

Abstract
Due to the fact that chronic inflammation as well as tumorigenesis in the gut is crucially impacted by the fate of intestinal epithelial cells, our article provides a comprehensive overview of the composition, function, regulation and homeostasis of the gut epithelium. In particular, we focus on those aspects which were found to be altered in the context of inflammatory bowel diseases or colorectal cancer and also discuss potential molecular targets for a disease-specific therapeutic intervention.
Keywords: Intestinal epithelial cells, Inflammatory bowel diseases, Colorectal cancer, Colitis-associated cancer, Epithelial integrity
Introduction
The gastrointestinal tract forms the largest contact surface between the human body and the environment (more than 300 m2). This large size is indeed a physiological adaption to fulfil absorption/nutrition functions. In this context, the equilibrium between oral tolerance to food antigens and microflora and protective immune responses against pathogens and harmful substances is crucial for the maintenance of intestinal homeostasis. Accordingly, intestinal mucosa represents a complex system in which immune and non-immune cells work together in a tightly regulated manner. Within this system, intestinal epithelium seals the intestinal mucosa towards the lumen and builds up a primary physical and immune barrier [1]. Although cellular transport within intestinal epithelia cells (IECs) is required for absorption and nutrition, the tightness of intestinal epithelium has to be maintained to avoid invasion of luminal components which could otherwise activate immune cells in the subepithelial compartment and generate local immune responses. Consequently, epithelial barrier function plays a relevant role in intestinal homeostasis and impacts on the pathogenesis of gut disorders, such as inflammatory bowel diseases (IBD) and colorectal cancer (CRC).
Crohn’s disease (CD) and ulcerative colitis (UC) represent the two main forms of IBD and show an increasing incidence and high prevalence in Europe and North America [2]. Patients affected by this chronic relapsing disease develop clinical symptoms like diarrhea, abdominal pain, rectal bleeding, fever, weight loss, fatigue and a considerable loss of life quality. Despite intense scientific efforts in the field of gastroenterology and immunology, the exact etiology of IBD remains undefined. IBD represents a multifactorial disease and the characteristic breakdown of intestinal homeostasis is considered to arise from a complex interaction between immunological and environmental factors in a genetically predisposed individual [3, 4]. The complex interplay between dysregulated mucosal immune cells, bacterial flora and impaired epithelial barrier function in the inflamed gut makes it highly challenging to define molecular targets for optimized IBD therapy. While current therapeutic strategies mainly focus on the control of overwhelming immune cell activation, during the last years a growing attention has also been paid to mucosal healing and maintenance of the epithelial barrier integrity [5]. Several studies were able to describe promising approaches with a beneficial effect on epithelial permeability (vitamin D, AT1001, heparanoid compounds), mucus production or integrity of the epithelial monolayer (butyrate, TFF3, anti-TNF antibodies) [6–8]. Taking into account the increased colon cancer risk of IBD patients with a prolonged course of disease [9], restoration and maintenance of epithelial homeostasis becomes particularly important as inflammation-associated epithelial stress is strongly suggested to be a key driver of IBD-associated colorectal cancer [10]. Beside duration of disease, the risk of IBD patients for cancer development depends on the extent of disease at diagnosis, disease severity and efficacy of IBD treatment [11]. In general, intestinal cancer disease (including sporadic colon cancer and inflammation-associated colon cancer) represents one of the major causes of death in developed countries, and its incidence is growing over the last years. It is estimated that 1.4 million new cases of colorectal cancer and almost 694,000 disease-related deaths occurred for instance in 2012 [12]. According to the Fearon and Vogelstein’s model [13], colorectal tumorigenesis shows three key features: tumor development is initiated by clonal expansion of a small number of cells (monoclonal); a regulated sequence of common key genetic alterations provides tumor cells with growth and survival advantages over normal gut epithelial cells; the accumulation of mutations within a tumor determines clinical and histopathological tumor manifestations [14, 15]. In addition to well-described common genetic alterations, other low-frequency candidate mutations are known to contribute relevantly to the heterogeneity of colorectal cancer [16, 17]. Overall, the heterogeneity of involved tumor suppressors, oncogenes and low-frequency somatic mutations makes it highly sophisticating to therapeutically control intestinal tumor growth or metastasis [16–18]. Accordingly, an extensive scientific effort is currently ongoing to further improve our understanding of molecular events and signaling cascades involved in control of epithelial homeostasis in the gut, as well as to carefully describe the interplay between intestinal epithelial cells (IECs) and their surrounding environment.
Due to the fact that chronic inflammation as well as tumorigenesis in the gut is crucially impacted by the fate of intestinal epithelial cells, the following article will try to provide a comprehensive overview of the composition, function, regulation and homeostasis of the gut epithelium. In particular, we will focus on those aspects which were found to be altered in the context of inflammatory bowel diseases or colorectal cancer and will also discuss potential molecular targets for a disease-specific therapeutic intervention (Fig. 1).
Fig. 1.
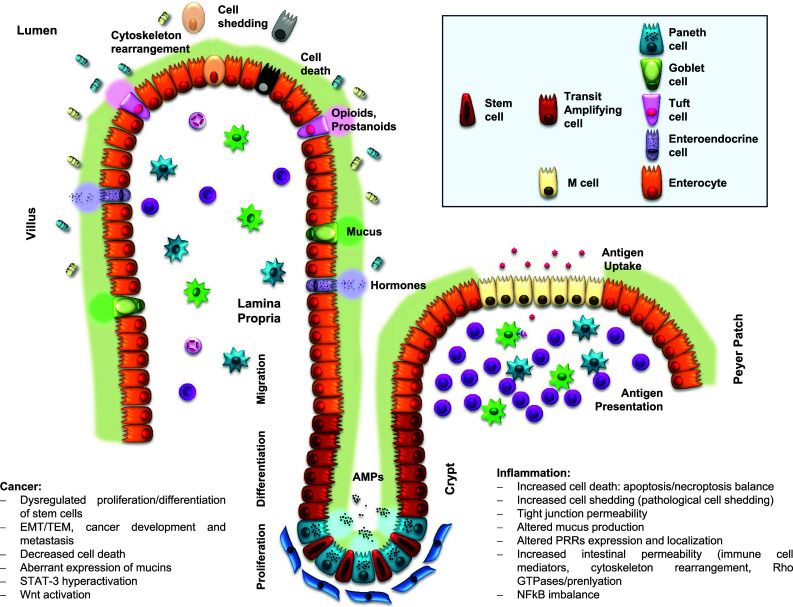
Intestinal epithelium: structure and key molecular events
Cellular composition and turnover of intestinal epithelium
The intestinal epithelium consists of a monolayer of polarized columnar epithelial cells. Cell composition of this epithelial layer is not homogenous, but contains undifferentiated stem cells, partially differentiated progenitors (transient-amplifying cells) and up to six subtypes of fully differentiated IECs. Mature and differentiated IECs can be functionally categorized in two lineages: enterocytes or absorptive IECs and secretory IECs [19]. Secretory IECs include Goblet cells, Paneth cells, enteroendocrine cells and Tuft cells [20, 21]. Finally, M cells represent specialized IECs which don’t fit in one of the before mentioned categories. M cells are located at the luminal surface of intestinal lymphoid structures and participate in the mucosal uptake of luminal antigens [22]. Beyond cell composition, the complex 3D structure of the intestinal surface significantly contributes to the uniqueness of gut epithelium. The epithelium in small intestine is folded and builds up finger-like structures and invaginations, denoted villus and crypts [23]. The described surface formation results in a large contact area between epithelium and luminal contents and thus allows highly efficient absorption of water and nutrients [24]. However, large intestine lacks villus structures and therefore provides optimized conditions for peristalsis and transport of faecal materials [25].
In adults, the intestinal epithelium represents the most robustly self-renewing tissue. Only 4–5 days are required for its complete renewal by a continuous cell migration from the crypt bottom to the villus tip. Stem cells located at the crypt base proliferate permanently and give rise to the transit-amplifying cells, partially differentiated cells with proliferative activity (progenitors). Due to the pressure of these upcoming cells at the crypt bottom, IECs move upwards along the villus, where the majority of differentiated and specialized IECs are located. At the surface of the villus tip, aged IECs will be released from the monolayer (epithelial cell shedding).
Under physiological conditions, the dynamic interaction between stem cell differentiation, cell migration from the crypt bottom to the villus tip, cell proliferation, cell death and shedding of aged cells is tightly regulated in order to enable a permanent renewal of gut epithelium without loss of barrier function and tissue homeostasis [26]. In the following paragraphs, we will focus on specific aspects of epithelial turnover in the gut and on their impact on intestinal pathology.
Stem cell proliferation and differentiation
Self-renewal of the intestinal epithelium is enabled by a permanent proliferation of stem cells located at the crypt, leading to the replenishment of the intestinal epithelium [27, 28].
Inserted between Paneth cells, crypt base columnar stem cells (CBCs; Lgr5+ cells) [29] give rise to rapidly dividing daughter cells, which will in turn differentiate into IECs of the secretory or absorptive lineage. Even ex vivo, Lgr5+ cells are able to generate organoids mimicking the architecture of the intestinal epithelium and containing all subtypes of differentiated IECs [30]. Multipotent Lgr5+ CBCs expand permanently and undergo a rapid random chromosome segregation, which increases the risks of genomic errors [31–33]. An alternative stem cell population is located in position +4 from the crypt, namely quiescent DNA label-retaining cells (LRCs) [34–36]. Bmi1+ LRCs are mitotic-inactive cells, whose multipotency has been demonstrated in vitro [30, 37] and in vivo [38]. LRCs can be activated by radiation and might play a role in recovery after injury [37]. However, their capacity to function as stem cells is still a matter of controversial discussions. There exists evidence which argues against an unidirectional and hierarchical pathway for differentiation of IECs, but rather supports the idea of a more complex plasticity and interconversion between these two multipotent cell populations depending on certain circumstances [39, 40]. Thus, Lgr5+ would work as main stem cells under physiological conditions and their function might be complemented or overtaken by LRCs in stress situations, such as tissue injury.
In general, stem cell differentiation is directed by various cell-extrinsic factors present in the intestinal stem cell niche located in the crypt area, which carefully control the balanced generation of different populations from identical precursors [41]. The majority of these factors are provided by neighboring cells located in close proximity to stem cells in the crypts. For instance, intestinal subepithelial myofibroblasts (ISEMFs) promote stem cell renewal and differentiation via secreted mediators (e.g. Wnt, TGF-β, growth factors and matrix metalloproteinases), but also by cell–cell interactions [42]. In addition, Paneth cells (or an analogous cell population in the colon) [43] are physically associated to Lgr5+ cells at the crypt bottom and produce essential niche signals for the proliferation and survival of these progenitors, such as EGF, TGF-α, Wnt3 and the Notch ligand Dll4 [44]. Interestingly, Paneth cells can also promote the differentiation of Bmi1+ LRCs [45]. Among these stem cell controlling factors, Wnt proteins fulfill a leading position [46, 47] and therefore the Wnt/β-catenin pathway and its impact on gut physiology will be discussed in a separate subchapter of this article.
The development of colorectal cancer is unavoidably linked to dysregulated proliferation and differentiation of stem cells in the crypt. Indeed, ISEMFs, which represent important regulators of stem cell differentiation, were found to be involved in the development of cancer [48]. However, there is evidence which supports the idea that any kind of epithelial cell is potentially able to drive neoplasia [49, 50], based on the recently proposed cellular plasticity processes [51]. Thus, as a consequence of key genetic alterations or external stimuli, differentiated somatic cells might be able to undergo reprogramming into induced pluripotent stem cells (iPSCs) and acquire the ability to proliferate in a stem cell-like manner [47]. Despite these observations, cancer stem cells represent the main drivers of colorectal cancer development and metastasis [52]. Accordingly, expression of stem cell markers like Lgr5, DCLKL1, CD133, CD44 and CD24 is increased in highly proliferating fractions of colorectal cancer [53]. One step further, the plasticity of stem cells allows the epithelial-mesenchymal transition (EMT), which crucially contributes to the growth and spread of primary tumors and represents a key process for the metastatic behavior of colorectal cancer [51]. Upregulation of EMT gene expression within human colorectal tumors could clearly be shown by transcriptomic analysis [54]. Furthermore, mesenchymal-epithelial transition (MET) is required for restoring the proliferative activity of metastatic tumor cells, which are located far away from the primary tumor site [51]. Summing up, the balance between EMT/MET is critical for the development and metastatic behavior of CRC.
Survival, apoptosis and necroptosis
In the intestinal epithelium, programmed cell death is a critical process which allows the elimination of non-functional (aged/damaged) cells and permits maintenance of epithelial integrity and consequently gut homeostasis [55]. Dysregulation of cell death within IECs was shown to be associated with severe gut pathology: increased cell death leads to breakdown of the epithelium barrier function, invasion of luminal content and subsequent inflammation, while decreased cell death contributes to the development of cancer [55–57].
Historically, cell death was differentiated into apoptosis and necrosis, or programmed and unregulated injury-like cell death, respectively [58]. This simplistic view has changed dramatically in the last 10 years, since new forms of cell death have been discovered and in particular the process of necroptosis has been defined [59]. Indeed, several studies demonstrated that these new players are crucial for the homeostasis of intestinal epithelium and development of gut pathologies, such as IBD and colorectal cancer [60, 61].
Described for the first time in 1970, apoptosis is defined as a programmed cell death induced by various stimuli and finally ending up in caspase activation. Apoptosis can occur via an intrinsic or an extrinsic pathway, depending on the triggering stimulus. Classically proposed as the antithesis of programmed cell death, necrosis is activated in response to external stress (infections, toxins, trauma, etc.) and results in the breakdown of the cell membrane and release of the intracellular content to the extracellular milieu. The immune system will be activated upon necrosis due to the production of pro-inflammatory cytokines by neighboring cells sensing this released material. On the interphase between apoptosis and necrosis, necroptosis was first described in 2003 as a new regulated necrotic-linked cell death pathway [62–65]. Although necroptosis is genetically programmed, regulated and activated by receptor ligation, necroptotic cells share the morphology of cells undergoing necrosis. Experiments in caspase-8 or FADD deficient mice nicely demonstrated that necroptotic cell death is negatively regulated by caspases and strictly depends on RIPK [66, 67]. Thus, inactivation of caspases inhibits apoptosis but simultaneously activates necroptosis, which is supposed to play a crucial role in maintenance of tissue homeostasis. Under physiological conditions, epithelial cell death activation in the gut can be observed in single IECs at the villus tip as well as at the crypt bottom. Those apoptotic IECs, which occur at the villus tip in small intestine or at the surface of the crypt in the colon, usually represent aged cells and are designated for shedding into the lumen. This specific process of programmed cell death, which is initiated by the loss of cell/matrix contact and involves caspase-3 activation, is called anoikis [68]. Although an improved understanding of the molecular link between activation of caspase-3 and subsequent cytoskeleton rearrangement in shedding IECs could be achieved recently, several aspects of this tightly regulated process are still a matter of discussion. Moreover, regulation of cell death in immature IECs at the crypt bottom remains even less defined. In general, epithelial apoptosis seems to be dispensable for the maintenance of gut architecture [69–74], but there exists strong evidence proving that dysregulated or excessive apoptosis within intestinal epithelium is associated with inflammation due to breakdown of the epithelial barrier function. The majority of genes associated with excessive IEC apoptosis belong to the NFκB pathway, such as NEMO [75], RELA [76], TAK1 [77], IKK1 [78] and IKK2 [79]. Another study showed the relevance of the transcription factor XBP1 for epithelial cell death, gut homeostasis and pathology in CD and UC patients [80, 81]. Finally, abrogation of STAT3 within IECs resulted in an increased rate of epithelial apoptosis and enhanced susceptibility to experimental colitis [10, 82]. All in all, these studies show the crucial role of apoptosis for the maintenance of epithelial homeostasis.
Apoptosis and necrosis have both been linked to IBD pathogenesis. A marked increase of epithelial cell death can be observed under chronic inflammatory conditions in the gut of IBD patients. UC patients show accumulation of apoptotic bodies in colon [83] and CD patients and individuals suffering from enteric infections are characterized by an increased number of apoptotic IECs [84, 85]. However, this excessive occurrence of programmed cell death in IECs seemed to be limited to inflamed gut areas [86] and therefore more likely represents a secondary phenomenon of inflammation than a causative effect underlying the inflammatory process. In contrast, there exists certain evidence for a causative role of non-apoptotic cell death in IECs in patients suffering from chronic intestinal inflammation. Increased numbers of dying cells in intestinal epithelium showing morphologic features of necrosis could be observed both in inflamed and uninflamed areas of the gut from CD patients and even in CD patients without any clinical sign of active disease [87, 88]. Interestingly, specific inhibition of apoptosis in IECs by inactivation of caspase-8 in mouse epithelium did not alter epithelial architecture but nevertheless went along with induction of inflammation in the terminal ileum [60, 89]. Several studies showed that the mechanism behind this phenomenon involves the induction of RIP3-dependent caspase-3-independent necroptotic death of Paneth cells. Although the location of Paneth cells is mainly restricted to the small intestine in healthy individuals, recent observational studies have shown the presence and/or metaplasia of Paneth cells in distal colon from IBD patients [90, 91]. Interestingly, the presence of Paneth cells directly correlated with the duration of the disease in UC patients, supporting the association between Paneth Cells and inflammation [90]. However, the lack of Paneth cells in healthy colon might indicate that the function of this cell type is not relevant for the initiation of colonic inflammation, but rather represents a consequence of the inflammatory milieu. Therefore, the potential impact of the described pathogenic mechanism and in particular of Paneth cell death on the development of colonic inflammation needs to be defined in future studies. However, a recent study described an increased expression of markers for necroptosis in the gut of pediatric IBD patients, while there were decreased levels of markers for apoptosis [92]. Furthermore, both isoforms of the master anti-apoptotic regulator cFLIP were found to be upregulated in gut tissue of CD and UC patients, implicating the inactivation of caspase-8 and activation of necroptosis in IBD [93].
Thus, an imbalance between apoptosis and necroptosis in intestinal epithelium can be suggested as key pathogenic factors in IBD.
Cell shedding
After differentiation and migration along the crypt-villus axis, aged IECs reach the villus tip, from where they are shed into the lumen. This continuous replacement of older and potentially damaged IECs by upcoming new cells enables the intestinal epithelium to maintain a constant barrier function and homeostasis of the tissue.
Considering epithelial cell extrusion as the final step in the process of epithelial turnover in the gut, it is important to keep in mind the relationship between cell death and shedding. For years, it was believed that activation of caspases occurs shortly before the completion of the shedding event (homeostatic caspase-dependent cell shedding) [94]. Importantly, in most of the underlying studies experimental cell shedding was initiated by administration of well-known inducers of cell death, such as LPS or TNF-α [95, 96]. More recently, it could be shown that cell shedding can also occur in a caspase-independent way, which was then defined as physiological cell shedding. In this process the activation of caspases occurs only secondary to the detachment of IECs from the monolayer and might represent a consequence of cytoskeleton rearrangement [97–99]. In conclusion, current data postulate the co-existence of a homeostatic caspase-independent cell shedding process important for physiological epithelial turnover and a caspase-dependent pathological cell shedding process, which often results in barrier dysfunction and inflammation.
In the process of pathological cell shedding, such as TNF-induced IEC extrusion, caspase activation initiates the following sequence of events: tight junction protein and actin redistribution; actin-myosin, microtubules and dynamin-dependent cell extrusion and closing of the epithelial gap by actin, ROCK, MLCK and dynamin [95]. Although numerous studies described mechanism involved in pathological or apoptosis-mediated cell extrusion [96, 100, 101], little is known about the recently identified process of physiological cell shedding. The latter is independent of caspase activation and occurs in epithelium of healthy tissues in order to maintain the balance between growth and cell death rates. Regarding the mechanism underlying physiological cell shedding, we can only assume that it is also dependent on cytoskeletal rearrangement and in particular involves the cytoskeletal protein ROCK, acto-myosin complexes and redistribution of tight junctions.
Under physiological conditions, the cell shedding event itself compromises the epithelial barrier function temporarily [102]. The immediate recovery of epithelial integrity is achieved by early redistribution of actin and tight junction proteins along the lateral membranes of shedding cells, a coordinated microfilament interaction and finally the sealing of the space left by the shedding cell by adjacent cells [95]. Under inflammatory conditions in IBD patients, the frequency of IEC shedding is markedly increased and subsequently the epithelial layer loses its capacity to maintain an effective barrier function via cytoskeletal rearrangement [103].
Cytoskeleton and tight junctions in gut epithelium
On a subcellular level, maintenance of epithelial cell shape, anchoring of cells to the basal membrane, homeostatic renewal of the intestinal epithelium and constant barrier function of the epithelial monolayer strongly depend on the complex and tightly regulated cytoskeleton network in IECs [26, 98, 104]. Cell membranes from individual epithelial cells act as an impermeable barrier, where solute passage is only possible via specific transporters. However, the space between neighboring cells (paracellular space) represents a vulnerable point within this physical barrier. Several intercellular junctions are therefore needed in order to maintain epithelial integrity: tight junctions, adherens junctions, desmosomes and gap junctions [104]. Tight junctions consist of transmembrane proteins, like for instance claudins, junctional adhesion molecules (JAM) and occludins, which intimately associate with cytoplasmic actin and myosin networks via several adaptor molecules or plaque proteins, such as ZO-1 or TJP1 [105, 106]. Actin cytoskeleton or intermediate filaments guarantee the mechanical strength of these intercellular junctions. Contraction of a perijunctional actomyosin ring further regulates the permeability of intestinal epithelium in a myosin light-chain kinase (MLCK) dependent manner [107]. In addition, the described intercellular junctions efficiently block the intramembranal diffusion of membrane components and thereby enable the apical-basolateral polarity of IECs [108]. Beside tight junctions, adherens junctions (AJs, e.g. E-cadherin, nectins) are assumed to form dynamic connection between epithelial cells, participating in actin polymerization [109, 110]. Finally, desmosomes (desmoglein, desmocolin) bind to keratin intermediate filaments and provide additional structural strength [111]. Tight junctions are known to seal the paracellular pathway, whereas adherens junctions and desmosomes rather provide the mechanical adhesive strength and thereby maintain cellular contacts and allow tight junction assembly. As a functionally unique intercellular junction, gap junctions (connexins, pannexins) act as direct communication sites (connexons) between neighboring cells and allow the exchange of intracellular material (ions, small molecules) [112].
As mentioned before, intercellular junctions are physically and functionally linked to the cytoskeleton of IECs [113, 114] and this association seems to be bidirectional [26]. Therefore, epithelial integrity and barrier function depends not only on the regulation of tight junctions or single components within the system of intercellular junctions, but rather on a dynamic and complex interaction between several of these components. Breakdown of epithelial integrity has been observed after disruption of intercellular junctions and cytoskeleton rearrangement, e.g. in the context of infection or inflammation [115–117]. Another important molecular player in this cellular network are small GTPases [118, 119], which will be discussed later in this article.
Loss of epithelial integrity and increased tight junction permeability is well known to be associated with development of gut inflammatory disorders, such as IBD [120–122]. In CD patients, a significant correlation between increased intestinal permeability and the presence of disease markers or clinical relapse could be shown [123, 124]. It still remains unclear whether this disease-associated loss of epithelial barrier function represents a cause or a consequence of intestinal inflammation. Performed studies in tight junction-deficient mice implicated that increased intestinal permeability due to the lack of single tight junction proteins can be efficiently compensated and so far only claudin-15 deficiency resulted in a pathological intestinal phenotype [125–128]. Rather than intrinsic dysregulation of specific cytoskeletal components, a potential impact of extrinsic factors on the interplay between intercellular junctions and cytoskeleton might underlie the loss of epithelial integrity in IBD patients, like for instance the pro-inflammatory cytokines IL-6 [129], IL-13 [130] and TNF [131, 132]. Indeed, release of TNF in inflamed mucosa triggers an increased expression and activity of myosin light chain kinase activity in IBD [133, 134]. Subsequently, an increased phosphorylation of myosin light-chain-II and the resulting overwhelming contraction of the perijunctional ring might explain the breakdown of epithelial barrier function in those patients [107]. However, in vivo confocal laser endomicroscopy in IBD patients during remission phase was able to demonstrate that loss of epithelial barrier function often precedes the clinical relapse of disease [101, 103, 135]. The latter argues for a causative role of disrupted permeability in IBD pathogenesis. Furthermore, even healthy relatives of Crohn’s disease patients showed an increased intestinal permeability [136–138].
Role of intestinal epithelial cells in immune surveillance
The described barrier function of the intestinal epithelium in the interphase between the gut lumen and the lamina propria of the mucosa also implicates a permanent exposition of IECs to extrinsic microbial as well as immune cell derived stimuli. Accordingly, there are multiple interactions between IECs, gut microbiota and lamina propria immune cells, which crucially impact on IEC function and intestinal homeostasis [139, 140].
After the initiation of postnatal enteral feeding, the gut is colonized by a large amount of microorganisms (microbiota or microflora). The majority of bacteria in the gut accumulate in the ileum and large intestine and a number of 1011 bacteria per gram of tissue is estimated [141, 142]. The mutually beneficial symbiotic relationship between the microbiota and the host supports host nutrition, contributes to the maturation of the host immune system and prevents intestinal mucosa from colonization with enteric pathogens. On the other hand, bacteria from the microbiota obtain a niche and adequate nutrients for their survival. However, bacteria colonization also involves a permanent risk for activation of local immune responses. Therefore, the intestinal epithelium fulfils a lifelong crucial role in immune surveillance, impairing the invasion of luminal content and thereby maintaining tissue homeostasis. Several studies have demonstrated the association between dysbiosis or imbalance of the gut microbiota and IBD [143–145]. Although several bacterial species, such as Mycobacterium paratuberculosis [146, 147], Salmonella [148] or Campylobacter [147], have been discussed as potential causative triggers of IBD development, there is a broad consensus that mainly bacteria from the physiological gut flora are responsible for the initiation of overwhelming intestinal immune responses in IBD. Indeed, experiments with germ-free mice supported the hypothesis that participation of normal microflora components in genetically predisposed individuals contributes to the development of chronic intestinal inflammation [149, 150].
Mucus and antimicrobial peptide production
As an additional physical and chemical outer barrier between luminal microorganisms and the epithelium, highly glycosylated mucins secreted by goblet cells give rise to a gel-like film covering the apical epithelial surface. The production of mucins by Goblet cells contributes to the equilibrium between intestinal epithelium and commensal flora. Being the most abundant component of this mucus layer, MUC2 protein represents an indispensable factor for maintenance of intestinal homeostasis in the colon. Accordingly, MUC2 deficient mice spontaneously develop colitis and are highly predisposed to inflammation-associated tumor disease [151, 152]. Besides MUC2, additional components, such as trefoil factors (TFF) or resistin-like molecules (RELM), significantly impact on the barrier function of the mucus layer. For instance, TFF3 contributes to the integrity of the mucus layer, promotes epithelial repair and resistance to apoptosis [7], while RELMβ drives mucin secretion, regulates inflammation and protects against parasite infection [153, 154].
The protective effect of the mucus layer is further supported by its ability to retain antimicrobial peptides (AMP) which are able to target and kill luminal bacteria [155]. AMP represent endogenous antibiotics which are able to recognize essential and highly conserved bacterial patterns and thereby control the composition of the gut microflora. The majority of AMP, such as defensins, cathelicidins and lysozyme, are secreted by Paneth cells in the crypt base of the small intestine, although certain AMP (e.g. RegIIIγ and β-defensins) are produced by absorptive enterocytes in small and large intestine [156]. Accumulation of secreted AMP within the mucus layer results in the formation of an efficient innate barrier which relevantly limits the direct contact between gut microbiota and the gut epithelium [21, 157]. Data from animal and human studies demonstrated that an impaired antimicrobial defense promotes the development of chronic inflammatory diseases such as IBD [158]. For instance, lack of cathelecidin in mice lead to increased severity of DSS-induced colitis [159]. In humans, CD patient with an ileal disease manifestation were found to be characterized by an decreased expression of α-defensins in intestinal epithelium and a subsequently diminished epithelial barrier function [158, 160]. Interestingly, decreased expression levels of α-defensins in CD patients turned out to be independent on inflammation [160, 161] and might therefore represent a very early step within the pathogenesis of this disease. Regarding the underlying mechanisms which are responsible for the observed pathologic lack of α-defensins, a compromised Paneth cell function and the inability of monocytes to provide adequate amounts of WNT ligands for Paneth cell stimulation was described in CD patients [160, 161]. Interestingly, in UC patients the expression of α-defensins is upregulated [162]. However, the antimicrobial defense might still be compromised due to the observed decreased thickness of the mucus layer in those patients [163]. Concerning CRC, the potential impact of AMP on tumor development has not been studied extensively. However, some studies claim that the ability of AMP to orchestrate adaptive immune responses might play a role in tumorigenesis [164, 165], but this has not been investigated in the context of CRC. Therefore, further research is needed in order to improve our understanding of this interesting aspect.
Pattern recognition receptors
Beyond a simple physical barrier, the intestinal epithelium fulfills an important innate immune function by recognizing microbes via pattern recognition receptors (PRRs) [139]. Belonging to the PRR family, Toll-like receptors (TLRs), NOD-like receptors (NLRs) and RIG-like receptors (RLRs) expressed on the surface of IECs act as frontline sensors of microbes and are able to initiate antimicrobial as well as immunoregulatory responses. TLR binding of respective ligands results in dimerization of the TLR, recruitment of specific adaptor proteins and activation of subsequent signaling pathways. For instance, Mal or (TRAM)/TRIF represent important TLR associated adaptor proteins, which are linked to downstream NFκB or IRF signaling, respectively [166, 167]. Microbiota-driven TLR signaling impacts on IEC proliferation and differentiation, IgA production, maintenance of epithelial tight junctions and release of antimicrobial peptides and thus represents an important control element in the convoluted coexistence of gut microflora and host organism [168, 169].
The crucial involvement of TLRs in IEC biology implicates that TLR signaling also impacts on intestinal pathology. Indeed, TLR expression was found to be upregulated in polarized intestinal epithelial cells in the gut of IBD patients (107). This phenomenon is most probably driven by an increased presence of proinflammatory cytokines, such as IFNγ [170], TNF [171] or IL-13 [172]. As TLRs on the apical surface of IECs are assumed to be tolerant against luminal stimuli, breakdown of epithelial integrity under inflammatory conditions might further trigger TLR signaling by allowing basolateral stimulation [173–175]. Interestingly, some TLRs show a disease-specific alteration of their intracellular distribution pattern: TLR3 expression is shifted to the basolateral surface in colon epithelial cells of ulcerative colitis patients [176] and TLR4 accumulates at the apical side in Crohn’s disease patients [176]. In addition to their altered expression and subcellular localization, the functional relevance of TLRs in gut epithelium was also found to be modified under inflammatory conditions. Although TLR4 and the TLR adaptor protein MYD88 are dispensable for bacterial-regulated proliferation of IECs under physiological conditions, this turned out to be not true in the context of experimental colitis in dextran sulfate sodium (DSS) exposed mice. Decreased IEC proliferation and increased IEC apoptosis could be detected in the inflamed colon of MYD88 and TLR4 deficient mice. Regarding the underlying mechanism, TLR4 signaling was suggested to contribute to IEC proliferation by inducing a COX-2/PGE2 dependent expression of epithelial growth factors (EGFR) [177] and by supporting the recruitment of mesenchymal stromal cells (fibroblast, macrophages) [178]. Although activation of other TLRs, such as TLR2, TLR3 or TLR9, was also described to be beneficial in the context of colitis, these effects were probably not mediated by a direct initiation of IEC intrinsic signaling cascades [179–181]. Finally, there exist intracellular negative regulators of TLR function which are responsible for the tolerance of epithelial TLRs against physiological gut microbiota. Altered function of these regulatory molecules goes along with breakdown of intestinal homeostasis and subsequent development of inflammation [182, 183]. Interestingly, patients suffering from IBD show a defective function of some of these TLR negative regulators, such as TOLLIP [184].
However, the capacity of epithelial TLRs to sense injury in the intestine and to limit the extent of damage by inhibition of IEC apoptosis, dampening of proinflammatory pathways and induction of IEC proliferation also has a negative side. Studies performed in experimental models of colorectal cancer clearly demonstrated that intensive TLR signaling is able to drive tumorigenesis and promote cancer development [185]. Interestingly, a protective effect in cancer development was described for some NLRs. This beneficial effect of NLRs was mediated by different mechanisms, such as regulation of proliferation and cell death of transformed IECs or IL-18-dependent tissue repair [183, 186, 187].
Lympho-epithelial interactions
IEC function and epithelial integrity is relevantly regulated by lamina propria immune cells and surrounding stromal cells [139]. Thus, a number of extrinsic factors, like pro-inflammatory cytokines or pro-apoptotic stimuli, are well known to impact on barrier function of the epithelial layer in the gut. Vice versa, IECs themselves produce soluble compounds and express specific surface proteins that in turn regulate the function of immune cells in the epithelial environment in order to generate tolerance, limit inflammation and support adequate immune responses. IECs constitutively express major histocompatibility complex (MHC) class I and II molecules and a diverse set of non-classical MHC class I molecules on their surface [188]. Thereby, IECs are able to act as non-professional antigen-presenting cells and to process and present luminal antigens to antigen-specific lymphocytes within the lamina propria [189]. Although IECs lack classical co-stimulatory molecules, the expression of non-classical MHC class I molecules and molecules of the B7 family [190] enables them to fulfill a costimulatory function on immune cells. Interestingly, the expression of epithelial MHC-II in the gut is upregulated in response to pro-inflammatory signals [191, 192] and intestinal inflammation in IBD patients significantly impacts on MHC-I and II compartments in intestinal epithelial cells [193]. A recently performed in vivo study showed that abrogation of the inflammation-induced MHCII expression on IECs in the context of experimental colitis resulted in a more severe course of disease and in an accumulation of CD4+ Th1 cells in the gut, implicating a relevant involvement of MHC-II expressing IECs in T cell-dependent mechanisms of intestinal mucosal tolerance [194].
Among those IEC derived factors which relevantly impact on T cell fate, tumour necrosis factor (TNF) represents one of the most intensively studied and best characterized players in intestinal homeostasis.
Members of the tumour necrosis factor superfamily (TNFSF; including 19 ligands and 29 receptors) display a broad spectrum of activities within intestinal mucosa and impact both on intestinal epithelium and immune cells. TNF proteins initiate signaling pathways related to cell survival, proliferation, differentiation and apoptosis and trigger production of inflammatory mediators. In brief, the ligand binds to its corresponding receptor, leading to the recruitment of adaptor proteins and initiation of subsequent signaling pathways [195]. The overall consequence of TNF signaling in intestinal mucosa strongly depends on the cellular context and on involved signaling pathways. Despite the well-established role of TNF members in T cell-mediated immune responses and autoimmune diseases [196, 197], TNF signaling also represent a key event in the maintenance of epithelial integrity [198] with relevant impact on IBD pathogenesis.
Tumour necrosis factor-alpha (TNF-α, or TNFSF2) represents the best known member of the TNF family. There exist two forms of TNF-α, a transmembrane and a soluble protein. While transmembrane TNF-α preferentially binds to TNF receptor 2 (TNFR2), soluble TNF-α predominantly interacts with TNFR1. TNF-α expression occurs in various cell types and is upregulated by various proinflammatory factors [199]. TNF gene was identified as a susceptible IBD locus [200–202] and expression of TNF-α was found to be elevated in IBD patients in several tissue compartments: TNF-α mRNA in colonic tissue [203, 204]; TNF-α protein in serum [205–207], stool [208], intestinal lamina propria and submucosa [209–212]. However, there also exist few studies which were not able to confirm increased TNF-α levels in IBD [213, 214]. Expression levels of TNFR are also elevated in serum [215], intestinal epithelial cells [216] and CD4+ cells [217] of IBD patients. Furthermore, regarding functional aspects of TNF-α signaling in the gut, experimental overexpression of TNF-α in mice resulted in the development of intestinal pathology resembling Crohn’s disease [218]. Finally, the highly successful use of anti-TNF-α agents in the treatment of IBD patients provides an unequivocal proof of the crucial functional role of this pathway in the pathogenesis of CD and UC. In addition to its beneficial effects on immune cells, therapeutic blockade of TNF-α is assumed to impact on rearrangement and expression of tight junction proteins and to protect epithelial barrier function [219–221].
In particular under inflammatory conditions, IECs are able to express TNFR1,TNFR2 and the pro-inflammatory cytokine TNF-α [216, 222, 223], which is also produced by invading immune cells and stroma cells in the gut mucosa. Accordingly, there is a well-established role of TNF-α in epithelial barrier regulation in the gut. As a first line of defense, TNF significantly contributes to a balanced mucus production by favoring IEC differentiation into the secretory lineage [224] and inducing MUC2 mRNA expression [225], but also by induction of cell death in Goblet cells [8]. Regarding intercellular junctions and epithelial cytoskeleton, TNF-α is able to modulate the expression of several tight junction proteins [171, 226] and, as already mentioned, triggers expression and activation of myosin light-chain kinase [107, 133, 227, 228]. Redistribution of tight junction and adherens junction proteins in the presence of TNF-α results in the induction of IEC shedding and thereby further impacts on epithelial integrity [95, 101, 198]. Interestingly, several studies implicated an association between hyperpermeability of intestinal epithelium and regulation of TNF sheddase enzymes, which catalyze shedding of the membrane-bound TNF-α protein from the cell surface [132, 229]. Finally, TNF-α represents an important regulator of IEC survival. On the one hand, TNF signaling is able to directly activate cell death within IECs [230–232] or induce the expression of the pro-apoptotic protein p53 [231] and thereby contributes to barrier defects. On the other hand, TNF-α protects from intestinal apoptosis and promotes wound healing in a ErbB- and Wnt-dependent pathway [233–235]. Thus, the overall consequence of TNF-α signaling in the intestinal epithelium is context-dependent and is determined by the selected intracellular signaling cascade which is triggered by TNFR ligation. Beside TNF-α, other TNF family members were found to be associated with dysfunction of epithelial barrier function in the context of IBD. These are for instance TL1A [236, 237], FasL [238–240], LIGHT [241], TRAIL [242] or TWEAK [131].
Although TNF might be considered as the main extrinsic regulator of IEC function, it is not the only immune cell derived factor involved in epithelial homeostasis in the gut. In particular, IL-6 and IL-22 should be taken into account in this context. IL-22 is produced predominantly by T cells and natural killer cells, while the IL-22 receptor is expressed solely on non-hematopoietic cells including intestinal epithelial cells [243]. In Crohn’s disease patients, lamina propria T cells and macrophages could be identified as major cellular source of intensively expressed IL-6 [244]. Most of IL-6 and IL-22 initiated effects on IECs are mediated via activation of STAT-3 and finally result in induction of IEC proliferation and inhibition of apoptosis. Due to the key importance of the STAT/JAK pathway in the intestinal epithelium of IBD patients, this pathway will be described in a separate subchapter in this article.
Master molecular regulators and signaling pathways in intestinal epithelium
As described before, stability, function and maintenance of the intestinal epithelial monolayer strictly depends on a balanced interplay between IEC differentiation, proliferation, survival and extrusion as well as on the capacity of IECs to respond to multiple epithelial-extrinsic triggers, like for instance components of gut microflora or immune cell derived mediators. The fate of IECs within this complex setting is tightly regulated by an intracellular network of key molecular factors organized in different signaling pathways. Selected candidates of this network, their disease-specific alteration and their applicability as potential therapeutic targets will be discussed in the following paragraphs.
NFκB signaling
The transcription factor NFκB controls the expression of a variety of genes critically involved in IEC homeostasis [245]. The NFκB/Rel family consists of 5 peptides (p50, p52, RelA, RelB, c-Rel) which form homo or heterodimers [246]. Inactive dimers are localized in the cytosol, where they are retained by an inhibitor protein (IκB). Ubiquitination and proteosomal degradation of IkB finally enables nuclear translocation and subsequent activation of NFκB [247]. NFκB target genes can be classified in four main groups: inflammation related genes, genes involved in cell cycle, anti-apoptotic mediators and negative regulators of NFκB signaling [248, 249].
Numerous studies clearly demonstrated a crucial involvement of NFκB in the pathogenesis of IBD. Increased levels of activated NFκB could be detected in the inflamed gut in different mouse models of IBD and, moreover, specific blockade of NF-kB signaling was able to ameliorate severity of experimental colitis [250–252]. An increased NFκB activation could also be shown in ex vivo stimulated IECs, strongly implicating a functional relevance of this pathway for gut epithelium [253, 254]. In the context of human disease, macrophages and IECs in the intestinal mucosa of IBD patients are characterized by an increased expression and activation of NFκB [250, 255]. With regard to immune cells, activation of NFκB leads to induction of proinflamamtory cytokines like IL-1, IL-2, IL-6 or TNF-α, which in turn contribute to the perpetuation of inflammation. Although NFκB is involved in the regulation of cytokines in gut epithelium as well, in the last ten years a growing attention has been paid to the protective effects of NFκB on epithelial barrier function. Two parallel studies in genetically modified mice strains demonstrated that inhibition of NFκB within IECs results in extensive intestinal inflammation [75, 256]. Interestingly, lack of NFκB activation led to increased apoptosis in IECs and thereby caused a disruption of barrier function and uncontrolled bacterial translocation. The observed protective role of NFκB signaling could further be confirmed by subsequent studies which focused on the role of other components of the NFκB pathway for maintenance of epithelial homeostasis in the gut. In particular, these studies were able to identify a key role of RelA [76], TAK1 [77], IKK1 and IKK2 [78, 79] in IEC biology. Overall, NFκB signaling represents an attractive therapeutic target in the context of IBD, although the opposite quality of NFκB mediated effects in IECs (epithelial protective) and in immune cells (pro-inflammatory) should be taken into account, which makes it necessary to target NFκB signaling in a cell type specific manner.
STAT-3
Signal transducer and activators of transcription (STAT) proteins represent critical components within cytokine induced signaling pathways. Among the 7 members of the STAT family, STAT3 is a pleiotropic mediator that can be activated by a broad panel of cytokines, among which IL-6, IL-11 and IL-22 have been extensively characterized. In general, interaction of the respective cytokine receptor (e.g. gp130) with its ligand results in receptor dimerization and activation of JAK by transphosphorylation. JAK thereupon catalyzes tyrosine phosphorylation of the receptor, which enables binding of STAT proteins. STAT is then activated by a double phosphorylation. First, JAK-mediated phosphorylation allows STAT dimerization, nuclear translocation and DNA binding. In addition, a maximal transcriptional activity of STAT is achieved by MAPK-mediated STAT phosphorylation. IEC relevant target genes of STAT-3 mainly include proteins involved in cell survival and proliferation [10, 257].
In the context of IBD, STAT-3 gene represents a well-known susceptible locus for CD and UC [258, 259], and increased IL-6 levels could be detected in serum and mucosa of affected patients [260]. The consequence of STAT-3 modulation for gut homeostasis depends on the cytokine which triggers STAT-3 activation and on the involved cell type [261–264]. Focusing on the intestinal epithelium, STAT-3 is a decisive factor in the transition between chronic colitis and colitis associated cancer (CAC). Accordingly, increased levels of STAT-3 activation in gut epithelium could be detected in IECs from ulcerative colitis patients with and without diagnosed dysplasia or colon cancer [265]. Conditional knockout mice carrying an IEC specific STAT-3 deficiency are characterized by a disturbed IL-22-induced wound healing and, subsequently, are highly susceptible to development of colitis [82]. Due to the well described impact of IL-22 triggered STAT-3 activation on the expression of mucins and anti-microbial peptides [266], STAT-3 signaling is also crucially involved in the epithelial defense against toxic and infectious agents [266, 267]. Despite the essential role of permanent epithelial turnover for maintenance of epithelial barrier function and wound healing, uncontrolled and overwhelming IEC proliferation represents a hallmark of intestinal cancer development, which might also be driven by activated STAT-3 proteins. While transient activation of STAT-3 is tightly regulated under physiological conditions, cancer cells are characterized by a persistent STAT-3 activation resulting in unlimited promotion of cell growth and inhibition of apoptosis. Accordingly, direct or indirect inhibition of STAT-3 signaling in IECs went along with a reduced intestinal tumor growth in the experimental azoxymethane/DSS model of colitis-associated cancer [10, 257]. Mechanistically, this observation might be explained by an increased induction of apoptosis in colorectal cancer cells in the absence of STAT-3 signaling [268] or by the recently postulated capacity of epithelial STAT-3 to promote gut homing of CD8 T cells and inhibit intestinal accumulation of regulatory T cells [269]. Moreover, the STAT-3 triggering cytokine IL-22 could be identified as a potent inducer of IEC hyperproliferation and driver of colorectal cancer progression in a bacteria-induced model [270]. Interestingly, an increased risk for the development of colon cancer was found to be associated with IL-22 gene polymorphisms [271].
Wnt/β-catenin pathway
Beyond its role during embryogenesis and in maintenance of tissue homeostasis, Wnt pathway is essential for epithelial replenishment and repair of epithelial injury in the gut. Under physiological conditions, the function and growth of intestinal stem cells is tightly regulated by multiple factors. Within this complex interplay, inhibition of the Wnt pathway results in crypt loss and a marked inhibition of epithelial proliferation [272]. Accordingly, the Wnt/β-catenin pathway was found to be altered in the vast majority of colorectal tumors [273].
Briefly, Wnt pathway activation is driven by intracellular accumulation of its main effector β-catenin. In the absence of Wnt, phosphorylated cytosolic β-catenin is included in a protein complex (destruction complex) which facilitates its ubiquitination and proteosomal degradation [274, 275]. Beside others, Axin and Adenomatous polyposis coli (APC) represent functionally important components of this destruction complex. Binding of the Wnt ligand to its respective receptor results in the release of β-catenin from the complex [276]. Subsequently, β-catenin accumulates in the cytosol and translocates into the nucleus, where it forms an active transcriptional complex (TCF/LEF) leading to the expression of Wnt target genes [277, 278]. The list of important Wnt/β-catenin target genes include regulators of cell migration (e.g. EPH), inducers of proliferative signals (e.g. c-myc, cyclin D1) and stem cell and cancer cell markers (e.g. Lgr5, Bmi1) [277, 278]. Regarding the ISC marker Lrg5, it could be shown that binding of R-spondin to Lrg5 is able to promote Wnt signaling [29, 279, 280]. In addition to the before described β-catenin dependent canonical Wnt signaling pathways, Wnt can alternatively affect cell fate via β-catenin independent noncanonical pathways. For instance, noncanonical signaling pathways initiated by Wnt-2 are able to induce a pro-invasive phenotype in cancer cells. Interestingly, recently performed whole-exome sequencing analyses of IBD-associated colon tumors implicated a predominant involvement of noncanonical Wnt pathways in colitis-associated tumorigenesis [281].
The majority of colorectal tumors are classified as nonhypermutated microsatellite stable (MSS) tumors. The sequence of tumorigenic events in MSS tumors was nicely described by the Fearon and Vogelstein’s model [13], which indeed demonstrated the crucial role of Wnt activation in this context. Frameshift or non-sense mutations in the APC gene (tumor suppressor) initiate tumor development, while tumor growth and progression are determined by the accumulation of somatic mutations, affecting genes like KRAS, PI3 K, TGFB, p53 and/or SMAD4 [282, 283]. On a molecular level, loss of APC function impairs the β-catenin destructive protein complex, subsequently blocks β-catenin ubiquitination and results in an uncontrolled activation of the Wnt/β-catenin pathway. Accordingly, APC mutations, both in mouse and human, result in the development of multiple intestinal adenomas [284, 285]. On the other hand, 10–15 % of colorectal tumors are classified as hypermutated microsatellite instable (MSI) tumors, which arise from defects in DNA mismatch repair mechanisms. Among other oncogenes or tumor suppressor genes, proteins involved in Wnt pathway are often mutated in MSI CRC (e.g. APC, β-catenin or AXIN2). Moreover, an epigenetic silencing of several negative regulators of this master pathway could be observed in colorectal cancer [286, 287].
Another important link between Wnt pathway and intestinal epithelial homeostasis arises from the bidirectional regulation between mucin expression and Wnt activation. This is of particular interest because several mucins are known to be aberrantly expressed or downregulated in cancer [288]. In the gut, MUC5AC is exclusively expressed in tumor tissue [289], while expression of MUC1 [290], MUC4 [291, 292] or MUC2 [293, 294] turned out to be decreased in colorectal tumors. Via their cytoplasmic tail, mucins are able to interact with cytosolic β-catenin. Dependent on the specific mucin type and on the cellular context, this interaction might either promote nuclear translocation of β-catenin or result in an accumulation of membrane-bound β-catenin. Vice versa, several studies strongly implicated that β-catenin might directly impact on the transcription of various mucin genes [295]. Subsequently it was postulated that alterations of the mucin expression pattern in colorectal cancer might occur as a consequence of Wnt activation [296–298]. All in all, activation of Wnt pathway in IECs has to be considered as master regulator of colorectal cancer development.
Rho GTPases
Small GTPases of the Ras superfamily act as molecular switches transducing extracellular signals into the intracellular compartment. The more than 50 members of this protein family are characterized by a low molecular weight (between 20 and 29 kDa) and by the presence of GTP binding domains. Five different subfamilies can be distinguished (Ras, Rho, Rab, Sar/ARF and Ran), which participate in different biological processes, such as cell differentiation, cell proliferation, cell migration, cytoskeleton rearrangement and vesicular trafficking. Overall, the maintenance of epithelial barrier integrity relies on a GTPase-mediated fine-tuning of junctional and cytoskeletal dynamics [299]. The activation status of small GTPases is regulated by the transition from inactive GDP-bound into active GTP-bound state [300]. Guanosine exchange factors (GEF), guanosine activating triphosphatases (GAP) and guanosine dissociation inhibitors (GDI) are responsible for a balanced regulation of this transition [301]. GTP-bound proteins interact with effector proteins in order to activate downstream pathways. Activation of small GTPases as well as the capacity to initiate downstream signaling cascades crucially depends on their subcellular localization [302].
Rho-A (Ras homology family member A), together with Rac-1/2, and Cdc42 represent the best described members of the Rho family; although growing attention has also been paid to other family members, like Rho-B, Rho-C and Rho-H. Impaired small GTPase function in intestinal epithelium is associated with junctional and cytoskeletal dysfunction as it is often observed in the gut of IBD patients [303, 304]. A wide number of in vitro studies indeed defined a relevant role of Rho proteins for epithelial integrity, cytoskeleton regulation, cell morphology and IEC migration [305–310]. Despite these numerous publications supporting the role of Rho proteins in maintenance of cytoskeleton in epithelium, it still remained unclear whether Rho inhibition, activation or both would modify epithelial integrity and permeability [311]. Moreover, there exist only very few data about the complex in vivo regulation of epithelial Rho proteins so far.
In the context of gastrointestinal tract and inflammation, few in vivo studies tried to clarify the functional role of Rho GTPases within IECs. Genetic deletion of Cdc42 in mice caused a defect in IEC proliferation and differentiation and resulted in an intestinal phenotype which resembled human microvillus inclusion disease [312, 313]. Similarly, epithelial cell differentiation turned out to be affected after mutations or abrogation of Rac-1 expression and interfered with physiological development of the intestine [314, 315]. A potential role of Rho-A in IBD pathogenesis was first suggested in 2003 when it was observed that experimental colitis as well as intestinal inflammation in IBD patients goes along with increased Rho-A activation in intestinal mucosa. Interestingly, systemic pharmacological inhibition of Rho-A was able to interfere with NFκB signaling and to ameliorate experimental colitis [316]. Supporting the hypothesis that dysregulated Rho-A function, both activation or inhibition, might negatively impact on epithelial integrity, altered Rho-A function could be observed in the epithelium of IBD patients [317]. Inflamed areas in the gut of Crohn’s disease or ulcerative colitis patients depicted an altered subcellular localization of Rho-A within IECs and showed a marked cytosolic accumulation of the presumably inactive small GTPase. On a functional level, IEC-restricted lack of Rho-A resulted in the development of spontaneous colitis [317].
With regard to tumorigenesis, Rho-A expression is upregulated in different kinds of cancer and has therefore been discussed as a potential cancer biomarker [318]. Several studies showed that Rac-1 and Cdc42-mediated regulation of IEC differentiation impacts on cancer development. For instance, activation of these two proteins is required for a Wnt-dependent mechanism of tumor initiation [315, 319, 320]. However, most of these studies used experimental models of sporadic tumors and did not consider inflammation-associated tumor diseases like colitis-associated cancer. Interestingly, recent investigations based on whole-exome sequencing were able to identify a potential key involvement of the Rho/Rac pathway in the pathogenesis of colitis-associated cancer [281]. Although some genetic alterations in IBD-associated colon tumors turned out to be similar to sporadic tumors (e.g. Ras, p53, TGF, Wnt) [273], tumors in IBD patients showed less alterations in APC and Ras and could instead be characterized by new pathways and mutations. These pathways were mainly related to cell communication, cell–cell signaling and adhesion. Interestingly, 50 % of IBD-associated tumors possessed at least a single mutation in genes related to the family of Rho GTPases. Somehow, alterations of Rho/Rac signaling could be observed more frequently in UC patients than in CD patients.
All in all, we conclude that small GTPases represent important mediators of epithelial homeostasis in the gut and significantly impact on the development of colitis and colitis-associated colon cancer. Further research in this area is needed in order to achieve a full understanding of their complex pathogenic involvement and their applicability as potential therapeutic targets.
Prenylation
In order to allow their association with cellular membranes, small GTPases first have to undergo prenylation. Prenylation represents a post-translational process consisting of the attachment of hydrophobic isoprenoid residues to the C-terminal CAAX motif of target proteins and is of particular importance for the intracellular localization and function of small GTPases. The attached hydrophobic residue finally enables the GTPase to anchor to the lipophilic cell membrane and to achieve its full activation status [302, 321]. Indeed, GEF-mediated GTPase activation preferentially takes place when GTPases are associated with cellular membranes [302, 322]. Dependent on the type of GTPase, it is either a geranylgeranyl pyrophosphate residue (e.g. Rac, Rho, Cdc42, Rab) or a farnesyl pyrophosphate residue (e.g. Ras), which is covalently attached to the target GTPase [323, 324]. These enzymatic reactions are catalyzed by respective prenyltransferases, namely geranylgeranyltransferases (GGTase-I and GGT-ase-II) and farnesyltransferase.
The initial idea of a potential link between prenylation and inflammation and/or cancer development arose from the identification of K-Ras as proto-oncogene [325]. Based on this observation and on the dependency of Ras function on appropriate prenylation, inhibitors of prenylation have been investigated in preclinical and clinical studies as anticancer agents and showed promising results [326–329]. In the context of inflammation, it was described that therapeutic use of statins in patients with atherosclerosis resulted in anti-inflammatory effects which were mediated via inhibition of prenylation [330, 331]. Based on this observation, the therapeutic use of statins as potent inhibitors of isoprenoid precursor synthesis has also been discussed in the context of intestinal inflammation and other inflammatory disorders not primary related to lipid metabolism [332–334]. Moreover, nitrogen-containing bisphosphonates, which are known to interfere with prenylation by inhibition of isoprenoid synthesis, could be identified as effective anti-inflammatory agents in several experimental models of colitis [335, 336]. Taken together, these observations convincingly argued for a functional impact of prenylation on intestinal homeostasis, chronic gut inflammation and cancer development.
In line with this hypothesis, a recent study clearly identified the indispensable role of geranylgeranylation within IECs for maintenance of intestinal homeostasis. Mice lacking GGTase-Iβ expression in intestinal epithelium develop a severe intestinal disease with increased intestinal permeability and marked intestinal inflammation [317]. The mechanism behind this striking phenomenon involves the destruction of intestinal architecture due to dramatic cytoskeleton rearrangement within IECs. In the absence of adequate epithelial geranylgeranylation, Rho-A dysfunction might alter cytoskeleton arrangement leading to dysregulation of cell shedding, increased epithelial permeability and breakdown of barrier function. Interestingly, enteral application of a synthetic Rho activator was able to partly compensate the lack of epithelial geranylgeranylation and to ameliorate gut pathology [317]. Thus, geranylgeranylation of Rho-A represents a crucial epithelium-intrinsic mechanism to maintain intestinal barrier integrity and gut homeostasis. The clinical transferability of these findings was further supported by the observation that gut epithelium of IBD patients with active disease is characterized by decreased GGTase-I expression levels [317].
In summary, we can postulate that prenylation represents a novel relevant pathway for maintenance of gut homeostasis and epithelial integrity. Regarding involved target proteins of prenylation, a particular role of Rho-A in IEC biology could be identified. In line with this, we can hypothesize that other proteins belonging to Rho family, which are also regulated by prenylation, might be important in this context as well. Future studies are needed in order to further elucidate the molecular events which are affected by Rho-A and other targets of prenylation within intestinal epithelium.
Conclusions and future perspectives
The here reviewed aspects of IEC biology and function clearly indicate that the maintenance of epithelial integrity and homeostasis should be considered as a key target in the therapeutic and/or even preventive clinical management of IBD and CRC.
However, due to the vast number of different mechanisms regulating epithelial homeostasis in the intestine and the complexity of the structure and cellular composition of gut tissue, profound collaborative scientific efforts are required in order to allow a detailed understanding of the interplay between involved molecular pathways. During the last years, an enormous advance has been achieved in this field and enabled us to gain interesting new insights into the role of IECs in gut homeostasis and pathology. In the context of inflammation, special attention has been paid to Paneth cell biology and necroptotic cell death, mucus composition, as well as AMP and epithelial-microbiom interactions, while stem cell proliferation/differentiation, Wnt pathway and STAT-3 signaling represent important candidates in colorectal tumorigenesis. Moreover, the recently identified crucial role of prenylation and small GTPases in the maintenance of epithelial architecture introduced further potential new target structures for an optimized treatment or early diagnosis of IEC-derived gut diseases. In future studies, it will now be important to translate these findings into innovative therapeutic strategies and pave the way for a clinical applicability and, subsequently, an optimized therapy of IBD and CRC patients.
Abbreviations
- AJs
Adherens junctions
- AMPs
Antimicrobial peptides
- APC
Adenomatous polyposis coli
- Bmi
B cell-specific Moloney murine leukemia virus integration site
- CAC
Colitis-associated cancer
- CBCs
Columnar stem cells
- CD
Crohn’s disease
- cFLIP
Cellular FLICE-inhibitory protein
- COX-2
Cyclooxygenase-2
- CRC
Colorectal cancer
- DCLKL
Doublecortin like kinase 1
- DNA
Deoxyribonucleic acid
- DSS
Dextran sodium sulfate
- EGF
Epidermal growth factor
- EMT
Epithelial-mesenchymal transition
- FADD
Fas-associated protein with death domain
- FasL
Fas ligand
- GAP
Guanosine activating triphosphatases
- GDI
Guanosine dissociation inhibitors
- GDP
Guanosine diphosphate
- GEF
Guanosine exchange factors
- GGTase
Geranylgeranyltransferase
- GTP
Guanosine triphosphate
- IBD
Inflammatory bowel diseases
- IECs
Intestinal epithelia cells
- IFN
Interferon
- IL
Interleukin
- IKK
IκB kinase
- iPSCs
Induced pluripotent stem cells
- IRF
Interferon regulatory factors
- ISC
Intestinal stem cell
- ISEMFs
Intestinal subepithelial myofibroblasts
- JAK
Janus kinase
- JAM
Junctional adhesion molecules
- Lgr
Leucine-rich repeat-containing G-protein-coupled receptor
- LIGHT
TNF ligand superfamily member 14
- LPS
Lipopolysaccharides
- LRCs
Label-retaining cells
- Mal
MyD88-adapter-like
- MAPK
Mitogen-activated protein kinases
- MET
Mesenchymal-epithelial transition
- MHC
Major histocompatibility complex
- MLCK
Myosin light-chain kinase
- MSI
Microsatellite instable
- MSS
Microsatellite stable
- MUC2
Mucin 2
- MYD88
Myeloid differentiation primary response gene 88
- NFκB
Nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells
- NLR
NOD-like receptor
- PGE2
Prostaglandin E2
- PI3K
Phosphatidylinositol-4,5-bisphosphate 3-kinase
- PRR
Pattern recognition receptor
- R
Receptor
- RELM
Resistin-like proteins
- Rho
Ras homology family member
- RIPK
Receptor-interacting protein kinases
- RLR
RIG-like receptor
- ROCK
Rho associated kinase
- STAT
Signal transducer and activators of transcription
- Tak
Tat-associated kinase
- TFF3
Trefoil factor 3
- TGF
Transforming growth factor
- Th
T helper cell
- TJP1
Tight junction protein 1
- TL1A
TNF-like ligand 1A
- TLR
Toll-like receptor
- TNF
Tumor Necrosis Factor
- TOLLIP
TOLL interacting protein
- TRAIL
Tumor necrosis factor related apoptosis inducing ligand
- TRIF
TIR-domain-containing adapter-inducing interferon-β
- TWEAK
TNF-related weak inducer of apoptosis
- UC
Ulcerative colitis
- Wnt
Wingless-type MMTV integration site family member
- XBP1
X-box binding protein 1
- ZO-1
Zona occuldens protein 1
References
- 1.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 2.Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142:46–54 e42. doi: 10.1053/j.gastro.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Investig. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 5.Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics. Front Immunol. 2013;4:280. doi: 10.3389/fimmu.2013.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kong J, Zhang Z, Musch MW, Ning G, Sun J, Hart J, et al. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am J Physiol Gastrointest Liver Physiol. 2008;294:G208–G216. doi: 10.1152/ajpgi.00398.2007. [DOI] [PubMed] [Google Scholar]
- 7.Taupin DR, Kinoshita K, Podolsky DK. Intestinal trefoil factor confers colonic epithelial resistance to apoptosis. Proc Natl Acad Sci USA. 2000;97:799–804. doi: 10.1073/pnas.97.2.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McElroy SJ, Prince LS, Weitkamp JH, Reese J, Slaughter JC, Polk DB. Tumor necrosis factor receptor 1-dependent depletion of mucus in immature small intestine: a potential role in neonatal necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol. 2011;301:G656–G666. doi: 10.1152/ajpgi.00550.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lennard-Jones JE, Melville DM, Morson BC, Ritchie JK, Williams CB. Precancer and cancer in extensive ulcerative colitis: findings among 401 patients over 22 years. Gut. 1990;31:800–806. doi: 10.1136/gut.31.7.800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15:91–102. doi: 10.1016/j.ccr.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 11.Lakatos PL, Lakatos L. Risk for colorectal cancer in ulcerative colitis: changes, causes and management strategies. World J Gastroenterol WJG. 2008;14:3937–3947. doi: 10.3748/wjg.14.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 13.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-I. [DOI] [PubMed] [Google Scholar]
- 14.Fearon ER, Hamilton SR, Vogelstein B. Clonal analysis of human colorectal tumors. Science. 1987;238:193–197. doi: 10.1126/science.2889267. [DOI] [PubMed] [Google Scholar]
- 15.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 16.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med. 2009;361:2449–2460. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 18.Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717–728. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 19.van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Ann Rev Physiol. 2009;71:241–260. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- 20.Specian RD, Oliver MG. Functional biology of intestinal goblet cells. Am J Physiol. 1991;260:C183–C193. doi: 10.1152/ajpcell.1991.260.2.C183. [DOI] [PubMed] [Google Scholar]
- 21.Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol. 2011;9:356–368. doi: 10.1038/nrmicro2546. [DOI] [PubMed] [Google Scholar]
- 22.Ohno H. Intestinal M cells. J Biochem. 2016;159:151–160. doi: 10.1093/jb/mvv121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marshman E, Booth C, Potten CS. The intestinal epithelial stem cell. BioEssays News Rev Mole Cell Develop Biol. 2002;24:91–98. doi: 10.1002/bies.10028. [DOI] [PubMed] [Google Scholar]
- 24.Kvietys PR, Granger DN. Role of intestinal lymphatics in interstitial volume regulation and transmucosal water transport. Ann N Y Acad Sci. 2010;1207(Suppl 1):E29–E43. doi: 10.1111/j.1749-6632.2010.05709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vereecke L, Beyaert R, van Loo G. Enterocyte death and intestinal barrier maintenance in homeostasis and disease. Trends Mol Med. 2011;17:584–593. doi: 10.1016/j.molmed.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 26.Turner JR. Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am J Pathol. 2006;169:1901–1909. doi: 10.2353/ajpath.2006.060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bjerknes M, Cheng H. The stem-cell zone of the small intestinal epithelium. III. Evidence from columnar, enteroendocrine, and mucous cells in the adult mouse. Am J Anat. 1981;160:77–91. doi: 10.1002/aja.1001600107. [DOI] [PubMed] [Google Scholar]
- 28.Bjerknes M, Cheng H. The stem-cell zone of the small intestinal epithelium. I. Evidence from Paneth cells in the adult mouse. Am J Anat. 1981;160:51–63. doi: 10.1002/aja.1001600105. [DOI] [PubMed] [Google Scholar]
- 29.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 30.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 31.Barker N, Tan S, Clevers H. Lgr proteins in epithelial stem cell biology. Development. 2013;140:2484–2494. doi: 10.1242/dev.083113. [DOI] [PubMed] [Google Scholar]
- 32.Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
- 33.Schepers AG, Vries R, van den Born M, van de Wetering M, Clevers H. Lgr5 intestinal stem cells have high telomerase activity and randomly segregate their chromosomes. EMBO J. 2011;30:1104–1109. doi: 10.1038/emboj.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barker N, van de Wetering M, Clevers H. The intestinal stem cell. Genes Dev. 2008;22:1856–1864. doi: 10.1101/gad.1674008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Potten CS, Booth C, Pritchard DM. The intestinal epithelial stem cell: the mucosal governor. Int J Exp Pathol. 1997;78:219–243. doi: 10.1046/j.1365-2613.1997.280362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pinto LH, Pak WL. Light-induced changes in photoreceptor membrane resistance and potential in Gecko retinas. II. Preparations with active lateral interactions. J Gen Physiol. 1974;64:49–69. doi: 10.1085/jgp.64.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan KS, Chia LA, Li X, Ootani A, Su J, Lee JY, et al. The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci USA. 2012;109:466–471. doi: 10.1073/pnas.1118857109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sangiorgi E, Capecchi MR. Bmi1 is expressed in vivo in intestinal stem cells. Nat Genet. 2008;40:915–920. doi: 10.1038/ng.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buczacki SJ, Zecchini HI, Nicholson AM, Russell R, Vermeulen L, Kemp R, et al. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature. 2013;495:65–69. doi: 10.1038/nature11965. [DOI] [PubMed] [Google Scholar]
- 40.Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, et al. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature. 2011;478:255–259. doi: 10.1038/nature10408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bissell MJ, Labarge MA. Context, tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell. 2005;7:17–23. doi: 10.1016/j.ccr.2004.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miyamoto S, Rosenberg DW. Role of Notch signaling in colon homeostasis and carcinogenesis. Cancer Sci. 2011;102:1938–1942. doi: 10.1111/j.1349-7006.2011.02049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rothenberg ME, Nusse Y, Kalisky T, Lee JJ, Dalerba P, Scheeren F, et al. Identification of a cKit(+) colonic crypt base secretory cell that supports Lgr5(+) stem cells in mice. Gastroenterology. 2012;142(1195–205):e6. doi: 10.1053/j.gastro.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469:415–418. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roth S, Franken P, Sacchetti A, Kremer A, Anderson K, Sansom O, et al. Paneth cells in intestinal homeostasis and tissue injury. PLoS One. 2012;7:e38965. doi: 10.1371/journal.pone.0038965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marson A, Foreman R, Chevalier B, Bilodeau S, Kahn M, Young RA, et al. Wnt signaling promotes reprogramming of somatic cells to pluripotency. Cell Stem Cell. 2008;3:132–135. doi: 10.1016/j.stem.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 48.De Wever O, Demetter P, Mareel M, Bracke M. Stromal myofibroblasts are drivers of invasive cancer growth. Int J Cancer. 2008;123:2229–2238. doi: 10.1002/ijc.23925. [DOI] [PubMed] [Google Scholar]
- 49.Cole JW, McKalen A. Studies on the morphogenesis of adenomatous polyps in the human colon. Cancer. 1963;16:998–1002. doi: 10.1002/1097-0142(196308)16:8<998::AID-CNCR2820160806>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 50.Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152:25–38. doi: 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 51.Nieto MA, Cano A. The epithelial-mesenchymal transition under control: global programs to regulate epithelial plasticity. Semin Cancer Biol. 2012;22:361–368. doi: 10.1016/j.semcancer.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 52.Vaiopoulos AG, Kostakis ID, Koutsilieris M, Papavassiliou AG. Colorectal cancer stem cells. Stem Cells. 2012;30:363–371. doi: 10.1002/stem.1031. [DOI] [PubMed] [Google Scholar]
- 53.Todaro M, Francipane MG, Medema JP, Stassi G. Colon cancer stem cells: promise of targeted therapy. Gastroenterology. 2010;138:2151–2162. doi: 10.1053/j.gastro.2009.12.063. [DOI] [PubMed] [Google Scholar]
- 54.Loboda A, Nebozhyn MV, Watters JW, Buser CA, Shaw PM, Huang PS, et al. EMT is the dominant program in human colon cancer. BMC Med Genomics. 2011;4:9. doi: 10.1186/1755-8794-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dagenais M, Douglas T, Saleh M. Role of programmed necrosis and cell death in intestinal inflammation. Curr Opin Gastroenterol. 2014;30:566–575. doi: 10.1097/MOG.0000000000000117. [DOI] [PubMed] [Google Scholar]
- 56.Gunther C, Neumann H, Neurath MF, Becker C. Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut. 2013;62:1062–1071. doi: 10.1136/gutjnl-2011-301364. [DOI] [PubMed] [Google Scholar]
- 57.Watson AJ. Review article: manipulation of cell death—the development of novel strategies for the treatment of gastrointestinal disease. Aliment Pharmacol Ther. 1995;9:215–226. doi: 10.1111/j.1365-2036.1995.tb00376.x. [DOI] [PubMed] [Google Scholar]
- 58.Kanduc D, Mittelman A, Serpico R, Sinigaglia E, Sinha AA, Natale C, et al. Cell death: apoptosis versus necrosis (review) Int J Oncol. 2002;21:165–170. [PubMed] [Google Scholar]
- 59.Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22:263–268. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seneviratne D, Ma J, Tan X, Kwon YK, Muhammad E, Melhem M, et al. Genomic instability causes HGF gene activation in colon cancer cells, promoting their resistance to necroptosis. Gastroenterology. 2015;148(181–91):e17. doi: 10.1053/j.gastro.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chan FK, Shisler J, Bixby JG, Felices M, Zheng L, Appel M, et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278:51613–51621. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- 65.Teng X, Degterev A, Jagtap P, Xing X, Choi S, Denu R, et al. Structure-activity relationship study of novel necroptosis inhibitors. Bioorg Med Chem Lett. 2005;15:5039–5044. doi: 10.1016/j.bmcl.2005.07.077. [DOI] [PubMed] [Google Scholar]
- 66.Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471:373–376. doi: 10.1038/nature09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grossmann J. Molecular mechanisms of “detachment-induced apoptosis—Anoikis”. Apoptosis Int J Program Cell Death. 2002;7:247–260. doi: 10.1023/A:1015312119693. [DOI] [PubMed] [Google Scholar]
- 69.Brinkman BM, Hildebrand F, Kubica M, Goosens D, Del Favero J, Declercq W, et al. Caspase deficiency alters the murine gut microbiome. Cell Death Dis. 2011;2:e220. doi: 10.1038/cddis.2011.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–918. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 71.Colussi PA, Kumar S. Targeted disruption of caspase genes in mice: what they tell us about the functions of individual caspases in apoptosis. Immunol Cell Biol. 1999;77:58–63. doi: 10.1046/j.1440-1711.1999.00788.x. [DOI] [PubMed] [Google Scholar]
- 72.Watson AJ, Pritchard DM. Lessons from genetically engineered animal models VII Apoptosis in intestinal epithelium: lessons from transgenic and knockout mice. Am J Physiol Gastrointest Liver Physiol. 2001;278:G1–G5. doi: 10.1152/ajpgi.2000.278.1.G1. [DOI] [PubMed] [Google Scholar]
- 73.Nakayama K, Nakayama K, Negishi I, Kuida K, Sawa H, Loh DY. Targeted disruption of Bcl-2 alpha beta in mice: occurrence of gray hair, polycystic kidney disease, and lymphocytopenia. Proc Natl Acad Sci USA. 1994;91:3700–3704. doi: 10.1073/pnas.91.9.3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nakayama K, Nakayama K, Negishi I, Kuida K, Shinkai Y, Louie MC, et al. Disappearance of the lymphoid system in Bcl-2 homozygous mutant chimeric mice. Science. 1993;261:1584–1588. doi: 10.1126/science.8372353. [DOI] [PubMed] [Google Scholar]
- 75.Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- 76.Steinbrecher KA, Harmel-Laws E, Sitcheran R, Baldwin AS. Loss of epithelial RelA results in deregulated intestinal proliferative/apoptotic homeostasis and susceptibility to inflammation. J Immunol. 2008;180:2588–2599. doi: 10.4049/jimmunol.180.4.2588. [DOI] [PubMed] [Google Scholar]
- 77.Kajino-Sakamoto R, Inagaki M, Lippert E, Akira S, Robine S, Matsumoto K, et al. Enterocyte-derived TAK1 signaling prevents epithelium apoptosis and the development of ileitis and colitis. J Immunol. 2008;181:1143–1152. doi: 10.4049/jimmunol.181.2.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eckmann L, Nebelsiek T, Fingerle AA, Dann SM, Mages J, Lang R, et al. Opposing functions of IKKbeta during acute and chronic intestinal inflammation. Proc Natl Acad Sci USA. 2008;105:15058–15063. doi: 10.1073/pnas.0808216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 80.Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Glimcher LH. XBP1: the last two decades. Ann Rheum Dis. 2010;69(Suppl 1):i67–i71. doi: 10.1136/ard.2009.119388. [DOI] [PubMed] [Google Scholar]
- 82.Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Iwamoto M, Koji T, Makiyama K, Kobayashi N, Nakane PK. Apoptosis of crypt epithelial cells in ulcerative colitis. J Pathol. 1996;180:152–159. doi: 10.1002/(SICI)1096-9896(199610)180:2<152::AID-PATH649>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 84.Hagiwara C, Tanaka M, Kudo H. Increase in colorectal epithelial apoptotic cells in patients with ulcerative colitis ultimately requiring surgery. J Gastroenterol Hepatol. 2002;17:758–764. doi: 10.1046/j.1440-1746.2002.02791.x. [DOI] [PubMed] [Google Scholar]
- 85.Edelblum KL, Yan F, Yamaoka T, Polk DB. Regulation of apoptosis during homeostasis and disease in the intestinal epithelium. Inflamm Bowel Dis. 2006;12:413–424. doi: 10.1097/01.MIB.0000217334.30689.3e. [DOI] [PubMed] [Google Scholar]
- 86.Di Sabatino A, Ciccocioppo R, Luinetti O, Ricevuti L, Morera R, Cifone MG, et al. Increased enterocyte apoptosis in inflamed areas of Crohn’s disease. Dis Colon Rectum. 2003;46:1498–1507. doi: 10.1007/s10350-004-6802-z. [DOI] [PubMed] [Google Scholar]
- 87.Dourmashkin RR, Davies H, Wells C, Shah D, Price A, O’Morain C, et al. Epithelial patchy necrosis in Crohn’s disease. Hum Pathol. 1983;14:643–648. doi: 10.1016/S0046-8177(83)80207-X. [DOI] [PubMed] [Google Scholar]
- 88.Barkla DH, Gibson PR. The fate of epithelial cells in the human large intestine. Pathology. 1999;31:230–238. doi: 10.1080/003130299105043. [DOI] [PubMed] [Google Scholar]
- 89.Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330–334. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- 90.Bedini OA, Naves A, San Miguel P, Quispe A, Guida C. Metaplasic Paneth cells in ulcerative colitis. Acta Gastroenterol Latinoam. 2014;44:285–289. [PubMed] [Google Scholar]
- 91.Simmonds N, Furman M, Karanika E, Phillips A, Bates AW. Paneth cell metaplasia in newly diagnosed inflammatory bowel disease in children. BMC Gastroenterol. 2014;14:93. doi: 10.1186/1471-230X-14-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pierdomenico M, Negroni A, Stronati L, Vitali R, Prete E, Bertin J, et al. Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am J Gastroenterol. 2014;109:279–287. doi: 10.1038/ajg.2013.403. [DOI] [PubMed] [Google Scholar]
- 93.Caprioli F, Stolfi C, Caruso R, Fina D, Sica G, Biancone L, et al. Transcriptional and post-translational regulation of Flip, an inhibitor of Fas-mediated apoptosis, in human gut inflammation. Gut. 2008;57:1674–1680. doi: 10.1136/gut.2008.149286. [DOI] [PubMed] [Google Scholar]
- 94.Bullen TF, Forrest S, Campbell F, Dodson AR, Hershman MJ, Pritchard DM, et al. Characterization of epithelial cell shedding from human small intestine. Lab Invest J Tech Methods Pathol. 2006;86:1052–1063. doi: 10.1038/labinvest.3700464. [DOI] [PubMed] [Google Scholar]
- 95.Marchiando AM, Shen L, Graham WV, Edelblum KL, Duckworth CA, Guan Y, et al. The epithelial barrier is maintained by in vivo tight junction expansion during pathologic intestinal epithelial shedding. Gastroenterology. 2011;140(1208–18):e1–e2. doi: 10.1053/j.gastro.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Williams JM, Duckworth CA, Watson AJ, Frey MR, Miguel JC, Burkitt MD, et al. A mouse model of pathological small intestinal epithelial cell apoptosis and shedding induced by systemic administration of lipopolysaccharide. Disease Models Mech. 2013;6:1388–1399. doi: 10.1242/dmm.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Eisenhoffer GT, Loftus PD, Yoshigi M, Otsuna H, Chien CB, Morcos PA, et al. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature. 2012;484:546–549. doi: 10.1038/nature10999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guan Y, Watson AJ, Marchiando AM, Bradford E, Shen L, Turner JR, et al. Redistribution of the tight junction protein ZO-1 during physiological shedding of mouse intestinal epithelial cells. Am J Physiol Cell Physiol. 2011;300:C1404–C1414. doi: 10.1152/ajpcell.00270.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Marinari E, Mehonic A, Curran S, Gale J, Duke T, Baum B. Live-cell delamination counterbalances epithelial growth to limit tissue overcrowding. Nature. 2012;484:542–545. doi: 10.1038/nature10984. [DOI] [PubMed] [Google Scholar]
- 100.Rosenblatt J, Raff MC, Cramer LP. An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr Biol CB. 2001;11:1847–1857. doi: 10.1016/S0960-9822(01)00587-5. [DOI] [PubMed] [Google Scholar]
- 101.Kiesslich R, Goetz M, Angus EM, Hu Q, Guan Y, Potten C, et al. Identification of epithelial gaps in human small and large intestine by confocal endomicroscopy. Gastroenterology. 2007;133:1769–1778. doi: 10.1053/j.gastro.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 102.Watson AJ, Chu S, Sieck L, Gerasimenko O, Bullen T, Campbell F, et al. Epithelial barrier function in vivo is sustained despite gaps in epithelial layers. Gastroenterology. 2005;129:902–912. doi: 10.1053/j.gastro.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 103.Kiesslich R, Duckworth CA, Moussata D, Gloeckner A, Lim LG, Goetz M, et al. Local barrier dysfunction identified by confocal laser endomicroscopy predicts relapse in inflammatory bowel disease. Gut. 2012;61:1146–1153. doi: 10.1136/gutjnl-2011-300695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Koch S, Nusrat A. Dynamic regulation of epithelial cell fate and barrier function by intercellular junctions. Ann N Y Acad Sci. 2009;1165:220–227. doi: 10.1111/j.1749-6632.2009.04025.x. [DOI] [PubMed] [Google Scholar]
- 105.Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol. 2007;127:2525–2532. doi: 10.1038/sj.jid.5700865. [DOI] [PubMed] [Google Scholar]
- 106.Gumbiner B. Structure, biochemistry, and assembly of epithelial tight junctions. Am J Physiol. 1987;253:C749–C758. doi: 10.1152/ajpcell.1987.253.6.C749. [DOI] [PubMed] [Google Scholar]
- 107.Cunningham KE, Turner JR. Myosin light chain kinase: pulling the strings of epithelial tight junction function. Ann N Y Acad Sci. 2012;1258:34–42. doi: 10.1111/j.1749-6632.2012.06526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Anderson JM, Van Itallie CM, Fanning AS. Setting up a selective barrier at the apical junction complex. Curr Opin Cell Biol. 2004;16:140–145. doi: 10.1016/j.ceb.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 109.Perez-Moreno M, Jamora C, Fuchs E. Sticky business: orchestrating cellular signals at adherens junctions. Cell. 2003;112:535–548. doi: 10.1016/S0092-8674(03)00108-9. [DOI] [PubMed] [Google Scholar]
- 110.Gates J, Peifer M. Can 1000 reviews be wrong? Actin, alpha-Catenin, and adherens junctions. Cell. 2005;123:769–772. doi: 10.1016/j.cell.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 111.Dusek RL, Godsel LM, Green KJ. Discriminating roles of desmosomal cadherins: beyond desmosomal adhesion. J Dermatol Sci. 2007;45:7–21. doi: 10.1016/j.jdermsci.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 112.Mese G, Richard G, White TW. Gap junctions: basic structure and function. J Invest Dermatol. 2007;127:2516–2524. doi: 10.1038/sj.jid.5700770. [DOI] [PubMed] [Google Scholar]
- 113.Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol. 2010;177:512–524. doi: 10.2353/ajpath.2010.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rodgers LS, Fanning AS. Regulation of epithelial permeability by the actin cytoskeleton. Cytoskeleton. 2011;68:653–660. doi: 10.1002/cm.20547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nusrat A, von Eichel-Streiber C, Turner JR, Verkade P, Madara JL, Parkos CA. Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect Immun. 2001;69:1329–1336. doi: 10.1128/IAI.69.3.1329-1336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Betanzos A, Javier-Reyna R, Garcia-Rivera G, Banuelos C, Gonzalez-Mariscal L, Schnoor M, et al. The EhCPADH112 complex of Entamoeba histolytica interacts with tight junction proteins occludin and claudin-1 to produce epithelial damage. PLoS One. 2013;8:e65100. doi: 10.1371/journal.pone.0065100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shifflett DE, Clayburgh DR, Koutsouris A, Turner JR, Hecht GA. Enteropathogenic E. coli disrupts tight junction barrier function and structure in vivo. Lab Invest J Tech Methods Pathol. 2005;85:1308–1324. doi: 10.1038/labinvest.3700330. [DOI] [PubMed] [Google Scholar]
- 118.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 119.Menke A, Giehl K. Regulation of adherens junctions by Rho GTPases and p120-catenin. Arch Biochem Biophys. 2012;524:48–55. doi: 10.1016/j.abb.2012.04.019. [DOI] [PubMed] [Google Scholar]
- 120.Hollander D. Permeability in Crohn’s disease: altered barrier functions in healthy relatives? Gastroenterology. 1993;104:1848–1851. doi: 10.1016/0016-5085(93)90668-3. [DOI] [PubMed] [Google Scholar]
- 121.Ukabam SO, Clamp JR, Cooper BT. Abnormal small intestinal permeability to sugars in patients with Crohn’s disease of the terminal ileum and colon. Digestion. 1983;27:70–74. doi: 10.1159/000198932. [DOI] [PubMed] [Google Scholar]
- 122.Mankertz J, Schulzke JD. Altered permeability in inflammatory bowel disease: pathophysiology and clinical implications. Curr Opin Gastroenterol. 2007;23:379–383. doi: 10.1097/MOG.0b013e32816aa392. [DOI] [PubMed] [Google Scholar]
- 123.Yacyshyn BR, Meddings JB. CD45RO expression on circulating CD19+ B cells in Crohn’s disease correlates with intestinal permeability. Gastroenterology. 1995;108:132–137. doi: 10.1016/0016-5085(95)90017-9. [DOI] [PubMed] [Google Scholar]
- 124.D’Inca R, Di Leo V, Corrao G, Martines D, D’Odorico A, Mestriner C, et al. Intestinal permeability test as a predictor of clinical course in Crohn’s disease. Am J Gastroenterol. 1999;94:2956–2960. doi: 10.1016/S0002-9270(99)00500-6. [DOI] [PubMed] [Google Scholar]
- 125.Tamura A, Kitano Y, Hata M, Katsuno T, Moriwaki K, Sasaki H, et al. Megaintestine in claudin-15-deficient mice. Gastroenterology. 2008;134:523–534. doi: 10.1053/j.gastro.2007.11.040. [DOI] [PubMed] [Google Scholar]
- 126.Khounlotham M, Kim W, Peatman E, Nava P, Medina-Contreras O, Addis C, et al. Compromised intestinal epithelial barrier induces adaptive immune compensation that protects from colitis. Immunity. 2012;37:563–573. doi: 10.1016/j.immuni.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schulzke JD, Gitter AH, Mankertz J, Spiegel S, Seidler U, Amasheh S, et al. Epithelial transport and barrier function in occludin-deficient mice. Biochim Biophys Acta. 2005;1669:34–42. doi: 10.1016/j.bbamem.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 128.Pope JL, Bhat AA, Sharma A, Ahmad R, Krishnan M, Washington MK, et al. Claudin-1 regulates intestinal epithelial homeostasis through the modulation of Notch-signalling. Gut. 2014;63:622–634. doi: 10.1136/gutjnl-2012-304241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Al-Sadi R, Ye D, Boivin M, Guo S, Hashimi M, Ereifej L, et al. Interleukin-6 modulation of intestinal epithelial tight junction permeability is mediated by JNK pathway activation of claudin-2 gene. PLoS One. 2014;9:e85345. doi: 10.1371/journal.pone.0085345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 131.Kawashima R, Kawamura YI, Oshio T, Son A, Yamazaki M, Hagiwara T, et al. Interleukin-13 damages intestinal mucosa via TWEAK and Fn14 in mice-a pathway associated with ulcerative colitis. Gastroenterology. 2011;141(2119–29):e8. doi: 10.1053/j.gastro.2011.08.040. [DOI] [PubMed] [Google Scholar]
- 132.Franze E, Caruso R, Stolfi C, Sarra M, Cupi ML, Ascolani M, et al. High expression of the “A Disintegrin And Metalloprotease” 19 (ADAM19), a sheddase for TNF-alpha in the mucosa of patients with inflammatory bowel diseases. Inflamm Bowel Dis. 2013;19:501–511. doi: 10.1097/MIB.0b013e31828028e8. [DOI] [PubMed] [Google Scholar]
- 133.Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest J Tech Methods Pathol. 2006;86:191–201. doi: 10.1038/labinvest.3700373. [DOI] [PubMed] [Google Scholar]
- 134.Wang F, Schwarz BT, Graham WV, Wang Y, Su L, Clayburgh DR, et al. IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology. 2006;131:1153–1163. doi: 10.1053/j.gastro.2006.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Schmitz H, Barmeyer C, Fromm M, Runkel N, Foss HD, Bentzel CJ, et al. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology. 1999;116:301–309. doi: 10.1016/S0016-5085(99)70126-5. [DOI] [PubMed] [Google Scholar]
- 136.Irvine EJ, Marshall JK. Increased intestinal permeability precedes the onset of Crohn’s disease in a subject with familial risk. Gastroenterology. 2000;119:1740–1744. doi: 10.1053/gast.2000.20231. [DOI] [PubMed] [Google Scholar]
- 137.Peeters M, Geypens B, Claus D, Nevens H, Ghoos Y, Verbeke G, et al. Clustering of increased small intestinal permeability in families with Crohn’s disease. Gastroenterology. 1997;113:802–807. doi: 10.1016/S0016-5085(97)70174-4. [DOI] [PubMed] [Google Scholar]
- 138.Katz KD, Hollander D, Vadheim CM, McElree C, Delahunty T, Dadufalza VD, et al. Intestinal permeability in patients with Crohn’s disease and their healthy relatives. Gastroenterology. 1989;97:927–931. doi: 10.1016/0016-5085(89)91499-6. [DOI] [PubMed] [Google Scholar]
- 139.Wells JM, Rossi O, Meijerink M, van Baarlen P. Epithelial crosstalk at the microbiota-mucosal interface. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4607–4614. doi: 10.1073/pnas.1000092107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wittkopf N, Neurath MF, Becker C. Immune-epithelial crosstalk at the intestinal surface. J Gastroenterol. 2014;49:375–387. doi: 10.1007/s00535-013-0929-4. [DOI] [PubMed] [Google Scholar]
- 141.Finegold SM, Attebery HR, Sutter VL. Effect of diet on human fecal flora: comparison of Japanese and American diets. Am J Clin Nutr. 1974;27:1456–1469. doi: 10.1093/ajcn/27.12.1456. [DOI] [PubMed] [Google Scholar]
- 142.Baumler AJ, Sperandio V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature. 2016;535:85–93. doi: 10.1038/nature18849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.DeGruttola AK, Low D, Mizoguchi A, Mizoguchi E. Current understanding of dysbiosis in disease in human and animal models. Inflamm Bowel Dis. 2016;22:1137–1150. doi: 10.1097/MIB.0000000000000750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2014;63:1275–1283. doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- 145.Walker AW, Sanderson JD, Churcher C, Parkes GC, Hudspith BN, Rayment N, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011;11:7. doi: 10.1186/1471-2180-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Thayer WR, Jr, Coutu JA, Chiodini RJ, Van Kruiningen HJ, Merkal RS. Possible role of mycobacteria in inflammatory bowel disease. II. Mycobacterial antibodies in Crohn’s disease. Dig Dis Sci. 1984;29:1080–1085. doi: 10.1007/BF01317079. [DOI] [PubMed] [Google Scholar]
- 147.Lamhonwah AM, Ackerley C, Onizuka R, Tilups A, Lamhonwah D, Chung C, et al. Epitope shared by functional variant of organic cation/carnitine transporter, OCTN1, Campylobacter jejuni and Mycobacterium paratuberculosis may underlie susceptibility to Crohn’s disease at 5q31. Biochem Biophy Res Commun. 2005;337:1165–1175. doi: 10.1016/j.bbrc.2005.09.170. [DOI] [PubMed] [Google Scholar]
- 148.Szilagyi A, Gerson M, Mendelson J, Yusuf NA. Salmonella infections complicating inflammatory bowel disease. J Clin Gastroenterol. 1985;7:251–255. doi: 10.1097/00004836-198506000-00013. [DOI] [PubMed] [Google Scholar]
- 149.Contractor NV, Bassiri H, Reya T, Park AY, Baumgart DC, Wasik MA, et al. Lymphoid hyperplasia, autoimmunity, and compromised intestinal intraepithelial lymphocyte development in colitis-free gnotobiotic IL-2-deficient mice. J Immunol. 1998;160:385–394. [PubMed] [Google Scholar]
- 150.Taurog JD, Richardson JA, Croft JT, Simmons WA, Zhou M, Fernandez-Sueiro JL, et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med. 1994;180:2359–2364. doi: 10.1084/jem.180.6.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Velcich A, Yang W, Heyer J, Fragale A, Nicholas C, Viani S, et al. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science. 2002;295:1726–1729. doi: 10.1126/science.1069094. [DOI] [PubMed] [Google Scholar]
- 152.Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131:117–129. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 153.Artis D, Wang ML, Keilbaugh SA, He W, Brenes M, Swain GP, et al. RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc Natl Acad Sci USA. 2004;101:13596–13600. doi: 10.1073/pnas.0404034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nair MG, Guild KJ, Du Y, Zaph C, Yancopoulos GD, Valenzuela DM, et al. Goblet cell-derived resistin-like molecule beta augments CD4+ T cell production of IFN-gamma and infection-induced intestinal inflammation. J Immunol. 2008;181:4709–4715. doi: 10.4049/jimmunol.181.7.4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang S, Thacker PA, Watford M, Qiao S. Functions of antimicrobial peptides in gut homeostasis. Curr Protein Pept Sci. 2015;16:582–591. doi: 10.2174/1389203716666150630135847. [DOI] [PubMed] [Google Scholar]
- 156.Cunliffe RN, Mahida YR. Expression and regulation of antimicrobial peptides in the gastrointestinal tract. J Leukoc Biol. 2004;75:49–58. doi: 10.1189/jlb.0503249. [DOI] [PubMed] [Google Scholar]
- 157.Gersemann M, Wehkamp J, Stange EF. Innate immune dysfunction in inflammatory bowel disease. J Intern Med. 2012;271:421–428. doi: 10.1111/j.1365-2796.2012.02515.x. [DOI] [PubMed] [Google Scholar]
- 158.Wehkamp J, Koslowski M, Wang G, Stange EF. Barrier dysfunction due to distinct defensin deficiencies in small intestinal and colonic Crohn’s disease. Mucosal Immunol. 2008;1(Suppl 1):S67–S74. doi: 10.1038/mi.2008.48. [DOI] [PubMed] [Google Scholar]
- 159.Koon HW, Shih DQ, Chen J, Bakirtzi K, Hing TC, Law I, et al. Cathelicidin signaling via the Toll-like receptor protects against colitis in mice. Gastroenterology. 2011;141(1852–63):e1–e3. doi: 10.1053/j.gastro.2011.06.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci USA. 2005;102:18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Courth LF, Ostaff MJ, Mailander-Sanchez D, Malek NP, Stange EF, Wehkamp J. Crohn’s disease-derived monocytes fail to induce Paneth cell defensins. Proc Natl Acad Sci USA. 2015;112:14000–14005. doi: 10.1073/pnas.1510084112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wehkamp J, Harder J, Weichenthal M, Mueller O, Herrlinger KR, Fellermann K, et al. Inducible and constitutive beta-defensins are differentially expressed in Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2003;9:215–223. doi: 10.1097/00054725-200307000-00001. [DOI] [PubMed] [Google Scholar]
- 163.Strugala V, Dettmar PW, Pearson JP. Thickness and continuity of the adherent colonic mucus barrier in active and quiescent ulcerative colitis and Crohn’s disease. Int J Clin Pract. 2008;62:762–769. doi: 10.1111/j.1742-1241.2007.01665.x. [DOI] [PubMed] [Google Scholar]
- 164.Rohrl J, Geissler EK, Hehlgans T. Friend or foe: a novel role of beta-defensins in tumor development. Oncoimmunology. 2012;1:1159–1160. doi: 10.4161/onci.20825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rohrl J, Huber B, Koehl GE, Geissler EK, Hehlgans T. Mouse beta-defensin 14 (Defb14) promotes tumor growth by inducing angiogenesis in a CCR6-dependent manner. J Immunol. 2012;188:4931–4939. doi: 10.4049/jimmunol.1102442. [DOI] [PubMed] [Google Scholar]
- 166.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 167.Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol. 2014;14:546–558. doi: 10.1038/nri3713. [DOI] [PubMed] [Google Scholar]
- 168.Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. 2010;10:131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 169.Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141–153. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- 170.Madara JL, Stafford J. Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J Clin Investig. 1989;83:724–727. doi: 10.1172/JCI113938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, et al. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–G376. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 172.Ceponis PJ, Botelho F, Richards CD, McKay DM. Interleukins 4 and 13 increase intestinal epithelial permeability by a phosphatidylinositol 3-kinase pathway. Lack of evidence for STAT 6 involvement. J Biol Chem. 2000;275:29132–29137. doi: 10.1074/jbc.M003516200. [DOI] [PubMed] [Google Scholar]
- 173.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol. 2001;167:1882–1885. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- 174.Rhee SH, Im E, Riegler M, Kokkotou E, O’Brien M, Pothoulakis C. Pathophysiological role of Toll-like receptor 5 engagement by bacterial flagellin in colonic inflammation. Proc Natl Acad Sci USA. 2005;102:13610–13615. doi: 10.1073/pnas.0502174102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lee J, Mo JH, Katakura K, Alkalay I, Rucker AN, Liu YT, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol. 2006;8:1327–1336. doi: 10.1038/ncb1500. [DOI] [PubMed] [Google Scholar]
- 176.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68:7010–7017. doi: 10.1128/IAI.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology. 2007;133:1869–1881. doi: 10.1053/j.gastro.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci USA. 2005;102:99–104. doi: 10.1073/pnas.0405979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Vijay-Kumar M, Wu H, Aitken J, Kolachala VL, Neish AS, Sitaraman SV, et al. Activation of toll-like receptor 3 protects against DSS-induced acute colitis. Inflamm Bowel Dis. 2007;13:856–864. doi: 10.1002/ibd.20142. [DOI] [PubMed] [Google Scholar]
- 180.Rachmilewitz D, Katakura K, Karmeli F, Hayashi T, Reinus C, Rudensky B, et al. Toll-like receptor 9 signaling mediates the anti-inflammatory effects of probiotics in murine experimental colitis. Gastroenterology. 2004;126:520–528. doi: 10.1053/j.gastro.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 181.Vijay-Kumar M, Aitken JD, Sanders CJ, Frias A, Sloane VM, Xu J, et al. Flagellin treatment protects against chemicals, bacteria, viruses, and radiation. J Immunol. 2008;180:8280–8285. doi: 10.4049/jimmunol.180.12.8280. [DOI] [PubMed] [Google Scholar]
- 182.Xiao H, Gulen MF, Qin J, Yao J, Bulek K, Kish D, et al. The Toll–interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity. 2007;26:461–475. doi: 10.1016/j.immuni.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 183.Vereecke L, Sze M, Mc Guire C, Rogiers B, Chu Y, Schmidt-Supprian M, et al. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J Exp Med. 2010;207:1513–1523. doi: 10.1084/jem.20092474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Steenholdt C, Andresen L, Pedersen G, Hansen A, Brynskov J. Expression and function of toll-like receptor 8 and Tollip in colonic epithelial cells from patients with inflammatory bowel disease. Scand J Gastroenterol. 2009;44:195–204. doi: 10.1080/00365520802495529. [DOI] [PubMed] [Google Scholar]
- 185.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124–127. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- 186.Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci USA. 2010;107:21635–21640. doi: 10.1073/pnas.1016814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zaki MH, Vogel P, Malireddi RK, Body-Malapel M, Anand PK, Bertin J, et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell. 2011;20:649–660. doi: 10.1016/j.ccr.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lin XP, Almqvist N, Telemo E. Human small intestinal epithelial cells constitutively express the key elements for antigen processing and the production of exosomes. Blood Cells Mol Dis. 2005;35:122–128. doi: 10.1016/j.bcmd.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 189.Hershberg RM, Cho DH, Youakim A, Bradley MB, Lee JS, Framson PE, et al. Highly polarized HLA class II antigen processing and presentation by human intestinal epithelial cells. J Clin Investig. 1998;102:792–803. doi: 10.1172/JCI3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Nakazawa A, Dotan I, Brimnes J, Allez M, Shao L, Tsushima F, et al. The expression and function of costimulatory molecules B7H and B7-H1 on colonic epithelial cells. Gastroenterology. 2004;126:1347–1357. doi: 10.1053/j.gastro.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 191.Hershberg RM, Framson PE, Cho DH, Lee LY, Kovats S, Beitz J, et al. Intestinal epithelial cells use two distinct pathways for HLA class II antigen processing. J Clin Investig. 1997;100:204–215. doi: 10.1172/JCI119514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Buning J, Schmitz M, Repenning B, Ludwig D, Schmidt MA, Strobel S, et al. Interferon-gamma mediates antigen trafficking to MHC class II-positive late endosomes of enterocytes. Eur J Immunol. 2005;35:831–842. doi: 10.1002/eji.200425286. [DOI] [PubMed] [Google Scholar]
- 193.Bar F, Sina C, Hundorfean G, Pagel R, Lehnert H, Fellermann K, et al. Inflammatory bowel diseases influence major histocompatibility complex class I (MHC I) and II compartments in intestinal epithelial cells. Clin Exp Immunol. 2013;172:280–289. doi: 10.1111/cei.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Thelemann C, Eren RO, Coutaz M, Brasseit J, Bouzourene H, Rosa M, et al. Interferon-gamma induces expression of MHC class II on intestinal epithelial cells and protects mice from colitis. PLoS One. 2014;9:e86844. doi: 10.1371/journal.pone.0086844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 196.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Ann Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 197.Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9:271–285. doi: 10.1038/nri2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Watson AJ, Hughes KR. TNF-alpha-induced intestinal epithelial cell shedding: implications for intestinal barrier function. Ann N Y Acad Sci. 2012;1258:1–8. doi: 10.1111/j.1749-6632.2012.06523.x. [DOI] [PubMed] [Google Scholar]
- 199.Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. 2012;119:651–665. doi: 10.1182/blood-2011-04-325225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hampe J, Shaw SH, Saiz R, Leysens N, Lantermann A, Mascheretti S, et al. Linkage of inflammatory bowel disease to human chromosome 6p. Am J Hum Genet. 1999;65:1647–1655. doi: 10.1086/302677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dechairo B, Dimon C, van Heel D, Mackay I, Edwards M, Scambler P, et al. Replication and extension studies of inflammatory bowel disease susceptibility regions confirm linkage to chromosome 6p (IBD3) Eur J Human Genet EJHG. 2001;9:627–633. doi: 10.1038/sj.ejhg.5200687. [DOI] [PubMed] [Google Scholar]
- 202.Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, McLeod RS, Griffiths AM, et al. Genomewide search in Canadian families with inflammatory bowel disease reveals two novel susceptibility loci. Am J Hum Genet. 2000;66:1863–1870. doi: 10.1086/302913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Dionne S, Hiscott J, D’Agata I, Duhaime A, Seidman EG. Quantitative PCR analysis of TNF-alpha and IL-1 beta mRNA levels in pediatric IBD mucosal biopsies. Dig Dis Sci. 1997;42:1557–1566. doi: 10.1023/A:1018895500721. [DOI] [PubMed] [Google Scholar]
- 204.Matsuda R, Koide T, Tokoro C, Yamamoto T, Godai T, Morohashi T, et al. Quantitive cytokine mRNA expression profiles in the colonic mucosa of patients with steroid naive ulcerative colitis during active and quiescent disease. Inflamm Bowel Dis. 2009;15:328–334. doi: 10.1002/ibd.20759. [DOI] [PubMed] [Google Scholar]
- 205.Komatsu M, Kobayashi D, Saito K, Furuya D, Yagihashi A, Araake H, et al. Tumor necrosis factor-alpha in serum of patients with inflammatory bowel disease as measured by a highly sensitive immuno-PCR. Clin Chem. 2001;47:1297–1301. [PubMed] [Google Scholar]
- 206.Murch SH, Lamkin VA, Savage MO, Walker-Smith JA, MacDonald TT. Serum concentrations of tumour necrosis factor alpha in childhood chronic inflammatory bowel disease. Gut. 1991;32:913–917. doi: 10.1136/gut.32.8.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Maeda M, Watanabe N, Neda H, Yamauchi N, Okamoto T, Sasaki H, et al. Serum tumor necrosis factor activity in inflammatory bowel disease. Immunopharmacol Immunotoxicol. 1992;14:451–461. doi: 10.3109/08923979209005404. [DOI] [PubMed] [Google Scholar]
- 208.Braegger CP, Nicholls S, Murch SH, Stephens S, MacDonald TT. Tumour necrosis factor alpha in stool as a marker of intestinal inflammation. Lancet. 1992;339:89–91. doi: 10.1016/0140-6736(92)90999-J. [DOI] [PubMed] [Google Scholar]
- 209.MacDonald TT, Hutchings P, Choy MY, Murch S, Cooke A. Tumour necrosis factor-alpha and interferon-gamma production measured at the single cell level in normal and inflamed human intestine. Clin Exp Immunol. 1990;81:301–305. doi: 10.1111/j.1365-2249.1990.tb03334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Reimund JM, Wittersheim C, Dumont S, Muller CD, Baumann R, Poindron P, et al. Mucosal inflammatory cytokine production by intestinal biopsies in patients with ulcerative colitis and Crohn’s disease. J Clin Immunol. 1996;16:144–150. doi: 10.1007/BF01540912. [DOI] [PubMed] [Google Scholar]
- 211.Breese EJ, Michie CA, Nicholls SW, Murch SH, Williams CB, Domizio P, et al. Tumor necrosis factor alpha-producing cells in the intestinal mucosa of children with inflammatory bowel disease. Gastroenterology. 1994;106:1455–1466. doi: 10.1016/0016-5085(94)90398-0. [DOI] [PubMed] [Google Scholar]
- 212.Murch SH, Braegger CP, Walker-Smith JA, MacDonald TT. Location of tumour necrosis factor alpha by immunohistochemistry in chronic inflammatory bowel disease. Gut. 1993;34:1705–1709. doi: 10.1136/gut.34.12.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Stevens C, Walz G, Singaram C, Lipman ML, Zanker B, Muggia A, et al. Tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-6 expression in inflammatory bowel disease. Dig Dis Sci. 1992;37:818–826. doi: 10.1007/BF01300378. [DOI] [PubMed] [Google Scholar]
- 214.Hyams JS, Treem WR, Eddy E, Wyzga N, Moore RE. Tumor necrosis factor-alpha is not elevated in children with inflammatory bowel disease. J Pediatr Gastroenterol Nutr. 1991;12:233–236. doi: 10.1097/00005176-199102000-00016. [DOI] [PubMed] [Google Scholar]
- 215.Owczarek D, Cibor D, Glowacki MK, Ciesla A, Mach P. TNF-alpha and soluble forms of TNF receptors 1 and 2 in the serum of patients with Crohn’s disease and ulcerative colitis. Pol Arch Med Wewn. 2012;122:616–623. doi: 10.20452/pamw.1537. [DOI] [PubMed] [Google Scholar]
- 216.Mizoguchi E, Mizoguchi A, Takedatsu H, Cario E, de Jong YP, Ooi CJ, et al. Role of tumor necrosis factor receptor 2 (TNFR2) in colonic epithelial hyperplasia and chronic intestinal inflammation in mice. Gastroenterology. 2002;122:134–144. doi: 10.1053/gast.2002.30347. [DOI] [PubMed] [Google Scholar]
- 217.Holtmann MH, Douni E, Schutz M, Zeller G, Mudter J, Lehr HA, et al. Tumor necrosis factor-receptor 2 is up-regulated on lamina propria T cells in Crohn’s disease and promotes experimental colitis in vivo. Eur J Immunol. 2002;32:3142–3151. doi: 10.1002/1521-4141(200211)32:11<3142::AID-IMMU3142>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 218.Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/S1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 219.Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, et al. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn’s disease. Am J Gastroenterol. 2002;97:2000–2004. doi: 10.1111/j.1572-0241.2002.05914.x. [DOI] [PubMed] [Google Scholar]
- 220.Fries W, Muja C, Crisafulli C, Costantino G, Longo G, Cuzzocrea S, et al. Infliximab and etanercept are equally effective in reducing enterocyte APOPTOSIS in experimental colitis. Int J Med Sci. 2008;5:169–180. doi: 10.7150/ijms.5.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Fischer A, Gluth M, Pape UF, Wiedenmann B, Theuring F, Baumgart DC. Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-alpha on tight junction proteins and signaling pathways in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2013;304:G970–G979. doi: 10.1152/ajpgi.00183.2012. [DOI] [PubMed] [Google Scholar]
- 222.Lala S, Ogura Y, Osborne C, Hor SY, Bromfield A, Davies S, et al. Crohn’s disease and the NOD2 gene: a role for paneth cells. Gastroenterology. 2003;125:47–57. doi: 10.1016/S0016-5085(03)00661-9. [DOI] [PubMed] [Google Scholar]
- 223.Bazzoni F, Beutler B. The tumor necrosis factor ligand and receptor families. N Engl J Med. 1996;334:1717–1725. doi: 10.1056/NEJM199606273342607. [DOI] [PubMed] [Google Scholar]
- 224.Novotny-Smith CL, Zorbas MA, McIsaac AM, Irimura T, Boman BM, Yeoman LC, et al. Down-modulation of epidermal growth factor receptor accompanies TNF-induced differentiation of the DiFi human adenocarcinoma cell line toward a goblet-like phenotype. J Cell Physiol. 1993;157:253–262. doi: 10.1002/jcp.1041570207. [DOI] [PubMed] [Google Scholar]
- 225.Iwashita J, Sato Y, Sugaya H, Takahashi N, Sasaki H, Abe T. mRNA of MUC2 is stimulated by IL-4, IL-13 or TNF-alpha through a mitogen-activated protein kinase pathway in human colon cancer cells. Immunol Cell Biol. 2003;81:275–282. doi: 10.1046/j.1440-1711.2003.t01-1-01163.x. [DOI] [PubMed] [Google Scholar]
- 226.Freour T, Jarry A, Bach-Ngohou K, Dejoie T, Bou-Hanna C, Denis MG, et al. TACE inhibition amplifies TNF-alpha-mediated colonic epithelial barrier disruption. Int J Mol Med. 2009;23:41–48. [PubMed] [Google Scholar]
- 227.Su L, Nalle SC, Shen L, Turner ES, Singh G, Breskin LA, et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology. 2013;145:407–415. doi: 10.1053/j.gastro.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409–419. doi: 10.1016/S0002-9440(10)62264-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Cesaro A, Abakar-Mahamat A, Brest P, Lassalle S, Selva E, Filippi J, et al. Differential expression and regulation of ADAM17 and TIMP3 in acute inflamed intestinal epithelia. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1332–G1343. doi: 10.1152/ajpgi.90641.2008. [DOI] [PubMed] [Google Scholar]
- 230.Van Hauwermeiren F, Armaka M, Karagianni N, Kranidioti K, Vandenbroucke RE, Loges S, et al. Safe TNF-based antitumor therapy following p55TNFR reduction in intestinal epithelium. J Clin Investig. 2013;123:2590–2603. doi: 10.1172/JCI65624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Goretsky T, Dirisina R, Sinh P, Mittal N, Managlia E, Williams DB, et al. p53 mediates TNF-induced epithelial cell apoptosis in IBD. Am J Pathol. 2012;181:1306–1315. doi: 10.1016/j.ajpath.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Piguet PF, Vesin C, Guo J, Donati Y, Barazzone C. TNF-induced enterocyte apoptosis in mice is mediated by the TNF receptor 1 and does not require p53. Eur J Immunol. 1998;28:3499–3505. doi: 10.1002/(SICI)1521-4141(199811)28:11<3499::AID-IMMU3499>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 233.Hilliard VC, Frey MR, Dempsey PJ, Peek RM, Jr, Polk DB. TNF-alpha converting enzyme-mediated ErbB4 transactivation by TNF promotes colonic epithelial cell survival. Am J Physiol Gastrointest Liver Physiol. 2011;301:G338–G346. doi: 10.1152/ajpgi.00057.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Frey MR, Edelblum KL, Mullane MT, Liang D, Polk DB. The ErbB4 growth factor receptor is required for colon epithelial cell survival in the presence of TNF. Gastroenterology. 2009;136:217–226. doi: 10.1053/j.gastro.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hobbs SS, Goettel JA, Liang D, Yan F, Edelblum KL, Frey MR, et al. TNF transactivation of EGFR stimulates cytoprotective COX-2 expression in gastrointestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2011;301:G220–G229. doi: 10.1152/ajpgi.00383.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Bamias G, Corridoni D, Pizarro TT, Cominelli F. New insights into the dichotomous role of innate cytokines in gut homeostasis and inflammation. Cytokine. 2012;59:451–459. doi: 10.1016/j.cyto.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Meylan F, Richard AC, Siegel RM. TL1A and DR3, a TNF family ligand-receptor pair that promotes lymphocyte costimulation, mucosal hyperplasia, and autoimmune inflammation. Immunol Rev. 2011;244:188–196. doi: 10.1111/j.1600-065X.2011.01068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Ueyama H, Kiyohara T, Sawada N, Isozaki K, Kitamura S, Kondo S, et al. High Fas ligand expression on lymphocytes in lesions of ulcerative colitis. Gut. 1998;43:48–55. doi: 10.1136/gut.43.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Yukawa M, Iizuka M, Horie Y, Yoneyama K, Shirasaka T, Itou H, et al. Systemic and local evidence of increased Fas-mediated apoptosis in ulcerative colitis. Int J Colorectal Dis. 2002;17:70–76. doi: 10.1007/s003840100340. [DOI] [PubMed] [Google Scholar]
- 240.Souza HS, Tortori CJ, Castelo-Branco MT, Carvalho AT, Margallo VS, Delgado CF, et al. Apoptosis in the intestinal mucosa of patients with inflammatory bowel disease: evidence of altered expression of FasL and perforin cytotoxic pathways. Int J Colorectal Dis. 2005;20:277–286. doi: 10.1007/s00384-004-0639-8. [DOI] [PubMed] [Google Scholar]
- 241.Cohavy O, Zhou J, Ware CF, Targan SR. LIGHT is constitutively expressed on T and NK cells in the human gut and can be induced by CD2-mediated signaling. J Immunol. 2005;174:646–653. doi: 10.4049/jimmunol.174.2.646. [DOI] [PubMed] [Google Scholar]
- 242.Begue B, Wajant H, Bambou JC, Dubuquoy L, Siegmund D, Beaulieu JF, et al. Implication of TNF-related apoptosis-inducing ligand in inflammatory intestinal epithelial lesions. Gastroenterology. 2006;130:1962–1974. doi: 10.1053/j.gastro.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 243.Witte E, Witte K, Warszawska K, Sabat R, Wolk K. Interleukin-22: a cytokine produced by T, NK and NKT cell subsets, with importance in the innate immune defense and tissue protection. Cytokine Growth Factor Rev. 2010;21:365–379. doi: 10.1016/j.cytogfr.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 244.Atreya R, Neurath MF. Involvement of IL-6 in the pathogenesis of inflammatory bowel disease and colon cancer. Clin Rev Allergy Immunol. 2005;28:187–196. doi: 10.1385/CRIAI:28:3:187. [DOI] [PubMed] [Google Scholar]
- 245.Lenardo MJ, Baltimore D. NF-kappa B: a pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989;58:227–229. doi: 10.1016/0092-8674(89)90833-7. [DOI] [PubMed] [Google Scholar]
- 246.Gloire G, Dejardin E, Piette J. Extending the nuclear roles of IkappaB kinase subunits. Biochem Pharmacol. 2006;72:1081–1089. doi: 10.1016/j.bcp.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 247.Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242:540–546. doi: 10.1126/science.3140380. [DOI] [PubMed] [Google Scholar]
- 248.Greten FR, Karin M. The IKK/NF-kappaB activation pathway-a target for prevention and treatment of cancer. Cancer Lett. 2004;206:193–199. doi: 10.1016/j.canlet.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 249.Huxford T, Malek S, Ghosh G. Structure and mechanism in NF-kappa B/I kappa B signaling. Cold Spring Harb Symp Quant Biol. 1999;64:533–540. doi: 10.1101/sqb.1999.64.533. [DOI] [PubMed] [Google Scholar]
- 250.Neurath MF, Pettersson S, Meyer zum Buschenfelde KH, Strober W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat Med. 1996;2:998–1004. doi: 10.1038/nm0996-998. [DOI] [PubMed] [Google Scholar]
- 251.Conner EM, Brand S, Davis JM, Laroux FS, Palombella VJ, Fuseler JW, et al. Proteasome inhibition attenuates nitric oxide synthase expression, VCAM-1 transcription and the development of chronic colitis. J Pharmacol Exp Ther. 1997;282:1615–1622. [PubMed] [Google Scholar]
- 252.Herfarth H, Brand K, Rath HC, Rogler G, Scholmerich J, Falk W. Nuclear factor-kappa B activity and intestinal inflammation in dextran sulphate sodium (DSS)-induced colitis in mice is suppressed by gliotoxin. Clin Exp Immunol. 2000;120:59–65. doi: 10.1046/j.1365-2249.2000.01184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Jobin C, Panja A, Hellerbrand C, Iimuro Y, Didonato J, Brenner DA, et al. Inhibition of proinflammatory molecule production by adenovirus-mediated expression of a nuclear factor kappaB super-repressor in human intestinal epithelial cells. J Immunol. 1998;160:410–418. [PubMed] [Google Scholar]
- 254.Jobin C, Hellerbrand C, Licato LL, Brenner DA, Sartor RB. Mediation by NF-kappa B of cytokine induced expression of intercellular adhesion molecule 1 (ICAM-1) in an intestinal epithelial cell line, a process blocked by proteasome inhibitors. Gut. 1998;42:779–787. doi: 10.1136/gut.42.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Rogler G, Brand K, Vogl D, Page S, Hofmeister R, Andus T, et al. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology. 1998;115:357–369. doi: 10.1016/S0016-5085(98)70202-1. [DOI] [PubMed] [Google Scholar]
- 256.Zaph C, Troy AE, Taylor BC, Berman-Booty LD, Guild KJ, Du Y, et al. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature. 2007;446:552–556. doi: 10.1038/nature05590. [DOI] [PubMed] [Google Scholar]
- 257.Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Lees AB, Goldstine HH. Communication channels. Science. 1961;134:527–529. doi: 10.1126/science.134.3478.527-b. [DOI] [PubMed] [Google Scholar]
- 259.Cenit MC, Alcina A, Marquez A, Mendoza JL, Diaz-Rubio M, de las Heras V, et al. STAT3 locus in inflammatory bowel disease and multiple sclerosis susceptibility. Genes Immun. 2010;11:264–268. doi: 10.1038/gene.2010.10. [DOI] [PubMed] [Google Scholar]
- 260.Neurath MF, Finotto S. IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 2011;22:83–89. doi: 10.1016/j.cytogfr.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 261.Wirtz S, Finotto S, Kanzler S, Lohse AW, Blessing M, Lehr HA, et al. Cutting edge: chronic intestinal inflammation in STAT-4 transgenic mice: characterization of disease and adoptive transfer by TNF- plus IFN-gamma-producing CD4+ T cells that respond to bacterial antigens. J Immunol. 1999;162:1884–1888. [PubMed] [Google Scholar]
- 262.Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/S1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 263.Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med. 2000;6:583–588. doi: 10.1038/75068. [DOI] [PubMed] [Google Scholar]
- 264.Kobayashi M, Kweon MN, Kuwata H, Schreiber RD, Kiyono H, Takeda K, et al. Toll-like receptor-dependent production of IL-12p40 causes chronic enterocolitis in myeloid cell-specific Stat3-deficient mice. J Clin Investig. 2003;111:1297–1308. doi: 10.1172/JCI17085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Li Y, de Haar C, Chen M, Deuring J, Gerrits MM, Smits R, et al. Disease-related expression of the IL6/STAT3/SOCS3 signalling pathway in ulcerative colitis and ulcerative colitis-related carcinogenesis. Gut. 2010;59:227–235. doi: 10.1136/gut.2009.184176. [DOI] [PubMed] [Google Scholar]
- 266.Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Investig. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Backert I, Koralov SB, Wirtz S, Kitowski V, Billmeier U, Martini E, et al. STAT3 activation in Th17 and Th22 cells controls IL-22-mediated epithelial host defense during infectious colitis. J Immunol. 2014;193:3779–3791. doi: 10.4049/jimmunol.1303076. [DOI] [PubMed] [Google Scholar]
- 268.Du W, Hong J, Wang YC, Zhang YJ, Wang P, Su WY, et al. Inhibition of JAK2/STAT3 signalling induces colorectal cancer cell apoptosis via mitochondrial pathway. J Cell Mol Med. 2012;16:1878–1888. doi: 10.1111/j.1582-4934.2011.01483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Nguyen AV, Wu YY, Liu Q, Wang D, Nguyen S, Loh R, et al. STAT3 in epithelial cells regulates inflammation and tumor progression to malignant state in colon. Neoplasia. 2013;15:998–1008. doi: 10.1593/neo.13952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, Szabady RL, et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med. 2013;210:917–931. doi: 10.1084/jem.20122308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Thompson CL, Plummer SJ, Tucker TC, Casey G, Li L. Interleukin-22 genetic polymorphisms and risk of colon cancer. Cancer Causes Control CCC. 2010;21:1165–1170. doi: 10.1007/s10552-010-9542-5. [DOI] [PubMed] [Google Scholar]
- 272.Pinto D, Gregorieff A, Begthel H, Clevers H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 2003;17:1709–1713. doi: 10.1101/gad.267103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, et al. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/S0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 275.Krausova M, Korinek V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014;26:570–579. doi: 10.1016/j.cellsig.2013.11.032. [DOI] [PubMed] [Google Scholar]
- 276.Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, et al. Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell. 2012;149:1245–1256. doi: 10.1016/j.cell.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 277.Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 278.Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, et al. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–399. doi: 10.1016/S0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 279.de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H, et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature. 2011;476:293–297. doi: 10.1038/nature10337. [DOI] [PubMed] [Google Scholar]
- 280.Glinka A, Dolde C, Kirsch N, Huang YL, Kazanskaya O, Ingelfinger D, et al. LGR4 and LGR5 are R-spondin receptors mediating Wnt/beta-catenin and Wnt/PCP signalling. EMBO Rep. 2011;12:1055–1061. doi: 10.1038/embor.2011.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Robles AI, Traverso G, Zhang M, Roberts NJ, Khan MA, Joseph C, et al. Whole-exome sequencing analyses of inflammatory bowel disease-associated colorectal cancers. Gastroenterology. 2016;150:931–943. doi: 10.1053/j.gastro.2015.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 283.Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 284.Gaspar C, Fodde R. APC dosage effects in tumorigenesis and stem cell differentiation. Int J Develop Biol. 2004;48:377–386. doi: 10.1387/ijdb.041807cg. [DOI] [PubMed] [Google Scholar]
- 285.Fearon ER. Molecular genetics of colorectal cancer. Ann Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 286.Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36:417–422. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- 287.Rawson JB, Manno M, Mrkonjic M, Daftary D, Dicks E, Buchanan DD, et al. Promoter methylation of Wnt antagonists DKK1 and SFRP1 is associated with opposing tumor subtypes in two large populations of colorectal cancer patients. Carcinogenesis. 2011;32:741–747. doi: 10.1093/carcin/bgr020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Andrianifahanana M, Moniaux N, Schmied BM, Ringel J, Friess H, Hollingsworth MA, et al. Mucin (MUC) gene expression in human pancreatic adenocarcinoma and chronic pancreatitis: a potential role of MUC4 as a tumor marker of diagnostic significance. Clin Cancer Res Off J Am Assoc Cancer Res. 2001;7:4033–4040. [PubMed] [Google Scholar]
- 289.Renaud F, Mariette C, Vincent A, Wacrenier A, Maunoury V, Leclerc J, et al. The serrated neoplasia pathway of colorectal tumors: identification of MUC5AC hypomethylation as an early marker of polyps with malignant potential. Int J Cancer. 2016;138:1472–1481. doi: 10.1002/ijc.29891. [DOI] [PubMed] [Google Scholar]
- 290.Kesari MV, Gaopande VL, Joshi AR, Babanagare SV, Gogate BP, Khadilkar AV. Immunohistochemical study of MUC1, MUC2 and MUC5AC in colorectal carcinoma and review of literature. Indian J Gastroenterol Off J Indian Soc Gastroenterol. 2015;34:63–67. doi: 10.1007/s12664-015-0534-y. [DOI] [PubMed] [Google Scholar]
- 291.Biemer-Huttmann AE, Walsh MD, McGuckin MA, Ajioka Y, Watanabe H, Leggett BA, et al. Immunohistochemical staining patterns of MUC1, MUC2, MUC4, and MUC5AC mucins in hyperplastic polyps, serrated adenomas, and traditional adenomas of the colorectum. J Histochem Cytochem Off J Histochem Soc. 1999;47:1039–1048. doi: 10.1177/002215549904700808. [DOI] [PubMed] [Google Scholar]
- 292.Shanmugam C, Jhala NC, Katkoori VR, Wan W, Meleth S, Grizzle WE, et al. Prognostic value of mucin 4 expression in colorectal adenocarcinomas. Cancer. 2010;116:3577–3586. doi: 10.1002/cncr.25095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Ogata S, Uehara H, Chen A, Itzkowitz SH. Mucin gene expression in colonic tissues and cell lines. Cancer Res. 1992;52:5971–5978. [PubMed] [Google Scholar]
- 294.Imai Y, Yamagishi H, Fukuda K, Ono Y, Inoue T, Ueda Y. Differential mucin phenotypes and their significance in a variation of colorectal carcinoma. World J Gastroenterol WJG. 2013;19:3957–3968. doi: 10.3748/wjg.v19.i25.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Pai P, Rachagani S, Dhawan P, Batra SK. Mucins and Wnt/beta-catenin signaling in gastrointestinal cancers: an unholy nexus. Carcinogenesis. 2016;37:223–232. doi: 10.1093/carcin/bgw005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.van de Wetering M, Oving I, Muncan V, Pon Fong MT, Brantjes H, van Leenen D, et al. Specific inhibition of gene expression using a stably integrated, inducible small-interfering-RNA vector. EMBO Rep. 2003;4:609–615. doi: 10.1038/sj.embor.embor865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–250. doi: 10.1016/S0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 298.Caderni G, Femia AP, Giannini A, Favuzza A, Luceri C, Salvadori M, et al. Identification of mucin-depleted foci in the unsectioned colon of azoxymethane-treated rats: correlation with carcinogenesis. Cancer Res. 2003;63:2388–2392. [PubMed] [Google Scholar]
- 299.Citalan-Madrid AF, Garcia-Ponce A, Vargas-Robles H, Betanzos A, Schnoor M. Small GTPases of the Ras superfamily regulate intestinal epithelial homeostasis and barrier function via common and unique mechanisms. Tissue Barr. 2013;1:e26938. doi: 10.4161/tisb.26938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013;93:269–309. doi: 10.1152/physrev.00003.2012. [DOI] [PubMed] [Google Scholar]
- 301.Garcia-Mata R, Boulter E, Burridge K. The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011;12:493–504. doi: 10.1038/nrm3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Benoit YD, Lussier C, Ducharme PA, Sivret S, Schnapp LM, Basora N, et al. Integrin alpha8beta1 regulates adhesion, migration and proliferation of human intestinal crypt cells via a predominant RhoA/ROCK-dependent mechanism. Biol Cell/ 2009;101:695–708. doi: 10.1042/BC20090060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Hammar E, Tomas A, Bosco D, Halban PA. Role of the Rho-ROCK (Rho-associated kinase) signaling pathway in the regulation of pancreatic beta-cell function. Endocrinology. 2009;150:2072–2079. doi: 10.1210/en.2008-1135. [DOI] [PubMed] [Google Scholar]
- 304.Bruewer M, Hopkins AM, Hobert ME, Nusrat A, Madara JL. RhoA, Rac1, and Cdc42 exert distinct effects on epithelial barrier via selective structural and biochemical modulation of junctional proteins and F-actin. Am J Physiol Cell Physiol. 2004;287:C327–C335. doi: 10.1152/ajpcell.00087.2004. [DOI] [PubMed] [Google Scholar]
- 305.Chandhoke SK, Mooseker MS. A role for myosin IXb, a motor-RhoGAP chimera, in epithelial wound healing and tight junction regulation. Mol Biol Cell. 2012;23:2468–2480. doi: 10.1091/mbc.E11-09-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Terry SJ, Zihni C, Elbediwy A, Vitiello E, Leefa Chong San IV, Balda MS, et al. Spatially restricted activation of RhoA signalling at epithelial junctions by p114RhoGEF drives junction formation and morphogenesis. Nat Cell Biol. 2011;13:159–166. doi: 10.1038/ncb2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Babbin BA, Jesaitis AJ, Ivanov AI, Kelly D, Laukoetter M, Nava P, et al. Formyl peptide receptor-1 activation enhances intestinal epithelial cell restitution through phosphatidylinositol 3-kinase-dependent activation of Rac1 and Cdc42. J Immunol. 2007;179:8112–8121. doi: 10.4049/jimmunol.179.12.8112. [DOI] [PubMed] [Google Scholar]
- 308.Espejo R, Rengifo-Cam W, Schaller MD, Evers BM, Sastry SK. PTP-PEST controls motility, adherens junction assembly, and Rho GTPase activity in colon cancer cells. Am J Physiol Cell Physiol. 2010;299:C454–C463. doi: 10.1152/ajpcell.00148.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Chen P, Kartha S, Bissonnette M, Hart J, Toback FG. AMP-18 facilitates assembly and stabilization of tight junctions to protect the colonic mucosal barrier. Inflamm Bowel Dis. 2012;18:1749–1759. doi: 10.1002/ibd.22886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Elbediwy A, Zihni C, Terry SJ, Clark P, Matter K, Balda MS. Epithelial junction formation requires confinement of Cdc42 activity by a novel SH3BP1 complex. J Cell Biol. 2012;198:677–693. doi: 10.1083/jcb.201202094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Hopkins AM, Walsh SV, Verkade P, Boquet P, Nusrat A. Constitutive activation of Rho proteins by CNF-1 influences tight junction structure and epithelial barrier function. J Cell Sci. 2003;116:725–742. doi: 10.1242/jcs.00300. [DOI] [PubMed] [Google Scholar]
- 312.Sakamori R, Das S, Yu S, Feng S, Stypulkowski E, Guan Y, et al. Cdc42 and Rab8a are critical for intestinal stem cell division, survival, and differentiation in mice. J Clin Investig. 2012;122:1052–1065. doi: 10.1172/JCI60282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Melendez J, Liu M, Sampson L, Akunuru S, Han X, Vallance J, et al. Cdc42 coordinates proliferation, polarity, migration, and differentiation of small intestinal epithelial cells in mice. Gastroenterology. 2013;145:808–819. doi: 10.1053/j.gastro.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Stappenbeck TS, Gordon JI. Rac1 mutations produce aberrant epithelial differentiation in the developing and adult mouse small intestine. Development. 2000;127:2629–2642. doi: 10.1242/dev.127.12.2629. [DOI] [PubMed] [Google Scholar]
- 315.Myant KB, Scopelliti A, Haque S, Vidal M, Sansom OJ, Cordero JB. Rac1 drives intestinal stem cell proliferation and regeneration. Cell Cycle. 2013;12:2973–2977. doi: 10.4161/cc.26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Segain JP, Raingeard de la Bletiere D, Sauzeau V, Bourreille A, Hilaret G, Cario-Toumaniantz C, et al. Rho kinase blockade prevents inflammation via nuclear factor kappa B inhibition: evidence in Crohn’s disease and experimental colitis. Gastroenterology. 2003;124:1180–1187. doi: 10.1016/S0016-5085(03)00283-X. [DOI] [PubMed] [Google Scholar]
- 317.Lopez-Posadas R, Becker C, Gunther C, Tenzer S, Amann K, Billmeier U, et al. Rho-A prenylation and signaling link epithelial homeostasis to intestinal inflammation. J Clin Investig. 2016;126:611–626. doi: 10.1172/JCI80997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Fritz G, Just I, Kaina B. Rho GTPases are over-expressed in human tumors. Int J Cancer. 1999;81:682–687. doi: 10.1002/(SICI)1097-0215(19990531)81:5<682::AID-IJC2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 319.Sakamori R, Yu S, Zhang X, Hoffman A, Sun J, Das S, et al. CDC42 inhibition suppresses progression of incipient intestinal tumors. Cancer Res. 2014;74:5480–5492. doi: 10.1158/0008-5472.CAN-14-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Myant KB, Cammareri P, McGhee EJ, Ridgway RA, Huels DJ, Cordero JB, et al. ROS production and NF-kappaB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell. 2013;12:761–773. doi: 10.1016/j.stem.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Ray RM, Vaidya RJ, Johnson LR. MEK/ERK regulates adherens junctions and migration through Rac1. Cell Motil Cytoskelet. 2007;64:143–156. doi: 10.1002/cm.20172. [DOI] [PubMed] [Google Scholar]
- 322.Olofsson B. Rho guanine dissociation inhibitors: pivotal molecules in cellular signalling. Cell Signal. 1999;11:545–554. doi: 10.1016/S0898-6568(98)00063-1. [DOI] [PubMed] [Google Scholar]
- 323.Xiao H, Qin X, Ping D, Zuo K. Inhibition of Rho and Rac geranylgeranylation by atorvastatin is critical for preservation of endothelial junction integrity. PLoS One. 2013;8:e59233. doi: 10.1371/journal.pone.0059233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Pechlivanis M, Kuhlmann J. Hydrophobic modifications of Ras proteins by isoprenoid groups and fatty acids—more than just membrane anchoring. Biochim Biophys Acta. 2006;1764:1914–1931. doi: 10.1016/j.bbapap.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 325.Shen WP, Aldrich TH, Venta-Perez G, Franza BR, Jr, Furth ME. Expression of normal and mutant ras proteins in human acute leukemia. Oncogene. 1987;1:157–165. [PubMed] [Google Scholar]
- 326.Lu J, Chan L, Fiji HD, Dahl R, Kwon O, Tamanoi F. In vivo antitumor effect of a novel inhibitor of protein geranylgeranyltransferase-I. Mol Cancer Ther. 2009;8:1218–1226. doi: 10.1158/1535-7163.MCT-08-1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Sebti SM, Hamilton AD. Farnesyltransferase and geranylgeranyltransferase I inhibitors in cancer therapy: important mechanistic and bench to bedside issues. Expert Opin Investig Drugs. 2000;9:2767–2782. doi: 10.1517/13543784.9.12.2767. [DOI] [PubMed] [Google Scholar]
- 328.Rao PV, Peterson YK, Inoue T, Casey PJ. Effects of pharmacologic inhibition of protein geranylgeranyltransferase type I on aqueous humor outflow through the trabecular meshwork. Invest Ophthalmol Vis Sci. 2008;49:2464–2471. doi: 10.1167/iovs.07-1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Lerner EC, Qian Y, Blaskovich MA, Fossum RD, Vogt A, Sun J, et al. Ras CAAX peptidomimetic FTI-277 selectively blocks oncogenic Ras signaling by inducing cytoplasmic accumulation of inactive Ras-Raf complexes. J Biol Chem. 1995;270:26802–26806. doi: 10.1074/jbc.270.45.26802. [DOI] [PubMed] [Google Scholar]
- 330.Blanco-Colio LM, Villa A, Ortego M, Hernandez-Presa MA, Pascual A, Plaza JJ, et al. 3-Hydroxy-3-methyl-glutaryl coenzyme A reductase inhibitors, atorvastatin and simvastatin, induce apoptosis of vascular smooth muscle cells by downregulation of Bcl-2 expression and Rho A prenylation. Atherosclerosis. 2002;161:17–26. doi: 10.1016/S0021-9150(01)00613-X. [DOI] [PubMed] [Google Scholar]
- 331.Park HJ, Kong D, Iruela-Arispe L, Begley U, Tang D, Galper JB. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors interfere with angiogenesis by inhibiting the geranylgeranylation of RhoA. Circ Res. 2002;91:143–150. doi: 10.1161/01.RES.0000028149.15986.4C. [DOI] [PubMed] [Google Scholar]
- 332.Abe Y, Murano M, Murano N, Morita E, Inoue T, Kawakami K, et al. Simvastatin attenuates intestinal fibrosis independent of the anti-inflammatory effect by promoting fibroblast/myofibroblast apoptosis in the regeneration/healing process from TNBS-induced colitis. Dig Dis Sci. 2012;57:335–344. doi: 10.1007/s10620-011-1879-4. [DOI] [PubMed] [Google Scholar]
- 333.Lee JY, Kim JS, Kim JM, Kim N, Jung HC, Song IS. Simvastatin inhibits NF-kappaB signaling in intestinal epithelial cells and ameliorates acute murine colitis. Int Immunopharmacol. 2007;7:241–248. doi: 10.1016/j.intimp.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 334.Ikeda M, Takeshima F, Isomoto H, Shikuwa S, Mizuta Y, Ozono Y, et al. Simvastatin attenuates trinitrobenzene sulfonic acid-induced colitis, but not oxazalone-induced colitis. Dig Dis Sci. 2008;53:1869–1875. doi: 10.1007/s10620-007-0102-0. [DOI] [PubMed] [Google Scholar]
- 335.Ballester I, Daddaoua A, Lopez-Posadas R, Nieto A, Suarez MD, Zarzuelo A, et al. The bisphosphonate alendronate improves the damage associated with trinitrobenzenesulfonic acid-induced colitis in rats. Br J Pharmacol. 2007;151:206–215. doi: 10.1038/sj.bjp.0707227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Sassa S, Okabe H, Nemoto N, Kikuchi H, Kudo H, Sakamoto S. Ibadronate may prevent colorectal carcinogenesis in mice with ulcerative colitis. Anticancer Res. 2009;29:4615–4619. [PubMed] [Google Scholar]