

Abstract
The composition of the gut microbiota is in constant flow under the influence of factors such as the diet, ingested drugs, the intestinal mucosa, the immune system, and the microbiota itself. Natural variations in the gut microbiota can deteriorate to a state of dysbiosis when stress conditions rapidly decrease microbial diversity and promote the expansion of specific bacterial taxa. The mechanisms underlying intestinal dysbiosis often remain unclear given that combinations of natural variations and stress factors mediate cascades of destabilizing events. Oxidative stress, bacteriophages induction and the secretion of bacterial toxins can trigger rapid shifts among intestinal microbial groups thereby yielding dysbiosis. A multitude of diseases including inflammatory bowel diseases but also metabolic disorders such as obesity and diabetes type II are associated with intestinal dysbiosis. The characterization of the changes leading to intestinal dysbiosis and the identification of the microbial taxa contributing to pathological effects are essential prerequisites to better understand the impact of the microbiota on health and disease.
Keywords: Bacteria, Cytokine, Mucin, Oxidative stress, Bacteriophage, Bacteriocins, Necrotizing enterocolitis, Cancer
Introduction
The gut microbiota can be viewed as an actual body organ contributing to the well-being of the host organism. The trillions of microbes colonizing the gastrointestinal tract influence local and systemic processes such as nutrient transformation [1], vitamin supply [2], maturation of mucosal immunity [3, 4], gut-to-brain communication [5], and even tumor progression [6]. Like other organs, the proper function of the gut microbiota relies on a stable cellular composition, which in the case of the human microbiota consists mainly of bacteria from the phyla Bacteroidetes, Firmicutes, Actinobacteria, and to a lesser extent Proteobacteria [7]. Large shifts in the ratio between these phyla or the expansion of new bacterial groups lead to a disease-promoting imbalance, which is often referred to as dysbiosis. A reduction of microbial diversity and outgrowth of Proteobacteria are cardinal features of dysbiosis [8, 9]. A growing number of diseases is associated with intestinal dysbiosis, which in some cases contributes to disease development or severity. Dysbiosis is a hallmark of inflammatory bowel diseases (IBD) such as ulcerative colitis and Crohn’s disease [10], but also metabolic disorders [11], autoimmune diseases [12], and neurological disorders [13]. Dysbiosis can trigger disease in the first weeks of life as observed in necrotizing enterocolitis [14], during adulthood through the promotion of colorectal cancer [15], or in elderly people as exemplified by Clostridium difficile-associated diarrhea [16].
Unlike infectious microbes, the pathogenicity of specific intestinal bacteria cannot be established through the application of Koch’s postulates given that a major fraction of the microbiota cannot be isolated as pure culture. Therefore, the pathogenic implication of specific microbes in a disease largely relies first on the identification of shifted bacterial populations based on high-throughput DNA sequencing of conserved 16S rRNA genes [17]. The replication of a disease through the transplantation of the gut microbiota from a diseased animal to a healthy one is often used in a second step to confirm the contribution of intestinal dysbiosis to disease. Microbiota transplantation demonstrated the contribution of intestinal microbes, among others, to obesity [18] and atherosclerosis [19] in mice. Nevertheless, despite strong evidence gained from 16S rRNA sequencing and microbiota transplantation, the culpability of specific bacterial groups enriched in a disease state often remains circumstantial. Instead of being true offenders, suspected bacteria can just be bystanders to real pathogens that remain below the threshold of current detection techniques. The extensive mutualism prevailing in the intestinal microbiota strengthens connections between offenders and bystanders. Bacteria producing a broad range of digestive enzymes frequently cross-feed other bacteria harboring limited foraging abilities [20]. Increased liberation of nutrients may promote the parallel outgrowth of harmless and harmful bacteria. Furthermore, the borderline between good and evil is often blurred given that some symbiotic bacteria may become pathogenic when present in larger numbers in the gut. Such bacteria, referred to as pathobionts [21], may be difficult to recognize when their expansion occurs simultaneously to other changes in the gut microbial composition. Beyond the assignment of guilt by association, the discovery of the mechanisms underlying the shifts of microbial groups is instrumental to understand the processes leading to dysbiosis. Accordingly, the identification of factors causing strong shifts in the gut microbiota is pivotal to devise strategies aimed at preventing intestinal dysbiosis.
Between resilience and fragility
Several exogenous and endogenous factors affect the microbial composition of the intestine. The resulting effects range from transient to long-lasting and these effects can scale from harmless to harmful. Often, a single factor is not sufficient to induce dysbiosis as the gut microbiota has an intrinsic resilience, a capacity to adapt to variations in nutrient availability and changing environmental conditions. The combined actions of several factors by contrast can move microbial groups to a tipping point, which eventually burst to vast shifts of pathological significance. The main factors influencing the composition of the gut microbiota are the diet, various drugs, the intestinal mucosa, the immune system, and the microbiota itself. Moderate shifts in microbial composition can then provide a window of opportunity for other aggravating factors to amplify changes in specific bacterial groups to the point of imbalance. Oxidative stress, bacteriophages, and bacteriocins are typical factors exacerbating shifts of the microbiota to the point of dysbiosis (Fig. 1).
Fig. 1.

Factors contributing to intestinal dysbiosis. The gut microbiota is subject to natural variations induced by the changing supply of nutrients, drugs, the immune system, and the intestinal mucosa. The action of stress factors such as oxidative stress, the induction of bacteriophages, and secretion of bacteriocins amplify the changes in microbial composition leading to decreased diversity and outgrowth of specific bacterial taxa
The threshold required to trigger dysbiosis largely depends on the bacterial groups affected. Broad shifts in the main phyla Bacteroidetes and Firmicutes may remain without pathologic consequence, whereas increased amounts of marginal groups may wreak havoc. For example, Enterobacteriaceae normally represent a minor fraction of the gut microbiota [22]. Bacteria from the family of Enterobacteriaceae can expand rapidly consecutive to changes in oxidative conditions of the gut such as occurring during inflammation [9, 23]. Because of the pyrogenicity of Enterobacteriaceae lipopolysaccharide (LPS), outgrowth of this bacterial family usually intensifies an ongoing inflammatory response.
In addition to the main factors introduced here above, additional parameters such as temperature, atmospheric pressure, and oxygen partial pressure also influence the microbial composition of the gut. For example, the exposure of mice to a low temperature of 6 °C increased intestinal Firmicutes levels at the cost of Bacteroidetes and the resulting cold-adapted microbiota increased energy expenditure [24]. The relative abundance of Firmicutes was also increased in human subjects living at high altitudes [25], although the impact of altitude on the microbiota may be difficult to distinguish from the effects of lower ambient temperatures and different dietary habits. A group of mice embarked on a 13-day space flight on the Space Shuttle Atlantis yielded the first data about the gut microbiota in micro-gravity. The analysis of microbial composition revealed only little changes at the phylum level, but some variations in Clostridiales and Lactobacillales at the order level [26]. Space flight also caused loss of body weight and decreased water intake, meaning that these alterations may also account for the changes in the gut microbiota reported.
Nutrition
The diet is a major element affecting the intestinal microbiota. Natural variations in food intake cause transient changes in microbial composition, although predominant components such as meat, fish, and fibers have durable effects on the microbiota and leave typical signatures characterized by shifts in specific bacterial groups [27]. Changing food composition as well as food shortage or oversupply affect the gut microbiota. The absence of nutrients in the gut occurring in parenteral feeding increases the levels of Proteobacteria, which promote inflammation at the mucosal wall and eventually cause a breakdown of the epithelial barrier [28]. Excess supply of nutrients leads to obesity, which is associated with dysbiosis and inflammatory metabolic disorders. Obesity is characterized by decreased microbial diversity [29] and over-representation of Firmicutes (Fig. 2) as observed in ob/ob mice [18, 30] and in obese humans [31, 32]. A lower ratio of Bacteroidetes to Firmicutes results in a higher release of LPS into the circulation [33]. Higher LPS levels contribute to a state of chronic low-grade inflammation occurring in obesity (Fig. 2a). In mice, elevated levels of circulating LPS initiate weight gain and up-regulate markers of inflammation to a similar extent as a high-fat diet [34]. Metabolic endotoxemia is further enhanced by increased permeability of the gut wall that is caused by a high-fat/high-sugar diet through increasing levels of adherent-invasive Escherichia coli, which infiltrates the intestinal epithelium thereby decreasing mucus thickness [35, 36]. The diet usually is a combination of protein, fat and carbohydrates, and therefore, the isolated effect of each macronutrient on the microbiota in vivo is not easily determined. But diets rich in one or two of these types of food provide valuable clues about their respective influences.
Fig. 2.
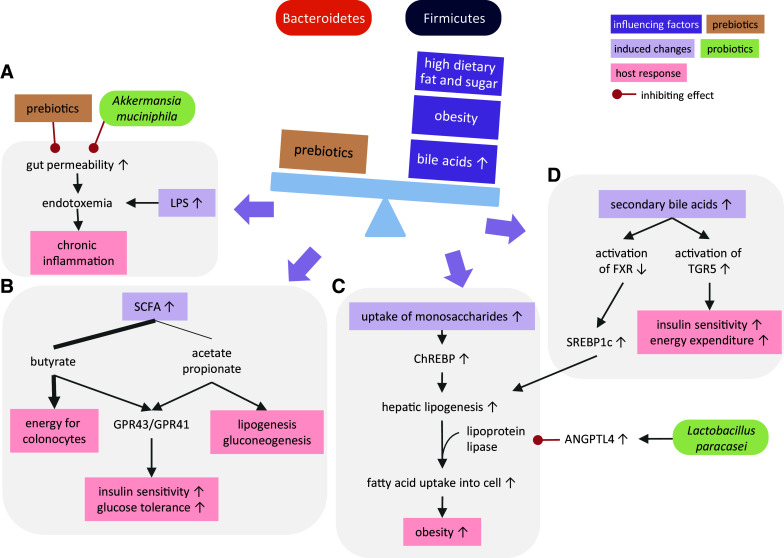
Consequences of nutritionally induced imbalance between Firmicutes and Bacteroidetes. Obesity, high dietary fat and sugar intake and an enlarged bile acid pool decrease the Bacteroidetes to Firmicutes ratio. Changes in this ratio affect chronic inflammation, and metabolic changes related to energy supply to colonocytes, lipogenesis, gluconeogenesis, insulin sensitivity and thereby glucose tolerance. Bacterial LPS (a), SCFA (b), increased monosaccharide uptake (c) and secondary bile metabolisms (d) are key mediators of such metabolic adaptations. ANGPTL4 angiopoietin-like factor IV, FXR farnesoid X receptor, TGR5 G-protein-coupled bile acid receptor, SCFA short chain fatty acid, GPR43/GPR41 G-protein-coupled receptors 43/41, LPS lipopolysaccharide, ChREBP carbohydrate response element-binding protein, SREBP1c sterol regulatory element-binding protein 1c
Proteins
In the long term, high uptake of animal proteins, amino acids and fats increases the relative amounts of Bacteroides, whereas low protein and elevated carbohydrate ingestion raises Prevotella levels [37, 38]. But short-term bursts of high-protein intake do not necessarily yield the same effects. In obese men, the consumption of a protein-rich diet did not affect the abundance of Bacteroides, but the Roseburia/Eubacterium rectale group of bacteria was reduced probably due to lower carbohydrate intake [39]. In rats, feeding with a high-protein diet is associated with lower contents of Clostridium species and Faecalibacterium prausnitzii, while Bacteroides do not increase in parallel [40]. Whereas the microbial changes induced by high-protein consumption are rather moderate, the changes in fermentation products are more evident. A high-protein diet increases the production of branched-chain fatty acids, but also the production of potentially toxic substances such as sulfide, ammonia and N-nitroso compounds [27, 39, 41]. With an excess dietary intake of protein and amino acids, also the synthesis of nitric oxide increases [42]. This antimicrobial product strongly influences the gut microbiota, and increased NO levels measured in obese patients likely contribute to the development of an obesity-associated microbiota [42–44].
Fats
A high fat intake induces remarkable changes in the gut microbiota composition. The overall diversity decreases together with the relative abundance of Bacteroidetes, whereas the relative abundance of Firmicutes increases [45]. Even structural features such as the degree of fatty acid saturation imprint the microbiota. Feeding unsaturated fats to mice increased Actinobacteria, lactic acid bacteria and Akkermansia muciniphila creating a microbial composition that protected from weight gain and white adipose tissue inflammation [46]. Interestingly, feeding mice with saturated fat resulted in a higher production of LPS and higher activation of Toll-like receptor (TLR)-4 and TLR2 than feeding with unsaturated fat [46].
High-fat diet also influences the gut microbiota indirectly by increasing the pool of bile acids. After emulsification of dietary lipids, the majority of bile acids is reabsorbed in the distal ileum. Non-absorbed bile acids strongly influence the microbial growth by creating an environment of low pH and strong antimicrobial activity [47]. Feeding rats with cholic acid leads to a microbial composition resembling the obesity pattern of low Bacteroidetes to Firmicutes ratio [48]. Considering the stimulatory effect of high-fat diet on bile acids in the large intestine in mice [49], bile acids likely contribute to the impact of high fat intake on obesity-related dysbiosis. Furthermore, bile acids are signaling molecules binding to the nuclear hormone farnesoid X receptor (FXR) and the G-protein-coupled bile acid receptor TGR5. Binding to FXR not only regulates bile acid synthesis but also influences lipid, glucose and energy homeostasis [50]. In the liver, FXR inhibits the induction of the sterol regulatory element-binding protein SREBP1c, thus inhibiting lipogenesis and decreasing the risk of steatosis. TGR5 signaling induces the production of glucagon-like peptide (GLP)-1 in the intestine which improves insulin sensitivity. By increasing mitochondrial activity in brown adipose tissue and oxidative phosphorylation in muscle, TGR5 activation also elevates energy expenditure [51, 52]. Gut bacteria regulate bile acid receptor signaling by converting primary bile acids into secondary bile acids that show different binding affinities. Especially bacteria of the phylum Firmicutes have 7α-dehydroxylation activity to turn cholic and chenodeoxycholic acid into deoxycholic and lithocholic acids, which have a lower binding affinity for FXR, but a higher affinity for TGR5 [53] (Fig. 2d).
Fibers
Fibers have a direct effect on the microbiota by reaching the colon due to their indigestibility and feeding microbial fermentation. A diet rich in plant polysaccharides promotes the growth of Bacteroidetes over Firmicutes [54]. Interestingly, a gut microbiota with an increased Firmicutes to Bacteroidetes ratio has a higher capacity to extract energy from the diet by providing more enzymes for the breakdown of dietary polysaccharides [18, 55], thereby increasing the uptake of monosaccharides and short chain fatty acids (SCFA) by the intestinal mucosa. This process maximizes nutrient utilization but in case of excess food supply also maximizes energy storage. Microbially released monosaccharides are transferred to the liver via the portal vein and activate the carbohydrate response element-binding protein ChREBP, leading to increased transcription of several genes involved in de novo hepatic lipogenesis [56, 57], thus augmenting lipid transfer to fat stores in peripheral tissues (Fig. 2c). The increased intestinal absorption of the SCFA butyrate, acetate and propionate provides additional energy for diverse tissues (Fig. 2b). Butyrate is mainly used by the colonocytes and stimulates their proliferation and differentiation [58]. Acetate fuels lipogenesis in peripheral tissues, especially muscle, whereas propionate enters gluconeogenesis in the liver [58]. A higher production of SCFA by obesity-associated microbiota might be one factor contributing to higher triglyceride deposition in fat tissues as well as in the liver [59]. In addition to their caloric contribution SCFA activate metabolic pathways by acting as ligands to the G-protein-coupled receptors GPR41 and GPR43 (also known as free fatty acid receptors 3 and 2) [60, 61]. GPR41 and GPR43 activation is associated with adipose tissue expansion and inflammatory processes, although the outcome of this activation as being protective or causative remains unclear (as reviewed in [62]). Activation of GPR41 and GPR43 also elevates leptin levels in adipocytes, which results in increased insulin sensitivity and higher satiety [63, 64]. GPR43 signaling in intestinal L-cells increases production of GLP-1 that improves glucose tolerance [65]. Acetate and propionate are the main ligands activating GPR43 in adipose tissue and immune cells as butyrate mainly serves as energy source for colonocytes and relatively small amounts reach the periphery [66]. In dysbiosis related to obesity, SCFA profiles change consecutive to decrease in the ratio between Bacteroidetes, producing high amounts of acetate and propionate, and Firmicutes, mainly producing butyrate [67]. Therefore, decreased acetate and propionate production by the microbiota likely reduces GPR43 signaling.
The question raises whether a balanced microbiota can be restored through prebiotic and probiotic supplementation. Prebiotics directly modulate the microbiota and entail reduced gut permeability and endotoxemia, thus reducing inflammation [47, 68, 69]. These changes are linked to higher levels of GLP-2 which reduces gut permeability [69]. An intake of the prebiotic oligofructose shifts the composition of the gut microbiota towards a lean pattern by increasing Bacteroidetes and reducing Firmicutes in ob/ob mice and in rats genetically prone to develop obesity and insulin resistance [70, 71]. Probiotics that induce the secretion or lower the suppression of angiopoietin-like factor IV (ANGPTL4, also known as fasting-induced adipose factor) have a beneficial effect on the lipid metabolism in adipocytes. ANGPTL4 inhibits lipoprotein lipase, which hydrolyses triglycerides from lipoproteins for the fatty acid uptake into the cell. Interestingly, germ-free mice deficient in ANGPTL4 lose their protection from diet-induced obesity [72]. Supplementation of mice with the probiotic Lactobacillus paracasei increases circulating levels of ANGPTL4 and reduces body fat [73] (Fig. 2c). Akkermansia muciniphila is another species that proved to reduce obesity when supplemented to mice [74, 75]. While this species might cause increased severity in colitis models [76, 77], it has a protective effect in obese mice by thickening the mucus layer, thereby decreasing gut permeability, reducing endotoxemia and preventing inflammation [75] (Fig. 2a).
Carbohydrates
The processing of complex plant polysaccharides, such as pectins, xylans and fructans, requires a battery of endo- and exoglycosidases featuring activities capable of releasing monosaccharides such as rhamnose, galacturonic acid, arabinose, xylose, fructose and glucose [78]. By contrast, the utilization of intestinal mucin glycans requires different activities consisting of galactosidases, N-acetylglucosaminidases, N-acetylgalactosaminidases, fucosidases and sialidases. The structural differences between dietary carbohydrates and intestinal glycans and the corresponding need for different processing machineries have pushed bacteria to specialize for the utilization of limited subsets of carbohydrates. The processing of complex carbohydrates often relies on cooperative actions between distinct bacterial taxa. In addition to enabling mutualistic interactions, the cleavage of complex carbohydrates and release of monosaccharides in the gut lumen also generate opportunities for bacteria, which lack carbohydrate-processing enzymes. For example, E. coli does not express any glycosidase capable of degrading complex carbohydrates, but it is an avid consumer of the monosaccharides N-acetylglucosamine, N-acetylneuraminic acid (Neu5Ac) and fucose [79]. Accordingly, intestinal E. coli and other Enterobacteriaceae respond to the presence of specific monosaccharides by increasing proliferation and changing the expression of virulence factors [80, 81].
The capacity to cleave sialic acid, such as Neu5Ac and N-glycolylneuraminic acid (Neu5Gc), is restricted to a limited number of bacterial taxa [82]. Nan gene clusters encoding sialidases, transporters and catabolic enzymes enable the release of sialic acid from intestinal glycans and its utilization as carbon source. Some Bacteroides species, such as Bacteroides fragilis, express fully operational nan clusters, whereas others, such as Bacteroides thetaiotaomicron [83] only express sialidases but lack transporters mediating the uptake of free sialic acid. Sialic acid liberated in this way is accessed by other bacteria that express transporters enabling the uptake of the sugar. This type of cross-feeding is a common mechanism prevailing in the intestinal environment. Monosaccharides released from intestinal glycans can therefore be utilized by bacteria devoid of glycosidases and mediate a strong proliferating response thereby leading to dysbiosis. Antibiotic treatment has been shown to disturb microbiota and lead to increased liberation of sialic acid, which fuels the expansion of the pathogens Salmonella enterica serovar Typhimurium and Clostridium difficile in a mouse model [84]. Similarly, the outgrowth of E. coli and exacerbation of intestinal inflammation occurring after dextran sulfate sodium ingestion was shown to depend on the release of sialic acid from intestinal α2,3-linked sialylated glycans [85]. As mentioned in the introduction to this review, the intricate interactions between bacterial taxa ranging from mutualistic to parasitic networks complicate the identification of the mechanisms underlying dysbiosis.
Drugs
Oral administration is the most frequently applied route of uptake for drugs. The convenience of this path enables the regular uptake of drugs without medical intervention, increasing the exposure of the gut microbiota to drugs and thereby promoting dysbiosis.
Non-steroidal anti-inflammatory drugs
Conventional non-steroidal anti-inflammatory drugs, such as aspirin, ibuprofen and naproxen, affect the intestinal microbial composition when taken daily over months, as shown by increased abundance of Bacteroidaceae and Enterobacteriaceae [86]. Because non-steroidal anti-inflammatory drugs cause stomach ulcers, proton-pump inhibitors are often prescribed in combination to alleviate these side effects on the gastric and small intestinal mucosa. Proton-pump inhibitors themselves have been reported to alter the gut microbiota, which contributes to increased risk for C. difficile-associated diarrhea [87] and hepatic encephalopathy in cirrhotic patients [88]. The impact of drugs on intestinal microbes underlines the confounding importance of medications when associating diseases with intestinal dysbiosis. For example, the hepatic gluconeogenesis inhibitor metformin is a standard medication used in the treatment of type 2 diabetes. As shown recently, the uptake of metformin affects the composition of the gut microbiota by elevating E. coli levels [89]. Accordingly, it is essential to take in account the impact of medications on intestinal microbes when addressing possible correlations between changes in the gut microbiota in chronic disorders.
Antibiotics
Through their antibacterial activity antibiotic drugs have an intrinsic potential in promoting intestinal dysbiosis. Most orally administered antibiotics will alter the gut microbiota, albeit transiently for the duration of treatment. Some antibiotics however induce long-lasting changes in the gut microbiota. Whereas several antibiotics, such as amoxicillin, do not have any significant long-term impact on the gut microbiota, treatment of children with macrolide antibiotics lead to long-lasting decrease in Firmicutes and Actinobacteria with concomitant increase in Bacteroidetes and Proteobacteria [90]. Similarly, treatment of adults with ciprofloxacin decreases gut microbial diversity transiently but also leaves a long-lasting signature characterized by increased abundance of Gram-positive aerobes [91]. Repeated exposure to antibiotics can destabilize the gut microbiota and promote the outgrowth of antibiotic-resistant pathogenic bacteria, as observed through the development of C. difficile-associated diarrhea in elderly people [92]. In addition to their expected antibiotic effects, some antibiotic drugs also exert an eubiotic action [93] by promoting the expansion of beneficial bacteria through the suppression of pathobionts. Such an eubiotic effect is typical for rifaximin, which contributes to increasing the gut microbial diversity in IBD patients [94] and also improves symptoms of irritable bowel syndrome [95].
Microbial regulation of drugs
As outlined here above, several drugs affect the gut microbiota. The reverse is true, too. Intestinal bacteria can metabolize drugs and thereby modify their bioavailability to the host [96]. Just to name few examples, the cholesterol-lowering drug simvastatin [97] and the glucocorticoid agonist prednisolone [98] are modified through multiple bacterially encoded enzymes present in the gut. The topoisomerase I inhibitor irinotecan applied in cancer chemotherapy is inactivated by glucuronidation in the liver. This modification is reversed in the gut through the action of bacterial glucuronidases, which re-activate the drug and increase intestinal toxicity [99]. Beyond their interactions with the gut microbiota, several drugs also affect the intestinal mucosa and its barrier function [100]. The complex interplay between drugs, the microbiota, the intestinal mucosa, and the immune system underline the importance of a comprehensive approach when unravelling the mechanisms underlying intestinal dysbiosis.
Intestinal mucosa
Mucins
The gastrointestinal tract is lined with mucus secreted by goblet cells, thereby protecting the epithelium from a direct contact with the microbiota. In addition to building a physical barrier, the intestinal mucus is a source of nutrients for intestinal bacteria that can liberate carbohydrates from the glycan chains of mucins. Several bacterial groups, including for example Akkermansia muciniphila and Bacteroides thetaiotaomicron, express carbohydrate hydrolases as part of polysaccharide-utilization loci, which confer the ability to extract and metabolize carbohydrates from the intestinal mucus. The composition and thickness of this mucus varies along the intestinal tract, being thin and patchy in the ileum, but thick and stratified in the colon where the bulk of the microbiota resides. Glycoproteins of the mucin family are the main constituents of the intestinal mucus. Mucins carry dense arrays of O-linked glycan chains featuring fucosylated and sialylated structures. The glycosylation pattern of mucins varies along the intestinal segments, with fucosylation being prominent in the ileum and decreasing in the colon, whereas the extent of sialylation increases from the ileum to the distal colon [101]. The distribution of mucins also varies along the gastrointestinal tract. Especially the gel-forming mucins MUC2, MUC5AC, MUC5B and MUC6, which represent the main constituents of the intestinal mucus in humans, are differentially expressed, with MUC5AC and MUC6 being mainly found in the stomach mucus and MUC2 being mainly found in the colon [102].
The human colon secretes about 200 ml of mucus daily. This amount is largely controlled through the transcriptional regulation of MUC2 expression. Multiple factors including the bacterial products LPS and lipoteichoic acid, cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-4 and IL-13, and hormones such as vasoactive intestinal peptide increase MUC2 transcription [103, 104]. Mucin glycosylation also changes under the influence of hormones and cytokines produced during inflammation [105]. Bacterial LPS and the cytokine IL-23 induce the expression of the fucosyltransferase FUT2 in the small intestine, which increases the fucosylation of intestinal mucins [81, 106]. The resulting changes in glycosylation alter the supply of carbohydrates available to bacteria utilizing mucin glycans as carbon source, thereby changing the microbiota composition. Increased availability of specific carbohydrates also affects the expression of virulence factors by pathogens, as shown by the repression of LEE virulence genes by enterohaemorrhagic E. coli mediated by increased concentration of free fucose in the colon [80].
Glycans
The ability to extract carbohydrates from mucin glycans is concentrated in bacterial groups, which express a vast set of hydrolase enzymes and transporters enabling the utilization of monosaccharides as carbon sources. Among the bacterial phyla of the human gut, Bacteroidetes express the largest carbohydrate-fermenting machineries. Several Firmicutes, such as Ruminococcus intestinalis, R. gnavus and R. flavefaciens also express more than 100 carbohydrate-degrading enzymes per genome and are capable of digesting mucin glycans [107]. By contrast, members of Proteobacteria, such as Enterobacteriaceae have very limited ability to degrade intestinal mucins. Among Actinobacteria, several Bifidobacterium spp. are specialized at fermenting complex fucosylated oligosaccharides [108], which explains their prominence in the gut of breastfed infants. Akkermansia muciniphila, a member of the Verrucomicrobiota phylum, commonly found in the gut microbiota is another microbe specialized in the utilization of intestinal mucins as carbon source [109]. Carbohydrate-fermenting machineries are decisive for the maintenance of specific bacterial groups in the gut, as demonstrated by the dramatic drop in fitness of a mutant B. thetaiotaomicron lacking polysaccharide-utilization loci involved in the metabolism of mucin O-glycans [110].
Adhesion to intestinal glycans
Bacterial adhesion is another parameter influenced by variations of the carbohydrate landscape in the intestinal mucosa. Several bacteria express adhesins, fimbriae and pili carrying carbohydrate-binding domains. Lactobacilli for example rely on a family of mucus-binding proteins to colonize the gastrointestinal tract [111]. In addition to commensals, pathogenic bacteria such as Campylobacter jejuni express adhesins that bind to fucosylated epitopes, such as blood group antigens H2, Lewis-b, Lewis-y and Lewis-x [112], which are exposed on intestinal mucins and epithelial cells. Changes in the density of fucosylated glycans or the passage of soluble fucosylated molecules, such as milk oligosaccharides [113] in the intestinal lumen alter the adhesion of C. jejuni to the intestinal mucosa and force its elimination from the intestinal tract. The importance of intestinal carbohydrates in regulating the binding of microbes and even controlling their tissue and animal tropism is illustrated by the binding specificity of E. coli K99. This strain expresses fimbriae, which recognize gangliosides terminated with α2,3-linked Neu5Gc [114]. This ligand is richly expressed in the intestinal mucosa of young piglets, which are the common targets of E. coli K99, whereas adult pigs, expressing gangliosides terminated with α2,3-linked Neu5Ac, are resistant to E. coli K99 infection. The role of glycosylation in conferring binding to the intestinal mucosa is not limited to bacteria. Several bacterial toxins of type AB5, such as cholera, shiga and pertussis toxins, enter cells after binding to surface carbohydrates. The SubAB toxin secreted by Shiga toxigenic E. coli for example binds to Neu5Gc-containing glycans on human gut epithelial cells [115]. Human cells cannot synthesize Neu5Gc, but this carbohydrate is incorporated on human glycans after ingestion of food rich in Neu5Gc such as red meat [116]. The modulation of SubAB susceptibility through the assimilation of dietary Neu5Gc shows that nutrients affect the composition of the intestinal mucosa and thereby the risk for disease. Changes in intestinal glycosylation can also alter the local distribution of bacteriophages, which express carbohydrate-binding proteins [117]. In addition to the diet, genetic polymorphisms related to intestinal glycans also have profound effects on the composition of the gut microbiota. For example, hypomorphic alleles leading to differential expression of the α1–2 fucosyltransferase FUT2 enzyme confer susceptibility to Crohn’s disease [118]. Finally, the microbiota itself also regulates intestinal mucin secretion and glycosylation, as outlined by the different glycosylation of MUC2 produced in conventionally raised mice and germ-free mice [119].
Immunity
The immune system enables a symbiotic relationship with commensal microbiota by maintaining a non-inflammatory homeostasis. This state of tolerance relies on multiple mechanisms such as a physical mucus barrier minimizing the contact to the epithelium, and the secretion of antimicrobial proteins and immunoglobulin A [120]. Despite the absence of inflammation, the immune system constantly senses and contains the gut microbiota. Each component of the immune system exerts pressure on portions of the gut microbiota (Fig. 3). For example, the absence of immunoglobulin A yields a strong expansion of anaerobic bacteria, especially mucosa-adherent segmented filamentous bacteria (SFB) of the phylum Firmicutes [121] (Fig. 3d). Components of the innate immune system, such as TLR, nucleotide oligomerization domain (NOD) proteins, and the inflammasome also affect the bacterial composition of the gut.
Fig. 3.
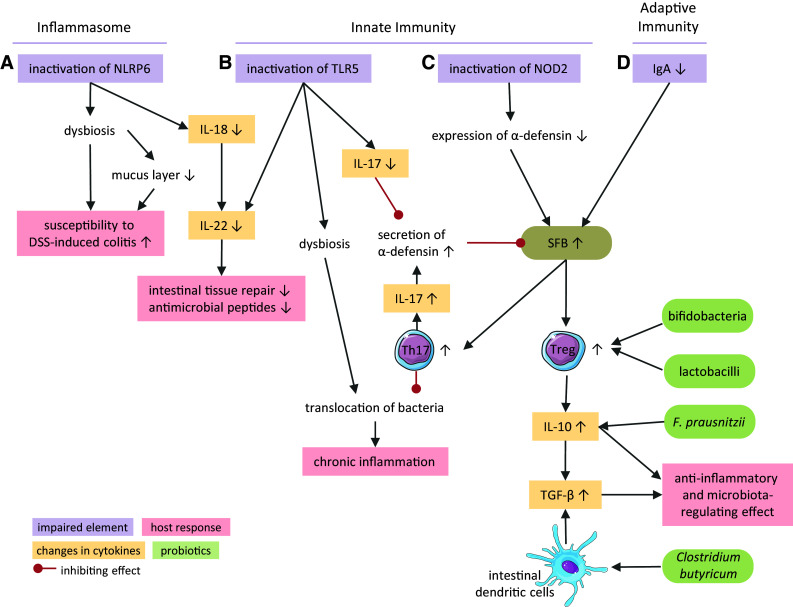
Mechanisms of immune regulation of the gut microbiota. Elements of the inflammasome (a), the innate (b, c) and adaptive immune systems (d) control the gut microbiota composition. Interplay between cytokines, immune cells, bacterial groups, and the intestinal environment affects inflammation, tissue repair, and secretion of antimicrobial peptides. TLR5 Toll-like receptor 5, NOD2 nucleotide oligomerization domain 2 receptor, NLRP6 NOD-like receptor family pyrin domain containing 6, IgA immunoglobulin A, IL interleukin, DSS dextran sulfate sodium, SFB segmented filamentous bacteria, Th17 cells T helper 17 cells, Treg cells regulatory T cells, TGF-β transforming growth factor beta
Inflammasome
Inactivation of the NOD-like receptor family pyrin domain containing 6 (NLRP6) protein, which is a component of the inflammasome complex, results in the expansion of Prevotella spp. and TM7 bacteria [122] (Fig. 3a). These microbial changes render mice more susceptible to dextran sulfate sodium-induced colitis and intestinal infections. The higher susceptibility is enhanced by impaired mucus secretion from goblet cells in NLRP6-deficient mice leading to a reduced mucus layer [123]. Interestingly, mucus reduction is directly caused by the altered microbiota and not by NLRP6-deficiency as it is transferable to wild-type mice by co-housing. NLRP6 activation mediates the secretion of IL-18 via caspase-1. The role of IL-18 in intestinal homeostasis is still controversial. It acts as a pro-inflammatory cytokine that suppresses mucin production by inhibiting the maturation of goblet cells, thus promoting colitis as typically seen in ulcerative colitis [124, 125]. In contrast, IL-18 also down-regulates IL-22-binding protein, which enables IL-22 to induce intestinal tissue repair and expression of antimicrobial peptides [126, 127].
Innate immunity
TLR5, the pattern recognition receptor that recognizes flagellin on the epithelial surface, plays a major role in maintaining the balance of the microbiota. Apart from stimulating IL-8 and TNFα secretion in epithelial cells and monocytes [128, 129], TLR5 signaling also induces the expression of IL-22 and IL-17 in the mucosa [130]. Upon inactivation of TLR5, dysbiosis with altered abundances of more than 100 phylotypes develops (Fig. 3b), thereby promoting several features of the metabolic syndrome including obesity and insulin resistance [131]. Microbial transplantation from TLR5-deficient mice to wild type mice confirmed the causative role of dysbiosis in the development of the metabolic syndrome. TLR5 deficiency can also cause a bloom in Enterobacteriaceae, especially E. coli, which results in spontaneous colitis [132, 133].
The NOD2 receptor, expressed in monocytes and Paneth cells [134], regulates commensal gut community by restricting the number of bacteria and the colonization by pathogens, especially in the terminal ileum [135]. Crohn’s disease is associated with polymorphisms in the NOD2 gene [136, 137]. Defects in the NOD2 receptor lower the expression of α-defensin in Paneth cells [138] (Fig. 3c). Loss of α-defensin increases the ratio of Firmicutes to Bacteroidetes. In line with this result, expression of human α-defensin in mice decreases the abundance of SFB, which belong to the phylum Firmicutes, and decreases the numbers of IL-17-producing Th17 cells in the lamina propria [136]. In the colon, SFB are located close to the epithelium [139] and are instrumental in initiating antimicrobial defense, for example by promoting the development of Th17 cells. The expression of IL-17 in turn increases α-defensin secretion, which inhibits the expansion of SFB. Accordingly, deletion of the IL-17 receptor leads to a similar dysbiosis as seen with defects in the NOD2 receptor [140]. SFB also induce the development of regulatory T (Treg) cells [141]. Treg cells maintain a mutualistic interaction with the microbiota by secreting anti-inflammatory IL-10 and transforming growth factor-β (TGF-β) [142]. IL-10 has a profound effect on the microbiota composition. Mice deficient in IL-10 have increased numbers of Verrucomicrobia, Bacteroidetes, and Proteobacteria as characterized by a 100-fold increase in E. coli [143]. These bacterial shifts are accompanied by inflammation in the caecum and colon [144]. The beneficial influence of probiotics like lactobacilli and bifidobacteria comprises the ability to induce Treg cells and thereby IL-10 secretion [145]. Also Faecalibacterium prausnitzii, which is less abundant in Crohn’s disease patients than in healthy subjects, exerts its anti-inflammatory effects partially via the elevation of IL-10 production [146]. TGF-β also suppresses an inflammatory response and mediates immune tolerance. Its production is not limited to Treg cells, but occurs in various cells of the intestinal mucosa including intestinal dendritic cells (DC) [147]. As IL-10, TGF-β maintains homeostasis of the gut microbiota by regulating microbial composition. In the absence of DC-specific TGF-β signaling, members of Enterobacteriaceae, especially E. coli, are significantly enriched [148]. The probiotic Clostridium butyricum is able to induce TGF-β signaling in DC, which in turn induces Treg cell generation [147].
Immune modulation by dysbiosis
While a dysbiotic gut community is a hallmark of several inflammatory diseases, dysbiosis in turn also triggers mechanisms that unbalance the intestinal homeostasis and cause inflammation. The translocation of bacteria across the gut epithelium increases in dysbiosis [149]. Small numbers of translocated commensal bacteria, as they occur in a healthy human gut, are removed by the action of Th1 and Th17 cells that are particularly induced by polysaccharides of Bacteroides spp. [150]. and mucosa-adherent SFB [151]. But high numbers of invading bacteria continuously activate TLRs and elicit an overexpression of pro-inflammatory cytokines, which damage the gut epithelium and lead to chronic intestinal inflammation [152]. Chronic inflammation is associated with several metabolic disorders such as autoimmune diabetes. Strikingly, higher SFB levels as found in MyD88-deficient mice protect mice of a diabetic genotype from developing the disease indicating that microbiota exert both inhibiting and promoting effects [153–156].
A disturbed microbiota also affects the maturation of the innate immune system as gut bacteria per se are a driving force in that process. Without microbiota, the function of neutrophils and DC is impaired, displaying reduced killing of pathogens and reduced secretion of type I interferons (IFN-I) and IL-15, respectively [157, 158]. Already the development of myeloid cells in the bone marrow is delayed in the absence of microbiota [159]. This delay impairs the clearance of systemic infections and increases the susceptibility to allergies [158–160]. Disturbances in the microbial community can have a similar detrimental effect. Mice treated with antibiotics during early development have an increased production of IL-4 and lower numbers of Treg cells, and later in life, are more susceptible to colitis and airway hyper-reactivity [161]. Persistent alterations caused by antibiotic treatment in early human life correlate with IBD, asthma and atopic dermatitis in later life [162–164]. The state of non-inflammatory homeostasis in the gut can be shaken up by both the host immune system and the intestinal microbiota. Imbalance of their interplay increases the risk for immune-related diseases.
Oxidative stress
Oxidative stress occurring during inflammation is a factor amplifying dysbiosis by strongly decreasing the microbial diversity in the gut and by promoting the outgrowth of specific bacterial taxa. Leukocyte infiltration is a hallmark of intestinal inflammation, which is accompanied by generation of reactive oxygen and nitrogen species. The resulting oxidative stress exerts a manifest antimicrobial action, especially targeting strictly anaerobic bacteria that are susceptible to oxygen intoxication. The amount of microbes drops dramatically upon onset of inflammation, leading to the depletion of close to 80% of the microbiota in some models [9]. In addition to killing anaerobic residents, reactive oxygen species also promote the selective growth of bacterial groups through nitrate and tetrathionate respiration [165]. Sulfate-reducing bacteria are widespread in the gut microbiota [166] and produce hydrogen sulfide (H2S) and thiosulfate (S2O3 2−), which can be oxidized to tetrathionate (S4O6 2−) in the presence of reactive oxygen species. Elevation of tetrathionate in the gut promotes the growth of certain Enterobacteriaceae including Salmonella and Citrobacter, which can use tetrathionate as a respiratory electron acceptor [167]. The reaction of nitric oxide with superoxide anion yields peroxynitrile (ONOO−), which is a strong reactive product of the respiratory burst of macrophages [168]. Peroxynitrile isomerizes to nitrate (NO3 −), which can be utilized by E. coli through nitrate respiration, thereby favoring its growth during inflammation. The importance of nitric oxide and nitrate respiration in conferring a growth advantage to E. coli was confirmed by inhibiting the nitric oxide synthase iNOS with aminoguanidine hydrochloride during colitis in a mouse model [169]. The ability to utilize nitrate as respiratory electron acceptor is a factor contributing to dysbiosis. Interestingly, nitrate respiration can be boosted in S. enterica ser. Typhimurium through expression of the bacteriophage-transmitted virulence gene sopE, which stimulates iNOS expression in the intestinal mucosa [170]. This example shows that oxidative stress and bacteriophages can synergize to promote dysbiosis.
Bacteriophages
The bacteriophage fraction of the gut microbiota is like the dark matter of the universe. Bacteriophages probably play a major role in the homeostasis of the gut microbiota, but their true contribution is difficult to establish given the challenging identification of bacteriophage signatures within microbial genomes. Unlike bacteria identified by 16S rRNA sequencing, bacteriophage genomes lack conserved regions enabling their simple classification. A recent metagenomics survey aimed at identifying bacteriophages in the human gut found 44 bacteriophage groups, of which about a fifth was found in the majority of the samples analyzed. A group of 23 bacteriophages, mainly representing members of the order of Caudovirales and family of Microviridae, was even found in more than 50% of healthy individuals [171]. Because lysogenic bacteriophages dominate the human gut [172, 173], phage sequences are mainly embedded as prophage DNA in bacterial chromosomes. The difficulty in distinguishing viral open reading frames from bacterially encoded genes likely results in the under-estimation of the bacteriophage diversity in the gut. The newly discovered adaptive immune system of bacteria consisting of captured foreign DNA fragments into bacterial chromosomes is a valuable source of information to recognize bacteriophage infections. The clustered regularly interspaced short palindromic repeats (CRISPRs) represent an archive of past infections in bacterial genomes and their sequencing reveals the history of phages encountered by bacterial hosts. The analysis of CRISPRs in the gut microbiota from 124 European subjects revealed close to 1000 bacteriophages, of which 78% were shared by at least two individuals [173]. The sequences of the DNA spacers flanking phage fragments enabled the assignment of 11 bacterial hosts for 31 assembled phage contigs, showing that 14 of these phages target bacteria of the families of Bacteroides and Parabacteroides [173]. The analysis of bacteriophage occurrence in IBD confirmed the diversity of bacteriophages in ulcerative colitis and Crohn’s disease. Bacteriophage richness, as defined by the number of taxa per sample, was increased in these diseases, whereas bacterial richness was concomitantly decreased [174]. Whether bacteriophages indeed contribute to disease development remains, however, unclear at this stage.
Environmental stress imposed by inflammation and antibiotics can activate the lytic cycle of integrated prophages, thereby leading to a rapid elimination of bacterial hosts. In addition to a sudden change in the abundance of some bacterial taxa, the lytic action of bacteriophages liberates intracellular toxins [175] as well as cell wall fragments, lipids and nucleic acids, which are recognized as pathogen-associated molecular patterns activating innate immunity. The interlaced stimulation of the immune response and activation of phage lytic cycles fuel each other, which amplifies dysbiosis occurring during gut inflammation. Beyond their impact on the gut microbiota consecutive to environmental challenges, phages also contribute to the long-term shape of the gut microbiome through their action as vectors for the horizontal transfer of resistance genes.
The intestinal mucosa is another factor influencing the interactions between bacteriophages and their bacterial hosts. Several bacteriophages express proteins featuring C-type lectin folds and immunoglobulin-like domains [176], which interact with the heavily O-glycosylated mucin MUC2 [177, 178] in the colon. For example, the highly antigenic outer capsid protein of the bacteriophage T4 preferentially binds to O-glycan chains found on mucins. Adhesion to intestinal glycoproteins increases the bacteriophage density in the mucus layer, which acts as a protective barrier for the host by killing mucus-penetrating bacteria [117]. Changes in mucosal glycosylation, as occurring during intestinal inflammation [81], can alter the local abundance of bacteriophages and affect the proliferation or eradication of specific bacterial groups, thereby promoting dysbiosis.
Bacteriocins
The prevalent competition for nutrients in the colon drives the development of strategies enabling bacteria to outcompete or eliminate their competitors. One of these strategies is illustrated by the secretion of bacteriocins, which are toxic proteins and peptides targeting related taxa competing for the same resources. The family of bacteriocins covers colicins in E. coli, pyocins in Pseudomonas, pesticins in Pasteurella pestis and Yersinia pestis among others [179]. Bacteriocins also include microcins, which are short antimicrobial peptides [180]. The bacterial strains producing bacteriocins also express immunity proteins that protect them against the toxic effect of their own bacteriocins. Most bacteriocins kill by forming pores in membranes or by cleaving nucleic acids. Stress conditions such as oxidative and genotoxic stress induce the expression of bacteriocins [181], thus underlining the significance of bacteriocins in the mechanisms amplifying shifts in bacterial composition during inflammation-related oxidative stress. The expression of microcins in Enterobacteriaceae is also induced in conditions of nutrient shortage. For example, E. coli Nissle 1917 secrete microcins [182] preventing the growth of other E. coli strains when iron availability is limited, for example during inflammation. In fact, supplementation of mice with iron during intestinal inflammation decreases the production of microcins, which results in the proliferation of competing E. coli thereby restricting the growth of E. coli Nissle 1917 [183]. Of note, E. coli Nissle 1917 is the only probiotic recommended by the European Crohn’s and Colitis Organization as an alternative to the non-steroid anti-inflammatory drug mesalazine in the treatment of ulcerative colitis, as underlined in recent meta-analyses [184, 185]. Niche competition in the intestine has also been described for Gram-positive bacteria, such as members of the Enterococcus genus. Enterococcus faecalis produces a bacteriocin transmitted through plasmid conjugation, which disrupts the proliferation of other enterococci [186]. Beyond their contribution to the development of dysbiosis during inflammation, bacteriocins represent interesting candidate drugs aiming at the selective inhibition of pathogenic bacteria resistant to conventional antibiotics, such as C. difficile [187] and methicillin-resistant Staphylococcus aureus [188].
Dysbiosis and disease
As outlined in the previous sections of this review, the mechanisms destabilizing the gut microbiota are plentiful. Equally numerous are the diseases, which intestinal dysbiosis influences the course and severity. Typical examples including IBD [189], type 1 diabetes [190], celiac disease [191], and cardiovascular disorders [192] have been covered extensively in other reviews. We here focus our discussion on three diseases affecting human beings at different stages of life, namely necrotizing enterocolitis in newborns, colorectal cancer in adults, and C. difficile-associated diarrhea in elderly people.
Necrotizing enterocolitis
Necrotizing enterocolitis is a fulminant gut inflammation that is most frequent in premature newborns, affecting up to 10% of infants with a birthweight below 1500 g. Mortality can be as high as 30% [14]. The first signs of necrotizing enterocolitis are usually a distended abdomen and bloody stool. As reflected by these unspecific symptoms, the pathogenesis of necrotizing enterocolitis is unclear. Several risk factors, including enteral feeding, bottle-feeding, immature immunity, and altered microbiota increase the incidence of necrotizing enterocolitis. Conversely, breastfeeding decreases the occurrence of necrotizing enterocolitis by at least sixfold in comparison with bottle-feeding [193]. This large impact has led the American Academy of Pediatrics to recommend feeding premature babies with breast milk immediately after birth [194]. The molecular nature of the protection conferred by breast milk remains however elusive. Breast milk lactoferrin and immunoglobulins have been investigated as possible protective compounds but found to be ineffective at decreasing the incidence and severity of necrotizing enterocolitis [195]. Given that pasteurized breast milk is as protective as fresh milk, heat-resistant compounds such as milk oligosaccharides are likely to contribute to the protective effect. Oral supplementation with the prebiotics galacto-oligosaccharide, fructo-oligosaccharide and lactulose nevertheless did not influence the course of necrotizing enterocolitis, although they mediated a relative increase of bifidobacteria and lactobacilli levels in the treated newborns [196]. Supplementation with the probiotic Bifidobacterium breve BBG-001 also failed to improve the survival rate of infants with necrotizing enterocolitis [197]. Despite the unclear etiology, several findings converge towards a central role of the gut microbiota in triggering necrotizing enterocolitis. A sudden rise of Proteobacteria and a concomitant fall of Firmicutes levels has been found to precede the onset of the disease. Enterobacteriaceae, which are prominent members of the Proteobacteria phylum, express hexacylated LPS that are strong pyrogens and induce a robust inflammatory response mediated through TLR4 signaling. The mechanisms underlying the increase in Proteobacteria remain unclear. Is the proliferation of facultative anaerobic bacteria such as Enterobacteriaceae facilitated by the presence of oxygen in the newborn colon? As outlined in the present review, multiple mechanisms account for the development of dysbiosis. Given the resilience of the gut microbiota in response to changes, the occurrence of dysbiosis in necrotizing enterocolitis is likely the result of a chain of events combining an inadequate supply of protective nutrients and prebiotics, an immature immune system and an insufficient secretion of intestinal mucus.
Colorectal cancer
Dysbiosis of the gut microbiota can entail severe consequences. It is a primary driving force of inflammation and is unequivocally linked to the development of colorectal cancer [15]. Around 15% of all cases of cancer are linked to a viral or bacterial infection [198]. Infectious agents, especially viruses, can initiate or enhance tumor growth by inducing chronic inflammation, transferring active oncogenes into the host genome or by promoting immunosuppression. Microbial pathogens can influence tumorigenesis either directly by substances that lead to DNA damage, such as nitric oxide or reactive oxygen species, or indirectly by creating a pro-inflammatory microenvironment [199]. For example, an infection with oncogenic Helicobacter pylori results in chronic inflammation with dysregulated β-catenin signaling in epithelial cells fostering malignant transformations in the stomach [200]. Also in the colon, risk for adenocarcinoma is increased with H. pylori infection [201]. Especially strains positive for the virulence factor cytotoxin-associated gene A (CagA) are linked to carcinoma development [201, 202]. But what role do members of the commensal microbiota play and which shifts in the microbiota are linked to tumor development? The microbiota composition is significantly different in colorectal cancer patients compared to healthy individuals. Colorectal cancer is associated with increased abundance of the phyla Firmicutes and Fusobacteria [203, 204]. Strikingly, Fusobacteria constitute around 10% of the gut bacteria in colorectal cancer patients, but less than 0.1% in healthy individuals. These shifts in the microbiota can create a gut community with higher genotoxic and carcinogenic potential. Fusobacterium nucleatum, a species highly abundant in tumor tissues [205], expresses the virulence factor Fusobacterium adhesin A (FadA) [206]. This adhesion molecule increases epithelial permeability and invasion of microbes into the cells [207]. FadA also activates proliferation and growth of normal and adenoma cells via β-catenin signaling [208].
A dysbiotic gut community may trigger tumor development via innate immune responses, more precisely by activation of MyD88. In a mouse model of spontaneous intestinal tumorigenesis, signaling through MyD88 was necessary for extensive tumor growth [209]. By contrast, mice with chemically induced colitis had more intestinal tumors without MyD88 signaling [210]. The second branch of MyD88 signaling, via inflammasome-derived IL-18, might explain the contradictory effects of MyD88 signaling in the different cancer models. The lack of protective and tissue-repairing IL-18 and the resulting inability to heal chemically induced epithelial damages might enhance the mutation rate and adenoma formation in epithelial cells, thereby outbalancing the protective effect of MyD88 deficiency [211–214]. In contrast to MyD88, TLR4 signaling showed consistent tumor-promoting effects in several cancer models [215]. TLR4 signaling is increased in colorectal cancer patients [216, 217]. The LPS-mediated increase in prostaglandin E2 that activates epidermal growth factor receptors [215, 218, 219] is needed to promote proliferation of epithelial cells and their protection against apoptosis. However, the same mechanism might also promote the formation and growth of colorectal tumors when LPS stimulation exceeds the normal level and elicits chronic TLR4 activation [219, 220]. Interestingly, an increased inflammatory state in obese individuals correlates with higher risk of colorectal cancer [221]. Elevated TLR4 activation is also observed in IBD [222]. The two main forms of IBD, ulcerative colitis and Crohn’s disease, are risk factors for colitis-associated colorectal cancer [223, 224]. The TLR/MyD88 pathway is in fact of major importance for the initiation of colitis-associated cancer. In the absence of MyD88, mice presenting spontaneous colitis induced by IL-10-deficiency fail to develop carcinogen-induced tumors [225].
Specific shifts in the microbiota facilitate the formation of colorectal cancer. High consumption of red meat, a rich source of thiol-containing amino acids, increases the number of sulfate-reducing bacteria (e.g., Desulfovibrio spp., Desulfobacter spp.) in the intestine. These bacteria generate H2S which decreases mucus formation, inhibits methylation of DNA and increases the generation of reactive oxygen species [226]. Also, single bacterial species can contribute to tumor growth. Enterotoxigenic Bacteroides fragilis stimulates cell proliferation via increased β-catenin nuclear signaling [227] and damages DNA through reactive oxygen species [228]. Colitogenic E. coli overrepresented in the context of inflammation in IL-10 deficient mice promotes the development of invasive carcinoma by synthesizing the genotoxin colibactin [143]. Colibactin causes DNA double-strand breaks and incomplete DNA repair resulting in genomic instability [229].
But microbiota can as well confer protection against colorectal cancer and prevent carcinogenesis. The microbial metabolite butyrate activates the receptor GPR109a that triggers production of cytoprotective IL-18 and induces differentiation of Treg cells through IL-10, while inhibiting formation of pro-inflammatory Th17 cells [230]. Probiotics such as bifidobacteria and lactobacilli create a favorable microenvironment that decreases not only inflammatory conditions but also the emergence of colorectal cancer. Especially in combination with prebiotics, Bifidobacterium and Lactobacillus genera were shown to reduce aberrant crypt foci occurrence in mice and in rats [226].
Clostridium difficile-associated diarrhea
Clostridium difficile is a spore-forming strictly anaerobic Gram-positive bacterium that is often found in asymptomatic subjects, including more than 50% of children and 15% of healthy adults. The mere presence of toxicogenic C. difficile in a host is not a predictive marker for intestinal inflammation [231]. Progression to disease requires in fact vegetative growth of C. difficile and secretion of toxins such as the TcdA and TcdB enzymes, which are glycosyltransferases modifying cytoplasmic Rho GTPases, thereby impairing cytoskeleton integrity [232]. The activity of TcdA and TcdB toxins is sufficient to trigger disease when released in the host intestinal tract [233]. The germination of C. difficile is facilitated by some bile acids found in the duodenum, such as taurocholate and deoxycholate [234]. The gut microbiota plays an essential role in suppressing the vegetative growth of C. difficile in asymptomatic subjects, although the mechanisms of this inhibition remain unclear. A group of microbes may prevent the proliferation of C. difficile by exhausting nutrients essential for its growth. Carbohydrates, such as N-acetylglucosamine and Neu5Ac derived from intestinal mucins, are important nutrients supporting the growth of C. difficile [84]. These carbohydrates are preferentially metabolized by other gut microbes, thereby limiting the expansion of C. difficile and consequently disease development [235]. Besides sequestering nutrients away from C. difficile, some microbes transform bile acids, thereby reducing the rate of germination of clostridial spores in the gut [236]. The importance of gut microbes in mediating resistance to C. difficile growth is illustrated by the impact of antibiotics on promoting C. difficile-associated diarrhea. The study of C. difficile infection in conjunction with antibiotic treatments linked the expansion of C. difficile to decreased Lachnospiraceae and increased Enterobacteriaceae levels in animal models [237–239]. Elevated levels of Enterobacteriaceae were also noted in elderly human subjects presenting with C. difficile-associated diarrhea [240, 241]. Finally, the astonishing success of fecal microbial transplantation definitively demonstrated the role of the gut microbiota at keeping C. difficile at bay. The treatment of patients with refractory C. difficile infection by infusion with microbiota derived from healthy donors cured more than 90% of cases, whereas the traditional treatment with vancomycin only improved 30% of cases [242]. The incidence of C. difficile infection increases with age, probably reflecting the progressive decreased microbial diversity and the loss of microbes conferring resistance to C. difficile. As recently presented, the expansion of C. difficile may be related to decreased bacteriocins secreted by bacteria that normally keep the pathogen at bay in asymptomatic subjects [187]. The identification of bacteriocins targeting C. difficile would represent a valuable alternative or at least a complementary approach to fecal microbial transplantation.
Concluding remarks
The gut microbiota is an inherent component of animal physiology that reacts to internal and environmental changes, while playing important roles in regulating multiple host functions. As outlined in the present review, the contribution of dysbiosis to diseases is undisputed, but the mechanisms in play and the assignment of truly pathogenic microbes often remains circumstantial, if not speculative. The recent development of high-throughput sequencing techniques linked to the establishment of reliable microbial 16S rRNA sequence databases resulted in an explosion of reports documenting the importance of the gut microbiota in regulating health and disease. Despite the wealth of information unraveled through past studies, the taxonomic identification of intestinal bacteria only clarifies a single variable of the equation explaining gut ecology. Metagenomic approaches documenting the global genetic diversity of the intestinal ecosystem are gaining momentum as sequencing technologies and bioinformatic analysis constantly improve [243]. The determination of biochemical parameters beyond the classical survey of SCFA profiles adds further dimensions to the characterization of metabolic pathways at play in microbial communities [244]. Finally, the integration of all data through heuristic algorithms [245] will not only facilitate the interpretation of experimental models but also enable the recognition of novel mutualistic networks among the gut microbiota. The comprehensive appreciation of gut ecology will allow a better control of intestinal dysbiosis and thereby lead to a significant health improvement across a broad range of inflammatory and metabolic conditions afflicting our modern society.
Acknowledgements
This work is funded by the Swiss National Science Foundation Grant CRSII3_154488/1.
References
- 1.Sonnenburg JL, Backhed F. Diet-microbiota interactions as moderators of human metabolism. Nature. 2016;535(7610):56–64. doi: 10.1038/nature18846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.LeBlanc JG, et al. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol. 2013;24(2):160–168. doi: 10.1016/j.copbio.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. 2016;535(7610):75–84. doi: 10.1038/nature18848. [DOI] [PubMed] [Google Scholar]
- 4.Thaiss CA, et al. The microbiome and innate immunity. Nature. 2016;535(7610):65–74. doi: 10.1038/nature18847. [DOI] [PubMed] [Google Scholar]
- 5.Mayer EA, Tillisch K, Gupta A. Gut/brain axis and the microbiota. J Clin Invest. 2015;125(3):926–938. doi: 10.1172/JCI76304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zitvogel L, et al. Cancer and the gut microbiota: an unexpected link. Sci Transl Med. 2015;7(271):271ps1. doi: 10.1126/scitranslmed.3010473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arumugam M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker AW, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011;11:7. doi: 10.1186/1471-2180-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lupp C, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2(2):119–129. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 10.Wlodarska M, Kostic AD, Xavier RJ. An integrative view of microbiome–host interactions in inflammatory bowel diseases. Cell Host Microbe. 2015;17(5):577–591. doi: 10.1016/j.chom.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerard P. Gut microbiota and obesity. Cell Mol Life Sci. 2016;73(1):147–162. doi: 10.1007/s00018-015-2061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knip M, Siljander H. The role of the intestinal microbiota in type 1 diabetes mellitus. Nat Rev Endocrinol. 2016;12(3):154–167. doi: 10.1038/nrendo.2015.218. [DOI] [PubMed] [Google Scholar]
- 13.Tremlett H, et al. The gut microbiome in human neurological disease: a review. Ann Neurol. 2017;81(3):369–382. doi: 10.1002/ana.24901. [DOI] [PubMed] [Google Scholar]
- 14.Neu J, Walker WA. Necrotizing enterocolitis. N Engl J Med. 2011;364(3):255–264. doi: 10.1056/NEJMra1005408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013;13(11):800–812. doi: 10.1038/nrc3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seekatz AM, Young VB. Clostridium difficile and the microbiota. J Clin Invest. 2014;124(10):4182–4189. doi: 10.1172/JCI72336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox MJ, Cookson WO, Moffatt MF. Sequencing the human microbiome in health and disease. Hum Mol Genet. 2013;22(R1):R88–R94. doi: 10.1093/hmg/ddt398. [DOI] [PubMed] [Google Scholar]
- 18.Turnbaugh PJ, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 19.Gregory JC, et al. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem. 2015;290(9):5647–5660. doi: 10.1074/jbc.M114.618249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rakoff-Nahoum S, Foster KR, Comstock LE. The evolution of cooperation within the gut microbiota. Nature. 2016;533(7602):255–259. doi: 10.1038/nature17626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chow J, Tang H, Mazmanian SK. Pathobionts of the gastrointestinal microbiota and inflammatory disease. Curr Opin Immunol. 2011;23(4):473–480. doi: 10.1016/j.coi.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tenaillon O, et al. The population genetics of commensal Escherichia coli . Nat Rev Microbiol. 2010;8(3):207–217. doi: 10.1038/nrmicro2298. [DOI] [PubMed] [Google Scholar]
- 23.Stecher B, et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007;5(10):2177–2189. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chevalier C, et al. Gut microbiota orchestrates energy homeostasis during cold. Cell. 2015;163(6):1360–1374. doi: 10.1016/j.cell.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Zhao X. Comparative analyses of fecal microbiota in Tibetan and Chinese Han living at low or high altitude by barcoded 454 pyrosequencing. Sci Rep. 2015;5:14682. doi: 10.1038/srep14682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ritchie LE, et al. Space environmental factor impacts upon murine colon microbiota and mucosal homeostasis. PLoS One. 2015;10(6):e0125792. doi: 10.1371/journal.pone.0125792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scott KP, et al. The influence of diet on the gut microbiota. Pharmacol Res. 2013;69(1):52–60. doi: 10.1016/j.phrs.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 28.Demehri FR, Barrett M, Teitelbaum DH. Changes to the intestinal microbiome with parenteral nutrition: review of a murine model and potential clinical implications. Nutr Clin Pract. 2015;30(6):798–806. doi: 10.1177/0884533615609904. [DOI] [PubMed] [Google Scholar]
- 29.Le Chatelier E, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500(7464):541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 30.Ley RE, et al. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102(31):11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ley RE, et al. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 32.Kasai C, et al. Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. BMC Gastroenterol. 2015;15:100. doi: 10.1186/s12876-015-0330-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caricilli AM, et al. Gut microbiota is a key modulator of insulin resistance in TLR 2 knockout mice. PLoS Biol. 2011;9(12):e1001212. doi: 10.1371/journal.pbio.1001212. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Cani PD, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 35.Festi D, et al. Gut microbiota and metabolic syndrome. World J Gastroenterol. 2014;20(43):16079–16094. doi: 10.3748/wjg.v20.i43.16079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez-Medina M, et al. Western diet induces dysbiosis with increased E. coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut. 2014;63(1):116–124. doi: 10.1136/gutjnl-2012-304119. [DOI] [PubMed] [Google Scholar]
- 37.Wu GD, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shankar V et al (2017) Differences in gut metabolites and microbial composition and functions between Egyptian and U.S. children are consistent with their diets. mSystems 2(1) [DOI] [PMC free article] [PubMed]
- 39.Russell WR, et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am J Clin Nutr. 2011;93(5):1062–1072. doi: 10.3945/ajcn.110.002188. [DOI] [PubMed] [Google Scholar]
- 40.Liu X, et al. High-protein diet modifies colonic microbiota and luminal environment but not colonocyte metabolism in the rat model: the increased luminal bulk connection. Am J Physiol Gastrointest Liver Physiol. 2014;307(4):G459–G470. doi: 10.1152/ajpgi.00400.2013. [DOI] [PubMed] [Google Scholar]
- 41.Magee EA, et al. Contribution of dietary protein to sulfide production in the large intestine: an in vitro and a controlled feeding study in humans. Am J Clin Nutr. 2000;72(6):1488–1494. doi: 10.1093/ajcn/72.6.1488. [DOI] [PubMed] [Google Scholar]
- 42.Alemany M. The problem of nitrogen disposal in the obese. Nutr Res Rev. 2012;25(1):18–28. doi: 10.1017/S0954422411000163. [DOI] [PubMed] [Google Scholar]
- 43.Zahedi Asl S, Ghasemi A, Azizi F. Serum nitric oxide metabolites in subjects with metabolic syndrome. Clin Biochem. 2008;41(16–17):1342–1347. doi: 10.1016/j.clinbiochem.2008.08.076. [DOI] [PubMed] [Google Scholar]
- 44.Dykhuizen RS, et al. Antimicrobial effect of acidified nitrite on gut pathogens: importance of dietary nitrate in host defense. Antimicrob Agents Chemother. 1996;40(6):1422–1425. doi: 10.1128/aac.40.6.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang C, et al. Structural resilience of the gut microbiota in adult mice under high-fat dietary perturbations. ISME J. 2012;6(10):1848–1857. doi: 10.1038/ismej.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caesar R, et al. Crosstalk between gut microbiota and dietary lipids aggravates WAT inflammation through TLR signaling. Cell Metab. 2015;22(4):658–668. doi: 10.1016/j.cmet.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bell DS. Changes seen in gut bacteria content and distribution with obesity: causation or association? Postgrad Med. 2015;127(8):863–868. doi: 10.1080/00325481.2015.1098519. [DOI] [PubMed] [Google Scholar]
- 48.Islam KB, et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology. 2011;141(5):1773–1781. doi: 10.1053/j.gastro.2011.07.046. [DOI] [PubMed] [Google Scholar]
- 49.Murakami Y, Tanabe S, Suzuki T. High-fat diet-induced intestinal hyperpermeability is associated with increased bile acids in the large intestine of mice. J Food Sci. 2016;81(1):H216–H222. doi: 10.1111/1750-3841.13166. [DOI] [PubMed] [Google Scholar]
- 50.Thomas C, et al. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7(8):678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 51.Thomas C, Auwerx J, Schoonjans K. Bile acids and the membrane bile acid receptor TGR5-connecting nutrition and metabolism. Thyroid. 2008;18(2):167–174. doi: 10.1089/thy.2007.0255. [DOI] [PubMed] [Google Scholar]
- 52.Watanabe M, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439(7075):484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 53.Fiorucci S, Distrutti E. Bile acid-activated receptors, intestinal microbiota, and the treatment of metabolic disorders. Trends Mol Med. 2015;21(11):702–714. doi: 10.1016/j.molmed.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 54.De Filippo C, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA. 2010;107(33):14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.DiBaise JK, et al. Gut microbiota and its possible relationship with obesity. Mayo Clin Proc. 2008;83(4):460–469. doi: 10.4065/83.4.460. [DOI] [PubMed] [Google Scholar]
- 56.Backhed F, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101(44):15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanders FW, Griffin JL. De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose. Biol Rev Camb Philos Soc. 2016;91(2):452–468. doi: 10.1111/brv.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guarner F, Malagelada JR. Gut flora in health and disease. Lancet. 2003;361(9356):512–519. doi: 10.1016/S0140-6736(03)12489-0. [DOI] [PubMed] [Google Scholar]
- 59.Arslan N. Obesity, fatty liver disease and intestinal microbiota. World J Gastroenterol. 2014;20(44):16452–16463. doi: 10.3748/wjg.v20.i44.16452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Le Poul E, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem. 2003;278(28):25481–25489. doi: 10.1074/jbc.M301403200. [DOI] [PubMed] [Google Scholar]
- 61.Brown AJ, et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. 2003;278(13):11312–11319. doi: 10.1074/jbc.M211609200. [DOI] [PubMed] [Google Scholar]
- 62.Ang Z, Ding JL. GPR41 and GPR43 in obesity and inflammation—protective or causative? Front Immunol. 2016;7:28. doi: 10.3389/fimmu.2016.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zaibi MS, et al. Roles of GPR41 and GPR43 in leptin secretory responses of murine adipocytes to short chain fatty acids. FEBS Lett. 2010;584(11):2381–2386. doi: 10.1016/j.febslet.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 64.Xiong Y, et al. Short-chain fatty acids stimulate leptin production in adipocytes through the G protein-coupled receptor GPR41. Proc Natl Acad Sci USA. 2004;101(4):1045–1050. doi: 10.1073/pnas.2637002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brandsma E, et al. The immunity-diet-microbiota axis in the development of metabolic syndrome. Curr Opin Lipidol. 2015;26(2):73–81. doi: 10.1097/MOL.0000000000000154. [DOI] [PubMed] [Google Scholar]
- 66.Cummings JH, et al. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut. 1987;28(10):1221–1227. doi: 10.1136/gut.28.10.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Macfarlane S, Macfarlane GT. Regulation of short-chain fatty acid production. Proc Nutr Soc. 2003;62(1):67–72. doi: 10.1079/PNS2002207. [DOI] [PubMed] [Google Scholar]
- 68.Walker AW, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011;5(2):220–230. doi: 10.1038/ismej.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cani PD, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. 2009;58(8):1091–1103. doi: 10.1136/gut.2008.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Everard A, et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes. 2011;60(11):2775–2786. doi: 10.2337/db11-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parnell JA, Reimer RA. Prebiotic fibres dose-dependently increase satiety hormones and alter Bacteroidetes and Firmicutes in lean and obese JCR:LA-cp rats. Br J Nutr. 2012;107(4):601–613. doi: 10.1017/S0007114511003163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Backhed F, et al. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA. 2007;104(3):979–984. doi: 10.1073/pnas.0605374104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aronsson L et al (2010) Decreased fat storage by Lactobacillus paracasei is associated with increased levels of angiopoietin-like 4 protein (ANGPTL4). PLoS One 5(9) [DOI] [PMC free article] [PubMed]
- 74.Cani PD, Everard A. Akkermansia muciniphila: a novel target controlling obesity, type 2 diabetes and inflammation? Med Sci (Paris) 2014;30(2):125–127. doi: 10.1051/medsci/20143002003. [DOI] [PubMed] [Google Scholar]
- 75.Everard A, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA. 2013;110(22):9066–9071. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Malik A, et al. IL-33 regulates the IgA-microbiota axis to restrain IL-1alpha-dependent colitis and tumorigenesis. J Clin Invest. 2016;126(12):4469–4481. doi: 10.1172/JCI88625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kang CS, et al. Extracellular vesicles derived from gut microbiota, especially Akkermansia muciniphila, protect the progression of dextran sulfate sodium-induced colitis. PLoS One. 2013;8(10):e76520. doi: 10.1371/journal.pone.0076520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gilbert HJ. The biochemistry and structural biology of plant cell wall deconstruction. Plant Physiol. 2010;153(2):444–455. doi: 10.1104/pp.110.156646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fabich AJ, et al. Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect Immun. 2008;76(3):1143–1152. doi: 10.1128/IAI.01386-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pacheco AR, et al. Fucose sensing regulates bacterial intestinal colonization. Nature. 2012;492(7427):113–117. doi: 10.1038/nature11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pickard JM, et al. Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature. 2014;514(7524):638–641. doi: 10.1038/nature13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vimr ER, et al. Diversity of microbial sialic acid metabolism. Microbiol Mol Biol Rev. 2004;68(1):132–153. doi: 10.1128/MMBR.68.1.132-153.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marcobal A, et al. Bacteroides in the infant gut consume milk oligosaccharides via mucus-utilization pathways. Cell Host Microbe. 2011;10(5):507–514. doi: 10.1016/j.chom.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ng KM, et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature. 2013;502(7469):96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang YL, et al. Sialic acid catabolism drives intestinal inflammation and microbial dysbiosis in mice. Nat Commun. 2015;6:8141. doi: 10.1038/ncomms9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rogers MA, Aronoff DM. The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin Microbiol Infect. 2016;178(2):e1–e9. doi: 10.1016/j.cmi.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Freedberg DE, et al. Proton pump inhibitors alter specific taxa in the human gastrointestinal microbiome: a crossover trial. Gastroenterology. 2015;149(4):883–885. doi: 10.1053/j.gastro.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tsai CF, et al. Proton pump inhibitors increase risk for hepatic encephalopathy in patients with cirrhosis in a population study. Gastroenterology. 2017;152(1):134–141. doi: 10.1053/j.gastro.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 89.Forslund K, et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature. 2015;528(7581):262–266. doi: 10.1038/nature15766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Korpela K, et al. Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nat Commun. 2016;7:10410. doi: 10.1038/ncomms10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Furuya-Kanamori L, et al. Comorbidities, exposure to medications, and the risk of community-acquired Clostridium difficile infection: a systematic review and meta-analysis. Infect Control Hosp Epidemiol. 2015;36(2):132–141. doi: 10.1017/ice.2014.39. [DOI] [PubMed] [Google Scholar]
- 93.Galdston I. Eubiotic medicine. Science. 1944;100(2587):76. doi: 10.1126/science.100.2587.76. [DOI] [PubMed] [Google Scholar]
- 94.Guslandi M. Rifaximin in the treatment of inflammatory bowel disease. World J Gastroenterol. 2011;17(42):4643–4646. doi: 10.3748/wjg.v17.i42.4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pimentel M, et al. Rifaximin therapy for patients with irritable bowel syndrome without constipation. N Engl J Med. 2011;364(1):22–32. doi: 10.1056/NEJMoa1004409. [DOI] [PubMed] [Google Scholar]
- 96.Swanson HI. Drug metabolism by the host and gut microbiota: a partnership or rivalry? Drug Metab Dispos. 2015;43(10):1499–1504. doi: 10.1124/dmd.115.065714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aura AM, et al. Drug metabolome of the simvastatin formed by human intestinal microbiota in vitro. Mol Biosyst. 2011;7(2):437–446. doi: 10.1039/C0MB00023J. [DOI] [PubMed] [Google Scholar]
- 98.Yadav V, et al. Colonic bacterial metabolism of corticosteroids. Int J Pharm. 2013;457(1):268–274. doi: 10.1016/j.ijpharm.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 99.Wallace BD, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330(6005):831–835. doi: 10.1126/science.1191175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lim YJ, Yang CH. Non-steroidal anti-inflammatory drug-induced enteropathy. Clin Endosc. 2012;45(2):138–144. doi: 10.5946/ce.2012.45.2.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Robbe C, et al. Evidence of regio-specific glycosylation in human intestinal mucins: presence of an acidic gradient along the intestinal tract. J Biol Chem. 2003;278(47):46337–46348. doi: 10.1074/jbc.M302529200. [DOI] [PubMed] [Google Scholar]
- 102.Corfield AP. Mucins: a biologically relevant glycan barrier in mucosal protection. Biochim Biophys Acta. 2015;1850(1):236–252. doi: 10.1016/j.bbagen.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 103.Ahn DH, et al. TNF-alpha activates MUC2 transcription via NF-kappaB but inhibits via JNK activation. Cell Physiol Biochem. 2005;15(1–4):29–40. doi: 10.1159/000083636. [DOI] [PubMed] [Google Scholar]
- 104.Hokari R, et al. Vasoactive intestinal peptide upregulates MUC2 intestinal mucin via CREB/ATF1. Am J Physiol Gastrointest Liver Physiol. 2005;289(5):G949–G959. doi: 10.1152/ajpgi.00142.2005. [DOI] [PubMed] [Google Scholar]
- 105.Larsson JM, et al. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm Bowel Dis. 2011;17(11):2299–2307. doi: 10.1002/ibd.21625. [DOI] [PubMed] [Google Scholar]
- 106.Goto Y, et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science. 2014;345(6202):1254009. doi: 10.1126/science.1254009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Png CW, et al. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol. 2010;105(11):2420–2428. doi: 10.1038/ajg.2010.281. [DOI] [PubMed] [Google Scholar]
- 108.Sela DA, et al. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc Natl Acad Sci USA. 2008;105(48):18964–18969. doi: 10.1073/pnas.0809584105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Derrien M, et al. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol. 2004;54(Pt 5):1469–1476. doi: 10.1099/ijs.0.02873-0. [DOI] [PubMed] [Google Scholar]
- 110.Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 2008;4(5):447–457. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Etzold S, Juge N. Structural insights into bacterial recognition of intestinal mucins. Curr Opin Struct Biol. 2014;28C:23–31. doi: 10.1016/j.sbi.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 112.Mahdavi J, et al. A novel O-linked glycan modulates Campylobacter jejuni major outer membrane protein-mediated adhesion to human histo-blood group antigens and chicken colonization. Open Biol. 2014;4:130202. doi: 10.1098/rsob.130202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ruiz-Palacios GM, et al. Campylobacter jejuni binds intestinal H(O) antigen (Fuc alpha 1, 2Gal beta 1, 4GlcNAc), and fucosyloligosaccharides of human milk inhibit its binding and infection. J Biol Chem. 2003;278(16):14112–14120. doi: 10.1074/jbc.M207744200. [DOI] [PubMed] [Google Scholar]
- 114.Kyogashima M, Ginsburg V, Krivan HC. Escherichia coli K99 binds to N-glycolylsialoparagloboside and N-glycolyl-GM3 found in piglet small intestine. Arch Biochem Biophys. 1989;270(1):391–397. doi: 10.1016/0003-9861(89)90042-8. [DOI] [PubMed] [Google Scholar]
- 115.Byres E, et al. Incorporation of a non-human glycan mediates human susceptibility to a bacterial toxin. Nature. 2008;456(7222):648–652. doi: 10.1038/nature07428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tangvoranuntakul P, et al. Human uptake and incorporation of an immunogenic nonhuman dietary sialic acid. Proc Natl Acad Sci USA. 2003;100(21):12045–12050. doi: 10.1073/pnas.2131556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Barr JJ, et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc Natl Acad Sci USA. 2013;110(26):10771–10776. doi: 10.1073/pnas.1305923110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.McGovern DP, et al. Fucosyltransferase 2 (FUT2) non-secretor status is associated with Crohn’s disease. Hum Mol Genet. 2010;19(17):3468–3476. doi: 10.1093/hmg/ddq248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Johansson ME, et al. Normalization of host intestinal mucus layers requires long-term microbial colonization. Cell Host Microbe. 2015;18(5):582–592. doi: 10.1016/j.chom.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10(3):159–169. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- 121.Suzuki K, et al. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc Natl Acad Sci USA. 2004;101(7):1981–1986. doi: 10.1073/pnas.0307317101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Elinav E, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145(5):745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wlodarska M, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156(5):1045–1059. doi: 10.1016/j.cell.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nowarski R, et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell. 2015;163(6):1444–1456. doi: 10.1016/j.cell.2015.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gersemann M, et al. Differences in goblet cell differentiation between Crohn’s disease and ulcerative colitis. Differentiation. 2009;77(1):84–94. doi: 10.1016/j.diff.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 126.Huber S, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491(7423):259–263. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zheng Y, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14(3):282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 128.McSorley SJ, et al. Bacterial flagellin is an effective adjuvant for CD4+ T cells in vivo. J Immunol. 2002;169(7):3914–3919. doi: 10.4049/jimmunol.169.7.3914. [DOI] [PubMed] [Google Scholar]
- 129.McDermott PF, et al. High-affinity interaction between gram-negative flagellin and a cell surface polypeptide results in human monocyte activation. Infect Immun. 2000;68(10):5525–5529. doi: 10.1128/IAI.68.10.5525-5529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Van Maele L, et al. TLR5 signaling stimulates the innate production of IL-17 and IL-22 by CD3(neg)CD127+ immune cells in spleen and mucosa. J Immunol. 2010;185(2):1177–1185. doi: 10.4049/jimmunol.1000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vijay-Kumar M, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328(5975):228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vijay-Kumar M, et al. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest. 2007;117(12):3909–3921. doi: 10.1172/JCI33084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Carvalho FA, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe. 2012;12(2):139–152. doi: 10.1016/j.chom.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kobayashi KS, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307(5710):731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 135.Petnicki-Ocwieja T, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci USA. 2009;106(37):15813–15818. doi: 10.1073/pnas.0907722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Salzman NH, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. 2010;11(1):76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ogura Y, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411(6837):603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 138.Wehkamp J, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci USA. 2005;102(50):18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Davis CP, Savage DC. Habitat, succession, attachment, and morphology of segmented, filamentous microbes indigenous to the murine gastrointestinal tract. Infect Immun. 1974;10(4):948–956. doi: 10.1128/iai.10.4.948-956.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kumar P, et al. Intestinal interleukin-17 receptor signaling mediates reciprocal control of the gut microbiota and autoimmune inflammation. Immunity. 2016;44(3):659–671. doi: 10.1016/j.immuni.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gaboriau-Routhiau V, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31(4):677–689. doi: 10.1016/j.immuni.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 142.Sakaguchi S, et al. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 143.Arthur JC, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338(6103):120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wohlgemuth S, et al. Reduced microbial diversity and high numbers of one single Escherichia coli strain in the intestine of colitic mice. Environ Microbiol. 2009;11(6):1562–1571. doi: 10.1111/j.1462-2920.2009.01883.x. [DOI] [PubMed] [Google Scholar]
- 145.Di Giacinto C, et al. Probiotics ameliorate recurrent Th1-mediated murine colitis by inducing IL-10 and IL-10-dependent TGF-beta-bearing regulatory cells. J Immunol. 2005;174(6):3237–3246. doi: 10.4049/jimmunol.174.6.3237. [DOI] [PubMed] [Google Scholar]
- 146.Sokol H, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105(43):16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kashiwagi I, et al. Smad2 and Smad3 inversely regulate TGF-beta autoinduction in clostridium butyricum-activated dendritic cells. Immunity. 2015;43(1):65–79. doi: 10.1016/j.immuni.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 148.Ihara S, et al. TGF-beta signaling in dendritic cells governs colonic homeostasis by controlling epithelial differentiation and the luminal microbiota. J Immunol. 2016;196(11):4603–4613. doi: 10.4049/jimmunol.1502548. [DOI] [PubMed] [Google Scholar]
- 149.Sato J, et al. Gut dysbiosis and detection of “live gut bacteria” in blood of Japanese patients with type 2 diabetes. Diabetes Care. 2014;37(8):2343–2350. doi: 10.2337/dc13-2817. [DOI] [PubMed] [Google Scholar]
- 150.Mazmanian SK, Kasper DL. The love-hate relationship between bacterial polysaccharides and the host immune system. Nat Rev Immunol. 2006;6(11):849–858. doi: 10.1038/nri1956. [DOI] [PubMed] [Google Scholar]
- 151.Ivanov II, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Karczewski J, et al. The effects of the microbiota on the host immune system. Autoimmunity. 2014;47(8):494–504. doi: 10.3109/08916934.2014.938322. [DOI] [PubMed] [Google Scholar]
- 153.Wen L, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455(7216):1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Burrows MP, et al. Microbiota regulates type 1 diabetes through Toll-like receptors. Proc Natl Acad Sci USA. 2015;112(32):9973–9977. doi: 10.1073/pnas.1508740112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Larsson E, et al. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut. 2012;61(8):1124–1131. doi: 10.1136/gutjnl-2011-301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kriegel MA, et al. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci USA. 2011;108(28):11548–11553. doi: 10.1073/pnas.1108924108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Clarke TB, et al. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med. 2010;16(2):228–231. doi: 10.1038/nm.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ganal SC, et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity. 2012;37(1):171–186. doi: 10.1016/j.immuni.2012.05.020. [DOI] [PubMed] [Google Scholar]
- 159.Khosravi A, et al. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe. 2014;15(3):374–381. doi: 10.1016/j.chom.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hill DA, et al. Commensal bacteria-derived signals regulate basophil hematopoiesis and allergic inflammation. Nat Med. 2012;18(4):538–546. doi: 10.1038/nm.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zeissig S, Blumberg RS. Life at the beginning: perturbation of the microbiota by antibiotics in early life and its role in health and disease. Nat Immunol. 2014;15(4):307–310. doi: 10.1038/ni.2847. [DOI] [PubMed] [Google Scholar]
- 162.Ng SC, et al. Geographical variability and environmental risk factors in inflammatory bowel disease. Gut. 2013;62(4):630–649. doi: 10.1136/gutjnl-2012-303661. [DOI] [PubMed] [Google Scholar]
- 163.Murk W, Risnes KR, Bracken MB. Prenatal or early-life exposure to antibiotics and risk of childhood asthma: a systematic review. Pediatrics. 2011;127(6):1125–1138. doi: 10.1542/peds.2010-2092. [DOI] [PubMed] [Google Scholar]
- 164.Flohr C, Pascoe D, Williams HC. Atopic dermatitis and the ‘hygiene hypothesis’: too clean to be true? Br J Dermatol. 2005;152(2):202–216. doi: 10.1111/j.1365-2133.2004.06436.x. [DOI] [PubMed] [Google Scholar]
- 165.Winter SE, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella . Nature. 2010;467(7314):426–429. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Scanlan PD, Shanahan F, Marchesi JR. Culture-independent analysis of desulfovibrios in the human distal colon of healthy, colorectal cancer and polypectomized individuals. FEMS Microbiol Ecol. 2009;69(2):213–221. doi: 10.1111/j.1574-6941.2009.00709.x. [DOI] [PubMed] [Google Scholar]
- 167.Hensel M, et al. The genetic basis of tetrathionate respiration in Salmonella typhimurium . Mol Microbiol. 1999;32(2):275–287. doi: 10.1046/j.1365-2958.1999.01345.x. [DOI] [PubMed] [Google Scholar]
- 168.Pryor WA, Squadrito GL. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am J Physiol. 1995;268(5 Pt 1):L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 169.Winter SE, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. 2013;339(6120):708–711. doi: 10.1126/science.1232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lopez CA et al (2012) Phage-mediated acquisition of a type III secreted effector protein boosts growth of salmonella by nitrate respiration. MBio 3(3) [DOI] [PMC free article] [PubMed]
- 171.Manrique P, et al. Healthy human gut phageome. Proc Natl Acad Sci USA. 2016;113(37):10400–10405. doi: 10.1073/pnas.1601060113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Minot S, et al. The human gut virome: inter-individual variation and dynamic response to diet. Genome Res. 2011;21(10):1616–1625. doi: 10.1101/gr.122705.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Stern A, et al. CRISPR targeting reveals a reservoir of common phages associated with the human gut microbiome. Genome Res. 2012;22(10):1985–1994. doi: 10.1101/gr.138297.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Norman JM, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015;160(3):447–460. doi: 10.1016/j.cell.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhang X, et al. Quinolone antibiotics induce Shiga toxin-encoding bacteriophages, toxin production, and death in mice. J Infect Dis. 2000;181(2):664–670. doi: 10.1086/315239. [DOI] [PubMed] [Google Scholar]
- 176.Fraser JS, Maxwell KL, Davidson AR. Immunoglobulin-like domains on bacteriophage: weapons of modest damage? Curr Opin Microbiol. 2007;10(4):382–387. doi: 10.1016/j.mib.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 177.Arike L, Hansson GC. The densely O-glycosylated MUC2 mucin protects the intestine and provides food for the commensal bacteria. J Mol Biol. 2016;428(16):3221–3229. doi: 10.1016/j.jmb.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kim YS, Ho SB. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep. 2010;12(5):319–330. doi: 10.1007/s11894-010-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Cotter PD, Hill C, Ross RP. Bacteriocins: developing innate immunity for food. Nat Rev Microbiol. 2005;3(10):777–788. doi: 10.1038/nrmicro1273. [DOI] [PubMed] [Google Scholar]
- 180.Rebuffat S. Microcins in action: amazing defence strategies of Enterobacteria. Biochem Soc Trans. 2012;40(6):1456–1462. doi: 10.1042/BST20120183. [DOI] [PubMed] [Google Scholar]
- 181.Ghazaryan L, et al. The role of stress in colicin regulation. Arch Microbiol. 2014;196(11):753–764. doi: 10.1007/s00203-014-1017-8. [DOI] [PubMed] [Google Scholar]
- 182.Patzer SI, et al. The colicin G, H and X determinants encode microcins M and H47, which might utilize the catecholate siderophore receptors FepA, Cir, Fiu and IroN. Microbiology. 2003;149(9):2557–2570. doi: 10.1099/mic.0.26396-0. [DOI] [PubMed] [Google Scholar]
- 183.Sassone-Corsi M, et al. Microcins mediate competition among Enterobacteriaceae in the inflamed gut. Nature. 2016;540(7632):280–283. doi: 10.1038/nature20557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Losurdo G, et al. Escherichia coli Nissle 1917 in Ulcerative colitis treatment: systematic review and meta-analysis. J Gastrointestin Liver Dis. 2015;24(4):499–505. doi: 10.15403/jgld.2014.1121.244.ecn. [DOI] [PubMed] [Google Scholar]
- 185.Scaldaferri F, et al. Role and mechanisms of action of Escherichia coli Nissle 1917 in the maintenance of remission in ulcerative colitis patients: an update. World J Gastroenterol. 2016;22(24):5505–5511. doi: 10.3748/wjg.v22.i24.5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kommineni S, et al. Bacteriocin production augments niche competition by enterococci in the mammalian gastrointestinal tract. Nature. 2015;526(7575):719–722. doi: 10.1038/nature15524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gebhart D et al (2015) A modified R-type bacteriocin specifically targeting Clostridium difficile prevents colonization of mice without affecting gut microbiota diversity. MBio 6(2) [DOI] [PMC free article] [PubMed]
- 188.Xin B, et al. Thusin, a novel two-component lantibiotic with potent antimicrobial activity against several gram-positive pathogens. Front Microbiol. 2016;7:1115. doi: 10.3389/fmicb.2016.01115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cammarota G, et al. The involvement of gut microbiota in inflammatory bowel disease pathogenesis: potential for therapy. Pharmacol Ther. 2015;149:191–212. doi: 10.1016/j.pharmthera.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 190.Scott FW, et al. Where genes meet environment-integrating the role of gut luminal contents, immunity and pancreas in type 1 diabetes. Transl Res. 2017;179:183–198. doi: 10.1016/j.trsl.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 191.Losurdo G, et al. The interaction between celiac disease and intestinal microbiota. J Clin Gastroenterol. 2016;50(Suppl 2):S145–S147. doi: 10.1097/MCG.0000000000000682. [DOI] [PubMed] [Google Scholar]
- 192.Tang WH, Hazen SL. The contributory role of gut microbiota in cardiovascular disease. J Clin Invest. 2014;124(10):4204–4211. doi: 10.1172/JCI72331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lucas A, Cole TJ. Breast milk and neonatal necrotising enterocolitis. Lancet. 1990;336(8730):1519–1523. doi: 10.1016/0140-6736(90)93304-8. [DOI] [PubMed] [Google Scholar]
- 194.Gartner LM, et al. Breastfeeding and the use of human milk. Pediatrics. 2005;115(2):496–506. doi: 10.1542/peds.2004-2491. [DOI] [PubMed] [Google Scholar]
- 195.Foster JP, Seth R, Cole MJ. Oral immunoglobulin for preventing necrotizing enterocolitis in preterm and low birth weight neonates. Cochrane Database Syst Rev. 2016;4:CD001816. doi: 10.1002/14651858.CD001816.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Srinivasjois R, Rao S, Patole S. Prebiotic supplementation in preterm neonates: updated systematic review and meta-analysis of randomised controlled trials. Clin Nutr. 2013;32(6):958–965. doi: 10.1016/j.clnu.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 197.Costeloe K, et al. Bifidobacterium breve BBG-001 in very preterm infants: a randomised controlled phase 3 trial. Lancet. 2016;387(10019):649–660. doi: 10.1016/S0140-6736(15)01027-2. [DOI] [PubMed] [Google Scholar]
- 198.Kuper H, Adami HO, Trichopoulos D. Infections as a major preventable cause of human cancer. J Intern Med. 2000;248(3):171–183. doi: 10.1046/j.1365-2796.2000.00742.x. [DOI] [PubMed] [Google Scholar]
- 199.Irrazabal T, et al. The multifaceted role of the intestinal microbiota in colon cancer. Mol Cell. 2014;54(2):309–320. doi: 10.1016/j.molcel.2014.03.039. [DOI] [PubMed] [Google Scholar]
- 200.Franco AT, et al. Activation of beta-catenin by carcinogenic Helicobacter pylori . Proc Natl Acad Sci USA. 2005;102(30):10646–10651. doi: 10.1073/pnas.0504927102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wang F, et al. Helicobacter pylori infection and normal colorectal mucosa-adenomatous polyp-adenocarcinoma sequence: a meta-analysis of 27 case–control studies. Colorectal Dis. 2014;16(4):246–252. doi: 10.1111/codi.12290. [DOI] [PubMed] [Google Scholar]
- 202.Hatakeyama M, Higashi H. Helicobacter pylori CagA: a new paradigm for bacterial carcinogenesis. Cancer Sci. 2005;96(12):835–843. doi: 10.1111/j.1349-7006.2005.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Gao Z, et al. Microbiota disbiosis is associated with colorectal cancer. Front Microbiol. 2015;6:20. doi: 10.3389/fmicb.2015.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ahn J, et al. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst. 2013;105(24):1907–1911. doi: 10.1093/jnci/djt300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Castellarin M, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22(2):299–306. doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Han YW, et al. Identification and characterization of a novel adhesin unique to oral fusobacteria. J Bacteriol. 2005;187(15):5330–5340. doi: 10.1128/JB.187.15.5330-5340.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Fardini Y, et al. Fusobacterium nucleatum adhesin FadA binds vascular endothelial cadherin and alters endothelial integrity. Mol Microbiol. 2011;82(6):1468–1480. doi: 10.1111/j.1365-2958.2011.07905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Rubinstein MR, et al. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14(2):195–206. doi: 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317(5834):124–127. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- 210.Salcedo R, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med. 2010;207(8):1625–1636. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hu B, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci USA. 2010;107(50):21635–21640. doi: 10.1073/pnas.1016814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zaki MH, et al. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol. 2010;185(8):4912–4920. doi: 10.4049/jimmunol.1002046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Chen GY, et al. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol. 2011;186(12):7187–7194. doi: 10.4049/jimmunol.1100412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol. 2011;11(1):9–20. doi: 10.1038/nri2891. [DOI] [PubMed] [Google Scholar]
- 215.Fukata M, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133(6):1869–1881. doi: 10.1053/j.gastro.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wang EL, et al. High expression of Toll-like receptor 4/myeloid differentiation factor 88 signals correlates with poor prognosis in colorectal cancer. Br J Cancer. 2010;102(5):908–915. doi: 10.1038/sj.bjc.6605558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Doan HQ, et al. Toll-like receptor 4 activation increases Akt phosphorylation in colon cancer cells. Anticancer Res. 2009;29(7):2473–2478. [PMC free article] [PubMed] [Google Scholar]
- 218.Fukata M, et al. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: role in proliferation and apoptosis in the intestine. Gastroenterology. 2006;131(3):862–877. doi: 10.1053/j.gastro.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Fukata M, Abreu MT. TLR4 signalling in the intestine in health and disease. Biochem Soc Trans. 2007;35(6):1473–1478. doi: 10.1042/BST0351473. [DOI] [PubMed] [Google Scholar]
- 220.Yu LC, et al. Host-microbial interactions and regulation of intestinal epithelial barrier function: from physiology to pathology. World J Gastrointest Pathophysiol. 2012;3(1):27–43. doi: 10.4291/wjgp.v3.i1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4(8):579–591. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 222.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68(12):7010–7017. doi: 10.1128/IAI.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Bernstein CN, et al. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer. 2001;91(4):854–862. doi: 10.1002/1097-0142(20010215)91:4<854::AID-CNCR1073>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 224.Lukas M. Inflammatory bowel disease as a risk factor for colorectal cancer. Dig Dis. 2010;28(4–5):619–624. doi: 10.1159/000320276. [DOI] [PubMed] [Google Scholar]
- 225.Uronis JM, et al. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One. 2009;4(6):e6026. doi: 10.1371/journal.pone.0006026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Azcarate-Peril MA, Sikes M, Bruno-Barcena JM. The intestinal microbiota, gastrointestinal environment and colorectal cancer: a putative role for probiotics in prevention of colorectal cancer? Am J Physiol Gastrointest Liver Physiol. 2011;301(3):G401–G424. doi: 10.1152/ajpgi.00110.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wu S, et al. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology. 2003;124(2):392–400. doi: 10.1053/gast.2003.50047. [DOI] [PubMed] [Google Scholar]
- 228.Goodwin AC, et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc Natl Acad Sci USA. 2011;108(37):15354–15359. doi: 10.1073/pnas.1010203108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Cuevas-Ramos G, et al. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci USA. 2010;107(25):11537–11542. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Singh N, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40(1):128–139. doi: 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Alasmari F, et al. Prevalence and risk factors for asymptomatic Clostridium difficile carriage. Clin Infect Dis. 2014;59(2):216–222. doi: 10.1093/cid/ciu258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Carter GP, Rood JI, Lyras D. The role of toxin A and toxin B in the virulence of Clostridium difficile . Trends Microbiol. 2012;20(1):21–29. doi: 10.1016/j.tim.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 233.Kelly CP, et al. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J Clin Invest. 1994;93(3):1257–1265. doi: 10.1172/JCI117080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Sorg JA, Sonenshein AL. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol. 2008;190(7):2505–2512. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Wilson KH, Perini F. Role of competition for nutrients in suppression of Clostridium difficile by the colonic microflora. Infect Immun. 1988;56(10):2610–2614. doi: 10.1128/iai.56.10.2610-2614.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Theriot CM, et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun. 2014;5:3114. doi: 10.1038/ncomms4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Buffie CG, et al. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect Immun. 2012;80(1):62–73. doi: 10.1128/IAI.05496-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Lawley TD, et al. Targeted restoration of the intestinal microbiota with a simple, defined bacteriotherapy resolves relapsing Clostridium difficile disease in mice. PLoS Pathog. 2012;8(10):e1002995. doi: 10.1371/journal.ppat.1002995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Reeves AE, et al. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile infection. Gut Microbes. 2011;2(3):145–158. doi: 10.4161/gmic.2.3.16333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Hopkins MJ, Macfarlane GT. Changes in predominant bacterial populations in human faeces with age and with Clostridium difficile infection. J Med Microbiol. 2002;51(5):448–454. doi: 10.1099/0022-1317-51-5-448. [DOI] [PubMed] [Google Scholar]
- 241.Rea MC, et al. Clostridium difficile carriage in elderly subjects and associated changes in the intestinal microbiota. J Clin Microbiol. 2012;50(3):867–875. doi: 10.1128/JCM.05176-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.van Nood E, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile . N Engl J Med. 2013;368(5):407–415. doi: 10.1056/NEJMoa1205037. [DOI] [PubMed] [Google Scholar]
- 243.Garza DR, Dutilh BE. From cultured to uncultured genome sequences: metagenomics and modeling microbial ecosystems. Cell Mol Life Sci. 2015;72(22):4287–4308. doi: 10.1007/s00018-015-2004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Thiele I, Palsson BO. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010;5(1):93–121. doi: 10.1038/nprot.2009.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Chen J, et al. Improving metabolic flux estimation via evolutionary optimization for convex solution space. Bioinformatics. 2007;23(9):1115–1123. doi: 10.1093/bioinformatics/btm050. [DOI] [PubMed] [Google Scholar]