wimo A, Winblad B, Rosengren L, Schott JM, Blennow K, Eriksdotter M, Zetterberg H. CSF neurofilament light differs in neurodegenerative diseases and predicts severity and survival. Neurology. 2014;83(21):1945–1953. doi: 10.1212/wnl.0000000000001015. [DOI] [PubMed] [Google Scholar]
- 219.Idland AV, Sala-Llonch R, Borza T, Watne LO, Wyller TB, Braekhus A, Zetterberg H, Blennow K, Walhovd KB, Fjell AM. CSF neurofilament light levels predict hippocampal atrophy in cognitively healthy older adults. Neurobiol Aging. 2017;49:138–144. doi: 10.1016/j.neurobiolaging.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 220.Zetterberg H, Skillbäck T, Mattsson N, et al. Association of cerebrospinal fluid neurofilament light concentration with alzheimer disease progression. JAMA Neurol. 2016;73(1):60–67. doi: 10.1001/jamaneurol.2015.3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.De Jong D, Jansen RWMM, Pijnenburg YAL, van Geel WJA, Borm GF, Kremer HPH, Verbeek MM. CSF neurofilament proteins in the differential diagnosis of dementia. J Neurol Neurosurg Psychiatry. 2007;78(9):936–938. doi: 10.1136/jnnp.2006.107326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Scherling CS, Hall T, Berisha F, Klepac K, Karydas A, Coppola G, Kramer JH, Rabinovici G, Ahlijanian M, Miller BL, Seeley W, Grinberg LT, Rosen H, Meredith J, Jr, Boxer AL. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol. 2014;75(1):116–126. doi: 10.1002/ana.24052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Rosengren LE, Karlsson J-E, Sjögren M, Blennow K, Wallin A. Neurofilament protein levels in CSF are increased in dementia. Neurology. 1999;52(5):1090. doi: 10.1212/wnl.52.5.1090. [DOI] [PubMed] [Google Scholar]
- 224.Burgoyne RD, Weiss JL. The neuronal calcium sensor family of Ca2+-binding proteins. Biochem J. 2001;353(1):1–12. doi: 10.1042/bj3530001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Groblewska M, Muszynski P, Wojtulewska-Supron A, Kulczynska-Przybik A, Mroczko B. The role of visinin-like protein-1 in the pathophysiology of Alzheimer’s disease. J Alzheimer’s Dis. 2015;47(1):17–32. doi: 10.3233/jad-150060. [DOI] [PubMed] [Google Scholar]
- 226.Bernstein H-G, Baumann B, Danos P, Diekmann S, Bogerts B, Gundelfinger ED, Braunewell K-H. Regional and cellular distribution of neural visinin-like protein immunoreactivities (VILIP-1 and VILIP-3) in human brain. J Neurocytol. 1999;28(8):655–662. doi: 10.1023/a:1007056731551. [DOI] [PubMed] [Google Scholar]
- 227.Spilker C, Dresbach T, Braunewell KH. Reversible translocation and activity-dependent localization of the calcium-myristoyl switch protein VILIP-1 to different membrane compartments in living hippocampal neurons. J Neurosci. 2002;22(17):7331–7339. doi: 10.1523/JNEUROSCI.22-17-07331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Spilker C, Gundelfinger ED, Braunewell KH. Evidence for different functional properties of the neuronal calcium sensor proteins VILIP-1 and VILIP-3: from subcellular localization to cellular function. Biochim Biophys Acta. 2002;1600(1–2):118–127. doi: 10.1016/S1570-9639(02)00452-1. [DOI] [PubMed] [Google Scholar]
- 229.Marambaud P, Dreses-Werringloer U, Vingtdeux V. Calcium signaling in neurodegeneration. Mol Neurodegener. 2009;4:20. doi: 10.1186/1750-1326-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Arispe N, Pollard HB, Rojas E. Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1-40)] in bilayer membranes. Proc Natl Acad Sci USA. 1993;90(22):10573–10577. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Braunewell KH. The visinin-like proteins VILIP-1 and VILIP-3 in Alzheimer’s disease-old wine in new bottles. Front Mol Neurosci. 2012;5:20. doi: 10.3389/fnmol.2012.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Braunewell K, Riederer P, Spilker C, Gundelfinger ED, Bogerts B, Bernstein HG. Abnormal localization of two neuronal calcium sensor proteins, visinin-like proteins (vilips)-1 and -3, in neocortical brain areas of Alzheimer disease patients. Dement Geriatr Cogn Disord. 2001;12(2):110–116. doi: 10.1159/000051244. [DOI] [PubMed] [Google Scholar]
- 233.Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol. 2001;58(9):1395–1402. doi: 10.1001/archneur.58.9.1395. [DOI] [PubMed] [Google Scholar]
- 234.Gomez-Isla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci. 1996;16(14):4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kirkwood CM, MacDonald ML, Schempf TA, Vatsavayi AV, Ikonomovic MD, Koppel JL, Ding Y, Sun M, Kofler JK, Lopez OL, Yates NA, Sweet RA (2016) Altered levels of Visinin-like protein 1 correspond to regional neuronal loss in Alzheimer disease and frontotemporal lobar degeneration. J Neuropathol Exp Neurol. 10.1093/jnen/nlv018 [DOI] [PMC free article] [PubMed]
- 236.Schnurra I, Bernstein HG, Riederer P, Braunewell KH. The neuronal calcium sensor protein VILIP-1 is associated with amyloid plaques and extracellular tangles in Alzheimer’s disease and promotes cell death and tau phosphorylation in vitro: a link between calcium sensors and Alzheimer’s disease? Neurobiol Dis. 2001;8(5):900–909. doi: 10.1006/nbdi.2001.0432. [DOI] [PubMed] [Google Scholar]
- 237.Lee JM, Blennow K, Andreasen N, Laterza O, Modur V, Olander J, Gao F, Ohlendorf M, Ladenson JH. The brain injury biomarker VLP-1 is increased in the cerebrospinal fluid of Alzheimer disease patients. Clin Chem. 2008;54(10):1617–1623. doi: 10.1373/clinchem.2008.104497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Babic Leko M, Borovecki F, Dejanovic N, Hof PR, Simic G. Predictive value of cerebrospinal fluid Visinin-like protein-1 levels for Alzheimer’s disease early detection and differential diagnosis in patients with mild cognitive impairment. J Alzheimer’s Dis. 2016;50(3):765–778. doi: 10.3233/jad-150705. [DOI] [PubMed] [Google Scholar]
- 239.Luo X, Hou L, Shi H, Zhong X, Zhang Y, Zheng D, Tan Y, Hu G, Mu N, Chan J, Chen X, Fang Y, Wu F, He H, Ning Y. CSF levels of the neuronal injury biomarker visinin-like protein-1 in Alzheimer’s disease and dementia with Lewy bodies. J Neurochem. 2013;127(5):681–690. doi: 10.1111/jnc.12331. [DOI] [PubMed] [Google Scholar]
- 240.Mroczko B, Groblewska M, Zboch M, Muszynski P, Zajkowska A, Borawska R, Szmitkowski M, Kornhuber J, Lewczuk P. Evaluation of visinin-like protein 1 concentrations in the cerebrospinal fluid of patients with mild cognitive impairment as a dynamic biomarker of Alzheimer’s disease. J Alzheimer’s Dis. 2015;43(3):1031–1037. doi: 10.3233/jad-141050. [DOI] [PubMed] [Google Scholar]
- 241.Tarawneh R, D’Angelo G, Macy E, Xiong C, Carter D, Cairns NJ, Fagan AM, Head D, Mintun MA, Ladenson JH, Lee JM, Morris JC, Holtzman DM. Visinin-like protein-1: diagnostic and prognostic biomarker in Alzheimer disease. Ann Neurol. 2011;70(2):274–285. doi: 10.1002/ana.22448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Tarawneh R, Head D, Allison S, Buckles V, Fagan AM, Ladenson JH, Morris JC, Holtzman DM. Cerebrospinal fluid markers of neurodegeneration and rates of brain atrophy in early Alzheimer disease. JAMA Neurol. 2015;72(6):656–665. doi: 10.1001/jamaneurol.2015.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Tarawneh R, Lee JM, Ladenson JH, Morris JC, Holtzman DM. CSF VILIP-1 predicts rates of cognitive decline in early Alzheimer disease. Neurology. 2012;78(10):709–719. doi: 10.1212/WNL.0b013e318248e568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Melah KE, Lu SY, Hoscheidt SM, Alexander AL, Adluru N, Destiche DJ, Carlsson CM, Zetterberg H, Blennow K, Okonkwo OC, Gleason CE, Dowling NM, Bratzke LC, Rowley HA, Sager MA, Asthana S, Johnson SC, Bendlin BB. Cerebrospinal fluid markers of Alzheimer’s disease pathology and microglial activation are associated with altered white matter microstructure in asymptomatic adults at risk for Alzheimer’s disease. J Alzheimer’s Dis. 2016;50(3):873–886. doi: 10.3233/jad-150897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Réus GZ, Fries GR, Stertz L, Badawy M, Passos IC, Barichello T, Kapczinski F, Quevedo J. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience. 2015;300:141–154. doi: 10.1016/j.neuroscience.2015.05.018. [DOI] [PubMed] [Google Scholar]