

Abstract
Background
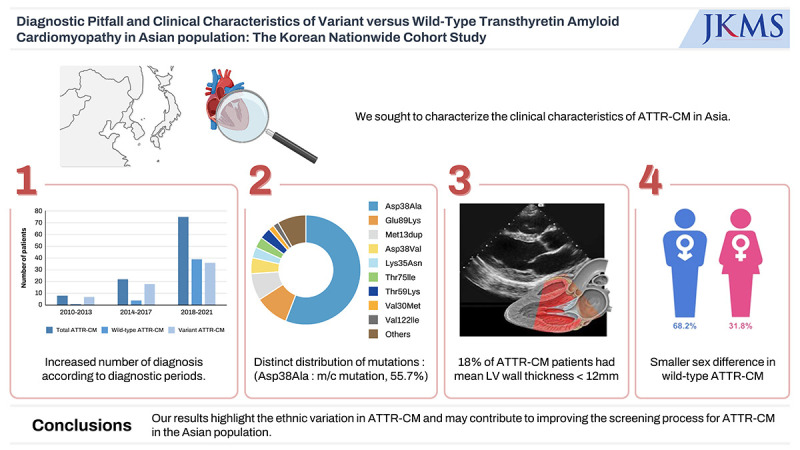
Transthyretin amyloidosis cardiomyopathy (ATTR-CM) is an under-recognized cause of heart failure (HF) with clinical phenotypes that vary across regions and genotypes. We sought to characterize the clinical characteristics of ATTR-CM in Asia.
Methods
Data from a nationwide cohort of patients with ATTR-CM from six major tertiary centres in South Korea were analysed between 2010 and 2021. All patients underwent clinical evaluation, biochemical laboratory tests, echocardiography, and transthyretin (TTR) genotyping at the time of diagnosis. The study population comprised 105 Asian ATTR-CM patients (mean age: 69 years; male: 65.7%, wild-type ATTR-CM: 41.9%).
Results
Among our cohort, 18% of the patients had a mean left ventricular (LV) wall thickness < 12 mm. The diagnosis of ATTR-CM increased notably during the study period (8 [7.6%] during 2010–2013 vs. 22 [21.0%] during 2014–2017 vs. 75 [71.4%] during 2018–2021). Although the duration between symptom onset and diagnosis did not differ, the proportion of patients with HF presenting mild symptoms increased during the study period (25% NYHA class I/II between 2010–2013 to 77% between 2018–2021). In contrast to other international registry data, male predominance was less prominent in wild-type ATTR-CM (68.2%). The distribution of TTR variants was also different from Western countries and from Japan. Asp38Ala was the most common mutation.
Conclusion
A nationwide cohort of ATTR-CM exhibited less male predominance, a proportion of patients without increased LV wall thickness, and distinct characteristics of genetic mutations, compared to cohorts in other parts of the world. Our results highlight the ethnic variation in ATTR-CM and may contribute to improving the screening process for ATTR-CM in the Asian population.
Keywords: Cardiac Amyloidosis, Diagnosis, Nationwide Registry, Transthyretin
Graphical Abstract
INTRODUCTION
Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive and fatal cardiomyopathy, which is an under-recognised cause of heart failure (HF).1,2,3,4,5,6 Because of the recent development of disease-modifying drugs and non-invasive diagnostic tools, the diagnosis of ATTR-CM is increasing.7,8,9,10,11,12 One contemporary estimate of cardiac amyloidosis in the United States reported an incidence of 17 per 100,000 persons in hospitalized patients > 65 years.13 ATTR-CM has been observed in 10.1% men presenting with HF with preserved ejection fraction (HFpEF) with a higher prevalence in older populations.14,15 Although physicians are now becoming aware of ATTR-CM as a possible etiology of HF, rather than a “rare disease”,13,16 a significant knowledge gap persists in the epidemiology of ATTR-CM.6,17,18,19,20
Most registries have included Caucasian and male patients, and whether the prevalence and clinical phenotypes are similar in the Asian population is not well known. The Asian population is under-represented in the Transthyretin Amyloidosis Outcomes Survey (THAOS) registry (251/5,894, 4.3%), which is the largest ongoing, global, longitudinal observational study of patients with ATTR-CM.13,14,15,16,17,18,19,20,21 As such, it remains unclear whether current estimates of disease prevalence and observed clinical phenotypes would be similar in Asian population. In this study, we aimed to investigate the clinical characteristics of ATTR-CM in Asian patients with multi-center nationwide Korean registry.
METHODS
We retrospectively analysed 105 patients with confirmed ATTR-CM from six tertiary hospitals in South Korea between 2010 and 2021. To minimize selection bias, all investigators thoroughly conducted a comprehensive search for patients with suspected ATTR-CM throughout the study period, adhering strictly to enrolment criteria. All patients who had a confirmatory diagnosis of ATTR-CM were validated through electric medical records. The diagnosis was established based on either endomyocardial biopsy or non-invasive bone scintigraphy grade 2 or 3 uptakes with no evidence of light-chain amyloidosis. All patients underwent clinically indicated transthoracic echocardiography and transthyretin (TTR) genotyping, as a part of diagnosis. All patients underwent comprehensive echocardiography in accordance with the guidelines.22 The protocol for cardiac bone scintigraphy, when used, consisted of a planar whole-body acquisition and single-photon emission computed tomography of the thorax, both performed 3 hours after injection of the radionucleotide tracer (99mTc-pyrophosphate and 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid). The intensity of myocardial uptake on cardiac bone scintigraphy was categorised as 0–3 according to the Perugini grading system.11
The clinical characteristics, echocardiography data, and pertinent laboratory data of the patients at the time of the diagnosis were collected. For temporal trend analysis, the patients were divided into 4-year periods according to the time of diagnosis in three time periods (2010–2013, 2014–2017, and 2018–2021). Over the time, there was increasing awareness of ATTR-CM due to advent of bone scintigraphy and the development of disease-modifying therapies.8,9 Symptom onset was adjudicated by the investigators based on medical record review for any cardiac or non-cardiac symptoms consistent with ATTR amyloidosis by the investigator.
Statistical analysis
Descriptive statistics are presented as percentages for categorical variables and as medians with interquartile ranges (IQRs) for continuous variables. All continuous variables were tested for normal distribution using the Shapiro–Wilk test and are presented as means ± standard deviations or medians with IQRs. The independent t-test was performed to analyse normally-distributed data in each group to compare means. When assumptions for t-test were not met, its nonparametric equivalent (Mann–Whitney U test) was used to compare the distribution between the two groups. Categorical data are presented as absolute numbers and frequencies (%) and compared using the χ2 test. All statistical analyses were performed using IBM SPSS Statistics Version 25 (IBM, Armonk, NY, USA).
Ethics statement
This study was reviewed and approved by the Institutional Review Board (IRB) of Samsung Medical Center and the requirement for informed consents was waived (IRB 2021-06-074, 2021-06-075) due to the retrospective nature of the study. This study followed the principles of the Declaration of Helsinki.
RESULTS
Baseline characteristics
Between 2010 and 2021, 105 patients with ATTR-CM were enrolled. Among these patients, 44 (41.9%) patients were diagnosed with wild-type ATTR-CM and 61 patients were diagnosed with variant ATTR-CM (58.1%). Among variant ATTR-CM, the most common variant was Asp38Ala (n = 34, 55.7%). The rest of the patients with variant ATTR-CM had the following mutations: Glu89Lys (n = 6, 9.8%), Met13dup (n = 5, 8.2%), Asp38Val (n = 3, 4.9%), Lys35Asn (n = 2, 3.3%), Thr75Ile (n = 2, 3.3%), Thr59Lys (n = 2, 3.3%), Val30Met (n = 1, 1.6%), Val122lle (n = 1, 1.6%), and others (n = 5, 8.2%).
The comparison of the clinical characteristics between wild-type and variant ATTR-CM is shown in Table 1. Patients with wild-type ATTR-CM were significantly older than those with variant ATTR-CM (mean age: 81.0 vs. 60.9, P < 0.001). The proportion of male was comparable between two groups (68.2% vs. 63.9%, P = 0.405). The proportion of patients with wild-type ATTR-CM associated with coronary artery disease, hypertension, stroke, and dyslipidemia was significantly higher than that of patients with variant ATTR-CM. The mean durations of symptoms before diagnosis were comparable between the two groups (1.6 ± 2.8 vs. 2.8 ± 3.3 years, P = 0.063). Most common ECG finding was presence of any block (38.1%). Prevalence of atrial fibrillation of higher in wild-type ATTR-CM compared to variant type ATTR-CM, possibly due to older age.
Table 1. Baseline characteristics of study population.
| Characteristics | Total (N = 105) | Wild-type ATTR-CM (n = 44) | Variant ATTR-CM (n = 61) | P value | |
|---|---|---|---|---|---|
| Age, yr | 69.3 ± 13.4 | 81.0 ± 8.8 | 60.9 ± 9.3 | < 0.001 | |
| Male | 69 (65.7) | 30 (68.2) | 39 (63.9) | 0.405 | |
| Body mass index, kg/m2 | 22.3 ± 3.8 | 23.3 ± 3.8 | 21.6 ± 3.8 | 0.027 | |
| Body surface area, /m2 | 1.61 ± 0.21 | 1.61 ± 0.18 | 1.16 ± 0.22 | 0.973 | |
| CAD | 15 (14.3) | 12 (27.3) | 3 (4.9) | 0.002 | |
| Diabetes mellitus | 18 (17.1) | 14 (31.8) | 4 (6.6) | 0.001 | |
| Hypertension | 39 (37.1) | 27 (61.4) | 12 (19.7) | < 0.001 | |
| Stroke | 11 (10.5) | 8 (18.2) | 3 (4.9) | 0.049 | |
| Chronic kidney disease | 24 (22.9) | 17 (38.6) | 7 (11.5) | 0.002 | |
| Dyslipidemia | 28 (58.3) | 18 (40.9) | 10 (16.4) | 0.007 | |
| Duration of symptoms before diagnosis, yr | 2.3 ± 3.3 | 1.6 ± 2.8 | 2.8 ± 3.3 | 0.063 | |
| Diagnostic method | |||||
| Cardiac biopsy | 74 (70.5) | 27 (61.4) | 47 (77.0) | 0.089 | |
| Non cardiac biopsy | 15 (14.3) | 3 (6.8) | 12 (19.7) | 0.090 | |
| Bone scan (non-invasive) | 83 (79.0) | 39 (88.6) | 44 (72.1) | 0.052 | |
| Six minute walk test, m | 400 ± 160 | 378 ± 184 | 462 ± 54 | 0.470 | |
| Systolic blood pressure, mmHg | 109 ± 16 | 114 ± 14 | 105 ± 17 | 0.005 | |
| Diastolic blood pressure, mmHg | 66 ± 11 | 67 ± 10 | 67 ± 12 | 0.763 | |
| NT-proBNP, pg/mL | 1,780 (614–3,452) | 2,695 (989–5,285) | 1,551 (571–2,703) | 0.022 | |
| Troponin T, pg/mL | 52 (27–74) | 71.5 (52.3–91.5) | 41 (25–62) | 0.001 | |
| Estimated GFR, mL/min/1.73 m2 | 77.3 ± 24.6 | 62.4 ± 19.0 | 88.3 ± 22.4 | < 0.001 | |
| ECG | |||||
| Pseudo-infarct ECG | 11 (10.5) | 3 (6.8) | 8 (13.1) | 0.352 | |
| Any blocks | 40 (38.1) | 15 (34.1) | 25 (41.0) | 0.544 | |
| Low/decreased QRS voltage | 15 (14.3) | 5 (11.4) | 10 (16.4) | 0.577 | |
| Atrial fibrillation | 15 (14.3) | 10 (22.7) | 5 (8.2) | 0.048 | |
| Cardiac device | |||||
| Permanent pacemaker | 16 (15.2) | 7 (15.9) | 9 (14.8) | 0.999 | |
| ICD | 3 (2.9) | 3 (4.9) | 0 (0.0) | 0.263 | |
| CRT | 1 (1.0) | 1 (2.3) | 0 (0.0) | 0.419 | |
| Extra-cardiac symptoms | |||||
| Polyneuropathy | 74 (70.5) | 20 (45.5) | 54 (88.5) | < 0.001 | |
| Autonomic neuropathy | 55 (52.4) | 10 (22.7) | 45 (73.8) | < 0.001 | |
| Bilateral CTS | 38 (36.2) | 15 (34.1) | 23 (37.7) | 0.837 | |
| Lumbar spinal stenosis | 28 (26.7) | 13 (29.5) | 15 (24.6) | 0.656 | |
| Rotator cuff tendon tear | 2 (1.9) | 1 (2.3) | 1 (1.6) | 0.999 | |
Values are presented as number (%) for categorical variables and mean ± standard deviation or median (interquartile range) for continuous variables.
ATTR-CM = transthyretin amyloidosis cardiomyopathy, CAD = coronary artery disease, NT-proBNP = N-terminal pro-B-type natriuretic peptide, GFR = glomerular filtration rate, ECG = electrocardiogram, ICD = implantable cardiac defibrillator, CRT = cardiac resynchronization therapy, CTS = carpal tunnel syndrome.
Regarding extra-cardiac symptoms, significantly more patients with variant ATTR-CM had polyneuropathy (88.5% vs. 45.5%, P < 0.001) and autonomic neuropathy (73.8% vs. 22.7%, P < 0.001), compared to those with wild-type ATTR-CM. The incidence of spinal stenosis and rotator cuff tendon tear were similar in both groups. Regarding baseline echocardiographic parameters, pericardial effusion was presented in 55% of patients. A total of 94 (89.5%) patient had preserved ejection fraction (50%) and 85 (81%) had increased LV filling pressure defined as E/e’ ≥ 15. Patients with wild-type ATTR-CM had significantly larger left atrial (LA) volume index (61.1 ± 20.0 vs. 47.7 ± 17.5 mL/m2, P = 0.002) and smaller left ventricular (LV) mass index (158.1 ± 36.0 vs. 182.3 ± 63.9 g/m2, P = 0.039) at the time of diagnosis than those with variant ATTR-CM (Table 2).
Table 2. Baseline echocardiographic characteristics.
| Characteristics | Total (N = 105) | Wild-type ATTR-CM (n = 44) | Variant ATTR-CM (n = 61) | P value |
|---|---|---|---|---|
| LV EDD, mm | 46.4 ± 5.1 | 46.8 ± 5.2 | 46.1 ± 5.1 | 0.481 |
| LV ESD, mm | 32.4 ± 6.4 | 32.9 ± 6.5 | 32.1 ± 6.4 | 0.548 |
| LV EDV, mL | 94.1 ± 27.9 | 92.8 ± 28.9 | 99.8 ± 27.3 | 0.288 |
| LV ESV, mL | 44.5 ± 21.2 | 43.4 ± 19.3 | 45.1 ± 22.4 | 0.760 |
| LV posterior wall thickness, mm | 13.6 ± 2.8 | 13.1 ± 2.0 | 13.8 ± 3.2 | 0.080 |
| LV septal wall thickness, mm | 14.5 ± 3.2 | 13.8 ± 2.2 | 14.9 ± 3.7 | 0.054 |
| Mean LV wall thickness | 14.1 ± 2.9 | 13.5 ± 1.9 | 14.4 ± 3.4 | 0.125 |
| LA volume index, mL/m2 | 52.4 ± 19.4 | 61.1 ± 20.0 | 47.7 ± 17.5 | 0.002 |
| LV ejection fraction, % | 56.9 ± 10.4 | 56.1 ± 9.4 | 57.5 ± 11.2 | 0.620 |
| LV mass index, mg/m2 | 172.5 ± 55.5 | 158.1 ± 36.0 | 182.3 ± 63.9 | 0.039 |
| E/e’ | 21.8 ± 8.8 | 22.2 ± 7.6 | 21.4 ± 9.5 | 0.667 |
| RV systolic pressure, mmHg | 36.7 ± 13.1 | 39.2 ± 12.8 | 34.9 ± 13.1 | 0.128 |
| Aortic stenosis ≥ moderate | 2 (1.9) | 2 (4.5) | 0 (0.0) | 0.173 |
| Pericardial effusion | 58 (55.2) | 23 (52.3) | 35 (57.4) | 0.158 |
Values are presented as number (%) for categorical variables and mean ± standard deviation for continuous variables.
LV = left ventricular, EDD = end-diastolic dimension, ESD = end-systolic dimension, EDV = end-diastolic volume, ESV = end-systolic volume, LA = left atrium, RV = right ventricular.
Temporal trends of confirmed cases
The number of patients diagnosed with ATTR-CM, whether wild-type or variant, increased over each 4-year interval (8 [7.6%] during 2010–2013 vs. 22 [21.0%] during 2014–2017 vs. 75 [71.4%] during 2018–2021) (Fig. 1). Compared to variant ATTR-CM, the diagnosis of wild-type ATTR-CM demonstrated a steeper increase. Between 2010 and 2013, only 1 (2.3%) patient was diagnosed with wild-type ATTR-CM, whereas 4 (9.1%) and 39 (88.6%) patients were diagnosed between 2014 and 2017 and between 2018 and 2021, respectively. The largest increase in the total number of patients diagnosed with ATTR-CM occurred in the most recent period, between 2018 and 2021. The number of patients diagnosed with wild-type ATTR-CM and variant type ATTR-CM during this period was 38-fold and 4-fold higher, respectively, than those diagnosed between 2010 and 2013. However, duration between symptom onset and diagnosis did not differ among diagnostic time periods (Supplementary Fig. 1). The increase in the average age at diagnosis observed in successive eras is consistent with increased diagnosis of the wild-type form of ATTR-CM over time (Supplementary Fig. 2).
Fig. 1. Numbers of patients according to diagnostic time periods.

ATTR-CM = transthyretin amyloidosis cardiomyopathy.
Clinical characteristics of ATTR-CM patients were compared according to diagnostic periods (Table 3). The proportion of patients with New York Heart Association (NYHA) functional class III/IV HF symptoms decreased significantly in recent time periods (75% during time between 2010 and 2013 vs. 23% during time between 2018 and 2021) (Supplementary Fig. 3). The use of cardiac bone scintigraphy significantly increased over time (25% vs. 32% vs. 99%, P < 0.001). The proportion of endomyocardial and non-cardiac biopsies decreased during the recent period (2018–2021); however, significant proportions of patients still underwent either cardiac or non-cardiac biopsies (100% vs.100% vs. 79%) (Supplementary Fig. 4).
Table 3. Comparisons of phenotypes of ATTR-CM according to diagnostic eras.
| Characteristics | 2010–2013 (n = 8) | 2014–2017 (n = 22) | 2018–2021 (n = 75) | P value | |
|---|---|---|---|---|---|
| Age, yr | 57.2 ± 11.9 | 65.2 ± 10.5 | 71.8 ± 13.5 | < 0.001 | |
| Male | 8 (100.0) | 12 (54.5) | 49 (65.3) | 0.067 | |
| Wild-type ATTR-CM | 1 (12.5) | 4 (18.2) | 39 (52.0) | 0.004 | |
| Variant ATTR-CM | 7 (87.5) | 18 (81.8) | 36 (48.0) | 0.004 | |
| Body mass index, kg/m2 | 19.2 ± 3.3 | 20.8 ± 3.6 | 23.1 ± 3.6 | 0.002 | |
| Body surface area, /m2 | 1.6 ± 0.20 | 1.57 ± 0.23 | 1.63 ± 0.20 | 0.532 | |
| CAD | 0 (0.0) | 1 (4.5) | 14 (18.7) | 0.122 | |
| Diabetes mellitus | 0 (0.0) | 2 (9.1) | 16 (21.3) | 0.166 | |
| Hypertension | 1 (12.5) | 4 (18.2) | 34 (45.3) | 0.022 | |
| Stroke | 0 (0.0) | 2 (9.1) | 9 (12.0) | 0.558 | |
| Chronic kidney disease | 2 (25.0) | 5 (22.7) | 17 (22.7) | 0.989 | |
| Dyslipidemia | 0 (0.0) | 2 (9.1) | 26 (34.7) | 0.012 | |
| Duration of symptoms before diagnosis, yr | 3.0 ± 1.2 | 2.8 ± 2.9 | 2.1 ± 3.6 | 0.571 | |
| NYHA class | |||||
| I/II | 2 (25.0) | 19 (86.4) | 44 (58.7) | 0.005 | |
| III/IV | 6 (75.0) | 3 (13.6) | 17 (22.7) | 0.002 | |
| Diagnosis | |||||
| Cardiac biopsy | 7 (87.5) | 19 (86.4) | 48 (64.0) | 0.071 | |
| Non cardiac biopsy | 1 (12.5) | 3 (13.6) | 11 (14.7) | 0.982 | |
| Bone scan (non-invasive) | 2 (25.0) | 7 (31.8) | 74 (98.7) | < 0.001 | |
| Systolic blood pressure, mmHg | 92.0 ± 8.5 | 101.1 ± 12.8 | 113.4 ± 15.9 | < 0.001 | |
| Diastolic blood pressure, mmHg | 60.3 ± 5.3 | 66.1 ± 9.4 | 67.4 ± 12.1 | 0.230 | |
| NT-proBNP, pg/mL | 3,322 (470–10,471) | 2,048 (1,726–2,803) | 1,502 (492–3,452) | 0.242 | |
| Troponin T, pg/mL | 87.0 (69.0–NA) | 44.0 (30.5–28.3) | 54.0 (26.3–75.5) | 0.504 | |
| Estimated GFR, mL/min/1.73 m2 | 87.6 ± 33.6 | 83.5 ± 28.8 | 74.5 ± 21.9 | 0.156 | |
| Cardiac device | |||||
| Permanent pacemaker | 3 (37.5) | 5 (22.7) | 8 (10.7) | 0.073 | |
| ICD | 0 (0.0) | 1 (4.5) | 2 (2.7) | 0.790 | |
| CRT | 0 (0.0) | 0 (0.0) | 1 (1.3) | 0.817 | |
| ECG | |||||
| Pseudo-infarct ECG | 2 (25.0) | 5 (22.7) | 4 (5.3) | 0.024 | |
| Any blocks | 4 (50.0) | 13 (59.1) | 23 (30.7) | 0.042 | |
| Low/decreased QRS voltage | 1 (12.5) | 3 (13.6) | 11 (14.7) | 0.982 | |
| Atrial fibrillation | 0 (0.0) | 4 (18.2) | 11 (14.7) | 0.446 | |
| Extra-cardiac symptoms | |||||
| Polyneuropathy | 7 (87.5) | 17 (77.3) | 50 (66.7) | 0.345 | |
| Autonomic neuropathy | 6 (75.0) | 13 (59.1) | 36 (48.0) | 0.270 | |
| Bilateral CTS | 1 (12.5) | 6 (27.3) | 31 (41.3) | 0.169 | |
| Lumbar spinal stenosis | 2 (25.0) | 7 (31.8) | 19 (25.3) | 0.828 | |
| Rotator cuff tendon tear | 0 (0.0) | 1 (4.5) | 1 (1.3) | 0.575 | |
Values are presented as number (%) for categorical variables and mean ± standard deviation or median (interquartile range) for continuous variables.
ATTR-CM = transthyretin amyloid cardiomyopathy, CAD = coronary artery disease, NYHA = New York Heart Association, NT-proBNP = N-terminal pro-B-type natriuretic peptide, NA = not applicable, GFR = glomerular filtration rate, ICD = implantable cardioverter defibrillator, CRT = cardiac resynchronization therapy, ECG = electrocardiogram, CTS = carpal tunnel syndrome.
Comparison of clinical characteristics at diagnosis between wild-type vs. Asp38Ala variant ATTR-CM
When the genotypes were analysed, patients with the most common mutation (Asp38Ala) were significantly younger and had significantly lower systolic blood pressure than those with wild-type ATTR-CM (Supplementary Table 1).
The mean N-terminal pro-B-type natriuretic peptide (NT-proBNP) and troponin T values were significantly lower in patients with Asp38Ala genotype than in those with wild-type ATTR-CM, as reflected by milder HF symptoms in patients with Asp38Ala genotype. Patients with Asp38Ala genotype ATTR-CM had higher mean LV wall thickness (13.5 ± 2.4 vs. 15.1 ± 3.5 mm, P = 0.016) and higher LV mass index (191.3 ± 70.3 vs. 158.0 ± 36.0 g/m2, P = 0.015) but smaller LA volume index (47.2 ± 18.5 vs. 61.1 ± 20.0 mL/m2, P = 0.006) than patients with wild-type ATTR-CM. Patients with Asp38Ala genotype ATTR-CM had significantly more autonomic (76.5% vs. 40.9%, P = 0.003) and peripheral neuropathy (91.2% vs. 45.5%, P < 0.001) symptoms than patients with wild-type ATTR-CM. Comparisons of clinical characteristics between Asp38Ala variant ATTR-CM and the other variant ATTR-CM are summarized in Supplementary Table 2.
Patients with mean LV wall thickness in normal range (< 12 mm)
A total of 19 (18.1%) patients had mean LV wall thickness {(Septal Wall + Posterior Wall)/2} less than 12 mm. Among these patients, 9 (47.4%) were men and 10 (52.6%) were women. The clinical presentation of ATTR-CM patients with LV wall thickness < 12 mm are described in Supplementary Table 3. The proportion of sex and body surface area (BSA) were comparable between patients with mean LV wall thickness ≥ 12 mm and < 12 mm (male: 69.8% vs. 47.4%, P = 0.107; BSA: 1.63 ± 0.21 vs. 1.55 ± 0.19, P = 0.128). Patients with mean LV wall thickness < 12 mm had significantly higher systolic blood pressure, lower NT-proBNP and higher LV ejection fraction, compared to those with mean LV wall thickness ≥ 12 mm (Table 4). However, the severity of HF symptoms was similar between two groups. LV mass index was significantly smaller in patients with mean LV wall thickness < 12 mm (116.8 ± 25.7 vs. 186.1 ± 52.3 g/m2, P < 0.001). Other parameters including age, sex, and, proportions of NYHA Fc III/IV were similar between patients with mean LV wall thickness ≥ 12 mm and < 12 mm. Extra-cardiac symptoms were similar, except higher incidence of trigger finger in patients with mean LV wall thickness < 12 mm (16.7% vs. 6.2%, P = 0.047). In our cohort, 6 male patients and 3 female patients did not meet the criteria of LV hypertrophy (female: relative wall thickness > 0.42 & LV mass index > 95 mg/m2, male: relative wall thickness > 0.42 & LV mass index > 115 g/m2). When we used sex adjusted reference ranges of LV mass index from the current guideline,22 5 male patients and 1 female patient had LV mass index of normal ranges (male: 50–102 g/m2, female: 44–88 g/m2).
Table 4. Comparison of characteristics between ATTR-CM patient with wall thickness over and less than 12 mm.
| Characteristics | Mean LV wall thickness ≥ 12 mm (n = 86) | Mean LV wall thickness < 12 mm (n = 19) | P value | |
|---|---|---|---|---|
| Age, yr | 68.4 ± 13.4 | 73.6 ± 12.9 | 0.129 | |
| Male | 60 (69.8) | 9 (47.4) | 0.107 | |
| NYHA Fc I/II | 56 (65.1) | 9 (47.4) | 0.193 | |
| NYHA Fc III/IV | 20 (24.7) | 6 (25.0) | 0.999 | |
| Body mass index, kg/m2 | 22.1 ± 3.8 | 23.2 ± 3.7 | 0.262 | |
| Body surface area, m2 | 1.63 ± 0.21 | 1.55 ± 0.19 | 0.128 | |
| Duration from symptom to diagnosis, mon | 30.9 ± 37.7 | 33.0 ± 51.4 | 0.829 | |
| Wild-type ATTR-CM | 34 (39.5) | 10 (41.7) | 0.316 | |
| Variant ATTR-CM | 52 (60.5) | 9 (37.5) | 0.316 | |
| Permanent pacemaker | 15 (17.4) | 1 (5.3) | 0.293 | |
| Systolic blood pressure, mmHg | 107.3 ± 15.1 | 117.6 ± 15.1 | 0.013 | |
| Diastolic blood pressure, mmHg | 66.1 ± 11.5 | 68.9 ± 10.1 | 0.321 | |
| NT-proBNP, pg/mL | 1,993 (798–3,725) | 614 (198–1,704) | 0.003 | |
| Troponin T, pg/mL | 50 (27–78) | 52 (30–63) | 0.523 | |
| ECG | ||||
| Pseudo-infarct ECG | 10 (11.6) | 1 (5.3) | 0.684 | |
| Any blocks | 38 (44.2) | 2 (10.5) | 0.008 | |
| Low/decreased QRS voltage | 12 (14.0) | 3 (15.8) | 0.733 | |
| Atrial fibrillation | 12 (14.0) | 3 (15.8) | 0.733 | |
| Echocardiographic parameters | ||||
| Mean wall thickness, mm | 14.8 ± 2.5 | 10.5 ± 1.3 | < 0.001 | |
| LV mass index, g/m2 | 186.1 ± 52.3 | 116.8 ± 25.7 | < 0.001 | |
| LV ejection fraction, % | 54.8 ± 9.8 | 62.8 ± 10.3 | 0.007 | |
| LA volume index, mL/m2 | 52.5 ± 17.1 | 52.3 ± 28.0 | 0.970 | |
| RV systolic pressure, mmHg | 36.3 ± 12.5 | 38.3 ± 15.9 | 0.580 | |
| Extracardiac symptoms | ||||
| Polyneuropathy | 63 (73.3) | 11 (57.9) | 0.265 | |
| Autonomic neuropathy | 57 (66.3) | 9 (47.4) | 0.188 | |
| Bilateral CTS | 34 (29.5) | 4 (21.1) | 0.188 | |
| Lumbar spinal stenosis | 23 (26.7) | 5 (26.3) | 0.999 | |
| Rotator cuff tendon tear | 2 (2.3) | 0 (0) | 0.999 | |
| Trigger finger | 5 (6.2) | 4 (16.7) | 0.047 | |
Values are presented as number (%) for categorical variables and mean ± standard deviation or median (interquartile range) for continuous variables.
ATTR-CM = transthyretin amyloid cardiomyopathy, LV = left ventricular, NYHA = New York Heart Association, NT-proBNP = N-terminal pro-B-type natriuretic peptide, ECG = electrocardiogram, LA = left atrium, RV = right ventricular, CTS = carpal tunnel syndrome.
DISCUSSION
This study comprehensively analyzed clinical characteristics of ATTR-CM patients from six major tertiary centres in South Korea over a 12-year period. The major findings of this study are as follows: 1) a substantial increase in the diagnosis of ATTR-CM was observed over the study period, 2) distinct genotype distribution in South Korea; Asp38Ala genotype was the most commonly observed; and 3) in our cohort, 18% of the patients with ATTR-CM had a mean LV wall thickness < 12 mm.
The number of patients diagnosed with ATTR-CM increased every 4-year periods, likely indicative of increased awareness, ease of non-biopsy diagnosis, and available effective therapy with tafamidis. As additional evidence of increased awareness, significantly more patients were diagnosed with milder HF symptoms in the more recent time periods. This is a clinically important observation because patients with less symptomatic ATTR-CM have better outcomes with tafamidis.9
The diagnosis of wild-type ATTR-CM is exponentially increasing, and wild-type ATTR-CM is expected to be the most common type of cardiac amyloidosis worldwide. Although the usage of bone scintigraphy significantly increased during the most recent time period, in our cohort, only 41.9% (n = 44) of the patients were diagnosed with wild-type ATTR-CM. According to the recent data using the THAOS registry, the number of patients with wild-type ATTR-CM was almost double that of patients with variant ATTR-CM (1,069 vs. 525).23 It is likely that a significant number of patients with wild-type ATTR-CM remain unrecognized, and our results may describe ongoing real-world barriers to be addressed, including diagnostic delay and physicians’ lack of awareness, especially for wild-type ATTR-CM.
Male predominance is prevalent in ATTR-CM patients. Male prevalence in variant ATTR-CM in our cohort was 64%. This is similar to the previous analysis of the THAOS data; male prevalence in patients with hereditary ATTR amyloidosis ranged from 50.6–73.2% in the main genotype groups.24 Interestingly, the male higher prevalence of male sex was predominant in cardiac phenotype in variant ATTR-CM. The male predominance was balanced in the entire cohort including asymptomatic carrier (59% male vs. 41% female). Two recent national studies from the United Kingdome25 and Spain26 reported female sex to be associated with presence of TTR mutation among older (age ≥ 70 years) patients diagnosed with ATTR-CM. Possibility of slower disease evolution, misdiagnosis biased by lack of sex-specific cut-off or non-indexed parameters, or cardioprotective effect of estrogen may be associated with male predominance in variant ATTR-CM despite autosomal dominant inheritance.24
Unlike variant type, male predominance was less pronounced in wild-type ATTR-CM, when compared to previous studies.27,28 Among wild-type ATTR-CM patients, 31.8% were female, which is significantly higher than the proportion of women enrolled in THAOS registry (5.4%).23 Although, true sex difference in prevalence of wild-type ATTR-CM cannot be determined due to under-diagnosis, the smaller sex difference observed in our registry was also noted in a Japanese database.29 This suggests male predominance may not be as much prominent as in THAOS registry. Therefore, diagnostic studies for ATTR-CM should be initiated in elderly HFpEF patients with red flag sign and symptoms for cardiac amyloidosis, regardless of sex.
It is notable that 18.1% of our cohort had mean LV wall thickness < 12 mm at the time of diagnosis. Moreover, according to age, sex specific echocardiographic criteria, six patients (5.7%) did not have LV hypertrophy. Among 44 patients with wild-type ATTR-CM, 10 (22.7%) had a mean LV wall thickness < 12 mm. This suggests that the current screening criterion of LV wall thickness ≥ 12 mm may not be sensitive enough for diagnosing Asian patients with ATTR-CM. Although direct comparison is not possible, the mean values of BSA (1.61 /m2) and body mass index (22.3 kg/m2) in our cohort were numerically smaller than those reported in THAOS (mean body mass index: 27 kg/m2)27 or other national registry data (UK National Amyloidosis Center; mean BSA 1.89–1.91 m2).28 The fact that patients were diagnosed with ATTR-CM despite not meeting criteria for increased LV wall thickness suggests that cardiologists at major tertiary hospitals with interest in and dedication to ATTR-CM have a higher awareness which results in increased diagnosis. Of note, patients with a mean LV wall thickness < 12 mm were diagnosed at an earlier stage, as indicated by significantly lower NT-proBNP levels than those observed in patients with a mean LV wall thickness ≥ 12 mm, further evidence of increased awareness resulting in earlier diagnosis with important prognostic implications. Our findings suggest that screening for ATTR-CM should not be limited to those with HF with an LV wall thickness > 12 mm, at least for Asians, particularly when other cardiac and non-cardiac symptom are present. If a diagnostic work up is started only in patients with increased LV wall thickness, patients in earlier stage would not be diagnosed, especially in Asian population. If elderly HFpEF patients present with extracardiac symptoms, including polyneuropathy, autonomic neuropathy, bilateral carpal tunnel syndrome or lumbar stenosis, suspicion for ATTR-CM should be raised. Increased LV wall thickness should prompt the diagnostic work up for ATTR-CM, however, normal range of LV wall should not halt further work up for ATTR-CM in the presence of red flag signs and symptoms.
Other notable distinction of ATTR-CM in Korean population was distinct genotype characteristics in South Korea. Asp38Ala genotype was the most frequently observed mutation. Distribution of genetic mutation is different among ethnicity and geographic regions. Val122Ile is the most common TTR mutation which occurs in 3.4% of African Americans and Val30Met is most frequently reported mutation in Japan and Portugal.3 All variant ATTR-CM in our cohort had mixed phenotype.
Although we analysed data from patients confirmed with ATTR-CM at major tertiary medical centres in South Korea, our data may not capture all patients diagnosed with ATTR-CM. The participating investigators were all cardiologists, which may have resulted in selection bias. Diagnostic patterns in Korea may differ from those in the rest of the world because of differences in reimbursement policy, as reflected by high number of biopsies performed. Since the pathologic evidence of the presence of amyloid is required to meet the criteria for co-payment policy for rare diseases in South Korea, a majority of patients underwent biopsy. Comparison analyses between clinical and echocardiographic parameters between wild-type vs. variant ATTR-CM were not age-adjusted, and differences observed could be due to the older age of wild-type ATTR-CM patients. Our study lacks outcome data, including mortality and hospitalization for HF.
In conclusion, the diagnosis of ATTR-CM is increasing in South Korea, with a recent sharp increase observed, especially in the diagnosis of wild-type ATTR-CM. Asp38Ala mutation was the most prevalent mutation in patients with variant ATTR-CM. The screening criterion of LV wall thickness > 12 mm may not be sensitive enough for Asian patients, given their smaller BSA. Therefore, an LV wall thickness < 12 mm should not discourage physicians from initiating a diagnostic work-up for ATTR-CM. Our findings may contribute to improving the screening process for ATTR-CM in the Asian population. Further studies are needed to confirm these findings and determine the most effective screening strategies for this patient population.
Footnotes
Funding: This work was supported by Pfizer (X9001301), by the Catholic Medical Center Research Foundation (2023), and by the research fund of Seoul St. Mary’s Hospital, The Catholic University of Korea.
Disclosure: The authors have no potential conflicts of interest to disclose.
- Data curation: Youn JC, Lee HW, Oh J, Son JW, Cho HJ, Lee S.
- Formal analysis: Kim D.
- Investigation: Kim D.
- Resources: Lee HW, Oh J, Son JW, Cho HJ.
- Supervision: Youn JC.
- Writing - original draft: Kim D.
- Writing - review & editing: Youn JC, Shah NR, Kittleson MM, Jeon ES.
SUPPLEMENTARY MATERIALS
Comparison of wild-type ATTR-CM and Asp38Ala variant ATTR-CM subjects
Comparison of Asp38Ala variant ATTR-CM and the other variant ATTR-CM
Presentation of ATTR-CM with mean LV wall thickness < 12 mm
Time between symptom onset and diagnosis according to time periods.
Mean age at the time of diagnosis according to diagnostic time periods.
Severity of HF symptom at the time of diagnosis according to diagnostic time periods.
Diagnostic methods according to diagnostic time periods.
References
- 1.Writing Committee. Kittleson MM, Ruberg FL, Ambardekar AV, Brannagan TH, Cheng RK, et al. 2023 ACC Expert Consensus decision pathway on comprehensive multidisciplinary care for the patient with cardiac amyloidosis: a report of the American College of Cardiology Solution Set Oversight Committee. J Am Coll Cardiol. 2023;81(11):1076–1126. doi: 10.1016/j.jacc.2022.11.022. [DOI] [PubMed] [Google Scholar]
- 2.Kittleson MM, Maurer MS, Ambardekar AV, Bullock-Palmer RP, Chang PP, Eisen HJ, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020;142(1):e7–e22. doi: 10.1161/CIR.0000000000000792. [DOI] [PubMed] [Google Scholar]
- 3.Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73(22):2872–2891. doi: 10.1016/j.jacc.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yingchoncharoen T, Wu TC, Choi DJ, Ong TK, Liew HB, Cho MC. Economic burden of heart failure in asian countries with different healthcare systems. Korean Circ J. 2021;51(8):681–693. doi: 10.4070/kcj.2021.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park JJ, Lee CJ, Park SJ, Choi JO, Choi S, Park SM, et al. Heart failure statistics in Korea, 2020: a report from the Korean Society of Heart Failure. Int J Heart Fail. 2021;3(4):224–236. doi: 10.36628/ijhf.2021.0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee HH, Cho SM, Lee H, Baek J, Bae JH, Chung WJ, et al. Korea Heart Disease Fact Sheet 2020: analysis of nationwide data. Korean Circ J. 2021;51(6):495–503. doi: 10.4070/kcj.2021.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim D, Choi JO, Kim K, Kim SJ, Kim JS, Jeon ES. Clinical and prognostic implications of capillary density in patients with cardiac light chain amyloidosis. ESC Heart Fail. 2021;8(6):5594–5599. doi: 10.1002/ehf2.13604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–2412. doi: 10.1161/CIRCULATIONAHA.116.021612. [DOI] [PubMed] [Google Scholar]
- 9.Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. doi: 10.1056/NEJMoa1805689. [DOI] [PubMed] [Google Scholar]
- 10.Dasgupta NR, Rissing SM, Smith J, Jung J, Benson MD. Inotersen therapy of transthyretin amyloid cardiomyopathy. Amyloid. 2020;27(1):52–58. doi: 10.1080/13506129.2019.1685487. [DOI] [PubMed] [Google Scholar]
- 11.Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46(6):1076–1084. doi: 10.1016/j.jacc.2005.05.073. [DOI] [PubMed] [Google Scholar]
- 12.Hanna M, Ruberg FL, Maurer MS, Dispenzieri A, Dorbala S, Falk RH, et al. Cardiac scintigraphy with technetium-99m-labeled bone-seeking tracers for suspected amyloidosis: JACC Review Topic of the Week. J Am Coll Cardiol. 2020;75(22):2851–2862. doi: 10.1016/j.jacc.2020.04.022. [DOI] [PubMed] [Google Scholar]
- 13.Gilstrap LG, Dominici F, Wang Y, El-Sady MS, Singh A, Di Carli MF, et al. Epidemiology of cardiac amyloidosis-associated heart failure hospitalizations among fee-for-service Medicare beneficiaries in the United States. Circ Heart Fail. 2019;12(6):e005407. doi: 10.1161/CIRCHEARTFAILURE.118.005407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.AbouEzzeddine OF, Davies DR, Scott CG, Fayyaz AU, Askew JW, McKie PM, et al. Prevalence of transthyretin amyloid cardiomyopathy in heart failure with preserved ejection fraction. JAMA Cardiol. 2021;6(11):1267–1274. doi: 10.1001/jamacardio.2021.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanskanen M, Peuralinna T, Polvikoski T, Notkola IL, Sulkava R, Hardy J, et al. Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: a population-based autopsy study. Ann Med. 2008;40(3):232–239. doi: 10.1080/07853890701842988. [DOI] [PubMed] [Google Scholar]
- 16.Lane T, Fontana M, Martinez-Naharro A, Quarta CC, Whelan CJ, Petrie A, et al. Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation. 2019;140(1):16–26. doi: 10.1161/CIRCULATIONAHA.118.038169. [DOI] [PubMed] [Google Scholar]
- 17.Jang SY, Kim D, Choi JO, Jeon ES. Incidence, cause of death, and survival of amyloidosis in Korea: a retrospective population-based study. Int J Heart Fail. 2021;3(3):172–178. doi: 10.36628/ijhf.2021.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim D, Choi JO, Kim K, Kim SJ, Jeon ES. Untangling amyloidosis: recent advances in cardiac amyloidosis. Int J Heart Fail. 2020;2(4):231–239. doi: 10.36628/ijhf.2020.0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jung MH, Chang S, Han EJ, Youn JC. Multimodal imaging and biomarkers in cardiac amyloidosis. Diagnostics (Basel) 2022;12(3):627. doi: 10.3390/diagnostics12030627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ravichandran S, Lachmann HJ, Wechalekar AD. Epidemiologic and survival trends in amyloidosis, 1987-2019. N Engl J Med. 2020;382(16):1567–1568. doi: 10.1056/NEJMc1917321. [DOI] [PubMed] [Google Scholar]
- 21.Dispenzieri A, Coelho T, Conceição I, Waddington-Cruz M, Wixner J, Kristen AV, et al. Clinical and genetic profile of patients enrolled in the Transthyretin Amyloidosis Outcomes Survey (THAOS): 14-year update. Orphanet J Rare Dis. 2022;17(1):236. doi: 10.1186/s13023-022-02359-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr. 2015;28(1):1–39.e14. doi: 10.1016/j.echo.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 23.Nativi-Nicolau J, Siu A, Dispenzieri A, Maurer MS, Rapezzi C, Kristen AV, et al. Temporal trends of wild-type transthyretin amyloid cardiomyopathy in the Transthyretin Amyloidosis Outcomes Survey. JACC CardioOncol. 2021;3(4):537–546. doi: 10.1016/j.jaccao.2021.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caponetti AG, Rapezzi C, Gagliardi C, Milandri A, Dispenzieri A, Kristen AV, et al. Sex-related risk of cardiac involvement in hereditary transthyretin amyloidosis: insights from THAOS. JACC Heart Fail. 2021;9(10):736–746. doi: 10.1016/j.jchf.2021.05.005. [DOI] [PubMed] [Google Scholar]
- 25.Porcari A, Razvi Y, Masi A, Patel R, Ioannou A, Rauf MU, et al. Prevalence, characteristics and outcomes of older patients with hereditary versus wild-type transthyretin amyloid cardiomyopathy. Eur J Heart Fail. 2023;25(4):515–524. doi: 10.1002/ejhf.2776. [DOI] [PubMed] [Google Scholar]
- 26.Maestro-Benedicto A, Vela P, de Frutos F, Mora N, Pomares A, Gonzalez-Vioque E, et al. Frequency of hereditary transthyretin amyloidosis among elderly patients with transthyretin cardiomyopathy. Eur J Heart Fail. 2022;24(12):2367–2373. doi: 10.1002/ejhf.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey) J Am Coll Cardiol. 2016;68(2):161–172. doi: 10.1016/j.jacc.2016.03.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ioannou A, Patel RK, Razvi Y, Porcari A, Sinagra G, Venneri L, et al. Impact of earlier diagnosis in cardiac ATTR amyloidosis over the course of 20 years. Circulation. 2022;146(22):1657–1670. doi: 10.1161/CIRCULATIONAHA.122.060852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winburn I, Ishii T, Sumikawa T, Togo K, Yasunaga H. Estimating the prevalence of transthyretin amyloid cardiomyopathy in a large in-hospital database in Japan. Cardiol Ther. 2019;8(2):297–316. doi: 10.1007/s40119-019-0142-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of wild-type ATTR-CM and Asp38Ala variant ATTR-CM subjects
Comparison of Asp38Ala variant ATTR-CM and the other variant ATTR-CM
Presentation of ATTR-CM with mean LV wall thickness < 12 mm
Time between symptom onset and diagnosis according to time periods.
Mean age at the time of diagnosis according to diagnostic time periods.
Severity of HF symptom at the time of diagnosis according to diagnostic time periods.
Diagnostic methods according to diagnostic time periods.