

Abstract
Activated protein C (APC) is a natural anticoagulant with strong anti-inflammatory, anti-apoptotic, and barrier stabilizing properties. These cytoprotective properties of APC are thought to be exerted through its pathway involving the binding of APC to endothelial protein C receptor and cleavage of protease-activated receptors. In this study, we found that APC enhanced endothelial barrier integrity via a novel pathway, by binding directly to and activating Tie2, a transmembrane endothelial tyrosine kinase receptor. Binding assays demonstrated that APC competed with the only known ligands of Tie2, the angiopoietins (Angs). APC bound directly to Tie2 (Kd ~3 nM), with markedly stronger binding affinity than Ang2. After binding, APC rapidly activated Tie2 to enhance endothelial barrier function as shown by Evan’s blue dye transfer across confluent cell monolayers and in vivo studies. Blocking Tie2 restricted endothelial barrier integrity. This study highlights a novel mechanism by which APC binds directly to Tie2 to enhance endothelial barrier integrity, which helps to explain APC’s protective effects in vascular leakage-related pathologies.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-016-2440-6) contains supplementary material, which is available to authorized users.
Keywords: Endothelial barrier integrity, Protein C, Sepsis, RCR252, Protease-activated receptor, SDS-PAGE, Biacore, Immunoprecipitation, Solid-phase binding assay
Introduction
Activated protein C (APC) is a vitamin K-dependent serine protease that plays a key role in the regulation of blood coagulation but also exerts a broad range of cytoprotective actions on endothelium including suppression of inflammation and stabilization of endothelial barrier function [1–3]. APC-mediated cytoprotective signaling requires endothelial protein C receptor (EPCR) and cleavage of protease-activated receptor (PAR)1 at Arg46 [4], which may occur in caveolin-1-enriched lipid rafts [5]. In addition, other receptors, such as sphingosine-1-phosphate receptor 1 (S1P1), epidermal growth factor receptor (EGFR), PAR3, and Tie2, may independently or co-operatively contribute to APC-mediated protective effects on endothelium [2, 6–8]. For example, APC protects against endothelial barrier disruption by cross-activating S1P1 [2] or by stimulating Ang1 whilst inhibiting Ang2 to activate Tie2 [6]—both these mechanisms require signaling through EPCR and PAR1.
Tie2 is a transmembrane endothelial tyrosine kinase receptor that not only regulates vessel maturation and remodeling angiogenesis, but also controls endothelial inflammation and permeability [9, 10]. Tie2-deficient mice are not viable and have severe vascular abnormalities [11, 12]. The angiopoietin (Ang)s are the only known ligands for Tie2 with Ang1 and Ang2 being the most abundant and best studied. Ang1 acts as an agonist and Ang2 as an antagonist by blocking the stabilizing action of Ang1. Ang1 ligation to Tie2 results in tyrosine phosphorylation, which subsequently promotes vascular endothelial cell survival and migration, maintains vessel integrity, inhibits vascular leakage, suppresses inflammatory gene expression, and prevents recruitment and transmigration of leukocytes [13–15]. Ang1/Tie2 signaling benefits pathologies of vascular activation, such as sepsis, stroke, diabetic retinopathy, rheumatoid arthritis, and asthma [16]. Ang2 competes with the binding of Ang1–Tie2 and sensitizes the endothelium to inflammatory cytokines, resulting in injury and vascular leakage which directly contributes to the adverse outcomes in sepsis [17, 18]. The balance between Ang1 and Ang2 regulates baseline endothelial barrier function and its response to injury and is, therefore, considered a gatekeeper of endothelial activation [19].
In this study, we revealed that APC can act as novel ligand for Tie2 without requiring EPCR or PAR-1, to initiate downstream signaling pathways and maintain vascular integrity.
Experimental procedures
In vivo permeability assay
C57BL/6 male mice were obtained from the Kearns facility at Kolling Institute, The University of Sydney. Six-to-eight-week-old C57BL/6 male mice were used for this study. To block Tie2, Tie2 kinase inhibitor (Tie2-I, 25 mg/kg) (Calbiochem) was injected intraperitoneally (i.p). After 30 min, mice were treated with APC (2 mg/kg) [20] (Eli Lilly) by tail intravenous (i.v) injection. Hyper-permeability was induced by intraperitoneal injection of 1 ml lipopolysaccharide (LPS, derived 1 from Escherichia coli serotype 055:B5, 5 or 10 mg/kg), 5 min after APC administration [21–24]. Evan’s blue (EB) dye (Sigma-Aldrich) was dissolved at a concentration of 30 mg/ml in phosphate-buffered saline (PBS). To this solution, bovine serum albumin (BSA) was added to a final concentration of 40 mg/ml (Sigma-Aldrich), mixed well with a magnetic stirrer, and then sterile filtered through a 0.22 µm syringe filter to prepare evans blue albumin (EBA). One hundred µl EBA was injected i.v. into mice and allowed to circulate for 30 min. The dye was collected, measured, and normalized as previously described [25, 26]. Briefly, while under anesthesia, the mice were perfused with PBS through the portal vein. Immediately after perfusion, mice were terminated and organs (lungs and kidneys) were removed. The collected organs were dried before homogenising in formamide at 60 °C for 16 h to extract the dye and centrifuged at 12,000×g with 3 ml of PBS. Absorbance of supernatant was measured at 620 and 740 nm using a spectrophotometer. The organs of mice that were not injected with EBA were also harvested and similarly treated to calculate the tissue-specific turbidity correction factors.
Use of animals was approved by the Royal North Shore Hospital Animal Care and Ethics Committee in accordance with the National Health and Medical Research Council of Australia.
Cell isolation and culture
Human umbilical vein endothelial cells (HUVEC) were isolated and cultured as previously described [6]. In some experiments, cells were blocked for 1 h with 5 µg/ml Tie2 blocking antibody (aTie2) or 2 µg/ml soluble (s)Tie2 (R&D Systems) that blocked the activation of Tie2, RCR252 to block EPCR receptor, or PAR1 inhibitor (PAR-I, SCH79797, Abcam) blocking PAR1 receptor. Cells were then treated with tumor necrosis factor (TNF)-α (100 ng/ml), LPS (10 µg/ml), vascular endothelial growth factor 165 (VEGF) (200 ng/ml) (R&D Systems), and/or APC. Passage 1–4 cells were used in experiments.
Permeability assay
Endothelial permeability was assessed using transwell Evan’s blue albumin (EBA) as previously described [27] with modifications [6]. HUVEC were cultured on gelatin-coated membrane supports (0.45 μm pore, 12 mm diameter) until confluent. Medium was changed to Biorich plus 2% fetal calf serum (FCS) before treatment. Cells were treated with or without with a Tie2 or non-blocking isotype for 1 h and then treated with or without TNF-α (100 ng/ml), LPS (10 µg/ml), VEGF (200 ng/ml), or Dimethyl sulfoxide (DMSO) for 30 min before the addition or not (no APC control) of APC (1 μg/ml) for 24 h. After treatment, medium in upper luminal chamber was replaced with 0.5 ml of 2% Evans blue in Biorich medium with 4% BSA and lower abluminal chamber with 1.5 ml of Biorich medium. The volume of media added to both chambers allowed for the levels to be equal, thus avoiding formation of hydrostatic pressure which could result in movement of medium in one direction. Medium (75 μl) was then collected from the lower chamber at selected timepoints (up to 80 min) and dye concentration analyzed spectrophotometrically at 630 nm.
Immunoprecipitation and western blot
Immunoprecipitation was performed on HUVEC lysates using Protein A/G PLUS-Agarose (Santa Cruz), as described previously [28] with minor changes. Proteins were analyzed by immunoblotting [29] with rabbit anti-human Tie2 (aTie2, 1:1000, Santa Cruz), rabbit anti-human Phosphorylated (P)-Tie2 (1:500, R&D Systems), and mouse anti-human PC antibody (1:1000, Abcam).
Enzyme-linked immunosorbent assay (ELISA)
Commercial Duo Set ELISA kits for total (T)-Tie2 and P-Tie2 (R&D Systems) were used according to manufacturer’s instructions.
Biacore binding assay
The binding interaction between APC (1 µg/ml) and sTie2-Fc (25 µg/ml) was observed by surface plasmon resonance using a Biacore 3000 sensor (GE Healthcare). The surface of carboxymethylated dextran matrix of a CM5 sensor chip (Biacore AB) was prepared using Amine Coupling Kit (Biacore AB). sTie2-Fc (25 µg/ml) was immobilized on prepared sensor chip. APC was then injected at a flow rate of 10 µl/min over the sTie2-Fc for 6 min, and binding signals were recorded in response unit (RU) [30, 31].
SDS-PAGE-based binding assay
The binding of APC to the soluble extracellular domain of Tie2-Fc (R&D Systems) was analyzed using SDS-PAGE-based binding assay. APC (20 ng) was mixed with sTie2-Fc (100 ng) and incubated in 500 µl NaCl–Tris buffer (50 mM Tris, 100 mM NaCl, pH 7.4) containing 0.02% TritonX-100 at 4 °C. After 2 h, 20 µl of protein A/G Plus-Agarose was added and incubated for 1 h at 4 °C. The protein-A conjugated samples were washed with NaCl-Tris buffer containing 0.02% TritonX-100. Samples were eluted with non-reducing sample buffer, separated by 6% PAGE with 0.5% SDS, and analyzed with mouse anti-human protein C antibody.
Solid-phase binding assay
sTie2-Fc (2 µg/ml in PBS) was coated onto a 96-well plate overnight at 4 °C followed by six washings with PBST (PBS + 0.05% Tween 20). The coated wells were blocked with blocking buffer (Thermo Scientific) for 4 h at room temperature, and then washed with PBST. Ang1, Ang2 (R&D Systems), or APC in 100 µl of PBS was added to coated wells and incubated overnight at 4 °C [32, 33]. Ang1, Ang2, or APC bound to sTie2 was detected by incubating with their respective primary antibodies. To determine significance of calcium ion (Ca2+) in APC–Tie2 binding, we performed binding assay experiments with HEPES buffer (20 mM, pH 7.5, 140 mM NaCl) with 1 mM Ca2+, 1 mM Mg2+, and 1 mM Mn+2. All washing steps included 1 mM Mg2+ and 1 mM Mn2+ in HEPES buffer. For time-dependent binding interaction analysis, APC was incubated with sTie2 for 2 min, 1 h, and O/N in the presence and absence of Ca2+. The absorbance was measured at 450 nm, and the absorbance at 570 nm was used as correction factor. For the Ca2+ experiments, the substrate enzyme was incubated for 10 min rather than the 5 min used for other experiments. For the competition binding assays, the binding of APC to sTie2 immobilized on the 96-well plate was performed in the presence or absence of Ang1 or Ang2 [34, 35].
Statistics
Statistical analysis was performed with two-tailed Student’s t test between two groups. ANOVA was used to compare means among three or more independent groups followed by Newman–Keuls post hoc test. All results are presented as mean ± SEM. A P value of less than 0.05 was considered statistically significant.
Results
APC rapidly activates the Tie2 receptor
When HUVEC were treated with APC (0.01–10 µg/ml) for 1 h, P-Tie2 was dose-dependently increased, peaking at 1 µg/ml APC (Fig. 1a). APC also increased activation of Tie2 in a time-dependent manner, from ~twofold at 15 min to ~4.5-fold at 60 min (Fig. 1b). To determine if this early activation of Tie2 by APC requires EPCR or PAR1, cells were treated with their respective blocking antibodies, RCR252 or PAR1-I for 1 h prior to APC treatment and Tie2 activation was assessed at 2 and 20 min. Confirmation that RCR252 works under these conditions is shown in Supplementary Fig. 1. ELISA results showed that blocking EPCR or PAR1 receptors had no significant effect on the activation of Tie2 by APC either at 2 or 20 min (Fig. 1c), indicating that neither of these receptors were involved in activation of Tie2. We then compared APC-induced Tie2 phosphorylation to that induced by the native ligands of Tie2, Ang1, or Ang2. When used at 1 µg/ml, Ang1 was the most potent activator with APC and Ang2 showing comparable levels of Tie2 activation (Fig. 1d).
Fig. 1.
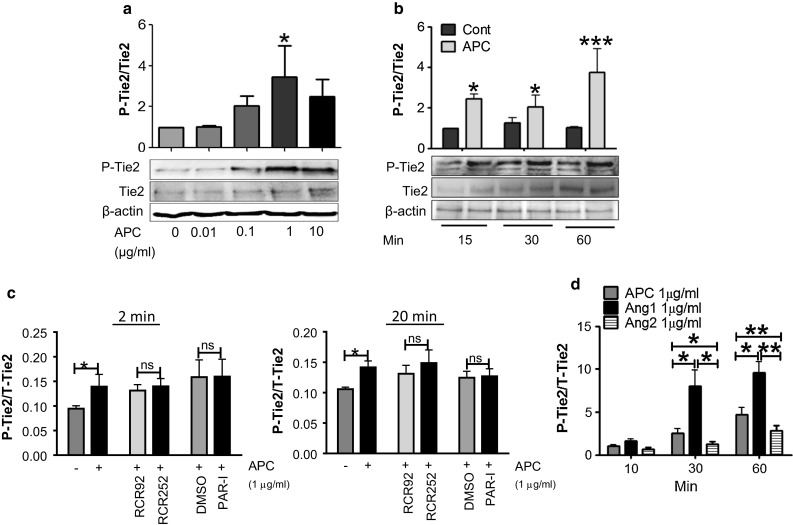
APC rapidly activates Tie2 in HUVEC. HUVEC were incubated with Biorich medium +2% FCS for 24 h before treatment. a Western blot for phosphorylated (P)-Tie2 and total(T)-Tie2 treated for 1 h with APC (1 μg/ml). Graph shows mean ± SD from n = 3 independent experiments. b HUVEC were treated with APC (1 μg/ml) for 15, 30, or 60 min, and cell lysates were assessed by western blotting. Graph shows mean ± SD from n = 4 independent experiments. c HUVEC were pre-incubated in Biorich medium +2% FCS for 1 h with blocking antibodies RCR252 (10 µg/ml) and PAR1-I (SCH79797) (1uM) and their respective controls (non-blocking antibody RCR92 and DMSO) then treated with (+) or without (−) APC for 2 (left panel) or 20 (right panel) min. Cell lysates were analyzed for P-Tie2 and T-Tie2 by ELISA. Graphs show mean ± SD from three separate experiments. d HUVEC were treated with APC, Ang1, or Ang2 for different times, and cell lysates were assessed for P/T-Tie2 by ELISA. Graph shows mean ± SD from n = 6 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001. ns not significant
Does APC bind to Tie2 receptor?
The above results raised the possibility of direct interaction between APC and Tie2. To investigate, we used several strategies. First, Biacore assay was used to detect the specificity of binding between APC and Tie2 in real time. APC showed specific binding to sTie2 compared to the control (blank chip surface). APC associated rapidly with sTie2 captured on sensor chip and dissociated slowly (Fig. 2a), confirming the specificity of binding between APC and Tie2. Second, co-immunoprecipitation was employed to examine whether APC can bind or associate with Tie2. Recombinant APC (~56 kDa) was co-incubated with sTie2 (~130 kDa) for 1 h and subjected to immunoblotting with mouse anti-human protein C antibody. Results showed a band at ~170 kDa (Fig. 2b), indicating that the molecules were bound together. To investigate whether APC binds to Tie2 in endothelial cells, APC was incubated with HUVEC and whole lysates were subjected to co-immunoprecipitation with rabbit anti-human Tie2 antibody, and then subjected to western blot detection with mouse anti-human protein C antibody. Again, a strong band at ~170 kDa was distinct, providing evidence that APC is associated with Tie2 in cells (Fig. 2c). Third, a solid-phase binding assay was performed to measure APC binding to Tie2 in the presence or absence of aTie2. On blocking the Tie2 receptor, binding between APC and Tie2 was inhibited significantly irrespective of APC concentration (0.1, 0.2, and 1 µg/ml) (Fig. 2d).
Fig. 2.
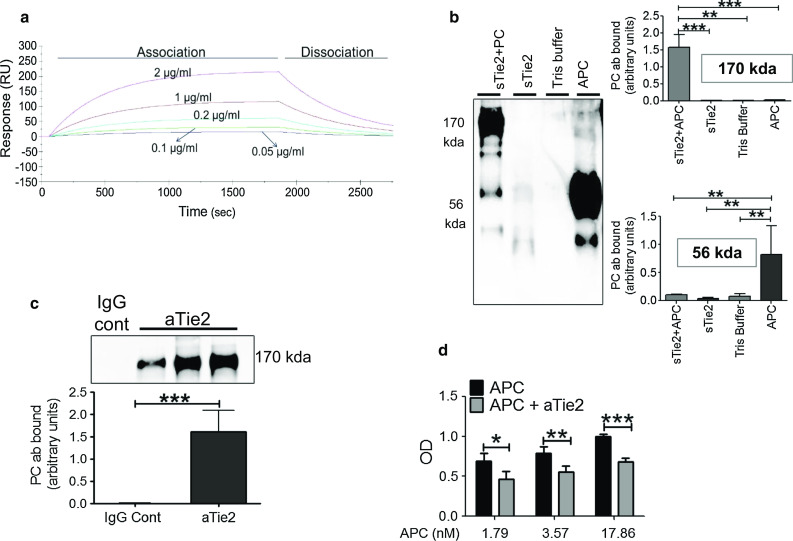
APC–Tie2 interaction. a Various doses of APC were passed over a Biacore sensor chip covalently coupled with Tie2-Fc (25 µg/ml). Binding is shown as Resonance Units (RU). b sTie2 (100 ng) and APC (20 ng) were co-incubated in Tris buffer and analyzed by immunoblotting (APC + sTie2) for protein C with mouse anti-human protein C antibody. sTie2 (100 ng) alone (in Tris buffer), Tris buffer alone, and APC (20 ng) alone (in Tris buffer) were treated similarly and used as controls. Graphs show PC antibody (ab) binding (y-axis) to APC + sTie2, sTie2, control Tris buffer, and APC for two molecular weights, 170 and 56 kDa. Y-axis is in arbitrary units (mean ± SD), for three separate experiments. c APC was incubated with HUVEC for 1 h, and then, whole lysates were subjected to co-immunoprecipitation with rabbit anti-human Tie2 antibody (anti-tie2) or IgG control, followed by western blot detection with mouse anti-human protein C antibody (PC). Graph shows arbitrary units (mean ± SD), for three separate experiments. d Tie2 receptor was blocked with a blocking antibody (aTie2, 8 µg/ml) for 1.5 h before incubating with different concentrations of APC. y-axis shows binding affinity as OD (450–570) nm. *P < 0.05, **P < 0.01, ***P < 0.001, n = 3
Binding affinity between APC and Tie2
A solid-phase saturation binding assay was performed between APC and Tie2 to determine half maximal binding concentration, referred to as BC50, and compared to Ang1 and Ang2 under similar binding conditions. The BC50 s for APC, Ang1, and Ang2 binding were ~3.14, 2.16, and 36.52 nM, respectively (Fig. 3a–c). Thus, compared to Ang1, ~1.5-fold higher concentration of APC was necessary to induce half maximal response in Tie2; however, compared to Ang2, ~11.6-fold lower level of APC was required.
Fig. 3.

Binding affinity between APC and Tie2. The graphs show BC50 for binding. a APC (n = 5); b Ang1 (n = 3); c Ang2 (n = 3), binding to Tie2, using different concentrations of ligands under similar conditions, as described in “Experimental procedures”. OD was measured by spectrophotometry at 450 and 570 nm. Graphs show mean ± SE. The graphs show saturation binding curve and scatchard analysis (inset) for: d APC and Tie2 (n = 5); e Ang1 and Tie2 (n = 3); and f Ang2 and Tie2 (n = 3)
Next, we determined binding affinity between Ang1, Ang2 or APC, and Tie2 by scatchard and saturation analysis, which produces values for maximum binding capacity (Bmax) and binding affinity (Kd) between interacting proteins, where lower Kd values signify stronger interaction. Figure 3d shows a specific binding analysis between APC and Tie2, where Bmax was 0.78 and Kd was 2.78 nM (Fig. 3d). Ang1–Tie2 interaction showed Bmax of 0.67 and Kd of 2.22 nM (Fig. 3e) and for Ang2–Tie2 Bmax was 1.03 and Kd was 33.26 nM (Fig. 3f). These results confirm the strong binding affinity between APC and Tie2, which is ~12 times higher than Ang2–Tie2. These results also confirm the previous findings that Ang1 binding to Tie2 is stronger than Ang2 [34].
To determine if additional Ca2+ is required for the interaction between APC and Tie2, binding assay was performed in the presence or absence of Ca2+. BC50 was approximately equal, i.e., ~3.54 and ~3.53 nM in the presence and absence of Ca2+ (Fig. 4a). The Kd value between APC and Tie2 in the presence of Ca2+ was 2.47 nM (Fig. 4b) and in its absence 2.78 nM (Fig. 3a). The protein–protein interaction remained unaffected in the presence of Ca2+ up to the 1 h timepoint (Fig. 4c). These results demonstrate that APC–Tie2 binding does not require additional Ca2+.
Fig. 4.

Binding affinity between APC and Tie2 in the presence of calcium ion (Ca2+). a Graph shows BC50 for APC binding to Tie2, using different concentrations of ligands in the absence (dashed line) or presence (full line) of Ca2+, as described in “Experimental procedures”. OD was measured by spectrophotometry at 450 and 570 nm and converted to % BC50. Graph represents mean ± SE from three separate experiments. b Graphs shows saturation binding curve and scatchard analysis. n = 3. c Graph represents binding between APC and soluble Tie2 in a time-dependent manner in the presence or absence of Ca2+. sTie2 and APC were incubated for different time periods [2 min (min), 1 h, and overnight (O/N)] and analyzed by an ELISA-based binding assay, as described in “Experimental procedures”. Graph shows OD (mean ± SD), for three separate experiments, with the no APC controls subtracted from all values. ns non-significant
Competitive binding between Tie2 ligands
To determine how the presence of either Ang1 or Ang2 affects the binding interaction between APC and Tie2, we performed competitive binding experiments. Approximate BC50 concentrations of each protein were used as determined previously in Fig. 3a–c, i.e., 1.75 nM Ang1, 35.09 nM Ang2, and 3.57 nM APC. The binding between APC and Tie2 was inhibited by ~66% in the presence of Ang1 and ~29% in the presence of Ang2 (Fig. 5a, b). When the concentration of APC was increased fivefold to 17.86 nM, inhibition of APC–Tie2 binding by Ang1 and Ang2 was reduced to ~58 and ~31%, respectively (Fig. 5c, d).
Fig. 5.

Competitive binding. a, b Competitive binding to Tie2 was assessed, as described in “Experimental procedures”, using BC50 concentrations of APC (3.57 nM), Ang1 (1.75 nM), and/or Ang2 (35.09 nM). c, d Similar to a and b, except APC was used in excess at a dose of 17.86 nM. a–d Graphs show binding affinity as OD (mean ± SD) from three separate experiments. The no APC control was subtracted from each data point. e Binding assays were conducted with increasing concentrations of APC and BC50 concentrations for Ang1 (full line) and Ang 2 (dashed line). Results using the stated concentration of competitor are expressed as a percentage inhibition of binding to Tie2 in the absence of competitor. Each curve represents results from three separate experiments. *P < 0.05, **P < 0.01, ***P < 0.001, n = 3. IC50 data on right was obtained from competitive binding assays and derived from three independent experiments, conducted in duplicate
Competitive binding was performed using different concentrations of APC (0.179–35.71 nM) and either 1.75 nM Ang1 or 35.09 nM Ang2. The binding of either Ang1 or Ang2–Tie2 was inhibited when co-incubated with APC (Fig. 5e). Half maximal inhibitory concentration (IC50) of APC was calculated by one site-fit logIC50 analysis (GraphPad Prism) to be 3.9 nM for inhibition of Ang1, or 4.0 nM for Ang2, binding to Tie2 (Fig. 5e). At a ten-fold higher concentration, APC inhibited Ang2 by ~90% and Ang1 by ~75%. Tie2 binding to Ang2 was inhibited comparatively more than Ang1, at any given concentration of APC.
APC rapidly suppresses endothelial permeability via Tie2
To determine whether APC induces permeability changes in endothelium directly via binding Tie2, hyper-permeability was induced in HUVEC with TNF-α (100 ng/ml) (Fig. 6a), LPS (10 µg/ml) (Fig. 6b), or vascular endothelial growth factor (VEGF) (200 ng/ml) (Fig. 6c) in the presence or absence of APC, with or without Tie2 blocking antibody and permeability was measured after 1 h using EBA dye transfer. As expected, APC significantly reduced permeability compared to TNF-α (1.6-fold), LPS (1.8-fold), or VEGF (2.3-fold) treatment alone. Pre-treatment of aTie2 resulted in a significant reversal of APC-induced endothelial integrity (Fig. 6a–c).
Fig. 6.

APC signals via Tie2 to reduce endothelial permeability. a–c HUVEC were incubated in the presence or absence (control) of 1 µg/ml APC for 1 h. Cells were pre-incubated for 1 h with aTie2 (5 µg/ml) and/or 30 min pre-treatment with a TNF-α (100 ng/ml), b LPS (10 µg/ml), c VEGF (200 ng/ml), or control non-blocking isotypes (IgG) or DMSO before APC treatment for 1 h. Permeability was measured using a modified Boyden chamber and results displayed as absorbance at 630 nm. Graphs show mean ± SD from three replicates for a and eight replicates for b and c. *P < 0.05, **P < 0.01 ***P < 0.001
APC signals through Tie2 to reverse vascular leakage in vivo
To determine whether Tie2 is required for APC’s inhibition of vascular leakage, mice were treated with Tie2 inhibitor(I) for 30 min prior to APC treatment in the presence or absence of LPS [36] and assessed at two timepoints. After 24 h, LPS (5 mg/kg) significantly increased Evan’s blue albumin (EBA) dye leakage by ~5.6-fold in the kidneys and ~twofold in the lungs, compared to control (PBS-treated). Pre-treatment with APC almost completely prevented this effect (Fig. 7a, b). Tie2-I alone did not affect EBA leakage into the kidneys or lungs; however, the protective effect of APC was significantly reversed by Tie2-I in all experiments (Fig. 7a, b), indicating APC signals through Tie2. To rule out possible effects of APC on promoting Ang1, which requires at least 16 h [6], we performed experiments over the shorter duration of 4 h. Inhibiting Tie2 with or without LPS (10 mg/kg) treatment resulted in increased vascular permeability and also prevented APC from exerting its protective effect (Fig. 7c, d), confirming the direct effects of Tie2 on APC’s actions.
Fig. 7.

APC prevents LPS-induced renal and pulmonary vascular injury in mice. Mice were injected with APC (2 mg/kg) before administering LPS (5 mg/kg for 24 h or 10 mg/kg for 4 h) or the same volume of PBS. The graphs show results for EBA amount extracted from kidneys and lungs of mice, treated with APC or PBS for: a, b 24 h and c, d 4 h. Mice were injected with Tie2 inhibitor (Tie2-I) (25 mg/kg) before treatment with APC (2 mg/kg) or equal volume of PBS for 24 h, plus or minus LPS (5 mg/kg for 24 h or 10 mg/kg for 4 h). Each group consisted of six mice. Statistical analysis using ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001
Discussion
This report shows that APC is a direct agonist for Tie2; it binds to and initiates rapid activation of the receptor which in turn manifests in barrier protection of endothelial cells. These actions of APC mimic those of Ang1, albeit less efficiently, and are supported by the functional similarities between Ang1 and APC. Mice that are completely deficient in protein C exhibit neonatal lethality in a similar fashion to Ang1 or Tie2 knockout mice [11, 37, 38]. Mice genetically predisposed to severe protein C deficiency exhibit exacerbated acute inflammation and reconstitution of low-protein C mice with recombinant APC reduces inflammation and extends survival after LPS challenge [38]. Recombinant Ang1 or genetically overexpressing Ang1 also protects against inflammation and/or vascular leakage and improves survival in endotoxin-induced shock or hyperoxia-induced lung injury [12, 39–41]. The anti-inflammatory properties of both APC and Ang1 signaling in endothelium are mediated, at least partly, via inhibition of NF-κB-mediated expression of leucocyte adhesion molecules, such as intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 [19, 42].
APC is well known for its cytoprotective actions on endothelial cells, most of which are mediated through EPCR-dependent cleavage and activation of PARs, particularly PAR1. For example, after binding EPCR and cleaving PAR-1, APC stimulates endothelial sphingosine kinase-1 which enhances S1P production, subsequently selectively activating Rac1 to exert the barrier-protective function by stabilizing the endothelial cytoskeleton [2, 3, 43]. Results from the current study suggest that the rapid activation of Tie2 by APC does not require EPCR or PAR1, rather that APC acts as a direct ligand for Tie2. Such a mechanism may provide an alternative protective means for APC’s actions when EPCR or PAR receptors are not available. For example, group V secretory phospholipase A2 (sPLA2V) inhibits EPCR anticoagulant and anti-apoptotic properties by accommodating lysophosphatidylcholine or platelet activating factor (PAF) in the hydrophobic groove and prevents APC binding EPCR on the endothelial surface [44]. The same phenomenon occurs in rheumatoid synovial fibroblasts [45] due to the inflammatory effects of sPLA2V [46, 47]. In such situations, APC would be free to bind Tie2 and exert cytoprotective signaling. However, our results should be interpreted with caution as the blocking antibody and antagonist only show that the effects of APC are independent of the actual binding sites on EPCR and PAR1, respectively, that the inhibitor blocks, and do not eliminate the possibility that these receptors are functionally involved in APC’s actions on Tie2, possibly via lateral associations.
In other cell types, APC has been found to directly bind and function through receptors other than EPCR and PAR1 to exert cytoprotective effects. In human monocytic-like U937 cells, binding to ApoER2 by APC signals via Dab1 phosphorylation and subsequent activation of PI3 K and Akt and inactivation of GSK3beta contributes to APC’s beneficial effects [48]. Cao et al. [49] have shown that the anti-inflammatory activity of APC on macrophages is dependent on it binding integrin CD11b/CD18, but not EPCR, within lipid rafts and facilitates APC cleavage and activation of PAR1, leading to enhanced production of S1P and suppression of the inflammation. APC directly binds to β1 and β3 integrins and inhibits neutrophil migration via its Arg-Gly-Asp (RGD) sequence, both in vitro and in vivo [50].
In vivo, the protective effect of APC in a mouse model of vascular leakage was reversed by Tie2-kinase inhibitor at 4 h. Considering that APC cannot induce Ang1 or suppress Ang2 synthesis over this short duration [6], these results suggest direct participation of Tie2 in APC’s actions. Taken together, our in vitro and animal studies show that APC signals directly and rapidly through Tie2 to induce vascular barrier integrity. However, there are at least three other mechanisms via which APC can activate Tie2. First, APC stimulates Ang1 and inhibits Ang2 to activate Tie2 and reduce endothelial permeability, with the first sign of protection occurring after 16 h [6]. Second, APC can enhance barrier integrity in human skin keratinocytes by a complex mechanism which requires binding to EPCR, cleaving, and activating PAR1 which causes intracellular transactivation of epidermal growth factor receptor (EGFR) followed by transactivation of Tie2 tyrosine kinase [51]. Whether this mechanism is functional in endothelium is unknown. Third, Stavenuiter and Mosnier [52] have recently shown that noncanonical cleavage of PAR3 by APC promotes a novel pathway for Tie2 activation and stabilization of vascular integrity. In addition, protein C inhibits rapid mobilization of Ang2 from Weibel–Palade bodies in endothelial cells which would effectively promote the actions of Ang1 and APC on Tie2 [53]. Considering Tie2’s vital role in barrier function, this apparent mechanistic redundancy of Tie2 activation by APC may serve to ensure continuous protection of endothelium from barrier disruption.
Solid-phase binding assays as well as scatchard and saturation analysis indicated strong binding affinity of APC–Tie2, which was less than that of Ang1 and Tie2, but remarkably ~12 times higher than that between Ang2 and Tie2. The higher Bmax value of APC–Tie2 binding analysis shows that larger numbers of binding sites are involved between APC and Tie2 compared to Ang1–Tie2. Although, Bmax for APC–Tie2 is less than Ang2–Tie2, the lower Kd for the former signifies that interaction between amino acids at these binding sites is more favorable. Competitive inhibition studies indicated that APC competes with both Ang1 and Ang2 for binding to Tie2, suggesting that APC and Angs share some similar binding sites to Tie2. It has been shown previously that APC and Angs require Ca2+ for structural stability and function [54, 55]; however, additional Ca2+ was not required for either Angs or APC to bind to Tie2 receptor. The exact nature of molecular interaction between APC and Tie2 awaits further structural analysis and in vitro molecular interaction analysis.
Ang2 can activate Tie2 in HUVEC and promote endothelial cell survival through the ERK1/2 and PI3K pathways [34, 56]. In our hands, Ang2 also partially induced Tie2 activation in HUVECs (Fig. 1d). However, Ang2 is generally considered as a non-signal-transducing ligand that inhibits Tie2 activation [57] and provokes inflammation and vascular hyper-permeability [58, 59]. Ang2 is elevated in sepsis, a very serious condition characterized physiologically by an aberrant systemic inflammatory response and microvascular dysfunction. Plasma levels of Ang2 correlate with the severity of illness and directly contribute to morbidity and mortality [59, 60]. Ang2 administration in healthy adult mice triggers sepsis-like changes, such as vascular leakage and lung injury [58, 59]. In sepsis models, Ang2 (+/−) mice exhibit reduced tissue inflammation, lower levels of both renal failure and lung injury, and show improved survival over wild-type litters [17]. Using mice engineered to inducibly overexpress Ang2, Ziegler et al. [61] showed that Ang2-mediated microvascular disintegration not only contributes to septic shock but that inhibition of the Ang2/Tie2 interaction during sepsis is a potential therapeutic target. Results here showing the competitive inhibition between APC and Ang2 together with the favorable binding affinity of APC to Tie2 suggest that APC could protect against Ang2-mediated disease by restoring Tie2 activation.
When endogenous protein C levels are lower than normal, endotoxic [62] and septic responses occur [63] and circulating protein C levels exhibit a strong inverse correlation with sepsis prognosis [64]. The PROWESS clinical trial demonstrated efficacy of APC in severe sepsis [65] and APC was subsequently FDA-approved (marketed as Xigris, Eli Lilly) to treat patients with severe sepsis. However, in 2011, APC was voluntarily withdrawn from the market by the supplier, due to questions on efficacy (PROWESS-SHOCK ClinicalTrials.gov, NCT00604214). A recent Cochrane review suggested that APC is not effective for treating patients with severe sepsis or septic shock and that it is associated with a risk of bleeding [66]. Nonetheless, reports continue to show that patients with septic shock who were treated with APC had a reduced in-hospital mortality compared with those not treated with APC [67]. Furthermore, in animal models, APC prevents endotoxin-induced sepsis by inhibiting activated leukocytes, cytokines, and vascular permeability [3, 68–70] (Fig. 7). The current study adds a new mechanism of action to the growing body of evidence that APC has potential to benefit sepsis and other diseases associated with vascular pathologies, such as rheumatoid arthritis and cancer.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Fig. 1. APC regulates Tie2 in HUVEC at least partially through EPCR at 4 h. HUVEC were incubated with Biorich medium +2% FCS for 24 h before treatment with APC (1 or 10 µg/ml) for 4 h. For some experiments, HUVEC were pre-incubated in Biorich medium + 2% FCS for 1 h with RCR52 (1 µg/ml), before treatment with APC (1 µg/ml) for 4 h. The cell lysates were analyzed for P-Tie2 by western blotting. Results were normalized with β-actin (PPTX 201 kb)
Acknowledgements
We thank Dr Anthony Ashton for helpful discussions and The Maternity unit at Royal North Shore Hospital for providing umbilical cords.
Compliance with ethical standards
Conflict of interest
CJ and MX have patents and commercial interests in molecular variants of APC and skin conditions.
References
- 1.Griffin JH, Fernandez JA, Mosnier LO, Liu D, Cheng T, Guo H, Zlokovic BV (2006) The promise of protein C. Blood cells Mol Dis 36 (2):211–216. doi:10.1016/j.bcmd.2005.12.023 [DOI] [PubMed]
- 2.Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105(8):3178–3184. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- 3.Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, Ye SQ, Garcia JG. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem. 2005;280(17):17286–17293. doi: 10.1074/jbc.M412427200. [DOI] [PubMed] [Google Scholar]
- 4.Mosnier LO, Sinha RK, Burnier L, Bouwens EA, Griffin JH. Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood. 2012;120(26):5237–5246. doi: 10.1182/blood-2012-08-452169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bae JS, Yang L, Manithody C, Rezaie AR. The ligand occupancy of endothelial protein C receptor switches the protease-activated receptor 1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood. 2007;110(12):3909–3916. doi: 10.1182/blood-2007-06-096651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minhas N, Xue M, Fukudome K, Jackson CJ. Activated protein C utilizes the angiopoietin/Tie2 axis to promote endothelial barrier function. FASEB J. 2010;24(3):873–881. doi: 10.1096/fj.09-134445. [DOI] [PubMed] [Google Scholar]
- 7.Gramling MW, Beaulieu LM, Church FC. Activated protein C enhances cell motility of endothelial cells and MDA-MB-231 breast cancer cells by intracellular signal transduction. Exp Cell Res. 2010;316(3):314–328. doi: 10.1016/j.yexcr.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burnier L, Mosnier LO. Novel mechanisms for activated protein C cytoprotective activities involving non-canonical activation of protease-activated receptor 3. Blood. 2013 doi: 10.1182/blood-2013-03-488957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiedler U, Augustin HG. Angiopoietins: a link between angiogenesis and inflammation. Trends Immunol. 2006;27(12):552–558. doi: 10.1016/j.it.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Brindle NP, Saharinen P, Alitalo K. Signaling and functions of angiopoietin-1 in vascular protection. Circ Res. 2006;98:1014–1023. doi: 10.1161/01.RES.0000218275.54089.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones N, Voskas D, Master Z, Sarao R, Jones J, Dumont DJ. Rescue of the early vascular defects in Tek/Tie2 null mice reveals an essential survival function. EMBO Rep. 2001;2(5):438–445. doi: 10.1093/embo-reports/kve093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCarter SD, Mei SH, Lai PF, Zhang QW, Parker CH, Suen RS, Hood RD, Zhao YD, Deng Y, Han RN, Dumont DJ, Stewart DJ. Cell-based angiopoietin-1 gene therapy for acute lung injury. Am J Respir Crit Care Med. 2007;175(10):1014–1026. doi: 10.1164/rccm.200609-1370OC. [DOI] [PubMed] [Google Scholar]
- 13.Fukuhara S, Sako K, Noda K, Zhang J, Minami M, Mochizuki N. Angiopoietin-1/Tie2 receptor signaling in vascular quiescence and angiogenesis. Histol Histopathol. 2010;25(3):387–396. doi: 10.14670/HH-25.387. [DOI] [PubMed] [Google Scholar]
- 14.van der Heijden M, van Nieuw Amerongen GP, Chedamni S, van Hinsbergh VW, Johan Groeneveld AB. The angiopoietin-Tie2 system as a therapeutic target in sepsis and acute lung injury. Expert Opin Ther Targets. 2009;13(1):39–53. doi: 10.1517/14728220802626256. [DOI] [PubMed] [Google Scholar]
- 15.Thurston G, Wang Q, Baffert F, Rudge J, Papadopoulos N, Jean-Guillaume D, Wiegand S, Yancopoulos GD, McDonald DM. Angiopoietin 1 causes vessel enlargement, without angiogenic sprouting, during a critical developmental period. Development. 2005;132(14):3317–3326. doi: 10.1242/dev.01888. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi H, Lin PC. Angiopoietin/Tie2 signaling, tumor angiogenesis and inflammatory diseases. Front Biosci. 2005;10:666–674. doi: 10.2741/1561. [DOI] [PubMed] [Google Scholar]
- 17.David S, Mukherjee A, Ghosh CC, Yano M, Khankin EV, Wenger JB, Karumanchi SA, Shapiro NI, Parikh SM. Angiopoietin-2 may contribute to multiple organ dysfunction and death in sepsis*. Crit Care Med. 2012;40(11):3034–3041. doi: 10.1097/CCM.0b013e31825fdc31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giuliano JS, Jr, Wheeler DS. Excess circulating angiopoietin-2 levels in sepsis: harbinger of death in the intensive care unit? Crit Care. 2009;13(1):114. doi: 10.1186/cc7685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lukasz A, Kumpers P, David S. Role of angiopoietin/tie2 in critical illness: promising biomarker, disease mediator, and therapeutic target? Scientifica (Cairo) 2012;2012:160174. doi: 10.6064/2012/160174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shibata M, Kumar SR, Amar A, Fernandez JA, Hofman F, Griffin JH, Zlokovic BV. Anti-inflammatory, antithrombotic, and neuroprotective effects of activated protein C in a Murine model of focal ischemic stroke. Circulation. 2001;103(13):1799–1805. doi: 10.1161/01.CIR.103.13.1799. [DOI] [PubMed] [Google Scholar]
- 21.Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, Covic L, Kuliopulos A. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol. 2007;8(12):1303–1312. doi: 10.1038/ni1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirschfield GM, Herbert J, Kahan MC, Pepys MB. Human C-reactive protein does not protect against acute lipopolysaccharide challenge in mice. J Immunol. 2003;171(11):6046–6051. doi: 10.4049/jimmunol.171.11.6046. [DOI] [PubMed] [Google Scholar]
- 23.Thompson WL, Karpus WJ, Van Eldik LJ. MCP-1-deficient mice show reduced neuroinflammatory responses and increased peripheral inflammatory responses to peripheral endotoxin insult. J Neuroinflammation. 2008;5:35. doi: 10.1186/1742-2094-5-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye X, Ding J, Zhou X, Chen G, Liu SF. Divergent roles of endothelial NF-{kappa}B in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med. 2008;205(6):1303–1315. doi: 10.1084/jem.20071393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han Lee ED, Pappalardo E, Scafidi J, Davis AE. Approaches toward reversal of increased vascular permeability in C1 inhibitor deficient mice. Immunol Lett. 2003;89(2–3):155–160. doi: 10.1016/S0165-2478(03)00130-5. [DOI] [PubMed] [Google Scholar]
- 26.Moitra J, Sammani S, Garcia JG. Re-evaluation of Evans Blue dye as a marker of albumin clearance in murine models of acute lung injury. Transl Res. 2007;150(4):253–265. doi: 10.1016/j.trsl.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Patterson CE, Rhoades RA, Garcia JG. Evans blue dye as a marker of albumin clearance in cultured endothelial monolayer and isolated lung. J Appl Phys. 1992;72(3):865–873. doi: 10.1063/1.351760. [DOI] [PubMed] [Google Scholar]
- 28.Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci. 1998;111(Pt 13):1853–1865. doi: 10.1242/jcs.111.13.1853. [DOI] [PubMed] [Google Scholar]
- 29.Xue M, Campbell D, Sambrook PN, Fukudome K, Jackson CJ. Endothelial protein C receptor and protease-activated receptor-1 mediate induction of a wound-healing phenotype in human keratinocytes by activated protein C. J Invest Dermatol. 2005;125(6):1279–1285. doi: 10.1111/j.0022-202X.2005.23952.x. [DOI] [PubMed] [Google Scholar]
- 30.Stitt TN, Conn G, Gore M, Lai C, Bruno J, Radziejewski C, Mattsson K, Fisher J, Gies DR, Jones PF, et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell. 1995;80(4):661–670. doi: 10.1016/0092-8674(95)90520-0. [DOI] [PubMed] [Google Scholar]
- 31.Davis JQ, Lambert S, Bennett V. Molecular composition of the node of Ranvier: identification of ankyrin-binding cell adhesion molecules neurofascin (mucin+/third FNIII domain−) and NrCAM at nodal axon segments. J Cell Biol. 1996;135(5):1355–1367. doi: 10.1083/jcb.135.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Y, Liu YJ, Yu Q. Angiopoietin-3 is tethered on the cell surface via heparan sulfate proteoglycans. J Biol Chem. 2004;279(39):41179–41188. doi: 10.1074/jbc.M400292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hartig PC, Cardon MC, Blystone CR, Gray LE, Jr, Wilson VS. High throughput adjustable 96-well plate assay for androgen receptor binding: a practical approach for EDC screening using the chimpanzee AR. Toxicol Lett. 2008;181(2):126–131. doi: 10.1016/j.toxlet.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 34.Yuan HT, Khankin EV, Karumanchi SA, Parikh SM. Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Mol Cell Biol. 2009;29(8):2011–2022. doi: 10.1128/MCB.01472-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grazzini E, Guillon G, Mouillac B, Zingg HH. Inhibition of oxytocin receptor function by direct binding of progesterone. Nature. 1998;392(6675):509–512. doi: 10.1038/33176. [DOI] [PubMed] [Google Scholar]
- 36.Lamping N, Dettmer R, Schroder NW, Pfeil D, Hallatschek W, Burger R, Schumann RR. LPS-binding protein protects mice from septic shock caused by LPS or gram-negative bacteria. J Clin Invest. 1998;101(10):2065–2071. doi: 10.1172/JCI2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87(7):1171–1180. doi: 10.1016/S0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 38.Lay AJ, Donahue D, Tsai MJ, Castellino FJ. Acute inflammation is exacerbated in mice genetically predisposed to a severe protein C deficiency. Blood. 2007;109(5):1984–1991. doi: 10.1182/blood-2006-07-037945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thurston G. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med. 2000;6:460–463. doi: 10.1038/74725. [DOI] [PubMed] [Google Scholar]
- 40.Witzenbichler B, Westermann D, Knueppel S, Schultheiss HP, Tschope C. Protective role of angiopoietin-1 in endotoxic shock. Circulation. 2005;111(1):97–105. doi: 10.1161/01.CIR.0000151287.08202.8E. [DOI] [PubMed] [Google Scholar]
- 41.Thurston G. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286:2511–2514. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- 42.Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene expression profile of antithrombotic protein C defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276(14):11199–11203. doi: 10.1074/jbc.C100017200. [DOI] [PubMed] [Google Scholar]
- 43.Russo A, Soh UJ, Paing MM, Arora P, Trejo J. Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc Natl Acad Sci USA. 2009;106(15):6393–6397. doi: 10.1073/pnas.0810687106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopez-Sagaseta J, Puy C, Tamayo I, Allende M, Cervero J, Velasco SE, Esmon CT, Montes R, Hermida J. sPLA2-V inhibits EPCR anticoagulant and antiapoptotic properties by accommodating lysophosphatidylcholine or PAF in the hydrophobic groove. Blood. 2012;119(12):2914–2921. doi: 10.1182/blood-2011-05-353409. [DOI] [PubMed] [Google Scholar]
- 45.Xue M, Shen K, McKelvey K, Li J, Chan YK, Hatzis V, March L, Little CB, Tonkin M, Jackson CJ. Endothelial protein C receptor-associated invasiveness of rheumatoid synovial fibroblasts is likely driven by group V secretory phospholipase A2. Arth Res Ther. 2014;16(1):R44. doi: 10.1186/ar4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thwin MM, Douni E, Arjunan P, Kollias G, Kumar PV, Gopalakrishnakone P. Suppressive effect of secretory phospholipase A2 inhibitory peptide on interleukin-1beta-induced matrix metalloproteinase production in rheumatoid synovial fibroblasts, and its antiarthritic activity in hTNFtg mice. Arth Res Ther. 2009;11(5):R138. doi: 10.1186/ar2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bostrom MA, Boyanovsky BB, Jordan CT, Wadsworth MP, Taatjes DJ, de Beer FC, Webb NR. Group v secretory phospholipase A2 promotes atherosclerosis: evidence from genetically altered mice. Arterioscler Thromb Vasc Biol. 2007;27(3):600–606. doi: 10.1161/01.ATV.0000257133.60884.44. [DOI] [PubMed] [Google Scholar]
- 48.Yang XV, Banerjee Y, Fernandez JA, Deguchi H, Xu X, Mosnier LO, Urbanus RT, de Groot PG, White-Adams TC, McCarty OJ, Griffin JH. Activated protein C ligation of ApoER2 (LRP8) causes Dab1-dependent signaling in U937 cells. Proc Natl Acad Sci USA. 2009;106(1):274–279. doi: 10.1073/pnas.0807594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao C, Gao Y, Li Y, Antalis TM, Castellino FJ, Zhang L. The efficacy of activated protein C in murine endotoxemia is dependent on integrin CD11b. J Clin Invest. 2010;120(6):1971–1980. doi: 10.1172/JCI40380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elphick GF, Sarangi PP, Hyun YM, Hollenbaugh JA, Ayala A, Biffl WL, Chung HL, Rezaie AR, McGrath JL, Topham DJ, Reichner JS, Kim M. Recombinant human activated protein C inhibits integrin-mediated neutrophil migration. Blood. 2009;113(17):4078–4085. doi: 10.1182/blood-2008-09-180968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xue M, Chow SO, Dervish S, Chan YK, Julovi SM, Jackson CJ. Activated protein C enhances human keratinocyte barrier integrity via sequential activation of epidermal growth factor receptor and Tie2. J Biol Chem. 2011;286(8):6742–6750. doi: 10.1074/jbc.M110.181388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stavenuiter F, Mosnier LO. Noncanonical PAR3 activation by factor Xa identifies a novel pathway for Tie2 activation and stabilization of vascular integrity. Blood. 2014;124(23):3480–3489. doi: 10.1182/blood-2014-06-582775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bae JS, Rezaie AR. Thrombin upregulates the angiopoietin-Tie2 Axis: endothelial protein C receptor occupancy prevents the thrombin mobilization of angiopoietin 2 and P-selectin from Weibel-Palade bodies. J Thromb Haemost. 2010;8(5):1107–1115. doi: 10.1111/j.1538-7836.2010.03812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mather T, Oganessyan V, Hof P, Huber R, Foundling S, Esmon C, Bode W. The 2.8-Angstrom crystal structure of gla-domainless activated protein c. EMBO J. 1996;15(24):6822–6831. [PMC free article] [PubMed] [Google Scholar]
- 55.Barton WA, Tzvetkova D, Nikolov DB. Structure of the angiopoietin-2 receptor binding domain and identification of surfaces involved in Tie2 recognition. Structure. 2005;13(5):825–832. doi: 10.1016/j.str.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 56.Harfouche R, Hussain SN. Signaling and regulation of endothelial cell survival by angiopoietin-2. Am J Physiol Heart Circ Physiol. 2006;291(4):H1635–H1645. doi: 10.1152/ajpheart.01318.2005. [DOI] [PubMed] [Google Scholar]
- 57.Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N, Daly TJ, Davis S, Sato TN, Yancopoulos GD. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277(5322):55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 58.Roviezzo F, Tsigkos S, Kotanidou A, Bucci M, Brancaleone V, Cirino G, Papapetropoulos A. Angiopoietin-2 causes inflammation in vivo by promoting vascular leakage. J Pharmacol Exp Ther. 2005;314(2):738–744. doi: 10.1124/jpet.105.086553. [DOI] [PubMed] [Google Scholar]
- 59.Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA, Sukhatme VP. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med. 2006;3(3):e46. doi: 10.1371/journal.pmed.0030046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ganter MT, Cohen MJ, Brohi K, Chesebro BB, Staudenmayer KL, Rahn P, Christiaans SC, Bir ND, Pittet JF. Angiopoietin-2, marker and mediator of endothelial activation with prognostic significance early after trauma? Ann Surg. 2008;247(2):320–326. doi: 10.1097/SLA.0b013e318162d616. [DOI] [PubMed] [Google Scholar]
- 61.Ziegler T, Horstkotte J, Schwab C, Pfetsch V, Weinmann K, Dietzel S, Rohwedder I, Hinkel R, Gross L, Lee S, Hu J, Soehnlein O, Franz WM, Sperandio M, Pohl U, Thomas M, Weber C, Augustin HG, Fassler R, Deutsch U, Kupatt C. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J Clin Invest. 2013 doi: 10.1172/JCI66549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Levi M, Dorffler-Melly J, Reitsma P, Buller H, Florquin S, van der Poll T, Carmeliet P. Aggravation of endotoxin-induced disseminated intravascular coagulation and cytokine activation in heterozygous protein-C-deficient mice. Blood. 2003;101(12):4823–4827. doi: 10.1182/blood-2002-10-3254. [DOI] [PubMed] [Google Scholar]
- 63.Ganopolsky JG, Castellino FJ. A protein C deficiency exacerbates inflammatory and hypotensive responses in mice during polymicrobial sepsis in a cecal ligation and puncture model. Am J Path. 2004;165(4):1433–1446. doi: 10.1016/S0002-9440(10)63401-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fisher CJ, Jr, Yan SB. Protein C levels as a prognostic indicator of outcome in sepsis and related diseases. Crit Care Med. 2000;28(9 Suppl):S49–S56. doi: 10.1097/00003246-200009001-00011. [DOI] [PubMed] [Google Scholar]
- 65.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ. Efficacy and safety of recombinant human activated protein C for severe sepsis. New Eng J Med. 2001;344(10):699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 66.Marti-Carvajal AJ, Sola I, Lathyris D, Cardona AF (2011) Human recombinant activated protein C for severe sepsis. Coch Data Syst Rev 4:CD004388 [DOI] [PubMed]
- 67.Sadaka F, O’Brien J, Migneron M, Stortz J, Vanston A, Taylor RW. Activated protein C in septic shock: a propensity-matched analysis. Crit Care. 2011;15(2):R89. doi: 10.1186/cc10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Murakami K, Okajima K, Uchiba M, Johno M, Nakagaki T, Okabe H, Takatsuki K. Activated protein C attenuates endotoxin-induced pulmonary vascular injury by inhibiting activated leukocytes in rats. Blood. 1996;87(2):642–647. [PubMed] [Google Scholar]
- 69.Murakami K, Okajima K, Uchiba M, Johno M, Nakagaki T, Okabe H, Takatsuki K. Activated protein C prevents LPS-induced pulmonary vascular injury by inhibiting cytokine production. Am J Physiol Lung Cell Mol Physio. 1997;272(2):L197–L202. doi: 10.1152/ajplung.1997.272.2.L197. [DOI] [PubMed] [Google Scholar]
- 70.Gupta A, Rhodes GJ, Berg DT, Gerlitz B, Molitoris BA, Grinnell BW (2007) Activated protein C ameliorates LPS-induced acute kidney injury and downregulates renal INOS and angiotensin 2. Am J Physio Ren Physio 293 (1):F245–F254. doi:10.1152/ajprenal.00477.2006 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. APC regulates Tie2 in HUVEC at least partially through EPCR at 4 h. HUVEC were incubated with Biorich medium +2% FCS for 24 h before treatment with APC (1 or 10 µg/ml) for 4 h. For some experiments, HUVEC were pre-incubated in Biorich medium + 2% FCS for 1 h with RCR52 (1 µg/ml), before treatment with APC (1 µg/ml) for 4 h. The cell lysates were analyzed for P-Tie2 by western blotting. Results were normalized with β-actin (PPTX 201 kb)