

Abstract
Accumulation of misfolded/unfolded aggregated proteins in the brain is a hallmark of many neurodegenerative diseases affecting humans and animals. Dysregulation of calcium (Ca2+) and disruption of fast axonal transport (FAT) are early pathological events that lead to loss of synaptic integrity and axonal degeneration in early stages of neurodegenerative diseases. Dysregulated Ca2+ in the brain is triggered by accumulation of misfolded/unfolded aggregated proteins in the endoplasmic reticulum (ER), a major Ca2+ storing organelle, ultimately leading to neuronal dysfunction and apoptosis. Calcineurin (CaN), a Ca2+/calmodulin-dependent serine/threonine phosphatase, has been implicated in T cells activation through the induction of nuclear factor of activated T cells (NFAT). In addition to the involvement of several other signaling cascades, CaN has been shown to play a role in early synaptic dysfunction and neuronal death. Therefore, inhibiting hyperactivated CaN in early stages of disease might be a promising therapeutic strategy for treating patients with protein misfolding diseases. In this review, we briefly summarize the structure of CaN, inhibition mechanisms by which immunosuppressants inhibit CaN, role of CaN in maintaining neuronal and synaptic integrity and homeostasis and the role played by CaN in protein unfolding/misfolding neurodegenerative diseases.
Keywords: Calcineurin regulation, Microglial activation, Tacrolimus, Neuroinflammation, Phosphorylated cAMP response element-binding protein (pCREB) and phosphorylated Bcl2-associated death promoter (pBAD)
Introduction
Protein misfolding disorders are a group of diseases with devastating effects ranging from abstract thinking and movement abnormalities to loss of mental function and dementia. Protein misfolding/unfolding is involved in the pathogenesis of several neurological diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), dementia with Lewy bodies, frontotemporal dementia (FTD), Huntington’s disease (HD), Creutzfeldt–Jacob disease (CJD), kuru, mad-cow disease, and scrapie [1–3]. Accumulation of amyloid beta (Aβ) and tau protein is a major causative factor in AD, whereas accumulation of alpha-synuclein and tau is associated with PD, SOD1 accumulation is associated with ALS, PolyQ and huntingtin accumulation is associated with HD, and prion protein accumulation is associated with CJD, kuru, mad-cow disease, and scrapie [1, 2, 4]
Folding and processing of secretary and transmembrane proteins occur in the endoplasmic reticulum (ER), a multifunctional organelle. Correct protein folding and processing in the ER are dependent upon balanced ER protein load and folding capacity. Under pathological conditions, the balance between folding load and capacity in the ER is disrupted, leading to accumulation of misfolded/unfolded proteins and ER stress [5]. ER stress activates an adaptive response known as the unfolded protein response (UPR) to correct misfolded/unfolded proteins and, thus, reduce ER stress, but long-term and severe ER stress dysregulates Ca2+ homeostasis and causes cytotoxic, synaptotoxic, and behavioral changes mediated by calcineurin (CaN) (also known as calcium (Ca2+)/calmodulin (CaM)-dependent serine/threonine protein phosphatase) [6–10].
In human and rodent models of neurodegenerative diseases, several regions of the brain show CaN immunoreactivity in neurons, but not in glia [11, 12]. CaN is localized in the cell soma, dendrites (including the postsynaptic density), dendritic spines, axons, and axon terminals [11]. The activity of CaN is regulated by the Ca2+-CaM complex. Important targets of CaN include the Bcl2-associated death promoter (BAD), a proapoptotic member of the Bcl2 gene family, and cAMP response element-binding (CREB), a transcription factor. Hyperactivated CaN dephosphorylates BAD, leading to dissociation of BAD from scaffolding protein 14-3-3 and interaction of BAD with mitochondrial membrane-associated protein Bcl-xL or other Bcl2 family proteins, followed by release of cytochrome c from the mitochondria, caspase activation, and neuronal apoptosis [13]. In addition, hyperactivated CaN inhibits translocation of CREB into the nucleus, suppressing gene expression and inducing synaptic dysfunction [14]. More recently, Gan and Silverman studied the molecular events in early stage of AD, suggesting that Ca2+ dysregulation and subsequent hyperactivated CaN resulted in spatiotemporal transport defects in dendrites prior to axons. Furthermore, these spatiotemporal events were independent of Tau protein [15]. Also, Pineda and coworkers found that BDNF transport defects observed in HD can be restored by CaN inhibition via FK506 in primary neuron cultures of mutant huntingtin and mouse cortical neurons [16].
All major protein misfolding diseases, including AD, PD, HD, ALS, and prion diseases, have devastating effects on humans and animals, because of the lack of effective therapeutic strategies and presymptomatic diagnostic tools. Thus, there is a dire need for novel therapeutic intervention strategies that are effective during the early stages of protein misfolding diseases.
Calcium dysregulation and calcineurin-mediated signaling in neurodegenerative diseases
Recent studies on protein misfolding diseases suggest that aggregation of misfolded/unfolded proteins in the ER caused by the failure of the folding machinery to overcome the accumulation of misfolded/unfolded aggregated proteins is the sole cause of sustained ER stress 3, 17–19]. ER and mitochondria are the main organelles contributing towards important cell survival processes such as Ca2+ homeostasis and lipid biosynthesis [20, 21]. The immediate adverse effect of ER stress on cells is the disruption of Ca2+ homeostasis [22]. Dysregulated Ca2+ signaling has been reported in patients afflicted with all major protein misfolding/unfolding neurodegenerative diseases, including AD, PD, HD, ALS, and prion diseases [19–21].
Ca2+ plays a vital role as a secondary messenger in many signaling pathways involved in maintaining cellular homeostasis. Neuronal activity requires an optimal Ca2+ concentration; therefore, the harmful effects of Ca2+dysregulation are observed more often in neurons than in other cell types [23]. Neuronal function and survival are dependent on maintenance of the proper Ca2+ concentration in the cytoplasm. Ca2+ homeostasis in the ER and cytoplasm is maintained via Ca2+ channels located on the membrane of the ER and adenosine triphosphate (ATP)-driven sarcoplasmic endoplasmic reticulum (SERCA)-associated Ca2+ pumps [23]. The ER is a dynamic Ca2+ storage site that contains a high concentration of Ca2+ (0.2–2 mM) in comparison with that of the cytoplasm (50–100 nM). Ca2+ uptake into the ER is mediated by SERCA pumps, while release of Ca2+ from the ER occurs via inositol 1,4,5-triphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) located on the ER membrane [10, 21, 24].
Recent studies on neurodegenerative diseases suggest that aggregation of misfolded/unfolded proteins leads to ER stress and a subsequent increase in cytoplasmic Ca2+ [19, 21, 22]. In the early stages of AD and prion diseases, Ca2+ released from ER-bound Ca2+ channels during ER stress induced by oligomeric Aβ and prion peptides 106–126 perturbs Ca2+ homeostasis in the ER, resulting in Ca2+ uptake by mitochondria. The increased Ca2+ concentration in the mitochondria leads to reactive oxygen species (ROS) production and release of cytochrome c and caspase-3, which initiate apoptosis [25]. Indeed, Ca2+ release from ER-resident Ca2+ channels is thought to be one of the first adaptive responses in cell lines infected with prion proteins [26]. Inhibition of signaling associated with the adaptive UPR through disruption of activating transcription factor 6 (ATF6) has recently been demonstrated in the SH-SY5Y cell model [27].
Activation of misfolded/unfolded aggregated protein-mediated pathways via ER stress dysregulates Ca2+ homeostasis and leads to changes in the activity levels of kinases and phosphatases related to normal cellular functioning and the cell cycle 21].
In recent studies, Medina and colleagues provided a new perspective on the relationship of CaN activity with dysregulated Ca2+ in organelles other than the ER, thus revealing the role of lysosomes in post-translational processing and transcriptional regulation [28, 29]. For decades, lysosomes were thought to be involved only in catabolic product processing at the terminal end of the phagocytic pathway, but Medina and colleagues showed that lysosomes regulate the transcription of transcription factor EB (TFEB), a substrate of CaN. Following Ca2+ dysregulation in lysosomes, TFEB is dephosphorylated and translocated to the nucleus, where it activates TFEB-related genes. The present review is focused on the role of CaN in the pathogenesis of neurodegenerative diseases.
Basic structure of calcineurin
CaN is the only known Ca2+-binding signaling transducer protein calmodulin (CaM)-dependent serine/threonine phosphatase and is involved in many cellular functions, such as controlling the cell cycle and regulating signal transduction by protein kinases. While numerous protein kinases have been studied using biochemical and structural assays, relatively few numbers of phosphatases (compared to kinases) achieve the specific dephosphorylation of thousands of phosphoprotein substrates. The specificity is achieved through the regulatory subunits [30–33]. CaN is a heterodimer consisting of a 59-kDa catalytic subunit known as calcineurin A (CaNA) and a 19-kDa regulatory subunit known as calcineurin B (CaNB) (Fig. 1). The crystalline structure of unligated CaN has been elucidated [34]. In addition, complexes of CaN with immunosuppressant tacrolimus [34, 35] and binding proteins FKBP12 and cyclophilin A (CyPA) have also been resolved [36, 37].
Fig. 1.
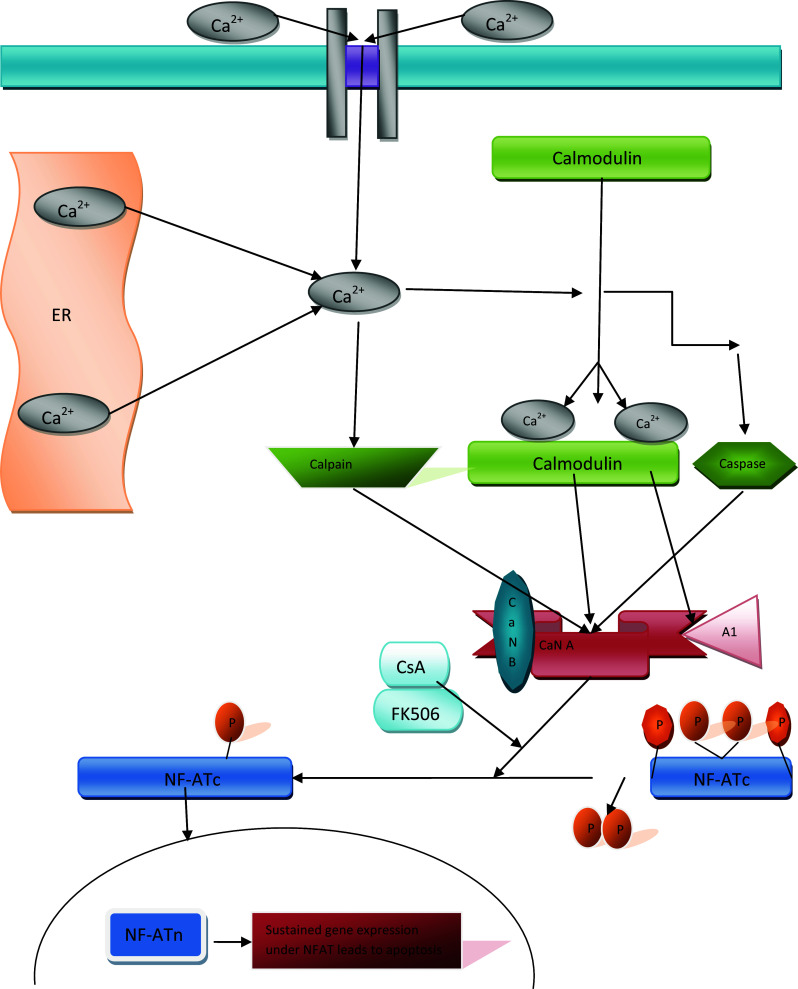
Schematic presentation of reversible and irreversible activation of calcineurin (CaN). ER stress induced by accumulation of misfolded/unfolded aggregated proteins in the ER leads to Ca2+ release from the ER. Dysregulated Ca2+ homeostasis activates CaN. Reversible CaN hyperactivation may induce a CaN activity level 15-fold higher than that induced by irreversible hyperactivation. Irreversible activation of CaN is observed after proteolytic cleavage of the CaNAI region and calmodulin (CaM) binding domain. The cleaved CaNAI and CaM binding domains are not sensitive to Ca2+/CaM and are thus constantly activated. Hyperactivation of calcineurin results in dephosphorylation and nuclear translocation of cytoplasmic NFAT (NFATc), upon which nuclear NFAT (NFATn) mediates sustained gene expression leading to apoptotic cell death. Immunosuppressant drugs such as tacrolimus (FK506) and cyclosporin A (CsA) inhibit CaN hyperactivation and block NFAT dephosphorylation, reducing apoptosis
The catalytic subunit of CaNA consists of 521 residues. Residues 1 through 346 comprise a phosphatase domain, residues 347 through 373 comprise a CaNB-binding helical (BBH) domain, residues 390 through 414 comprise a CaM binding region, and residues 469–486 comprise an autoinhibitory (AI) region. Structural analysis of the phosphatase domain of CaNA revealed the presence of two beta sheets flanked by a mixture of alpha and beta sheets on one side and alpha helices on the other side, in addition to hydrophobic residues on the inner core of the beta sandwich. The Ser/Thr protein phosphatase-1 (PPase-1) catalytic subunit is structurally similar to the phosphatase domains of CaNA [38, 39], bacterial phage k Ser/Thr protein phosphatase (k-PPase) [40], and purple acid phosphatase (PAPase) [41]. Indeed, PPase-1 and PPase-2A have more than 40 % sequence identity with CaNA [42]. However, Das and colleagues reported that PPase-2C, another member of the Ser/Thr protein phosphatase family, is structurally similar to the alpha-to-beta sandwich-type structure of the CaNA catalytic domain, but the topological secondary structures of PPase-2C and CaNA show clear differences [43]. In contrast to other PPases, PPase-2C does not show significant sequence similarity or homology with CaN or PPase-1.
CaNB-binding helix BBH is a multifunctional helix that interacts with CaNB on one side and forms a complex with immunophilin and immunosuppressants on the other side. The formation of a hydrophobic core between the residues of BBH and CaNB occurs when BBH hydrophobic residues Val349, Val357, Val364, Val368, Leu354, Leu365, and Leu369 combine with CaNB hydrophobic residues Val54, Val57, Val156, Leu91, Phe81, Phe93, Ile97, and Ile106. In contrast, a mixture of hydrophobic and hydrophilic residues is present on the opposite side of BBH. Thus, a surface is formed over CaNB where drug/receptor complexes such as FKBP12–FK506 and CyPA–CsA can bind.
There are two linearly arranged Ca2+ binding domains along the BBH domain. The first Ca2+ binding domain is made up of residues 1 through 84, while the second Ca2+ binding domain consists of residues 86–169 [39]. Each of the Ca2+ binding domains on the BBH domain is made up of two Ca2+ binding EF hand motifs that are similar to those found in CaM. Five clearly defined co-ordinations are observed for the four Ca2+ ions bound by the EF hand motifs. EF1 has two oxygen chains: the side chain oxygen atoms at Asp30, Asp32, Ser34, and Glu41, as well as the carbonyl oxygen chain at Ser36. EF2 has two oxygen chains comprising the side chain oxygen atoms at Asp62, Asp64, Asn66, and Glu73, as well as the carbonyl chain oxygen at Gly68. EF3 has two oxygen chains: the side chain of oxygen atom at Asp99, Asp101, Asp103, and Glu110, as well as the carbonyl oxygen chain of Tyr105. EF4 has two oxygen chains: the side chain of oxygen atoms at Asp140, Asp142, Asp144, and Glu151, as well as the carbonyl oxygen chain at Arg146. Ca2+/CaM complex plays a vital role in enzymatic activation of CaN. Kinetic studies have revealed a reversible 15-fold increase in the enzymatic activity of CaN in the presence of the Ca2+/CaM complex (Fig. 1). CaM has the unique capacity to bind to phosphorylated and non-phosphorylated CaN [44]. In addition to activation through the Ca2+/CaM complex, CaN is activated through two other irreversible proteolytic pathways (Fig. 1), the first of which is a caspase-mediated pathway that is activated by cleavage of the AI region and CaM-binding domain of CaN and is insensitive to the Ca2+/CaM complex [45]; however, this pathway of CaN activation is controversial because it is activated during the initiation of downstream apoptotic signaling. The second irreversible proteolytic pathway of CaN activation is mediated by partial proteolysis by Ca2+-dependent cysteine protease calpain (Fig. 1) [46]. CaN is split into three truncated forms after calpain cleavage: 45, 48, and 57-kDa fragments. The 45-kDa domain lacks the Ca2+ and CaM complex domain and AI domain. A57-kDa fragment that is sensitive to the Ca2+ and CaM complex and missing only the AI domain has been found in the brains of human AD patients [47].
Inhibition of CaN by immunophilins and immunosuppressants
CaN inhibitor tacrolimus binds to a site at the BBH-CaNB domain that is approximately 25 amino acids away from the active site of CaN. FKBP12 is the most probable binding site for 12 kDa size tacrolimus and CyPA–CsA and FKBP12–FK506 complexes block the phosphatase activity of CaN [35, 48, 49]. However, the CaN activity induced by small substrate para-nitrophenylphosphate suggests that activation of CaN by the immunophilin–cyclosporin complex is not as simple as predicted by the spatial blockade model. Structural studies of the CyPA–CsA–CaN complex revealed that catalytic residue Arg122 of CaNA forms a hydrogen bond with Arg148 of CyPA. These astonishing observations about residue Arg122 of CaNA lead us presume that direct regulation of CaN activity by CyPA–CsA can occur. Furthermore, structural studies of the unligated form of CaN and the CaN–CyPA–CsA–FK506 complex showed that the autoinhibitory loop of CaN interacts with CyPA and FKBP12, suggesting that the most probable consequence of immunophilin–drug complexes is conformational alteration of the autoinhibitory fragment of CaN. Conformational changes in the autoinhibitory loop might favor smaller substrates and prevent large substrates from binding.
There are several CaN inhibitors available in the market but two most commonly used CaN inhibitors are tacrolimus (FK506) and cyclosporin A (CsA). Tacrolimus (trade name Prograf) is a product of the bacterium Streptomyces tsukubaensis. It is a macrolide lactone and acts by inhibiting calcineurin. The drug is used primarily in liver and kidney transplantations, although in some clinics it is used in heart, lung, and heart/lung transplantations. It binds to the immunophilin FKBP1A, followed by the binding of the complex to calcineurin and the inhibition of its phosphatase activity. In this way, it prevents the cell from transitioning from the G0 into G1 phase of the cell cycle [50]. Cyclosporin is thought to bind to the cytosolic protein cyclophilin (an immunophilin) of immunocompetent lymphocytes, especially T-lymphocytes. This complex of cyclosporin and cyclophilin inhibits the phosphatase calcineurin, which under normal circumstances induces the transcription of interleukin-2. The drug also inhibits lymphokine production and interleukin release, leading to a reduced function of effector T-cells. Tacrolimus is more potent and specific than CsA and has less pronounced side-effects [51].
Calcineurin and neurodegeneration
Maintaining neuronal integrity and homeostasis
CaN is the only Ca2+-dependent phosphatase found abundantly in the cytosol and presynaptic and postsynaptic terminals of neurons. CaN plays a major role in maintaining cellular homeostasis when Ca2+ levels are altered. Several research groups have focused on the putative role of CaN in neuronal activity [21, 52, 53]. Ca2+ influx in the neuronal cytosol in response to accumulation of misfolded/unfolded protein aggregates activates several proteins that initiate downstream signaling. CaN is promptly activated by cytosolic Ca2+ influx because of the affinity of CaN (0.1–1 nM) for Ca2+/CaM and colocalization of CaN with the N-methyl-d-aspartate (NMDA) receptors [54, 55]. Activated CaN inhibits further Ca2+ influx into the cytosol through multiple mechanisms. CaN inhibits Ca2+ efflux through the ER membrane by suppressing voltage-gated Ca2+ channel activity and inhibiting Ca2+-induced Ca2+ release from the ER by negatively regulating inositol triphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) [56–60].
In addition to its role in controlling Ca2+ influx into the cytosol under ER stress, CaN is an important regulator of gene expression. CaN directly controls cAMP response element-binding protein (CREB), an important neuronal transcription factor that plays a vital role in cognition and memory [7, 32, 55, 61]. CREB is phosphorylated by Ca2+-dependent and Ca2+-independent protein kinases, ultimately leading to nuclear translocation and activation of CREB-dependent gene expression [61]. Expression of CREB-target genes is controlled by CaN through dephosphorylation of CREB [9, 52, 53, 61]. Another important CaN-regulated transcription factor, nuclear factor of activated T-cells (NFAT), plays a crucial role in axonal outgrowth. Dephosphorylation of NFAT4 by CaN leads to nuclear translocation and neutrophilin- and netrin-dependent gene expression [62, 63]. In addition, myocyte enhancer factor 2 (MEF2), a pro-survival transcription factor that plays an important role in cell differentiation, cell division, and cell death, has also been shown to be modulated by CaN [64, 65]. MEF2 is activated following sumoylation and acetylation induced by CaN-dependent dephosphorylation. MEF activation leads to expression of numerous genes, including activity-regulated cytoskeletal-associated protein (Arc) and synaptic Ras GTPase activating protein (SYNGAP), followed by inhibition of dendritic claw development during neural morphogenesis [63, 64]. Phosphorylation of MEF2 at the CaN target site (Ser408) is implicated in neurotoxin-induced apoptosis [66]. CaN also dephosphorylates actin filament phosphoprotein filamin in 293FT cells [67]. CaNβ protects brain after injury by activation of the PERK-dependent pathway of the UPR in mice model of traumatic brain injury [68]. Kim and colleagues shown that CaN inhibition through FK506 rescued impaired synaptic plasticity in presenilin 1 M146 V mutant mice model of AD; furthermore, FK506 stabilized GluA1 phosphorylation to improve synaptic trafficking via AMPA receptors [69]. Calpastatin overexpression reduced oxidative stress-induced impairment and cell death in SHSY5Y cell line by decreasing calpain and CaN activity and induction of mitochondrial fission [70].
Maintaining synaptic integrity
Although the final outcome of all protein unfolding/misfolding diseases is neuronal apoptosis in response to ER stress, earlier pathological anomalies, such as altered axonal transport and synaptic dysregulation, occur well before neuronal apoptosis [71–74]. Under normal physiological conditions, CaN plays a vital role in maintaining normal synaptic integrity and function under the regulatory influence of Ca2+ [52]. CaN is relatively abundant in neuronal presynaptic and postsynaptic terminals. In presynaptic terminals, neurotransmitter release and uptake via exocytosis and endocytosis are dependent on CaN (Fig. 2) [53, 75]. In postsynaptic terminals, CaN regulates voltage-gated sodium and potassium ion channels [76].
Fig. 2.
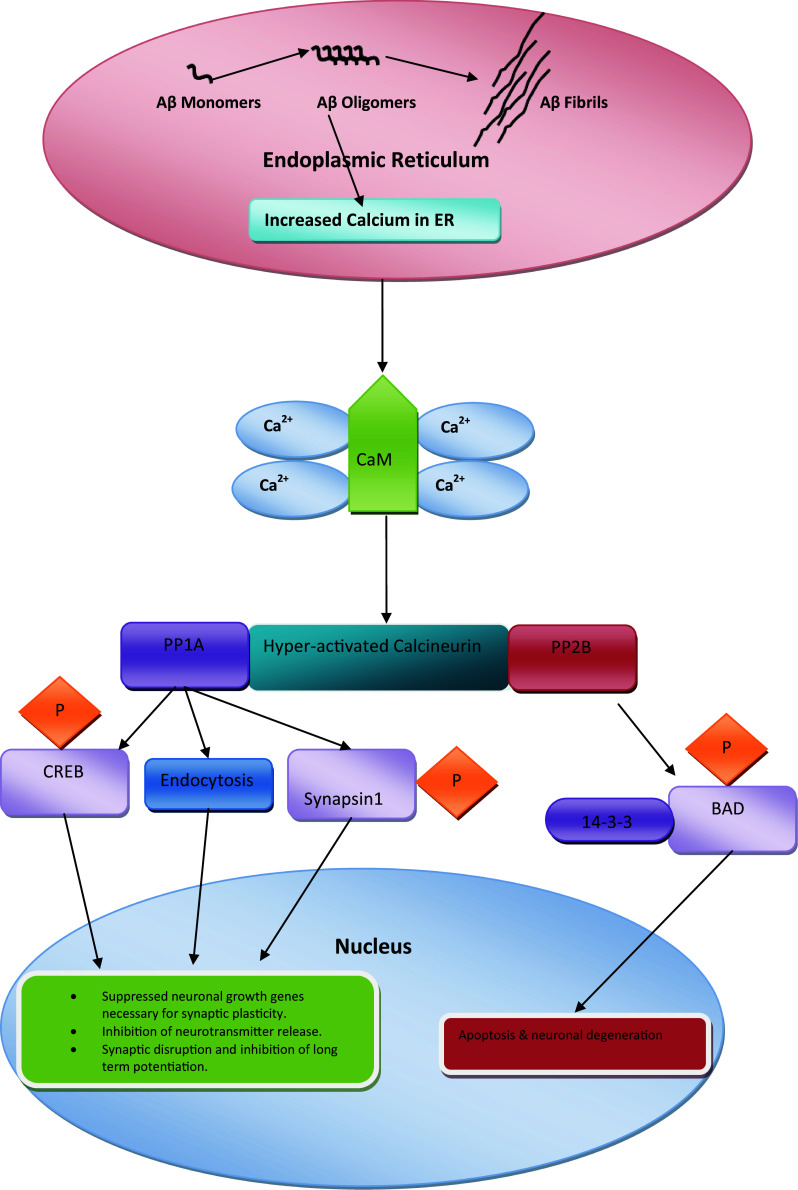
Schematic presentation of ER stress-mediated reversible (synaptic dysfunction) and irreversible (neuronal apoptosis) signaling pathways upon misfolded/unfolded protein accumulation in the ER. Hyperactivation of the PP1A domain of calcineurin following dysregulation of Ca2+ homeostasis results in three distinct anomalies: (1) phosphorylation of CREB suppresses expression of neuronal growth genes necessary for plasticity; (2) endocytosis of AMPA-bound Aβ oligomers results in synaptic disruption and inhibition of long-term potentiation; (3) phosphorylation of synapsin-1 results in inhibition of neurotransmitter release. Activation of the irreversible pathway by the PP2B domain of hyperactivated calcineurin leads to detachment of BAD from scaffolding protein 14-3-3 and BAD phosphorylation
Several studies in transgenic mice with altered forms of CaN and knockout mice lacking CaN have shown that long-term potentiation (LTP) and cognition are dependent on normal levels of CaN [21, 77–79].The role of CaN hyperactivation in Alzheimer’s disease (AD) has been extensively studied. Endocytosis of the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor was found to be mediated by hyperactivated CaN in Aβ-induced synaptic dysfunction and memory loss associated with inhibition of LTP [80]. In this model, AMPA receptors on dendritic spines bind to Aβ oligomers, after which endocytosis and subsequent internalization of Aβ oligomers bound to AMPA receptors are mediated by CaN [80]. Aβ oligomers bound to AMPA receptors co-localize with glucarpidase 2 (cpg2), which plays an important role in activity-dependent glutamate receptor endocytosis, at the postsynaptic endocytic zone of excitatory synapses. CaN inhibitors such as tacrolimus and CsA enhance memory by preventing Aβ oligomer-induced loss of surface AMPA receptors and dendritic spines. An independent study conducted using a mouse model of AD showed that soluble Aβ-mediated loss of dendritic spines was correlated with chronic activation of CaN [81]. Inhibition of CaN reverses medium- and long-term deficits in cognition and memory [82]. Zhang and colleagues used PS1-M146V KI mouse model (KI) to study the structural plasticity of hippocampal slices, showing calcineurin inhibition through tacrolimus rescued mushroom spine maintenance defects [21]. The interaction of phosphatases PP1, PP1A and PP2B with adenosine receptor 1 (A1R) results in reduced adenosine-induced persistent synaptic depression (APSD) and glutamine A1 (GluA1) release in rat hippocampal slices, but PP2A inhibition selectively prevents glutamine A2 (GluA2) endocytosis [83].
CaN plays a vital role in regulating synaptic function by post-translational modification of target proteins and controls expression of genes necessary for synaptic plasticity. As mentioned earlier, CREB is a major target of CaN (Fig. 2). Neuronal growth and survival require expression of CREB-target genes that control various proteins, including BDNF and its receptor tropomyosin-related kinase B (trkB) [84–88]. CREB is activated by phosphorylation and nuclear translocation (Fig. 2). Hyperactivated CaN leads to dephosphorylation and inactivation of CREB, ultimately suppressing CREB-dependent gene expression and resulting in synaptic dysfunction and memory loss [61]. Synaptic plasticity associated with cognitive impairment has been reported in the presence of misfolded Aβ and prions [2, 7, 89–91]. Experiments using mice showed that CaN inhibition resulted in increased dendritic branching and spine density [92]. Yamamoto-Sasaki and coworkers showed that immunoreactivity of pCREB was significantly decreased in dementia of the AD type and proposed that impaired cAMP signaling may contribute to the pathophysiology of AD [93]. More recently, Taglialatela and colleagues showed that solid organ transplant patients chronically treated with CaN inhibitor FK506 to suppress rejection had a lower incidence of dementia and AD in comparison with the normal population [94]. Mukherjee and colleagues found that rescue of optimum CREB phosphorylation by CaN inhibition slowed the progression of behavioral abnormalities in prion-infected mice at the clinical stage of prion disease [7]. However, maintaining the basal level of CaN activity is required for CREB-dependent gene expression [95, 96]. An early pathological abnormality associated with all neurodegenerative diseases is focal bead-like swelling in dendrites and axons. Interestingly, in a study using an AD model, Aβ-induced neuritic beading was rescued by CaN inhibition, which also rescued structural disruption of neuronal networks in the presence of Aβ [97].
CaN also regulates a range of proteins involved in neurotransmitter release, including synaptobrevin, synapsin, rabphilin2A, synaptotagmin, and dephosphins [98]. Vesicular transport is controlled by a major synaptic protein called synapsin I (Fig. 2), which is attached to the actin cytoskeleton under normal resting conditions. During neuronal excitation, kinases phosphorylate synapsin-I, which detaches from vesicles, facilitating neurotransmitter release [99]. Ubiquitously expressed enzymes CaN and PP2A prevent neurotransmitter release via dephosphorylation of the synaptic protein synapsin-I [100]. Recently, it is demonstrated that CaN proteolysis in activated astrocytes may be the cause of impaired synaptic functions in neurodegenerative diseases and injury models [101]. The results described above show that CaN is involved in maintenance of synaptic structure and function; therefore, hyperactivation of CaN amplifies synaptic dysfunction, whereas maintaining CaN activity at an optimum level could prevent synaptic dysfunction, an early event in a range of neurodegenerative disorders.
Calcineurin and protein misfolding/unfolding neurodegenerative diseases
Overactivation of CaN in neurons results in early, reversible damage and late, irreversible damage. Reversal of neuronal injury is only possible in the early stages of disease; if treatment is not started early, proapoptotic Bcl-2 family proteins are activated, leading to apoptosis [2, 23, 102]. Chronically increased cytoplasmic Ca2+ triggers CaN hyperactivation, suppressing phosphorylation of pro-apoptotic protein BAD [13–104]. Scaffolding protein 14-3-3 plays a crucial role in phosphorylation of BAD under normal conditions (Fig. 2). Dephosphorylation of BAD leads to dissociation of BAD from 14-3-3 and interaction between BAD, Bcl-x, and other Bcl-2 family proteins located on the mitochondrial membrane [13, 103, 105]. Interaction of BAD with Bcl-x and Bcl-2 family proteins suppresses the anti-apoptotic activity of BAD (Fig. 2) [105]. Low levels of phosphorylated BAD have been reported in several cell lines exposed to misfolded/unfolded protein aggregates, as well as in the brains of mice serving as models of several types of neurodegenerative diseases [89, 90]. Mukherjee and colleagues showed that pharmacological inhibition of CaN by FK506 in the clinical stage of mice serving as a prion disease model restored BAD phosphorylation and inhibited apoptosis. Inhibition of CaN hyperactivation significantly prevented neuronal loss in prion-infected mice, while increasing their survival rate [7].
Recently, it has been implicated that CaN hyperactivation results in triggering the NFAT signaling cascade leading to Aβ-induced cell death in AD patients (Fig. 1) [106]. NFAT proteins are a family of transcription factors that are important mediators of the immune response. The NFAT complex consists of at least two different components [107], the inducible nuclear transcription factor component, known as NFATn, and the phosphorylated cytoplasmic NFAT protein located in the cytoplasm, known as NFATc. Hyperactivated CaN leads to translocation of NFATc into the nucleus and formation of a complex consisting of NFATc and NFATn, which triggers target gene expression (Fig. 1). Aβ-mediated chronic activation of CaN results in activation of a neuronal isoform of NFAT known as NFATc4 [106]. Continuous NFAT activation and nuclear signaling result in morphological neurodegenerative abnormalities, including neuritic dystrophy, dendrite simplification, and dendritic spine loss [106]. Ni and coworkers showed that isofurane-induced cognitive impairments in aged rats can be rescued with CsA; furthermore, NFATc4 nuclear translocation was inhibited with CsA treatment [108], while transcriptional profiling of neural stem cells identified CaN-NFATc4 signaling as major regulator of neural stem cell biology [109]. CaN inhibition in transgenic mouse model successfully prevented neuritic beading and calcium overload [110].
Tau phosphorylation is regulated by a balance between tau kinase and phosphatase activities. Disruption of this equilibrium is suggested to be the origin of abnormal tau phosphorylation and thereby that might contribute to tau aggregation in AD [111]. It has been shown that the regulatory and catalytic subunits of CaN bind with tau protein, and tau is dephosphorylated; the balance between phosphorylation and dephosphorylation of tau is influenced by calmodulin which tries to impair the binding between CaN subunits and tau [112]. SHSY5Y cells treated with CaN inhibitors produced reduced calcineurin activity and a corresponding increase in extracellular ptau181 [113]. Similarly, Luo and colleagues showed that FK506 significantly enhanced the phosphorylation of tau at Ser-262 (12E8 site), Ser-198, Ser-199, and/or Ser-202 (Tau-1 site) and Ser-396 and/or Ser-404 (PHF-1 site), without affecting total tau [114]. A study using A53T transgenic mice as a model of PD showed that α-synuclein increased CaN phosphatase activity and facilitated NFATc3 translocation to the nucleus in midbrain dopaminergic neurons and cell lines, ultimately leading to apoptosis. Inhibition of CaN with cyclosporin A ameliorated α-synuclein-induced loss of neurons and increased the survival rate of dopaminergic neurons through the suppression of CaN and NFAT signaling pathway [115]. One must be very careful when devising a strategy to inhibit hyperactivated CaN, as reduction of CaN lower than optimum can be neurotoxic; Caraveo and colleagues demonstrated that CaN activity must be delicately balanced to ensure survival when neurons are exposed to high levels of α-synuclein; complete inhibition of CaN resulted in neurotoxicity, while moderate CaN inhibition provided a neuroprotective effect in PD patients [116]. Similarly, CaN downregulation systematically via Cys A or locally through shRNA produced anxiety and depression-like behavior in C57BL/6J mouse. These results suggest that chronic administration of CsA in transplant patients could have significant effects on anxiety and mood and this should be recognized as a potential clinical consequence of treatment to prevent transplant rejection [117].
Some research groups have suggested that CaN inhibition could be used as a therapeutic strategy to treat patients with HD (Fig. 2) [16]. Phosphorylated huntingtin (Htt) positively regulates vesicular transport of neurotrophins such as brain-derived neuronal growth factor (BDNF). In HD patients, compromised Htt protein function results in reduced neurotrophic support and neuronal death [118]. Hyperactivation of CaN results in aberrant dephosphorylation of Htt, dysregulated BDNF transport, and neurodegeneration. Recently, Gratuze and colleagues work using mouse models of HD and cell lines demonstrated that accumulation of mutant Htt protein resulted in tau hyperphosphorylation via down-regulation of CaN [119]. Genetic and pharmacological inhibition of CaN activity prevents neuronal death in mouse models of HD [118, 119]. Furthermore, mutant Htt toxicity was minimized when RCAN1-IL, a CaN regulator, was overexpressed in the ST14A cell line, a model of HD [120].
Takehiro and colleagues found that CaN inhibitor FK506 strongly inhibited accumulation of the scrapie-associated form of prion protein (PrPSc) in both prion-infected cells and mice. Furthermore, CaN inhibitor FK506 treatment of PrPSc infected cells resulted in the elevation of all major autophagy-related proteins such as LC3-II, ATG7 and the complex of ATG5 and ATG12, while autolysosome formation was significantly increased [121]. Adenosine 1 receptor (A1R) has been studied recently in hyphoxic/ischemic model where reduced level of PP2B leads to neuroprotection via α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (also known as AMPA receptor, AMPAR, or quisqualate receptor) [83]. The results described above indicate that CaN plays an important role in neurodegeneration.
Other than immunophilins and immunosuppressants, CaN activity can be inhibited through the genetic manipulation of endogenous regulator of calcineurin RCAN1. Kishi and colleagues have shown that phosphorylation of the RCAN1 leads to the activation of CaN phosphatase activity [122]. The inhibitory activity of RCAN3 overexpression in jurkat cells has been also documented [123]. Gelpi and coworkers shown that genetic inhibition of CaN inhibited the development of LV hypertrophy in TGZ mice [124]; similarly, genetic and pharmacological inhibition of calcineurin corrected the BDNF transport defect in Huntington’s disease mice model [16]. Adenoviral vectored overexpression of RCAN1-4 reduced the cytokines profile in cerebral ischemia mice model [125].
Individuals with Down syndrome (DS) develop the neuropathology of AD, suggesting that the overexpression of genes on chromosome 21, like RCAN1, plays a role in AD. RCAN1 is under the control of a stress response promoter. More than 50 % individuals affected with DS show the neuropathology of AD when they reach their middle age. Individuals with DS display early onset of AD neuropathology and provide an opportunity to research genetic factors associated with AD [126]. Martin and coworkers showed that RCAN1 gene overexpression causes deficits in hippocampal-dependent short- and long-term memory in RCAN1-TG mice [126]. Similarly, RCAN1 overexpression leads to deficits in memory and synaptic plasticity, tau pathology and dysregulation of mitochondrial dynamics in mice [127]. Patel and coworkers demonstrated that RCAN1 overexpression resulted in impaired neurotrophin trafficking via the inhibition of NGF receptors endocytosis. Genetic correction of RCAN1 rescued the NGF-dependent trafficking, neuronal survival and innervations in mice model of DS [128].
Conclusions and future perspectives
Prompt and efficient treatment of major protein unfolding/misfolding-related neurodegenerative disorders is only possible with thorough understanding of the mechanisms underlying neurodegeneration caused by unfolded/misfolded proteins. It is clear from previous investigations that accumulation of misfolded/unfolded proteins results in ER stress and dysregulation of calcium homeostasis. To correct protein processing in the ER, reduce accumulation of unfolded/misfolded proteins, and restore normal cellular function, cells engage an adaptive response known as the UPR. Sustained long-term ER stress leads to early neurodegenerative anomalies such as synaptic dysfunction and hampered axonal transport, which are followed by neuronal death. Prolonged ER stress causes calcium to be released from the ER into the cytoplasm via calcium channels, activating apoptotic signaling pathways. Among the many proteins affected by dysregulated Ca2+ homeostasis is CaN, a key phosphatase in the brain. Recent investigations of neurodegenerative diseases have implicated altered CaN activity in the molecular mechanisms leading to synaptic dysfunction and neuronal apoptosis. Administration of CaN inhibitors such as tacrolimus and cyclosporin A to mice serving as models of various neurodegenerative diseases appears to be therapeutically beneficial. CaN inhibitors have been widely tested as therapeutic drugs in mouse models of AD; the work of Takehiro and colleagues on prion-infected mice and cell line provided encouraging results. Enhanced degradation of PrPsc was achieved through increased autophagic molecules, although the molecular basis of this effect remains unknown. Medina and colleagues showed that inhibition of hyperactivated CaN suppresses TFEB activity and, vice versa, in Hela cells, providing new directions for neurodegeneration research, as TFEB is involved in autophagy and apoptosis signaling pathways originating from lysosomes. Furthermore, Caraveo and colleagues demonstrated that fine tuning the level of CaN in α-synuclein-exposed cells is crucial, because a minor alteration in CaN abundance may change the role of CaN from neuroprotective to neurotoxic in patients with PD, as could also be the case for other neurodegenerative diseases. CaN inhibitor tacrolimus has been widely used to prevent organ rejection by solid organ transplant patients, but recent evidence showing reduced rates of dementia and AD in solid organ transplant patients chronically treated with tacrolimus demonstrated the potential of this drug as a treatment for patients with neurodegenerative diseases [93]. Kim and coworkers demonstrated that the inhibition CaN rescues impaired synaptic plasticity in presenilin 1 M146 V mutant hippocampal neuron cultures [69]. That could prove as a beacon of hope for neurodegenerative disease patients, particularly those affected by AD. Latest studies employing yeast model showed that plasma membrane protein Rch1 negatively regulates cytosolic calcium homeostasis and positively regulated by calcium/calcineurin signaling pathway which might be the case in humans [129]. Further research to investigate the molecular basis of neurodegeneration is required for the development of effective therapeutic strategies for patients with neurodegenerative diseases.
Acknowledgments
The authors are grateful to Professor Giulio Taglialatela, Director, Mitchell Center for Neurodegenerative Diseases, University of Texas Medical Branch for helpful comments during preparation of the manuscript.
Abbreviations
- FAT
Fast axonal transport
- ER
Endoplasmic reticulum
- CaN
Calcineurin
- NFAT
Nuclear factor of activated T cells
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
- ALS
Amyotrophic lateral sclerosis
- FTD
Frontotemporal dementia
- HD
Huntington’s disease
- CJD
Creutzfeldt-Jacob disease
- BAD
Bcl2-associated death promoter
- CREB
cAMP response element-binding
- SERCA
Sarcoplasmic endoplasmic reticulum Calcium adenosine
- CaM
Calmodulin
- IP3R
Inositol 1,4,5-triphosphate receptors
- RyR
Ryanodine receptors
- TFEB
Transcription factor EB
- AMPA
α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid
- BDNF
Brain-derived neuronal growth factor
- UPR
Unfolded protein response
- GluA1
Glutamine A1
- GluA2
Glutamine A2
- trkB
Tropomyosin-related kinase B
- Aβ
Amyloid beta
- MEF2
Myocyte enhancer factor 2
Compliance with ethical standards
Ethics approval and consent to participate
According to the journal’s requirement we hereby declare that it is a review article where no animal or human experiments were conducted; we followed the strict ethical rules according to compilation of already published data on the current topic.
Conflict of interest
All the authors declare that there are no competing interests amongst them.
Funding source
This work was supported by the Natural Science Foundation of China (Project Nos. 31272532 and 31172293).
References
- 1.Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci. 2003;4:49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]
- 2.Mukherjee A, Soto C. Role of calcineurin in neurodegeneration produced by misfolded proteins and endoplasmic reticulum stress. Curr Opin Cell Biol. 2011;23:223–230. doi: 10.1016/j.ceb.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah SZA, Zhao D, Khan SH, Yang L. Unfolded response pathways in neurodegenerative diseases. J Mol Neurosci. 2015;57(4):529–537. doi: 10.1007/s12031-015-0633-3. [DOI] [PubMed] [Google Scholar]
- 4.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 5.Beriault DR, Werstuck GH. Detection and quantification of endoplasmic reticulum stress in living cells using the fluorescent compound, Thioflavin T. Biochimica et Biophysica Acta. 1833;2013:2293–2301. doi: 10.1016/j.bbamcr.2013.05.020. [DOI] [PubMed] [Google Scholar]
- 6.Dineley KT, Kayed R, Neugebauer V, Fu Y, Zhang W, Reese LC, Taglialatela G. Amyloid-beta oligomers impair fear conditioned memory in a calcineurin-dependent fashion in mice. J Neurosci Res. 2010;88(2923–2932):70. doi: 10.1002/jnr.22445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukherjee A, Morales-Scheihing D, Gonzalez-Romero D, Green K, Taglialatela G, Soto C. Calcineurin inhibition at the clinical phase of prion disease reduces neurodegeneration, improves behavioral alterations and increases animal survival. PLoS Pathog. 2010;6(10):e1001138. doi: 10.1371/journal.ppat.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hudry E, Wu HY, Arbel-Ornath M, Hashimoto T, Matsouaka R, Fan Z, Spires-Jones TL, Betensky RA, Bacskai BJ, Hyman BT. Inhibition of the NFAT pathway alleviates amyloid beta neurotoxicity in a mouse model of Alzheimer’s disease. J Neurosci. 2012;32:3176–3192. doi: 10.1523/JNEUROSCI.6439-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cavallucci V, Berretta N, Nobili A, Nisticò R, Mercuri NB, D’Amelio M. Calcineurin inhibition rescues early synaptic plasticity deficits in a mouse model of Alzheimer’s disease. Neuromol Med. 2013;15:541–548. doi: 10.1007/s12017-013-8241-2. [DOI] [PubMed] [Google Scholar]
- 10.Shah SZA, Zhao D, Khan SH, Yang L. Regulatory mechanisms of endoplasmic reticulum resident IP3 receptors. J Mol Neurosci. 2015;56(4):938–948. doi: 10.1007/s12031-015-0551-4. [DOI] [PubMed] [Google Scholar]
- 11.Goto S, Matsukado Y, Mihara Y, Inoue N, Miyamoto E. The distribution of calcineurin in rat brain by light and electron microscopic immunohistochemistry and enzyme-immunoassay. Brain Res. 1986;397:161–172. doi: 10.1016/0006-8993(86)91381-8. [DOI] [PubMed] [Google Scholar]
- 12.Goto S, Matsukado Y, Mihara Y, Inoue N, Miyamoto E. Calcineurin in human brain and its relation to extrapyramidal system. Immunohistochemical study on postmortem human brains. Acta Neuropathol. 1986;72:150–156. doi: 10.1007/BF00685977. [DOI] [PubMed] [Google Scholar]
- 13.Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+ induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- 14.Groth RD, Dunbar RL, Mermelstein PG. Calcineurin regulation of neuronal plasticity. Biochem Biophys Res Commun. 2003;311:1159–1171. doi: 10.1016/j.bbrc.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 15.Gan KJ, Silverman MA. Dendritic and axonal mechanisms of Ca2+ elevation impair BDNF transport in Aβ oligomer-treated hippocampal neurons. Mol Biol Cell. 2015;26:1058–1071. doi: 10.1091/mbc.E14-12-1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pineda JR, Pardo R, Zala D, Yu H, Humbert S, Saudou F. Genetic and pharmacological inhibition of calcineurin corrects the BDNF transport defect in Huntington’s disease. Mol Brain. 2009;2:33. doi: 10.1186/1756-6606-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lehotsky J, Kaplan P, Babusikova E, Strapkova A, Murin R. Molecular pathways of endoplasmic reticulum dysfunctions: possible cause of cell death in the nervous system. Physiol Res. 2003;52:269–274. [PubMed] [Google Scholar]
- 18.Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 19.Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–392. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- 20.Manfredi G, Kawamata H. Mitochondria and endoplasmic reticulum crosstalk in amyotrophic lateral sclerosis. Neurobiol Dis. 2016;90:35–42. doi: 10.1016/j.nbd.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang H, Liu J, Sun S, Pchitskaya E, Popugaeva E, Bezprozvanny I. Calcium signaling, excitability, and synaptic plasticity defects in a mouse model of Alzheimer’s disease. J Alzheimer’s Dis. 2015;45:561–580. doi: 10.3233/JAD-142427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hetz CA, Soto C. Stressing out the ER: a role of the unfolded protein response in prion-related disorders. Curr Mol Med. 2006;6:37–43. doi: 10.2174/156652406775574578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27:6407–6418. doi: 10.1038/onc.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zalk R, Lehnart SE, Marks AR. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem. 2007;76:367–385. doi: 10.1146/annurev.biochem.76.053105.094237. [DOI] [PubMed] [Google Scholar]
- 25.Ferreiro E, Resende R, Costa R, Oliveira CR, Pereira CMF. An endoplasmic-reticulum-specific apoptotic pathway is involved in prion and amyloid-beta peptides neurotoxicity. Neurobiol Dis. 2006;23:669–678. doi: 10.1016/j.nbd.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 26.Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003;22:5435–5445. doi: 10.1093/emboj/cdg537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Credle JJ, Forcelli PA, Delannoy M, Oaks AW, Permaul E, Berry DL, Duka V, Wills J, Sidhu A. α-Synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson’s disease. Neurobiology of Disease. 2015;76:112–125. doi: 10.1016/j.nbd.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 28.Medina DL, Paola SD, Peluso I, Armani A, Stefani DD, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, Ade Matteis M, Ballabio A. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Bio. 2015;17(3):288–299. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tong Y, Song F. Intracellular calcium signaling regulates autophagy via calcineurin mediated TFEB dephosphorylation. Autophagy. 2015;11(7):1192–1195. doi: 10.1080/15548627.2015.1054594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klee CB, Draetta GF, Hubbard MJ. Calcineurin. Adv Enzymol Relat Areas Mol Biol. 1988;61:149–200. doi: 10.1002/9780470123072.ch4. [DOI] [PubMed] [Google Scholar]
- 31.Aramburu J, Rao A, Klee CB. Calcineurin: from structure to function. Curr Top Cell Regul. 2000;36:237–295. doi: 10.1016/S0070-2137(01)80011-X. [DOI] [PubMed] [Google Scholar]
- 32.Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80:1483–1521. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- 33.Molkentin JD. Calcineurin, mitochondrial membrane potential, and cardiomyocyte apoptosis. Circ Res. 2001;88:1220–1222. doi: 10.1161/hh1201.093159. [DOI] [PubMed] [Google Scholar]
- 34.Kissinger CR, Parge NE, Knighton DR, Lewis CT, Pelletier LA, Tempczyk A, Kallish VJ, Tucker KD, Showalter RE, Moomaw EW, Gastinel LN, Habuka N, Chen X, Maldonado F, Barker JE, Bacquet R, Villafranca E. Crystal structure of human calcineurin and the human FKBP12–FK506–calcineurin complex. Nature. 1995;378:641–644. doi: 10.1038/378641a0. [DOI] [PubMed] [Google Scholar]
- 35.Griffith JP, Kim JL, Kim BE, Simchak ND, Thomson JA, Fitzgibbon MJ, Flaming MA, Caron PR, Haiao K, Navia MA. X-ray structure of calcineurin inhibited by the immunophilin—immunosuppressant FKBP12–FK506 complex. Cell. 1995;82:507–522. doi: 10.1016/0092-8674(95)90439-5. [DOI] [PubMed] [Google Scholar]
- 36.Huai Q, Kim HY, Liu Y, Zhao Y, Mondragon A, Liu J, Ke H. Crystal structure of calcineurin–cyclophilin–cyclosporine shows common but distinct recognition of immunophilin–drug complexes. Proc Natl Acad Sci USA. 2002;99:12037–12042. doi: 10.1073/pnas.192206699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin L, Harrison SC. Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin. Proc Natl Acad Sci USA. 2002;99:13522–13526. doi: 10.1073/pnas.212504399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.J Goldberg, H. Huang, Y. Kwon, P. Greengard, A.C. Nairn, J. Kuriyan. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1, Nature 376 (1995) 745–753 [DOI] [PubMed]
- 39.Wang HL, Du YW, Xiang BQ, Lin WL, Wei Q. The regulatory domains of CNA have different effects on the inhibition of CN activity by FK506 and CsA. IUBMB Life. 2007;59(6):388–393. doi: 10.1080/15216540701370721. [DOI] [PubMed] [Google Scholar]
- 40.Voegtli WC, White DJ, Reiter NJ, Rusnak F, Rosenzweig AC. Structure of the bacteriophage Lambda Ser/Thr protein phosphatase with sulfate ion bound in two coordination modes. Biochemistry. 2000;39:15365. doi: 10.1021/bi0021030. [DOI] [PubMed] [Google Scholar]
- 41.Klabunde T, Strater N, Frohlich R, Witzel H, Krebs B. Mechanism of Fe(III)–Zn(II) purple acid phosphatase based on crystal structures. J Mol Biol. 1996;259:737–748. doi: 10.1006/jmbi.1996.0354. [DOI] [PubMed] [Google Scholar]
- 42.Ito A, Hashimoto T, Hirai M, Takeda T, Shuntoh H, Kuno T, Tanaka C. The complete primary structure of calcineurin A, a calmodulin binding protein homologous with protein phosphatases1 and 2. Biochem Biophys Res Commun. 1989;163:1492–1497. doi: 10.1016/0006-291X(89)91148-0. [DOI] [PubMed] [Google Scholar]
- 43.Das AK, Helps NR, Cohen PTW, Barford D. Crystal structure of the protein serine/threonine phosphatase 2C at 2.0 A resolution. EMBO J. 1996;15:6798–6809. [PMC free article] [PubMed] [Google Scholar]
- 44.Hashimoto Y, King MM, Soderling TR. Regulatory interactions of calmodulin-binding proteins: phosphorylation of calcineurin by autophosphorylated Ca2+/calmodulin-dependent protein kinase II. Proc Natl AcadSci USA. 1988;85:7001–7005. doi: 10.1073/pnas.85.18.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mukerjee N, McGinnis KM, Park YH, Gnegy ME, Wang KK. Caspase-mediated proteolytic activation of calcineurin in thapsigargin-mediated apoptosis in SH-SY5Y neuroblastoma Cells. Arch Biochem Biophys. 2000;379:337–343. doi: 10.1006/abbi.2000.1889. [DOI] [PubMed] [Google Scholar]
- 46.Wu HY, Tomizawa K, Matsui H. Calpain-calcineurin signaling inthe pathogenesis of calcium-dependent disorder. Acta Med Okayama. 2007;61:123–137. doi: 10.18926/AMO/32905. [DOI] [PubMed] [Google Scholar]
- 47.Liu F, Grundke-Iqbal I, Iqbal K, Oda Y, Tomizawa K, Gong CX. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem. 2005;280:37755–37762. doi: 10.1074/jbc.M507475200. [DOI] [PubMed] [Google Scholar]
- 48.Winder DG, Mansuy IM, Osman M, Moallem TM, Kandel ER. Genetic and pharmacological evidence for a novel intermediate phase of long-term potentiation suppressed by calcineurin. Cell. 1998;92:25–37. doi: 10.1016/S0092-8674(00)80896-X. [DOI] [PubMed] [Google Scholar]
- 49.Swanson SK, Born T, Zydowsky LD, Cho H, Chang HY, Walsh CT, Rusnak F. Cyclosporin-mediated inhibition of bovine calcineurin by cyclophilins A and B. Proc Natl Acad Sci USA. 1992;89:3741–3745. doi: 10.1073/pnas.89.9.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyata S, Ohkubo Y, Mutoh S. A review of the action of tacrolimus (FK506) on experimental models of rheumatoid arthritis. Inflamm Res. 2005;54:1–9. doi: 10.1007/s00011-004-1318-5. [DOI] [PubMed] [Google Scholar]
- 51.Tedesco D, Haragsim L. Cyclosporine: a review. J Transplant. 2012;230386:1–7. doi: 10.1155/2012/230386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shibasaki F, Hallin U, Uchino H. Calcineurin as a multifunctional regulator. J Biochem. 2002;131:1–15. doi: 10.1093/oxfordjournals.jbchem.a003063. [DOI] [PubMed] [Google Scholar]
- 53.Mansuy IM. Calcineurin in memory and bidirectional plasticity. Biochem Biophys Res Commun. 2003;311:1195–1208. doi: 10.1016/j.bbrc.2003.10.046. [DOI] [PubMed] [Google Scholar]
- 54.Klee CB, Crouch TH, Krinks MH. Calcineurin: a calcium- and calmodulin-binding protein of the nervous system. Proc Natl Acad Sci USA. 1979;76:6270–6273. doi: 10.1073/pnas.76.12.6270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hubbard MJ, Klee CB. Calmodulin binding by calcineurin ligand-induced renaturation of protein immobilized on nitrocellulose. J Biol Chem. 1987;262:15062–15070. [PubMed] [Google Scholar]
- 56.Armstrong DL. Calcium channel regulation by calcineurin, a Ca2+-activated phosphatase in mammalian brain. Trends Neurosci. 1989;12:117–122. doi: 10.1016/0166-2236(89)90168-9. [DOI] [PubMed] [Google Scholar]
- 57.Zhu Y, Yakel JL. Calcineurin modulates G protein-mediated inhibition of N-type calcium channels in rat sympathetic neurons. J Neurophysiol. 1997;78:1161–1165. doi: 10.1152/jn.1997.78.2.1161. [DOI] [PubMed] [Google Scholar]
- 58.Burley JR, Sihra TS. A modulatory role for protein phosphatase 2B (calcineurin) in the regulation of Ca2+ entry. Eur J Neurosci. 2000;12:2881–2891. doi: 10.1046/j.1460-9568.2000.00178.x. [DOI] [PubMed] [Google Scholar]
- 59.Day M, Olson PA, Platzer J, Striessnig J, Surmeier DJ. Stimulation of 5-HT(2) receptors in prefrontal pyramidal neurons inhibits Ca(v)1.2 L type Ca(2+) currents via a PLCbeta/IP3/calcineurin signaling cascade. J Neurophysiol. 2002;87:2490–2504. doi: 10.1152/jn.00843.2001. [DOI] [PubMed] [Google Scholar]
- 60.Heindorff K, Baumann O. Calcineurin is part of a negative feedback loop in the InsP3/Ca2+ signalling pathway in blowfly salivary glands. Cell Calcium. 2014;56:215–224. doi: 10.1016/j.ceca.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 61.Haruhiko Bito, Deisseroth Karl, Tsien Richard W. CREB phosphorylation and dephosphorylation: a Ca2+ and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/S0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- 62.Graef IA, Wang F, Charron F, Chen L, Neilson J, Tessier-Lavigne M, Crabtree GR. Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell. 2003;113:657–670. doi: 10.1016/S0092-8674(03)00390-8. [DOI] [PubMed] [Google Scholar]
- 63.Groth RD, Mermelstein PG. Brain-derived neurotrophic factor activation of NFAT (nuclear factor of activated T-cells)-dependent transcription: a role for the transcription factor NFATc4 in neurotrophin-mediated gene expression. J Neurosci. 2003;23:8125–8134. doi: 10.1523/JNEUROSCI.23-22-08125.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shalizi A, Gaudilliere B, Yuan Z, Stegmuller J, Shirogane T, Ge Q, Tan Y, Schulman B, Harper JW, Bonni A. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science. 2006;311:1012–1017. doi: 10.1126/science.1122513. [DOI] [PubMed] [Google Scholar]
- 65.Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- 66.Gong X, Tang X, Wiedmann M, Wang X, Peng J, Zheng D, Blair LA, Marshall J, Mao Z. Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron. 2003;38:33–46. doi: 10.1016/S0896-6273(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 67.García E, Stracher A, Jay D. Calcineurin dephosphorylates the C-terminal region of lamin in an important regulatory site: a possible mechanism for lamin mobilization and cell signaling. Arch Biochem Biophys. 2006;446:140–150. doi: 10.1016/j.abb.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 68.Chen Y, Holstein DM, Aime S, Bollo M, Lechleiter JD. Calcineurin β protects brain after injury by activating the unfolded protein response. Neurobiol Dis. 2016;94:139–156. doi: 10.1016/j.nbd.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim S, Violette CJ, Ziff EB. Reduction of increased calcineurin activity rescues impaired homeostatic synaptic plasticity in presenilin 1 M146V mutant. Neurobiol Aging. 2015;36:3239–3246. doi: 10.1016/j.neurobiolaging.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tangmansakulchai K, Abubakar Z, Kitiyanant N, Suwanjang W, Leepiyasakulchai C, Govitrapong P, Chetsawang B. Calpastatin overexpression reduces oxidative stress-induced mitochondrial impairment and cell death in human neuroblastoma SH-SY5Y cells by decreasing calpain and calcineurin activation, induction of mitochondrial fission and destruction of mitochondrialfusion. Mitochondrion. 2016 doi: 10.1016/j.mito.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 71.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 72.Wishart TM, Parson SH, Gillingwater TH. Synaptic vulnerability in neurodegenerative disease. J Neuropathol ExpNeurol. 2006;65:733–739. doi: 10.1097/01.jnen.0000228202.35163.c4. [DOI] [PubMed] [Google Scholar]
- 73.Mallucci GR. Prion neurodegeneration: starts and stops at the synapse. Prion. 2009;3:195–201. doi: 10.4161/pri.3.4.9981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y, Zhao D, Pan B, Song Z, Shah SZA, Yin X, Zhou X, Yang L. Death receptor 6 and caspase-6 regulate prion peptide-induced axonal degeneration in rat spinal neurons. J Mol Neurosci. 2015;56(4):966–976. doi: 10.1007/s12031-015-0562-1. [DOI] [PubMed] [Google Scholar]
- 75.Karsten Baumgartel, Mansuy Isabelle M. Neural functions of calcineurin in synaptic plasticity and memory. Learn Memory. 2012;19:375–384. doi: 10.1101/lm.027201.112. [DOI] [PubMed] [Google Scholar]
- 76.Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- 77.Mansuy IM, Mayford M, Jacob B, Kandel ER, Bach ME. Restricted and regulated overexpression reveals calcineurin as a keycomponent in the transition from short-term to long-termmemory. Cell. 1998;92:39–49. doi: 10.1016/S0092-8674(00)80897-1. [DOI] [PubMed] [Google Scholar]
- 78.Malleret G, Haditsch U, Genoux D, Jones MW, Bliss TV, Vanhoose AM, Weitlauf C, Kandel ER, Winder DG, Mansuy IM. Inducible and reversible enhancement of learning, memory and long-term potentiation by genetic inhibition of calcineurin. Cell. 2001;104:675–686. doi: 10.1016/S0092-8674(01)00264-1. [DOI] [PubMed] [Google Scholar]
- 79.Zeng H, Chattarji S, Barbarosie M, Rondi-Reig L, Philpot BD, Miyakawa T, Bear MF, Tonegawa S. Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell. 2001;107:617–629. doi: 10.1016/S0092-8674(01)00585-2. [DOI] [PubMed] [Google Scholar]
- 80.Zhao WQ, Santini F, Breese R, Ross D, Zhang XD, Stone DJ, Ferrer M, Townsend M, Wolfe AL, Seager MA, et al. Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption. J Biol Chem. 2010;285:7619–7632. doi: 10.1074/jbc.M109.057182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Taglialatela G, Hogan D, Zhang WR, Dineley KT. Intermediate and long-term recognition memory deficits in Tg2576 mice are reversed with acute calcineurin inhibition. Behav Brain Res. 2009;200:95–99. doi: 10.1016/j.bbr.2008.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stockwell J, Chen Z, Niazi M, Nosib S, Cayabyab FS. Protein phosphatase role in adenosine A1 receptor-induced AMPA receptor trafficking and rat hippocampal neuronal damage in hypoxia/reperfusion injury. Neuropharmacology. 2016;102:254–265. doi: 10.1016/j.neuropharm.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 84.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/S0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 85.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 86.Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/S0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 87.Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- 88.Benito E, Barco A. CREB’s control of intrinsic and synapticplasticity: implications for CREB-dependent memory models. Trends Neurosci. 2010;33:230–240. doi: 10.1016/j.tins.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 89.Agostinho P, Lopes JP, Velez Z, Oliveira CR. Overactivation of calcineurin induced by amyloid-beta and prion proteins. Neurochem Int. 2008;52:1226–1233. doi: 10.1016/j.neuint.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 90.Reese LC, Zhang W, Dineley KT, Kayed R, Taglialatela G. Selective induction of calcineurin activity and signaling by oligomeric amyloid beta. Aging Cell. 2008;7:824–835. doi: 10.1111/j.1474-9726.2008.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dineley KT, Kayed R, Neugebauer V, Fu Y, Zhang W, Reese LC, Taglialatela G. Amyloid beta oligomers impair fear conditioned memory in a calcineurin-dependent fashion in mice. J Neurosci Res. 2010;88(13):2923–2932. doi: 10.1002/jnr.22445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Spires-Jones TL, Kay K, Matsouka R, Rozkalne A, Betensky RA, Hyman BT. Calcineurin inhibition with systemic FK506 treatment increases dendritic branching and dendritic spine density in healthy adult mouse brain. Neurosci Lett. 2011;487:260–263. doi: 10.1016/j.neulet.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yamamoto-Sasaki M, Ozawa H, Saito T, Rösler M, Riederer P. Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Res. 1999;824:300–303. doi: 10.1016/S0006-8993(99)01220-2. [DOI] [PubMed] [Google Scholar]
- 94.Taglialatela G, Cristiana R, Luca C. Reduced incidence of dementia in solid organ transplant patients treated with calcineurin inhibitors. J Alzheimer’s Dis. 2015;47:329–333. doi: 10.3233/JAD-150065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kingsbury TJ, Bambrick LL, Roby CD, Krueger BK. Calcineurin activity is required for depolarization-induced CREB dependentgene transcription in cortical neurons. J Neurochem. 2007;103:761–770. doi: 10.1111/j.1471-4159.2007.04801.x. [DOI] [PubMed] [Google Scholar]
- 96.Lam BY, Zhang W, Enticknap N, Haggis E, Cader MZ, Chawla S. Inverse regulation of plasticity-related immediate early genes by calcineurin in hippocampal neurons. J Biol Chem. 2009;284:12562–12571. doi: 10.1074/jbc.M901121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphor proteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- 99.Hosaka M, Hammer RE, Sudhof TC. A phospho-switch controls the dynamic association of synapsins with synaptic vesicles. Neuron. 1999;24:377–387. doi: 10.1016/S0896-6273(00)80851-X. [DOI] [PubMed] [Google Scholar]
- 100.Jovanovic JN, Sihra TS, Nairn AC, Hemmings HC, Jr, Greengard P, Czernik AJ. Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J Neurosci. 2001;21:7944–7953. doi: 10.1523/JNEUROSCI.21-20-07944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pleiss MM, Sompol P, Kraner SD, Abdul HM, Furman JL, Guttmann RP, Wilcock DM, Nelson PT, Norris CM. Calcineurin proteolysis in astrocytes: Implications for impaired synaptic function. Biochimica et Biophysica Acta. 1862;2016:1521–1532. doi: 10.1016/j.bbadis.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Klumpp S, Krieglstein J. Serine/threonine protein phosphatases in apoptosis. Curr Opin Pharmacol. 2002;2:458–462. doi: 10.1016/S1471-4892(02)00176-5. [DOI] [PubMed] [Google Scholar]
- 103.Shou Y, Li L, Prabhakaran K, Borowitz JL, Isom GE. Calcineurin mediated Bad translocation regulates cyanide-induced neuronal apoptosis. Biochem J. 2004;379:805–813. doi: 10.1042/bj20031107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang L, Omori K, Suzukawa J, Inagaki C. Calcineurin-mediated BAD Ser155 dephosphorylation in ammonia-induced apoptosis of cultured rat hippocampal neurons. Neurosci Lett. 2004;357:73–75. doi: 10.1016/j.neulet.2003.12.032. [DOI] [PubMed] [Google Scholar]
- 105.Strasser A, O’Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- 106.Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, et al. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nguyen T, Di Giovanni S. NFAT signaling in neural development and axon growth. Int J Dev Neurosci. 2008;26:141–145. doi: 10.1016/j.ijdevneu.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ni C, Li Z, Qian M, Zhou Y, Wang J, Guo X. Isoflurane induced cognitive impairment in aged rats through hippocampal calcineurin/NFAT signaling. Biochem Biophys Res Commun. 2015;460:889–895. doi: 10.1016/j.bbrc.2015.03.083. [DOI] [PubMed] [Google Scholar]
- 109.Moreno M, Fernández V, Monllau JM, Borrell V, Lerin C, de la Iglesia N. Transcriptional profiling of hypoxic neural stem cells identifies calcineurin-NFATc4 signaling as a major regulator of neural stem cell biology. Stem Cell Rep. 2015;5:157–165. doi: 10.1016/j.stemcr.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sun X, Wu Y, Herculano B, Song W. RCAN1 overexpression exacerbates calcium overloading-induced neuronal apoptosis. PLoS One. 2014;9(4):e95471. doi: 10.1371/journal.pone.0095471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Martina L, Latypova X, Wilsona CM, Magnaudeix A, Perrin M-L, Terro F. Tau protein phosphatases in Alzheimer’s disease: The leading role of PP2A. Ageing Res Rev. 2013;12:39–49. doi: 10.1016/j.arr.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 112.Yu D, Tong L, Song G, Lin W, Zhang L, Bai W, Gong H, Yin Y, Wei Q. Tau binds both subunits of calcineurin, and binding is impaired by calmodulin. Biochimica et Biophysica Acta. 2008;1783:2255–2261. doi: 10.1016/j.bbamcr.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 113.Karch CM, Jeng AT, Goate AM. Calcium phosphatase calcineurin influences tau metabolism. Neurobiol Aging. 2013;34:374–386. doi: 10.1016/j.neurobiolaging.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Luo J, Ma J, Da-Yu Y, Fan B, Zhang W, Ling-Hui T, Wei Q. Infusion of FK506, a specific inhibitor of calcineurin, induces potent tau hyperphosphorylation in mouse brain. Brain Res Bull. 2008;76:464–468. doi: 10.1016/j.brainresbull.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 115.Luo J, Sun L, Lin X, Liu G, Yu J, Parisiadou L, Xie C, Ding J, Cai H. A calcineurin- and NFAT-dependent pathway is involved in α-synuclein-induced degeneration of midbrain dopaminergic neurons. Hum Mol Genet. 2014;23(24):6567–6574. doi: 10.1093/hmg/ddu377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Caraveo G, Auluck PK, Whitesell L, Chung CY, Baru V, Mosharov EV, Yan X, Ben-Johny MU, Soste M, Picotti P, Kim H, Caldwell KA, Caldwell GA, Sulzer D, Yue DT, Lindquist S. Calcineurin determines toxic versus beneficial responses to α synuclein. Proc Natl Acad Sci. 2014;111(34):E3544–E3552. doi: 10.1073/pnas.1413201111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mineur YS, Taylor SR, Picciotto MR. Calcineurin downregulation in the amygdala is sufficient to induce anxiety-like and depression-like behaviors in C57BL/6J male mice. Biol Psychiatry. 2014;75:991–998. doi: 10.1016/j.biopsych.2014.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Humbert S, Saudou F. Huntington’s disease: intracellular signaling pathways and neuronal death. J Soc Biol. 2005;199(3):247–251. doi: 10.1051/jbio:2005026. [DOI] [PubMed] [Google Scholar]
- 119.Gratuze M, Noel A, Julien C, Cisbani G, Millot-Rousseau P, Morin F, Dickler M, Goupil C, Bezeau F, Poitras I, Bissonnette S, Whittington RA, Hebert SS, Ciccechetti F, Parker JA, Samadi P, Planel E. Tau hyperphosphorylation and deregulation of calcineurin in mouse model of huntington’s disease. Hum Mol Genet. 2014;24(1):86–99. doi: 10.1093/hmg/ddu456. [DOI] [PubMed] [Google Scholar]
- 120.Ermak G, Hench KJ, Chang KT, Sachdev S, Davies KJ. Regulator of calcineurin (RCAN1-1L) is deficient in Huntington disease and protective against mutant huntingtin toxicity in vitro. J Biol Chem. 2009;284:11845–11853. doi: 10.1074/jbc.M900639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Takehiro N, Satoh K, Ishibashi D, Fuse T, Sano K, Kamatari YO, Kuwata K, Shigematsu K, Iwamaru Y, Takenouchi T, Kitani H, Atarashi NNR. FK506 reduces abnormal prion protein through the activation of autolysosomal degradation and prolongs survival in prion-infected mice. Autophagy. 2013;9(9):1386–1394. doi: 10.4161/auto.25381. [DOI] [PubMed] [Google Scholar]
- 122.Kishi T, Ikeda A, Nagao R, Koyama N. The SCFCdc4 ubiquitin ligase regulates calcineurin signaling through degradation of phosphorylated Rcn1, an inhibitor of calcineurin. Proc Natl Acad Sci. 2007;104(44):17418–17423. doi: 10.1073/pnas.0704951104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mulero MC, Aubareda A, Schlüter A, Pérez-Riba M. RCAN3, a novel calcineurin inhibitor that down-regulates NFAT-dependent cytokine gene expression. Biochimica et Biophysica Acta. 2007;1773:330–341. doi: 10.1016/j.bbamcr.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 124.Gelpi RJ, Gao S, Zhai P, Yan L, Hong C, Danridge LMA, Ge H, Maejima Y, Donato M, Yokota M, Molkentin JD, Vatner DE, Vatner SF, Sadoshima J. Genetic inhibition of calcineurin induces diastolic dysfunction in mice with chronic pressure overload. Am J Physiol Heart Circ Physiol. 2009;297:H1814–H1819. doi: 10.1152/ajpheart.00449.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sobrado M, Ramirez BG, Neria F, Lizasoain I, Arbones ML, Minami T, Redondo JM, Moro MÁ, Cano E. Regulator of calcineurin 1 (Rcan1) has a protective role in brain ischemia/reperfusion injury. J Neuroinflamm. 2012;9(48):1–13. doi: 10.1186/1742-2094-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Martin KR, Corlett A, Dubach D, Mustafa T, Coleman HA, Parkington HC, Merson TD, Bourne JA, Porta S, Arbonés ML, Finkelstein DI, Pritchard MA. Over-expression of RCAN1 causes Down syndrome-like hippocampal deficits that alter learning and memory. Hum Mol Genet. 2012;21(13):3025–3041. doi: 10.1093/hmg/dds134. [DOI] [PubMed] [Google Scholar]
- 127.Wong H, Levenga J, Cain P, Rothermel B, Klann E, Hoeffer C. RCAN1 overexpression promotes age-dependent mitochondrial dysregulation related to neurodegeneration in Alzheimer’s diseas. Acta Neuropathol. 2015;130:829–843. doi: 10.1007/s00401-015-1499-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Patel A, Yamashita N, Ascan M, Bodmer D, Boehm E, Bodkin-Clarke C, Ryu YK, Kuruvilla R. RCAN1 links impaired neurotrophin trafficking to aberrant development of the sympathetic nervous system in down syndrome. Nat Commun. 2015 doi: 10.1038/ncomms10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhao Y, Yan H, Happeck R, Peiter-Volk T, Xu H, Zhang Y, Peiter E, van Oostende Triplet C, Whiteway M, Jiang L. The plasma membrane protein Rch1 is a negative regulator of cytosolic calcium homeostasis and positively regulated by the calcium/calcineurin signaling pathway in budding yeast. Eur J Cell Biol. 2016;95(3–5):164–174. doi: 10.1016/j.ejcb.2016.01.001. [DOI] [PubMed] [Google Scholar]