

Abstract
Toxins have been shown to have many biological functions and to constitute a rich source of drugs and biotechnological tools. We focus on toxins that not only have a specific activity, but also contain residues responsible for transmembrane penetration, which can be considered bioportides—a class of cell-penetrating peptides that are also intrinsically bioactive. Bioportides are potential tools in pharmacology and biotechnology as they help deliver substances and nanoparticles to intracellular targets. Bioportides characterized so far are peptides derived from human proteins, such as cytochrome c (CYCS), calcitonin receptor (camptide), and endothelial nitric oxide synthase (nosangiotide). However, toxins are usually disregarded as potential bioportides. In this review, we discuss the inclusion of some toxins and molecules derived thereof as a new class of bioportides based on structure activity relationship, minimization, and biological activity studies. The comparative analysis of the amino acid residue composition of toxin-derived bioportides and their short molecular variants is an innovative analytical strategy which allows us to understand natural toxin multifunctionality in vivo and plan novel pharmacological and biotechnological products. Furthermore, we discuss how many bioportide toxins have a rigid structure with amphiphilic properties important for both cell penetration and bioactivity.
Keywords: Bioportide, Cargo delivery, Cell-penetrating peptides, Chlorotoxin, Crotamine, Toxins
Introduction
The study of toxins has traditionally focused on their primary pharmacological targets, such as phospholipases, proteases, and ion channel modulators. Venom and poison action usually occurs in solution (blood, extracellular matrix, i.e., interfering with coagulation and degrading proteins) or at the cell surface, i.e., ion channels. However, recent studies have shown that many toxins are multifunctional—have several biological targets—which may or may not be associated with their toxic role in host protection and prey catching. Furthermore, there is an increasing list of peptide toxins that can penetrate into cells, targeting intracellular pathways.
The cell penetration property of certain toxins is important in the development of new drugs and drug carriers. Cell penetration sequences occur as independent entities that can be fused or complexed with—and help deliver—bioactive sequences. We also discuss that, unlike most common targeting sequences so far characterized in biology, whereby the targeting domain consists of a continuous sequence of amino acid residues associated in tandem with the transported (cargo) sequence, i.e., peptide signals or nuclear targeting sequences, toxin transmembrane transporter sequences can also be found interspersed in the peptide sequence, overlapping other functional domains that can trigger, for instance, cell death or activation of intracellular pathways. This type of conformation has been found in peptides derived from many human proteins, such as cytochrome c, the calcitonin receptor, and endothelial nitric oxide synthase, described as a class of “bioportides”.
We believe that venoms are an abundant source of cell-penetrating peptides (CPPs) with intracellular activities and can be considered bioportides. This concept may aid in the understanding of toxin biological activity, multifunctionality, and potential druggability.
Cell-penetrating peptides—overall concept
CPPs are a class of biomolecules that can translocate through cellular membranes into the intracellular environment. We define CPPs, for the purposes of this review, as cationic peptides that enter cells in a non-toxic and receptor-independent way. Regarding toxins that can penetrate into cells, this concept excludes antimicrobial cationic peptides (that have intrinsic cytotoxicity due to membrane damage), toxins that enter cells via formation of pores or by causing membrane damage (such as phospholipases), and toxins that depend on specific receptor binding for endocytosis (such as several plant and microbial-derived toxins). In spite of all these exceptions, the CPP class defined here is associated with many peptides that penetrate non-specifically into cells using common transport mechanisms. These peptides are very attractive biotechnological tools for delivery of molecular cargos, such as nanoparticles, DNA and other molecules of high chemical complexity—and otherwise membrane impermeable—into cells both in vitro and in vivo [1–4].
Many researchers develop CPPs which act solely as transporters, without any other intrinsic activity [5], fusing or crosslinking them with active proteins/peptides/nanoparticles in tandem. Recent approaches for identifying CPPs, however, have shown that several of these can have both cell-penetrating and biological activities associated with discontinuous (overlapping) sequences. Hällbrink et al. [6] used a quantitative structure activity relationship (QSAR) algorithm to identify new CPPs. QSAR uses databases that contain a set of variables associated with each amino acid residue, such as charge, bulk of side chain, presence of heavy atoms (C, N, S, and O), hydrogen donors or acceptors, etc., and considers the effect of each amino acid residue on the properties of the overall peptide. Hällbrink et al. fed the algorithm with a training set of cationic known CPPs and a negative control set of non-functional CPP analogs that do not penetrate into cells. They used the overall properties of amino acids and found that it is possible to predict CPPs with a success rate of 90 %. CPP sequences of existing proteins and of randomly generated peptides were synthesized and successfully found to penetrate into cells, confirming the in silico prediction [6].
Cronican et al. [7] also studied cationic peptides. They had found that engineered green fluorescent proteins (GFPs), when highly charged, efficiently penetrate into cells. Based on the charge-to-peptide length of their modified GFPs, these authors searched human protein databases and found proteins with a high charge-to-length ratio, which proved to have cell-penetrating properties both in vivo and in vitro, although their previously known functions were not associated with this property. Among the proteins predicted and proved to penetrate cells are (full length or domains of): defensin 3 (an extracellular antimicrobial protein), HRX, c-Jun and N-DEK (nuclear DNA-interacting proteins); HBEGF and N-HGF (extracellular growth factors); and eotaxin (a chemotactic protein). The authors analyzed the whole human proteome and estimate that around 3 % are potential CPPs [7].
Original primary CPP sequences, also called protein transduction domains (PTDs), were isolated from both insect and viral transcription factors (TF) that cross the nuclear membrane to perform their biological role [8–11]. CPPs have since been detected among human TFs and other human proteins [12–15].
Usually, CPPs adopt an amphipathic secondary structure, which is favorable for their interaction with the cell membrane lipid bilayer. The majority of studies show that cell-penetrating mechanisms rely on the interaction between positively charged CPP residues and negatively charged molecules present on the cell surface, such as glycosaminoglycans (GAGs) and/or phospholipids [16]. This interaction is followed by CPP intracellular penetration via endocytosis-mediated translocation or direct translocation through the membrane through transitory structures such as inverted micelles [17–19].
The positive charge of CPPs is due to the presence of basic amino acid residues, mainly arginine (R) and lysine (K). The guanidine head group of arginine can form hydrogen bonds with the negatively charged phosphate and sulfate groups on the cell surface, favoring peptide internalization under physiological pH. Lysine does not have a guanidine group and exhibits less cell-penetrating efficiency [20]. Furthermore, tryptophan (W) has appeared to be an important aromatic residue for membrane translocation and its involvement in GAG binding and in endocytic pathways has been demonstrated [21, 22]. Tryptophan integration in CPP sequences is associated with the efficiency of internalization because of its strong interaction with negatively charged GAGs [4]. In addition, as shown in previous studies, CPP sequences that contain tryptophan and arginine, such as (R/W)9 and (R/W)16, are able to remodel the actin cytoskeleton [21]. Therefore, the biochemical characteristics of each residue that comprises the peptide primary sequence exhibit fundamental importance in facilitating peptide movement across the cell membrane [5, 12, 23].
Bioportides
Recently, the term ‘bioportide’ has been employed to designate a class of CPPs which, besides their cell-penetrating capacity, also possess intrinsic biological activity. Bioportides can have both cell-penetrating and pharmacological activities either by in tandem or discontinued association of cell localization and bioactive sequences (as exemplified in Fig. 1), although the in tandem structure has been the most studied so far. Bioportides present wide distribution throughout the body, being deposited in the intracellular compartment of target organs or tissues. It is noteworthy that, although there are a growing number of bioportides being identified, especially by John Howl’s and Sarah Jones’ groups, each with its intrinsic biological activity [23, 24], toxins have not yet been included in this list.
Fig. 1.

Discontinuous (rhegnylogic) or continuous (sychnologic) forms of organization of cell-penetrating toxins. Based on Portoghese et al. [27] and Howl et al. [23], proteins and peptides can be organized according to the domains (1) responsible for transport through membranes and (2) capable of binding and activating receptors, in two general ways: either with a discontinuous organization of both these domain sequences, here considered ‘rhegnylogic,’ or by a continuous, or ‘in tandem’ organization of these domains, here called ‘sychnologic.’ In the first case, cell penetration and activation of receptors are inseparable and are often even overlapped as illustrated by a coiled region at the C-terminus of the left protein structure. Moreover, sequences must be very tightly organized and loss of structural organization can jeopardize protein activity both regarding cell penetration and activation of receptors. Toxins frequently guarantee structure maintenance via disulfide bridges. Indeed, toxin reduction and cleavage of disulfide bridges often causes loss of biological activity. The discovery of proteins with such organization usually requires complex computational analyses of overall properties of amino acid residues and their relationship with other residues of the sequence (QSAR). On the other hand, as shown on the right side of the figure, proteins or peptides can have the domains responsible for cell penetration and receptor activation in separate and independent portions, which can be dissociated and combined with other proteins, maintaining their activity. We have indicated an intermediate joining region that can be thought of as a ‘hinge’ portion that can be particularly flexible, without compromising the independent activities of the transport and receptor binding domains. The binding of transport domains with nanoparticles or receptor binding domains is frequently explored in biotechnology and pharmacology to allow delivery into cells. In this review, we show how peptide sequences derived from toxins, such as NrTPs, SyLop-1 or MCaUF1-9, are used as vectors both by their combination with otherwise non-penetrating sequences or by cross-linking with larger particles
Dr. John Howl’s and Sarah Jones’ groups first defined the term ‘bioportide’ in 2008 [25], using Robert Schwyzer’s [26] terms “sychnologic” (continuous) and “rhegnylogic” (dispersed) that originally designate two types of hormones and their interaction with receptors: (a) receptors for adrenocorticotropic hormone and opioid peptides, wherein part of the receptor binding sequence is conserved between different agonists and is important to trigger receptor signaling and another part is the receptor binding sequence, found in tandem with the first, but without any necessary steric constraint in relation to the first, which has the function of addressing different agonists to different receptor subclasses; (b) in opposition, hormones organized in a “rhegnylogic” fashion have discontinuous amino acids residues that bind to the receptor and must be organized in a rigid structure for adequate signaling, as exemplified by insulin [27] (Fig. 1).
Whereas initial CPPs were engineered to be mere cargo transporters linked in tandem to pharmacophores, now, with the use of QSAR-based algorithms, it is possible to study CPPs with bioactivity associated with discontinued sequences.
Human protein-derived CPPs as bioportides
Cytochrome c (CYCS) as a bioportide
CYCS77–101 was identified using the QSAR-based algorithm [25, 28]. The rightmost structures in Fig. 2 present, in blue residues, the cryptic CPP CYCS77–101 that can mimic the trafficking events and apoptogenic activity of the native CYCS (peptide sequence presented in Table 1). CYCS77–101 accumulates at specific intracellular sites, has an efficient uptake and a wide distribution inside brain tumor cells, thus producing high cumulative and cytotoxic effects [25]. These data suggest that CYCS77–101 binds to several intracellular targets, triggering a caspase 3-dependent cell death [29]. For example, when CYCS binds to cardiolipin (target), it gains a peroxidase activity that contributes to the triggering of apoptosis [30]. The conjugation of CYCS77–101 with the nuclear localization sequence (NLS) from NUP153 forms NUP153–CYCS77–101 which has an enhanced bioportide activity compared with CYCS77–101 and increased nuclear penetration and apoptosis induction ability. In fact, four peptides derived from CYCS demonstrate CPP capacities, however, distinct intracellular localization (cytoplasm, nucleus, endoplasmic reticulum), thus suggesting the potential existence of different intracellular targets (Table 1). Since CYCS86–101 is a shorter variant of CYCS77–101, it seems that the ability of CYCS77–101 to enter into the nucleus is abolished due to the presence of additional amino acids and redistribution of positive charge (Table 1). Furthermore, both of these peptides demonstrate apoptogenic activity in tumor cells, thus mimicking the role of CYCS as a key regulator of programmed cell death. In contrast, another two cytochrome c-derived peptides: CYCS79–92 and CYCS79–88, which are inserts of CYCS77–101, are minimally toxic [25, 31] (Table 1).
Fig. 2.

Cytochrome c and the derived CYCS77–101. Tridimensional surfaces of cytochrome c (CYCS UniProt structure identifier 3ZCF [98]); lower line of molecules corresponds to opposite face of the molecule depicted on first line; in blue, in rightmost two structures, segment of residues 77–101, which has bioportide properties (cell penetration and cytotoxicity). Electrostatic surface colored according to Coulombic electrostatic potential, e = 4r, thresholds ± 5 kcal mol−1 e−1 at 298 K. Hydrophobicity surface colored following the Hessa and von Heijne hydropathic scale thresholds (dark orange most hydrophobic; white 0; aquamarine most hydrophilic) as described by Hessa et al. [99]. Molecular graphics and analyses performed using the UCSF Chimera package software [100]
Table 1.
Human and toxin-derived bioportides
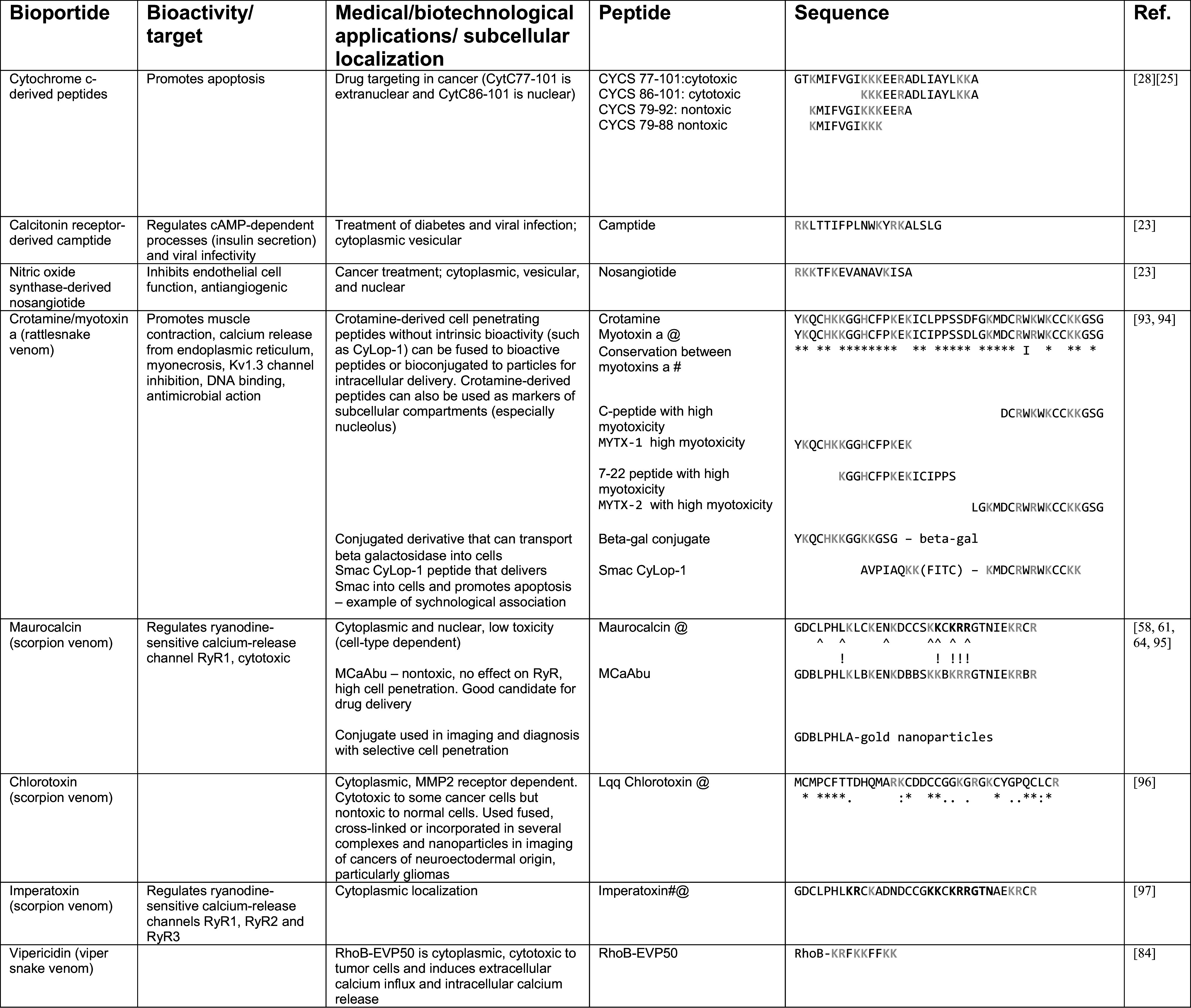
Toxin alignments regarding crotamine and chlorotoxin show that relevant residues are spread throughout the sequences. They also indicate the high conservation of cysteines. Indeed, disulfide bridges are important for toxin function (indicated by @). Myotoxin a most bioactive peptides are found in the N- and C-termini, as are nuclear localization sequences important for intracellular transport (please refer to Fig. 3)
Letters in gray indicate basic residues, which are frequently found in CPPs
An * (asterisk) indicates positions which have a single, fully conserved residue. A : (colon) indicates conservation between groups of strongly similar properties—scoring >0.5 in the Gonnet PAM 250 matrix. A. (period) indicates conservation between groups of weakly similar properties—scoring = <0.5 in the Gonnet PAM 250 matrix
# Alignment data not scored using Gonnet matrix, only full conservation indicated (overall identity >76 %). This reference shows alignment between 14 myotoxin a sequences. There are many more currently in public databases
^ and ! below sequences indicate residues most important in cell penetration and RyR modulation by maurocalcine, respectively. B in the maurocalcine-derived peptide indicates aminobutyric acid
# In bold, residues important in Imperatoxin A regulation of ryanodine receptors (GDCLPHLKRCKADNDCCGKKCKRRGTNAEKRCR)
Camptide and nosangiotide as bioportides
The QSAR model was also employed to identify putative bioportides within the primary sequences of the human type 1 calcitonin receptor (CALCR) [6] and a calmodulin binding domain of endothelial nitric oxide synthase (NOS3) [23, 32]. Camptide is derived from a portion of CALCR with vesicular/cytoplasmic distribution (Table 1). It has been shown to modulate insulin secretion and viral infectivity. In addition, camptide stimulates cAMP synthesis and increases the initial rate of GTP binding to trimeric G-proteins (GTP binding proteins).
Another example of bioportide is nosangiotide, which is derived from NOS3—an enzyme that generates the vasoprotective molecule nitric oxide and is the major endothelial cell weapon to fight vascular disease. Nosangiotide shows vesicular (lysosomal/mitochondrial), cytoplasmic, nuclear distribution (Table 1) [23]. It has a potent antiangiogenic activity in vitro, inhibiting proliferation, chemotaxis, and the tube-forming ability of primary bovine aortic endothelial cells induced by fibroblast growth factor 2 (FGF2). In vivo, nosangiotide also revokes the ability of FGF2 to induce revascularization in the chorioallantoic membrane assay [23].
Natural toxin-derived CPPs as bioportides
Although the bioportides described so far in the literature derive from human proteins, many toxins can also be considered bioportides, since they can enter cells and bind to intracellular targets.
Crotamine
Crotamine is a toxin of the family of myotoxins alpha, or a, found in the venom of the South American rattlesnake Crotalus durissus terrificus. Mature crotamine has stretches of positively charged residues concentrated at N- and C-termini that seem to correspond to two putative nuclear localization sequences (NLS2–18/NLS27–39), shown in green in Fig. 3a [33]. Crotamine has similar structural properties as human protein-derived bioportides, being rhegnylogically organized and intrinsically bioactive (Table 1). Positively charged sites are distributed throughout the crotamine structure and optimize the electrostatic interaction with negative charges on the cell surface (Fig. 3c; Table 1). Crotamine penetrates into cells where it has vesicular, cytoplasmic, and nuclear distribution [34]. It accumulates in lysosomes of tumor and metastatic cells in a murine melanoma model in which it has a major cytotoxic effect, especially in vivo [35–37]. Although several studies concerning the mechanisms of action of crotamine are still ongoing, the current landscape of knowledge presented above assures that crotamine is an authentic bioportide.
Fig. 3.

Crotamine, myotoxin a and peptides derived thereof a. Mature primary sequence of crotamine (UniProt structure identifier Q9PWF3) is aligned with MYTX (UniProtKB/Swiss-Prot sequence P01476.1) and three crotamine-derived CPPs. The disulfide bridge pattern is shown in green lines. In gray, conserved sequences between crotamine and MYTX, in pea-green basic stretches forming the putative NLS2–18 and NLS27–39, in turquoise, putative DNA interacting residues (DNA31–35); in pink and purple, nuclear localization signals that, combined, generate NrTP-1 [46] and NrTP-6 [101] (purple indicates the conserved sequence between the three peptides); in blue, remaining CyLop-1 sequence [48]. b, c The secondary structure and tridimensional surfaces of crotamine are shown for two opposite surfaces, in b the distribution of hydrophobic and hydrophilic sequences is shown [102], having been based on UniProt structure 1Z99 [102]. The vertical dashed line delimitates hydrophobic surfaces of the toxin. The dipole moment was calculated using default parameters with the Protein Dipole Moments Server [103], based on Peigneur et al. [41] and Sabatier and De Waard [104]. In c, the distribution of electrostatic charge is shown. Electrostatic surface colored according to Coulombic electrostatic potential, e = 4r, thresholds ± 5 kcal mol−1 e−1 at 298 K. Hydrophobicity surface colored following the Hessa and von Heijne hydropathic scale thresholds (dark orange most hydrophobic; white 0; aquamarine most hydrophilic) as described by Hessa et al. [44]. Secondary structure: beta-strand depicted in yellow, alpha-helix in red, coil in pale gray and disulfide bonds, in green. Molecular graphics and analyses for three-dimensional structures were performed with the UCSF Chimera package software [100]
In addition, crotamine is capable of binding electrostatically to negatively charged DNA allowing its delivery into cells [33, 35, 38]. Inspection of the in silico three-dimensional structure of crotamine suggests that residues Arg31 to Lys35 (RW 4-K) could serve as potential DNA-binding sites (Fig. 3a). An hexapeptide derived from crotamine containing this sequence has lower DNA binding affinity compared to the full-length crotamine, indicating that added interactions within the structure are necessary for activity, a typical characteristic of rhegnylogic-associated functions [39].
We demonstrated that the cytotoxic effect of crotamine in tumor cells involves rapid intracellular calcium release and loss of mitochondrial membrane potential, selectively triggering tumor cell death [40]. Crotamine is also a potent blocker of the mitochondrial voltage-gated potassium channel (KV1.3) in non-tumoral cells [41], and has the dyad formed by an aromatic residue and a basic residue that potentially bind and block the pore of voltage gated channels [41, 42].
Although no mutagenesis studies have been carried out with crotamine to investigate the mechanism of inhibition of Kv1.3 channels, a lot can be inferred from its structure. Structure–activity correlation studies have verified that a given venom peptide can be active on several channels through diverse interacting surfaces [43]. Similarly, many scorpion toxins with the α/β cysteine stabilized fold, like the knottin toxins, act on KV channels through binding sites primarily involving their β-sheet structures, although they may also interact with small conductance Ca2+-activated K+ channels (SK) through surfaces opposite to their α-helical structures, which shows why these small toxins can have a large selectivity profile with multifunctional activities [43]. Likewise, in Huys et al. [44], it was proposed that two separate functional faces might exist on the scorpion BmTx3 molecule, responsible for the two different K+-current-blocking functions. Another well-established example of multiple interacting surfaces can be found in the Janus-faced atracotoxins, which are a family of insect-specific excitatory neurotoxins isolated from the venom of Australian funnel-web spiders. They contain a strikingly asymmetric distribution of charged residues, from which their name is derived. The toxins are selective for insect voltage-gated potassium channels [45]. These characteristics again indicate the rhegnylogic nature of toxins, in which discontinuous amino acid residues must be folded into a specific conformation to have activity.
Structure-guided reconstruction of crotamine was proposed in an attempt to define a minimal crotamine motif fundamental for membrane translocation and internalization. A crotamine-derived peptide was obtained fusing two portions of crotamine: N-terminal residues Y01–G09 and C-terminal residues K38–G42 (Fig. 3a, highlighted in pink and purple). This so-called nucleolar targeting peptide (NrTP1) is also characterized by the presence of a Lys-rich palindromic hexad (KKGGKK) [35, 46]. The motifs associated with membrane translocation, which are composed mostly of basic and hydrophilic domains (Fig. 3a–c), were further used for the design of NrTP1 derivatives, for instance, NrTP6, in which there is a substitution of Cys 04 by Ser. In contrast to crotamine, NrTP1 demonstrates preferential nucleolar homing, and although NrTP1 shows efficient penetrating capacity in cancer cells, it does not present any toxicity [38]. Being non-toxic at the micromolar range, NrTP6 is also an excellent candidate for drug transport in the style of traditional CPP carriers (sychnologically, or sequentially, organized), as has already been shown by the intracellular transport of conjugated NrTP6 and β-Galactosidase (Table 1) into HeLa cells [47]. Since β-Galactosidase is a very large molecule (116.5 kDa) that exceeds the size of many therapeutically relevant cargos such as antibodies (~150 kDa), it is likely that NrTP6 is an important candidate for the intracellular transport of multiple drugs and even nanoparticles.
CyLoP-1 is a typical peptide designed to transport tandem-associated cargo into cells. It is a crotamine derivative discovered using structure–activity relationship (SAR) studies (Fig. 3a) and is composed by the segment 30–39 (depicted in blue and purple in Fig. 3a) of the crotamine core, which is highly hydrophobic, comprises the tryptophan-rich DNA binding motif (indicated as DNA31–35 in Fig. 3a) and has cell-penetrating properties [48]. CyLoP-1 localizes to the cytoplasm and nucleus. To verify the capability of CyLoP-1 to transport biologically active molecules across cellular membranes, the pro-apoptotic peptide sequence AVPIAQK (SmacN7) was attached to CyLoP-1 (Table 1). AVPIAQK is derived from the N-terminus of the second mitochondria-derived activator of caspase (Smac) protein. Induction of caspase-3 activity was observed after the treatment of HeLa cells with SmacN7-K (FITC)-CyLoP-1 in comparison with K(FITC)-CyLoP-1 or even with SmacN7-K(FITC)-Tat (49–57), which is derived from Tat, one of the best characterized and explored CPPs. CyLoP-1 or Smac-N7, as well as the Tat derivative peptide, failed to increase the caspase-3 activity alone. These results indicate that CyLoP-1 at low micromolar concentrations is nontoxic, while presenting the superior ability to deliver bioactive cargos into the cytosol. The efficient cargo delivery mediated by CyLoP-1 is due to the presence of the (RW)4-K molecular motif, which is known to optimize the interaction of positively charged CPPs with negatively charged GAGs [49].
Myotoxin a
Myotoxin a (MYTX) is a small basic polypeptide isolated from the venom of the prairie rattlesnake (Crotalus viridis viridis) (Fig. 3a; Table 1). MYTX is closely related to crotamine and also demonstrates CPP activity [50]. Several studies carried out in the 1980s and 1990s using radiolabeled MYTX showed that it induces Ca2+ release from the sarcoplasmic reticulum (SR) of skeletal muscle cells [51–53], probably by binding to calsequestrin [54], ATP/ADP translocase [55] and inhibiting Ca2+-ATPase [56]. The attachment of MYTX to Ca2+-ATPase is believed to cause uncoupling of the calcium pump [56]. Five molecular variants of MYTX were obtained by chemical cleavage and only two of them YKQCHKKGGHCFPKEK (MYTX-1) and LGKMDCRWKWKCCKKGSG (MYTX-2) (Table 1) inhibited 45Ca uptake into isolated SR and bound to Ca2+-ATPase, as well as induced weak skeletal muscle vacuolization similar to that caused by native MYTX and increased serum creatine kinase activity [57]. These molecular variants correspond to the N-terminal and C-terminal portions of MYTX, respectively, while other peptides, which did not show any activity or were less active, correspond to the center of the MYTX sequence [56, 57]. This study demonstrates that molecular variants from both the N-terminal and the C-terminal regions of the primary sequence of MYTX are required to express its biological activity. MYTX-1 and MYTX-2 sequences can also be found in crotamine (Table 1), suggesting that such crotamine-derived molecular variants may have similar biological activity as those of MYTX, although this must still be verified. On the other hand, considering the literature on crotamine, it is possible to conclude that MYTX-1 may possess nuclear, while MYTX-2, cytoplasmic, translocation properties, which also remains to be experimentally confirmed. In regard to the purposes of this review, both of these MYTX molecular variants are bioportides, because they are translocated into cells and are intrinsically bioactive.
The myotoxin a family has many other toxins derived from other rattlesnake venoms with high identity with myotoxin a and crotamine. Table 1 shows that areas of high conservation are found throughout the sequence though tend to be greatest in the N and C-termini. Cysteine residues are highly conserved as well, indicating the importance of the three disulfide bridges found in this protein family. These data support the rhegnylogic organization of myotoxin a: biological activity and cell penetration residues are found throughout the sequence and require specific protein folding to be functional.
Maurocalcine
Maurocalcine (MCa) is a toxin from the venom of the scorpion Scorpio maurus palmatus that folds in a classical inhibitor cysteine knot (ICK). It is able to penetrate into cells and shows cytoplasmic localization [58, 59] (Fig. 4a). MCa induces Ca2+ release from intracellular stores by directly binding to the skeletal muscle isoform of the ryanodine receptor 1 (RyR-1), a calcium channel that mediates Ca2+ release from the sarcoplasmic reticulum into the cytoplasm and, thereby, plays a key role in triggering muscle contraction following depolarization of T-tubules. The residues interacting with RyR-1 are showed in pale-purple in Fig. 4a and were determined according to their similarity to imperatoxin-A, another CPP, as discussed below [60]. Single residue mutations of maurocalcine also show key residues important in RyR modulation and cell penetration, which partially coincide (Table 1), indicating its rhegnylogic organization [61]. MCa is non-toxic by intravenous injection, despite its pharmacological activity on RyR-1, but is extremely toxic when injected into the intracerebroventricule of mice [59]. Several analogs of MCa have been developed that lack its characteristic neurotoxic activity without affecting its vector efficiency, like MCaUF1–9 and d-maurocalcin. MCa mutations on Arg23 and Arg24 yield efficient vectors devoid of toxicity [59, 61, 62]. In addition, a disulfide-less mutant called MCaAbu has been produced, by replacing the six-cysteine residues of MCa with the L-α-aminobutyric acid residue (Table 1). This peptide does not have any effect on RyR-1, whereas its penetration activity is retained [63]. Maurocalcine derivatives have also been conjugated to gold nanoparticles to enhance their delivery into cells and show promise in the development of imaging techniques [64].
Fig. 4.

Structure and function of maurocalcine and chlorotoxin. a, b The primary, secondary and tridimensional surfaces of maurocalcine (GeneBank P60254; UniProt 1C6W [105]) and chlorotoxin (GeneBank P45639, UniProt 1CHL [106]) are shown, respectively. Gray conserved sequences; purple ryanodine receptor binding sequences (RyR1), based on [97]. The upper part of a corresponds to the alignment of maurocalcine (UniProtKB/Swiss-Prot: P60254.1) and insectotoxin I5A from the venom of the Mesobuthus eupeus scorpion (UniProtKB/Swiss-Prot sequence P15222.2). The upper part of b corresponds to the alignment of chlorotoxin (UniProtKB/Swiss-Prot sequence P45639.1) and imperatoxin-A from Pandinus imperator scorpion (UniProtKB/Swiss-Prot: P59868.1). The disulfide bridge pattern is shown in green. Lower portions of a and b indicate opposite surfaces of maurocalcine and chlorotoxin, with the distribution of hydrophobic and hydrophilic regions and charge distribution, based on [62, 104, 107]. Electrostatic surface colored according to Coulombic electrostatic potential, e = 4r, thresholds ± 5 kcal mol−1 e−1 at 298 K. Hydrophobicity surface colored following the Hessa and von Heijne hydropathic scale thresholds (dark orange most hydrophobic; white 0; aquamarine most hydrophilic) as described by Hessa et al. [99]. The dashed line shows peptide anisotropy, with highly hydrophobically charged residues above and positively charged sequences in the lower half. Secondary structure: beta-strand is depicted in yellow, alpha-helix in red, coil in pale gray and disulfide bonds are depicted in green. Molecular graphics and analyses for three-dimensional structures were performed with the UCSF Chimera package software [100]
Chlorotoxin
Chlorotoxin (CTX) is a neuropeptide from the venom of the deathstalker scorpion (Leiurus quinquestriatus) with considerable sequence homology to small insectotoxins [65]. It also has the ICK fold, and is able to paralyze insects and crayfish, albeit is non-toxic to mammals (Fig. 4b; Table 1). Since the initial discovery of chlorotoxin, many similar toxins have been identified from other scorpion species, shared biological activity. Table 1 shows the most conserved residues amongst several chlorotoxin members, including cysteines important in protein folding. The study of the mechanism of action of chlorotoxin has been challenging, because its cell surface receptor remains under investigation—several other receptors have been claimed besides chloride channels—after which it was called—matrix metalloprotease MMP-2 in complex with αvβ3 integrin and a chloride channel [66, 67] and Annexin A2 [68–70]. CTX binds specifically to the glioma cell surface as a specific chloride channel and matrix MMP-2 blocker, leading to the inhibition of cell migration and blocking chloride currents [71]. A synthetically cyclized CTX derivative, TM-601, translocates cell membranes and localizes near the trans-Golgi in gliomas, lung carcinoma, and normal vascular endothelial cells [72, 73]. In contrast, it shows cytoplasmic localization in astrocytes and normal human dermal fibroblasts [74]. As mentioned above, CTX binds to a protein complexes formed by chloride channels; MMP-2; αvβ3 integrin; MT1-MMP and TIMP-2, and this binding produces internalization of the entire protein complex, thereby leading to reduction of the activity of both the chloride channel and MMP-2 [66]. This dependence of chlorotoxin on receptor binding renders it uncharacteristic as compared to other cationic CPPs that usually do not require specific receptor binding for translocation. The conjugation of iron nanoparticles with multiple CTX molecules deactivates membrane-bound MMP-2, produces receptor-mediated endocytosis, and inhibits cell volume changes enhancing inhibition of invasion in vitro compared to monomeric CTX [75]. DeBin et al. [76] showed that CTX acts as a blocker of small conductance Cl− channels that are widely expressed throughout the central nervous system. This has led to many applications of CTX and derived peptides in cancer-targeting for imaging, including conjugation to many types of nanoparticles and anticancer drugs (reviewed in [77]) for which there already are human clinical trials [78]. The many examples chlorotoxin conjugates being currently tested points out the importance of development of toxin-derived CPPs [77].
It should be reminded, however, that the precise mechanism of CTX action is far from fully elucidated, and its effect as a chlorine blocker is still controversial: (1) patch-clamp reports have shown that, in spite of CTX-mediated chloride current block, the functional inhibition of the channel occurs with a lower affinity than expected from binding experiments (600 nM) [79]; (2) CTX has no effect on the proliferative rate of C6 glioma cells in vitro [80]; and (3) CTX does not inhibit volume-regulated, Ca-activated nor cyclic AMP-activated Cl− channels [81]. Hence, while CTX may bind to Cl− channels, it does not necessarily block them, and the real pharmacological target for CTX probably still needs to be discovered and described.
Imperatoxin
Imperatoxin A (IpTxa) is purified from the venom of the African scorpion Pandinus imperator. It stimulates intracellular calcium release upon binding to RyRs [82]. Like many of the aforementioned toxins, IpTxa has a very tightly determined structure; mostly, hydrophobic, but also polarized with positive ions, which researchers hypothesized could interact with lipid membranes. The activity of IpTxa on RyR has been known for many years, but it was not known if the effect was by direct or indirect binding. Studies carried out with perfused intact mouse ventricular myocytes and fluorescently labeled IpTxa show that the toxin penetrates into cells, localizing, preferably, to the cytoplasm, where it binds to RyR [82] (Table 1).
Cryptic vipericidin peptides
The analyses of proteomes have shown that many active proteins derive from proteolysis of precursor proteins, besides other forms of cleavage [83, 84]. These peptides have been called “cryptides,” since they are hidden within the precursor protein. Such cryptides may have the same function as the precursor or entirely different functions [83]. Venom toxins are stored in a precursor form in animal glands and are activated upon inoculation into the target organism [84]. Indeed, venom contains not only toxin precursors, but also several proteases. Furthermore, the tissue of the target may react to the venom releasing and activating another set of proteases, such as those associated with inflammation. Therefore, the analysis of venom proteomes should include the prediction of proteolytic products of toxins by action of venom- and target-derived proteases, since all these byproducts may have important biological roles and are potentially new pharmacological agents. When such cryptides are cell-penetrating and target intracellular sites, they constitute another class of bioportides.
Such approach was used by Wang et al. [84] in the study of the vipericidins lachesicidin, a toxin derived from the South American pit viper Lachesis muta rhombeata (Genebank accession number: AGS36142.1) and batroxicidin, from Bothrops atrox (UniProtKB/Swiss-Prot: U5KJC9.1). The authors synthesized peptides derived from the lysine-rich, α-helical, carboxyl-terminal of the toxins, known to have an antimicrobial action and be rich in potential protease cleavage sites. They screened for toxicity of rhodamine B-labeled derivatives against zebrafish larvae and detected a highly toxic peptide named RhoB-EVP50. Further studies of this nine amino acid-residue peptide showed that it penetrates into the cytoplasm of mammalian cells, and at higher doses, that it is cytotoxic and induces calcium influx and intracellular calcium release in tumoral cell lines. RhoB-EVP50 (Table 1), therefore, also constitutes a bioportide.
Structural organization of natural toxins and bioportidebility
Many venom peptides exhibit electric charge anisotropy and hydropathicity. Figure 3c shows the calculated dipole moment for crotamine, which is represented by the purple arrow trace pointing from the most negative (tip) toward the most positive region of the molecule (top). The electrostatic anisotropy guides and orients the peptide within the electrostatic field of the membrane receptor target [41, 85]. Characteristically, crotamine exhibits an amphipathic surface with a central hydrophobic patch that circumvents the entire molecule center, splitting the surface in two distinctive hydrophilic lobules (left and right delineated by dashed vertical lines, Fig. 3b).
MCa and CTX display a remarkable anisotropy: one face is highly basic, while the opposite face is relatively hydrophobic (Fig. 4a, b). Splitting the molecules into a hydrophobic domain on top and a hydrophilic basic face at bottom, denoted by the horizontal dashed line of Fig. 4a, b, possibly contributes to the ability that these toxins have to interact with the cell membrane and translocate into cells [86]. MCa can be schematically represented by three domains: one hydrophobic head at the top of the peptide, a larger second face, mainly basic, and a third side domain that contains the pharmacophore, composed by the residues that interact with RyR-1 [62]. Unfolded reduced MCa is slightly less efficient in cell penetration than the folded/oxidized MCa, indicating that the correct positioning in space of the various structural determinants is important for optimization of MCa cell penetration [62], which is a common feature of rhegnylogically organized (discontinuous) domains of many bioportides. Unfolded truncated MCaUF1–9 is a small CPP derived from the hydrophobic domain of MCa and is very efficient in membrane translocation in comparison with Tat, penetratin, polyarginine or unfolded MCa [62]. Surprisingly, the net charge of MCaUF1–9 is neutral, but at acidic pH (lower than 6), the protonation of the histidine residue at position 6 increases its ability to be retained at polarized ends of CHO cell membranes [62]. Several scorpion toxins with knottin folds, such as hadrucalcin, imperatoxin-A, hemicalcin, and opicalcins, share high similarity at the hydrophobic domain with MCa, and one can predict that they also might behave as efficient CPPs [87]. Given the importance of the knottin fold in cell penetration, authors explore this property in search of independent cell-penetrating domains, modified to have low toxicity and to be bound in tandem (sychnologically) with bioactive domains, in the form of conventional CPPs.
Structure–activity relationship (SAR) studies show that a given venom peptide can be active on several targets through diverse interacting surfaces. Many scorpion toxins with α/β cysteine stabilized knottin like fold act on KV channels through binding sites primarily involving their β-sheet structures. Several families exist with knottin folds and differing biological activities—part of the sequences, associated with a certain portion of the folded protein, enables cell penetration. Other sequences are associated with other protein surfaces and biological activities such as interaction with SK channels. Such combination of effects due to different interacting surfaces allows for multifunctionality even in these very small toxins [88]. Most studies by SAR have been teasing apart the domains responsible for transmembrane transport and carrying out sequence changes to decrease cytotoxicity to use such sequences as conventional carrier CPPs. Computational approaches may now allow this type of toxin to be optimized as a bioportide: selecting and modifying sequences, so that key amino acid residues discontinuously distributed can fold and carry out pharmacological effects in conjunction with membrane permeability.
Bioportidebility of natural toxins versus of human protein-derived CPPs
As mentioned earlier in this text, bioportides have been found, so far, exclusively in human-derived peptides. However, in this review, we suggest that bioportides can occur in toxins and toxin-derived peptides (as in other proteomes), opening a new field for search for biotechnological tools and drugs with improved pharmacokinetic properties.
As occurs with human CYCS, proteomimetic CPPs of crotamine and MYTX collectively target different cell organelles, including lysosomes, SR, mitochondria, and the nucleus. Furthermore, the multifunctional role of CYCS-derived sequences requires them to translocate numerous biological membranes, including, in the case of CYCS77–101, the cytoplasmic-nuclear interface, for which CYCS77–101 is expected to have a NLS [89–91]. Regarding crotamine, at least two proteomimetic sequences, one responsible for cytoplasmic membrane translocation, CyLop-1, while the other, NrTP1, for nuclear membrane translocation, have been identified, though they appear to be specifically associated with targeting, having no other biological activity as yet identified. Like CYCS77–101, full-length CTX and crotamine are cytotoxic to some cell types. Moreover, CTX and crotamine have selective cytotoxicity against melanoma cancer cells in vitro and in vivo, inhibiting cell proliferation, growth, and cancer cell invasion [75]. Therefore, there are both non-toxic and toxic forms of CYCS and toxin-derived peptides, which can be useful depending on the aim: simple transport (traditional CPPs) or transport associated with cytotoxicity (bioportide)—as in the case of targeted tumor treatment. In this last case, some toxin-derived CPPs actually present an advantage over CYCS77–101, in that they do not require the addition of tumor homing sequences. Crotamine and CTX do not have this limitation, since they naturally recognize and home to tumors, although the mechanism of this event is still unknown.
Conclusion
We hereby describe several toxins and peptides derived thereof that can enter mammalian cells and have intracellular targets. The cell-penetrating activity of the toxins presented in this review is, in itself, nontoxic, i.e., membranes remain intact after peptide transport, whereas other features of the peptides confer biological activity, such as mobilization of intracellular calcium stores and induction of apoptosis.
Many other toxins with intracellular targets have been discovered primarily in plants and bacteria. These include ribosome inactivating proteins saporin α-CD7 and ricin, from plants and several bacterial-derived toxins, such as derivatives of hemolysin from enterohemorrhagic Escherichia coli, diphtheria toxin, Pseudomonas Exotoxin A and Anthrax toxin. All of these bind to specific cell receptors, entering the cell by endocytosis. They also fit the definition of bioportides. Given their high cytotoxicity, these toxins have been used to treat cancer cells. However, to decrease side-effects, they have been modified to disable their receptor-binding domains, responsible for cell targeting and endocytosis. Instead, they are associated with other target molecules, more specific for cancer cells (reviewed in [92]). Unlike the CPPs discussed in this review, which are highly cationic, can be predicted based on computational algorithms and are found quite frequently in the human genome, receptor-dependent toxins must be discovered in a more specific approach, based on the receptor of interest.
Since peptides and proteins are usually incapable of transposing membranes, traditionally peptide toxins have not been studied regarding intracellular targets, except for some receptor-dependent toxins, as discussed above. Here, we argue that this approach restricts the field of toxin research and its potential in the discovery of new pharmacological and biotechnological agents. Furthermore, it allows the “natural” biological role of toxins to be better characterized and may indicate whole new mechanisms of venom toxicity and its co-evolution with that of toxin resistance by target organisms.
We also discuss in this review that some toxin families have cell permeability associated with channel receptor binding domains: highly polarized structures whereby one portion of the molecule is hydrophobic and another is cationic. This is important in both positioning and binding to channel receptors on the extracellular cell surface and initial binding, followed by transmembrane transport, regarding membrane penetration ability. It is probable that the same type of domain also allows peptide interaction with intracellular channels. In this case, intracellular transport and intracellular biological activity are not associated with independent peptide sequences (sychnologically organized), but can be overlapping (rhegnylogically organized), which differs from the traditional knowledge of targeting portions of proteins, such as secretion signal peptides and nuclear localization domains.
Given that toxins evolve to have a high biological activity in target organisms—including mammals—and that there is a large toxin proteome (from animals, plants, and microorganisms), the fact that many toxins (or peptides derived thereof) can have intracellular targets opens a new broad avenue for applied toxin research. Moreover, QSAR studies with mammalian proteomes suggest that cell penetration and intracellular biological activities can occur within larger sequences with different “natural/original” biological activities—these can be identified computationally, synthesized, and can generate important pharmacological peptides. It is probable that these intrinsic/internal sequences also occur within toxin sequences. Indeed, one approach for the discovery of such internal sequences is the study of cryptic toxins, as was shown for vipericidin.
The development of high throughput DNA and protein sequencing technologies, of potent in silico algorithms (and computers) and of techniques that require only very small samples for venom characterization are currently enabling toxinologists to explore toxins much more efficiently. Dwindling biodiversity has put pressure on identification and sequencing of venoms and venom databases are quickly becoming available and will probably increase exponentially over the next few years. As mentioned before, QSAR use for identification of cell-penetrating toxins with intracellular targets has not been used to systematically study toxin libraries to date, but, as shown here, has a high potential for the discovery of new drugs and therapeutic tools.
Acknowledgments
We would like to thank GSK (Glaxo Smith Kline) for financing this publication, Grant 2015/50040-4, São Paulo Research Foundation (FAPESP). J. T. was supported by grants GOE3414N (F.W.O.-Vlaanderen) and IUAP 7/10 (Inter-University Attraction Poles Program, Belgian State, Belgian Science Policy).
Compliance with ethical standards
Conflict of interest
None of the authors have conflict of interest with the subject matter or materials discussed in the text.
Contributor Information
Irina Kerkis, Phone: + 5511 26279705, Email: irina.kerkis@butantan.gov.br.
Paulo L. de Sá Junior, Phone: + 5511 26279705, Email: paulsaj2001@yahoo.com.br
References
- 1.Mae M, Langel U. Cell-penetrating peptides as vectors for peptide, protein and oligonucleotide delivery. Curr Opin Pharmacol. 2006;6:509–514. doi: 10.1016/j.coph.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 2.Heitz F, Morris MC, Divita G. Twenty years of cell-penetrating peptides: from molecular mechanisms to therapeutics. Br J Pharmacol. 2009;157:195–206. doi: 10.1111/j.1476-5381.2009.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Järver P, Mäger I, Langel Ü. In vivo biodistribution and efficacy of peptide mediated delivery. Trends Pharmacol Sci. 2010;31:528–535. doi: 10.1016/j.tips.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 4.Bechara C, Sagan S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013;587:1693–1702. doi: 10.1016/j.febslet.2013.04.031. [DOI] [PubMed] [Google Scholar]
- 5.Howl J, Jones S. Insights into the molecular mechanisms of action of bioportides: a strategy to target protein-protein interactions. Expert Rev Mol Med. 2015;17:e1. doi: 10.1017/erm.2014.24. [DOI] [PubMed] [Google Scholar]
- 6.Hällbrink M, Kilk K, Elmquist A, et al. Prediction of cell-penetrating peptides. Int J Pept Res Ther. 2005;11:249–259. doi: 10.1007/s10989-005-9393-1. [DOI] [Google Scholar]
- 7.Cronican JJ, Thompson DB, Beier KT, et al. Potent delivery of functional proteins into Mammalian cells in vitro and in vivo using a supercharged protein. ACS Chem Biol. 2010;5:747–752. doi: 10.1021/cb1001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 9.Derossi D, Calvet S, Trembleau A, et al. Cell internalization of the third helix of the antennapedia homeodomain is receptor-independent. J Biol Chem. 1996;271:18188–18193. doi: 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- 10.Elliott G, O’Hare P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell. 1997;88:223–233. doi: 10.1016/S0092-8674(00)81843-7. [DOI] [PubMed] [Google Scholar]
- 11.Schwarze SR, Hruska KA, Dowdy SF. Protein transduction: unrestricted delivery into all cells? Trends Cell Biol. 2000;10:290–295. doi: 10.1016/S0962-8924(00)01771-2. [DOI] [PubMed] [Google Scholar]
- 12.Milletti F. Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov Today. 2012;17:850–860. doi: 10.1016/j.drudis.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Futaki S, Suzuki T, Ohashi W, et al. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 14.Duchardt F, Ruttekolk IR, Verdurmen WPR, et al. A cell-penetrating peptide derived from human lactoferrin with conformation-dependent uptake efficiency. J Biol Chem. 2009;284:36099–36108. doi: 10.1074/jbc.M109.036426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harreither E, Rydberg HA, Åmand HL, et al. Characterization of a novel cell penetrating peptide derived from human Oct4. Cell Regen. 2014;3:2. doi: 10.1186/2045-9769-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koren E, Torchilin VP. Cell-penetrating peptides: breaking through to the other side. Trends Mol Med. 2012;18:385–393. doi: 10.1016/j.molmed.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 17.Madani F, Lindberg S, Langel Ü, et al. Mechanisms of cellular uptake of cell-penetrating peptides. J Biophys. 2011;2011:1–10. doi: 10.1155/2011/414729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, Tsui T, Ma W. Intracellular delivery of molecular cargo using cell-penetrating peptides and the combination strategies. Int J Mol Sci. 2015;16:19518–19536. doi: 10.3390/ijms160819518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Pisa M, Chassaing G, Swiecicki J-M. Translocation mechanism(s) of cell-penetrating peptides: biophysical studies using artificial membrane bilayers. Biochemistry. 2015;54:194–207. doi: 10.1021/bi501392n. [DOI] [PubMed] [Google Scholar]
- 20.Gupta B, Levchenko T, Torchilin V. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv Drug Deliv Rev. 2005;57:637–651. doi: 10.1016/j.addr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Delaroche D, Aussedat B, Aubry S, et al. Tracking a new cell-penetrating (W/R) nonapeptide, through an enzyme-stable mass spectrometry reporter tag. Anal Chem. 2007;79:1932–1938. doi: 10.1021/ac061108l. [DOI] [PubMed] [Google Scholar]
- 22.Walrant A, Correia I, Jiao C-Y, et al. Different membrane behaviour and cellular uptake of three basic arginine-rich peptides. Biochim Biophys Acta Biomembr. 2011;1808:382–393. doi: 10.1016/j.bbamem.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 23.Howl J, Matou-Nasri S, West DC, et al. Bioportide: an emergent concept of bioactive cell-penetrating peptides. Cell Mol Life Sci. 2012;69:2951–2966. doi: 10.1007/s00018-012-0979-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lukanowska M, Howl JJS. Bioportides: bioactive cell-penetrating peptides that modulate cellular dynamics. Biotechnol J. 2013;8:918–930. doi: 10.1002/biot.201200335. [DOI] [PubMed] [Google Scholar]
- 25.Howl J, Jones S. Proteomimetic cell penetrating peptides. Int J Pept Res Ther. 2008;14:359–366. doi: 10.1007/s10989-008-9135-2. [DOI] [Google Scholar]
- 26.Schwyzer R. ACTH: a short introductory review. Ann N Y Acad Sci. 1977;297:3–26. doi: 10.1111/j.1749-6632.1977.tb41843.x. [DOI] [PubMed] [Google Scholar]
- 27.Portoghese PS. Bivalent ligands and the message-address concept in the design of selective opioid receptor antagonists. Trends Pharmacol Sci. 1989;10:230–235. doi: 10.1016/0165-6147(89)90267-8. [DOI] [PubMed] [Google Scholar]
- 28.Jones S, Östlund P, Langel Ü, et al. A rhegnylogic strategy for the synthesis of signal transduction modulatory, cell penetrating peptides. In: Rolka K, Rekowski P, Silberring J, et al., editors. peptides. Geneva: Kenes International; 2006. pp. 430–443. [Google Scholar]
- 29.Jones S, Holm T, Mäger I, et al. Characterization of bioactive cell penetrating peptides from human cytochrome c: protein mimicry and the development of a novel apoptogenic agent. Chem Biol. 2010;17:735–744. doi: 10.1016/j.chembiol.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 30.Kagan VE, Tyurin VA, Jiang J, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 31.Jones S, Holm T, Mäger I, et al. Characterization of bioactive cell penetrating peptides from human cytochrome c: protein mimicry and the development of a novel apoptogenic agent. Chem Biol. 2010;17:735–744. doi: 10.1016/j.chembiol.2010.05.018. [DOI] [PubMed] [Google Scholar]
- 32.Aoyagi M. Structural basis for endothelial nitric oxide synthase binding to calmodulin. EMBO J. 2003;22:766–775. doi: 10.1093/emboj/cdg078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerkis A, Kerkis I, Rádis-Baptista G, et al. Crotamine is a novel cell-penetrating protein from the venom of rattlesnake Crotalus durissus terrificus . FASEB J. 2004;18:1407–1409. doi: 10.1096/fj.03-1459fje. [DOI] [PubMed] [Google Scholar]
- 34.Kerkis A, Kerkis I, Radis-Baptista G, et al. Crotamine is a novel cell-penetrating protein from the venom of rattlesnake Crotalus durissus terrificus . FASEB J. 2004;18:1407. doi: 10.1096/fj.03-1459fje. [DOI] [PubMed] [Google Scholar]
- 35.Hayashi MAF, Nascimento FD, Kerkis A, et al. Cytotoxic effects of crotamine are mediated through lysosomal membrane permeabilization. Toxicon. 2008;52:508–517. doi: 10.1016/j.toxicon.2008.06.029. [DOI] [PubMed] [Google Scholar]
- 36.Kerkis I, Silva F de S, Pereira A, et al. Biological versatility of crotamine—a cationic peptide from the venom of a South American rattlesnake. Expert Opin Investig Drugs. 2010;19:1515–1525. doi: 10.1517/13543784.2010.534457. [DOI] [PubMed] [Google Scholar]
- 37.Kerkis I, Hayashi MAF, Prieto ARB, et al. State of the art in the studies on crotamine, a cell penetrating peptide from South American rattlesnake. Biomed Res. 2014 doi: 10.1155/2014/675985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Radis-Baptista G, Kerkis I. Crotamine, a small basic polypeptide myotoxin from rattlesnake venom with cell-penetrating properties. Curr Pharm Des. 2011;17:4351–4361. doi: 10.2174/138161211798999429. [DOI] [PubMed] [Google Scholar]
- 39.Chen PC, Hayashi MA, Oliveira EB, Karpel RL. DNA-interactive properties of crotamine, a cell-penetrating polypeptide and a potential drug carrier. PLoS One. 2012;7:e48913. doi: 10.1371/journal.pone.0048913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nascimento FD, Sancey L, Pereira A, et al. The natural cell-penetrating peptide crotamine targets tumor tissue in vivo and triggers a lethal calcium-dependent pathway in cultured cells. Mol Pharm. 2012;9:211–221. doi: 10.1021/mp2000605. [DOI] [PubMed] [Google Scholar]
- 41.Peigneur S, Orts DJB, Prieto da Silva AR, et al. Crotamine pharmacology revisited: novel insights based on the inhibition of K-V channels. Mol Pharmacol. 2012;82:90–96. doi: 10.1124/mol.112.078188. [DOI] [PubMed] [Google Scholar]
- 42.Leanza L, Biasutto L, Managò A, et al. Intracellular ion channels and cancer. Front Physiol. 2013;4:1–7. doi: 10.3389/fphys.2013.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mouhat S, Andreotti N, Jouirou B, Sabatier JM. Animal toxins acting on voltage-gated potassium channels. Curr Pharm Des. 2008;14:2503–2518. doi: 10.2174/138161208785777441. [DOI] [PubMed] [Google Scholar]
- 44.Huys I, Xu C-Q, Wang C-Z, et al. BmTx3, a scorpion toxin with two putative functional faces separately active on A-type K+ and HERG currents. Biochem J. 2004;378:745–752. doi: 10.1042/bj20031324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Connor M, Smith R, et al. Discovery and characterization of a family of insecticidal neurotoxins with a rare vicinal disulfide bridge. Nat Struct Biol. 2000;7:505–513. doi: 10.1038/75921. [DOI] [PubMed] [Google Scholar]
- 46.Radis-Baptista G, de la Torre BG, Andreu D. A novel cell-penetrating peptide sequence derived by structural minimization of a snake toxin exhibits preferential nucleolar localization. J Med Chem. 2008;51:7041–7044. doi: 10.1021/jm8009475. [DOI] [PubMed] [Google Scholar]
- 47.Rádis-Baptista G, de la Torre BG, Andreu D. Insights into the uptake mechanism of NrTP, a cell-penetrating peptide preferentially targeting the nucleolus of tumour cells. Chem Biol Drug Des. 2012;79:907–915. doi: 10.1111/j.1747-0285.2012.01377.x. [DOI] [PubMed] [Google Scholar]
- 48.Jha D, Mishra R, Gottschalk S, et al. CyLoP-1: a novel cysteine-rich cell-penetrating peptide for cytosolic delivery of cargoes. Bioconjug Chem. 2011;22:319–328. doi: 10.1021/bc100045s. [DOI] [PubMed] [Google Scholar]
- 49.Bechara C, Sagan S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013;587:1693–1702. doi: 10.1016/j.febslet.2013.04.031. [DOI] [PubMed] [Google Scholar]
- 50.Tu AT, Morita M. Attachment of rattlesnake venom myotoxin a to sarcoplasmic reticulum: peroxidase conjugated method. Br J Exp Pathol. 1983;64:633–637. [PMC free article] [PubMed] [Google Scholar]
- 51.Furukawa K, Funayama K, Ohkura M, et al. Ca2+ release induced by myotoxin alpha, a radio-labellable probe having novel Ca2+ release properties in sarcoplasmic reticulum. Br J Pharmacol. 1994;113:233–239. doi: 10.1111/j.1476-5381.1994.tb16199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohkura M, Furukawa K, Oikawa K, Ohizumi Y. The properties of specific binding site of 125I-radioiodinated myotoxin a, a novel Ca++ releasing agent, in skeletal muscle sarcoplasmic reticulum. J Pharmacol Exp Ther. 1995;273:934–939. [PubMed] [Google Scholar]
- 53.Katagiri C, Ishikawa HH, Ohkura M, et al. Properties of specific binding site of myotoxin a, a powerful convulsant, in brain microsomes. Can J Physiol Pharmacol. 1998;76:395–400. doi: 10.1139/y98-035. [DOI] [PubMed] [Google Scholar]
- 54.Ohkura M, Furukawa K, Tu AT, Ohizumi Y. Calsequestrin is a major binding protein of myotoxin alpha and an endogenous Ca2+ releaser in sarcoplasmic reticulum. Eur J Pharmacol. 1994;268:R1–R2. doi: 10.1016/0922-4106(94)90126-0. [DOI] [PubMed] [Google Scholar]
- 55.Hirata Y, Nakahata N, Ohkura M, Ohizumi Y. Identification of 30 kDa protein for Ca(2+) releasing action of myotoxin a with a mechanism common to DIDS in skeletal muscle sarcoplasmic reticulum. Biochim Biophys Acta. 1999;1451:132–140. doi: 10.1016/S0167-4889(99)00082-8. [DOI] [PubMed] [Google Scholar]
- 56.Utaisincharoen P, Baker B, Tu AT. Binding of myotoxin a to sarcoplasmic reticulum Ca(2+)-ATPase: a structural study. Biochemistry. 1991;30:8211–8216. doi: 10.1021/bi00247a017. [DOI] [PubMed] [Google Scholar]
- 57.Baker B, Utaisincharoen P, Tu AT. Structure-function relationship of myotoxin a using peptide fragments. Arch Biochem Biophys. 1992;298:325–331. doi: 10.1016/0003-9861(92)90418-V. [DOI] [PubMed] [Google Scholar]
- 58.Boisseau S, Mabrouk K, Ram N, et al. Cell penetration properties of maurocalcine, a natural venom peptide active on the intracellular ryanodine receptor. Biochim Biophys Acta Biomembr. 2006;1758:308–319. doi: 10.1016/j.bbamem.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 59.Fajloun Z, Kharrat R, Chen L, et al. Chemical synthesis and characterization of maurocalcine, a scorpion toxin that activates Ca(2+) release channel/ryanodine receptors. FEBS Lett. 2000;469:179–185. doi: 10.1016/S0014-5793(00)01239-4. [DOI] [PubMed] [Google Scholar]
- 60.Lee J-Y, Choi Y-S, Suh J-S, et al. Cell-penetrating chitosan/doxorubicin/TAT conjugates for efficient cancer therapy. Int J Cancer. 2011;128:2470–2480. doi: 10.1002/ijc.25578. [DOI] [PubMed] [Google Scholar]
- 61.Mabrouk K, Ram N, Boisseau S, et al. Critical amino acid residues of maurocalcine involved in pharmacology, lipid interaction and cell penetration. Biochim Biophys Acta Biomembr. 2007;1768:2528–2540. doi: 10.1016/j.bbamem.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 62.Tisseyre C, Bahembera E, Dardevet L, et al. Cell penetration properties of a highly efficient mini maurocalcine Peptide. Pharmaceuticals (Basel) 2013;6:320–339. doi: 10.3390/ph6030320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ram N, Weiss N, Texier-Nogues I, et al. Design of a disulfide-less, pharmacologically inert, and chemically competent analog of maurocalcine for the efficient transport of impermeant compounds into cells. J Biol Chem. 2008;283:27048–27056. doi: 10.1074/jbc.M804727200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khamehchian S, Nikkhah M, Madani R, Hosseinkhani S. Enhanced and selective permeability of gold Nanoparticles functionalized with cell penetrating peptide derived from maurocalcine animal toxin. J Biomed Mater Res A. 2016 doi: 10.1002/jbm.a.35806. [DOI] [PubMed] [Google Scholar]
- 65.DeBin JA, Maggio JE, Strichartz GR. Purification and characterization of chlorotoxin, a chloride channel ligand from the venom of the scorpion. Am J Physiol. 1993;264:C361–C369. doi: 10.1152/ajpcell.1993.264.2.C361. [DOI] [PubMed] [Google Scholar]
- 66.McFerrin MB, Sontheimer H. A role for ion channels in glioma cell invasion. Neuron Glia Biol. 2006;2:39–49. doi: 10.1017/S1740925X06000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deshane J, Garner CC, Sontheimer H. Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. J Biol Chem. 2003;278:4135–4144. doi: 10.1074/jbc.M205662200. [DOI] [PubMed] [Google Scholar]
- 68.Kesavan K, Ratliff J, Johnson EW, et al. Annexin A2 is a molecular target for TM601, a peptide with tumor-targeting and anti-angiogenic effects. J Biol Chem. 2010;285:4366–4374. doi: 10.1074/jbc.M109.066092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tatenhorst L, Rescher U, Gerke V, Paulus W. Knockdown of annexin 2 decreases migration of human glioma cells in vitro. Neuropathol Appl Neurobiol. 2006;32:271–277. doi: 10.1111/j.1365-2990.2006.00720.x. [DOI] [PubMed] [Google Scholar]
- 70.Jacoby DB, Dyskin E, Yalcin M, et al. Potent pleiotropic anti-angiogenic effects of TM601, a synthetic chlorotoxin peptide. Anticancer Res. 2010;30:39–46. [PubMed] [Google Scholar]
- 71.Qin C, He B, Dai W, et al. Inhibition of metastatic tumor growth and metastasis via targeting metastatic breast cancer by chlorotoxin-modified liposomes. Mol Pharm. 2014;11:3233–3241. doi: 10.1021/mp400691z. [DOI] [PubMed] [Google Scholar]
- 72.Hockaday DC, Shen S, Fiveash J, et al. Imaging glioma extent with 131I-TM-601. J Nucl Med. 2005;46:580–586. [PubMed] [Google Scholar]
- 73.Dardevet L, Rani D, Aziz T, et al. Chlorotoxin: a helpful natural scorpion peptide to diagnose glioma and fight tumor invasion. Toxins (Basel) 2015;7:1079–1101. doi: 10.3390/toxins7041079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wiranowska M, Colina LO, Johnson JO. Clathrin-mediated entry and cellular localization of chlorotoxin in human glioma. Cancer Cell Int. 2011;11:27. doi: 10.1186/1475-2867-11-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Veiseh O, Gunn JW, Kievit FM, et al. Inhibition of tumor-cell invasion with chlorotoxin-bound superparamagnetic nanoparticles. Small. 2008;5:256–264. doi: 10.1002/smll.200800646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.DeBin JA, Strichartz GR. Chloride channel inhibition by the venom of the scorpion Leiurus quinquestriatus. Toxicon. 1991;29:1403–1408. doi: 10.1016/0041-0101(91)90128-E. [DOI] [PubMed] [Google Scholar]
- 77.Ojeda PG, Wang CK, Craik DJ. Chlorotoxin: structure, activity, and potential uses in cancer therapy. Biopolymers. 2016;106:25–36. doi: 10.1002/bip.22748. [DOI] [PubMed] [Google Scholar]
- 78.Stroud MR, Hansen SJ, Olson JM. In vivo bio-imaging using chlorotoxin-based conjugates. Curr Pharm Des. 2011;17:4362–4371. doi: 10.2174/138161211798999375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ullrich N, Bordey A, Gillespie GY, Sontheimer H. Expression of voltage-activated chloride currents in acute slices of human gliomas. Neuroscience. 1998;83:1161–1173. doi: 10.1016/S0306-4522(97)00456-9. [DOI] [PubMed] [Google Scholar]
- 80.Rouzaire-Dubois B, Milandri JB, Bostel S, Dubois JM. Control of cell proliferation by cell volume alterations in rat C6 glioma cells. Pflugers Arch. 2000;440:881–888. doi: 10.1007/s004240000371. [DOI] [PubMed] [Google Scholar]
- 81.Maertens C, Wei L, Tytgat J, et al. Chlorotoxin does not inhibit volume-regulated, calcium-activated and cyclic AMP-activated chloride channels. Br J Pharmacol. 2000;129:791–801. doi: 10.1038/sj.bjp.0703102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gurrola GB, Capes EM, Zamudio FZ, et al. Imperatoxin A, a cell-penetrating peptide from scorpion venom, as a probe of Ca-release channels/ryanodine receptors. Pharmaceuticals (Basel) 2010;3:1093–1107. doi: 10.3390/ph3041093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pimenta DC, Lebrun I. Cryptides: buried secrets in proteins. Peptides. 2007;28:2403–2410. doi: 10.1016/j.peptides.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 84.Wang L, Chan JYW, Rêgo JV, et al. Rhodamine B-conjugated encrypted vipericidin nonapeptide is a potent toxin to zebrafish and associated with in vitro cytotoxicity. Biochim Biophys Acta. 2015;1850:1253–1260. doi: 10.1016/j.bbagen.2015.02.013. [DOI] [PubMed] [Google Scholar]
- 85.Jouirou B, Mouhat S, Andreotti N, et al. Toxin determinants required for interaction with voltage-gated K+ channels. Toxicon. 2004;43:909–914. doi: 10.1016/j.toxicon.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 86.Estève E, Mabrouk K, Dupuis A, et al. Transduction of the scorpion toxin maurocalcine into cells. Evidence that the toxin crosses the plasma membrane. J Biol Chem. 2005;280:12833–12839. doi: 10.1074/jbc.M412521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schwartz EF, Capes EM, Diego-García E, et al. Characterization of hadrucalcin, a peptide from Hadrurus gertschi scorpion venom with pharmacological activity on ryanodine receptors. Br J Pharmacol. 2009;157:392–403. doi: 10.1111/j.1476-5381.2009.00147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Andreotti N, Sabatier J-M. The deciphered genome of Mesobuthus martensii uncovers the resistance mysteries of scorpion to its own venom and toxins at the ion channel level. Toxins (Basel) 2013;5:2209–2211. doi: 10.3390/toxins5112209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ruiz-Vela A, González de Buitrago G, Martínez-A C. Nuclear Apaf-1 and cytochrome c redistribution following stress-induced apoptosis. FEBS Lett. 2002;517:133–138. doi: 10.1016/S0014-5793(02)02607-8. [DOI] [PubMed] [Google Scholar]
- 90.Nur-E-Kamal A, Gross SR, Pan Z, et al. Nuclear translocation of cytochrome c during apoptosis. J Biol Chem. 2004;279:24911–24914. doi: 10.1074/jbc.C400051200. [DOI] [PubMed] [Google Scholar]
- 91.Jones S, Martel C, Belzacq-Casagrande A-S, et al. Mitoparan and target-selective chimeric analogues: membrane translocation and intracellular redistribution induces mitochondrial apoptosis. Biochim Biophys Acta Mol Cell Res. 2008;1783:849–863. doi: 10.1016/j.bbamcr.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 92.Shapira A, Benhar I. Toxin-based therapeutic approaches. Toxins (Basel) 2010;2:2519–2583. doi: 10.3390/toxins2112519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ponce-Soto LA, Martins-de-Souza D, Marangoni S. Structural and pharmacological characterization of the crotamine isoforms III-4 (MYX4_CROCu) and III-7 (MYX7_CROCu) isolated from the Crotalus durissus cumanensis venom. Toxicon. 2010;55:1443–1452. doi: 10.1016/j.toxicon.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 94.Baker B, Tu AT, Middlebrook JL. Binding of myotoxin a to cultured muscle cells. Toxicon. 1993;31:271–284. doi: 10.1016/0041-0101(93)90145-9. [DOI] [PubMed] [Google Scholar]
- 95.Ram N, Weiss N, Texier-Nogues I, et al. Design of a disulfide-less, pharmacologically inert, and chemically competent analog of maurocalcine for the efficient transport of impermeant compounds into cells. J Biol Chem. 2008;283:27048–27056. doi: 10.1074/jbc.M804727200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ali SA, Alam M, Abbasi A, et al. Structure–activity relationship of chlorotoxin-like peptides. Toxins (Basel) 2016;8:36. doi: 10.3390/toxins8020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee CW, Lee EH, Takeuchi K, et al. Molecular basis of the high-affinity activation of type 1 ryanodine receptors by imperatoxin A. Biochem J. 2004;377:385–394. doi: 10.1042/bj20031192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rajagopal BS, Edzuma AN, Hough MA, et al. The hydrogen-peroxide-induced radical behaviour in human cytochrome c-phospholipid complexes: implications for the enhanced pro-apoptotic activity of the G41S mutant. Biochem J. 2013;456:441–452. doi: 10.1042/BJ20130758. [DOI] [PubMed] [Google Scholar]
- 99.Hessa T, Kim H, Bihlmaier K, et al. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature. 2005;433:377–381. doi: 10.1038/nature03216. [DOI] [PubMed] [Google Scholar]
- 100.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 101.Rodrigues M, de la Torre BG, Radis-Baptista G, et al. Efficient cellular delivery of beta-galactosidase mediated by NrTPs, a new family of cell-penetrating peptides. Bioconjug Chem. 2011;22:2339–2344. doi: 10.1021/bc200421z. [DOI] [PubMed] [Google Scholar]
- 102.Fadel V, Bettendorff P, Herrmann T, et al. Automated NMR structure determination and disulfide bond identification of the myotoxin crotamine from Crotalus durissus terrificus . Toxicon. 2005;46:759–767. doi: 10.1016/j.toxicon.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 103.Felder CE, Prilusky J, Silman I, Sussman JL. A server and database for dipole moments of proteins. Nucleic Acids Res. 2007;35:W512–W521. doi: 10.1093/nar/gkm307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sabatier J-M, De Waard M. Handbook of biologically active peptides. Handb Biol Act Pept. 2013 [Google Scholar]
- 105.Mosbah A, Kharrat R, Fajloun Z, et al. A new fold in the scorpion toxin family, associated with an activity on a ryanodine-sensitive calcium channel. Proteins. 2000;40:436–442. doi: 10.1002/1097-0134(20000815)40:3<436::AID-PROT90>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 106.Lippens G, Najib J, Wodak SJ, Tartar A. NMR sequential assignments and solution structure of chlorotoxin, a small scorpion toxin that blocks chloride channels. Biochemistry. 1995;34:13–21. doi: 10.1021/bi00001a003. [DOI] [PubMed] [Google Scholar]
- 107.Lukács B, Sztretye M, Almássy J, et al. Charged surface area of maurocalcine determines its interaction with the skeletal ryanodine receptor. Biophys J. 2008;95:3497–3509. doi: 10.1529/biophysj.107.120840. [DOI] [PMC free article] [PubMed] [Google Scholar]