

Abstract
Hedgehog signaling is an evolutionarily conserved pathway which is essential in embryonic and postnatal development as well as adult organ homeostasis. Abnormal regulation of Hedgehog signaling is implicated in many diseases including cancer. Consequently, substantial efforts have made in the past to develop potential therapeutic agents that specifically target the Hedgehog signaling for cancer treatment. Here, we review the therapeutic agents for inhibition of the Hedgehog signaling and their clinical advances in cancer treatment.
Keywords: Hedgehog signaling, Patched, Smothern, Cancer, Small molecules, Therapeutic, Drugs, Safety and clinic
Introduction
Hedgehog (HH) signaling is an evolutionarily conserved pathway that is indispensable for developmental patterning and adult tissue homeostasis. HH signaling was first identified in Drosophila Melanogaster for its essential role in early embryo patterning, and subsequently has been reported in variety of vertebrates [1, 2]. In mammalians, three HH paralogues, Sonic Hedgehog (sHH), Indian Hedgehog (iHH), and Desert Hedgehog (dHH), have been reported, and each of them displays unique expression patterns and functions [3–5]. For instance, sHH is essential for correct formation of the limbs, phallus, somites, and neural tube in early embryogenesis [6–8], whereas iHH is restricted to chondrocyte development and dHH is limited to spermatogenesis and nerve sheath formation in Schwann cells [9–12]. All the three HH orthologues mediate the HH signaling in primary cilia of mammalian cells in a similar way. In the absence of the HH ligands, a 12-span transmembrane protein Patched (PTCH) is positioned in cilia and catalytically inhibits Smoothened (SMO), preventing SMO accumulation to cilia (Fig. 1). Full-length GLI (GLI2/3) proteins are then phosphorylated by protein kinase A (PKA), glycogen synthase kinase 3β (GSK3β), and casein kinase 1 (CK1), leading to proteolytic process to generate repressor GLI (GLIR) for suppression of the HH target gene transcription [13–15] (Fig. 1). However, when extracellular HH ligands bind to PTCH, PTCH is displaced from cilia and inhibitory influence of PTCH on SMO is then removed. Activated SMO subsequently relocates to cilia to transduce the downstream signaling (Fig. 1). The complex of Suppressor of Fused (SUFU) and GLI is then disassociated within cilia, and the activated GLI proteins bypass proteolytic processing and translocate into nucleus to induce transcription of the HH target genes, including GLI1, PTCH1, SNAIL, and HH-interacting protein (HHIP), cyclin D1 (CCND1), c-Myc, BMI1 polycomb ring finger (BMI1) and B-cell CLL/lymphoma 2 (BCL2), etc [16–18]. SUFU is a negative regulator of the HH signaling by sequester GLI proteins in the cytoplasm to suppress GLI transcriptional activation [19, 20].
Fig. 1.
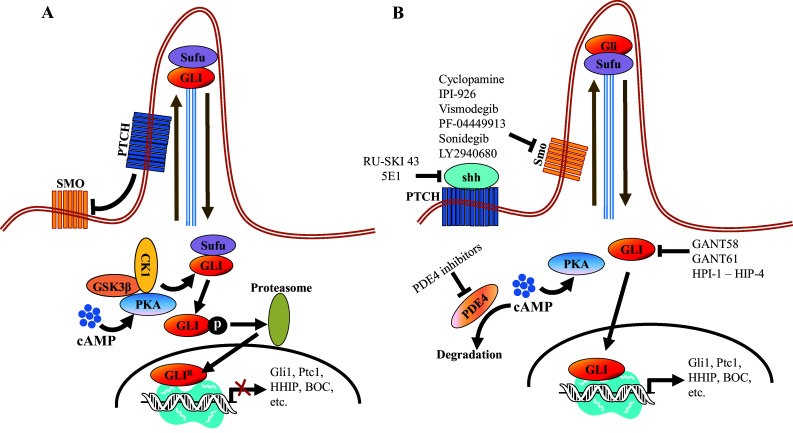
Schematic representation of the HH signaling pathway and the oncology-indication drug candidates discussed in the paper. a In the absence of HH ligands, PTCH positioned in cilia prevents SMO accumulation in cilia and inhibits SMO activity. PKA, CK1 and GSK3β phosphorylate GLI2/3, which subsequently is proteolytically processed to an NH2-terminal truncated GLI2/3 repressor form for suppression of the HH signaling target gene expression. b In the presence of HH ligands, HH ligands bind and inactivate PTCH, and activated SMO then translocates to cilia, leading to disassociation of SUFU-GLI complex. The activated GLI 2/3 then translocates into the nucleus to induce the HH signaling target gene transcription
Abnormal activation of HH signaling is implicated in many types of cancer [21]. In addition, increasing evidence supports that the HH signaling also plays a critical role in maintaining “stemness” of cancer stem cells (CSCs), a subpopulation of tumor cells that are believed to account for tumor initiation, growth, and recurrence as well as drug resistance [22–27]. Thus, eradication of CSCs by targeting the HH signaling represents a potential effective therapeutic strategy for cancer, and significant efforts have been made in the past decades to develop HH signaling inhibitors. In this article, we review development of therapeutic agents targeting the HH pathway and their clinical advances in cancer treatment.
Hedgehog signaling pathway in cancer and cancer stem cells
Implication of HH signaling in cancer was first suggested in malignant glioma by identification of overexpression of HH target gene GLI1 [28]. Further studies have led to proposal of three primary HH signaling models in cancer: ligand-independent mutation-driven signaling, ligand-dependent autocrine/juxtacrine signaling, and ligand-dependent paracrine signaling [18, 21]. In the ligand-independent mutation-driven signaling model, PTCH inactivating mutations were identified in patients with basal cell nevus syndrome, a rare autosomal dominant disorder with a high risk of basal cell carcinoma, medulloblastoma and rhabdomyosarcoma [29–33]. Moreover, activating mutations in SMO or inactivating mutations in SuFu that can constitutively activate the HH signaling in the absence of HH ligands were also reported in sporadic basal cell carcinoma, chondrosarcoma, and medulloblastoma [34–38]. However, in the ligand-dependent autocrine/juxtacrine signaling model, elevated HH ligand expression in the tumor cells constitutively activates the HH pathway in themselves or the adjacent tumor cells to support tumor growth and survival [39–41]. This signaling model has been observed in many cancers, including lung cancer, esophagus cancer, digestive tract cancer, pancreas cancer, prostate cancer, breast cancer, and liver and brain cancer [23, 39–44]. In contrast, in the ligand-dependent paracrine signaling, tumor-produced HH ligands activate the HH signaling in the stromal microenvironment which then feeds back and contributes to tumor progression [45–47].
Recently, increasing evidence supports that CSCs, a small subset of cancer cells with capability of self-renewal and differentiation into heterogeneous tumor cells, are responsible for tumor initiation, growth, and recurrence as well as drug resistance [48–50]. Further studies have supported that abnormal activation of HH signaling plays an essential role in CSC regulation and maintenance in various cancers including glioblastoma, lung squamous cell carcinoma, breast cancer, pancreatic adenocarcinoma, myeloma, and chronic myeloid leukemia (CML) [22–27]. For instance, activation of HH signaling enhanced multiple myeloma CSC expansion, whereas inhibition of the HH pathway effectively attenuated multiple myeloma CSC clonal expansion [51].
In summary, abnormal activation of HH signaling plays a critical role in tumorigenesis and CSC maintenance, and targeting the HH signaling pathway represents an important therapeutic strategy to treat cancer, and potentially disrupt CSCs’ stemness and functions to improve overall cancer treatment outcomes.
Therapeutic agents targeting Hedgehog signaling
Given the critical roles of abnormal activation of the HH signaling in various types of cancer and CSCs, substantial efforts have been made to develop therapeutic agents to inhibit HH signaling by targeting various key components in the pathway cascade.
Target HH ligands
As the ligand-depend HH signaling activation is associated with various cancers, therapeutic reagents targeting HH ligands to inhibit dysregulated HH signaling activation have been highly sought after for cancer treatments. Monoclonal antibody 5E1, which blocks binding of all three mammalian HH orthologues to PTCH for HH signaling inhibition, was generated with mouse hybridoma using the rat SHH N-terminal domain as the antigen [52, 53]. In the preclinical studies, 5E1 has been shown to suppress growth of medulloblastoma and pancreatic tumors in mouse models, respectively [54, 55]. In addition to the biological antibody, a small molecule RUSKI-43 has also been developed to specifically target the hedgehog acyltransferase [56] (Table 1 ). Hedgehog acyltransferase is an essential enzyme for the SHH palmitoylation, a critical step to significantly enhance sHH ligand potency during sHH processing before it binds to the PCTH receptor [56–59]. RUSKI-43 was shown to reduce proliferation and anchorage-independent growth of breast cancer cells as well as inhibit pancreatic tumor growth in animal models [60, 61]. Nevertheless, a recent study has indicated that RUSKI-43 possesses cytotoxic activity unrelated to canonical sHH signaling and the authors also reported a preferred small molecule RUSKI-201 which selectively inhibits catalytic function of Hedgehog acyltransferase [62]. To date, no clinical trials of 5E1, RUSKI-43, or RUSKI-201 have taken place yet.
Table 1.
Selected small molecules that inhibit the HH signaling
| Structure | Name | Target | References |
|---|---|---|---|
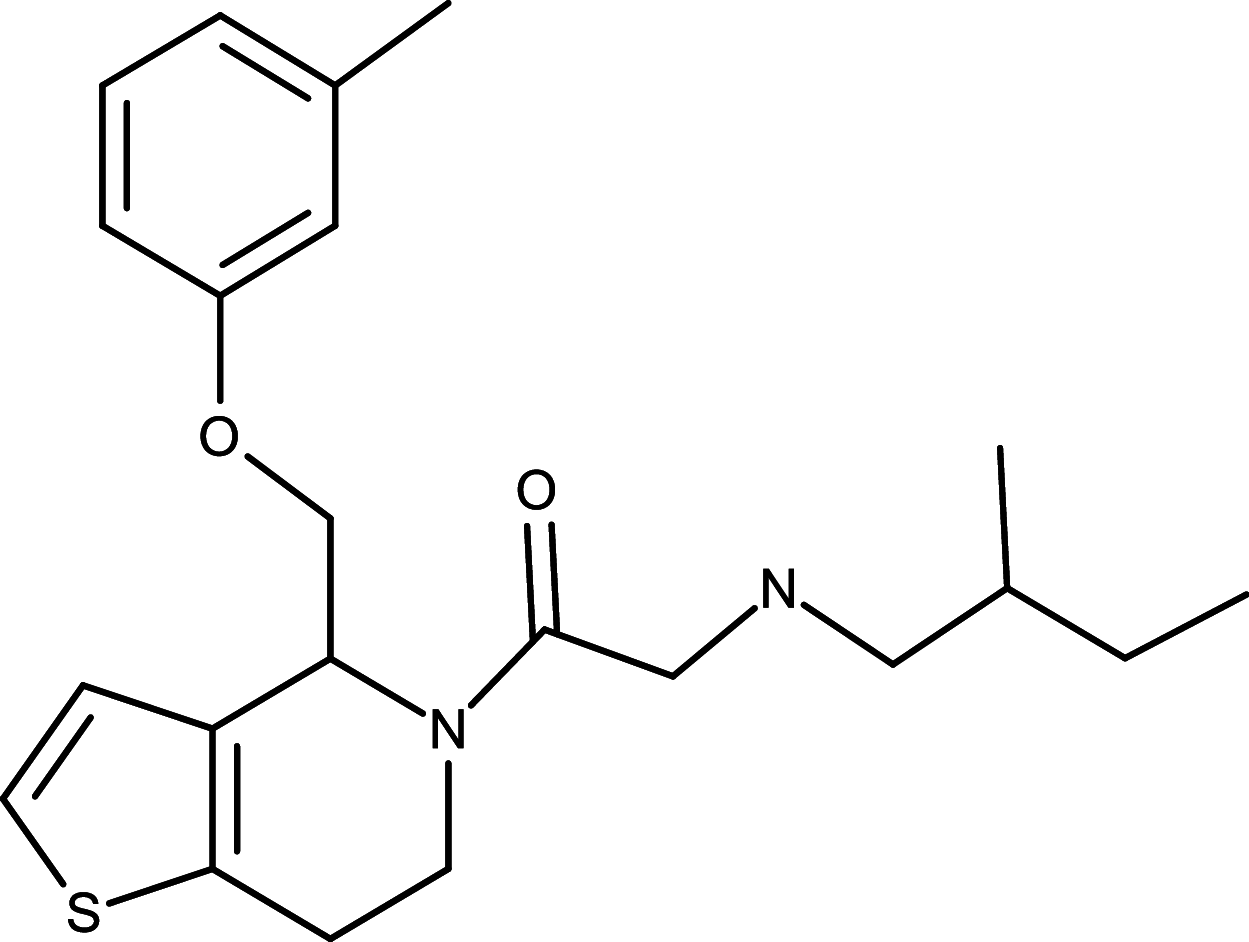
|
RUSKI-43 | SHH ligand | [56] |
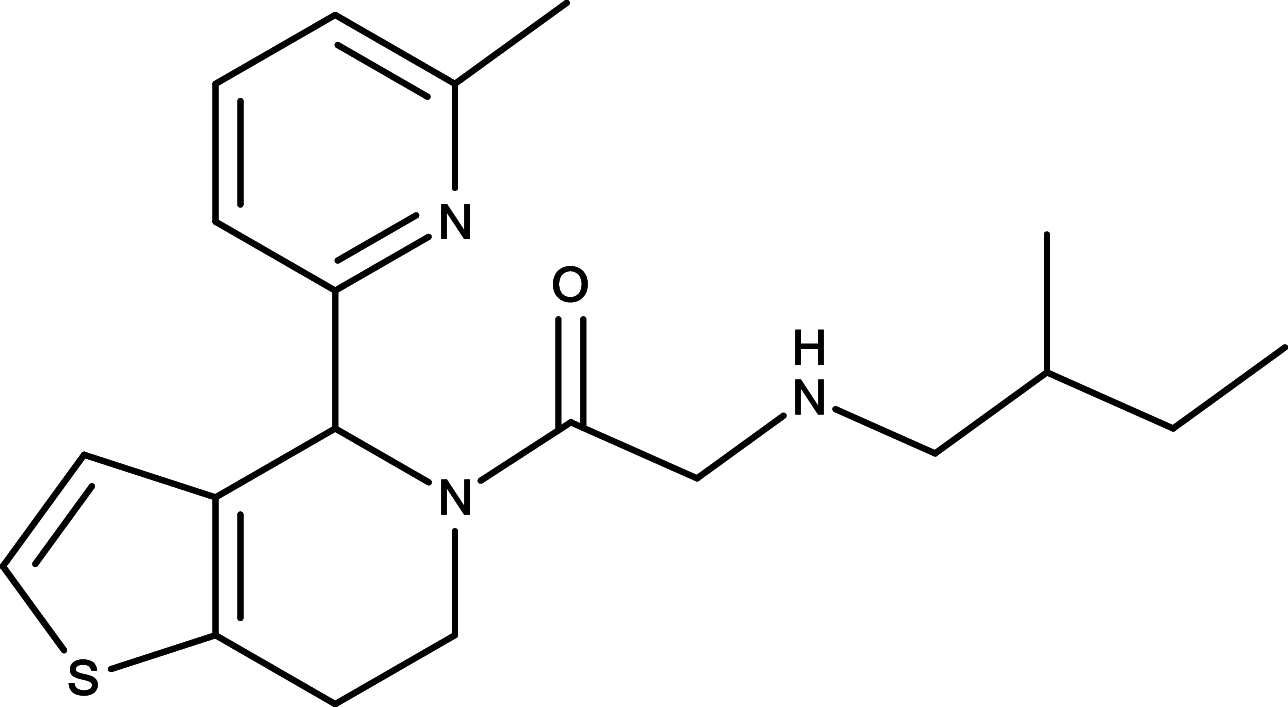
|
RUSKI-201 | SHH ligand | [62] |

|
Cyclopamine | SMO | [63] |

|
KAAD-cyclopamine | SMO | [64] |

|
IPI-926 (Saridegib) | SMO | [71] |
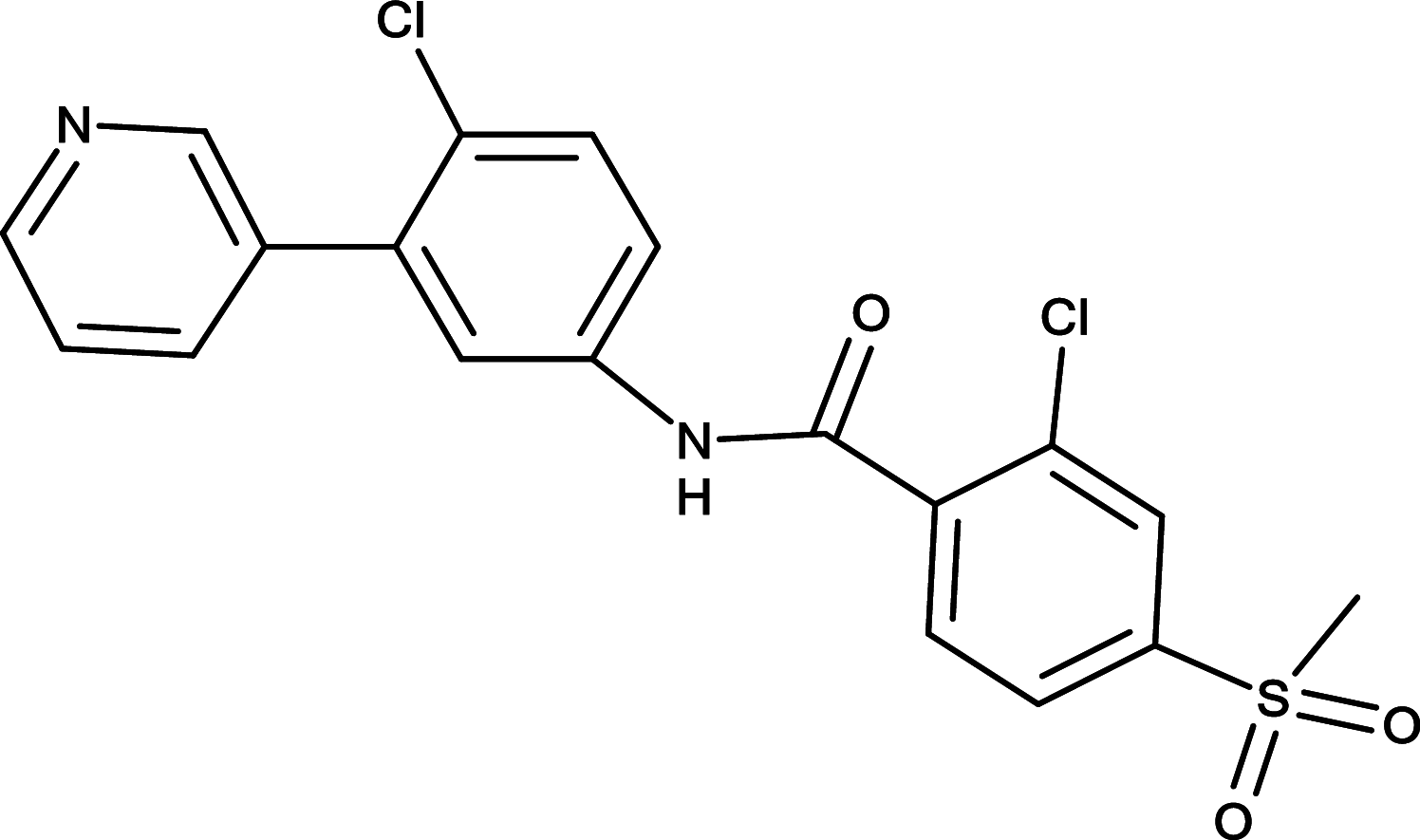
|
Vismodegib (GDC-0449) | SMO |
[76] [77] |
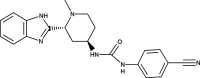
|
PF-04449913 (Glasdegib) | SMO | [81] |

|
LY2940680 (Taladegib) | SMO |
[86] [87] |

|
Sonidegib (LDE-225) | SMO |
[76] [77] |

|
GANT58 | GLI | [92] |
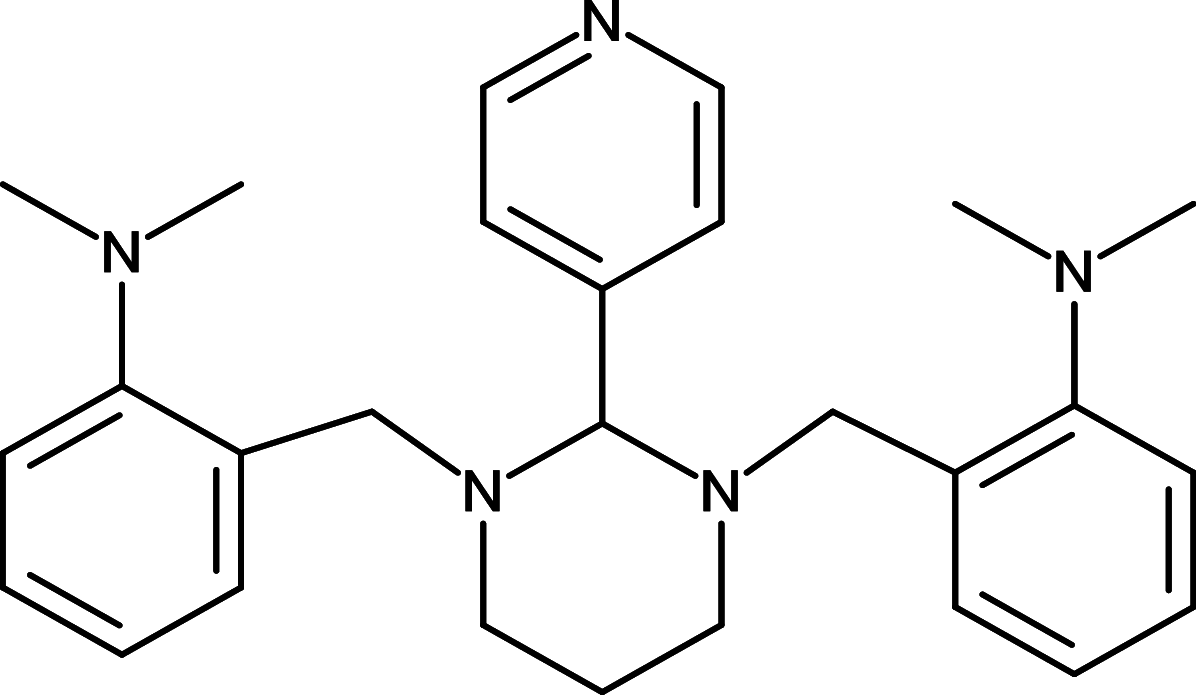
|
GANT61 | GLI | [92] |
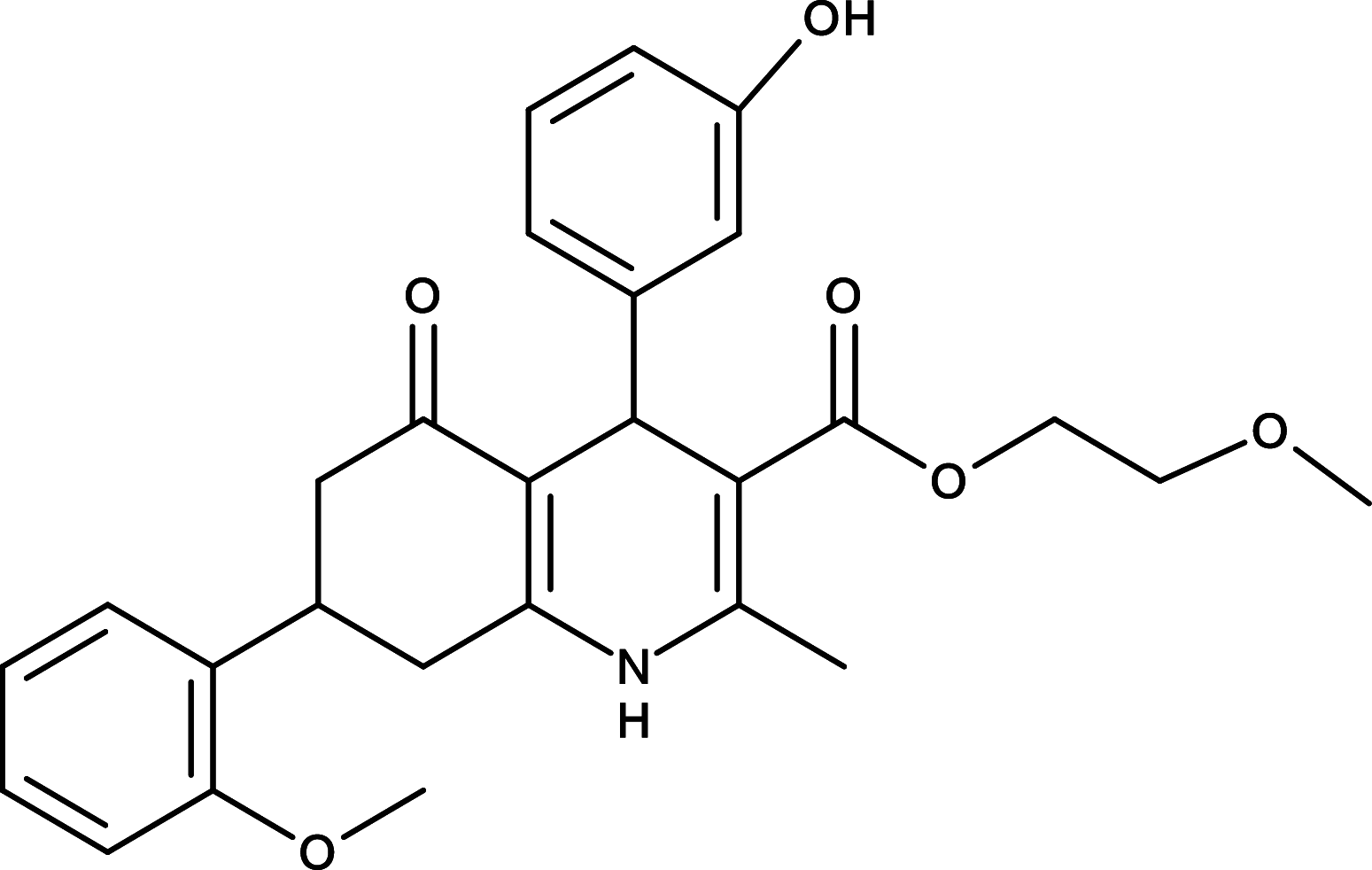
|
HPI-1 | GLI | [97] |
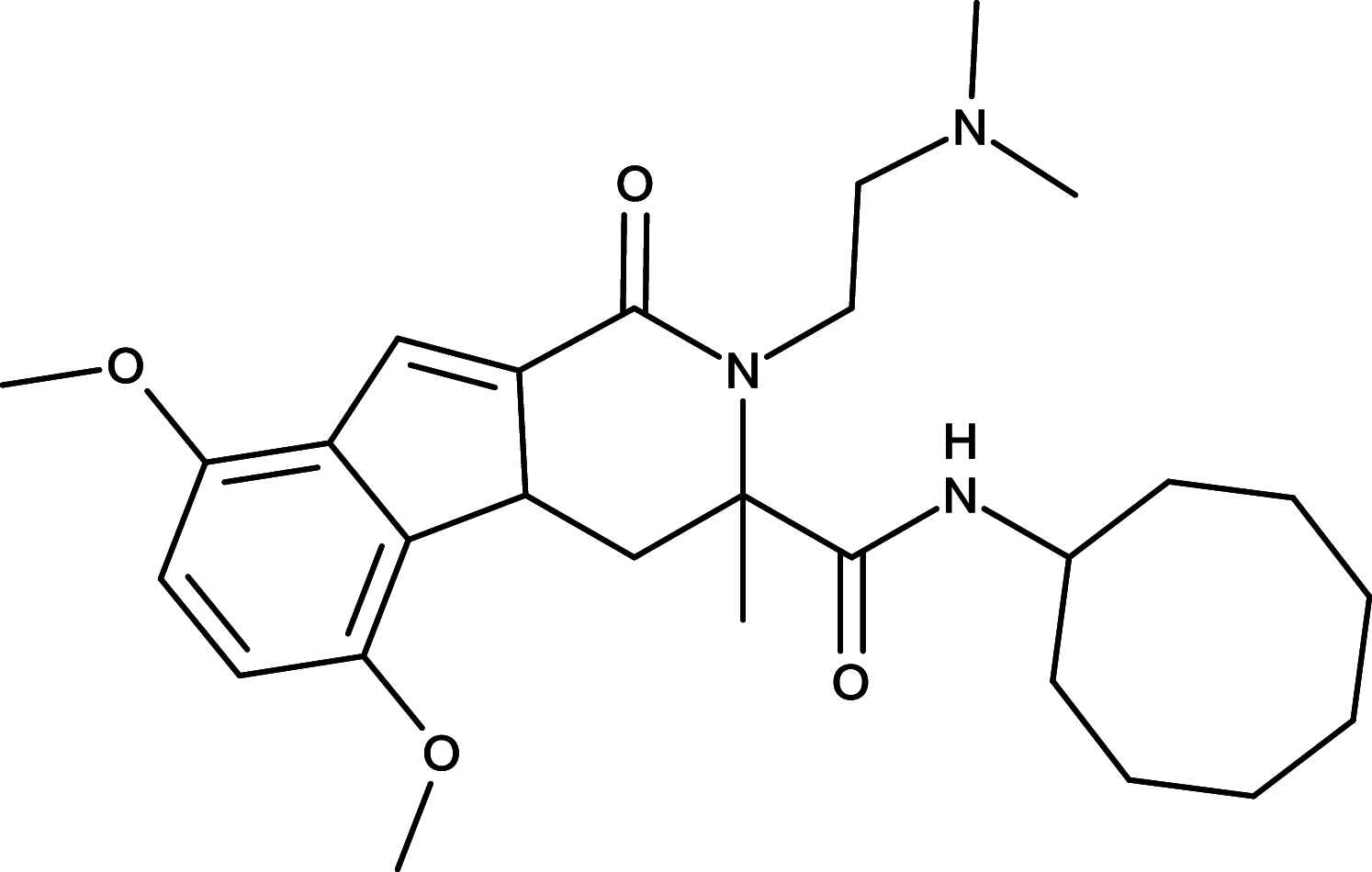
|
HPI-2 | GLI | [97] |
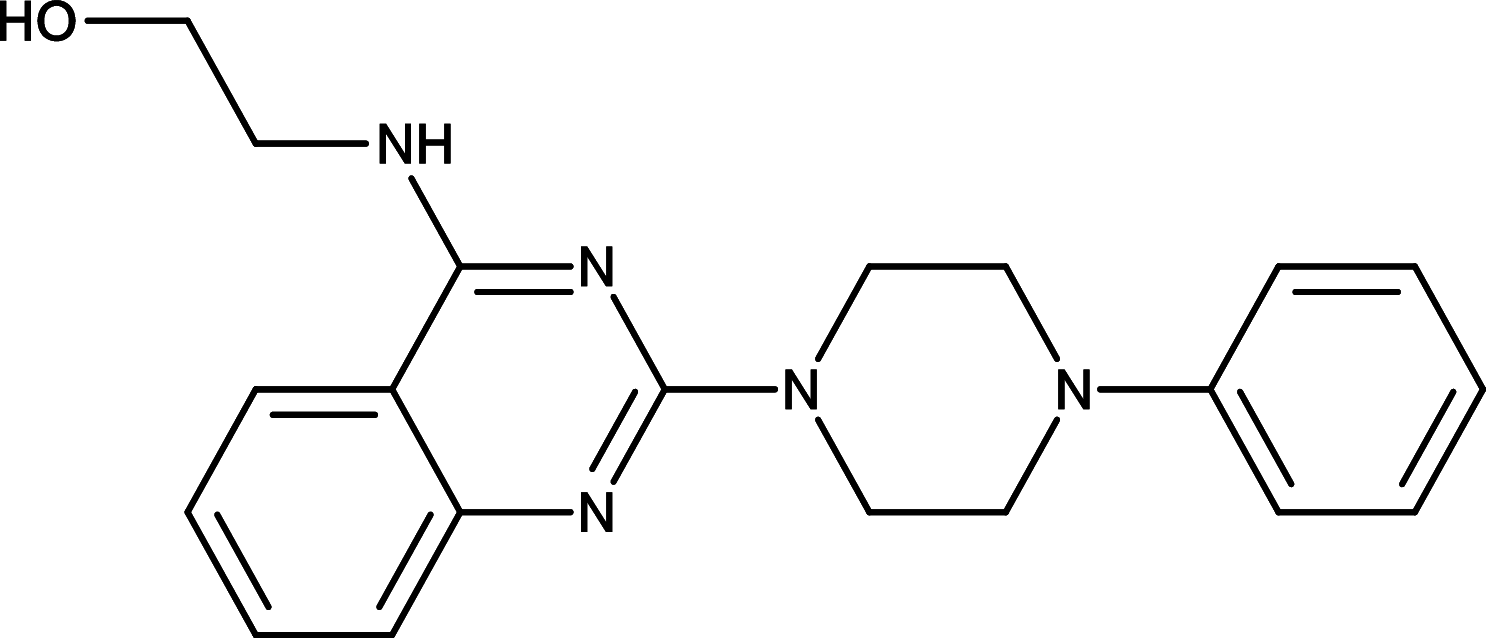
|
HPI-3 | GLI | [97] |

|
HPI-4 | Ciliogenesis | [97] |
|
|
Arsenic trioxide | GLI |
[98] [99] |
Target SMO
SMO has been a primary target in development of the HH signaling inhibitors for decades. To date, numerous SMO inhibitors have been developed and two of them (Vismodegib and Sonidegib) have been approved by FDA for basal cell carcinoma (BCC) treatment (Table 1).
Cyclopamine and its derivatives
Cyclopamine isolated from lily Veratrum Californicum is an alkaloid targeting SMO for HH signaling inhibition [63, 64] (Table 1). Intensive preclinical studies have demonstrated that cyclopamine effectively inhibits growth of tumors, including human glioma, melanoma, colon, pancreatic, prostate cancers, small cell lung cancer, and medulloblastoma [40, 43, 65–67]. In addition, a topical cream containing cyclopamine was shown to regress human basal cell carcinomas [68]. Nevertheless, therapeutic potential of cyclopamine as an HH signaling inhibitor for human cancers was limited by its side effects, low solubility in normal saline, and other physiological solutions as well as instability under acidic conditions (human stomach environment) [69, 70]. To overcome these issues, several cyclopamine derivatives that display more-drug like properties have been developed, including KAAD-cyclopamine and IPI-926 (Saridegib) [64, 71, 72] (Table 1). Particularly, IPI-926 displays improved metabolic stability, pharmacokinetics, and potency over cyclopamine [73]. In vivo studies in animal models further demonstrated that IPI-926 effectively attenuated tumor growth in medulloblastoma, chondrosarcoma, and pancreatic cancer [71, 72, 74]. Subsequently, IPI-926 entered into clinical trials for various cancers, and it was shown well tolerated in clinical trials [75]. However, further Phase 2 trials of IPI-926 were terminated for patients with pancreatic cancer or myelofibrosis for their safety and poor clinical benefit.
Vismodegib (GDC-0449) and Sonidegib (LDE-225)
The SMO inhibitor, Vismodegib, is the first HH pathway inhibitor approved by FDA for treatment of metastatic BCC, or patients with recurrent, locally advanced BCC who are not candidates for surgery or radiation therapy [76, 77] (Table 1). Currently, clinical trials of vismodegib as a monotherapy or in combination with other therapeutic drugs are ongoing for various cancers, including medulloblastoma, metastatic pancreatic cancer, metastatic prostate cancer, intracranial meningioma, advanced head/neck basal cell carcinoma, recurrent glioblastoma, and acute myeloid leukemia [78]. In 2015, Sonidegib became the second SMO inhibitor approved by FDA to treat patients with locally recurrent advanced BCC following surgery or radiation therapy and those who are not candidates for surgery or radiation therapy (Table 1). Both Vismodegib and Sonidegib have displayed positive initial response in patients with BCC. In addition, clinical Phase I/II trials of Vismodegib and Sonidegib to treat solid tumors and hematological malignancies have been conducted or are underway [78, 79]. For instance, clinical Phase I study of Sonidegib demonstrated its acceptable safety profile and antitumor activity in patients with various malignancies, including medulloblastoma, lung cancer BCC and advanced solid tumor [80].
Other SMO inhibitors and clinical challenges of the SMO inhibitors
In addition, a number of other SMO inhibitors have been developed and entered to clinical trials including PF-04449913 (Glasdegib) and LY2940680 (Taladegib) (Table 1). For instance, Munchhof et al. reported an SMO inhibitor PF-04449913 which displays excellent potency and drug properties [81], and PF-04449913 was shown to attenuate the leukemia-initiation potential of acute myeloid leukemia cells in a serial transplantation mouse model [82]. The initial Phase I study of PF-04449913 supported its safety, tolerance, and potential efficacy in acute myeloid leukemia, myelodysplastic syndrome, myelofibrosis, chronic myelomonocytic leukemia, and advanced solid tumors [83–85]. Subsequent phase II trials of PF-04449913 are underway for acute myeloid leukemia, high-risk myelodysplastic syndrome (NCT01546038), myelofibrosis previously treated with ruxolitinib (NCT02226172), and refractory/relapsed myelodysplastic syndrome or chronic myelomonocytic leukemia (NCT01842646). Moreover, LY2940680, another SMO inhibitor which binds to the extracellular end of the transmembrane-helix bundle of SMO for HH signaling inhibition, has been developed [86, 87]. Currently, LY2940680 is being tested in Phase I and Phase II trials for advanced solid tumors and esophageal cancers (NCT02530437).
Although development of SMO inhibitor-based drugs represents a great breakthrough in cancer treatments, they are also facing some formidable challenges. Other than their common clinical side effects, including fatigue, nausea, muscle cramps, and dysgeusia, therapeutic SMO inhibitors are often associated with the critical drug resistant problem [88–90]. For instance, Yauch et al. reported that medulloblastomas had a dramatic initial response to vismodegib, but subsequently acquired drug resistance through a new SMO mutation which can bypass the drug inhibition [88]. Similarly, two additional new SMO mutations that mediate resistance to vismodegib in BCC’s patients have also been detected [89]. The fact that SMO quickly acquires oncogenic mutations for the drug resistance argues long-term benefits of SMO inhibitor-based drugs, supporting the idea that targeting downstream components of the HH signaling cascade may overcome drug resistance associated with the SMO inhibitors in cancer treatment [91].
Target GLI
GLI is a critical transcription factor positioned in very downstream of the HH signaling, and targeting GLI therefore represents an effective therapeutics to overcome the acquired drug resistance for the SMO inhibitors (Fig. 1). Consequently, efforts have been made to develop reagents targeting GLI for the HH pathway inhibition. For instance, Lauth and colleagues have identified two small molecule GLI inhibitors, GANT58 and GANT61 in a cell-based screen [92] (Table 1). Both GANT58 and GANT61 block GLI1 expressions, and particularly GANT61 appears to prevent DNA binding to GLI1 or destabilize the GLI1–DNA complex. In an in vivo xenograft studies, GANT61 showed strong attenuation for the growth of prostate cancer and rhabdomyosarcoma [92, 93]. Further studies also demonstrated that GANT61 robustly suppressed proliferation of colon cancer cells, ovarian cancer cells, and canine osteosarcoma cells [94–96]. Moreover, Hyman and colleagues identified a new class of HH pathway inhibitors termed HPI-1, HPI-2, HPI-3, and HPI-4 in a high-throughput screening [97] (Table 1). It has been shown that HPI-1 inhibits HH signaling through a mechanism that is potentiated by GLI phosphorylation, whereas both HPI-2 and HPI-3 block the conversion of full-length GLI 2 proteins into its active form for the HH signaling. Different from the rest of HIPs (HIP-1, HIP-2, and HIP-3), HPI-4 was shown to inhibit the HH signaling by disrupting ciliogenesis, an essential ciliary processes for GLI function in mammalians [97]. Finally, arsenic trioxide (ATO) is another GLI inhibitor which directly binds to GLI1 and GLI2 to inhibit GLI transcriptional activity, thus decreasing expression of endogenous GLI target genes [98, 99] (Table 1). ATO showed inhibitory activity in models of medulloblastoma and Ewing’s sarcoma, osteosarcoma, acute promyelocytic leukemia, rhabdosarcoma, malignant pleural mesothelioma, prostate, and colon cancer cells [98–103]. In addition, ATO has been approved for treatment of acute promyelocytic leukemia [104], and currently, a few of clinical trials of ATO are underway for solid tumors and hematological malignancies.
Target phosphodiesterase-4 (PDE4)
PDE4 is an enzyme that specifically hydrolyzes cyclic AMP (cAMP), and PDE4 inhibitor-based dugs have previously been used to treat non-malignant diseases, such as depression, asthma, and pulmonary hypertension [105–107]. In addition, increasing evidence supports that PDE4 activation is also implicated in breast tumor, brain tumor, lung cancer and colorectal cancer, etc [108–111]. Nevertheless, the important role of PDE4 in the HH signaling was not recognized until very recently. In 2015, two groups demonstrated that PDE4 inhibition down-regulated the HH pathway to suppress the tumor growth [112, 113], suggesting that PDE4 may play a key role in the HH signaling. Subsequently, we elucidated mechanism of PDE4 inhibition for the HH signaling suppression by identification of Eggmanone (EGM), an extraordinarily selective PDE4 inhibitor in fibroblast cells and zebrafish models [114]. We showed that EGM specifically increases cAMP levels, resulting in activation of protein kinase K (PKA) which disrupts a process downstream of GLI ciliary trafficking for HH inhibition (Fig. 1) [114]. To date, despite some PDE4 inhibitor drugs have been approved by FDA, they are typically used for non-cancer treatments. For instance, Roflumilast was approved for treatment of severe chronic obstructive pulmonary disease and Otezla (apremilast) is used for treatments of active psoriatic arthritis and moderate-to-severe plaque psoriasis [115, 116]. Whether those PDE4 inhibitor drugs can benefit cancer patients remain unknown, and further clinical investigations are warranted. However, caution needs to be taken to target PDE4 for cancer treatment as PDE4 has four subtypes (PDE4A, PDE4B, PDE4C, and PDE4D), and understanding the critical roles of PDE4 subtypes in cancer progression is essential to develop selective subtype-specific therapies in cancer treatments.
Conclusion
Abnormal activation of the HH signaling is involved in various types of cancer and CSCs. Therefore, targeting the key components of the HH pathway has become an important therapeutic strategy for cancer treatment. In the past few years, numerous HH signaling inhibitors, particularly by targeting SMO, have been developed, and two SMO inhibitors (Vismodegib and Sonidegib) have been approved by FDA for treatment of advanced or metastatic BCC. Despite their initial efficacies in cancer treatment, those SMO inhibitors are often associated with the drug resistant problem as cancer exposed to those SMO inhibitors can quickly acquire new SMO mutations to circumvent the drug inhibition for the HH signaling. Therefore, it has been proposed that targeting the downstream SMO in the HH signaling cascade may represent a valid anticancer therapeutic strategy. Consequently, a number of small molecules targeting GLI have been developed, and some of them have showed promising outcomes in both preclinical and clinical studies. In addition, the important role of PDE4 in the HH signaling pathway has recently been recognized, and the PDE4 may represent a promising new target to inhibit the HH signaling for cancer treatment.
Acknowledgements
This work was supported by the seed fund of College of Veterinary Medicine at Western University of Health Sciences and Faculty Development Grant from Chinese American Faculty Association of Southern California (CAFA). The authors would like to acknowledge ChemAxon (http://www.chemaxon.com) for providing an academic license to their software.
Compliance with ethical standards
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- 1.Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287(5785):795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 2.Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15(23):3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 3.O’Hara WA et al (2011) Desert hedgehog is a mammal-specific gene expressed during testicular and ovarian development in a marsupial. BMC Dev Biol 11 [DOI] [PMC free article] [PubMed]
- 4.Kumar S, Balczarek KA, Lai ZC. Evolution of the hedgehog gene family. Genetics. 1996;142(3):965–972. doi: 10.1093/genetics/142.3.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pathi S, et al. Comparative biological responses to human Sonic, Indian, and Desert Hedgehog. Mech Dev. 2001;106(1–2):107–117. doi: 10.1016/S0925-4773(01)00427-0. [DOI] [PubMed] [Google Scholar]
- 6.Patten I, Placzek M. The role of Sonic Hedgehog in neural tube patterning. CMLS Cell Mol Life Sci. 2000;57(12):1695–1708. doi: 10.1007/PL00000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perron M, et al. A novel function for Hedgehog signalling in retinal pigment epithelium differentiation. Development. 2003;130(8):1565–1577. doi: 10.1242/dev.00391. [DOI] [PubMed] [Google Scholar]
- 8.Echelard Y, et al. Sonic-Hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of Cns polarity. Cell. 1993;75(7):1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- 9.Vortkamp A, et al. Regulation of rate of cartilage differentiation by Indian Hedgehog and PTH-related protein. Science. 1996;273(5275):613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 10.Szczepny A, Hime GR, Loveland KL. Expression of Hedgehog signalling components in adult mouse testis. Dev Dyn. 2006;235(11):3063–3070. doi: 10.1002/dvdy.20931. [DOI] [PubMed] [Google Scholar]
- 11.Mirsky R, et al. Schwann cell-derived desert Hedgehog signals nerve sheath formation. Ann N Y Acad Sci. 1999;883:196–202. doi: 10.1111/j.1749-6632.1999.tb08582.x. [DOI] [PubMed] [Google Scholar]
- 12.Parmantier E, et al. Schwann cell-derived Desert Hedgehog controls the development of peripheral nerve sheaths. Neuron. 1999;23(4):713–724. doi: 10.1016/S0896-6273(01)80030-1. [DOI] [PubMed] [Google Scholar]
- 13.Varjosalo M, et al. Application of active and kinase-deficient kinome collection for identification of kinases regulating Hedgehog signaling. Cell. 2008;133(3):537–548. doi: 10.1016/j.cell.2008.02.047. [DOI] [PubMed] [Google Scholar]
- 14.Varjosalo M, Taipale J. Hedgehog signaling. J Cell Sci. 2007;120(1):3–6. doi: 10.1242/jcs.03309. [DOI] [PubMed] [Google Scholar]
- 15.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22(18):2454–2472. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 16.Ryan KE, Chiang C. Hedgehog secretion and signal transduction in vertebrates. J Biol Chem. 2012;287(22):17905–17913. doi: 10.1074/jbc.R112.356006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Habib JG, O’Shaughnessy JA. The hedgehog pathway in triple-negative breast cancer. Cancer Med. 2016;5(10):2989–3006. doi: 10.1002/cam4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scales SJ, de Sauvage FJ. Sauvage, mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. 2009;30(6):303–312. doi: 10.1016/j.tips.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 19.Kogerman P, et al. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999;1(5):312–319. doi: 10.1038/13031. [DOI] [PubMed] [Google Scholar]
- 20.Stone DM, et al. Characterization of the human suppressor of fused, a negative regulator of the zinc-finger transcription factor Gli. J Cell Sci. 1999;112(Pt 23):4437–4448. doi: 10.1242/jcs.112.23.4437. [DOI] [PubMed] [Google Scholar]
- 21.Rubin LL, de FJ. Sauvage, Targeting the Hedgehog pathway in cancer. Nat Rev Drug Discov. 2006;5(12):1026–1033. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]
- 22.Merchant AA, Matsui W. Targeting Hedgehog–a cancer stem cell pathway. Clin Cancer Res. 2010;16(12):3130–3140. doi: 10.1158/1078-0432.CCR-09-2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clement V, et al. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17(2):165–172. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bar EE, et al. Cyclopamine-mediated Hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25(10):2524–2533. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dierks C, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell. 2008;14(3):238–249. doi: 10.1016/j.ccr.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 26.Zhao C, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458(7239):776–779. doi: 10.1038/nature07737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Justilien V, et al. The PRKCI and SOX2 oncogenes are Co-amplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell. 2014;25(2):139–151. doi: 10.1016/j.ccr.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kinzler KW, et al. Identification of an amplified, highly expressed gene in a human glioma. Science. 1987;236(4797):70–73. doi: 10.1126/science.3563490. [DOI] [PubMed] [Google Scholar]
- 29.Hahn H, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85(6):841–851. doi: 10.1016/S0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 30.Johnson RL, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272(5268):1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 31.Wolter M, et al. Mutations in the human homologue of the Drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1997;57(13):2581–2585. [PubMed] [Google Scholar]
- 32.Lam CW, et al. Novel mutations in the PATCHED gene in basal cell nevus syndrome. Mol Genet Metab. 2002;76(1):57–61. doi: 10.1016/S1096-7192(02)00021-5. [DOI] [PubMed] [Google Scholar]
- 33.Xie J, et al. Mutations of the PATCHED gene in several types of sporadic extracutaneous tumors. Cancer Res. 1997;57(12):2369–2372. [PubMed] [Google Scholar]
- 34.Xie J, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391(6662):90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 35.Yan T, et al. Patched-one or smoothened gene mutations are infrequent in chondrosarcoma. Clin Orthop Relat Res. 2008;466(9):2184–2189. doi: 10.1007/s11999-008-0332-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor MD, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002;31(3):306–310. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- 37.Brugieres L, et al. Incomplete penetrance of the predisposition to medulloblastoma associated with germ-line SUFU mutations. J Med Genet. 2010;47(2):142–144. doi: 10.1136/jmg.2009.067751. [DOI] [PubMed] [Google Scholar]
- 38.Slade I, et al. Heterogeneity of familial medulloblastoma and contribution of germline PTCH1 and SUFU mutations to sporadic medulloblastoma. Fam Cancer. 2011;10(2):337–342. doi: 10.1007/s10689-010-9411-0. [DOI] [PubMed] [Google Scholar]
- 39.Berman DM, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425(6960):846–851. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 40.Watkins DN, et al. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422(6929):313–317. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 41.Thayer SP, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425(6960):851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sicklick JK, et al. Dysregulation of the Hedgehog pathway in human hepatocarcinogenesis. Carcinogenesis. 2006;27(4):748–757. doi: 10.1093/carcin/bgi292. [DOI] [PubMed] [Google Scholar]
- 43.Karhadkar SS, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature. 2004;431(7009):707–712. doi: 10.1038/nature02962. [DOI] [PubMed] [Google Scholar]
- 44.Kubo M, et al. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004;64(17):6071–6074. doi: 10.1158/0008-5472.CAN-04-0416. [DOI] [PubMed] [Google Scholar]
- 45.Yauch RL, et al. A paracrine requirement for Hedgehog signalling in cancer. Nature. 2008;455(7211):406–410. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
- 46.Tian H, et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci USA. 2009;106(11):4254–4259. doi: 10.1073/pnas.0813203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nolan-Stevaux O, et al. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009;23(1):24–36. doi: 10.1101/gad.1753809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu G, et al. Analysis of gene expression and chemoresistance of CD133(+)cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. doi: 10.1186/1476-4598-5-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delude C. Tumorigenesis: testing ground for cancer stem cells. Nature. 2011;480(7377):S43–S45. doi: 10.1038/480S43a. [DOI] [PubMed] [Google Scholar]
- 50.Chen J, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488(7412):522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peacock CD, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci USA. 2007;104(10):4048–4053. doi: 10.1073/pnas.0611682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ericson J, et al. Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell. 1996;87(4):661–673. doi: 10.1016/S0092-8674(00)81386-0. [DOI] [PubMed] [Google Scholar]
- 53.Bosanac I, et al. The structure of SHH in complex with HHIP reveals a recognition role for the Shh pseudo active site in signaling. Nat Struct Mol Biol. 2009;16(7):691–697. doi: 10.1038/nsmb.1632. [DOI] [PubMed] [Google Scholar]
- 54.Coon V, et al. Molecular therapy targeting Sonic hedgehog and hepatocyte growth factor signaling in a mouse model of medulloblastoma. Mol Cancer Ther. 2010;9(9):2627–2636. doi: 10.1158/1535-7163.MCT-10-0486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang Q, et al. Tumor-stroma interaction in orthotopic primary pancreatic cancer xenografts during hedgehog pathway inhibition. Int J Cancer. 2013;133(1):225–234. doi: 10.1002/ijc.28006. [DOI] [PubMed] [Google Scholar]
- 56.Petrova E, et al. Inhibitors of Hedgehog acyltransferase block Sonic Hedgehog signaling. Nat Chem Biol. 2013;9(4):247–249. doi: 10.1038/nchembio.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buglino JA, Resh MD. Hhat is a palmitoylacyltransferase with specificity for N-palmitoylation of Sonic Hedgehog. J Biol Chem. 2008;283(32):22076–22088. doi: 10.1074/jbc.M803901200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mann RK, Beachy PA. Novel lipid modifications of secreted protein signals. Annu Rev Biochem. 2004;73:891–923. doi: 10.1146/annurev.biochem.73.011303.073933. [DOI] [PubMed] [Google Scholar]
- 59.Goetz JA, et al. A highly conserved amino-terminal region of sonic hedgehog is required for the formation of its freely diffusible multimeric form. J Biol Chem. 2006;281(7):4087–4093. doi: 10.1074/jbc.M511427200. [DOI] [PubMed] [Google Scholar]
- 60.Matevossian A, Resh MD (2015) Hedgehog Acyltransferase as a target in estrogen receptor positive, HER2 amplified, and tamoxifen resistant breast cancer cells. Molecular Cancer 14 [DOI] [PMC free article] [PubMed]
- 61.Petrova E, Matevossian A, Resh MD. Hedgehog acyltransferase as a target in pancreatic ductal adenocarcinoma. Oncogene. 2015;34(2):263–268. doi: 10.1038/onc.2013.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rodgers UR, et al. Characterization of Hedgehog acyltransferase inhibitors identifies a small molecule probe for Hedgehog signaling by cancer cells. ACS Chem Biol. 2016;11(12):3256–3262. doi: 10.1021/acschembio.6b00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cooper MK, et al. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280(5369):1603–1607. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]
- 64.Chen JK, et al. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002;16(21):2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Varnat F, et al. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med. 2009;1(6–7):338–351. doi: 10.1002/emmm.200900039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feldmann G, et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007;67(5):2187–2196. doi: 10.1158/0008-5472.CAN-06-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sanchez P, Altaba ARI. In vivo inhibition of endogenous brain tumors through systemic interference of Hedgehog signaling in mice. Mech Dev. 2005;122(2):223–230. doi: 10.1016/j.mod.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 68.Tabs S, Avci O. Induction of the differentiation and apoptosis of tumor cells in vivo with efficiency and selectivity. Eur J Dermatol. 2004;14(2):96–102. [PubMed] [Google Scholar]
- 69.Mimeault M, et al. Cytotoxic effects induced by docetaxel, gefitinib, and cyclopamine on side population and nonside population cell fractions from human invasive prostate cancer cells. Mol Cancer Ther. 2010;9(3):617–630. doi: 10.1158/1535-7163.MCT-09-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee ST, et al. Cyclopamine: from cyclops lambs to cancer treatment. J Agric Food Chem. 2014;62(30):7355–7362. doi: 10.1021/jf5005622. [DOI] [PubMed] [Google Scholar]
- 71.Tremblay MR, et al. Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926) J Med Chem. 2009;52(14):4400–4418. doi: 10.1021/jm900305z. [DOI] [PubMed] [Google Scholar]
- 72.Campbell VT et al (2011) Direct targeting of the Hedgehog pathway in primary chondrosarcoma xenografts with the Smoothened inhibitor IPI-926. Cancer Res 71
- 73.Goff RD, Thorson JS. Enhancement of cyclopamine via conjugation with nonmetabolic sugars. Org Lett. 2012;14(10):2454–2457. doi: 10.1021/ol300703z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Olive KP, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a Mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jimeno A, et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin Cancer Res. 2013;19(10):2766–2774. doi: 10.1158/1078-0432.CCR-12-3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sekulic A, et al. Efficacy and safety of the Hedgehog pathway inhibitor vismodegib in patients with advanced basal cell carcinoma (Bcc): 12-month erivance bcc study update. Ann Oncol. 2012;23:362–362. [Google Scholar]
- 77.Chang A, et al. Efficacy and safety of vismodegib in advanced basal cell carcinoma. J Invest Dermatol. 2012;132:S93–S93. [Google Scholar]
- 78.Rimkus TK et al (2016) Targeting the Sonic Hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers (Basel) 8(2) [DOI] [PMC free article] [PubMed]
- 79.Danial C, et al. An Investigator-initiated open-label trial of sonidegib in advanced basal cell carcinoma patients resistant to vismodegib. Clin Cancer Res. 2016;22(6):1325–1329. doi: 10.1158/1078-0432.CCR-15-1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ahnert JR et al (2010) A phase I dose-escalation study of LDE225, a smoothened (Smo) antagonist, in patients with advanced solid tumors. J Clin Oncol 28(15)
- 81.Munchhof MJ, et al. Discovery of PF-04449913, a potent and orally bioavailable inhibitor of smoothened. ACS Med Chem Lett. 2012;3(2):106–111. doi: 10.1021/ml2002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fukushima N, et al. Small-molecule Hedgehog inhibitor attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Sci. 2016;107(10):1422–1429. doi: 10.1111/cas.13019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jamieson C, et al. Phase 1 dose-escalation study of PF-04449913, an oral Hedgehog (Hh) inhibitor, in patients with select hematologic malignancies. Blood. 2011;118(21):195–196. [Google Scholar]
- 84.Wagner AJ, et al. A phase I study of PF-04449913, an oral Hedgehog inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21(5):1044–1051. doi: 10.1158/1078-0432.CCR-14-1116. [DOI] [PubMed] [Google Scholar]
- 85.Martinelli G, et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: a phase 1 safety and pharmacokinetics study. Lancet Haematol. 2015;2(8):E339–E346. doi: 10.1016/S2352-3026(15)00096-4. [DOI] [PubMed] [Google Scholar]
- 86.Wang C, et al. Structure of the human smoothened receptor bound to an antitumour agent. Nature. 2013;497(7449):338. doi: 10.1038/nature12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bender MH et al (2011) Identification and characterization of a novel smoothened antagonist for the treatment of cancer with deregulated Hedgehog signaling. Cancer Res 71
- 88.Yauch RL, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326(5952):572–574. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pricl S, et al. Smoothened (SMO) receptor mutations dictate resistance to vismodegib in basal cell carcinoma. Mol Oncol. 2015;9(2):389–397. doi: 10.1016/j.molonc.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Atwood SX, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27(3):342–353. doi: 10.1016/j.ccell.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gonnissen A, Isebaert S, Haustermans K. Targeting the Hedgehog signaling pathway in cancer: beyond Smoothened. Oncotarget. 2015;6(16):13899–13913. doi: 10.18632/oncotarget.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lauth M, et al. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci USA. 2007;104(20):8455–8460. doi: 10.1073/pnas.0609699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Srivastava RK, et al. GLI inhibitor GANT-61 diminishes embryonal and alveolar rhabdomyosarcoma growth by inhibiting Shh/AKT-mTOR axis. Oncotarget. 2014;5(23):12151–12165. doi: 10.18632/oncotarget.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mazumdar T, et al. Hedgehog signaling drives cellular survival in human colon carcinoma cells. Cancer Res. 2011;71(3):1092–1102. doi: 10.1158/0008-5472.CAN-10-2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen Q, et al. Down-regulation of Gli transcription factor leads to the inhibition of migration and invasion of ovarian cancer cells via integrin beta4-mediated FAK signaling. PLoS One. 2014;9(2):e88386. doi: 10.1371/journal.pone.0088386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shahi MH, Holt R, Rebhun RB. Blocking signaling at the level of GLI regulates downstream gene expression and inhibits proliferation of canine osteosarcoma cells. PLoS One. 2014;9(5):e96593. doi: 10.1371/journal.pone.0096593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hyman JM, et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc Natl Acad Sci USA. 2009;106(33):14132–14137. doi: 10.1073/pnas.0907134106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kim J, et al. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci USA. 2010;107(30):13432–13437. doi: 10.1073/pnas.1006822107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Beauchamp EM, et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J Clin Invest. 2011;121(1):148–160. doi: 10.1172/JCI42874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cai X, et al. Synergistic inhibition of colon carcinoma cell growth by Hedgehog-Gli1 inhibitor arsenic trioxide and phosphoinositide 3-kinase inhibitor LY294002. Onco Targets Ther. 2015;8:877–883. doi: 10.2147/OTT.S71034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nakamura S, et al. Arsenic trioxide prevents osteosarcoma growth by inhibition of GLI transcription via DNA damage accumulation. PLoS One. 2013;8(7):e69466. doi: 10.1371/journal.pone.0069466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang D, et al. Arsenic trioxide inhibits the Hedgehog pathway which is aberrantly activated in acute promyelocytic leukemia. Acta Haematol. 2013;130(4):260–267. doi: 10.1159/000351603. [DOI] [PubMed] [Google Scholar]
- 103.You M, et al. Targeting of the Hedgehog signal transduction pathway suppresses survival of malignant pleural mesothelioma cells in vitro. J Thorac Cardiovasc Surg. 2014;147(1):508–516. doi: 10.1016/j.jtcvs.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 104.List A, et al. Opportunities for Trisenox (arsenic trioxide) in the treatment of myelodysplastic syndromes. Leukemia. 2003;17(8):1499–1507. doi: 10.1038/sj.leu.2403021. [DOI] [PubMed] [Google Scholar]
- 105.Scott AIF, et al. Inpatient major depression—is rolipram as effective as amitriptyline. Eur J Clin Pharmacol. 1991;40(2):127–129. doi: 10.1007/BF00280065. [DOI] [PubMed] [Google Scholar]
- 106.Torphy TJ, Undem BJ. Phosphodiesterase inhibitors: new opportunities for the treatment of asthma. Thorax. 1991;46(7):512–523. doi: 10.1136/thx.46.7.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Izikki M, et al. Effects of roflumilast, a phosphodiesterase-4 inhibitor, on hypoxia- and monocrotaline-induced pulmonary hypertension in rats. J Pharmacol Exp Ther. 2009;330(1):54–62. doi: 10.1124/jpet.108.148742. [DOI] [PubMed] [Google Scholar]
- 108.Drees M, Zimmermann R, Eisenbrand G. 3′,5′-Cyclic nucleotide phosphodiesterase in tumor-cells as potential target for tumor-growth inhibition. Cancer Res. 1993;53(13):3058–3061. [PubMed] [Google Scholar]
- 109.Sengupta R, et al. Treating brain tumors with PDE4 inhibitors. Trends Pharmacol Sci. 2011;32(6):337–344. doi: 10.1016/j.tips.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pullamsetti SS, et al. Phosphodiesterase-4 promotes proliferation and angiogenesis of lung cancer by crosstalk with HIF. Oncogene. 2013;32(9):1121–1134. doi: 10.1038/onc.2012.136. [DOI] [PubMed] [Google Scholar]
- 111.Tsunoda T, et al. Inhibition of phosphodiesterase-4 (PDE4) activity triggers luminal apoptosis and AKT dephosphorylation in a 3-D colonic-crypt model. Mol Cancer. 2012;11:46. doi: 10.1186/1476-4598-11-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Powers GL, et al. Phosphodiesterase 4D inhibitors limit prostate cancer growth potential. Mol Cancer Res. 2015;13(1):149–160. doi: 10.1158/1541-7786.MCR-14-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ge X et al (2015) Phosphodiesterase 4D acts downstream of Neuropilin to control Hedgehog signal transduction and the growth of medulloblastoma. Elife 4, e07068 [DOI] [PMC free article] [PubMed]
- 114.Williams CH, et al. An in vivo chemical genetic screen identifies phosphodiesterase 4 as a pharmacological target for hedgehog signaling inhibition. Cell Rep. 2015;11(1):43–50. doi: 10.1016/j.celrep.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Giembycz MA, Field SK. Roflumilast: first phosphodiesterase 4 inhibitor approved for treatment of COPD. Drug Des Devel Ther. 2010;4:147–158. doi: 10.2147/dddt.s7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zerilli T, Ocheretyaner E. Apremilast (Otezla): a new oral treatment for adults with psoriasis and psoriatic arthritis. Pharm Ther. 2015;40(8):495–500. [PMC free article] [PubMed] [Google Scholar]