

Abstract
Cell death is a major determinant of inflammatory disease severity. Whether cells live or die during inflammation largely depends on the relative success of the pro-survival process of autophagy versus the pro-death process of apoptosis. These processes interact and influence each other during inflammation and there is a checkpoint at which cells irrevocably commit to either one pathway or another. This review will discuss the concept of the autophagy/apoptosis checkpoint and its importance during inflammation, the mechanisms of inflammation leading up to the checkpoint, and how the checkpoint is regulated. Understanding these concepts is important since manipulation of the autophagy/apoptosis checkpoint represents a novel opportunity for treatment of inflammatory diseases caused by too much or too little cell death.
Keywords: Beclin-1, Atg5, HMGB1, Caspase, Calpain
Introduction
Inflammation is a cellular response to stress, injury, or infection. The idea of inflammation dates to at least the first century AD when Celsus identified redness (rubor), heat (calor), swelling (tumor), and pain (dolor) as the cardinal signs of inflammation. However, we are just now beginning to understand the molecular basis of inflammation and the role that it plays in disease. In most cases, inflammation is a beneficial and physiologic process. Self-renewing tissues, such as the gastrointestinal tract or skin, are exposed to the external environment and undergo low-level inflammation constantly. Under these conditions inflammation acts to make cells resistant to infection through initiation of cellular defense mechanisms and readies tissues for repair by activating resident or infiltrating stem cells. When tissues become damaged, inflammation recruits professional immune cells to areas of damage and increases their numbers by activating proliferative programs. It also leads to limited, controlled cell death to remove infected or damaged cells and allow tissue healing.
The role of cell death in inflammation is particularly interesting since inappropriate cell death is linked to many different inflammatory diseases. Too little cell death can lead to neoplasia or chronic infection while too much can lead to organ failure or microbial invasion into normally sterile tissues. These factors mean that cell death during inflammation is a complicated and highly regulated process. Cellular survival and death mechanisms are simultaneously activated during inflammation and are extensively interconnected. These interconnections appear to be regulated leading to the concept that a “checkpoint” exists that commits cells to either survival or death. When this checkpoint malfunctions it can lead to inflammatory disease, but it also represents an opportunity to manipulate cell survival or death for treatment.
The importance of the autophagy/apoptosis checkpoint during inflammation
Macroautophagy, referred to as autophagy in this review, is a process in which intracellular contents are targeted, engulfed by a double membrane, and transported to lysosomes for destruction [1]. It is generally considered a pro-survival and anti-inflammatory process since autophagy removes damaged cellular components and destroys microbial invaders. Apoptosis is the most common and best characterized form of inflammation-induced cell death [2]. It is a process wherein stressed or damaged cells activate proteolytic signaling cascades that result in cell death. Both autophagy and apoptosis are activated by inflammatory stimuli and compete and interact within cells. The competition is for protein effectors that participate in both pathways and each pathway generates and destroys components that are required for or regulate the opposing pathway. In the course of this competition and interaction, the cells reach a tipping point or checkpoint where they commit to either autophagy or apoptosis with subsequent survival or death. Regulation of this checkpoint is essential since both autophagy and cell death have beneficial and detrimental effects during inflammation.
Beneficial and detrimental effects of autophagy
Autophagy performs many functions that are essential for cell survival during inflammation [3]. It preserves energy availability and removes damaged organelles through limited cellular catabolism [4]. Removal of damaged mitochondria is particularly important since they produce reactive oxygen species and contribute to cell stress and damage. Autophagy decreases pro-inflammatory signals by eliminating intracellular organisms, degrading pro-inflammatory signaling platforms, and by controlling cytokine production and release [5]. It also clears or limits the spread of infection since microorganisms free in the cytosol or encased in phagosomes can be captured and delivered to lysosomes by autophagy [6, 7]. The net effect of microbial capture by autophagy is usually anti-inflammatory through destruction of pro-inflammatory components of microbes. However, it also potentially amplifies inflammation through delivery of cytosolic or phagocytosed microbially derived components to endosomes for innate immune recognition and to endo-lysosomal compartments for antigen presentation [8–10]. Autophagy-dependent antigen presentation also regulates adaptive immune cells, which in turn depend on autophagy for a variety of specialized functions [11].
Though the net effect of autophagy is generally considered beneficial to cells, it can also have detrimental effects. For example, autophagy has been implicated in tumorigenesis and tumor proliferation due to aberrant survival of transformed cells [12, 13]. In addition, the autophagic membrane or completed autophagosomes can be co-opted by intracellular microbes leading to microbial persistence within infected cells or viral replication utilizing components of the autophagic machinery [14, 15].
Beneficial and detrimental effects of apoptosis
During infection, cell death can limit the spread of microbes through direct killing or by depriving them of cellular resources for survival and replication [16–20]. Microbes released by dying cells are also exposed to immune detection by professional immune cells and can be killed through innate or adaptive immune targeting. This is particularly important for defense against microbes that have evolved to evade intracellular killing mechanisms and survive inside cells [21]. Limited cell death removes damaged cells to allow healing with restoration of normal tissue or organ structure and function [22]. This includes programmed death of activated immune cells that are eliminated to reduce the numbers of potentially autoreactive cells and minimize the risk of immune-mediated disease [23]. Inflammation-induced cell death also has the potential to alert non-affected sites and ready them for assault [24].
While cell death has many beneficial effects during inflammation, it can also contribute to inflammatory disease. The death of cells lining epithelial surfaces can lead to compromised barriers between the organism and the extracellular environment [25]. This puts the organism at risk for microbial invasion, exposure to environmental toxins, or loss of nutrients across the damaged interface. Death of parenchymal or supporting cell populations within organs can threaten vital life-sustaining functions and three-dimensional integrity of those organs [26]. Some microbes have also found ways to use cell death to their advantage. Cell death releases intracellular microbes to infect new cells and death of infected or noninfected cells can remove barriers to systemic dissemination of microorganisms [27]. Inflammation-induced death during infection can also lead to elimination of specific cell populations that can potentially kill microbes and microorganisms can co-opt cellular death mechanisms for their own purposes [28–30]. Finally, cell death can release intracellular pro-inflammatory molecules which perpetuate inflammation and damage beyond the initial threat to lead to chronic inflammatory disease and pathologic tissue remodeling [31].
The potential for beneficial and detrimental effects of both autophagy and cell death means that the mechanisms that determine whether cells live, or die during inflammation are highly regulated. Autophagy and apoptosis are two of the major mechanisms influencing these decisions, so understanding how these processes are activated, how they proceed in cells during inflammation, and how they influence each other, is important in understanding cell death during inflammation.
Activation of autophagy and apoptosis during inflammation
Multiple inputs feed into the cell to activate autophagy and apoptosis during inflammation. The character and kinetics of each pathway are influenced by the nature of the initial inflammatory stimulus, the level and duration of inflammatory stimulation, and how the cell detects the stimulus. These factors allow differential cellular outcomes during the same inflammatory event since different cell populations may be exposed to different inflammatory stimuli based on their location within tissues or have different sets of receptors to sense inflammatory stimuli.
The inflammatory environment
Initial inflammatory stimuli experienced by cells can originate from invading microbes, the host, or both host and microbe. Inflammatory stimuli originating from microbes primarily consist of pathogen-associated molecular pattern molecules (PAMPs). Microbial PAMPs include di- and tri-acylated lipoproteins, components of peptidoglycan, nucleic acids, flagellin, lipopolysaccharide (LPS), zymosan, β-glycan, α-mannan, glycoinositolphospholipids, lipoteichoic acid and many others [32].
Upon encountering PAMPs, host cells produce and release their own pro-inflammatory signals. These include a huge array of cytokines and chemokines. A discussion of the role of cytokines in inflammation is beyond the scope of this review, but a small number of cytokines have been strongly linked to both autophagy and inflammation-induced cell death. These include tumor necrosis factor alpha (TNF-α), the interleukin-1 family (IL-1α and IL-1β), and interferon-gamma (IFN-γ) [33–40].
Cells that die during inflammation or certain immune cells also contribute pro-inflammatory stimuli through release of intracellular damage-associated molecular pattern molecules (DAMPs). This terminology covers a wide range of intracellular components that includes high mobility group box-1 (HMGB1), heat shock proteins, the S100 family of proteins, adenosine triphosphate (ATP), uric acid, and many others [41]. DAMPs provide pro-inflammatory stimuli in the absence of microbes or potentiate microbe-induced inflammation.
Host cells also produce death receptor ligands during inflammatory responses. The founding member of this family of pro-inflammatory/pro-death molecules was the cytokine TNF-α, but it now also includes Fas ligand (FasL/CD95L), TNF-related apoptosis-inducing ligand (TRAIL/Apo2L), and TNF-like ligand 1A (TL1A) [42].
The range of pro-inflammatory stimuli present during immune responses means that cells experience a complex program of inflammatory stimuli, not just a single pro-inflammatory factor. The pattern, intensity, and duration of stimuli imparts a range of information that includes the presence of invading microbes and the outcome of inflammation in other cells involved in the response.
Level and duration of inflammatory stimuli
The initial wave of inflammatory stimuli is characterized not only by the types of stimuli present, but also by the levels and the length of time that they are present in the inflammatory milieu. This is true for both microbial and host elements in the inflammatory environment. As the host response develops, increasing amounts of microbial PAMPs may be present in the environment due to microbial replication or increased killing. However, successful responses should lead to PAMP detection for a relatively short period of time. The long-term presence of PAMPs within the inflammatory environment indicates a potentially failed response and can change the nature of the cell types and host pro-inflammatory mediators present in the inflammatory environment. The number of host cells in the inflammatory environment also changes over the course of an inflammatory response. As the response continues, larger numbers of host cells are recruited into a response leading to increased amounts of cytokines, chemokines, and DAMPs in the environment. This also means that the proximity of cells to the epicenter of the inflammatory event can influence their response. This is particularly true of professional immune cells that migrate along chemotactic gradients to reach sites of inflammation [43].
Sensing extracellular-derived signals of inflammation
Microbial derived pattern molecules and DAMPs are sensed through a set of intracellular and cell surface expressed receptors called pattern recognition receptors. These receptors are classified into five families based on protein domain homology: toll-like receptors (TLR), nucleotide-oligomerization domain (NOD)-like receptors (NLR), absent in melanoma-2 (AIM2)-like receptors (ALRs), C-type lectin receptors (CLR), and the retinoic acid-inducible gene-I (RIG-1)-like receptors (RLR) [44].
Toll-like receptors are type I transmembrane proteins that contain leucine-rich repeats. They recognize bacterial, viral, fungal, and protozoal PAMPs at the cell surface (TLR1, TLR2, TLR4, TLR5, TLR6, and TLR11) or in endosomes (TLR3, TLR7, TLR8, TLR9, and TLR10) [41]. The array of TLR expressed in a given cell is dependent upon cell type and activation status [45]. Toll-like receptors are also thought to sense DAMPs, either alone or in complex with microbial components. After ligand binding, TLR homo- or hetero-dimerize and recruit intracellular Toll/interleukin-1 receptor (TIR) adaptor molecules via TIR–TIR interactions [44]. All of the TLR except TLR3 utilize the myeloid differentiation primary-response protein 88 (MyD88)-dependent pathway, which controls the activation of mitogen-activated protein kinases (MAPKs) and the transcription factor nuclear factor-kappa B (NF-κB) [46]. Toll-like receptor 3 and TLR4 utilize the TIR-domain-containing adaptor protein inducing IFN-β (TRIF/TICAM1)-dependent pathway, which mediates type I IFN production [47]. Activation of NF-κB and MAPK results in transcriptional upregulation of pro-inflammatory cytokines, chemokines, interferons and cell survival factors along with activation of non-transcriptional responses, such as phagocytosis, autophagy, cytokine processing, and sometimes death [44].
The NLR family consists of at least 22 members, including the NOD proteins (NOD1 and NOD2) and the inflammasome-associated NLR (NLRP1, NLRP3, NLRC4). These are cytosolic sensors that recognize PAMPs and DAMPs. NOD1 and NOD2 detect components of bacterial peptidoglycan whereas inflammasomes detect Bacillus anthracis lethal toxin, ATP, uric acid, fungi, bacteria, viruses, bacterial flagellin or certain bacterial type III secretion systems [41]. Signaling through the NOD1 and NOD2 receptors recruits the adaptor protein RIPK2/RICK which activates NF-κB and the MAPK pathway [48, 49]. This leads to pro- and anti-inflammatory changes in cells that are similar to TLR signaling. Signaling through inflammasome-associated NLR triggers the assembly of inflammasomes, multiprotein cytosolic complexes that cleave and activate pro-caspase 1 [50]. Active caspase-1 cleaves pro-IL-1β and pro-IL-18 into the active forms of these cytokines [51]. Production of these mature cytokines requires the coordinated efforts of multiple innate immune receptors since pro-IL-β and pro-IL-18 are produced in response to NF-κB activation. Inflammasome assembly and caspase-1 activation leads to a specific form of pro-inflammatory death, pyroptosis, in immune cells.
The ALR represents another type of inflammasome. The founding member of this family is AIM2, which detects cytosolic DNA [52]. Upon ligand binding AIM2 interacts with the adaptor ASC and triggers inflammasome assembly with caspase-1 activation and subsequent activation of the cytokines IL-1β and IL-18 [53]. As with other inflammasomes, caspase-1 activation through ALR leads to pyroptosis in immune cells.
C-type lectin receptors sense carbohydrates, such as high mannose, fucose, β-glucan, and α-mannans [54]. This group includes DC-SIGN, Langerin, Dectin-1, CLEC1 and CLEC2, Mincle, Mannose receptor, and DEC205. Ligand binding to CLR can lead to internalization and degradation of pathogens or receptor-mediated signaling, depending on the CLR [55]. C-type lectin receptors with signaling functions activate NF-κB, MAP kinases, and NFAT [44].
There are three members of the RLR family: RIG-I, melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2) [47]. These are DExH/D box helicases that detect foreign RNA in the cell cytosol [44]. Signaling through RLR leads to production of pro-inflammatory cytokines and type I IFN [41].
Cell death receptor ligands are sensed through receptors of the TNF-receptor superfamily. Some of these receptors contain a cytoplasmic death domain (DD) and so are classified as death receptors. In humans, these are: TNF-R1, CD95 (Fas/APO-1), TRAIL-R1 (DR4), TRAIL-R2 (APO-2/TRICK/DR5, KILLER), DR3 (TRAMP/APO-3), and DR6 [42]. These receptors fall into two categories based on their intracellular adaptor protein usage. TNF-R1 and DR3 bind TNF-receptor-associated death domain (TRADD) and ligand binding generally results in pro-inflammatory effects through activation of NF-κB and MAPK [56]. CD95, DR4, and DR5 bind Fas-associated DD (FADD) and activate the cell extrinsic pathway of apoptosis [56].
The types of inflammatory receptors present on cells are not uniform across different cell types. This means that different cells types can experience the same pattern of inflammatory stimuli in different ways. For example, different dendritic cell subsets express different components of the TLR repertoire which dictate their responses to PAMPs [43]. The cellular receptor repertoire may also be influenced by whether cells have been exposed to multiple waves of inflammation since cells that survive an initial exposure to inflammatory stimuli may change their receptor expression in response to that stimulus. For example, co-stimulation with TLR7 and TLR9 agonists decreases expression of both TLRs in endosomes of macrophages via induction of suppressor of cytokine signaling-1 (SOCS1); whereas LPS significantly up-regulates expression of TLR2 mRNA and protein in the mouse lung [57, 58]. The sequence in which cellular receptors are activated can also influence the character of the inflammatory response. For example, when cells are stimulated by IFN-γ and then TNFα, they have more robust NF-κB activation than when TNFα serves as the initial stimulus [59]. This is particularly true of cells that receive both extracellular and intracellular inflammatory stimuli. For instance, the ability of macrophages to clear infection with mycobacteria through autophagy is highly dependent on the extracellular cytokine environment. Autophagosome maturation is inhibited by IL-4 and IL-13, but IFN-γ increases autophagosome maturation and clearance [39, 60].
Sensing intracellular-derived signals of inflammation
Cells also sense and respond to intracellular changes during inflammatory responses. The endoplasmic reticulum (ER) is responsible for protein synthesis, folding, post-translational modification, and transport of the final protein product to other locations within the cell. It contains an array of chaperone systems, including glycosidases, Ca2+-dependent chaperones, and members of the protein disulfide isomerase (PDI) family [61]. Under normal physiologic conditions, ER chaperones and folding enzymes ensure that proteins are folded and exported correctly. This process requires precise control of lumenal Ca2+ concentration, redox homeostasis, and oxygen supply. When cells experience stress leading to changes in the conditions within the ER lumen, such as accumulation of large amounts of unfolded proteins, the ER initiates a stress response called the unfolded protein response (UPR) [62]. During the UPR, the intralumenal ER chaperone glucose-regulated protein 78 (GRP78/BiP/HSPA5) is released from its interaction with the intraluminal domains of the transmembrane proteins PKR-like ER kinase (PERK), inositol requiring enzyme 1 (IRE1), and activation transcription factor 6 (ATF6). This releases PERK, IRE1, and ATF6 from the ER membrane into the cytosol and allows them to initiate responses designed to alleviate ER stress [63].
Inflammation commonly leads to activation of the UPR through increased production of cellular defense and signaling proteins, such as cytokines or viral-derived proteins during infection [64, 65]. Additionally, pro-inflammatory signaling through TLR pathways can directly activate the ER UPR and suppresses UPR-induced apoptosis [66–68]. Conversely, the UPR can also activate or modulate intracellular pro-inflammatory signaling pathways [69].
The primary function of the UPR is to promote cell survival by decreasing overall protein synthesis and activating transcription factors which regulate expression of genes coding for chaperones, components of the ER-associated degradation (ERAD) system, and components of the autophagy machinery [70]. ER stress-induced increases in autophagic flux also promote cell survival [71]. However, prolonged ER stress responses can lead to activation of pro-apoptotic pathways and cell death [72].
Inflammation can also lead to oxidative stress through generation of increased reactive oxygen species (ROS). ROS are small, short-lived, and highly reactive chemical species derived from incomplete oxygen reduction. The major cellular reactive oxygen species are superoxide (O2 −), hydrogen peroxide (H2O2), hydroxyl anions (OH−), hydroxyl radicals (OH), and hypochlorous acid (HOCl) [73]. Mitochondria produce ROS as a by-product of respiration and the ER produces them as part of the normal protein folding process [74, 75]. During normal cellular metabolism, ROS are produced at low levels and act as cellular signaling molecules that promote proliferation and survival. Inflammation-induced increases in metabolic demands in the mitochondria and protein folding demands in the ER lead to increased production of ROS. ER stress leading to cytosolic calcium flux can further increase ROS production in the mitochondria [76]. Cells have nonenzymatic and enzymatic antioxidizing agents to counteract ROS, such as glutathione, thioredoxin, superoxide dismutase, catalase and peroxidases [77]. The transcription factor NF-E2-related factor 2 (Nrf2) is also stabilized under these conditions and translocates to the nucleus where it leads to up regulation of a network of cytoprotective and antioxidant genes [73]. When the production of ROS outpaces cellular antioxidant defenses, cells experience oxidative stress which leads to cellular damage and activates autophagic and apoptotic pathways [78].
Modulating immune activation
The inflammatory stimuli in the environment and the receptors present on or in a cell are the main determinants of inflammatory activation. However, homeostatic states within cells can also contribute to inflammatory activation through modulation of these responses. Previous exposure to low-level inflammatory stimuli, nutrient status, and oxidative stress all influence outcomes during inflammatory responses [52, 79–82]. These types of factors determine the intracellular availability of proteins involved in pro- and counter-inflammatory programs, the availability of energy necessary to execute these programs, and the activation state of the programs at the time that the cell encounters inflammatory stimuli.
When cells are exposed to inflammatory stimuli and activate NF-κB, one of the products of this pro-inflammatory pathway is the anti-inflammatory protein tumor necrosis factor, alpha-induced protein 3 (TNFAIP3/A20) [83–85]. Production of TNFAIP3 then restricts responses to subsequent NF-κB activating stimuli. Since NF-κB signaling influences both autophagy and apoptosis this influences how these two pathways proceed upon subsequent inflammatory stimulation.
Nutrient status can affect immune activation with respect to autophagy and apoptosis in several ways. Autophagy was originally described as a cell survival process activated during starvation. During starvation it can direct limited cell catabolism to maintain energy availability. This is regulated by the upstream deactivation of mammalian target of rapamycin (mTOR) [86]. Therefore, low energy states are more likely to favor higher autophagy activity at the time that cells encounter inflammatory stimuli with the potential to influence the outcome of the inflammatory response. This is exemplified by studies showing that mycobacterial clearance through autophagy is improved when cells are concurrently starved [39].
Starvation-induced autophagy also requires sphingolipids and is regulated by the cellular complement of sphingolipid metabolites present in the cell [87]. Sphingolipids are structural membrane lipids that are found in all eukaryotic cells [88]. They are synthesized de novo in the ER from condensation of serine and palmitoyl coenzyme A (CoA) by serine palmitoyl transferase. They can be degraded in lysosomes by glycosidases or acid sphingomyelinases to form ceramides, which can then be deacylated to generate sphingosine. Sphingosine can subsequently be recycled back to ceramide or phosphorylated to form sphingosine-1-phosphate (S1P). Interconversion of sphingolipid metabolites as well as their rapid turnover allows them to also act as signaling molecules during inflammation [89]. Pathogens, oxidative stress, and cytokines lead to increased levels of ceramide within cells, which promotes both autophagy and apoptosis. However, conversion of ceramide to S1P appears to favor autophagy over apoptosis [90].
Execution of autophagy and apoptosis during inflammation
Autophagy and apoptosis are both complex cascades of protein interactions that alter the cell, sometimes in ways that influence the function of the opposing pathway. Differences in activation can impart an advantage to either autophagy or apoptosis during inflammation, but the way these programs are executed is also important to determine the outcome at the autophagy/apoptosis checkpoint.
Autophagy
The process of autophagy requires the coordinated interactions of over 30 different proteins and is regulated by an even larger number of additional proteins [91]. It is initiated through release of Beclin-1 from its association with Bcl-2 and translocation of the ULK1-Atg13-FIP200-Atg101 complex to a source of lipid membrane [4]. The Beclin-1-Atg14-Ambra1-Vps15-Vps34 (PI3 K) complex then assembles on this membrane, leading to formation of an isolation membrane [3]. WIPI proteins are added and then the Atg5-Atg12-Atg16L1 complex acts to transform it into phagophore by conjugating LC3 to phosphatidylethanolamine (PE) in the membrane [4]. The LC3 lipidation reaction is a key event in autophagy that results from the concerted actions of a large number of autophagy-related (Atg) proteins in two ubiquitin-like conjugation pathways [92]. In the first of these pathways, Atg12 is activated by Atg7, an E1-like enzyme. The activated Atg12 is then transferred to Atg10, an E2-like enzyme, and finally it is covalently conjugated to Atg5 [93]. The Atg5-Atg12 conjugate then forms a complex with Atg16L1, which leads to formation of Atg5-Atg12-Atg16L1 multimers [94].
In the second ubiquitin-like reaction, LC3 is cleaved at its C-terminal glycine by Atg4 shortly after synthesis [95, 96]. It is then activated by Atg7, transferred to Atg3, and finally its exposed glycine forms an amide bond with PE via interaction with the Atg5-Atg12-Atg16L1 complex [97].
LC3 bound to the double membrane phagophore loads targeted cargo into the forming vesicle through interactions with autophagy adaptor proteins [98]. Then the double membrane elongates, is sealed to enclose the cargo, and the completed autophagosome is transported along microtubules to fuse with lysosomes [99]. In classical, degradative autophagy, this results in digestion of the inner membrane of the autophagosome and its contents which are released back into the cytosol as amino acids [92].
Under homeostatic conditions, autophagy is active at low or basal levels to perform housekeeping functions [100]. During inflammation, it is induced or upregulated by a large number of signals related to nutritional status and inflammation. Toll-like receptor signaling through TLR1, 2, 3, 4, 5, 6, 7, or 9 increases autophagy [101–106]. In this pathway, autophagy is initiated when the adaptor proteins MyD88 and TRIF bind to Beclin-1 and decrease its interaction with Bcl-2 [103]. In addition, TRAF6, an E3 ubiquitin ligase downstream of TLR4 signaling, can ubiquitinate Beclin-1 to release it from Bcl-2 [107]. NLR-related signaling through NOD1 and NOD2 or the inflammasomes NRLX1 and AIM2 also increases autophagy [108–111]. Finally, the cytokines associated with inflammation-induced death, TNFα, IL-1α, IL-1β, and IFN-γ, increase autophagy [35, 39, 112, 113].
Intracellular stress also promotes autophagy during inflammation. Endoplasmic reticulum stress and the UPR support autophagy through upregulation of genes coding for autophagy proteins, mTOR inactivation, ULK1/2 complex activation, and PI3K complex activation [114].
Likewise, stress-induced release of Ca2+ from the endoplasmic reticulum activates autophagy via a signaling pathway involving calmodulin-dependent kinase kinase beta (CaMKK-β), AMP kinase (AMPK), and mTOR [115, 116].
Apoptosis
Apoptosis was the first type of programmed cell death (PCD) described and is still the best characterized. Morphologically, it is distinguished by cell rounding and shrinkage, chromatin condensation, nuclear fragmentation, and membrane blebbing [117]. Apoptotic cells also undergo DNA fragmentation and externalize phosphatidylserine on their plasma membrane, although both the plasma membrane and lysosomal membranes remain essentially intact. After death, apoptotic cells are rapidly cleared by phagocytic cells in a process that is generally considered to be anti-inflammatory. However, if apoptotic cells are not cleared quickly, they can undergo secondary necrosis with cytoplasmic swelling, lysosomal membrane permeabilization, cytoplasmic membrane permeabilization, and release of active caspase-3 [118]. Secondary necrosis is then a pro-inflammatory form of cell death [119].
Apoptosis is initiated through the extrinsic or intrinsic apoptotic signaling pathways [120]. Binding of TNFα, FasL, or TRAIL to their cognate death domain receptors on the cell surface activates the extrinsic pathway. TNFα binding to TNFR1 initially leads to assembly of a pro-survival signaling complex called complex I. In complex I, TRADD, RIPK1, and the E3 ubiquitin ligases TNF-receptor-associated factor 2 (TRAF2), the cellular inhibitors of apoptosis (cIAP1 or cIAP2), and the linear ubiquitin chain assembly complex (LUBAC) assemble. This leads to ubiquitination of RIPK1 with recruitment of the TGF-activated kinase 1 (TAK1)-TAK1-binding protein (TAB) complex and activation of NF-κB and MAPK [121]. In sensitized or stressed cells, RIPK1 is deubiqutinated and complex I is destabilized and replaced with complex IIa [122]. Complex IIa consists of TRADD, RIPK1, FADD, cellular FLICE-like inhibitory protein (cFLIP), and an initiator caspase (pro-caspase-8 or -10). If cFLIP levels are low, the caspase undergoes activation and directs the cell toward apoptosis through cleavage of RIP1 and activation of effector caspases (caspase-3, -6, and -7). These caspases go on to cleave a number of structural and metabolic proteins leading to apoptosis. If caspase-8 or 10 activation is blocked or fails to activate in complex IIa then RIP3 and mixed lineage kinase domain-like (MLKL) are recruited into the complex to form complex IIb, which is also called the necrosome [123]. This complex directs the cell to undergo programmed cell death with features of necrosis in what is sometimes referred to as the extrinsic necroptosis pathway.
Activation of the Fas receptor by FasL or TRAIL-R1 or TRAIL-R2 by TRAIL leads to assembly of the membrane-associated death-inducing signaling complex (DISC) through the adaptor FADD. FADD recruits initiator caspases and cFLIP into the DISC. Initiator caspase activation then leads to activation of effector caspases and apoptosis. Downstream of Fas receptor activation, caspase-8 also cleaves BID and this truncated form (tBID) localizes to the mitochondria where it triggers mitochondrial outer membrane permeabilization (MOMP) and releases cytochrome c [124]. Cytochrome c interacts with apoptosis protease activating factor-1 (Apaf-1) to form the apoptosome and activate caspase-9 with subsequent cleavage and activation of executioner caspases [125].
The intrinsic apoptotic pathway is initiated by intracellular stress such as DNA damage, or growth factor withdrawal. This stress leads to activation of Bcl-2-homology 3 (BH3) only proteins that antagonize anti-apoptotic Bcl-2 family members. Bax and/or Bak are activated which causes them to oligomerize and form a channel in the mitochondrial outer membrane. Cytochrome c, second mitochondria-derived activator of caspase/direct IAP-binding protein with low PI (Smac/DIABLO) and the serine protease high-temperature requirement protein A2 (HtrA2/OMI) are released into the cytosol through this pore [126]. The cytochrome c associates with Apaf-1 and caspase-9 to form the apoptosome while Smac/DIABLO and HrtA2/OMI antagonize the inhibitor of apoptosis proteins (IAPs). The interaction between Smac/DIABLO and XIAP relieves its inhibition of caspase-3, -7, and -9 activation contributing to activation of the effector caspases and apoptosis [127].
Calpains are calcium-sensitive cysteine proteases that modify target protein functions through cleavage [128]. They participate in apoptosis through activation of the initiator caspase, caspase-8, and of the effector caspases, caspase-7 and -12 [129–133]. Activated calpain-1 also cleaves Bid and Bax to induce mitochondrial cytochrome c release and apoptosis [134–137]. Finally, calpain-1 activates caspase-3 and PARP during TNF-induced apoptosis, and cleaves Apaf after Ca2+ overload in cardiomyocytes [138].
Endoplasmic reticulum stress also plays a role in apoptosis. Activation of the UPR is generally considered a pro-survival response, but in cases where normal ER function cannot be restored the response is switched from survival to apoptosis. This involves calcium release from the ER with subsequent calpain and effector caspase activation, downregulation of Bcl-2, and increased generation and release of ROS into the cytoplasm [139].
Autophagic cell death
Autophagic cell death has also been identified as a mechanism of programmed cell death. However, whether this is death due to autophagy or death coincident with autophagy remains controversial [140]. Many of the original studies describing autophagy-induced death relied on the observation of autophagy in dying cells and did not examine autophagic flux. In autophagic flux studies, the kinetics of autophagy are examined, which allows distinctions between fully competent autophagy and accumulation of autophagosomes due to a failure of autophagy to go to completion. Many of these studies also relied on chemical inhibitors of various steps of the autophagic pathway, which are now known to have additional functions outside of autophagy [1]. Doubt that this type of death exists was also increased by a recent study that tested 1400 death-inducing compounds and found that none killed through autophagy [141]. Further studies into the relationship between autophagy and apoptosis may help to clarify whether this is an independent cause of death due to excessive autophagy or part of a continuum of cell death in cells undergoing autophagy.
Interactions between autophagy and apoptosis
While autophagy and apoptosis are often discussed as if they are two separate pathways, in reality they are extensively interconnected. Both are activated by inflammatory stimuli, though differences in the type, duration, or sensing of inflammation can lead to differences in the kinetics of the two pathways. Differences in the homeostatic state of the cells can also favor one pathway over another during the intracellular execution of inflammation. In the end, the competition between the two pathways determines whether a cell survives or undergoes programmed cell death.
Regulation of apoptosis by the process of autophagy
The process of autophagy acts to both increase and decrease apoptosis. Autophagy limits apoptosis through degradation of pro-apoptotic stimuli such as damaged mitochondria or cytotoxic protein aggregates [142–144]. It limits pyroptosis through destruction of components of the NF-κB signaling machinery, assembled inflammasomes, or pro-IL-1β [36, 38, 145–147]. It also degrades activated caspase-8 to limit TRAIL-induced apoptosis and prevents accumulation of the BH3-only protein NBK/Bik on ER membranes to prevent initiation of apoptosis [148, 149]. Alternately, autophagy promotes apoptosis by functioning as a platform for caspase-8 activation [150–152]. This may be a mechanism to activate cell death when autophagy is stalled and cannot go to completion.
Regulation of apoptosis by autophagic proteins
Autophagic proteins also act outside of the autophagic process to influence apoptosis. Atg12 is involved in the formation of autophagic membranes through interactions with Atg5 and Atg16L1. However, it also interacts with Bcl-2 and Mcl-1 to inhibit their anti-apoptotic activity and allow caspase activation [153]. Atg7 and Beclin-1 are also required for caspase-8-induced death, although the mechanism has not been elucidated [154]. Conversely, the autophagic protein UVRAG (UV irradiation resistance-associated gene) limits MOMP by preventing translocation of Bax to mitochondria [155].
Regulation of autophagy by apoptotic proteases
Cellular proteases involved in apoptosis are required for autophagy. Caspase-9 and Atg7 interact, and mutually influence each other. Atg7 inhibits the pro-apoptotic activity of caspase-9, whereas caspase-9 facilitates Atg7-dependent activation of LC3 for conjugation to autophagosomes [156, 157]. Calpain activity is also required for initiation of autophagy [158].
Pro-apoptotic proteases can cleave autophagy proteins. These cleavage events can lead to degradation and loss of cytoprotective autophagy or they can modify the proteins to change their function. Beclin-1, Atg3, Atg4, Atg5, Atg7, Atg9, p62, and AMBRA1 all undergo proteolytic cleavage [134, 159–169]. Cleavage of Beclin-1 and Atg5 is particularly interesting since these cleavage events switch both from pro-autophagic to pro-apoptotic proteins [160, 161]. After cleavage, the pro-apoptotic fragments of Beclin-1 and Atg5 translocate to the mitochondria where they participate in cytochrome c release and activation of the intrinsic pathway of apoptosis.
Apoptosis regulating proteins in autophagy
The autophagic functions of Beclin-1 are inhibited by interaction with the anti-apoptotic Bcl-2 family members Bcl-2, Bcl-xL, and Mcl-1 [170, 171]. Likewise, cFLIP inhibits autophagy by interfering with Atg3 and LC3 interactions [172]. Kinases involved in apoptotic pathways also play a role in autophagy. Death-associated protein kinase (DAPK) and DAPK-related proteins kinase (DRP)-1 promotes autophagy through phosphorylation of Beclin-1 to release it from its inhibitor Bcl-2 [173–175].
Autophagy and apoptosis are co-dependent
Although autophagy and apoptosis are antagonistic in terms of cell survival, the two processes are intertwined and dependent upon each other. Autophagy and apoptosis utilize an overlapping set of proteins. This means that availability of these proteins can affect both pathways simultaneously. Activation of both pathways, or at least a subset of proteins in each pathway, is necessary for either pathway to function. This is in part due to the fact that proteins modified in one pathway are often required to modify proteins for function in the opposing pathway. Transient or permanent protein modifications can also make proteins in this overlapping set function exclusively in one pathway. These factors suggest that autophagy and apoptosis promote and limit each other and that regulatory factors that control the cellular commitment to either autophagy or apoptosis are likely to exist.
Regulation of the autophagy/apoptosis checkpoint
Beclin-1 and Atg5 are core autophagy proteins that are also involved in apoptosis (Fig. 1). Beclin-1 is part of one of two protein kinase complexes involved in autophagy initiation, while Atg5 is involved in autophagosome membrane elongation. Beclin-1 is released from its complex with Bcl-2 to participate in autophagy by JNK phosphorylation of Bcl-2, DAPK-mediated phosphorylation of Beclin-1, translocation of the nuclear protein HMGB1 to the cell cytosol, or competition from other BH3-only proteins for Bcl-2 binding [176–183]. Atg5 must be complexed to Atg12 and Atg16L1 to participate in autophagy. Sequential reactions with Atg7 and Atg10 covalently conjugate Atg12 to Atg5 and this conjugate forms a complex with Atg16L1 [97]. If there are high levels of free Atg5 in the cell, Atg5 can interact with FADD to promote apoptosis [184].
Fig. 1.
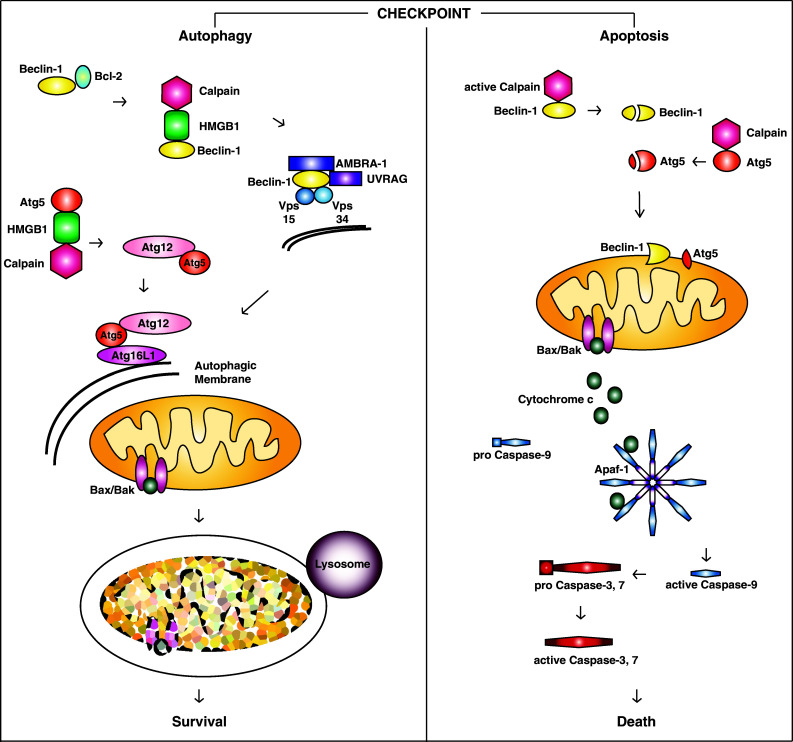
Autophagy/apoptosis checkpoint regulation. The presence of cytosolic HMGB1 pushes cells toward autophagy at the autophagy/apoptosis checkpoint. It blocks calpain-mediated cleavage of Beclin-1 and Atg5, which preserves their pro-autophagic functions and prevents their transformation into pro-apoptotic effectors. Under these conditions, Beclin-1 is released from Bcl-2 and forms a complex that initiates autophagosome membrane formation. Atg5 is conjugated to Atg12 and then this conjugate forms a complex with Atg16L1. The Atg5-Atg12-Atg16L1 complex participates in elongation of the autophagic membrane which surrounds targeted cargo, such as damaged mitochondria. After the autophagic membrane encloses the cargo, the completed vesicle fuses with lysosomes to destroy the contents of the autophagosome. This removes pro-apoptotic stimuli from the cell and allows it to survive inflammation. In the absence of cytosolic HMGB1, cells are directed toward apoptosis. Beclin-1 and Atg5 are cleaved and translocate to mitochondria where they participate in the intrinsic pathway of cell death. Bax/Bak opens pores in the mitochondrial outer membrane which releases cytochrome c into the cell cytosol. Cytochrome c localizes to the apoptosome where it interacts with Apaf-1 and pro-caspase-9, leading to activation of caspase-9. Active caspase-9 then activates the effector caspases, caspase-3 and caspase-7. The effector caspases cleave a number of structural and metabolic targets within the cell, leading to apoptotic death
Beclin-1 and Atg5 can also be cleaved by apoptotic proteases. Caspases-3 and -6 and calpain-1 cleave Beclin-1 and calpain-1 cleaves Atg5 [159–161]. Cleavage renders Beclin-1 and Atg5 unable to participate in autophagy and they are converted to active pro-apoptotic proteins. This constitutes passage of the autophagy/apoptosis checkpoint since this change shifts the cells to apoptosis without the opposition of autophagy. At this point, the cells are committed to death. Not surprisingly, this cleavage event is regulated. The nuclear, non-histone DNA-binding protein HMGB1 is translocated to the cell cytosol by the same stimuli that activate autophagy and apoptosis [185]. Once in the cytosol this protein is able to interact with both Beclin-1 and Atg5 and protect them from calpain-mediated cleavage conversion into pro-apoptotic proteins [186]. This prevents activation of apoptosis and preserves autophagy to favor selection of autophagy at the autophagy/apoptosis checkpoint.
Conclusions/perspectives
The presence of regulators that switch cells between autophagy and apoptosis explains how both processes can be activated, but one gains advantage over the other during inflammation. HMGB1-mediated protection of Beclin-1 and Atg5 from calpain cleavage likely represents only one checkpoint regulator in the decision making process between autophagy and apoptosis. The presence of regulators suggests that cells do not commit to autophagy or apoptosis when sensing inflammation, but rather during the execution of the inflammatory program. This allows the intracellular environment and the progress of cell survival efforts to influence whether cells undergo autophagy or apoptosis. It is also advantageous because pathogens attempting to circumvent cellular defenses are likely to ‘trip alarms’ and the cell can use this information while calculating whether to live or die.
The idea of an autophagy/apoptosis checkpoint is well-established and new data suggests that this checkpoint is directly regulated during inflammation. Additional research is now needed to identify new checkpoint regulators and the steps that they influence. One challenge with these future studies will be distinguishing autophagic and apoptotic functions for proteins that participate in both pathways. Genetic knockout of these proteins affects both autophagy and apoptosis. Studies using Beclin-1 and Atg5 deficient systems have illustrated this conundrum and suggest that studying specific targeted mutations affecting either autophagic or apoptotic functions of these proteins may be necessary to fully understand how they participate in these processes [173, 187–189].
Another goal of investigations into autophagy/apoptosis regulators is identification of small molecules or other treatments that could manipulate the checkpoint to favor cell survival or death. This could open up exciting new therapeutic options for a number of chronic diseases. For example, in diseases with massive inflammation and cell death, but low risk of intracellular infection, shifting the checkpoint to favor autophagy over apoptosis could be beneficial. Diseases in this category would potentially include inflammatory bowel disease and rheumatoid arthritis. Conversely, situations wherein disease was related to inappropriate cell survival or failure of PCD might benefit from shifting the checkpoint to favor apoptosis over autophagy. Diseases in this category might include chronic intracellular infections or neoplasia. Targeting a single regulator presents an opportunity for shifting the cells toward or away from death, depending on the desired outcome. Therefore, identifying proteins that regulate the autophagy/apoptosis checkpoint and therapies that affect their functions would be expected to lead to more effective and precise treatments for a number of chronic inflammatory diseases.
Acknowledgements
Funding was provided by Crohn’s and Colitis Foundation of America and National Institute of Diabetes and Digestive and Kidney Diseases (Grant Nos. http://dx.doi.org/10.13039/100001063, P30 DK42086).
Abbreviations
- PAMPs
Pathogen-associated molecular pattern molecules
- LPS
Lipopolysaccharide
- TNF-α
Tumor necrosis factor alpha
- IL-1
Interleukin-1
- IFN-γ
Interferon-gamma
- DAMPs
Damage-associated molecular pattern molecules
- HMGB1
High mobility group box-1
- ATP
Adenosine triphosphate
- FasL/CD95L
Fas ligand
- TRAIL/Apo2L
TNF-related apoptosis-inducing ligand
- TL1A
TNF-like ligand 1A
- TLR
Toll-like receptors
- NOD
Nucleotide-oligomerization domain
- NLR
NOD-like receptors
- AIM2
Absent in melanoma-2
- ALRs
AIM2-like receptors
- CLR
C-type lectin receptors
- RIG-1
Retinoic acid-inducible gene-I
- RLR
RIG-I-like receptors
- TIR
Toll/interleukin-1 receptor
- MyD88
Myeloid differentiation primary-response protein 88
- MAPKs
Mitogen-activated protein kinases
- NF-κB
Nuclear factor-kappa B
- TRIF/TICAM1
TIR-domain-containing adaptor protein inducing IFN-β
- MDA5
Melanoma differentiation-associated gene 5
- LGP2
Laboratory of genetics and physiology 2
- DD
Death domain
- TRADD
TNF-receptor-associated death domain
- FADD
Fas-associated DD
- SOCS1
Suppressor of cytokine signaling-1
- ER
Endoplasmic reticulum
- PDI
Protein disulfide isomerase
- UPR
Unfolded protein response
- GRP78/BiP/HSPA5
Glucose-regulated protein 78
- PERK
PKR-like ER kinase
- IRE1
Inositol requiring enzyme 1
- ATF6
Activation transcription factor 6
- ERAD
ER-associated degradation
- ROS
Reactive oxygen species
- Nrf2
NF-E2-related factor 2
- TNFAIP3/A20
Tumor necrosis factor, alpha-induced protein 3
- mTOR
Mammalian target of rapamycin
- CoA
Coenzyme A
- S1P
Sphingosine-1-phosphate
- PE
Phosphatidylethanolamine
- CaMKK-β
Calmodulin-dependent kinase kinase beta
- AMPK
AMP kinase
- PCD
Programmed cell death
- TRAF2
TNF-receptor-associated factor 2
- cIAP1 or cIAP2
Cellular inhibitors of apoptosis
- LUBAC
Linear ubiquitin chain assembly complex
- TAK1
TGF-activated kinase 1
- TAB
TAK1-binding protein
- cFLIP
Cellular FLICE-like inhibitory protein
- MLKL
Mixed lineage kinase domain-like
- DISC
Death-inducing signaling complex
- MOMP
Mitochondrial outer membrane permeabilization
- Apaf-1
Apoptosis protease activating factor-1
- BH3
Bcl-2-homology 3
- Smac/DIABLO
Second mitochondria-derived activator of caspase/direct IAP-binding protein with low PI
- HtrA2/OMI
Serine protease high-temperature requirement protein A2
- IAPs
Inhibitor of apoptosis proteins
- UVRAG
UV irradiation resistance-associated gene
- DAPK
Death-associated protein kinase
- DRP
DAPK-related proteins kinase
References
- 1.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 5.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyle KB, Randow F. The role of “eat-me” signals and autophagy cargo receptors in innate immunity. Curr Opin Microbiol. 2013 doi: 10.1016/j.mib.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 7.Sanjuan MA, Dillon CP, Tait SWG, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 8.Lee HK, Lund JM, Ramanathan B, et al. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007;315:1398–1401. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 9.Schmid D, Pypaert M, Münz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mintern JD, Macri C, Chin WJ, et al. Differential use of autophagy by primary dendritic cells specialized in cross-presentation. Autophagy. 2015;11:906–917. doi: 10.1080/15548627.2015.1045178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deretic V, Kimura T, Timmins G, et al. Immunologic manifestations of autophagy. J Clin Invest. 2015;125:75–84. doi: 10.1172/JCI73945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White E. The role for autophagy in cancer. J Clin Invest. 2015;125:42–46. doi: 10.1172/JCI73941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kenific CM, Debnath J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2015;25:37–45. doi: 10.1016/j.tcb.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong X, Levine B. Autophagy and viruses: adversaries or allies? J Innate Immun. 2013;5:480–493. doi: 10.1159/000346388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deretic V, Singh S, Master S, et al. Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell Microbiol. 2006;8:719–727. doi: 10.1111/j.1462-5822.2006.00705.x. [DOI] [PubMed] [Google Scholar]
- 16.Nogueira CV, Lindsten T, Jamieson AM, et al. Rapid pathogen-induced apoptosis: a mechanism used by dendritic cells to limit intracellular replication of Legionella pneumophila . PLoS Pathog. 2009;5:e1000478. doi: 10.1371/journal.ppat.1000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singhera GK, Chan TS, Cheng JY, et al. Apoptosis of viral-infected airway epithelial cells limit viral production and is altered by corticosteroid exposure. Respir Res. 2006;7:78. doi: 10.1186/1465-9921-7-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chaudhuri AR, Jeor SS, Maciejewski JP. Apoptosis induced by human cytomegalovirus infection can be enhanced by cytokines to limit the spread of virus. Exp Hematol. 1999;27:1194–1203. doi: 10.1016/S0301-472X(99)00044-2. [DOI] [PubMed] [Google Scholar]
- 19.Marriott HM, Bingle CD, Read RC, et al. Dynamic changes in Mcl-1 expression regulate macrophage viability or commitment to apoptosis during bacterial clearance. J Clin Invest. 2005;115:359–368. doi: 10.1172/JCI200521766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grassmé H, Kirschnek S, Riethmueller J, et al. CD95/CD95 ligand interactions on epithelial cells in host defense to Pseudomonas aeruginosa . Science. 2000;290:527–530. doi: 10.1126/science.290.5491.527. [DOI] [PubMed] [Google Scholar]
- 21.Flannagan RS, Cosío G, Grinstein S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol. 2009;7:355–366. doi: 10.1038/nrmicro2128. [DOI] [PubMed] [Google Scholar]
- 22.Wu Y-S, Chen S-N. Apoptotic cell: linkage of inflammation and wound healing. Front Pharmacol. 2014;5:1. doi: 10.3389/fphar.2014.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanaly ST, Nashleanas M, Hondowicz B, Scott P. TNF receptor p55 is required for elimination of inflammatory cells following control of intracellular pathogens. J Immunol. 1999;163:3883–3889. [PubMed] [Google Scholar]
- 24.Beattie L, d’El-Rei Hermida M, Moore JWJ, et al. A transcriptomic network identified in uninfected macrophages responding to inflammation controls intracellular pathogen survival. Cell Host Microbe. 2013;14:357–368. doi: 10.1016/j.chom.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vereecke L, Beyaert R, van Loo G. Enterocyte death and intestinal barrier maintenance in homeostasis and disease. Trends Mol Med. 2011;17:584–593. doi: 10.1016/j.molmed.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 26.Bantel H, Schulze-Osthoff K. Apoptosis in hepatitis C virus infection. Cell Death Differ. 2003;10(Suppl 1):S48–S58. doi: 10.1038/sj.cdd.4401119. [DOI] [PubMed] [Google Scholar]
- 27.Rangel SM, Diaz MH, Knoten CA, et al. The role of ExoS in dissemination of Pseudomonas aeruginosa during Pneumonia. PLoS Pathog. 2015;11:e1004945. doi: 10.1371/journal.ppat.1004945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Badley AD, Pilon AA, Landay A, Lynch DH. Mechanisms of HIV-associated lymphocyte apoptosis. Blood. 2000;96:2951–2964. [PubMed] [Google Scholar]
- 29.Selliah N, Finkel TH. Biochemical mechanisms of HIV induced T cell apoptosis. Cell Death Differ. 2001;8:127–136. doi: 10.1038/sj.cdd.4400822. [DOI] [PubMed] [Google Scholar]
- 30.Rodríguez-Grille J, Busch LK, Martínez-Costas J, Benavente J. Avian reovirus-triggered apoptosis enhances both virus spread and the processing of the viral nonstructural muNS protein. Virology. 2014;462–463:49–59. doi: 10.1016/j.virol.2014.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31:1334–1356. doi: 10.1183/09031936.00018908. [DOI] [PubMed] [Google Scholar]
- 32.Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22:240–273. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cha H-H, Hwang JR, Kim H-Y, et al. Autophagy induced by tumor necrosis factor α mediates intrinsic apoptosis in trophoblastic cells. Reprod Sci. 2014;21:612–622. doi: 10.1177/1933719113508816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jia G, Cheng G, Gangahar DM, Agrawal DK. Insulin-like growth factor-1 and TNF-alpha regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol. 2006;84:448–454. doi: 10.1111/j.1440-1711.2006.01454.x. [DOI] [PubMed] [Google Scholar]
- 35.Keller CW, Fokken C, Turville SG, et al. TNF-induces macroautophagy and regulates MHC class II expression in human skeletal muscle cells. J Biol Chem. 2010;286:3970–3980. doi: 10.1074/jbc.M110.159392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shi C-S, Shenderov K, Huang N-N, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Luca A, Smeekens SP, Casagrande A, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci USA. 2014;111:3526–3531. doi: 10.1073/pnas.1322831111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harris J, Hartman M, Roche C, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286:9587–9597. doi: 10.1074/jbc.M110.202911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gutierrez MG, Master SS, Singh SB, et al. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 40.Berchtold LA, Prause M, Størling J, Mandrup-Poulsen T (2016) Cytokines and Pancreatic β-Cell Apoptosis. pp 99–158 [DOI] [PubMed]
- 41.Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012;4:a006049. doi: 10.1101/cshperspect.a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walczak H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb Perspect Biol. 2013;5:a008698. doi: 10.1101/cshperspect.a008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 44.Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168:554–561. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]
- 46.Broz P, Monack DM. Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol. 2013;13:551–565. doi: 10.1038/nri3479. [DOI] [PubMed] [Google Scholar]
- 47.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 48.Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442:39–44. doi: 10.1038/nature04946. [DOI] [PubMed] [Google Scholar]
- 49.Girardin SE, Boneca IG, Viala J, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 50.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 51.Nunes T, de Souza HS. Inflammasome in intestinal inflammation and cancer. Mediat Inflamm. 2013;2013:654963. doi: 10.1155/2013/654963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol. 2012;28:137–161. doi: 10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- 54.van Kooyk Y, Rabinovich GA. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat Immunol. 2008;9:593–601. doi: 10.1038/ni.f.203. [DOI] [PubMed] [Google Scholar]
- 55.Geijtenbeek TBH, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. 2009;9:465–479. doi: 10.1038/nri2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol. 2009;10:348–355. doi: 10.1038/ni.1714. [DOI] [PubMed] [Google Scholar]
- 57.Lee H-J, Kim K-C, Han JA, et al. The early induction of suppressor of cytokine signaling 1 and the downregulation of toll-like receptors 7 and 9 induce tolerance in costimulated macrophages. Mol Cells. 2015;38:26–32. doi: 10.14348/molcells.2015.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsuda N, Yamazaki H, Takano K, et al. Priming by lipopolysaccharide exaggerates acute lung injury and mortality in responses to peptidoglycan through up-regulation of Toll-like receptor-2 expression in mice. Biochem Pharmacol. 2008;75:1065–1075. doi: 10.1016/j.bcp.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 59.Wesemann DR, Benveniste EN. STAT-1 alpha and IFN-gamma as modulators of TNF-alpha signaling in macrophages: regulation and functional implications of the TNF receptor 1:STAT-1 alpha complex. J Immunol. 2003;171:5313–5319. doi: 10.4049/jimmunol.171.10.5313. [DOI] [PubMed] [Google Scholar]
- 60.Harris J, De Haro SA, Master SS, et al. T helper 2 cytokines inhibit autophagic control of intracellular Mycobacterium tuberculosis. Immunity. 2007;27:505–517. doi: 10.1016/j.immuni.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 61.Schönthal AH. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica (Cairo) 2012;2012:857516. doi: 10.6064/2012/857516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jäger R, Bertrand MJM, Gorman AM, et al. The unfolded protein response at the crossroads of cellular life and death during endoplasmic reticulum stress. Biol Cell. 2012;104:259–270. doi: 10.1111/boc.201100055. [DOI] [PubMed] [Google Scholar]
- 64.Rath E, Haller D. Inflammation and cellular stress: a mechanistic link between immune-mediated and metabolically driven pathologies. Eur J Nutr. 2011;50:219–233. doi: 10.1007/s00394-011-0197-0. [DOI] [PubMed] [Google Scholar]
- 65.He B. Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ. 2006;13:393–403. doi: 10.1038/sj.cdd.4401833. [DOI] [PubMed] [Google Scholar]
- 66.Martinon F, Chen X, Lee A-H, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Messlik A, Schmechel S, Kisling S, et al. Loss of Toll-like receptor 2 and 4 leads to differential induction of endoplasmic reticulum stress and proapoptotic responses in the intestinal epithelium under conditions of chronic inflammation. J Proteome Res. 2009;8:4406–4417. doi: 10.1021/pr9000465. [DOI] [PubMed] [Google Scholar]
- 68.Woo CW, Cui D, Arellano J, et al. Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by toll-like receptor signalling. Nat Cell Biol. 2009;11:1473–1480. doi: 10.1038/ncb1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Janssens S, Pulendran B, Lambrecht BN. Emerging functions of the unfolded protein response in immunity. Nat Immunol. 2014;15:910–919. doi: 10.1038/ni.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deegan S, Saveljeva S, Gorman AM, Samali A. Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci. 2013;70:2425–2441. doi: 10.1007/s00018-012-1173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ogata M, Hino S, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mittal M, Siddiqui MR, Tran K, et al. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126–1167. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Adam-Vizi V, Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol Sci. 2006;27:639–645. doi: 10.1016/j.tips.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 76.Zhang K. Integration of ER stress, oxidative stress and the inflammatory response in health and disease. Int J Clin Exp Med. 2010;3:33–40. [PMC free article] [PubMed] [Google Scholar]
- 77.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 78.Zhang L, Wang K, Lei Y, et al. Redox signaling: potential arbitrator of autophagy and apoptosis in therapeutic response. Free Radic Biol Med. 2015;89:452–465. doi: 10.1016/j.freeradbiomed.2015.08.030. [DOI] [PubMed] [Google Scholar]
- 79.Schauvliege R, Vanrobaeys J, Schotte P, Beyaert R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J Biol Chem. 2002;277:41624–41630. doi: 10.1074/jbc.M207852200. [DOI] [PubMed] [Google Scholar]
- 80.Wang S, Miura M, Jung YK, et al. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell. 1998;92:501–509. doi: 10.1016/S0092-8674(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 81.Fontana L Neuroendocrine factors in the regulation of inflammation: excessive adiposity and calorie restriction. Exp Gerontol 44:41–45. doi:10.1016/j.exger.2008.04.005 [DOI] [PMC free article] [PubMed]
- 82.Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med. 2010;49:1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hitotsumatsu O, Ahmad R-C, Tavares R, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity. 2008;28:381–390. doi: 10.1016/j.immuni.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boone DL, Turer EE, Lee EG, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 85.Lee EG, Boone DL, Chai S, et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Russell RC, Yuan H-X, Guan K-L. Autophagy regulation by nutrient signaling. Cell Res. 2013;24:42–57. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li Y, Li S, Qin X, et al. The pleiotropic roles of sphingolipid signaling in autophagy. Cell Death Dis. 2014;5:e1245. doi: 10.1038/cddis.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Garcia-Ruiz C, Morales A, Fernández-Checa JC. Glycosphingolipids and cell death: one aim, many ways. Apoptosis. 2015;20:607–620. doi: 10.1007/s10495-015-1092-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature. 2014;510:58–67. doi: 10.1038/nature13475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lee J, Yeganeh B, Ermini L, Post M. Sphingolipids as cell fate regulators in lung development and disease. Apoptosis. 2015;20:740–757. doi: 10.1007/s10495-015-1112-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tanida I. Autophagy basics. Microbiol Immunol. 2011;55:1–11. doi: 10.1111/j.1348-0421.2010.00271.x. [DOI] [PubMed] [Google Scholar]
- 93.Ravikumar B, Sarkar S, Davies JE, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 94.Burman C, Ktistakis NT. Autophagosome formation in mammalian cells. Semin Immunopathol. 2010;32:397–413. doi: 10.1007/s00281-010-0222-z. [DOI] [PubMed] [Google Scholar]
- 95.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–2518. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hemelaar J, Lelyveld VS, Kessler BM, Ploegh HL. A single protease, Apg4B, is specific for the autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3, GABARAP, and Apg8L. J Biol Chem. 2003;278:51841–51850. doi: 10.1074/jbc.M308762200. [DOI] [PubMed] [Google Scholar]
- 97.Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. “Protein modifications: beyond the usual suspects” review series. EMBO Rep. 2008;9:859–864. doi: 10.1038/embor.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 99.Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct. 2008;33:109–122. doi: 10.1247/csf.08005. [DOI] [PubMed] [Google Scholar]
- 100.Eskelinen E-L, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793:664–673. doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 101.Delgado MA, Elmaoued RA, Davis AS, et al. Toll-like receptors control autophagy. EMBO J. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Anand PK, Tait SWG, Lamkanfi M, et al. TLR2 and RIP2 pathways mediate autophagy of Listeria monocytogenes via extracellular signal-regulated kinase (ERK) activation. J Biol Chem. 2011;286:42981–42991. doi: 10.1074/jbc.M111.310599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shi C-S, Kehrl JH. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem. 2008;283:33175–33182. doi: 10.1074/jbc.M804478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xu Y, Jagannath C, Liu X-D, et al. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shin D-M, Yuk J-M, Lee H-M, et al. Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell Microbiol. 2010;12:1648–1665. doi: 10.1111/j.1462-5822.2010.01497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Henault J, Martinez J, Riggs JM, et al. Noncanonical autophagy is required for type I interferon secretion in response to DNA-immune complexes. Immunity. 2012;37:986–997. doi: 10.1016/j.immuni.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shi C-S, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal. 2010;3:ra42. doi: 10.1126/scisignal.2000751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Travassos LH, Carneiro LAM, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 109.Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 110.Wang L-J, Huang H-Y, Huang M-P, et al. The microtubule-associated protein EB1 links AIM2 inflammasomes with autophagy-dependent secretion. J Biol Chem. 2014;289:29322–29333. doi: 10.1074/jbc.M114.559153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lei Y, Wen H, Yu Y, et al. The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity. 2012;36:933–946. doi: 10.1016/j.immuni.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pilli M, Arko-Mensah J, Ponpuak M, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37:223–234. doi: 10.1016/j.immuni.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Harris J. Autophagy and IL-1 Family Cytokines. Front Immunol. 2013;4:83. doi: 10.3389/fimmu.2013.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rashid H-O, Yadav RK, Kim H-R, Chae H-J. ER stress: autophagy induction, inhibition and selection. Autophagy. 2015;11:1956–1977. doi: 10.1080/15548627.2015.1091141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Høyer-Hansen M, Jäättelä M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007;14:1576–1582. doi: 10.1038/sj.cdd.4402200. [DOI] [PubMed] [Google Scholar]
- 116.Høyer-Hansen M, Bastholm L, Szyniarowski P, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-β, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 117.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. 2010;584:4491–4499. doi: 10.1016/j.febslet.2010.10.046. [DOI] [PubMed] [Google Scholar]
- 119.Haslett C. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am J Respir Crit Care Med. 1999;160:S5–S11. doi: 10.1164/ajrccm.160.supplement_1.4. [DOI] [PubMed] [Google Scholar]
- 120.Lamkanfi M, Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 2010;8:44–54. doi: 10.1016/j.chom.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 121.Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- 122.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/S0092-8674(03)00521-X. [DOI] [PubMed] [Google Scholar]
- 123.Han J, Zhong C-Q, Zhang D-W. Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat Immunol. 2011;12:1143–1149. doi: 10.1038/ni.2159. [DOI] [PubMed] [Google Scholar]
- 124.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/S0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 125.Ow Y-LP, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol. 2008;9:532–542. doi: 10.1038/nrm2434. [DOI] [PubMed] [Google Scholar]
- 126.Saelens X, Festjens N, Vande Walle L, et al. Toxic proteins released from mitochondria in cell death. Oncogene. 2004;23:2861–2874. doi: 10.1038/sj.onc.1207523. [DOI] [PubMed] [Google Scholar]
- 127.Duprez L, Wirawan E, Vanden Berghe T, Vandenabeele P. Major cell death pathways at a glance. Microbes Infect. 2009;11:1050–1062. doi: 10.1016/j.micinf.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 128.Łopatniuk P, Witkowski JM. Conventional calpains and programmed cell death. Acta Biochim Pol. 2011;58:287–296. [PubMed] [Google Scholar]
- 129.Gafni J, Cong X, Chen SF, et al. Calpain-1 cleaves and activates caspase-7. J Biol Chem. 2009;284:25441–25449. doi: 10.1074/jbc.M109.038174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Martinez JA, Zhang Z, Svetlov SI, et al. Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells. Apoptosis. 2010;15:1480–1493. doi: 10.1007/s10495-010-0526-4. [DOI] [PubMed] [Google Scholar]
- 131.Vaisid T, Barnoy S, Kosower NS. Calpain activates caspase-8 in neuron-like differentiated PC12 cells via the amyloid-beta-peptide and CD95 pathways. Int J Biochem Cell Biol. 2009;41:2450–2458. doi: 10.1016/j.biocel.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 132.Tan Y, Wu C, De Veyra T, Greer PA. Ubiquitous calpains promote both apoptosis and survival signals in response to different cell death stimuli. J Biol Chem. 2006;281:17689–17698. doi: 10.1074/jbc.M601978200. [DOI] [PubMed] [Google Scholar]
- 133.Weber H, Müller L, Jonas L, et al. Calpain mediates caspase-dependent apoptosis initiated by hydrogen peroxide in pancreatic acinar AR42J cells. Free Radic Res. 2013;47:432–446. doi: 10.3109/10715762.2013.785633. [DOI] [PubMed] [Google Scholar]
- 134.Shi M, Zhang T, Sun L, et al. Calpain, Atg5 and Bak play important roles in the crosstalk between apoptosis and autophagy induced by influx of extracellular calcium. Apoptosis. 2013;18:435–451. doi: 10.1007/s10495-012-0786-2. [DOI] [PubMed] [Google Scholar]
- 135.Wood DE, Thomas A, Devi LA, et al. Bax cleavage is mediated by calpain during drug-induced apoptosis. Oncogene. 1998;17:1069–1078. doi: 10.1038/sj.onc.1202034. [DOI] [PubMed] [Google Scholar]
- 136.Gao G, Dou QP. N-terminal cleavage of bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes bcl-2-independent cytochrome C release and apoptotic cell death. J Cell Biochem. 2000;80:53–72. doi: 10.1002/1097-4644(20010101)80:1<53::AID-JCB60>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 137.Chen M. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J Biol Chem. 2001;276:30724–30728. doi: 10.1074/jbc.M103701200. [DOI] [PubMed] [Google Scholar]
- 138.Smith MA, Schnellmann RG. Calpains, mitochondria, and apoptosis. Cardiovasc Res. 2012;96:32–37. doi: 10.1093/cvr/cvs163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004–1010. doi: 10.1038/nrm2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shen S, Kepp O, Kroemer G. The end of autophagic cell death? Autophagy. 2012;8:1–3. doi: 10.4161/auto.8.1.16618. [DOI] [PubMed] [Google Scholar]
- 142.Xue L, Fletcher GC, Tolkovsky AM. Mitochondria are selectively eliminated from eukaryotic cells after blockade of caspases during apoptosis. Curr Biol. 2001;11:361–365. doi: 10.1016/S0960-9822(01)00100-2. [DOI] [PubMed] [Google Scholar]
- 143.Zhang Y, Qi H, Taylor R et al The role of autophagy in mitochondria maintenance: characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy 3:337–346 [DOI] [PubMed]
- 144.Mitter SK, Song C, Qi X, et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy. 2014;10:1989–2005. doi: 10.4161/auto.36184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Paul S, Kashyap AK, Jia W, et al. Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-κB. Immunity. 2012;36:947–958. doi: 10.1016/j.immuni.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shibata Y, Oyama M, Kozuka-Hata H, et al. p47 negatively regulates IKK activation by inducing the lysosomal degradation of polyubiquitinated NEMO. Nat Commun. 2012;3:1061. doi: 10.1038/ncomms2068. [DOI] [PubMed] [Google Scholar]
- 147.Kimura T, Jain A, Choi SW, et al. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol. 2015 doi: 10.1083/jcb.201503023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hou W, Han J, Lu C, et al. Autophagic degradation of active caspase-8. Autophagy. 2014;6:891–900. doi: 10.4161/auto.6.7.13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chen S, Zhou L, Zhang Y, et al. Targeting SQSTM1/p62 induces cargo loading failure and converts autophagy to apoptosis via NBK/Bik. Mol Cell Biol. 2014;34:3435–3449. doi: 10.1128/MCB.01383-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Young MM, Takahashi Y, Khan O, et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287:12455–12468. doi: 10.1074/jbc.M111.309104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Laussmann MA, Passante E, Düssmann H, et al. Proteasome inhibition can induce an autophagy-dependent apical activation of caspase-8. Cell Death Differ. 2011;18:1584–1597. doi: 10.1038/cdd.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Deegan S, Saveljeva S, Logue SE, et al. Deficiency in the mitochondrial apoptotic pathway reveals the toxic potential of autophagy under ER stress conditions. Autophagy. 2014;10:1921–1936. doi: 10.4161/15548627.2014.981790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rubinstein AD, Eisenstein M, Ber Y, et al. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell. 2011;44:698–709. doi: 10.1016/j.molcel.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 154.Yu L, Alva A, Su H, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 155.Yin X, Cao L, Kang R, et al. UV irradiation resistance-associated gene suppresses apoptosis by interfering with BAX activation. EMBO Rep. 2011;12:727–734. doi: 10.1038/embor.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Han J, Hou W, Goldstein LA, et al. A Complex between Atg7 and Caspase-9: a novel mechanism of cross-regulation between autophagy and apoptosis. J Biol Chem. 2014;289:6485–6497. doi: 10.1074/jbc.M113.536854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jeong H-S, Choi HY, Lee E-R, et al. Involvement of caspase-9 in autophagy-mediated cell survival pathway. Biochim Biophys Acta. 2011;1813:80–90. doi: 10.1016/j.bbamcr.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 158.Demarchi F, Schneider C. The calpain system as a modulator of stress/damage response. Cell Cycle. 2007;6:136–138. doi: 10.4161/cc.6.2.3759. [DOI] [PubMed] [Google Scholar]
- 159.Norman JM, Cohen GM, Bampton ETW. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy. 2010;6:1042–1056. doi: 10.4161/auto.6.8.13337. [DOI] [PubMed] [Google Scholar]
- 160.Yousefi S, Perozzo R, Schmid I, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 161.Wirawan E, Vande Walle L, Kersse K, et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010;1:e18. doi: 10.1038/cddis.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.You M, Savaraj N, Kuo MT, et al. TRAIL induces autophagic protein cleavage through caspase activation in melanoma cell lines under arginine deprivation. Mol Cell Biochem. 2012;374:181–190. doi: 10.1007/s11010-012-1518-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rohn TT, Wirawan E, Brown RJ, et al. Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer’s disease brain. Neurobiol Dis. 2011;43:68–78. doi: 10.1016/j.nbd.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Russo R, Berliocchi L, Adornetto A, et al. Calpain-mediated cleavage of Beclin-1 and autophagy deregulation following retinal ischemic injury in vivo. Cell Death Dis. 2011;2:e144. doi: 10.1038/cddis.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pagliarini V, Wirawan E, Romagnoli A, et al. Proteolysis of Ambra1 during apoptosis has a role in the inhibition of the autophagic pro-survival response. Cell Death Differ. 2012;19:1495–1504. doi: 10.1038/cdd.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Betin VMS, Lane JD. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J Cell Sci. 2009;122:2554–2566. doi: 10.1242/jcs.046250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Betin VMS, MacVicar TDB, Parsons SF, et al. A cryptic mitochondrial targeting motif in Atg4D links caspase cleavage with mitochondrial import and oxidative stress. Autophagy. 2012;8:664–676. doi: 10.4161/auto.19227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Siddiqui MA, Mukherjee S, Manivannan P, Malathi K. RNase L cleavage products promote switch from autophagy to apoptosis by caspase-mediated cleavage of Beclin-1. Int J Mol Sci. 2015;16:17611–17636. doi: 10.3390/ijms160817611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Luo S, Rubinsztein DC. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ. 2010;17:268–277. doi: 10.1038/cdd.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Ciechomska IA, Goemans CG, Tolkovsky AM. Why doesn’t Beclin 1, a BH3-only protein, suppress the anti-apoptotic function of Bcl-2? Autophagy. 2009;5:880–881. doi: 10.4161/auto.9096. [DOI] [PubMed] [Google Scholar]
- 171.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–580. doi: 10.1038/cdd.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.R Safa A. Roles of c-FLIP in apoptosis, necroptosis, and autophagy. J Carcinog Mutagen. 2013 doi: 10.4172/2157-2518.S6-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gozuacik D, Bialik S, Raveh T, et al. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008;15:1875–1886. doi: 10.1038/cdd.2008.121. [DOI] [PubMed] [Google Scholar]
- 174.Inbal B, Bialik S, Sabanay I, et al. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol. 2002;157:455–468. doi: 10.1083/jcb.200109094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Levin-Salomon V, Bialik S, Kimchi A. DAP-kinase and autophagy. Apoptosis. 2014;19:346–356. doi: 10.1007/s10495-013-0918-3. [DOI] [PubMed] [Google Scholar]
- 176.Luo S, Rubinsztein DC. Atg5 and Bcl-2 provide novel insights into the interplay between apoptosis and autophagy. Cell Death Differ. 2007;14:1247–1250. doi: 10.1038/sj.cdd.4402149. [DOI] [PubMed] [Google Scholar]
- 177.Maiuri MC, Le Toumelin G, Criollo A, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Erlich S, Mizrachy L, Segev O et al Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 3:561–568 [DOI] [PubMed]
- 180.Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4:949–951. doi: 10.4161/auto.6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zalckvar E, Berissi H, Mizrachy L, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10:285–292. doi: 10.1038/embor.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zalckvar E, Berissi H, Eisenstein M, Kimchi A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy. 2009;5:720–722. doi: 10.4161/auto.5.5.8625. [DOI] [PubMed] [Google Scholar]
- 183.Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–892. doi: 10.1083/jcb.200911078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Pyo J-O, Jang M-H, Kwon Y-K, et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280:20722–20729. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- 185.Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol. 2012;8:195–202. doi: 10.1038/nrrheum.2011.222. [DOI] [PubMed] [Google Scholar]
- 186.Zhu X, Messer JS, Wang Y, et al. Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J Clin Invest. 2015;125:1098–1110. doi: 10.1172/JCI76344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lin HH, Lin S-M, Chung Y, et al. Dynamic involvement of ATG5 in cellular stress responses. Cell Death Dis. 2014;5:e1478. doi: 10.1038/cddis.2014.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lépine S, Allegood JC, Edmonds Y, et al. Autophagy induced by deficiency of sphingosine-1-phosphate phosphohydrolase 1 is switched to apoptosis by calpain-mediated autophagy-related gene 5 (Atg5) cleavage. J Biol Chem. 2011;286:44380–44390. doi: 10.1074/jbc.M111.257519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Shimizu S, Kanaseki T, Mizushima N, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]