

Abstract
Drug therapy is vital in cancer treatment. Accurate analysis of drug sensitivity for specific cancers can guide healthcare professionals in prescribing drugs, leading to improved patient survival and quality of life. However, there is a lack of web-based tools that offer comprehensive visualization and analysis of pancancer drug sensitivity. We gathered cancer drug sensitivity data from publicly available databases (GEO, TCGA and GDSC) and developed a web tool called Comprehensive Pancancer Analysis of Drug Sensitivity (CPADS) using Shiny. CPADS currently includes transcriptomic data from over 29 000 samples, encompassing 44 types of cancer, 288 drugs and more than 9000 gene perturbations. It allows easy execution of various analyses related to cancer drug sensitivity. With its large sample size and diverse drug range, CPADS offers a range of analysis methods, such as differential gene expression, gene correlation, pathway analysis, drug analysis and gene perturbation analysis. Additionally, it provides several visualization approaches. CPADS significantly aids physicians and researchers in exploring primary and secondary drug resistance at both gene and pathway levels. The integration of drug resistance and gene perturbation data also presents novel perspectives for identifying pivotal genes influencing drug resistance. Access CPADS at https://smuonco.shinyapps.io/CPADS/ or https://robinl-lab.com/CPADS.
Keywords: drug sensitivity, CPADS, pancancer, gene perturbation, Shiny, web tools
INTRODUCTION
Drug therapy remains a cornerstone in the treatment of many cancer patients, as it can significantly increase survival rates and improve quality of life [1]. However, resistance to antineoplastic drugs—caused by genetic mutations and other nongenetic factors—has become a major obstacle limiting treatment efficacy [2–7]. Growing clinical evidence reveals that intrinsic or acquired tumour drug resistance is often associated with genetic as well as epigenetic alterations. By comparing omics and experimental data from tumour samples (or cell lines) before and after treatment, or between responders and non-responders, researchers have explored the molecular mechanisms associated with drug resistance [8–10]. For example, Vokes et al. [11] found TP53 deletions caused rapid resistance to the EGFR-targeted drug tyrosine kinase inhibitors (TKIs) in lung cancer, reducing overall survival and progression-free survival in patients. Analysing The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) data, Yang et al. [12] linked m6A gene expression to lung cancer cell sensitivity to etoposide, afatinib and dasatinib. Pothuraju et al. [13] showed MUC5AC overexpression mediated colorectal cancer resistance to 5-fluorouracil and oxaliplatin via CD44/β-catenin/p53/p21 pathway activation. In summary, while cytotoxic and targeted therapies continue to be part of treatment paradigms, resistance to such regimens emerges, thus limiting patient survival. Ultimately, integrative omics profiling provides a comprehensive understanding of factors influencing drug response and resistance, thus guiding more personalized cancer therapy. Systematic utilization of an omics approach could help to elucidate mechanisms of resistance and inform clinicians of different drug combinations that may be considered to overcome such resistance and improve patient outcomes.
Gene perturbation techniques, including knockdown, knockout or overexpression utilizing Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR), transgenic models, RNA interference and other methods, are invaluable for studying cellular physiological activities, clarifying gene functions and elucidating genotype–phenotype relationships. In recent years, increasing evidence shows that gene perturbation can help researchers discover new therapeutic targets and elucidate mechanisms of tumorigenesis and drug resistance [14]. Braun et al. [15] found that transcriptional activation of Mgmt by fusing dCas9 with the transcriptional activator VP64 induced temozolomide resistance in acute B-lymphoblastic leukaemia (B-ALL) cells in vitro and in vivo. In addition, FN3K knockdown was found to restore sensitivity of hepatocellular and non-small cell lung cancer cells to erlotinib in vivo, suggesting FN3K inhibition could block NRF2’s cancer-promoting and drug resistance effects [16]. Moreover, numerous public databases and web tools, such as MSigDB and GPSAdb [17], provide high-quality gene perturbation data. Integration of gene perturbation with functional genomics datasets could be a promising strategy for identifying genetic vulnerabilities and genotype–phenotype relationships. By elucidating cancer cell dependencies, these approaches may systematically uncover novel anticancer drug target candidates.
Many public databases, such as GEO [18] Genomics of Drug Sensitivity in Cancer (GDSC) [19], containing drug sensitivity data, are available to support physicians and researchers. However, it is difficult for medical researchers without programming expertise to access and analyse drug sensitivity datasets and corresponding clinical information. To better assist in elucidating tumour drug resistance mechanisms, several web-based tools for drug sensitivity research have been developed, including canSAR [20], CellMiner [21], DrugBank [22], GSCALite [23], PharmacoDB [24] and DRESIS [25]. These web-based applications, which integrate current oncology therapeutics, enable analysis of drug sensitivity data from selected sources. For instance, canSAR presents extensive drug and protein data to predict tumour cell drug sensitivities and associated protein targets [20], and CellMiner enables analysis of miRNA, DNA methylation and drug sensitivity data from multiple cell lines, including the NCI-60 panel, enabling analyses of miRNA, DNA methylation and drug sensitivity [19]. Although facilitating investigations of tumour-related drugs to a certain extent, most of these web tools only utilize a single database (e.g. GDSC) rather than providing a comprehensive dataset from the many available sources for drug sensitivity studies. Therefore, extensive anti-tumour drug data in GEO and TCGA remains largely untapped. Furthermore, none of these web pages directly enables simultaneous analysis of gene perturbation data and pancancer drug resistance. In terms of the richness of analysis modules, these webpages often only provide basic information listings or simple graphical displays, which cannot meet the diverse data analysis and visualization needs of users. Finally, in terms of customization, these web-based applications usually only allow users to use fixed colour schemes and methodological parameters.
To address these limitations, we developed the Comprehensive Pancancer Analysis of Drug Sensitivity (CPADS), a user-friendly web-based tool for analysing drug sensitivity across cancer types. CPADS integrates data from several major public databases, encompassing diverse cancer types and drug classes. CPADS enables differential expression analysis, correlation analysis, pathway analysis and genetic perturbation analysis, with accurate, high-definition visualization of results in a customized manner. The intuitive interface and detailed guidelines make CPADS widely accessible to clinicians and researchers alike. Overall, CPADS harnesses extensive omics resources to deliver actionable insights into tumour drug response and resistance through flexible, interactive analytics and visualizations tailored to each user.
MATERIALS AND METHODS
Data collection
The data in CPADS cover 177 datasets from GEO, 33 datasets from TCGA, drug sensitivity information from the GDSC database, and gene perturbation data from GPSAdb [17] (Supplementary Table 1). The microarray data from GEO were downloaded using the downloaded GEOquery [26] package, and high-throughput data were obtained directly from the GEO database website (https://www.ncbi.nlm.nih.gov/geo/). TCGA data were obtained from the TCGAbiolinks [27] package, which downloaded RNA-seq data in count format. GDSC data were downloaded from the GDSC webpage (https://www.cancerrxgene.org/). The gene perturbation data were obtained via the GPSAdb web tool (https://www. gpsadb.com/) and from the Chemical and Genetic Perturbations (CGP) collection in MSigDB. The above data were further screened and cleaned based on the inclusion criteria: (i) sample source of human or mouse, (ii) complete clinical information and expression profile data (microarray/high-throughput sequencing), (iii) at least 10 samples with no less than 3 samples before and after drug use and (iv) samples with a single drug treatment (to better study the effect of drug treatment on gene expression, we temporarily did not include samples with multidrug combination treatment).
Data pre-processing and analysis
Data pre-processing
For GEO microarray data, the corresponding expression profile data obtained from the GEOquery package were normalized via the limma [28] package. Similarly, the remaining high-throughput sequencing data were normalized using the limma [28] package for expression profile data. Finally, all transcriptome data were log2 transformed. The delineation of sensitive versus resistant groups for data from GEO was based on the groupings in the clinical information form provided by the data uploader, and the delineation of data from GDSC and TCGA was based on the median of the IC50 predicted by the GDSC or using the pRRopheticR package.
Differentially expressed gene analysis
For the GEO dataset, all samples were divided into groups based on drug treatment status (before or after drug treatment) or drug resistance status (resistant or sensitive) based on clinical data. Subsequently, depending on the data type and data format, the limma [28] package or DESeq2 package [29] was used for gene difference analysis, i.e. microarray data. Some high-throughput sequencing data (FPKM and RPKM formats) were processed using the limma [28] package, while the remaining count-based high-throughput sequencing data were processed using the DESeq2 package. The samples from GDSC were divided into a sensitive group with a low half-maximal inhibitory concentration (IC50) and a resistant group with a high IC50 based on the IC50 values under different drug treatments provided on the GDSC website and were analysed for differences using the limma [28] package. Notably, CPADS does not provide a variance analysis function for TCGA because this database does not provide detailed drug IC50 information. The ridge regression model was used as a proxy to predict the corresponding IC50 information for TCGA samples, and CPADS does not use this value as a basis for differentiating between drug resistance and drug sensitivity. For visualization of the variance analysis results, the ggplot package was used to draw volcano plots and box plots, and the pheatmap package was used to construct heatmaps.
Correlation analysis
For normalized expression profile data, the Spearman and Pearson algorithms were applied to compute single gene and multigene correlation analyses (using the ggstatsplot and stats packages, respectively). For visualization of the correlation analysis results, we used the ggstatsplot package, the ggplot package and the ComplexHeatmap [30] package to draw correlation scatter plots and heatmaps.
Pathway analysis
A total of 13 632 commonly used tumour-associated gene sets from the MSigDB database, including Hallmark, C2: Canonical pathways (CP) and C5: Gene Ontology gene sets (GO), were downloaded (Supplementary Table 2). Three common pathway analysis algorithms, including Gene Set Enrichment Analysis (GSEA), Single Sample Gene Set Enrichment Analysis (ssGSEA) and Pathview, were implemented via the ClusterProfiler [31] package, GSVA [32] package and Pathview [33] package, respectively. For visualization of pathway analysis results, GSEA plots, dot plots, bar plots, ridge plots, enrichment plots and heatmaps were generated via the GseaVis package, ggplot package, enrichplot package and pheatmap package for implementation.
Drug analysis
All samples were divided into high- and low-expression groups based on the median expression of the genes. A box plot of drug IC50 values according to the corresponding grouping was generated via the ggplot package. The analysis between the two groups was calculated by the Wilcoxon rank sum test via the ggpubr package.
Gene perturbation analysis
This module incorporates 3405 gene perturbation data points from CGP and 6096 gene perturbation data points from GPSAdb in the MSigDB database [17]. The GPSAdb perturbation data are derived from RNAseq data of 3048 human cell lines with gene knockdown in the GEO, and this dataset enables the identification of causal candidate perturbation genes from the differentially expressed gene after individual knockdowns [17]. Relying on the ClusterProfiler [31] package and GSVA [32] packages, we performed both GSEA and ssGSEA on the above data. For visualization of the results of the enrichment analysis, users can choose from various plots: GSEA, point, bar, ridge and enrichment. Heatmaps are generated with various packages: GseaVis, ggplot, enrichplot and pheatmap.
Web server implementation
CPADS is a free web page open to all users, and no features require a login. CPADS is available at https://smuonco.shinyapps.io/CPADS/ or https://robinl-lab.com/CPADS. CPADS development is mainly based on R (v.4.1.3) and the Shiny package.
Statistical analysis
All analyses used were provided by R (v.4.1.3). Comparisons between two groups of variables were performed using the Wilcoxon rank sum test, and all correlation coefficients were calculated by Spearman’s method and/or Pearson’s method. P < .05 was considered to indicate statistical significance, and asterisks indicate different significance levels based on P-values: ‘*’: P < .05, ‘**’: P < .01, ‘***’: P < .001, ‘****’: P < .0001.
RESULTS
CPADS currently incorporates 177 datasets from three databases (TCGA, GEO and GDSC) comprising 969 tumour samples across 44 cancer types and 288 drugs (Figure 1A). The main modules include differential expression analysis, correlation analysis, pathway analysis, drug analysis, genetic perturbation analysis and an overview of sample selection. A key highlight is the genetic perturbation module for screening potential drug resistance genes, which can be validated using the differential expression and correlation modules to aid drug resistance gene discovery. All analysis visualizations are provided as high-resolution, publication-quality PDF images. Detailed analysis results, expression profiles and clinical summary tables are downloadable in Excel format from each analysis module or the Data section.
Figure 1.

Workflow of CPADS. (A) CPADS is a web platform for pancancer pharmacovigilance analysis. A total of 177 datasets with more than 29 000 tumour samples from three databases, GEO, GDSC and TCGA, involving 288 drugs and 44 cancer types, were collected. All data passed the standard preprocessing step. CPADS enables five main types of analysis: differentially expressed gene analysis, correlation analysis, pathway analysis, drug analysis and gene perturbation analysis. The web pages are built based on R language and Shiny, and all analysis results are available in PDF format with corresponding visualization and EXCEL format with result tables. (B) The workflow diagram shows how the gene perturbation analysis module can be combined with other modules to explore potential drug resistance targets. Gene perturbation analysis is used to screen the set of genes perturbed by gene perturbation analysis that are associated with drug resistance in tumour cells, followed by the use of differential analysis, correlation analysis and other modules to verify whether the relevant set of genes can be used as potential drug resistance targets and finally to identify the genes involved in the drug resistance process.
Differentially expressed gene analysis
This module enables the comparison of gene expression changes between conditions. CPADS provides volcano plots and heatmaps to visualize differential expression across 177 GEO datasets and 30 cancer types in the GDSC under various drug treatments. Specifically, CPADS allows differential expressed gene analysis for 177 GEO and 30 GDSC datasets under different drug treatments. These analyses can be implemented via the ‘Volcano Plot’ and ‘Heatmap’ sections based on the preferred visualization method. Upregulation and downregulation of genes are depicted in volcano plots (Figure 2A and B) and heatmaps. By default, plots are labelled with the top 10 most significantly up/downregulated genes. For customized needs, the left-side controls offer options, including gene names, significance criteria, P-value and fold change thresholds, colour schemes, etc. Detailed results are available in the ‘Data’ section, where upregulation and downregulation can be assessed by table shading and text hints. In the Heatmap section, CPADS compares differential gene expression between groups (control/treatment and sensitive/resistant) with significance marked on the graphs (Figure 2C).
Figure 2.
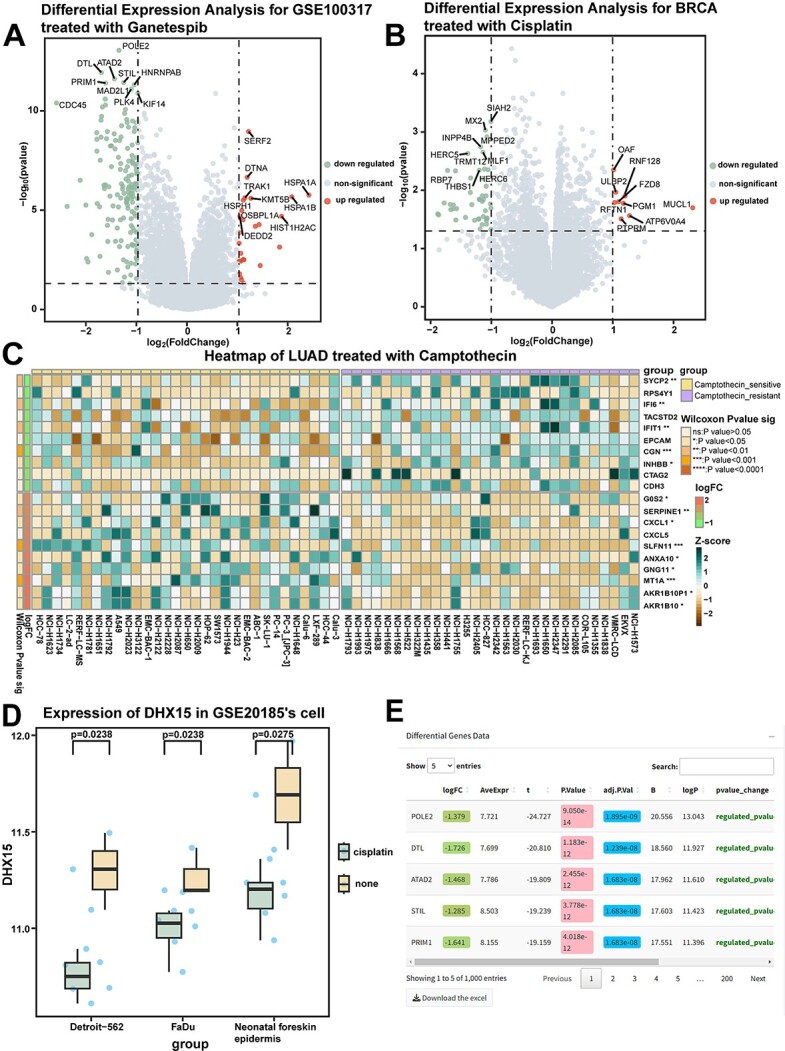
Differentially expressed gene analysis module: differential gene expression analysis in control/drug-treated groups (drug-sensitive/resistant groups). (A) The results of gene difference analysis between the ganetespib drug treatment group and the control group in GSE100317 are shown in the volcano plot. (B) The results of differentially expressed gene analysis between cisplatin-treated and control breast cancer cell lines in GDSC are shown in a volcano plot. BRCA, breast cancer. (C) Heatmap showing the results of differentially expressed gene analysis: between camptothecin-sensitive and drug-resistant lung adenocarcinoma cell lines in GDSC. P-values were calculated by the Wilcoxon rank sum test. ns: P > .05; *: P < .05; **: P < .01; ***: P < .001; ****: P < .0001. LUAD, Lung adenocarcinoma. (D) The box plot demonstrates the difference in DHX5 expression in different cell lines in GSE20185 before and after treatment with cisplatin. P-values were calculated by the Wilcoxon rank sum test. (E) The table shows the results of the analysis of the variance module.
Furthermore, the ‘Gene Analysis’ section enables differential expression analysis for 121 GEO datasets containing cell line, tissue or genotype information. This allows users to explore the expression differences of target genes between groups. Results are presented as box plots (Figure 2D). Users can also download refined visualizations and detailed result tables (Figure 2E).
Correlation analysis
In this module, CPADS provides two submodules for single-gene and multigene correlation analysis, along with visualization of results. Users can explore correlations in gene expression across cancers before and after drug treatments.
The single gene correlation module enables analysing correlations between two genes’ expression levels under different groupings (control/treatment or sensitive/resistant) for GEO data. This reveals drug effects on expression patterns. For GDSC and TCGA data, it allows correlating gene expression with IC50. Two methods (Pearson and Spearman) and multiple colour schemes are available. Figure 3A shows ZZZ3 and TP53 correlation after cerulenin treatment in GSE102791. Figure 3B shows FGD1 expression and imatinib IC50 correlation in pancreatic cancer.
Figure 3.

Genetic correlation analysis module: correlation analysis of multigene expression/single gene expression with drug IC50 before and after drug treatment. (A) The scatter plot demonstrates the correlation between ZZZ3 and TP53 before and after cerulenin treatment in GSE120791, and the R value was calculated by Spearman. (B) The scatter plot shows the correlation between the expression of the FGD1 gene and IC50 in pancreatic cancer cases treated with imatinib. The R value was calculated by the Spearman algorithm. (C) The bubble plots demonstrate the polygenic correlations in GSE100317 for the ganetespib treatment group compared to the control group. The upper right corner represents the correlation in the treatment group, and the lower left corner represents the correlation in the control group.
The multigene module allows multigene correlation analysis of up to 15 genes simultaneously for GEO and GDSC data. Correlation heatmaps are plotted (Figure 3C), with colour combinations visualizing correlations between target genes’ expression levels across groupings (control and treatment groups, sensitive and resistant groups). Customization of algorithms and colour schemes is enabled.
Pathway analysis
The GSEA module enables pathway enrichment analysis of differentially expressed genes from GEO data and GDSC data. Users can select specific pathway sets (including the common 18 803 tumour-associated gene sets in MSigDB) for visualization as classic GSEA plots (Figure 4A), dot plots (Figure 4B), bar plots (Figure 4C), ridge plots (Figure 4D) and enrichment plots (Figure 4E). Full enrichment results can be viewed in ‘Data’ to facilitate further screening of pathways of interest. For example, with GSE138863 (Figure 4A), the MYC-related HALLMARK_Myc_Targets_V1 gene set decreased after disulfiram treatment. This aligns with Meier et al., who determined MYC expression using qPCR and Western blotting after disulfiram treatment of SF188 cells and GPC16 cells. Further GSEA revealed disulfiram not only decreased MYC expression but also led to downregulation of MYC target genes [34]. After enrichment analysis using GSE130437 (Figure 4E), the palbociclib-treated group showed significant changes in cell cycle-related gene sets compared to control, consistent with Lanceta et al., who found palbociclib altered expression of cell cycle-related genes [35].
Figure 4.

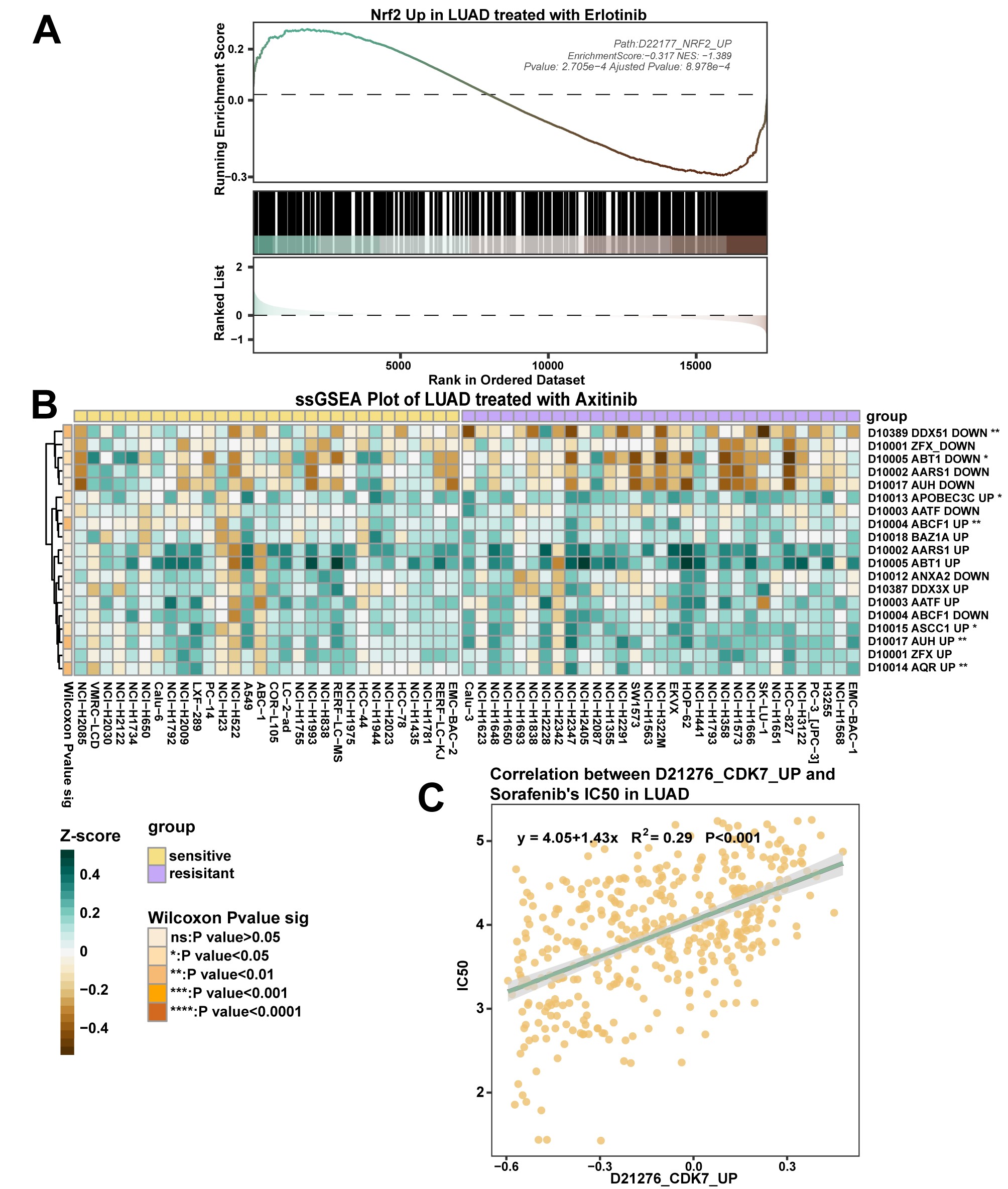
Pathway enrichment analysis module: enrichment analysis of differentially expressed genes in a specified set of genes before and after drug treatment. (A) The GSEA plot demonstrates the enrichment of GSE138863 in the MYC gene set in the treatment and control groups of disulfiram. (B–D) Dot, bar and ridge plots showing the significantly altered gene set sequencing of GSE103115 in the treated and control groups of cisplatin, respectively. (E) The enrichment analysis graph demonstrates the significantly altered gene set of GSE130437 in the palbociclib-treated and control groups. (F) The heatmap shows the results of ssGSEA of pathway enrichment in the GSE138942 JQ1 treatment and control groups. ns: P > .05, *: P < .05; **: P < .01; ***: P < .001; ****: P < .0001. (G) Pathview plots show the changes in the expression of individual genes in the ‘adherens junction’ gene set in GSE107707 after treatment with JQ1.
The ssGSEA module allows ssGSEA for 1–20 pathways of interest, and the calculated enrichment score results are presented in a heatmap form (Figure 4F). The Pathview module maps and integrates transcriptome data from drug-treated pathways of interest based on user-selected Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. It then visualizes the altered expression of individual genes within those pathways (Figure 4G).
Drug sensitivity analysis
CPADS shows the relationship between gene expression and IC50 values for 30 tumour types in GDSC and 33 types in TCGA, using box plots. The ‘By Drug’ submodule divides samples into the sensitive (low IC50) and the resistant (high IC50) groups based on the selected drug’s IC50. It plots the box diagram of differences in gene expression between high and low IC50 groups; for example, TCGA data for lung adenocarcinoma (LUAD) cases were divided into sorafenib-resistant and sensitive groups, showing high and low EGFR expression, respectively (Figure 5A).
Figure 5.

Drug analysis module: comparison of drug IC50 values under high and low gene expression groupings (or comparison of gene expression under high and low drug IC50). (A) Box plot showing EGFR gene expression in the sorafenib-sensitive and drug-resistant groups of LUAD cases in TCGA. P-values calculated by the Wilcoxon rank sum test. LUAD, lung adenocarcinoma. (B) Box plots show the IC50 of the high and low expression groups of the LUAD cell line DDIT4 gene dasatinib in GDSC. P-values were calculated by the Wilcoxon rank sum test. (C and D) The table shows the calculated results of batch screening for differentially expressed genes (C) and drugs (D). Darker background colour of logFC in green indicates a more significant downregulation of gene expression.
The ‘By Gene’ module groups cases by median expression of a selected gene, depicting IC50 value differences across drugs. For example, dividing LUAD in the TCGA by DDIT4 expression showed significantly different dasatinib IC50 values between high and low expression groups (Figure 5B), indicating that DDIT4 is a key for dasatinib resistance in LUAD cases, consistent with findings by Shi et al. [36] Notably, screening differentially expressed genes by sensitivity results or screening drugs by differentially expressed genes is very common but often laborious. To enable batch screening, P-values for all drug/gene groups are calculable at once (Figure 5C and D). Users can view full tables in ‘Data’ to obtain information for differentially expressed genes versus drug sensitivity.
Gene perturbation analysis
In this section, CPADS presents enrichment analysis results of GEO, GDSC and TCGA data in CGP and GPSAdb gene perturbation data, providing users with information on genes and pathways associated with drug resistance traits. The GSEA module performs enrichment analysis on GEO and GDSC differential gene data using 9501 perturbation gene sets from CGP and GPSAdb. Visualizations include classic GSEA plots (Supplementary Figure 1A), dot plots, bar plots, ridge plots and enrichment plots. Considering differential contributions of up/downregulated genes to pathway enrichment, we divided all genes into upregulated, downregulated and combined up/downregulated groups. Users can select from three groups according to the type of data they need to analyse. Moreover, all enrichment analysis results for the dataset are viewable in ‘Data’ for further investigation of genes or pathways of interest.
The ssGSEA module allows 1–20 pathways from GPSAdb and CGP selection for ssGSEA enrichment analysis, and the calculated enrichment scores can be presented in a heatmap form to show the high and low pathway scores and enrichment results across samples (Supplementary Figure 1B), and correlation scatter plots depict pathway score-IC50 correlations. For example, sorafenib IC50 positively correlated with the CDK7 pathway score in liver cancer (Supplementary Figure 1C), suggesting CDK7’s key role in sorafenib resistance, consistent with the findings of Wang et al. [37].
Case study: L1CAM as a new drug resistance target
By performing transcriptome analysis of cell lines both before and after drug treatment as well as gene perturbation (knockout/knockdown/overexpression), we obtained differentially expressed genes for the drug treatment versus gene perturbation conditions. Associations between drug sensitivity and individual genes can be explored with an enrichment analysis performed on both differential gene sets. The enrichment analysis results can be directly utilized to screen for key genes influencing drug sensitivity (Figure 6A). Enrichment analysis for the two groups (drug-treated and knockdown/knockout/overexpression-treated groups) of differentially expressed genes can be explored for an association of drug sensitivity with single genes. Results of this enrichment analysis can be used to correlate drug resistance with knockdown/knockout/overexpression treatments, which in turn can be used to screen for key genes affecting drug sensitivity (Figure 6A).
Figure 6.
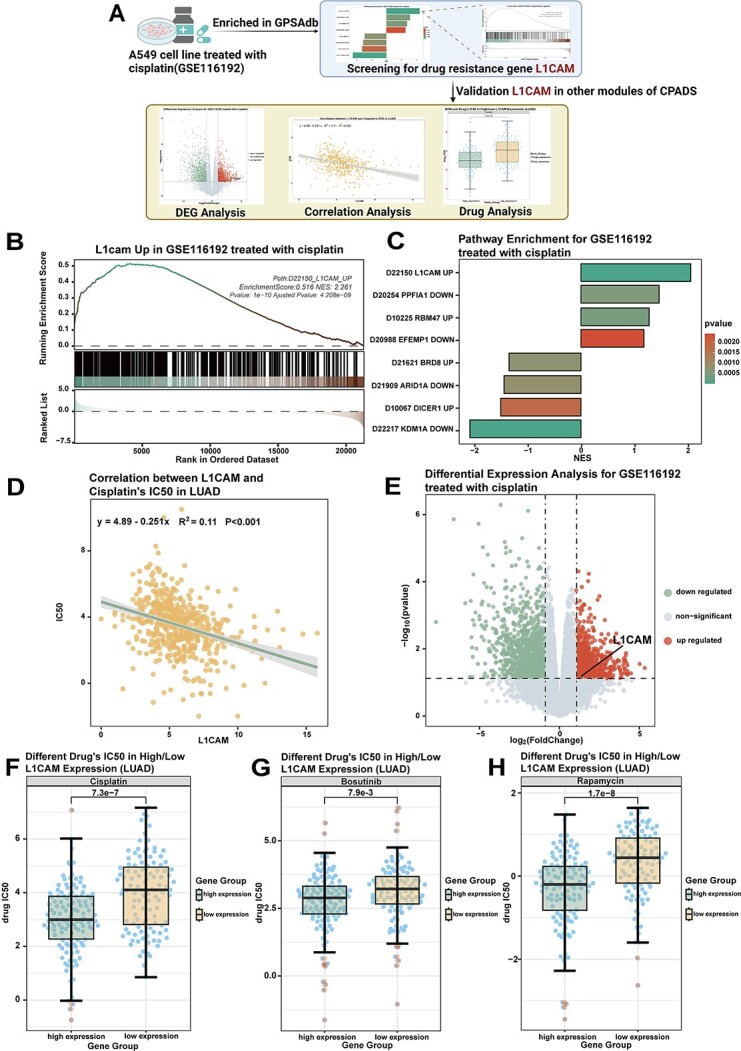
Screening for the drug resistance gene L1CAM using gene perturbation analysis. (A) Workflow diagram showing the process of screening for the drug resistance gene L1CAM by combining the gene perturbation module with other drug sensitivity analysis modules. (B) The GSEA graph shows the enrichment analysis of the differentially expressed gene of GSE116192 in the gene set ‘D22150_L1CAM_UP’. (C) The bar graph shows the enrichment of GSE116192 in GPSAdb. The colours represent the high and low P-values. (D) Volcano plot showing the results of the differential analysis performed by GSE116192. L1CAM gene expression was increased after cisplatin treatment. (E) Correlation scatter plot demonstrating the correlation between L1CAM gene expression and cisplatin IC50 in LUAD cases in TCGA. LUAD, lung adenocarcinoma, R value calculated by the Spearman algorithm. (F) The box plot demonstrates the difference in the IC50 of cisplatin between the high and low expression groups after grouping LUAD cases in TCGA according to L1CAM gene expression. P-values were calculated by the Wilcoxon rank sum test. (G) The box plot demonstrates the difference in the IC50 of bosutinib between the high and low expression groups after grouping LUAD cases in TCGA according to L1CAM gene expression. P-values were calculated by the Wilcoxon rank sum test. (E) The box plot demonstrates the difference in rapamycin IC50 between the high and low expression groups after grouping LUAD cases in the TCGA according to L1CAM gene expression. P-values were calculated by the Wilcoxon rank sum test.
As an example of this approach, we explored key genes for cisplatin resistance in non-small cell lung cancer. First, GSEA enrichment in the gene perturbation module of the cisplatin-treated dataset GSE116192 in GPSAdb showed the ‘D22150_L1CAM_UP’ gene set was significantly enriched (Figure 6B and C). This suggests overlap between genes differentially expressed after L1CAM overexpression and those altered by GSE116192 drug treatment. L1CAM overexpression may confer cisplatin resistance. Further verification in the differentially expressed gene analysis module revealed that L1CAM gene expression was upregulated with some significance after cisplatin treatment of the cell line in the GSE116192 dataset (Figure 6D). As seen in the correlation analysis, L1CAM expression is negatively correlated with cisplatin IC50 in LUAD from TCGA (Figure 6E). L1CAM high/low expression groups showed significantly different cisplatin IC50 in LUAD (Figure 6F). Differences were also seen for bosutinib/rapamycin (Figure 6G and H). These findings imply L1CAM overexpression associates with cisplatin resistance, and L1CAM may also mediate resistance to bosutinib, rapamycin, etc.
L1CAM is a 220-kDa transmembrane glycoprotein belonging to the neuronal immunoglobulin superfamily, originally described as a neural adhesion molecule involved in neuronal cell migration [36]. Outside the nervous system, aberrant L1CAM expression can promote tumour cell proliferation [37]. Previous studies demonstrate L1CAM expression associates with growth and metastasis in non-small cell lung carcinoma (NSCLC) [38]. L1CAM can confer cisplatin resistance by promoting phosphorylation of the extracellular signal kinase (ERK) and protein kinase B (AKT) pathways [39, 40]. These pathways also play key roles in NSCLC development, metastasis and drug resistance [7, 41–43]. These studies further validate the significance of L1CAM in NSCLC biology and its connection to cisplatin resistance.
DISCUSSION
CPADS is a publicly available, interactive web-based application that integrates extensive tumour drug resistance-related transcriptomic and clinical data from major public repositories, including GEO, TCGA and GDSC. By consolidating large volumes of gene expression and drug response data across diverse cancer types into one platform, CPADS aims to make pancancer analysis of drug sensitivity more convenient. The intuitive interface and detailed tutorials empower researchers and clinicians without programming expertise to easily understand transcriptomic changes before and after drug treatment for each cancer type. By elucidating these molecular alterations associated with drug response, CPADS facilitates the systematic exploration of genetic mechanisms conferring tumour drug resistance or sensitivity. These mechanistic insights can ultimately help inform evidence-based clinical decision-making around optimal therapeutic strategies for specific cancer subtypes and individual patients.
Compared with web-based tools developed in recent years to explore drug sensitivity (Table 1), CPADS has several key advantages in terms of the expansive breadth of integrated datasets, large sample size, diverse analysis methods and high degree of customizability. CPADS incorporates data from sources beyond just GDSC and TCGA—it also includes 177 manually curated datasets identified through comprehensive screening of the GEO repository and permits future GEO datasets to be easily added and integrated. By harnessing data from these three major public resources, CPADS currently amasses over 29 000 pharmacogenomic samples spanning 288 drugs and 44 cancer types, greatly expanding the richness of data compared to previous tools. Moreover, to empower multifaceted investigation, CPADS offers researchers and clinicians five analysis modules, including differential expression, correlation, pathway, drug sensitivity and genetic perturbation analysis. Each module provides multiple visualization options, including plots, heatmaps, enrichment graphs and more. Users can customize all visualizations by tuning parameters, like thresholds, colour schemes, significance criteria and more to meet their professional needs and research questions. This flexibility and depth of analytics enables the exploration of diverse hypotheses related to tumour drug responses. Overall, the unparalleled scale and diversity of integrated datasets, coupled with versatile analytics and customization, make CPADS a comprehensive and user-friendly portal for pancancer pharmacogenomic research.
Table 1.
Comparison of CPADS with other tumour drug sensitivity analysis web pages
| CPADS | canSAR [18] | CellMiner [19] | GSCALite [21] | PharmacoDB [22] | DRESIS [23] | |
|---|---|---|---|---|---|---|
| Dataset | TCGA, GDSC, GEO(including NCI-60) | TCGA, ICGC | GDSC, CCLE, CTRP, NCI-60 | GDSC, TCGA, GTEx | NCI-60, GDSC, PRISM, CCLE, gCSI | DrugBank, CCNSC, TTD |
| Sample | ~29 000 | ~25 000 | 1400 | 23 594 | 1757 | – |
| Cancer Type | 44 | 26 | 60 | 33 | 30 | 125 |
| Cancer Analysis Module | ①Differential expression gene analysis | Genomic data analysis | Correlation analysis | ①Genomic data analysis | ①Genomic data analysis | Resistance mechanisms analysis |
| ②Correlation analysis | ②Pathway activity | ②Correlation analysis | ||||
| ③Pathway analysis(GSEA, ssGSEA, Pathview) | ③miRNA network | ③Drug sensitivity | ||||
| ④Drug analysis | ④Drug sensitivity | |||||
| ⑤Gene perturbations | ||||||
| Parameters Customization | YES | NO | NO | NO | NO | NO |
| Colour Customization | YES | NO | NO | NO | NO | NO |
TCGA, The Cancer Genome Atlas; GDSC, The Cancer Genome Atlas; GEO, Gene Expression Omnibus; GSEA, Gene Set Enrichment Analysis; ssGSEA, Single sample gene set enrichment analysis; ICGC, International Cancer Genome Consortium; CCLE, Cancer Cell Line Encyclopedia; CTRP, Cancer Therapeutics Response Portal; NCI, National Cancer Institute; GTEx, Genotype-Tissue Expression; PRISM, Profiling Relative Inhibition Simultaneously in Mixtures; gCSI, Genentech Cell Line Screening Initiative; CCNSC, Cancer Chemotherapy National Service Center; TTD, Therapeutic Target Database.
While CPADS integrates extensive datasets, we acknowledge that there are still limitations in terms of the currently incorporated data sources. The initial release relies solely on drug sensitivity data from GEO, GDSC and TCGA. However, valuable pharmacogenomics data continue to emerge, such as from the Cancer Genome Project and continuously updated GEO repository. Our team actively monitors new datasets and literature to identify promising drug sensitivity resources. We plan to incorporate additional databases into future CPADS versions to enhance data breadth. Moreover, as research progresses, new techniques and tools for analysing drug responses will arise. Moreover, CPADS still has some limitations in predicting drug sensitivity, and there is still room for improvement in enhancing the specificity and sensitivity of prediction compared to existing tools that use algorithms, such as deep learning [44], graph convolutional networks (GCNs) and autoencoders (AEs) [45]. Next, we will further explore the integration of antitumour drug data and algorithms and enhance the prediction effect of CPADS. We will continue honing CPADS analysis modules to incorporate cutting-edge methods in the field, ensuring the tool leverages the latest advances in pancancer pharmacogenomics. We also aim to implement new modules to meet emerging user needs and apply state-of-the-art visualizations. We recognize the current limitations in data sources and analysis capabilities. User feedback will help guide optimal expansion of databases, analytical methods and features. We envision CPADS evolving as a one-stop resource for pancancer pharmacogenomics through iterative refinement.
We developed CPADS, a user-friendly web-based platform integrating diverse multi-omic datasets. CPADS provides an array of analytical modules, including differential expression, correlation, pathway and perturbation analysis, enabling rapid investigation of drug response across cancer types. Customizable parameters and visualizations allow tailored analysis to meet diverse research needs. Detailed guidelines make CPADS accessible to users without programming expertise. By harnessing pharmacogenomic resources into flexible, interactive analytics, CPADS delivers insights into tumour drug sensitivity and resistance. We envision CPADS evolving into a practical tool assisting in-depth exploration of tumour pharmacogenomics, guiding more effective cancer therapies.
Key Points
CPADS collects over 29 000 pharmacogenomic samples from GEO, GDSC and TCGA, covering 288 drugs and 44 cancer types, greatly expanding the richness of the data compared to previous tools.
CPADS offers researchers and clinicians five analysis modules, including differential expression, correlation, pathway, drug sensitivity and genetic perturbation analysis. It should be noted that CPADS provides a gene perturbation analysis function that covers both GPSA and CGP data, which facilitates users to better explore drug resistance genes and their mechanisms.
A wide range of analytical modules are available with flexible customisation options to meet the free and professional needs of the user.
Supplementary Material
{kind=link}
Author Biographies
Kexin Li is a researcher at the Department of Oncology, Zhujiang Hospital, Southern Medical University, Guangzhou, Guangdong, China. She is experienced in genome-wide data handling and biological network analysis.
Hong Yang is a researcher at the Department of Oncology, Zhujiang Hospital, Southern Medical University, Guangzhou, Guangdong, China. She is experienced in genome-wide data handling and biological network construction.
Anqi Lin is a researcher at the Department of Oncology, Zhujiang Hospital, Southern Medical University, Guangzhou, Guangdong, China. She is experienced in genome-wide data handling and biological network analysis.
Jiayi Xie is a researcher at the Department of Oncology, Zhujiang Hospital, Southern Medical University, Guangzhou, Guangdong, China. She is experienced in genome-wide data handling and biological network analysis.
Haitao Wang is a researcher at the Thoracic Surgery Branch Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA. He is experienced in bioinformatics in the detection, diagnosis and prediction of disease.
Jianguo Zhou is a researcher at the Second Affiliated Hospital of Zunyi Medical University, Zunyi, P. R. China. He is experienced in bioinformatics in the detection, diagnosis and prediction of disease.
Shamus R. Carr is a researcher at the Thoracic Surgery Branch Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA. He is experienced in bioinformatics in the detection, diagnosis and prediction of disease.
Zaoqu Liu is a master student at the Department of Interventional Radiology, The First Affiliated Hospital of Zhengzhou University. His research interests include bioinformatics, machine learning, precision medicine and cancer system biology.
Xiaohua Li is a researcher at the Department of Respiratory and Critical Care Medicine, Sixth People's Hospital of Chengdu, Chengdu, Sichuan, China. She is experienced in genome-wide data handling and biological network analysis.
Jian Zhang is a group leader at the Department of Oncology, Zhujiang Hospital, Southern Medical University, Guangzhou, Guangdong, China. His research is focused on high-throughput omics data analysis of multiple cancer types.
Quan Cheng is a researcher in the Department of Neurosurgery at Xiangya Hospital, China. His research is focused on high-throughput omics data analysis of central nervous system tumors and precision medicine for cancer.
David S. Schrump is a researcher at the Thoracic Surgery Branch Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA. His research is focused on high-throughput omics data analysis of multiple cancer types.
Peng Luo is a senior researcher at the Department of Oncology, Zhujiang Hospital, Southern Medical University, Guangzhou, Guangdong, China. He is experienced in genome-wide data handling and biological network analysis.
Ting Wei is a professor at the Department of Oncology, Zhujiang Hospital, Southern Medical University, Guangzhou, Guangdong, China. Her research interests are at the intersection of bioinformatics and genomics and their role in tumors.
Contributor Information
Kexin Li, Department of Oncology, Zhujiang Hospital, Southern Medical University, No. 253, Industrial Avenue Zhong, Guangzhou, Guangdong, China; The Second School of Clinical Medicine, Southern Medical University, No. 253, Industrial Avenue Zhong, Guangzhou, Guangdong, China.
Hong Yang, Department of Oncology, Zhujiang Hospital, Southern Medical University, No. 253, Industrial Avenue Zhong, Guangzhou, Guangdong, China.
Anqi Lin, Department of Oncology, Zhujiang Hospital, Southern Medical University, No. 253, Industrial Avenue Zhong, Guangzhou, Guangdong, China.
Jiayi Xie, Department of Oncology, Zhujiang Hospital, Southern Medical University, No. 253, Industrial Avenue Zhong, Guangzhou, Guangdong, China.
Haitao Wang, Thoracic Surgery Branch Center for Cancer Research, National Cancer Institute, National Institutes of Health, 9609 Medical Center Drive, Rockville, Maryland 20850, United States.
Jianguo Zhou, Department of Oncology, The Second Affiliated Hospital of Zunyi Medical University, Intersection of Xinlong Avenue and Xinpu Avenue, Honghuagang District, Zunyi City 563000, China.
Shamus R Carr, Thoracic Surgery Branch Center for Cancer Research, National Cancer Institute, National Institutes of Health, 9609 Medical Center Drive, Rockville, Maryland 20850, United States.
Zaoqu Liu, State Key Laboratory of Proteomics, Beijing Proteome Research Center, National Center for Protein Sciences (Beijing), Beijing Institute of Lifeomics, No. 33, Life Park Road, Zhongguancun, Changping District, Beijing 102206, China; State Key Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, Department of Pathophysiology, Peking Union Medical College, No. 1 Shuaifuyuan Wangfujing Dongcheng District, Beijing 100730, China.
Xiaohua Li, Department of Respiratory and Critical Care Medicine, Sixth People’s Hospital of Chengdu, No. 16, Jianjian South Street, Chengdu, Sichuan Province 610051, China.
Jian Zhang, Department of Oncology, Zhujiang Hospital, Southern Medical University, No. 253, Industrial Avenue Zhong, Guangzhou, Guangdong, China.
Quan Cheng, Department of Neurosurgery, National Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, No. 87 Xiangya Road, Changsha, Hunan, China; Xiangya Hospital, Central South University, No. 87 Xiangya Road, Changsha, Hunan, China.
David S Schrump, Thoracic Surgery Branch Center for Cancer Research, National Cancer Institute, National Institutes of Health, 9609 Medical Center Drive, Rockville, Maryland 20850, United States.
Peng Luo, Department of Oncology, Zhujiang Hospital, Southern Medical University, No. 253, Industrial Avenue Zhong, Guangzhou, Guangdong, China.
Ting Wei, Department of Oncology, Zhujiang Hospital, Southern Medical University, No. 253, Industrial Avenue Zhong, Guangzhou, Guangdong, China.
FUNDING
Natural Science Foundation of Guangdong Province (2018A030313846 and 2021A1515012593), the Science and Technology Planning Project of Guangdong Province (2019A030317020), the National Natural Science Foundation of China (81802257, 81871859, 81772457, 82172750 and 82172811), GuangDong Basic and Applied Basic Research Foundation (Guangdong - Guangzhou Joint Fouds) (2022A1515111212), Medical Science and Technology Project of Sichuan Provincial Health Commission (Number 21PJ153).
DATA AVAILABILITY
R Studio is a widely used language and environment for statistical computing (https://www.r-project.org/). Shiny, an open-source collaboration program, can be found on the GitHub repository (https://github.com/rstudio/shiny). For this study, TCGA data were acquired from the TCGA database (https://www.cancer.gov/tcga) using the TCGAbiolinks package (https://github.com/BioinformaticsFMRP/TCGAbiolinks). Gene expression data from GEO (NCBI’s Gene Expression Omnibus) (https://www.ncbi.nlm.nih.gov/geo) and GDSC (Genomics of Drug Sensitivity in Cancer) (https://www.cancerrxgene.org/) were also obtained. Furthermore, data from the GPSA were collected from the GPSA webpage (https://www.gpsadb.com/).
AUTHOR CONTRIBUTION
All authors were involved in the conceptualization and design of the study. Kexin Li, Hong Yang, Anqi Lin, Jiayi Xie and Haitao Wang collected the data, performed the data analysis, constructed the web pages, and wrote the manuscript. Jianguo Zhou, Zaoqu Liu, Xiaohua Li, Quan Cheng, Jian Zhang and Peng Luo reviewed the manuscript and supervised the work. All authors read and approved the final manuscript.
References
- 1. Bedard PL, Hyman DM, Davids MS, Siu LL. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 2020;395:1078–88. [DOI] [PubMed] [Google Scholar]
- 2. Boumahdi S, Sauvage FJ. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov 2020;19:39–56. [DOI] [PubMed] [Google Scholar]
- 3. Nussinov R, Tsai C-J, Jang H. Anticancer drug resistance: an update and perspective. Drug Resist Updat 2021;59:100796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu L, Xie Y, Yang H, et al. HPVTIMER: a shiny web application for tumor immune estimation in human papillomavirus-associated cancers. iMeta 2023;2:e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sammut S-J, Crispin-Ortuzar M, Chin S-F, et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature 2022;601:623–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang N, Kandalai S, Zhou X, et al. Applying multi-omics toward tumor microbiome research. iMeta 2023;2:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tan AC. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac Cancer 2020;11:511–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aissa AF, Islam ABMMK, Ariss MM, et al. Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer. Nat Commun 2021;12:1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vokes NI, Chambers E, Nguyen T, et al. Concurrent TP53 mutations facilitate resistance evolution in EGFR-mutant lung adenocarcinoma. J Thorac Oncol 2022;17:779–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang Y, Qian Z, Feng M, et al. Study on the prognosis, immune and drug resistance of m6A-related genes in lung cancer. BMC Bioinformatics 2022;23:437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pothuraju R, Rachagani S, Krishn SR, et al. Molecular implications of MUC5AC-CD44 axis in colorectal cancer progression and chemoresistance. Mol Cancer 2020;19:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu D, Zhao X, Tang A, et al. CRISPR screen in mechanism and target discovery for cancer immunotherapy. Biochim Biophys Acta Rev Cancer 2020;1874:188378. [DOI] [PubMed] [Google Scholar]
- 13. Braun CJ, Bruno PM, Horlbeck MA, et al. Versatile in vivo regulation of tumor phenotypes by dCas9-mediated transcriptional perturbation. Proc Natl Acad Sci U S A 2016;113:E3892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sanghvi VR, Leibold J, Mina M, et al. The oncogenic action of NRF2 depends on de-glycation by Fructosamine-3-kinase. Cell 2019;178:807–819.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo S, Xu Z, Dong X, et al. GPSAdb: a comprehensive web resource for interactive exploration of genetic perturbation RNA-seq datasets. Nucleic Acids Res 2023;51:D964–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002;30:207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang W, Soares J, Greninger P, et al. Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 2013;41:D955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mitsopoulos C, Di Micco P, Fernandez EV, et al. canSAR: update to the cancer translational research and drug discovery knowledgebase. Nucleic Acids Res 2021;49:D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Luna A, Elloumi F, Varma S, et al. CellMiner cross-database (CellMinerCDB) version 1.2: exploration of patient-derived cancer cell line pharmacogenomics. Nucleic Acids Res 2021;49:D1083–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018;46:D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu C-J, Hu F-F, Xia M-X, et al. GSCALite: a web server for gene set cancer analysis. Bioinformatics 2018;34:3771–2. [DOI] [PubMed] [Google Scholar]
- 22. Feizi N, Nair SK, Smirnov P, et al. PharmacoDB 2.0: improving scalability and transparency of in vitro pharmacogenomics analysis. Nucleic Acids Res 2022;50:D1348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun X, Zhang Y, Li H, et al. DRESIS: the first comprehensive landscape of drug resistance information. Nucleic Acids Res 2023;51:D1263–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Davis S, Meltzer PS. GEOquery: a bridge between the gene expression omnibus (GEO) and BioConductor. Bioinformatics 2007;23:1846–7. [DOI] [PubMed] [Google Scholar]
- 25. Colaprico A, Silva TC, Olsen C, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 2016;44:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016;32:2847–9. [DOI] [PubMed] [Google Scholar]
- 29. Yu G, Wang L-G, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013;29:1830–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meier S, Cantilena S, Niklison Chirou MV, et al. Alcohol-abuse drug disulfiram targets pediatric glioma via MLL degradation. Cell Death Dis 2021;12:785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lanceta L, O’Neill C, Lypova N, et al. Transcriptomic profiling identifies differentially expressed genes in Palbociclib-resistant ER+ MCF7 breast cancer cells. Genes (Basel) 2020;11:467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shi Y, Xu Y, Xu Z, et al. TKI resistant-based prognostic immune related gene signature in LUAD, in which FSCN1 contributes to tumor progression. Cancer Lett 2022;532:215583. [DOI] [PubMed] [Google Scholar]
- 35. Wang C, Jin H, Gao D, et al. A CRISPR screen identifies CDK7 as a therapeutic target in hepatocellular carcinoma. Cell Res 2018;28:690–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lindner J, Rathjen FG, Schachner M. L1 mono- and polyclonal antibodies modify cell migration in early postnatal mouse cerebellum. Nature 1983;305:427–30. [DOI] [PubMed] [Google Scholar]
- 37. Gavert N, Conacci-Sorrell M, Gast D, et al. L1, a novel target of beta-catenin signaling, transforms cells and is expressed at the invasive front of colon cancers. J Cell Biol 2005;168:633–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hai J, Zhu C-Q, Bandarchi B, et al. L1 cell adhesion molecule promotes tumorigenicity and metastatic potential in non-small cell lung cancer. Clin Cancer Res 2012;18:1914–24. [DOI] [PubMed] [Google Scholar]
- 39. Yoon H, Min J-K, Lee DG, et al. L1 cell adhesion molecule and epidermal growth factor receptor activation confer cisplatin resistance in intrahepatic cholangiocarcinoma cells. Cancer Lett 2012;316:70–6. [DOI] [PubMed] [Google Scholar]
- 40. Roberts CM, Tran MA, Pitruzzello MC, et al. TWIST1 drives cisplatin resistance and cell survival in an ovarian cancer model, via upregulation of GAS6, L1CAM, and Akt signalling. Sci Rep 2016;6:37652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen B, Song Y, Zhan Y, et al. Fangchinoline inhibits non-small cell lung cancer metastasis by reversing epithelial-mesenchymal transition and suppressing the cytosolic ROS-related Akt-mTOR signaling pathway. Cancer Lett 2022;543:215783. [DOI] [PubMed] [Google Scholar]
- 42. Song P, Song B, Liu J, et al. Blockage of PAK1 alleviates the proliferation and invasion of NSCLC cells via inhibiting ERK and AKT signaling activity. Clin Transl Oncol 2021;23:892–901. [DOI] [PubMed] [Google Scholar]
- 43. Pan Z, Wang K, Wang X, et al. Cholesterol promotes EGFR-TKIs resistance in NSCLC by inducing EGFR/Src/Erk/SP1 signaling-mediated ERRα re-expression. Mol Cancer 2022;21:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheng X, Dai C, Wen Y, et al. NeRD: a multichannel neural network to predict cellular response of drugs by integrating multidimensional data. BMC Med 2022;20:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang H, Dai C, Wen Y, et al. GADRP: graph convolutional networks and autoencoders for cancer drug response prediction. Brief Bioinform 2023;24:bbac501. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
R Studio is a widely used language and environment for statistical computing (https://www.r-project.org/). Shiny, an open-source collaboration program, can be found on the GitHub repository (https://github.com/rstudio/shiny). For this study, TCGA data were acquired from the TCGA database (https://www.cancer.gov/tcga) using the TCGAbiolinks package (https://github.com/BioinformaticsFMRP/TCGAbiolinks). Gene expression data from GEO (NCBI’s Gene Expression Omnibus) (https://www.ncbi.nlm.nih.gov/geo) and GDSC (Genomics of Drug Sensitivity in Cancer) (https://www.cancerrxgene.org/) were also obtained. Furthermore, data from the GPSA were collected from the GPSA webpage (https://www.gpsadb.com/).