

Abstract
CK2 denotes a ubiquitous and pleiotropic protein kinase whose holoenzyme is composed of two catalytic (α and/or α′) and two regulatory β subunits. The CK2 consensus sequence, S/T-x-x-D/E/pS/pT is present in numerous phosphosites, but it is not clear how many of these are really generated by CK2. To gain information about this issue, advantage has been taken of C2C12 cells entirely deprived of both CK2 catalytic subunits by the CRISPR/Cas9 methodology. A comparative SILAC phosphoproteomics analysis reveals that, although about 30% of the quantified phosphosites do conform to the CK2 consensus, only one-third of these are substantially reduced in the CK2α/α′(−/−) cells, consistent with their generation by CK2. A parallel study with C2C12 cells deprived of the regulatory β subunit discloses a role of this subunit in determining CK2 targeting. We also find that phosphosites notoriously generated by CK2 are not fully abrogated in CK2α/α′(−/−) cells, while some phosphosites unrelated to CK2 are significantly altered. Collectively taken our data allow to conclude that the phosphoproteome generated by CK2 is not as ample and rigidly pre-determined as it was believed before. They also show that the lack of CK2 promotes phosphoproteomics perturbations attributable to kinases other than CK2.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-017-2705-8) contains supplementary material, which is available to authorized users.
Keywords: Protein phosphorylation, Quantitative proteomics, Sequence logo, Kinase inhibitors, Cell signalling
Introduction
The acronym CK2 (derived from the misnomer “casein kinase-2”) denotes a ubiquitous and highly conserved protein kinase, related to the CMGC branch of the kinome, whose holoenzyme is composed of two catalytic (α and/or α′) and two non-catalytic β subunits. These latter are not sensu stricto regulatory subunits, exerting a strict control over enzymatic activity, as it happens with other protein kinases, but they deeply influence many properties of the holoenzyme, notably stability, interaction with partners and effectors and substrate specificity (reviewed in [1–5]). In other words, the catalytic subunits of CK2 are active either in the absence or in the presence of the β subunits. This observation in conjunction with the notion that its active conformation does not require any previous phosphorylation event supports the concept that CK2 is “constitutively active”.
This property neatly differentiates CK2 from the majority of “onco-kinases” whose pathogenic potential is due to mutations conferring unscheduled activity to enzymes that are otherwise inactive in the absence of specific stimuli. In the case of CK2 gain of function mutations of this kind have never been described; nevertheless, its implication in malignant transformation is soundly documented by both coincidental arguments and experimental evidence. In particular, CK2 is invariably high in tumours and in cancer cells [6], whose survival critically relies on its activity. Considering, moreover, that many of the cell biology phenomena associated with cancer are dependent on CK2 activity, and that CK2 dramatically enhances the transforming potential of oncogenes in transgenic mouse models [7, 8], the conclusion has been reached that quite a variety of tumours are “addicted” to abnormally high CK2 [9], fostering the search and development of cell permeable selective CK2 inhibitors (as reviewed in [10]). One of these, CX-4945, has entered clinical trials for the treatment of different kinds of cancer [11].
A part from its relevance to pathological situations, constitutive activity of CK2 raises the question of its raison d’etre. We know some of the “effects” of it, but what about the “reasons” that justify this property, unusual among signalling molecules like protein kinases? The commonest, albeit rather simplistic answer, is that, due to its “extraordinary” pleiotropy, this kinase is working not only in the presence of an ample variety of stimuli, but also under basal conditions. CK2 pleiotropy is indeed grounded on sound arguments: on one side its “lateral” (and in some cases apparently pleonastic) implication in so many signalling pathways, although, given its constitutive activity, it never plays a crucial hierarchical role in any of them; on the other the countless and ever increasing list of its targets [12]. Now-a-days, there are more than 700 phosphosites generated by CK2 retrieved in the PhosphoSitePlus database [13], but this may be just the top of an iceberg if we trust estimates based on the frequency of the CK2 consensus sequence among the phosphosites of the whole human phosphoproteome, where their proportion approaches 30% [14]. However, predictions merely based on the CK2 consensus sequence or even on the Sequence Logo of bona fide CK2 phosphosites would be misleading since the consensus sequence of CK2 (pS/pT-x-x-E/D/pS/pT) is also found inside numerous phosphosites notoriously generated by protein kinases different from CK2 [15]: these are almost 15,000 altogether and about 3000 of them also conform to the CK2 consensus, according to the PhosphoSitePlus database [13]. Consequently, the mere fulfilment of the CK2 consensus alone is far from being a sufficient criterion for assigning a given phosphosite to CK2 itself. To discriminate between “true” and “false” CK2 phosphosites advantage can be taken of cell permeable CK2 inhibitors, assuming that only the former but not the latter will decrease upon cell treatment with the inhibitor. However, as discussed elsewhere [16, 17], this approach is not satisfactory for a number of reasons, notably the off-target effects of the inhibitors whose selectivity for CK2 is never absolute, and the slow turnover of most CK2 phosphosites, whose occupancy is only marginally decreased in the interval of time spanning between the addition of the inhibitor and cell death induced by the inhibitor itself.
A novel strategy to address the problem has been recently made available by the generation of C2C12 cell clones entirely deprived of CK2 activity by CRISPR/Cas9 methodology [18]. The viability of these clones provided the demonstration that CK2 catalytic activity, albeit essential under special circumstances, e.g. embryonic development [19] and neoplastic growth [9], may be dispensable under normal conditions. CK2α/α′(−/−) cells, in conjunction with those lacking the non-catalytic β subunit, provide a powerful tool for the identification of bona fide CK2 targets if a quantitative phosphoproteomics analysis of these cells is performed in comparison to that of their wild type counterparts. Here, we describe the exploitation of this strategy for the identification of a number of phosphosites unambiguously generated by CK2. Collectively taken our results show that the phosphoproteome generated by CK2 is neither as ample as suspected before nor rigidly pre-determined, while being variably affected by the availability of the non-catalytic β subunit.
Materials and methods
Materials
Protease inhibitor cocktail was from Calbiochem (Darmstadt, Germany), while phosphatase inhibitor cocktails 2 and 3 were from Sigma-Aldrich (Dorset, UK). Anti-CK2α antibody was from Bio-Rad Laboratories (Hercules, CA, USA). Anti-CK2β, anti-phospho-Akt1 S129 and anti-phospho-Cdc37 S13 antibodies were from Abcam (Cambridge, UK). Anti-β-actin antibody was purchased from Sigma-Aldrich (Saint Louis, MO, USA). Anti-Akt1, anti-phospho-NF-kBp65 S529, anti-NF-kBp65, anti-Cdc37 antibodies were from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-phospho-CK2 substrate antibody was from Cell Signalling Technology (Danvers, MA, USA). Secondary antibodies towards rabbit and mouse IgG, conjugated to horseradish peroxidase, were from PerkinElmer (Waltham, MA, USA). Labelled amino acids for SILAC experiments, amino acids-free cell culture medium, and dialyzed foetal bovine serum were purchased from Silantes GmbH (Muchen, Germany). Not labelled amino acids (l-arginine, l-lysine, l-glutamine and l-proline) were purchased from Sigma-Aldrich.
Cell culture and SILAC experiments
C2C12 cells were maintained in 5% CO2 in DMEM supplemented with 10% FBS, 2 mM l-glutamine, 100 U/mL penicillin and 100 mM streptomycin, in an atmosphere containing 5% CO2. CK2 Knockout cells have been described in [18]. For SILAC experiments, cells were grown in DMEM containing either unlabelled l-arginine and l-lysine (Arg0, Lys0) (light) or equimolar amounts of l-[13C6]-arginine and l-[13C6]-lysine (Arg6, Lys6) (medium) or l-[13C6,15N4]-arginine and l-[13C6,15N2]-lysine (Arg10, Lys8) (heavy) supplemented with 2 mM l-glutamine, 1% penicillin/streptomycin, 10% dialyzed Foetal Bovine Serum (Silantes GmbH), and 200 mg/L-proline to prevent the conversion of arginine to proline [20]. Cells were grown in SILAC medium for seven cell doublings. Labelled cells were lysed by the addition of ice-cold buffer containing 20 mM HEPES (pH 8.0), 9 M urea, protease inhibitors EDTA-free (1 tablet/10 mL, Roche) and phosphatases inhibitors Cocktail 2 and 3 (Sigma) and sonicated. Cell debris was removed by centrifugation and SILAC-encoded samples were pooled at a ratio of 1:1:1, as specified in Fig. 1. In particular for the phosphoproteomics studies, we used wild type C2C12 cells and 2 clones (A and B) of CK2α/α′(−/−) cells and 2 clones (A and B) of CK2β(−/−) cells. Characterization of these cellular clones is detailed in [17]. Two biological replicates with a label-swap strategy were obtained for wild type vs CK2α/α′(−/−) cells and for wild type vs CK2β(−/−) cells.
Fig. 1.
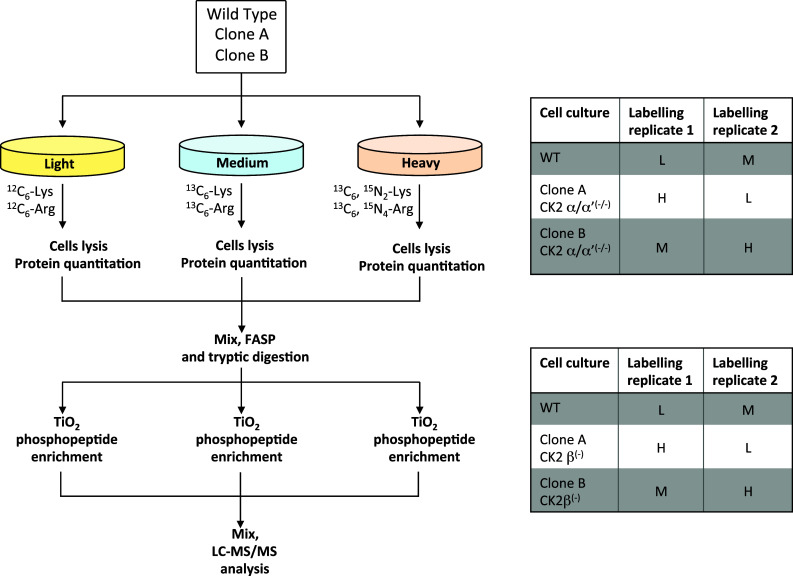
Workflow of the SILAC experiments. The figure summarises the experimental set-up that was used to compare the phosphoproteomes of C2C12 wild type cells with those relative to CK2α/α′(−/−) cells (clone A and clone B) and CK2β(−/−) cells (clone A and clone B). Two biological replicates with a label-swap strategy were performed for each of the triplex SILAC experiments. To reduce the variability associated to the phosphopeptide enrichment step, the TiO2 affinity purification was performed in triplicate for each biological replicate and phosphopeptides were then pooled before LC–MS/MS analysis
Cell lysis and western blotting analysis
Cells were detached, centrifuged, washed with PBS and lysed by the addition of ice-cold buffer containing 20 mM Tris–HCl (pH 8.0), 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 1% Triton X-100 (v/v), protease inhibitor cocktail Complete (Roche) and phosphatase inhibitor Cocktail 2 and 3 (Sigma). After 20 min incubation on ice, the lysate was centrifuged 10 min at 10,000×g at 4 °C. The supernatant was collected and protein concentration was determined by the Bradford method. Equal amounts of total protein extracts were loaded on 12% SDS-PAGE, blotted on Immobilon-P membranes (Millipore), processed by western blot with the indicated antibody, and detected by chemiluminescence on a Kodak Image Station 440MM PRO. Quantitation of the signal was obtained by analysis with the Kodak 1D Image software.
In vitro phosphorylation
Cell lysates were incubated in the radioactive mixture consisting in 50 mM Tris (pH 7.5), 100 μM ATP ([γ-33P]ATP ∼ 2000 cpm/pmol), 10 mM Mg2+, 100 mM NaCl in presence or absence of 500 nM CX-4945 at 37 °C for 10 min. The reaction was stopped with the addition of 2 × Laemmli sample buffer, and samples were subjected to SDS-PAGE. Gels were coomassie stained, dried, exposed overnight to a multipurpose storage phosphor screen and analysed using a cyclone storage phosphor system (Packard).
Protein digestion and phosphopeptide enrichment
Protein lysate from wild type and CK2α/α′(−/−) cells (clone A and clone B) was digested in-solution following the Filtered Aided Sample Preparation (FASP) protocol [21] using Vivacon 500 filters (Sartorius) with a molecular cut-off of 10 kDa. Briefly, 600 μg of SILAC coded proteins (200 μg per condition) were digested using 3 FASP filters (200 μg for each filter), centrifuged at 14,000 RPM for 15 min, washed twice with washing buffer (8 M urea, 100 mM Tris–HCl pH 8.5), and incubated with 50 mM dithiotreitol (Fluka) in washing buffer at 55 °C for 30 min. After centrifugation (14,000 RPM, 15 min), proteins were alkylated with 50 mM iodoacetamide (Sigma) in washing buffer for 20 min at room temperature in the dark. After centrifugation, 2 washing steps were performed with washing buffer, followed by 2 washes with 50 mM NH4HCO3, pH 8.0. Protein digestion was performed overnight at 37 °C using 85 μL of trypsin (47 ng/μL, Promega) in 50 mM NH4HCO3. Peptides were collected by 2 steps of centrifugation (14,000 RPM for 10 min) with 100 μL of 50 mM NH4HCO3. Samples from the 3 FASP filters were acidified by adding 1 μL of pure formic acid (Fluka) and diluted to 1 mL with 0.1% formic acid. Sample cleaning/desalting was performed using SepPak C18 cartridges (Waters), following manufacturer’s instructions. Eluted peptides from the three FASP preparations were finally pooled, dried under vacuum and then suspended in 150 μL of 80% acetonitrile (ACN) 6% trifluoroacetic acid (TFA). The sample was divided into three identical aliquots, each subjected to phosphopeptides enrichment using homemade micro-columns packed into Stage tips (C18 material, 200 μL volume, Proxeon) using 400 μg of TiO2, as already described in [16]. Briefly, samples were slowly loaded onto the micro-columns and washed twice with 50 μL of 50% ACN, 6% TFA, 200 mM NaCl, twice with 50 μL of 80% ACN, 6% TFA and twice with 50 μL of 0.1% TFA. Bound peptides were eluted with 50 μL of 10% NH4OH (pH ≈ 11) and 50 μL of 50% ACN, 0.1% formic acid from each of the three micro-columns, acidified with 5 μL of formic acid, pooled together into a single sample and dried under vacuum. The exact same procedure (FASP, protein digestion, phosphopeptide enrichment) was adopted for the SILAC analysis of wild type C2C12 cells vs CK2β(−/−) cells (clone A and clone B).
LC–MS/MS analysis
Phosphopeptides enriched samples obtained as described above were suspended in 60 μL of 3% ACN, 0.1% formic acid and LC–MS/MS analysis was performed using a LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) coupled with a nano-HPLC Ultimate 3000 (Dionex-Thermo Fisher Scientific). 8 μL of sample was loaded onto a trap column (C18, 300 μm I.D., 10 mm, Protecol, Analytical Technology) at a flow rate of 8 μL/min and peptides were separated by reverse phase chromatography using a homemade column packed into a pico-frit tip (75 μm I.D., 15 μm tip, 11 cm length, New Objective) with C18 material (Aeris peptide 3.6 μm XB-C18, Phenomenex). The separation was achieved with a linear gradient of ACN from 3 to 50% in 90 min at a flow rate of 250 nL/min. Ion source capillary temperature was set at 200 °C, the spray was optimised at 1.3 kV voltage and the instrument operated in data-dependent mode with a top-four acquisition method (a full scan at 60,000 resolution on the Orbitrap followed by the MS2 fragmentation in the linear trap of the 4 most intense ions). To increase the number of phosphopeptide identifications and spectra quality, each sample was analysed three times, using identical chromatographic conditions but different fragmentation methods: MS2, neutral loss triggered MS3, and multistage activation MSA, as described in Salvi et al. [22].
Data analysis
Raw files were analysed with the software Proteome Discoverer 1.4 (Thermo Fisher Scientific) linked to a Mascot search engine (version 2.2.4, Matrix Science) that matched the raw data against the mouse section of Uniprot database (version 2015.04.01; 53301 sequences). Search parameters were as follows: peptide and fragment tolerance were set at 10 ppm and 0.6 Da, respectively; trypsin was set as digesting enzyme with up to one missed-cleavage; carbamidomethyl cysteine was considered as static modification, while methionine oxidation and phosphorylation of threonine, serine and tyrosine residues were considered as variable modifications. The algorithm Percolator and a search against the corresponding randomised database were used to assess the reliability of peptide identifications and calculate the false discovery rate (FDR): only peptides identified with a q value < 0.01 (confidence > 99%) and rank 1 were considered in our analysis. SILAC quantification was performed directly by Proteome Discoverer software and was based on the quantification of only unique peptides, i.e. peptides not shared by more than one protein. The algorithm PhosphoRS [23] was used to help in the assignment of the phosphorylation sites. Data obtained from the three different analyses (MS2, neutral loss triggered MS3, and multistage activation MSA) were merged using a MudPIT protocol to produce a single output file. All identified peptides are reported along with all relevant parameters in Supplementary Table S1 (for wild type vs CK2α/α′(−/−) cells) and Supplementary Table S3 (for wild type vs CK2β(−/−) cells).
Statistical and bioinformatic analysis
The quantification of SILAC triplets was manually reviewed and integrated for those signals that the software could not automatically quantify because of the partial overlapping of labelled peptides. Using informatics tools developed in house based on Bash script and Python code, data were filtered to consider only phosphorylated peptides for which the algorithm PhosphoRS assigned the phosphorylation site(s) with a confidence ≥ 70%. The list of phosphopeptides was re-written (starting from the corresponding protein sequence) in a new format in which seven residues upstream and downstream flanked every phosphosite. All quantifications associated with a specific phosphosite were then averaged and only those phosphosites that were identified and quantified in both biological replicates were considered. Four lists of phosphopeptides were compiled: one relative to the phosphosites quantified in CK2α/α′(−/−) clone A vs wild type cells, one relative to the phosphosites quantified in CK2α/α′(−/−) clone B vs wild type cells, one relative to the phosphosites quantified in CK2β(−/−) clone A vs wild type cells, and finally one relative to the phosphosites quantified in CK2β(−/−) clone B vs wild type cells. For each of these lists, a two-tailed Z test was performed (considering the quantification values obtained from the 2 biological replicates) and standard deviations (SD) and coefficient of variations (CV %) were calculated. A p value ≤ 0.05 combined with a fold change ≥ 1.5 or ≤ −1.5 was used to highlight phosphopeptides which showed a significant change in their abundance in knocked out cells as compared to wild type cells. All other quantified phosphosites were re-evaluated by filtering away those characterised by a CV % ≥ 1.5 the average CV %. The final lists of phosphopeptides with their SILAC ratios and fold change in knocked out vs wild type cells are reported in Supplementary Tables S2 and S4.
Sequence Logo analysis was performed using a stand-alone platform for web logo [24, 25].
Results
Phosphoproteomics of CK2α/α′(−/−) cells
Quantitative phosphoproteomics analyses were performed on C2C12 cells either wild type or genetically deprived of both catalytic subunits of CK2, or lacking the regulatory subunit β of the kinase [18], according to the workflow reported in Fig. 1.
Wild type cells and two different CK2α/α′(−/−) cellular clones (named clone A and clone B) were compared using a SILAC approach. The experiment was performed on two biological replicates and with a label-swap strategy, as reported in Fig. 1 and already described in [17]. The same experimental setup was used to compare the phosphoproteome of wild type cells with that of two different cellular clones lacking the regulatory subunit β of CK2 (named CK2β(−/−) clone A and clone B).
The analysis of CK2α/α′(−/−) vs wild type cells led to the identification of 1930 phosphopeptides (see supplementary Table S1). To increase the robustness of quantification, only those phosphosites that were unambiguously identified (with a PhosphoRS probability score ≥ 70%) and quantified in both biological replicates were considered, leading to the final quantification of 471 non-redundant phosphosites, listed in Table S2, supplementary material (all details regarding the way data were processed are reported in the Methods section). As summarised in Fig. 2, obtained combining the data from the analysis of the single clones A and B (see Table 1) about one-third of these phosphosites (154 altogether) display the motif pS/pT-x-x-E/D/pS/pT, specifically recognised by CK2. Only a minority of these however (about 30%) are substantially decreased (fold change ≤ −1.5) in CK2α/α′(−/−) cells, as expected in the case of bona fide targets of CK2. The majority are not significantly altered, and a few phospho-sites are even substantially increased (fold change ≥ 1.5). See supplementary material (Table S2) for details about the individual sites and the extent of their alterations in α/α′(−/−) vs wild type cells.
Fig. 2.
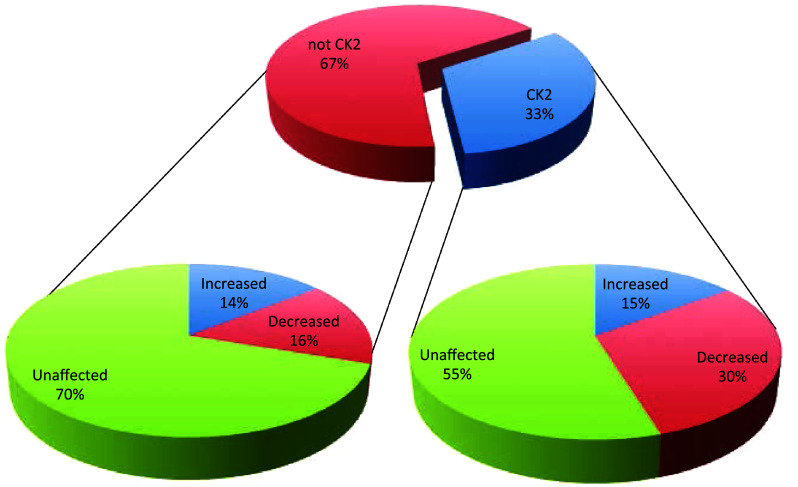
Pie charts summarising the results obtained from the SILAC experiment of wild type vs CK2α/α′(−/−) cells. The central pie chart shows the percentage of phosphosites (reliably quantified in the SILAC experiment of wild type vs CK2α/α′(−/−) cells) that either conform (CK2 consensus) or do not conform (not CK2 consensus) to the canonical consensus sequence of the kinase (pS/pT-X-X-E/D/pSpT). For each of these two categories, the percentage of phosphosites that are either significantly increased or decreased, or that do not show significant changes in their abundance are reported in the lateral pie charts
Table 1.
Synopsis of phosphosites quantified in CK2 knockout cells
| CK2α/α′(−/−) | CK2β(−/−) | Common between CK2α/α′(−/−) and CK2β(−/−) | |||
|---|---|---|---|---|---|
| Clone A | Clone B | Clone A | Clone B | Clone A + Clone B | |
| Non-redundant quantified phosphosites | 446 | 452 | 260 | 257 | 163 |
| Decreased phosphosites | 86 | 86 | 49 | 50 | 22 |
| Increased phosphosites | 62 | 52 | 39 | 39 | 11 |
| Unaffected phosphosites | 308 | 324 | 175 | 172 | 112 |
| Phosphosites with CK2 consensus | 148 | 146 | 59 | 58 | 41 |
| Decreased phosphosites with CK2 consensus | 45 | 48 | 11 | 15 | 9 |
| Increased phosphosites with CK2 consensus | 25 | 15 | 8 | 11 | 4 |
| Unaffected phosphosites with CK2 consensus | 85 | 89 | 42 | 34 | 22 |
| Phosphosites without CK2 consensus | 300 | 308 | 201 | 199 | 122 |
| Decreased phosphosites without CK2 consensus | 41 | 38 | 38 | 35 | 13 |
| Increased phosphosites without CK2 consensus | 38 | 37 | 31 | 28 | 7 |
| Unaffected phosphosites without CK2 consensus | 224 | 235 | 133 | 138 | 90 |
Phosphosites are sorted according to the presence of the CK2 consensus sequence and to their responsiveness to CK2 knockout
A parallel analysis on the quantified phosphosites that do not conform to the CK2 consensus, discloses a situation where the proportion of phosphosites unaffected by the suppression of CK2 catalytic subunits is largely predominant (70%) with a significant number of phosphosites, however, that are either decreased or increased (see Table 1 and Fig. 2).
Considering that our CK2α/α′(−/−) cells are entirely devoid of any trace of CK2 activity [18], our data provide the demonstration that only relatively few of the numerous phosphosites conforming to its consensus are entirely or predominantly generated by CK2 itself. This is not surprising considering that the CK2 consensus is also abundantly present in numerous phosphosites which are generated by all the most pleiotropic protein kinases, despite the fact that their consensuses are different from that of CK2, as illustrated in Fig. 3.
Fig. 3.

CK2 consensus is present in numerous phosphosites generated by the most pleiotropic protein kinases. The graph shows the total number of phosphosites reported in the kinase substrate database of PhosphoSitePlus for each of the most pleiotropic protein kinases. Red bars indicate the number of phosphosites that conform also to the canonical consensus sequence of CK2
The conclusion that the phosphosites conforming to the CK2 consensus decreased in α/α′(−/−) cells do represent bona fide endogenous targets of CK2 in C2C12 cells is also supported by their Sequence Logo (Fig. 4a) which is almost perfectly superimposable to that extracted from bona fide CK2 substrates conforming to the consensus retrieved in the PhosphoSitePlus database (see Fig. 4c). By sharp contrast, the Sequence Logo extracted from CK2 sites not conforming to the canonical consensus retrieved in the PhosphoSitePlus database (Fig. 4d) is sharply different from that extracted from phosphosites not conforming to the CK2 consensus but nevertheless substantially decreased in α/α′(−/−) cells (Fig. 4b): the former in fact is enriched in acidic residues at positions other than n + 3 which instead are lacking in the latter, where the neat predominance of a Pro at position n + 1 is symptomatic of phosphosites generated by Pro-directed protein kinases rather than by CK2. It can be concluded therefore that these phosphosites are not generated by CK2 despite the fact that they are down-regulated in CK2α/α′(−/−) cells.
Fig. 4.

Sequence logo analysis. Sequence Logos obtained from all phosphopeptides either conforming (panel A) or not conforming (panel B) to the CK2 consensus sequence and characterised by a significantly reduced phosphorylation level in CK2α/α′(−/−) with respect to wild type C2C12 cells. Panel C and D display the Sequence Logos obtained from all phosphosites attributed to CK2 in the PhosphoSitePlus database and that either conform (panel C) or do not conform (panel D) to the canonical consensus sequence of CK2. Sequence Logos obtained from all phosphopeptides either conforming (panel E) or not conforming (panel F) to the CK2 consensus sequence and characterised by a significantly reduced phosphorylation level in CK2β(−/−) with respect to wild type C2C12 cells
A plausible mechanism by which phosphosites not directly generated by CK2 are altered in CK2α/α′(−/−) cells would be that in these cells a number of protein kinases other than CK2 are up- and down-regulated. This possibility is documented by our previous proteomics analysis showing that, among the 25 protein kinases quantified in that study, a few were substantially increased or decreased in the CK2α/α′(−/−) cells as compared to wild type [18]. These can explain why a number of phosphosites not conforming to the CK2 consensus are nevertheless down-regulated in CK2α/α′(−/−) cells. On the other hand, if we take into account that many phosphosites generated by all the most pleiotropic kinases include the CK2 consensus as well (see Fig. 3), it appears quite possible that protein kinases whose expression is increased in CK2 null cells are responsible for the paradoxical increased occupancy of a number of phosphosites conforming to the CK2 consensus in CK2α/α′(−/−) cells. Another question arising from our data is why the phosphorylation of CK2 sites in cells where CK2 is lacking is not entirely suppressed, but appears to be only reduced to variable extents. A residual phosphorylation would be expectable if a kinase is pharmacologically inhibited, considering, on one side that inhibition is never absolute, and on the other that, before the kinase started being inhibited, all its phosphosites were loaded with phosphate and their dephosphorylation may be a slow process hardly reaching completion [16]. But in the case of clones, like ours, where a given kinase never existed before, one has to assume that residual phosphorylation of bona fide targets of the missing kinase is performed by other enzymes. Such an assumption has been adopted and discussed to explain the residual phosphorylation of secreted proteins in cells where the kinase committed to their phosphorylation, the Golgi “casein kinase” Fam20C was disrupted by the CRISPR/Cas9 technology [26].
To assess if a similar explanation applies to our C2C12 cells devoid of CK2 catalytic subunits, three well-established CK2 targets widely exploited as reporters of endogenous CK2 activity have been compared for their phosphorylation in CK2α/α′(−/−) vs wild type cells. These proteins were not identified in our proteomic analysis, probably due to their low abundance. Nevertheless, the results shown in Fig. 5 provide the clear-cut evidence that all these sites still undergo significant phosphorylation, albeit to variable extent, also in CK2α/α′(−/−) cells. Such a “residual” phosphorylation is just detectable in the case of Akt Ser-129 and NF-kBp65 Ser529 and more evident in Cdc37 Ser-13. This means that in C2C12 cells deprived of CK2 catalytic subunits, the phosphorylation of these proteins is, at least partially, ensured by other kinase(s). The contribution of these kinases to the phosphorylation of the same sites in wild type cells remains a matter of conjecture.
Fig. 5.

Western blot analysis of CK2-substrates in wild type and CK2-KO cells. a, b Cellular lysates of wild type (WT), two different CK2α/α′(−/−) clones (A and B), and two different CK2β(−/−) clones (A and B) were analysed by western blot with the antibodies that specifically recognise phosphosites generated by CK2, as indicated. β-actin was used as loading control. Figures are representative of three separate experiments
Phosphoproteomics of CK2β(−/−) cells
A vexed question concerning CK2 is whether in living organisms its catalytic subunits may work in the absence of the non-catalytic β subunit with which they tend to associate and form a very stable heterotetrameric holoenzyme. This is clearly possible in vitro, where the isolated catalytic subunits are active per se also in the absence of β, whose association with the catalytic subunits, however, has functional consequences regarding the stability, specificity and regulation of CK2 itself [27]. Despite several reports supporting the concept that the CK2 holoenzyme and its isolated catalytic subunits may play distinct roles also in the living cell, the actual occurrence of this kind of regulatory mechanism is still a matter of debate [27]. Pertinent to this is also the “symmetrical” question as to whether the β subunit might play roles which are independent of CK2, as it has been hypothesised sometimes [1].
In this respect, it should be mentioned that β has been reported to interact with and affect the activity of a number of kinases, notably A-Raf [28], c-Mos [29], Chk1 [30], and PAK [31]. The β subunit is synthesised in large excess over the catalytic subunits of CK2, undergoing a subsequent proteasomal degradation affecting only the free subunits which are not assembled with the catalytic ones [32, 33], nor presumably, with other protein partners. Consistent with this scenario, we have recently shown that in CK2α/α′(−/−) cells, the β subunit also tends to disappear not because its synthesis is reduced but because its degradation is increased [18]. It is also possible, however, that during their short lifespan, free β subunits are committed to some functions not related to CK2 itself. Intriguingly, we have observed that CK2α null cells express a higher level of CK2β, although they undergo some of the proteomics alterations observed in the CK2α/α′(−/−) cells [18].
Given this background we reasoned that CK2β(−/−) cells, where the overall catalytic activity of CK2, far from being decreased, is actually increased due to higher level of CK2α, can provide a good model to shed light on the functions of the isolated catalytic subunits and gain information about CK2 targets whose phosphorylation is variably affected by the β subunit.
To this purpose, a SILAC quantitative phosphoproteomics analysis of C2C12 cells deprived of CK2β has been performed whose outcome has been compared to that presented above for the CK2α/α′(−/−) cells.
In the case of C2C12 CK2β(−/−) cells, our analysis led to the identification of 1236 phosphopeptides (see Supplementary Table S3). Again, to increase the robustness of quantification, only the phosphosites that were unambiguously identified (with a PhosphoRS probability score ≥ 70%) and quantified in both biological replicates were considered, resulting in the final quantification of 273 non-redundant phosphosites (for details about the individual sites and the extent of their alterations in CK2β(−/−) vs wild type cells see supplementary material, Table S4). As shown in Fig. 6, obtained combining the data from the analysis of the single clones A and B as they are on display in Table 1, 23% of these sites conform to the consensus of CK2 with on average 22% of them undergoing a substantial decrease in the CK2β(−/−) cells. Both these figures are significantly lower than the corresponding ones in the CK2α/α′(−/−) cells (33 and 30%, respectively, see Table 1; compare Figs. 2, 6), consistent with the concept that the lack of the β subunit has a smaller impact on the phosphoproteome generated by CK2 than the lack of the catalytic subunits. To note, however, that also the Sequence Logo of the phosphosites conforming to the CK2 consensus and substantially reduced in the β null cells (Fig. 4e) is largely superimposable to that of bona fide CK2 phosphosites conforming to the CK2 consensus retrieved in the PhosphoSitePlus database (Fig. 4c), strengthening the conclusion that this subset of phosphosites, similar to those quantified in the CK2α/α′(−/−) cells, is truly generated by CK2. This does not apply to those sites whose phosphorylation is substantially reduced in β null cells but that do not conform to the CK2 consensus, whose Sequence Logo (Fig. 4f), similar to that of the analogous phosphosites of CK2α/α′(−/−) cells (Fig. 4b) has nothing to share with the Sequence Logo of bona fide CK2 phosphosites not conforming to the CK2 consensus, retrieved in the PhosphoSitePlus database (Fig. 4d). To sum up, the data of Fig. 6 are consistent with observations already available in the literature. In particular: (1) the proportion of reduced phosphosites conforming to the CK2 consensus smaller than that found in CK2α/α′(−/−) cells is expectable considering that only those sites critically dependent on β (class III, see below) are affected; (2) the finding that a number of phosphosites conforming to the CK2 consensus are actually increased can be explained considering that in CK2β(−/−) cells the overall catalytic activity is up-regulated [18], thus enhancing the targeting of those sites whose phosphorylation is either unaffected or even hampered by the β subunit (class I and II, respectively, see below); (3) also the remarkable proportion of phosphosites not conforming to the CK2 consensus which are nevertheless decreased (22%) may not come as a surprise if earlier reports are taken into account showing that CK2 β may activate other kinases besides CK2 (see above).
Fig. 6.
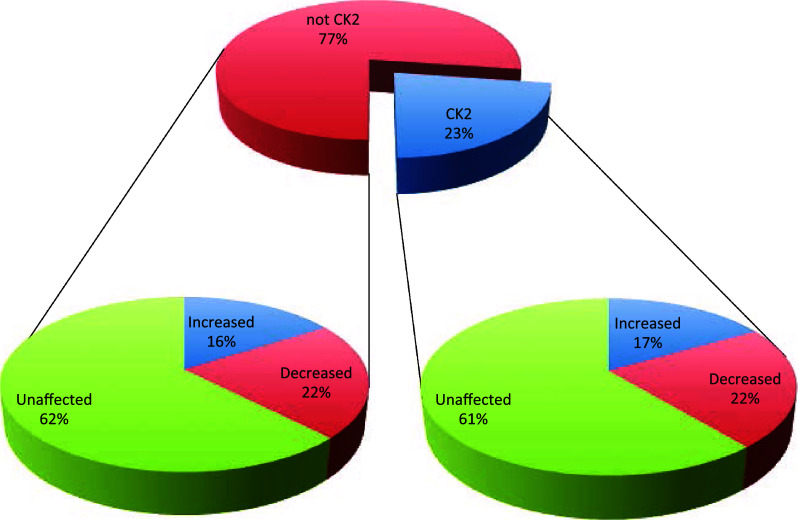
Pie charts summarising the results obtained from the SILAC experiment of wild type vs CK2β(−/−) cells. The central graph shows the percentage of phosphosites (reliably quantified in the SILAC experiment of wild type vs CK2β(−/−) cells) that either conform (CK2 consensus) or do not conform (not CK2 consensus) to the canonical consensus sequence of the kinase. For each of these two categories, the percentage of phosphosites that are either significantly increased or decreased, or that do not show significant changes in their abundance are reported in the lateral pie charts
Looking at the lists of the 471 and 273 non-redundant phosphosites confidently quantified in CK2α/α′(−/−) and in CK2β(−/−) cells (Tables S2, S4), respectively, it turns out that 163 of these are present in both. A comparative analysis of these common phosphosites provides useful information about functional correlations between the catalytic and the regulatory subunits of CK2 as well as about the possibility that the β subunit may play distinct role(s) independent of CK2 itself. In particular the scrutiny of common phosphosites which conform to the CK2 consensus and are substantially decreased in the CK2α/α′(−/−) cells will disclose endogenous protein substrates falling in the conventional classes I, III and II, respectively, as defined by in vitro experimentation with recombinant subunits of CK2 [2]. Class I includes substrates (exemplified by inhibitor-2 of protein phosphatase 1) whose phosphorylation is not substantially affected by the β subunit, while substrates of class II, typically represented by calmodulin, and of class III (exemplified by Rev) are those whose phosphorylation is either inhibited by or critically dependent on the β subunit, respectively (see ref [2] for additional details). As indicated in Table 2, where the 57 phosphosites conforming to the CK2 consensus which are substantially decreased in the CK2α/α′(−/−) cells are listed, 15 of these were also quantified in CK2β(−/−) cells (highlighted in bold in Table 2) nine of these being also reduced in CK2β(−/−) cells, while the others are not decreased, consistent with their targeting by the isolated catalytic subunits. These features reflect the behaviours expected for class III and I CK2 substrates, respectively [2]. Given the small number of these phosphosites it is impossible to extract reliable Sequence Logos and disclose local features eventually accounting for their different dependence on the β subunit. It should be mentioned in this connection that in the case of calmodulin, i.e. the typical representative of class II substrates, and of Rev, a representative of class III, their responsiveness to β is mostly lost if instead of the full length proteins, short peptides reproducing their phosphoacceptor sites are tested as CK2 substrates [34, 35] suggesting that the effects of β are mediated by docking sites remote from the phosphoacceptor residues. This is not the case of another class III substrate, however, eIF2β, whose dependence on the CK2β subunit for the phosphorylation of Ser-2 is observed with both the full size protein and with a peptide encompassing its N-terminal segment [36]. Clearly, additional work needs to be done to shed light on the structural features rendering the phosphorylation of CK2 substrates variably responsive to the β subunit.
Table 2.
Summary of quantified phosphosites attributable to CK2 based on their consensus sequence and decreased occupancy in CK2α/α′(−/−) cells
| Sequence | p-site | Accession | Gene name | Fold change CK2α/α′(−/−) vs WT | CK2β(−/−) vs WT | CK2 protein substrate | CK2 p-site |
|---|---|---|---|---|---|---|---|
| APKEELAsDLEEMAT | 229 | A2APD7 | Nop56 | − 1.56 | D | [41] | – |
| SPKEEVAsEPEEAAS | 252 | A2APD7 | Nop56 | − 1.74 | D | [41] | – |
| TKQSNASsDVEVEEK | 106 | A2BI12 | Psip1 | − 6.76 | – | – | [37] |
| AADEDWDsELEDDLL | 41 | D3YV48 | Rbm33 | − 1.73 | – | – | [37] |
| ELEKLQLsDEESVFE | 531 | D3Z069 | Phldb2 | − 15.15 | D | – | – |
| KGSAEGssDEEGKLV | 101 | E0CXA0 | Hdgf | − 41.67 | – | [14] | [14] |
| KKGSAEGssDEEGKL | 100 | E0CXA0 | Hdgf | − 41.67 | – | [14] | [14] |
| AKEEsEEsDEDMGFG | 307 | E9Q070 | Gm8730 | − 2.79 | – | [41, 42] | [41] |
| KAEAKEEsEEsDEDM | 304 | E9Q070 | Gm8730 | − 2.79 | – | [42] | – |
| KKEEsEEsEDDMGFG | 104 | E9Q3T0 | Gm10073 | − 5.35 | D | – | – |
| VEAKKEEsEEsEDDM | 101 | E9Q3T0 | Gm10073 | − 5.35 | D | – | – |
| YHLPDAEsDEDEDFK | 178 | E9Q3V6 | Sept2 | − 2.74 | U | [43] | [43] |
| DLKSSKAsLGsLEGE | 5522 | E9Q616 | Ahnak | − 2.00 | – | [37] | – |
| GHYEVTGsDDEAGKL | 5607 | E9Q616 | Ahnak | − 6.10 | – | [37] | [37] |
| RSSEVVLsGDDEDYQ | 116 | E9Q616 | Ahnak | − 10.42 | D | [37] | – |
| DLDLEPLsDLEEGLE | 1111 | E9Q8F1 | Kdm5a | − 1.78 | – | – | – |
| QEESLEDsDVDADFK | 60 | O35344 | Kpna3 | − 4.17 | – | [37] | [37] |
| VVEEHCAsPEEKTLE | 1307 | P14873 | Map1b | − 2.18 | – | [44] | – |
| KAVLMIRtPEELDDS | 634 | P26231 | Ctnna1 | − 1.58 | – | [45] | [45] |
| TPEELDDsDFETEDF | 641 | P26231 | Ctnna1 | − 1.60 | D | [45] | [45] |
| EEDGVTGsQDEEDSK | 563 | P35564 | Canx | − 6.33 | – | [46, 47] | [46] |
| KLEEKQKsDAEEDGV | 553 | P35564 | Canx | − 90.91 | – | [46, 47] | [46] |
| REEQTDTsDGESVTH | 51 | P36916 | Gnl1 | − 6.99 | – | [37] | [37] |
| RRGLLYDssEEDEER | 139 | P97310 | Mcm2 | − 5.24 | – | [37] | [37] |
| RRGLLYDsSEEDEER | 139 | P97310 | Mcm2 | − 1.56 | U | [37] | [37] |
| VTEPQEEsEEEVEEP | 149 | P97855 | G3bp1 | − 1.62 | – | [48] | – |
| KDEKKEEsEEsDDDM | 102 | P99027 | Rplp2 | − 2.42 | U | [49, 50] | [50] |
| KKEEsEEsDDDMGFG | 105 | P99027 | Rplp2 | − 2.42 | U | [49, 50] | [50] |
| PNSEAPLsGSEDADD | 137 | Q05D44 | Eif5b | − 1.98 | – | [37] | – |
| TSTKRPKsIDDSEME* | 271 | Q3TYK3 | Nfix | − 2.65 | – | – | – |
| RVLLAADsEEEGDFP | 168 | Q5PSV9 | Mdc1 | − 3.64 | – | [37] | [37] |
| GSVSEDNsEDEISNL | 29 | Q5SWU9 | Acaca | − 3.11 | D | [51] | – |
| DDSAKFDsNEEDTAS | 1453 | Q64511 | Top2b | − 4.29 | – | [37, 52] | – |
| KIVETINsDsDSEFG | 1509 | Q64511 | Top2b | − 3.62 | – | [37, 52] | – |
| KTSFDQDsDVDIFPS | 1568 | Q64511 | Top2b | − 3.00 | – | [37, 52] | – |
| VETINsDsDSEFGIP | 1511 | Q64511 | Top2b | − 3.62 | – | [37, 52] | [37] |
| ESQESLKsPEEEDQR | 819 | Q6P5H2 | Nes | − 1.79 | – | – | – |
| QHQESLRsLGEVEWE | 1216 | Q6P5H2 | Nes | − 1.50 | – | – | – |
| WEEGREDsEADELGE | 1541 | Q6P5H2 | Nes | − 1.93 | – | – | – |
| GEEPSEYtDEEDTKD | 205 | Q80UU9 | Pgrmc2 | − 2.35 | – | [53] | [53] |
| QRLVSPGsANETSSI | 2481 | Q80X90 | Flnb | − 1.51 | – | – | – |
| IEDGEEGsEDDAEWV | 438 | Q8BH64 | Ehd2 | − 4.48 | – | – | – |
| RHLEKVAsEEEEVPL | 19 | Q8CC35-3 | Synpo | − 3.18 | D | – | – |
| AKQKFHDsEGDDTEE | 393 | Q8K019-3 | Bclaf1 | − 1.65 | – | [37] | [37] |
| QEVLDYFsDKESAKQ | 381 | Q8K019-3 | Bclaf1 | − 1.70 | – | [37] | [37] |
| QFHPRSSsLGDLLRE | 395 | Q8K124 | Plekho2 | − 1.50 | U | – | – |
| FDRWLDEsDAEMELR | 114 | Q8K2C9 | Hacd3 | − 1.78 | – | – | – |
| SGREKPDsDDDLDIE | 660 | Q8R4U7 | Luzp1 | − 4.46 | – | – | – |
| RKGTGDCsDEEVDGK | 1943 | Q8VDD5 | Myh9 | − 2.46 | U | [37, 54] | [37, 54] |
| QQFDDGGsDEEDIWE | 571 | Q922D4-2 | Ppp6r3 | − 2.45 | – | [37] | [37] |
| VEDDIDLsDVELDDL | 428 | Q922R8 | Pdia6 | − 7.35 | – | [37] | [37] |
| RVAEPEEsEAEEPAA | 79 | Q9D1T2 | Mxra7 | − 5.32 | – | – | – |
| SRTARIAsDEEIQGT | 173 | Q9ET54-3 | Palld | − 1.81 | – | [37] | [37] |
| EIITEEPsEEEADMP | 118 | Q9JIK5 | Ddx21 | − 2.27 | – | [37] | [37] |
| GHRELALssPEDLTQ | 526 | Q9QYR6 | Map1a | − 2.45 | – | [44] | – |
| HRELALssPEDLTQD | 527 | Q9QYR6 | Map1a | − 2.45 | – | [44] | – |
| VEMKNEKsEEEQSSA | 228 | Q9Z204-2 | Hnrnpc | − 6.13 | – | [37] | [37] |
Table lists all phosphosites conforming to the CK2 consensus whose phosphorylation was significantly decreased in CK2α/α′(−/−) cells. SILAC fold changes are reported for each phosphorylation site. Moreover, if the protein or the same residue is already reported in literature as a CK2 target the reference is shown. Highlighted in bold phosphosites that were confidently quantified also in CK2β(−/−) cells and whose occupancy in these cells was found to be significantly decreased (D) or unchanged (U)
*Phosphosite NOT present in PhosphositePlus database
By repeating on the lysates of β null cells, the same WB quantification of bona fide CK2 phosphosites already performed on the lysates of CK2α/α′(−/−) cells, it turns out that the responsiveness of these sites to the absence of the β subunit closely parallels that observed in the CK2α/α′(−/−) cells (Fig. 5b), supporting the conclusion that in living cells the targeting of these sites by CK2 is performed by the holoenzyme, being dependent on the β subunit.
The variable dependence on the β subunit for the phosphorylation of CK2 substrates has been corroborated by two different experiments. On one side, advantage has been taken of commercially available antibodies developed to recognise phosphosites generated by CK2 (Fig. 7a): in general, all protein bands recognised by these antibodies in C2C12 cell lysates (with just one possible exception) display a reduced (in some cases almost absent) signal in CK2α/α′(−/−) cells, nicely confirming that CK2 indeed contributes to the generation of phosphosites recognised by these antibodies. The intensity variation of these bands in the lysates of CK2β(−/−) cells only partially parallels that observed in the CK2α/α′(−/−) ones: as highlighted in the figure in fact some bands are weakened to the same extent, but some other are not, confirming that the β subunit can be either dispensable or required for the targeting of CK2 substrates. It should be noted that also by this approach knocking out CK2 does not entirely nullify, but just variably reduces the recognition by CK2 phosphosite specific antibodies, consistent with phosphoproteomics analysis, thus reinforcing the concept that alternative kinases can be implicated in their generation.
Fig. 7.

Comparison of global CK2 substrate phosphorylation in CK2α/α′ and in CK2β knockout clones. a Cellular lysates of wild type (WT), CK2α/α′(−/−) and CK2β(−/−) were analysed by western blot with a specific antiphospho-CK2 substrate antibody, as described in the experimental section. β-actin was used as loading control. CK2 substrates that decrease their phosphorylation both in CK2α/α′(−/−) and CK2β(−/−) are denoted by two asterisks, while substrates that display a decreased phosphorylation level only in CK2α/α′(−/−) are denoted by a single asterisk. b In vitro phosphorylation of cell lysates. 15 μg of cell lysates were incubated in the presence of [γ-33P]ATP as described in the Methods section. Where indicated, 500 nM of CX-4945 (CX) was added to the phosphorylation reaction. After 10 min at 37 °C, proteins were separated by electrophoresis on SDS-PAGE, coomassie stained and analysed by PhosphorImager (l.e. means long exposition). Radioactive bands whose intensity decreases in CK2α/α′(−/−) are denoted by an asterisk. Figures are representative of at least four independent experiments
By a different approach, a number of protein bands whose phosphorylation is variably influenced by the β subunit have been detected by incubating with 33P-ATP the lysates of C2C12 cells, either WT, or lacking the CK2 catalytic or the regulatory subunits. As shown in Fig. 7b where the autoradiograms of the SDS-PAGE are on display, many bands either vanish or become fainter in the CK2α/α′(−/−) lysates, and are also reduced by CX-4945, symptomatic of their generation by CK2. Not all of these, however, are also fainter in the CK2β(−/−) lysate, the intensity of few of them being almost unaffected by the lack of CK2β, denoting that their phosphorylation does not require the CK2 holoenzyme, being catalysed equally well by the isolated catalytic subunits.
Discussion
Collectively taken our data show that neither conformation to the CK2 consensus (often embedded within sites generated by other kinases), nor decreased occupancy in CK2α/α′(−/−) cells (sometimes attributable to altered expression of other kinases) alone are sufficient criteria for assigning a given phosphosite to CK2. The concomitant occurrence of both criteria, however, led to the identification of a subset of 57 phosphosites—listed in Table 2—whose generation by CK2 is hardly disputable, also considering that their Sequence Logo is almost perfectly superimposable to that of analogous sites already listed as bona fide CK2 targets in the PhosphoSitePlus database (see Fig. 4). Intriguingly, none of these phosphosites are mentioned among those generated by CK2 according to PhosphoSitePlus, although all of them, with the only exception of NFix pS271, were already retrieved in the PhosphoSitePlus database. It should be noted, however, that the relatedness of many of these phosphosites to CK2 is supported by data already available in the literature, as referenced in Table 2: in some instances, the protein has been reported to be a target of CK2, although the direct implication of CK2 in the phosphorylation of that individual residue is still a matter of conjecture. Sometimes evidence is available that a given residue can be phosphorylated in vitro by CK2.
In this respect, it will be interesting to compare our present list of pS/pT-x-x-E/D/pS/pT phosphosites substantially decreased in CK2α/α′(−/−) C2C12 cells with repertoires of phosphosites attributed to CK2 in other studies based on different criteria. In particular, a repertoire of 988 CK2 phosphosites was compiled by Bian et al. [37] based on an in vitro approach where dephosphorylated proteins were phosphorylated by recombinant CK2 and the phosphosites generated were identified by mass spectrometry. 24 of these were also quantified in our study and 18 of them (75%) are among those listed in Table 2, whose occupancy is drastically decreased in our CK2 null cells, while the remaining are not, consistent with the view that the phosphorylation of these latter can be performed by kinases other than CK2, at least in C2C12 cells.
On the other hand, a reliable comparison with phosphosites significantly altered by short treatment of HEK-293T cells with the CK2 inhibitor quinalizarin [16] is not possible because, in addition to the different kinds of cells used, almost none of the phosphosites quantified in that study were also found in the present analysis. It should be stressed anyway that, as discussed elsewhere [18], the rationale of the two approaches is entirely different: the inhibitor in fact impinges on phosphosites that are already occupied and whose dephosphorylation after CK2 blockade may take a long time, especially in the case of slowly turning over CK2 sites; by contrast in the present study, any CK2 catalysed phosphorylation is fully prevented from the very beginning, due to the total absence of the kinase itself.
A legitimate question rising from our analysis is whether occupancy of a phosphosite in CK2α/α′(−/−) C2C12 cells comparable to that found in wild type control is or is not a sufficient criterion to rule out the implication of CK2 in its phosphorylation. Considering that conditions may deeply vary among different cell lines and depending on their metabolic status and that knocking out of CK2 catalytic subunits is causative of proteomics rearrangements [18] reflecting in alterations of signalling pathways, one cannot exclude that a phosphosite conforming to the CK2 consensus whose occupancy is not decreased in the CK2α/α′(−/−) C2C12 cells is nevertheless generated, at least in part, by CK2 in the wild type cells or, even more so in different cell lines. An incontrovertible conclusion that can be drawn, however, is that CK2 is dispensable for the generation of phosphosites behaving in this manner. Whether under different conditions CK2 may contribute to their phosphorylation remains a matter of conjecture, but it is undeniable that in the absence of CK2 their phosphorylation can be entirely accomplished by other kinase(s).
Pertinent to this is the observation that in the total absence of CK2 catalytic activity a residual phosphorylation of bona fide CK2 sites, notably of Cdc37 S13, is still going on. This may reflect compensatory mechanisms that ensure the occurrence of critical signalling steps whenever CK2, primarily committed to this function, is lacking.
On the other hand, the concept that an individual site may be targeted by more than one kinase should not come as a surprise in the light of data in the literature showing the contribution of two or more kinases to the generation of a given phosphosite. A paradigmatic example is provided by p53, harbouring numerous phosphosites some of which are targeted by up to 5 distinct protein kinases [38]. In particular one of these, S352, is susceptible to phosphorylation by CK2 as well as by two other protein kinases, PKR and p38. In a similar manner, α-synuclein S129, originally identified as a CK2 and CK1 target [39], is now recognised as a phosphosite potentially generated by a plethora of kinases such as GRKs (GRK1-3, 5, 6), Lrrk2 and Plks (Plk1-3) [40]. Moreover, considering all the 733 phosphosites attributed to CK2 in PhosphositePlus database, it turns out that 109 of these are also present in the database of phosphosites generated by other kinases.
15 phosphosites, among those listed in Table 2 have been also quantified in CK2β(−/−) cells where only the non-catalytic subunit of CK2 is lacking. Of these, 9 show a significantly decreased phosphorylation level in these cells (fold change ≤ −1.5), similar to what observed in CK2α/α′(−/−) cells, while the others are not significantly affected, consistent with the presence in C2C12 cells of subsets of CK2 substrates for the phosphorylation of which the β subunit is variably required. Such a conclusion has been also corroborated by alternative approaches where protein bands phosphorylated by CK2 were identified by two different criteria in lysates of C2C12 cells, either wild type or lacking both catalytic subunits or just the regulatory β subunit (see Fig. 7).
A limit of this study is that we cannot rule out the possibility that alterations attributed to variable occupancy of a given phosphosite might in some cases rather reflect changes in the concentration of the protein substrate. Attempts to normalise our data exploiting a quantitative proteomics analysis previously performed on the same cells [18] would be biased by the fact that the two sets of data have been generated in different experiments, not to say about the only modest overlapping of phosphosites quantified here with proteins quantified in that study. What we can say, however, is that by applying such a normalisation to phosphosites conforming to the CK2 consensus whenever possible (77 sites out of 154), the information provided by Fig. 2 was substantially confirmed with a proportion of phosphosites conforming to the CK2 consensus unaffected in CK2α/α′(−/−) almost identical either before or after normalisation (55 vs 53%).
In summary, our study has added 57 new phosphosites to the growing list of CK2 endogenous targets, providing at the same time the demonstration that many phosphosites conforming to the CK2 consensus are unaffected by knocking out CK2, which instead influences the phosphoproteomes generated by other kinases.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1. Table lists all (phospho)peptides identified in the triplex SILAC experiment (both biological replicates) of wild type vs CK2α/α’(-/-) cells. Peptide sequences, confidence level, protein group accession number, modifications, PhosphoRS site probabilities, SILAC ratios, q-values, Mascot scores, expectation values, number of missed cleavages, experimental m/z, Δppm are reported (XLSX 3273 kb)
Table S2. Table lists all phosphosites that were reliably quantified in the triplex SILAC experiment (both biological replicates) of wild type vs CK2α/α’(-/-) cells. Peptide sequences, protein group accession number, SILAC ratios, fold change, and protein description are reported (XLSX 69 kb)
Table S3. Table lists all (phospho)peptides identified in the triplex SILAC experiment (both biological replicates) of wild type vs CK2β(-/-) cells. Peptide sequences, confidence level, protein group accession number, modifications, PhosphoRS site probabilities, SILAC ratios, q-values, Mascot scores, expectation values, number of missed cleavages, experimental m/z, Δppm are reported (XLSX 2423 kb)
Table S4. Table lists all phosphosites that were reliably quantified in the triplex SILAC experiment (both biological replicates) of wild type vs CK2β(-/-) cells. Peptide sequences, protein group accession number, SILAC ratios, fold change, and protein description are reported (XLSX 44 kb)
Acknowledgements
This work was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC), Grant number IG 18756 (to L.A.P.). C. F. was supported by a Grant from the “Collegio Ghislieri”, Pavia. J.V. was supported by a Grant from the Fondazione per la Ricerca sulla Fibrosi Cistica (Grant #10/2016 adopted by Gruppo di Sostegno FFC di Seregno) (to M.S.). The authors wish to thank the Cassa di Risparmio di Padova e Rovigo (Cariparo) Holding for funding the acquisition of the LTQ-Orbitrap XL mass spectrometer.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Cinzia Franchin and Christian Borgo contributed equally to this work.
Contributor Information
Mauro Salvi, Email: mauro.salvi@unipd.it.
Giorgio Arrigoni, Email: giorgio.arrigoni@unipd.it.
Lorenzo A. Pinna, Email: lorenzo.pinna@unipd.it
References
- 1.Allende CC, Allende JE. Promiscuous subunit interactions: a possible mechanism for the regulation of protein kinase CK2. J Cell Biochem Suppl. 1998;30–31:129–136. doi: 10.1002/(SICI)1097-4644(1998)72:30/31+<129::AID-JCB17>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 2.Pinna LA. Protein kinase CK2: a challenge to canons. J Cell Sci. 2002;115(Pt 20):3873–3878. doi: 10.1242/jcs.00074. [DOI] [PubMed] [Google Scholar]
- 3.St-Denis NA, Litchfield DW. Protein kinase CK2 in health and disease: from birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol Life Sci. 2009;66(11–12):1817–1829. doi: 10.1007/s00018-009-9150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pinna LA. The raison d’etre of constitutively active protein kinases: the lesson of CK2. Acc Chem Res. 2003;36(6):378–384. doi: 10.1021/ar020164f. [DOI] [PubMed] [Google Scholar]
- 5.Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369(Pt 1):1–15. doi: 10.1042/bj20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ortega CE, Seidner Y, Dominguez I. Mining CK2 in cancer. PLoS One. 2014;9(12):e115609. doi: 10.1371/journal.pone.0115609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu X, Landesman-Bollag E, Channavajhala PL, Seldin DC. Murine protein kinase CK2: gene and oncogene. Mol Cell Biochem. 1999;191(1–2):65–74. doi: 10.1023/A:1006866412652. [DOI] [PubMed] [Google Scholar]
- 8.Channavajhala P, Seldin DC. Functional interaction of protein kinase CK2 and c-Myc in lymphomagenesis. Oncogene. 2002;21(34):5280–5288. doi: 10.1038/sj.onc.1205640. [DOI] [PubMed] [Google Scholar]
- 9.Ruzzene M. Pinna LA (2010) Addiction to protein kinase CK2: a common denominator of diverse cancer cells? Biochim Biophys Acta. 1804;3:499–504. doi: 10.1016/j.bbapap.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 10.Cozza G, Pinna LA. Casein kinases as potential therapeutic targets. Expert Opin Ther Targets. 2016;20(3):319–340. doi: 10.1517/14728222.2016.1091883. [DOI] [PubMed] [Google Scholar]
- 11.Pierre F, Chua PC, O’Brien SE, Siddiqui-Jain A, Bourbon P, Haddach M, Michaux J, Nagasawa J, Schwaebe MK, Stefan E, Vialettes A, Whitten JP, Chen TK, Darjania L, Stansfield R, Bliesath J, Drygin D, Ho C, Omori M, Proffitt C, Streiner N, Rice WG, Ryckman DM, Anderes K. Pre-clinical characterization of CX-4945, a potent and selective small molecule inhibitor of CK2 for the treatment of cancer. Mol Cell Biochem. 2011;356(1–2):37–43. doi: 10.1007/s11010-011-0956-5. [DOI] [PubMed] [Google Scholar]
- 12.Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17(3):349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 13.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salvi M, Sarno S, Cesaro L, Nakamura H, Pinna LA. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim Biophys Acta. 2009;1793(5):847–859. doi: 10.1016/j.bbamcr.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 15.Venerando A, Cesaro L, Pinna LA. From phosphoproteins to phosphoproteomes: a historical account. FEBS J. 2017 doi: 10.1111/febs.14014. [DOI] [PubMed] [Google Scholar]
- 16.Franchin C, Cesaro L, Salvi M, Millioni R, Iori E, Cifani P, James P, Arrigoni G, Pinna L. Quantitative analysis of a phosphoproteome readily altered by the protein kinase CK2 inhibitor quinalizarin in HEK-293T cells. Biochim Biophys Acta. 2015;1854(6):609–623. doi: 10.1016/j.bbapap.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 17.Franchin C, Salvi M, Arrigoni G, Pinna LA. Proteomics perturbations promoted by the protein kinase CK2 inhibitor quinalizarin. Biochim Biophys Acta. 2015;1854((10 Pt B)):1676–1686. doi: 10.1016/j.bbapap.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 18.Borgo C, Franchin C, Scalco S, Bosello-Travain V, Donella-Deana A, Arrigoni G, Salvi M, Pinna LA. Generation and quantitative proteomics analysis of CK2alpha/alpha’(−/−) cells. Sci Rep. 2017;7:42409. doi: 10.1038/srep42409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lou DY, Dominguez I, Toselli P, Landesman-Bollag E, O’Brien C, Seldin DC. The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol Cell Biol. 2008;28(1):131–139. doi: 10.1128/MCB.01119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bendall SC, Hughes C, Stewart MH, Doble B, Bhatia M, Lajoie GA. Prevention of amino acid conversion in SILAC experiments with embryonic stem cells. Mol Cell Proteomics. 2008;7(9):1587–1597. doi: 10.1074/mcp.M800113-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6(5):359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 22.Salvi M, Trashi E, Cozza G, Franchin C, Arrigoni G, Pinna L. Investigation on PLK2 and PLK3 substrate recognition. Biochim Biophys Acta. 2012;1824(12):1366–1373. doi: 10.1016/j.bbapap.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Taus T, Kocher T, Pichler P, Paschke C, Schmidt A, Henrich C, Mechtler K. Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res. 2011;10(12):5354–5362. doi: 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- 24.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18(20):6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tagliabracci VS, Wiley SE, Guo X, Kinch LN, Durrant E, Wen J, Xiao J, Cui J, Nguyen KB, Engel JL, Coon JJ, Grishin N, Pinna LA, Pagliarini DJ, Dixon JE. A single kinase generates the majority of the secreted phosphoproteome. Cell. 2015;161(7):1619–1632. doi: 10.1016/j.cell.2015.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bibby AC, Litchfield DW. The multiple personalities of the regulatory subunit of protein kinase CK2: CK2 dependent and CK2 independent roles reveal a secret identity for CK2beta. Int J Biol Sci. 2005;1(2):67–79. doi: 10.7150/ijbs.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hagemann C, Kalmes A, Wixler V, Wixler L, Schuster T, Rapp UR. The regulatory subunit of protein kinase CK2 is a specific A-Raf activator. FEBS Lett. 1997;403(2):200–202. doi: 10.1016/S0014-5793(97)00011-2. [DOI] [PubMed] [Google Scholar]
- 29.Chen M, Cooper JA. The beta subunit of CKII negatively regulates Xenopus oocyte maturation. Proc Natl Acad Sci USA. 1997;94(17):9136–9140. doi: 10.1073/pnas.94.17.9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guerra B, Issinger OG, Wang JY. Modulation of human checkpoint kinase Chk1 by the regulatory beta-subunit of protein kinase CK2. Oncogene. 2003;22(32):4933–4942. doi: 10.1038/sj.onc.1206721. [DOI] [PubMed] [Google Scholar]
- 31.Mentzel B, Jauch E, Raabe T. CK2beta interacts with and regulates p21-activated kinases in Drosophila. Biochem Biophys Res Commun. 2009;379(2):637–642. doi: 10.1016/j.bbrc.2008.12.136. [DOI] [PubMed] [Google Scholar]
- 32.Litchfield DW, Luscher B. Casein kinase II in signal transduction and cell cycle regulation. Mol Cell Biochem. 1993;127–128:187–199. doi: 10.1007/BF01076770. [DOI] [PubMed] [Google Scholar]
- 33.Luscher B, Litchfield DW. Biosynthesis of casein kinase II in lymphoid cell lines. Eur J Biochem. 1994;220(2):521–526. doi: 10.1111/j.1432-1033.1994.tb18651.x. [DOI] [PubMed] [Google Scholar]
- 34.Arrigoni G, Marin O, Pagano MA, Settimo L, Paolin B, Meggio F, Pinna LA. Phosphorylation of calmodulin fragments by protein kinase CK2. Mechanistic aspects and structural consequences. Biochemistry. 2004;43(40):12788–12798. doi: 10.1021/bi049365c. [DOI] [PubMed] [Google Scholar]
- 35.Marin O, Sarno S, Boschetti M, Pagano MA, Meggio F, Ciminale V, D’Agostino DM, Pinna LA. Unique features of HIV-1 Rev protein phosphorylation by protein kinase CK2 (‘casein kinase-2’) FEBS Lett. 2000;481(1):63–67. doi: 10.1016/S0014-5793(00)01971-2. [DOI] [PubMed] [Google Scholar]
- 36.Salvi M, Sarno S, Marin O, Meggio F, Itarte E, Pinna LA. Discrimination between the activity of protein kinase CK2 holoenzyme and its catalytic subunits. FEBS Lett. 2006;580(16):3948–3952. doi: 10.1016/j.febslet.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 37.Bian Y, Ye M, Wang C, Cheng K, Song C, Dong M, Pan Y, Qin H, Zou H. Global screening of CK2 kinase substrates by an integrated phosphoproteomics workflow. Sci Rep. 2013;3:3460. doi: 10.1038/srep03460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4(10):793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 39.Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T, Meijer L, Kahle PJ, Haass C. Constitutive phosphorylation of the Parkinson’s disease associated alpha-synuclein. J Biol Chem. 2000;275(1):390–397. doi: 10.1074/jbc.275.1.390. [DOI] [PubMed] [Google Scholar]
- 40.Xu Y, Deng Y, Qing H. The phosphorylation of alpha-synuclein: development and implication for the mechanism and therapy of the Parkinson’s disease. J Neurochem. 2015;135(1):4–18. doi: 10.1111/jnc.13234. [DOI] [PubMed] [Google Scholar]
- 41.Zhang M, Han G, Wang C, Cheng K, Li R, Liu H, Wei X, Ye M, Zou H. A bead-based approach for large-scale identification of in vitro kinase substrates. Proteomics. 2011;11(24):4632–4637. doi: 10.1002/pmic.201100339. [DOI] [PubMed] [Google Scholar]
- 42.Szyszka R. Protein kinases phosphorylating acidic ribosomal proteins from yeast cells. Folia Microbiol (Praha) 1999;44(2):142–152. doi: 10.1007/BF02816233. [DOI] [PubMed] [Google Scholar]
- 43.Huang YW, Surka MC, Reynaud D, Pace-Asciak C, Trimble WS. GTP binding and hydrolysis kinetics of human septin 2. FEBS J. 2006;273(14):3248–3260. doi: 10.1111/j.1742-4658.2006.05333.x. [DOI] [PubMed] [Google Scholar]
- 44.Diaz-Nido J, Serrano L, Mendez E, Avila J. A casein kinase II-related activity is involved in phosphorylation of microtubule-associated protein MAP-1B during neuroblastoma cell differentiation. J Cell Biol. 1988;106(6):2057–2065. doi: 10.1083/jcb.106.6.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ji H, Wang J, Nika H, Hawke D, Keezer S, Ge Q, Fang B, Fang X, Fang D, Litchfield DW, Aldape K, Lu Z. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-Catenin from beta-Catenin and transactivation of beta-Catenin. Mol Cell. 2009;36(4):547–559. doi: 10.1016/j.molcel.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong HN, Ward MA, Bell AW, Chevet E, Bains S, Blackstock WP, Solari R, Thomas DY, Bergeron JJ. Conserved in vivo phosphorylation of calnexin at casein kinase II sites as well as a protein kinase C/proline-directed kinase site. J Biol Chem. 1998;273(27):17227–17235. doi: 10.1074/jbc.273.27.17227. [DOI] [PubMed] [Google Scholar]
- 47.Cala SE, Ulbright C, Kelley JS, Jones LR. Purification of a 90-kDa protein (Band VII) from cardiac sarcoplasmic reticulum. Identification as calnexin and localization of casein kinase II phosphorylation sites. J Biol Chem. 1993;268(4):2969–2975. [PubMed] [Google Scholar]
- 48.Takeda E, Hieda M, Katahira J, Yoneda Y. Phosphorylation of RanGAP1 stabilizes its interaction with Ran and RanBP1. Cell Struct Funct. 2005;30(2):69–80. doi: 10.1247/csf.30.69. [DOI] [PubMed] [Google Scholar]
- 49.Thoen C, De Herdt E, Slegers H. Identification of the ribosomal proteins phosphorylated by the ribosome-associated casein kinase type II from cryptobiotic gastrulae of the brine shrimp Artemia sp. Biochem Biophys Res Commun. 1986;135(2):347–354. doi: 10.1016/0006-291X(86)90001-X. [DOI] [PubMed] [Google Scholar]
- 50.Hasler P, Brot N, Weissbach H, Parnassa AP, Elkon KB. Ribosomal proteins P0, P1, and P2 are phosphorylated by casein kinase II at their conserved carboxyl termini. J Biol Chem. 1991;266(21):13815–13820. [PubMed] [Google Scholar]
- 51.Witters LA, Bacon GW. Protein phosphatases active on acetyl-CoA carboxylase phosphorylated by casein kinase I, casein kinase II and the cAMP-dependent protein kinase. Biochem Biophys Res Commun. 1985;130(3):1132–1138. doi: 10.1016/0006-291X(85)91733-4. [DOI] [PubMed] [Google Scholar]
- 52.Escargueil AE, Plisov SY, Filhol O, Cochet C, Larsen AK. Mitotic phosphorylation of DNA topoisomerase II alpha by protein kinase CK2 creates the MPM-2 phosphoepitope on Ser-1469. J Biol Chem. 2000;275(44):34710–34718. doi: 10.1074/jbc.M005179200. [DOI] [PubMed] [Google Scholar]
- 53.Cahill MA. The evolutionary appearance of signaling motifs in PGRMC1. Biosci Trends. 2017;11(2):179–192. doi: 10.5582/bst.2017.01009. [DOI] [PubMed] [Google Scholar]
- 54.Wang D, Jang DJ. Protein kinase CK2 regulates cytoskeletal reorganization during ionizing radiation-induced senescence of human mesenchymal stem cells. Cancer Res. 2009;69(20):8200–8207. doi: 10.1158/0008-5472.CAN-09-1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Table lists all (phospho)peptides identified in the triplex SILAC experiment (both biological replicates) of wild type vs CK2α/α’(-/-) cells. Peptide sequences, confidence level, protein group accession number, modifications, PhosphoRS site probabilities, SILAC ratios, q-values, Mascot scores, expectation values, number of missed cleavages, experimental m/z, Δppm are reported (XLSX 3273 kb)
Table S2. Table lists all phosphosites that were reliably quantified in the triplex SILAC experiment (both biological replicates) of wild type vs CK2α/α’(-/-) cells. Peptide sequences, protein group accession number, SILAC ratios, fold change, and protein description are reported (XLSX 69 kb)
Table S3. Table lists all (phospho)peptides identified in the triplex SILAC experiment (both biological replicates) of wild type vs CK2β(-/-) cells. Peptide sequences, confidence level, protein group accession number, modifications, PhosphoRS site probabilities, SILAC ratios, q-values, Mascot scores, expectation values, number of missed cleavages, experimental m/z, Δppm are reported (XLSX 2423 kb)
Table S4. Table lists all phosphosites that were reliably quantified in the triplex SILAC experiment (both biological replicates) of wild type vs CK2β(-/-) cells. Peptide sequences, protein group accession number, SILAC ratios, fold change, and protein description are reported (XLSX 44 kb)