

Abstract
There is ample evidence that many proteins or regions of proteins lack a well-defined folded structure under native-like conditions. These are called intrinsically disordered proteins (IDPs) or intrinsically disordered regions (IDRs). Whether this intrinsic disorder is also their main structural characteristic in living cells has been a matter of intense debate. The structural analysis of IDPs became an important challenge also because of their involvement in a plethora of human diseases, which made IDPs attractive targets for therapeutic development. Therefore, biophysical approaches are increasingly being employed to probe the structural and dynamical state of proteins, not only in isolation in a test tube, but also in a complex biological environment and even within intact cells. Here, we survey direct and indirect evidence that structural disorder is in fact the physiological state of many proteins in the proteome. The paradigmatic case of α-synuclein is used to illustrate the controversial nature of this topic.
Keywords: Intrinsic disorder, In cell NMR, Intracellular protein delivery, Conformational ensemble
Introduction
The field of structural disorder in proteins rests on diverse experimental evidence provided by a broad range of biophysical and structural biology techniques, such as NMR, X-ray crystallography and many others [1]. Observations of the lack of a well-defined structure under native conditions provide the basis of including proteins and protein regions into our collection of disordered proteins [2]. Experimentally proven IDPs/IDRs form the basis of bioinformatics predictors, which are then used to predict structural disorder at the level of all UniProt proteins, providing the basis of our view of widespread structural disorder in the entire protein world [3]. It should not be neglected, however, that the majority of experimental evidence for structural disorder comes from in vitro studies of IDPs/IDRs. Very often, it is not taken into account that the intracellular milieu is highly crowded and contains all the potential physiological binding partners of the protein. Whereas this has given rise to much speculation with regard to the true physiological state of IDPs/IDRs, we reckon there is a broad range of observations and considerations that (in)directly prove that structural disorder is the intrinsic and functionally relevant state of these proteins. In this review, we outline and elaborate all these different lines of evidence to guide the reader towards the conclusion that structural disorder is the physiological and functional state for most IDPs/IDRs.
Indirect experimental evidence for structural disorder
There are various lines of indirect evidence, as well as functional considerations that argue convincingly for the flexible nature of proteins in vivo. Considerations on the structure–function relationship of proteins often points to the indispensability—and thus inherent nature—of structural disorder for protein functions, in a sense that it cannot be fulfilled by an ordered, rigid structure. These types of functions originate from the structurally heterogeneous and dynamic nature of IDPs, which may be rationalized in terms of the higher energy (in fact, higher entropy) conformational state of IDPs [4]. These functions are thus termed entropic chain functions [5, 6], and they are thought to emanate from the highly dynamic structural state best described by a structural ensemble [7]. To give a few examples, there is conclusive evidence that the PEVK region of titin is an entropic spring in muscle [8], which ensures, like a piece of rubber, elasticity to the tissue. The projection domain of MAP2 [9], a microtubule-associated protein, functions as an entropic bristle that provides spacing in the cytoskeleton. Of similar structural logic is the operation of the FG repeat regions of nucleoporins (NUPs), which regulate transport through the nuclear pore complex (NPC) by combining extremely rapid and specific recognition of transport proteins and entropic exclusion of large molecules [10, 11]. In all, function in these cases stems directly from the disordered state of the protein, i.e., their folded state in vivo is rather counterintuitive. Actually, forcing these proteins to fold by changing conditions may actually impair their function, as demonstrated in the case of NUPs [12]. In addition, protease sensitivity assays further demonstrated that most FG NUPs are intrinsically disordered in situ, within the NPCs of purified yeast nuclei [10]. These experiments are close to reporting the structural behavior of NUPs under true in vivo conditions, confirming the conclusion based on functional considerations.
Following this line of thought, the concept of in vivo structural dynamics can be directly concluded for extracellular IDPs/IDRs, which do not experience a similar crowded environment as compared to intracellular proteins. The most notable examples are: (i) caseins, which occur in milk and function as stabilizers of calcium phosphate and nutrients [13], (ii) salivary proline-rich glycoproteins, which function in the saliva and digestive tract, where they bind and neutralize polyphenolic plant compounds, tannins [14], and (iii) bacterial fibronectin-binding proteins, which are transmembrane proteins that protrude from the bacterial surface and mediate adherence to the extracellular matrix [5, 15].
One may also argue for the disordered physiological state if IDPs/IDRs by analyzing their geometry in the bound state, when their structure in complex with their partner is available in the Protein Data Bank (PDB) [16] (Fig. 1). For example, calpain is a protease that is regulated by an IDP inhibitor, calpastatin, which assumes an extended structure upon binding to the enzyme. The inhibitor wraps around the calpain surface with the concomitant formation of three α-helical segments, while two stretches of 21 and 9 residues remain disordered (even in the bound state) (Fig. 1). This is similar to the case of many other IDPs/IDRs, because disordered proteins often function by molecular recognition, when they transiently or permanently bind to a structured partner. In most of these cases, they assume an extended, open, structure in the bound state [17], i.e., it does not make much sense to assume their compact, folded, state prior to binding that would have to unfold to assume the structure seen in the complex. Closely related to this argument is the observation that IDPs/IDRs are frequently involved in the assembly of large complexes [18], which, due to steric clashes, cannot be disassembled from rigid components. Often, the disordered protein is intimately interdigitated with the partner, thus its inherent flexibility is essential for reaching the final structural state.
Fig. 1.

Crystal structure of calpastatin bound to its folded partner, calpain (PDB entry 3BOW). Calpain (surface representation) is composed of a large (80 kDa, yellow) and a small (30 kDa, orange) subunit. Calpastatin (red cartoon representation) binds in an extended conformation
In addition, a unique structural–functional feature of some IDPs/IDRs is that they can bind to distinct partners in a process termed binding promiscuity [19] or one-to-many signaling [20], and/or have been suggested to have different functions with different partners, termed moonlighting [21]. In several of these cases, when the structure in the partner-bound state is known, it can be observed that the IDP/IDR acquires different structures. For example, the C-terminal domain of RNAP II in complex with either RNA guanylyl transferase Cgt1 and peptidyl-proline isomerase Pin1 [22], or the HIF-1α interaction domain binding to either the TAZ1 domain of Creb-Binding-Protein [23] or the asparagine hydroxylase FIH [24]. Such structural malleability is much more compatible with the disordered state than a stable folded structural state, prior to binding.
Protein evolution also bears witness to physiological disorder
A solid argument for the lack of a compact, folded structure under physiological conditions derives from the high evolutionary rate of many IDPs/IDRs (Fig. 2) [25, 26]. Evolutionary changes in sequence are limited by constraints on residues involved in functional/structural interactions, which result in evolutionary conservation of folded domains enabling their classification into families in the Pfam database [27]. The homology of sequences points to their common evolutionary origin, which is the basis of transferring structural/functional information in genome annotation efforts [28]. Due to the lack of a folded structure, there are much less constraints on IDPs/IDRs, for example, the pairwise genetic distances within disordered and ordered regions of 26 protein families was found to differ significantly, disordered regions evolving significantly faster in 19 families, and more slowly in two families only [25]. Related studies on caseins have also pointed to their “anomalous” evolutionary behavior, because their translated regions have much higher mutation rates than their non-translated regions [29]. In all, the observation that IDPs/IDRs are subject to much less structural constraints in their native state than their structured counterparts, can be best interpreted in terms of the lack a well-defined structure in vivo.
Fig. 2.

Fast evolution of disordered sequences. Partial multiple sequence alignment (MSA) of RNase E orthologs from four different bacterial species. MSA was created by T-Coffee [150] for regions 300–720 of RNAse E sequences downloaded from the KEGG database (http://www.kegg.jp). The scheme shows part of the catalytic domain (first three lines), which is highly conserved and aligns well (indicated by pink color), and the intrinsically disordered C-terminal regulatory region, which shows very little recognizable homology (yellow and green)
Adapted from Ref. [151]
A more detailed comparative genomic analysis of the evolutionary variability of IDP/IDR sequences has actually suggested that structural disorder partitions into three distinct categories [26]: (i) regions where disorder is conserved but amino acid sequence evolves fast (termed flexible disorder), (ii) regions in which both structural disorder and amino acid sequences are conserved (termed constrained disorder), and (iii) regions in which neither structural disorder nor amino acid sequence is conserved (non-conserved disorder). In this regard, flexible disorder has the characteristics commonly attributed to disorder, and represents a strong case for the physiological disorder of IDPs/IDRs.
On the other hand, one might argue that constrained disorder, which has different functional attributes and is involved in RNA binding and protein chaperone functions, might be better interpreted in terms of a folded, structurally and functionally more constrained physiological state. This is not (always) the case, however, because genuine IDRs may have all the attributes of protein domains. Traditionally, protein domains have three operational definitions, being regarded as (i) autonomous structural/folding units, (ii) evolutionary modules, and (iii) functional elements of proteins [30]. Conserved sequence patterns longer than 20–30 residues can also be recognized in IDRs and these regions have been often termed domains as they represent evolutionary, structural and functional units of proteins. These intrinsically disordered domains (IDDs) thus conform to all three definitions outlined above. To give a few examples, the kinase-inhibitory domain (KID) is a sequence element of about 60 residues that can be recognized in cyclin-dependent kinase (CDK) inhibitors p21, p27 and p57. The short, 40-residue long Wiskott–Aldrich syndrome protein (WASP)-homology domain 2 (WH2) appears in several actin-binding proteins, such as thymosin-β4, Cordon-bleu, ciboulot and WASP, whereas the catenin-binding domain of about 25 residues can be found in the cadherin cytoplasmic tail and T cell factors LEF-1, Tcf3, and Tcf4 [30]. In all, bioinformatics predictions suggest that about 12% of Pfam domains have more than 50% predicted disorder and more than 4% of them are fully disordered, i.e., the conservation of many domains may not necessarily indicate that they assume a folded state under physiological conditions.
Further insight on the physiological structural state of IDPs/IDRs can be obtained by comparing the evolutionary dynamics of their sequences at the nucleotide and amino acid levels. Although sequence similarity is usually studied at the protein (amino acid) level, disregarding mutations that leave the amino acid sequence unchanged, the comparison of mutations that change the amino acid encoded (missense or nonsynonymous) to those that do not change it (sense or synonymous) is very informative with regards to the structure and function of a disordered region. More specifically, regions that are under high functional (and structural) constraint, are subject to purifying selection and thus their ratio of nonsynonymous (K A) to synonymous (K S) mutations is much lower, typically on the order of 0.1–0.2, than that of unconstrained regions [31]. For example, the Gln-rich transactivator domain of the sex-determining transcription factor, SRY, evolves much faster (K A/K S = 0.4–0.8) than its globular DNA-binding domain (K A/K S = 0.1–0.2) [32, 33]. This particular observation also argues that seemingly unconstrained evolution may be compatible with functional constraints: although the trans-activator domains of transcription factors are functionally indispensable, they often undergo rapid evolution, and their binding interactions are encoded in a distributed and dynamic manner, putting little recognizable constraint on their evolution. This creates a particularly challenging situation with aligning sequences of IDPs, because their homology often cannot be recognized based on sequence similarity, thus their genes may be missed in genomic searches. A particularly relevant example is securin, the inhibitor of separase, the protease responsible for triggering anaphase in mitosis. Securin is an IDP [34], and has been identified through functional analogy in yeast S. cerevisiae (Pds1p9), S. pombe (Cut210), D. melanogaster (pimples11), and H. sapiens (PTTG1). These proteins, by all means, are evolutionarily homologous, yet they are extremely variable in length and show practically no recognizable similarity in sequence, except for short segments in the N-terminus [34].
The unique evolutionary dynamics of IDPs/IDRs also have bearing on the predictability of structural disorder from sequence. As suggested, their sequence signatures often vary through evolution to a level that it cannot be recognized by homology searches, yet they have specific features embedded in their sequence that enables the highly dependable prediction of disorder by bioinformatics tools. As compared to globular proteins, IDPs/IDRs are enriched in disorder-promoting (mostly charged and polar) amino acids and are depleted in order-promoting (mostly hydrophobic) amino acids [35], which often manifests itself in a high net charge and low mean hydrophobicity [36]. Overall, their unique structural state can be reliably predicted from their amino acid sequence by dozens of predictors based on different principles, such as amino acid composition, sequence signatures, physical and chemical features, secondary structure propensity, and pairwise amino acid interaction energies [37], which underscores that IDPs/IDRs are physically different from ordered proteins, i.e., the lack of ability to fold is their intrinsic property.
Initiation of regulated protein degradation
Regulated degradation of proteins, which is primarily carried out in the cell by the ubiquitin–proteasome system (UPS) [38] is also linked with structural disorder. The UPS is a hierarchic system of interconnected enzymatic components, the action of which culminates in the buildup of a poly-ubiquitin chain on particular lysine residue(s) of the substrate. This chain is then recognized by the proteasome, a multi-subunit protease that degrades it to small peptides released for further processing [39]. The specific signals recognized by UPS are termed “degrons”, which are protein elements that confer metabolic instability on proteins [40]. Recently, it has been suggested that degrons are composed of several independent but interconnected elements [41, 42]. Successful degradation requires a recognition motif (primary degron), the Lys residues carrying the poly-ubiquitin chain (secondary degron), and a nearby intrinsically disordered initiation site (tertiary degron) for successful proteasomal engagement [43]. This arrangement has given rise to the suggestion of a “tripartite” degradation signal on proteins [44], the combinatorial complexity of which is required for highly coordinated and regulated protein degradation in the proteome.
Of immediate relevance to our point of the in vivo significance of structural disorder is that these findings posit that too stable proteins cannot be degraded by the proteasome, and the intrinsically disordered initiation site has to be present near the poly-ubiquitin chain for successful proteasomal engagement. Whereas this concept relies on in vitro observations, it would be unlikely that the in vivo situation was different. If IDPs/IDRs were folded in vivo, this would be inhibitory to their effective degradation, and actually many folded proteins might have disordered minor states serving regulated degradation in the cell.
Structural and molecular characterization of intrinsic disorder: to funnel or not to funnel
Traditionally, a comprehensive understanding of protein function is unimaginable without detailed structural knowledge. Although not trivial, this is also the case with disordered proteins. In their case it holds particularly true that solving and analyzing a three-dimensional structure per se is not sufficient, as such structures typically provide only a static description, while proteins are highly dynamic. This can be best appreciated via the landscape theory of protein structures. Comprehensive biophysical and structural studies of protein folding and stability leads to their representation in terms of energy landscapes [45]. Well-folded proteins typically display a single global energy minimum that represents their native state (Fig. 3a, d), while for IDPs/IDRs the conformational heterogeneity (in their unbound state) is reflected in a rather shallow energy landscape with multiple isoenergetic minima (Fig. 3b, e). In practical terms, this means that structurally disordered proteins in their native, unbound yet functional state cannot be adequately described by a single population-averaged three-dimensional structure. Rather, they sample continuously a multitude of different conformational states (the various local minima in the landscape) on a very short timescale (reflected by the fact that the local minima are separated by low energy barriers (Fig. 3b). Nevertheless, many IDPs/IDRs adopt more highly ordered conformations upon interactions with other cellular components in a process of binding-induced folding [46]. The structures of these bound states can be solved (e.g., calpain–calpastatin complex (Fig. 1) or the p27 KID–KIX complex (Fig. 3f). In the context of energy landscapes this can be represented by a discrete low energy minimum (Fig. 3c). Yet, some IDPs or IDRs remain noticeably disordered even when bound to their partner(s), resulting in the formation of heterogeneous, “fuzzy” complexes [47] (Fig. 3f). This residual disorder (also termed ‘fuzziness’) can also be recognized when IDRs are part of large multi-domain proteins [48]. This intrinsic disorder and concomitant conformational heterogeneity does not permit the unequivocal characterization of the structural behavior with the traditional methods. Rather, structural studies of intrinsic disorder aim usually to obtain experimental constraints on the ensemble of conformational states that is sampled by the IDP of interest (Fig. 3). These “fuzzy” functional states of IDPs/IDRs add a strong case for the existence of structural disorder within the functioning protein.
Fig. 3.
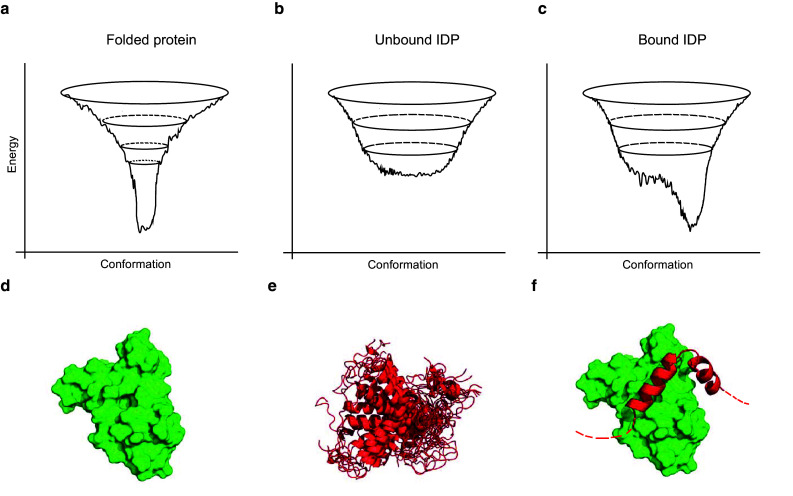
Conformational energy landscapes and structural representations of ordered and disordered proteins and their complexes. a The traditional view holds that the conformational energy of a protein can be described by a funnel-like surface (folding funnel), in which the global minimum corresponds to the native state. The funnel ensures an effective down-hill folding, even though kinetically trapped intermediates or misfolded states can be present in the landscape. b IDPs typically have less distinct energy landscapes, characterized by several nearly isoenergetic minima that represent the lowest-energy conformational states, which are separated from each other by low-energy barriers. Hence the IDP can easily adopt multiple different states upon changing the environment or external stimuli (such as altering quinary structure or post-translational modifications). c IDPs can reach a well-defined global energy minimum when they undergo binding-induced folding. The presence of a partner molecule can influence the energy landscape and stabilize the conformation that the IDP acquires upon binding to the partner surface. d The NMR structural ensemble (17 NMR conformers) of the well-folded KIX domain of CREB Binding protein (1KDX.pdb) represents a single, globular, compact equilibrium conformational state, depicted in the surface representation. e The conformational ensemble (each state is depicted in the cartoon representation) of the unbound p27 kinase inhibitory domain (p27KID) (PED entry PED2AAA) is described by molecular dynamics (MD) computations and solution NMR spectroscopy. The ensemble is represented here by 17 conformational states and depicts that p27-KID is highly disordered. f The p27KID–KIX complex (1KDX.pdb) reveals that p27KID undergoes a binding-induced folding event: it showcases a coil–helix transition upon binding to KIX, forming two α-helices. The dashed line represents the residue stretches that remain disordered in the bound state, an example to the phenomenon that is called fuzziness
Direct experimental evidence of IDPs/IDRs: to be challenged or not to be challenged
IDPs are associated with many human diseases when they are mutated or over- or underexpressed, or experience a changed functionality, an altered degradation, an impaired trafficking or loss of binding partners [49, 50]. Therefore, in analogy to the D3-concept that links protein intrinsic disorder to degenerative diseases [49], we introduced the F3-concept for flexible proteins since their “function follows flexibility” [51]. Some of the best-studied IDPs, such as α-synuclein, tau protein, and p53, play a prominent role in human diseases like Parkinson’s disease, Alzheimer’s dementia and cancer [52–54] (Table 1). As IDPs became attractive targets for therapeutic intervention, the structural description of their molecular behavior became an important challenge.
Table 1.
A selection of landmark studies that allowed the field of in cell studies of IDPs to develop into maturity
| Protein | Disease model | Biophysical technique | Cell type | Delivery method | References |
|---|---|---|---|---|---|
| Human superoxide dismutase 1 | Amyotrophic lateral sclerosis | NMR | Escherichia coli | Overexpression | [147] |
| Human superoxide dismutase 1 | Amyotrophic lateral sclerosis | NMR | Human embryonic kidney (HEK293T) cells | Overexpression | [80, 134] |
| Human superoxide dismutase 1 | Amyotrophic lateral sclerosis | NMR | HeLa cells | HIV-1 TAT cell penetrating peptide | [148] |
| α-Synuclein | Parkinson’s disease | NMR | HeLa cells | Streptolysin O toxin | [82] |
| α-Synuclein | Parkinson’s disease | NMR, EPR | HeLa cells | Electroporation | [54] |
| Thymosin-β4 | Human carcinomas | NMR | Human embryonic kidney (HEK293F) cells | Streptolysin O toxin | [83] |
| Tau protein | Alzheimer’s dementia | NMR | Xenopus laevis oocytes | Microinjection | [53] |
| Prothymosin-α | Diabetes | FRET | Xenopus laevis oocytes | Microinjection | [149] |
| FlgM | Salmonella typhimurium | NMR | Escherichia coli | Overexpression | [129, 130] |
| α-Synuclein | Parkinson’s disease | NMR | Escherichia coli | Overexpression | [108, 130] |
Various techniques, cell types and IDPs provided the experimental proof that also inside the cell the intrinsic protein disorder can be directly visualized. Different researchers have conducted these experiments often in the context of human diseases
The determination of a unique high-resolution structure is simply not possible for an isolated IDP, and a detailed structural and dynamic characterization of IDPs cannot typically be provided by a single biophysical technique. Therefore, accurate descriptions of IDPs and IDRs rely on a multiparametric approach that combines various biophysical methods that can provide information on the overall compactness of IDPs, their conformational stability, shape, residual secondary structure, transient long-range contacts, regions of restricted or enhanced mobility, etc. The current state-of-the-art characterization of IDPs happens by combining experimental data with computations in search of the functional repertoire of IDPs [7].
Direct experimental evidence for the intrinsic disorder of proteins is often provided by optical techniques such as far ultraviolet circular dichroism (CD) spectroscopy and fluorescence spectroscopy, but also Fourier transform infrared spectroscopy (FTIR), liquid state nuclear magnetic resonance (NMR), electron paramagnetic resonance (EPR, but also the more powerful double electron–electron resonance (DEER), Forster resonance energy transfer (FRET), small angle X-ray scattering (SAXS), dynamic light scattering (DLS), and even limited proteolysis [1, 55–58]. These primary techniques used to characterize IDPs, sometimes even at the single molecule level, are often combined and their results can be integrated with computational tools, thereby yielding conformational ensembles that are currently best describing the highly dynamic structural state of IDPs [59]. There are many excellent reviews available in literature that cover the use of in vitro biophysical characterization for IDPs and we refer the interested reader to these as a starting point for further exploration [1, 60, 61]. It is sufficient to state that these experimental techniques combined provide comprehensive and undisputable evidence for the structural disorder of many IDPs and IDRs under in vitro conditions. The current release of the database dedicated to these data, DisProt, holds more than 1200 IDP/IDR cases with solid experimental evidence [2].
Intracellular intrinsic disorder: fact or fiction?
Based on the foregoing considerations, arguments and observations, we can state that it is well-documented that IDPs behave fundamentally different from ordered proteins. “Non-believers” might still claim that IDPs are artefactual due to the controlled and artificial conditions under which they are studied. Therefore, the structural biology and cell biology communities launched an integrative approach to tackle protein structural behavior in the complex and challenging environment of the cell.
Even Anfinsen realized that the native conformation of proteins is conditional, by stating during his Nobel prize acceptance speech that: “the native conformation is determined by the totality of interatomic interactions and hence by the amino acid sequence, in a given environment” [62]. Several external factors, such as macromolecular crowding, co-factors, chemical modifications, self-assembly, and binding partners may change the free energy landscape of a protein drastically [63]. Although lots of information can be extracted from in vitro investigations, reality has many additional layers of complexity added to it. The cell is no longer viewed as a membrane-enclosed entity that contains molecules that diffuse and tumble around freely. Instead, it is a crowded environment (up to 400 mg/mL of macromolecules) that contains organelles (which can be membrane-less, e.g. formed via phase separation) and this compartmentalization is essential to expedite the essential processes of life [64, 65]. Since the complex energy landscape of IDPs/IDRs is particularly sensitive to such environment, there are two expedient strategies to address the intracellular effect on IDPs/IDRs: (i) mimicking crowding and confinement in vitro, or (ii) devising effective experimental strategies for studying IDPs in live cells.
Cellular mimicking: crowding and confinement
Amongst others, Allen Minton has pioneered the concept of macromolecular crowding and confinement to describe the interior of a cell [64, 66–68]. Despite often being used as synonyms, crowding is dynamic while confinement is static: the cell interior can be defined as being confining because of the relatively small cell volumes (10−9–10−4 µL) and the presence of subcellular compartments whose volumes can be limited to 10−14 µL [66]. To mimic this intracellular environment, which is extremely crowded with an almost complete lack of unoccupied space and a limited amount of free water, the crowding is often artificially modeled by concentrated solutions of various polymers. A systematic structural study of selected IDPs with such distinct crowding agents revealed that notwithstanding some local residual structure and/or a somewhat more compact ensemble of conformations, intrinsic structural disorder remains their most prominent feature under such experimental conditions [69]. Based on single molecule FRET studies, Ben Schuler and colleagues observed that polyethylene glycol-induced crowding lead to compaction of human prothymosin-α and the N-terminal domain of HIV-1 integrase [70, 71]. However, under similar in vitro crowding conditions other IDPs, like p27-KID and c-Fos transactivation domain, do not undergo any compaction [72]. While mounting evidence shows that with different types of crowders it is possible to fine-tune the effects on proteins, IDPs could be grouped into two classes, foldable and non-foldable, based on their response to the crowded environment [73, 74]. Theillet et al. present an excellent overview of studies that address the physicochemical implications of molecular crowding on IDP behavior [74].
In spite of the advances in our understanding, it is important to realize that the currently used artificial crowding agents do not adequately replicate all natural effects of the intracellular environment. For example, non-specific binding events (weak attractive forces) play a subtle role in the cell since natural macromolecules can induce both stabilization and destabilization when used as crowding agents [66, 74, 75]. Also the excluded volume and steric repulsion consequences of a simplified crowding model are insufficient to explain all the phenomenological observations with IDPs in crowded media. Hence, the transient and very weak interactions between proteins that are collectively known as “quinary structure” can add an additional layer of complexity in the attempts to mimic the cell interior inside a test tube [76]. The recognition of the quinary structure is another step in the right direction.
In cell NMR
Since it was first proposed in 2001, the importance of the direct measurement of multidimensional NMR spectra of proteins inside living cells, i.e. in cell NMR, can hardly be overestimated [77]. The term “in cell NMR” is used mainly to probe polypeptides inside living cells, while the term “in vivo NMR” has been coined specifically to the studies of small molecules inside cells and even living organisms [77]. Yet, the first indications that disordered polypeptides exist in intact cells came from in vivo proton NMR experiments [78].
The critical step for in cell NMR data acquisition comprises successful sample preparation by introduction of an isotope-labeled protein (labeled with 13C and 15N) into living cells. Practically all cell types can be used, i.e. prokaryotic and eukaryotic cells, as long as they remain viable and in suspension during the course of data collection. Isotope-labeled proteins can be introduced into cells either by endogenously over-expressing the target protein using an inducible plasmid or by “controlled” delivery of exogenously produced proteins from the outside (Fig. 4). Therefore, NMR is the technique par excellence to study IDPs inside cells, because if the protein-of-interest is selectively isotope-labelled, the ‘background’ environment is virtually invisible.
Fig. 4.
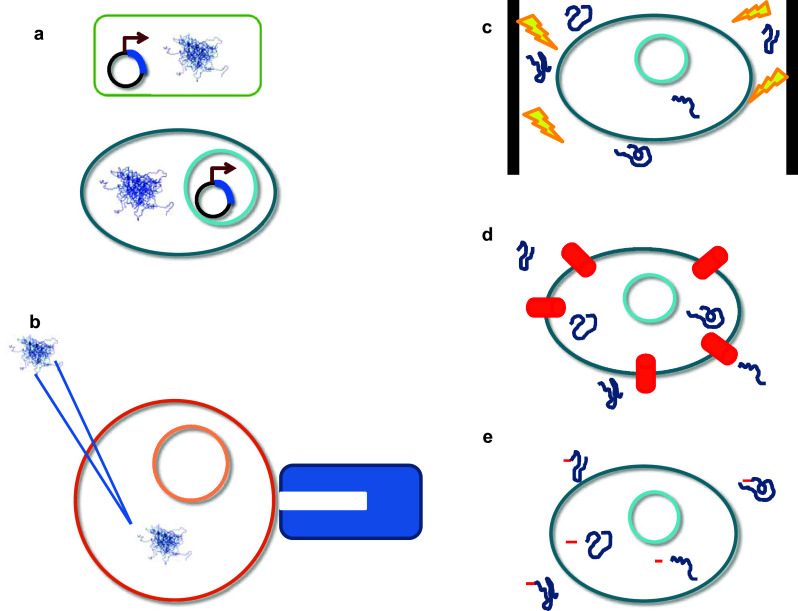
Overview of intracellular protein delivery methods for in cell structural analysis (mainly using NMR). a The IDP of interest can be overexpressed with recombinant DNA technology in prokaryotic or eukaryotic cells by growing the cells in unlabeled medium, followed by induction of expression in isotopically labelled medium. The cell suspension can be readily used for direct in cell NMR data collection. b Alternatively, the protein of interest is overexpressed using recombinant production systems, purified to homogeneity and introduced into (eukaryotic) cells via either microinjection [e.g. a Xenopus laevis oocyte is kept in place with a holding pipet (represented in blue) while the purified IDP is injected intracellularly]. c Electroporation causes transient permeability of the cell membrane to allow the protein of interest to enter. d Cell penetrating peptides (CPP, represented in red) that are covalently linked to the proteins allow entry of the protein inside the cells, and CPPs can be removed subsequently. e IDPs can be delivered through cell-permeabilizing toxins that form transient pores, which enable the protein to diffuse into the cell according to the concentration gradient. As an IDP example in a and b, the ensemble of monomeric α-synuclein is represented by ten different conformational states that were generated using molecular dynamics simulations restrained with inter-atomic distances derived from paramagnetic relaxation enhancement NMR (PED entry 9AAC)
The “simplest” in cell NMR approach is overexpression in living Escherichia coli cells, whereby the cell suspension is subjected to NMR studies (Fig. 4a). The cells are grown in isotope-enriched media to ensure that the overexpressed proteins are labeled with the isotopes and hence are NMR visible. As such, the NMR spectra contain atomic-level information about the structure and dynamics of the proteins (Table 1 and references therein). The drawback can be elevated background levels of isotope labeled endogenous protein material (compromising the signal-to-noise ratio) but also overexpression artefacts [79]. While induced over-expression of the protein of interest avoids any transfer or extracellular manipulation steps, often selecting specific promotors allows control of the intracellular protein concentration. Such overexpression strategy has also been used for in cell NMR in eukaryotic cells, including insect cells and human cell lines [80, 81]. For example, the human superoxide dismutase 1 (SOD1) was directly expressed in HEK293T cells and the in cell NMR study described the complete maturation and dynamic behavior of the protein.
Besides overexpression and/or the use of bacterial systems, technological progress has allowed us to handle eukaryotic cells such as HeLa and HEK293 cells for in cell NMR studies (Table 1). A number of techniques have been developed to achieve isotope labeling of the specific target protein and its subsequent intracellular delivery.
Removable tagging with a cell-penetrating peptide (CPP, e.g. a tag derived from the Tat protein of HIV-1) has been used to study different proteins in HeLa cells (Fig. 4). As explicitly demonstrated in the example of α-synuclein, it could be delivered to HeLa cells by linking it to the peptide tag via an oxidative, disulfide-coupling reaction [82]. Likewise, Bekei and colleagues describe the protocol to transfer α-synuclein into HeLa cells using the reversible pore forming properties of streptolysin O (Fig. 4). In contrast to CPP-mediated IDP uptake, toxins offer the advantage that cellular protein transduction does not rely on active biological processes like endocytosis, but on simple passive diffusion. Similarly, Ogino et al. introduced the isotope-labeled actin-sequestering IDP thymosin β4 in human embryonic kidney cells [83]. Also electroporation has been employed to successfully introduce α-synuclein in mammalian cells for in cell NMR [54]. This strategy allows the introduction of the proteins-of-interest in a large quantity of cells simultaneously, but might drastically affect their survival.
High throughput is still somewhat limited when microinjection is employed as a delivery method. Microinjection has the inherent limitation that it is ideally only suited to manipulate large cells such as Xenopus laevis oocytes. It offers the advantage of a more reliable protein concentration control. Transfer of proteins expressed in bacterial systems into X. laevis oocytes is frequently utilized for post-translational modification studies [53, 84]. Since E. coli does not handle most post-translational modifications (PTMs), a recombinantly isotope-labeled protein can be delivered into the eukaryotic cellular environment and studied in time-derived series to monitor the post-translational modifications.
Irrespective of the delivery method of isotope-labeled proteins into mammalian cells, the quality of the resulting in cell NMR samples needs to be assessed routinely by evaluating the overall cell viability, protein delivery efficiency, intracellular protein concentration, localization, and stability [85]. Of particular importance are quality control experiments that assess protein leakage from manipulated cells during the in cell NMR experiment [86, 87]. The study by Barnes and Pielak linked protein expression, protein leakage and in cell NMR in E. coli and revealed that the interference of protein leakage becomes likely when the protein being studied exceeds 20% of the total cellular protein content [88, 89].
The sensitivity of NMR spectroscopy has also limitations, and therefore, at least a minimum protein concentration (typically in the 10−5–10−3 molar range) is required to obtain good quality data with an acceptable signal-to-noise ratio. Such concentrations for a given protein do not always correspond to the normal physiology, and therefore, only proteins that are sufficiently abundant inside cells are the most eligible for such in cell NMR studies. So far, in cell NMR studies have confirmed the structural disorder of all studied IDPs and IDRs in live cells (Table 1).
Will in cellula crystallography or cryo-ET reveal protein disorder inside the cell?
In several protein crystal structures, intrinsic disorder can be recognized by the absence of electron density or by the presence of high temperature factors for the backbone atoms. Typically, crystallographic approaches begin with a pure, homogeneous sample, with as little conformational flexibility as possible. Yet, recent technological advances have allowed for investigating protein crystals in cellula by mounting frozen live cells directly in the X-ray beam [90]. With the use of a microfocus synchrotron X-ray beam, the structure of a viral protein could be successfully determined from microcrystals within cells, removing the preparatory step of sample isolation and maintaining a favourable biological environment [90]. It should not be missed, though, that the cells were treated with cryoprotectant and were flash-frozen at 100 K, which hardly can be considered to be physiologically relevant experimental conditions (that are needed to avoid radiation damage). This raises the question whether in similar in cellula crystallization experiments, it would be possible to identify intrinsic disorder upon solving the in cell crystal structure, but also showcases the absolute power of in cell NMR approaches: it can not only provide ‘atomic level views’ of IDPs but also allows conditional variations [e.g. apply heat stress (thermal shock for few minutes or prolonged time) to the cells in the NMR tube] and final testing of the cell integrity and viability upon finishing the experiments.
In contrast to crystallography, whereby the crystallization process typically selects a protein in a single conformation, cryo-electron microscopy (cryo-EM) is particularly well suited for obtaining structural and dynamical information for systems that exhibit multiple conformational or compositional states. Proteins in cryo-EM samples are free to move around until the moment of flash-freezing. Because cryo-EM is a single-particle technique, such conformational transitions can be captured and studied, and ultimately lead to deeper biological insights about protein function and mechanism [91]. For example, Yi and coworkers reported that the cryoEM study the p300 in different environments (alone or in complex with antibodies or coactivators) reflects its intrinsically disordered nature and conformational variability [92]. As many macromolecular complexes cannot be purified, while knowing their structure, dynamics and location is crucial for understanding their cellular function, cellular ultrastructure imaging using cryo-electron tomography (cryo-ET) provides an important link between light microscopy and in vitro structure determination methods. In a typical cryo-ET study, a biological sample (from a cell up to an entire organism) is flash frozen (the sample is preserved in a hydrated, close-to-native state under cryogenic conditions), thinned to an appropriate thickness, and then multiple images are captured using an electron microscope as the sample is tilted along an axis to finally reconstruct a three-dimensional picture, or tomogram. The NPC that is a classic example for the entropic chain function was the subject of such a paradigmatic cryo-ET study, which emphasizes the large heterogeneity between NPCs that were extracted from different cells, while NPCs within the same cell are more homogenous among themselves [93]. Cryo-ET may facilitate in situ structural biology on a proteomic scale and holds promise for revealing the molecular organization and for characterizing IDRs regions in unperturbed cellular environments [93, 94].
To be cleaved or not to be cleaved inside the cell?
Until recently, limited or controlled proteolysis has typically been applied to purified proteins for the identification of solvent-accessible and high mobility regions. Proteolytic modification, coupled with electrophoresis, has long been recognized as an effective method and low resolution tool to detect regions with disorder [57]. Recent developments enable now the large-scale analyses of protein conformational changes directly in their biological context [95]. Picotti and coworkers coupled limited proteolysis with a proteomics workflow to probe the structural and dynamical features of more than 1000 yeast proteins simultaneously upon a change of nutrients.
This proteome-wide application of limited proteolysis is an exciting development that can be used in both targeted and discovery experiments to investigate both subtle and pronounced structural changes in a biologically complex yet physiologically relevant background.
Upcoming biophysical techniques for in cell characterization: fast relaxation imaging and dynamic nuclear polarization NMR
Historically, fluorescence microscopy has been used to observe the localization of labeled proteins inside the cell. Since functional protein dynamics can span time scales from microseconds to hours, the fast end of this time scale is beyond conventional fluorescence imaging approaches. However, fast relaxation imaging (FReI) combines fluorescence microscopy and temperature jumps to probe biomolecular dynamics [96]. FReI allows cells to be imaged for minutes or hours, but also reveals processes in the cell that range from microseconds to seconds. By inducing fast processes though a laser T-jump, one can monitor how fast processes in the cell (e.g., protein folding or protein–protein binding) evolve in function of time upon changes in the cell due to the cell cycle, etc. One can also compare dynamics from cell to cell or among different subcellular compartments, even at the optical resolution limit (250 nm for conventional fluorescence imaging).
Another exciting development is dynamic nuclear polarization (DNP) that can dramatically enhance the sensitivity of NMR in a complex biological environment [97]. By overcoming the limits in instrumental sensitivity, such a DNP NMR strategy enables the structural analysis of IDPs at endogenous levels inside the cell. The paradigmatic experiment with yeast Sup53 confirms that the cellular environment alters the structural state of an IDP because of multiple direct interactions with cellular components [97].
Hence, both FReI and DNP NMR have a high potential for monitoring transient binding and conformational dynamics of IDPs/IDRs in live cells.
Phase transition: a new functional paradigm
In vitro and intracellular phase transitions supported by IDPs give a novel dimension to our arguments on the physiological relevance of the disordered state of proteins. Compartmentalization of cellular material achieves physical separation of different processes and components from one another. Physical compartmentalization of the cell is ensured by membrane-bound vesicles termed organelles, such as the nucleus, endoplasmic reticulum, endosomes or the Golgi apparatus, which are involved in organizing a great variety of key biological processes.
Recently, a stream of publications has suggested that organelle-like bodies can be formed without the support of membranes [98–100]. The formation of such membrane-less organelles may be termed liquid–liquid demixing (LLDM), liquid–liquid phase separation (LLPS), or sol–gel transition, which do not necessarily cover exactly the same physical process [100]. Membrane-less organelles usually, but not without exception, form by the combination of protein and RNA, and are generally referred to as ribonucleoprotein (RNP) bodies or granules. Long-known RNP bodies in the cytoplasm include stress granules and processing bodies, and in the nucleus, the nucleolus, Cajal bodies, and PML bodies. The importance of membrane-less subcellular structures is supported by their ubiquitous presence in cells and their (patho)physiological relevance [101]. Physically, these intracellular compartments are multicomponent, viscous liquid-like structures that form via spontaneous phase transitions driven by supersaturation of its components. The formation of RNA granules is usually responsive to changes in the concentration of RNA, salt, and of other proteins. The most frequently indicated protein components of granules are fused in sarcoma (FUS), heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1), TDP 43, and neurofilament proteins.
There is substantial evidence that the major and specific component proteins within these compartments are both necessary and sufficient for making the phase transition in vitro as well as in vivo [102]. These relevant proteins tend to have two major characteristics: (i) they contain multiple interaction domains/motifs often embedded in regions of low-complexity sequences (LCS), and (ii) large segments of them are intrinsically disordered. LCSs of phase-separating proteins are characterized by repeats and a low overall diversity of amino acids within the sequence [103]. The sequence features of IDR LCSs enable conformational heterogeneity and multivalency that are critical in driving phase transitions. It appears that the IDRs involved in the phase transitions are not only disordered in the isolated monomeric state, they are also disordered in the droplet phase, probably due to highly heterogeneous, multivalent, transient and very dynamic interactions with their partner proteins and RNA [104].
Most significantly, there is increasing evidence that phase transitions observed in vitro are in direct link with the formation of the liquid droplets in cells. For example, the disordered tails of Ddx4, a primary constituent of nuage or germ granules, form phase-separated organelles both in live cells and in vitro, and the observed growth of the droplets upon overexpression of the protein in cells can be fit to models of LLPS [105]. In the case of the protein FUS involved in amyotrophic lateral sclerosis (ALS), it was found that droplets appear in cells when the constituent protein pass a threshold concentration, and in an in vitro ‘‘aging’’ experiment its liquid droplets convert to an aggregated state, accelerated by disease-related mutations [106]. In addition, nucleoli were shown to grow by fusion events and through Ostwald ripening, in a manner expected for a liquidly demixed phase [107]. These examples demonstrate the parallels of the in vitro and in vivo observed phase transitions, underscoring the residual disorder in the droplet phase in cells, providing a strong indication that structural disorder of these particular class of proteins is relevant in vivo.
Antithesis: the α-synuclein paradox
As demonstrated in the previous sections, the concept of protein disorder is supported by a great variety of observations. That structural disorder is not an isolation artefact but the real physical state of (many) IDPs/IDRs, has been heavily debated nonetheless. To illustrate this, we describe the fascinating α-synuclein paradox, i.e., a variety of contradicting findings—and attempts to bring them to a common ground—on the structural state of this protein, over the past 10–15 years. In short, α-synuclein was initially described as an IDP [82, 108] but contrasting results have challenged this notion by describing this protein as a helical tetramer under physiological conditions [109–111].
α-Synuclein is localized within the presynaptic termini and nuclei of neuronal cells [112]. In 1993, it was discovered in amyloid deposits in the brain of patients suffering from Alzheimer’s disease and Parkinson’s disease [113]. This discovery was a breakthrough and many researchers addressed the physiological and structural characterization of α-synuclein.
Its first structural characterization came from CD, DLS, SAXS and FTIR experiments [114, 115]. CD analysis showed 2% α-helicity and 70% random coil structure, while DLS yielded a Stokes radius greater than the value expected for a globular protein, indicating an apparent molecular weight (M w) of 57 kDa, instead of 14.5 kDa. This result correlated with a SAXS gyration radius (R g) value of 41 Å, instead of the theoretical 15.1 Å. In agreement with these data, FTIR showed a peak at 1650 cm−1, which also support a high level of structural disorder. Finally, this high flexibility was confirmed in 2001 by NMR [116] as the 2D HSQC showed a narrow proton dispersion centered at about 8 ppm. Davidson et al. reported in 1998 that α-synuclein binds to small unilamellar vesicles (SUV) containing acidic phospholipids. They showed by CD that lipid binding increases α-helicity of the protein from 3 to 80% [117].
Despite all this evidence, Bartels et al. [109] showed that “native” purification (purification under mild conditions) of α-synuclein lead to a stable helix-rich tetramer. In fact, all the previous studies have used non-native purification protocols (heat precipitation or detergent-based purification). They found by native gel electrophoresis of samples from M17D, HEK, HeLa and COS cells expressing α-synuclein and also from mouse frontal cortex and human red blood cells, a band at 45–55 kDa, reacting on Western blot (WB) with anti-α-synuclein antibody, much higher than the expected Mw of monomeric α-synuclein.
These observations were correlated by scanning transmission electron microscopy (STEM) [109]. STEM could generate 1000 red blood cell (RBC)-derived α-synuclein particles under native conditions. The obtained distribution peak gave a value of 55 kDa, similarly to that observed by analytical ultracentrifugation. All these reported values are compatible with a tetrameric state. Surprisingly, mass spectrometry (MS) showed N-terminal acetylation on the tetramer but not on the recombinantly expressed α synuclein [109], and it was raised that this chemical modification may be the reason for experimental inconsistencies between different labs.
Concerning the secondary structure content, CD experiments showed an α-helix-rich spectrum that was not affected by the addition of small unilamellar vesicles (SUVs) of lipids. This result was in strong contradiction with previous experiments, where α-synuclein was found to shift from random coil fold to an α-helix-rich structure upon addition of lipids [117]. The authors suggested that lipids could stabilize the tetrameric structure although removal of lipids did not affect the CD signal. Interestingly, the apparent K D of lipid–tetramer interaction was two orders of magnitude smaller than that of the monomer [109, 118]. Another research group showed that upon removal of the glutathione S-transferase tag from α-synuclein, an α-helix-rich tetramer forms under native conditions: CD showed 65% helix and boiling of the sample led to the non-reversible formation of a random coil CD spectrum [110].
These results in the field of α-synuclein set the cat among the pigeons and seven laboratories joined forces to nail down the true nature of α-synuclein structure [119]. In a comprehensive study this consortium used diverse biological sources, purifications protocols and experimental techniques, such as NMR, native and non-native electrophoresis, CD, DLS, enzyme-linked immunosorbent assay, cross-linking, WB and gel filtration. This substantial body of data collectively gave unanimous support for the disordered monomeric state of α-synuclein, although it was not excluded by the authors that the tetramer might exist under physiological conditions.
Using the chemical shift assignment of the tetramer [110], Binolfi and colleagues [120] reconstructed an artificial 2D HSQC spectrum that was compared with the 2D HSQC spectrum of the monomeric form of α-synuclein that was purified under denaturing conditions and placed in the same buffer that was used for the original tetramer data acquisition. Surprisingly, the two spectra were identical, but two differences were observed for Tyr39 and Leu113. To go further in the analysis, the 2D HSQC spectrum of α-synuclein was determined inside E. coli after overexpression. Here again, the spectrum only showed cross-peaks corresponding to the monomeric IDP.
To overcome problems of overexpression and to investigate the in vivo oligomeric state of α-synuclein, Dettmer et al. [121] used a human erithroleukemia (HEL) cell line, which has a naturally high abundance in α-synuclein. Using disuccinimidyl glutarate as an amine-reactive cross-linker on intact cells, they could detect a predominant form at 60 kDa and two minor forms at 80 and 100 kDa. By applying the same approach, no tetrameric state could be detected in the cell lysates. They hypothesized that the inability to cross-link the tetrameric state of α-synuclein could be due to a decreased molecular crowding effect. In accord, at significantly higher total protein concentrations, the band at 60 kDa could again be detected. These results suggest that molecular crowding and/or other molecules have an impact on the state of oligomerization of α-synuclein.
As suggested, N-terminal acetylation of α-synuclein may play a role in the tetramerisation of the protein [109], whereas α-synuclein is not acetylated in E. coli. It has also been described that α-synuclein, which was isolated from deposits in post-mortem brains from patients suffering from dementia with Lewy bodies and Parkinson’s disease, was N-terminally acetylated [122]. Therefore, many laboratories started using the NatB acetyltransferase bacterial co-expression system to produce recombinant N-terminally acetylated protein [123]. Despite acetylation, three different labs showed a disordered state of α-synuclein [119, 124, 125]. Within the disordered state, N-terminal acetylation increases α-helicity for the first 12 residues, with helical tendency decreasing with distance from the acetylation site.
In accord, NMR with or without NatB showed a line broadening of the first ten residues, which suggests that this region is involved in protein–protein interaction(s), chemical change and/or structural rearrangements [125]. The spectra are clearly in agreement with a monomeric state of the protein (Fig. 5).
Fig. 5.

Nuclear magnetic resonance spectra of α-synuclein recorded in vitro and in vivo. a Proton–nitrogen correlation (HSQC) spectra of lysates from cells transfected with α-synuclein alone (black), α-synuclein and NatB (acetyltransferase) together (red), and purified recombinant α-synuclein (blue). Peaks present in spectra of lysates from cells transfected with NatB alone are indicated by boxes and do not originate from or report on α-synuclein. The resonances that change position upon co-transfection with NatB are indicated by arrows pointing from their position in the unmodified protein spectrum toward their position in the modified protein spectrum. Resonances for residues Met-1 and Asp-2, which are not typically observed for the unacetylated protein, are indicated. b Proton–nitrogen HSQC spectra for intact cells transfected with α-synuclein alone (black), α-synuclein and NatB together (red), and NatB alone (green)
Reprinted from Ref. [119]. Copyright 2012 American Society for Biochemistry and Molecular Biology
Recently, Iljina et al. [111] showed that arachidonic acid, which is very abundant in neuronal cells, induced α-helix rich oligomers. These oligomers could be formed at physiologically-relevant concentrations and pathological mutants of α-synuclein gave less oligomers than wild type. These oligomers showed a very close resemblance to the native α-helical tetramers previously described [109, 121]. They suggested that these oligomeric species are in equilibrium with the monomeric form, in analogy to what was proposed for with the native oligomers [126, 127]. This is the indication that the story has not yet come to a conclusion.
Should intrinsic disorder inside cells remain controversial and contested?
Another example that highlights the controversy that sometimes surrounds intrinsic disorder inside the cell is based on the work on the Salmonella typhimurium FlgM protein. This is a 97-residues-long transcription factor inhibitor that is intrinsically disordered as evidenced by in vitro NMR in dilute solution [128]. Yet, its C-terminus can form a transient helix, which can be induced by molecular crowders and apparently also in living cells [129]. By comparing the HSQC spectra of FlgM in dilute buffer solution and in living E. coli cells Dedmon and coworkers claimed that FlgM is actually structured inside E. coli based on the disappearance of the C-terminal cross-peaks. These findings were recently corrected when the Pielak group published the results of hydrogen–deuterium exchange (HDX) study conducted in E. coli with NMR [130]. Since their HDX study also comprised α-synuclein, the authors concluded that true intrinsic disorder can persist inside the crowded cellular interior and that weak interactions between proteins and macromolecules in cells do not necessarily affect the intrinsic rates of exchange.
The NMR characterization of the tubulin-associated unit (Tau) proved to be less controversial and in support for the thesis that intrinsic disorder can exist as such inside eukaryotic cells. Isolated Tau was one of the first recognized IDPs [131]. Despite its pathological involvement in Alzheimer’s disease and high resolution NMR studies for more than a decade of experiments, its exact functioning remains debated. Yet, the assignment of isolated Tau NMR spectra confirmed its intrinsically disordered nature [132, 133]. Recombinantly produced 15N-labeled Tau was also injected into X. laevis oocytes to a concentration of 5 µM by Lippens and coworkers [53]. Since many cross-peaks disappeared in the in cell HSQC spectra as compared to the in vitro HSQC spectra, this observation was attributed to binding events to tubulin and other cellular components. Novel posttranslational modifications (i.e. phosphorylation) could also be identified, although post-in cell NMR-homogenization of the oocytes improved the NMR data quality. Importantly, it was found that the in cell NMR spectra resemble the in vitro NMR data, which is characteristic for natively unfolded protein [53].
Besides the in cell characterization of α-synuclein, the in cell NMR field was also developed thanks to the work on superoxide dismutase 1 (SOD1) (Table 1). The work of Luchinat, Banci and coworkers exploited in cell NMR to characterize the protein folding and maturation of SOD1 mutants that are associated to familial cases of amyotrophic lateral sclerosis [134]. Admittedly, while the physiologically relevant state of SOD1 is a well-folded conformation, they observed the occurrence of an unstructured apo SOD1 conformation inside human cells thanks to an elegant experimental approach. This disordered state is likely the precursor for potentially toxic species [134]. Hence, also in this SOD1 case in cell NMR showcases its power to link intrinsic disorder to protein behavior in the intracellular context.
Dark proteome: “disordered is it?”
The difficulty to classify α-synuclein in the (dis)ordered protein universe paved the way to the recent introduction of the dark proteome. The term dark proteome refers to proteins whose structural and functional features are not well understood. Structural biologists have expected that a high degree of disorder of certain proteins could explain this dark side of the proteome [135] but computational analysis showed that most dark proteins have a low level of disorder, very similar to the light side [136].
SwissProt [137] contains about 546,000 meticulously curated sequences, whereas Aquaria [138] provides pre-calculated sequence-to-structure alignments that assist the search for matching structures. Perdigao et al. [136] used these alignments to map the dark proteome. Their definition is that any amino acid that aligns in Aquaria (with a PDB structure), will be categorized as a light residue, whereas an amino acid which does not, will qualify as a dark residue. They noted that with such a definition the dark proteome will be underestimated because Aquaria includes very remote homologies and uses even low-quality NMR and EM structures. Such a stringent definition of the dark proteome ensures that the mapping of Perdigao et al. does not contain any known structure. The proportion of dark residues was significantly smaller for archaea and bacteria (13–14%) as compared to eukaryotes and viruses (44–54%).
As mentioned before, it has been expected that the dark proteome could be explained by the high proportion of structural disorder in the proteome. To this end, structural disorder was calculated for each protein as the percentage of residues which are predicted to be disordered by IUPred [139] and plotted against the darkness score (the percentage of dark residues in a protein). Darkness was greater than disorder for almost all the eukaryotic proteins, implying that most of the dark residues are in fact not disordered. By the distribution of structural disorder, most of the dark proteome is very similar to the light one, and cannot be explained by structural disorder. Structural disorder is related to, but not synonymous with compositional bias (low complexity) of amino acid sequences [140]. Therefore, it was also raised that the explanation of the dark proteome might be amino acid compositional bias [141]. By comparing the distribution of compositionally biased residues to that of dark residues, however, it was again observed that darkness is higher than the bias for almost all the proteins, i.e., most of the dark proteome had very low compositional bias. In yet another effort for explaining the presence of darkness in the proteome, it was also related to transmembrane (TM) domains, which are very challenging to characterize structurally. A large fraction of TM residues were not dark and most dark proteins had no TM residues, i.e., TM also does not explain the darkness of the proteome.
In all, most of the dark proteome (45–70%) could not be explained by any of the previous structural factors, and they are described as “unknown unknowns”, because they are probably structured but have not yet been structurally characterized, for reasons not fully understood [136]. These dark proteome studies highlight that structural biologists await the daunting task to discover new conformations if one focuses on these unknown unknowns. It remains to be seen whether such an approach would lead to a breakthrough in our understanding of intrinsic disorder inside cells.
Future challenges and outlook
Structural disorder in proteins has been recognized for almost two decades now, during which it has gone a long way from an awkward and marginal idea to a mainstream concept in structural–molecular biology. It is not at all unwarranted to suggest that it has lived up to its original claim of transforming the structure–function paradigm of proteins. As indicated throughout this paper, even the very issue of the existence of structural disorder in live cells is often contested, many issues are still open and they will keep researchers busy for several years to come.
Our global structural depiction of IDPs [142] might miss functionally relevant local structural detail, and description of minor states may only be obtained from ensemble representations obtained by a combination of methods, such as NMR, SAXS, FRET and molecular dynamics simulations [143, 144]. How this level of descriptive structural detail can be extended to cellular conditions is not trivial to answer at the moment. Our most adequate approach is in cell NMR [145]. This technique, however, is rather insensitive and represents a special challenge to apply at physiologically relevant concentrations [146]. The novel sensitivity-enhancing technique of DNP NMR enables structural studies in biologically complex environments, and may bring the breakthrough in in cell studies of IDPs [97].
Although NMR spectroscopy is a powerful tool for structural and dynamics studies of IDPs in vitro and in cellula, obtaining a complete picture of conformational ensembles (and the effects of chemical modification (PTMs or proteolysis) or changing chemical environment) of IDPs requires the complementation with other biophysical and cell biology techniques. Therefore, we need continued efforts to bridge the gap between the in vitro structural biology approaches and in vivo cell biology. Within the structural biology field, the emergence of hybrid structure determination methods, which use a variety of biophysical, biochemical, and modelling techniques to determine the shapes of biologically relevant molecules, opens promising opportunities but also constitute major challenges for the management and representation of structural data [7]. Liquid state NMR, FRET, fluorescence correlation spectroscopy and protein delivery techniques are by far the most notable disruptive technologies that are driving this field by circumventing experimental obstacles.
Additional challenges that will need to be addressed from an experimental point of view are variables like cell-type specificity, tissue-specificity, subcellular localization (in situ and in organello studies…), proteostasis, the specificities during the cell cycle (i.e. different phases in the cell cycle). This will ultimately lead to tackle successfully the endeavor to generate a complete picture of IDPs/IDRs, their interaction network and quinary interactions and adaptability to ever changing conditions within the cell. Such a detailed description will require expertise from different angles in an integrated and multidisciplinary approach. Whereas there is a long way to go, we may conclude that the synergy of structural biology, cellular biology and cellular biophysics has already led to the development of highly sensitive techniques that converge toward confirming that intrinsic disorder does exist inside the cell and represents a unique and functionally indispensable structural state of proteins.
Acknowledgements
This work was supported by the Odysseus Grant G.0029.12 from Research Foundation Flanders (FWO). KP is the recipient of a FWO long-term postdoctoral fellowship. The authors thank Jesper Oemig, Katrien Willegems and Alexander Shkumatov for useful discussions.
Footnotes
Kris Pauwels and Pierre Lebrun contributed equally.
References
- 1.Eliezer D. Biophysical characterization of intrinsically disordered proteins. Curr Opin Struct Biol. 2009;19:23–30. doi: 10.1016/j.sbi.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Piovesan D, Tabaro F, Micetic I, Necci M, Quaglia F, Oldfield C, Aspromonte MC, Davey NE, Davidovic R, Dosztanyi Z, Elofsson A, Gasparini A, Hatos A, Kajava AV, Kalmar L, Leonardi E, Lazar T, Macedo-Ribeiro S, Macossay Castillo MM, Meszaros A, Minervini G, Murvai N, Pujols J, Roche DB, Salladini E, Schad E, Schramm A, Szabo B, Tantos A, Tonello F, Tsirigos KD, Veljkovic N, Ventura S, Vranken W, Warholm P, Uversky VN, Dunker AK, Longhi S, Tompa P, Tosatto SCE. DisProt 7.0: A major update of the database of disordered proteins. Nucleic Acids Res. 2017;45:D1123–D1124. doi: 10.1093/nar/gkw1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Potenza E, Di Domenico T, Walsh I, Tosatto SC. MobiDB 2.0: An improved database of intrinsically disordered and mobile proteins. Nucleic Acids Res. 2014;43:D315–D320. doi: 10.1093/nar/gku982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baruah A, Rani P, Biswas P. Conformational entropy of intrinsically disordered proteins from amino acid triads. Sci Rep. 2015;5:11740. doi: 10.1038/srep11740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. doi: 10.1016/S0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 6.van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, Kim PM, Kriwacki RW, Oldfield CJ, Pappu RV, Tompa P, Uversky VN, Wright PE, Babu MM. Classification of intrinsically disordered regions and proteins. Chem Rev. 2014;114:6589–6631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varadi M, Tompa P. The protein ensemble database. Adv Exp Med Biol. 2015;870:335–349. doi: 10.1007/978-3-319-20164-1_11. [DOI] [PubMed] [Google Scholar]
- 8.Trombitas K, Greaser M, Labeit S, Jin JP, Kellermayer M, Helmes M, Granzier H. Titin extensibility in situ: entropic elasticity of permanently folded and permanently unfolded molecular segments. J Cell Biol. 1998;140:853–859. doi: 10.1083/jcb.140.4.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mukhopadhyay R, Hoh JH. AFM force measurements on microtubule-associated proteins: the projection domain exerts a long-range repulsive force. FEBS Lett. 2001;505:374–378. doi: 10.1016/S0014-5793(01)02844-7. [DOI] [PubMed] [Google Scholar]
- 10.Denning DP, Patel SS, Uversky V, Fink AL, Rexach M. Disorder in the nuclear pore complex: the FG repeat regions of nucleoporins are natively unfolded. Proc Natl Acad Sci USA. 2003;100:2450–2455. doi: 10.1073/pnas.0437902100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Milles S, Mercadante D, Aramburu IV, Jensen MR, Banterle N, Koehler C, Tyagi S, Clarke J, Shammas SL, Blackledge M, Grater F, Lemke EA. Plasticity of an ultrafast interaction between nucleoporins and nuclear transport receptors. Cell. 2015;163:734–745. doi: 10.1016/j.cell.2015.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel SS, Belmont BJ, Sante JM, Rexach MF. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell. 2007;129:83–96. doi: 10.1016/j.cell.2007.01.044. [DOI] [PubMed] [Google Scholar]
- 13.Halwer M. Light-scattering study of effect of electrolytes on alpha- and beta-casein solutions. Arch Biochem Biophys. 1954;51:79–87. doi: 10.1016/0003-9861(54)90455-5. [DOI] [PubMed] [Google Scholar]
- 14.Pascal C, Pate F, Cheynier V, Delsuc MA. Study of the interactions between a proline-rich protein and a flavan-3-ol by NMR: residual structures in the natively unfolded protein provides anchorage points for the ligands. Biopolymers. 2009;91:745–756. doi: 10.1002/bip.21221. [DOI] [PubMed] [Google Scholar]
- 15.House-Pompeo K, Xu Y, Joh D, Speziale P, Hook M. Conformational changes in the fibronectin binding MSCRAMMs are induced by ligand binding. J Biol Chem. 1996;271:1379–1384. doi: 10.1074/jbc.271.3.1379. [DOI] [PubMed] [Google Scholar]
- 16.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tompa P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005;579:3346–3354. doi: 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 18.Hegyi H, Schad E, Tompa P. Structural disorder promotes assembly of protein complexes. BMC Struct Biol. 2007;7:65. doi: 10.1186/1472-6807-7-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kriwacki RW, Hengst L, Tennant L, Reed SI, Wright PE. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc Natl Acad Sci USA. 1996;93:11504–11509. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunker AK, Obradovic Z. The protein trinity—linking function and disorder. Nat Biotechnol. 2001;19:805–806. doi: 10.1038/nbt0901-805. [DOI] [PubMed] [Google Scholar]
- 21.Tompa P, Szasz C, Buday L. Structural disorder throws new light on moonlighting. Trends Biochem Sci. 2005;30:484–489. doi: 10.1016/j.tibs.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 22.Fabrega C, Shen V, Shuman S, Lima CD. Structure of an mRNA capping enzyme bound to the phosphorylated carboxy-terminal domain of RNA polymerase II. Mol Cell. 2003;11:1549–1561. doi: 10.1016/S1097-2765(03)00187-4. [DOI] [PubMed] [Google Scholar]
- 23.Dames SA, Martinez-Yamout M, De Guzman RN, Dyson HJ, Wright PE. Structural basis for Hif-1 alpha/CBP recognition in the cellular hypoxic response. Proc Natl Acad Sci USA. 2002;99:5271–5276. doi: 10.1073/pnas.082121399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ. Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1 alpha. J Biol Chem. 2003;278:1802–1806. doi: 10.1074/jbc.C200644200. [DOI] [PubMed] [Google Scholar]
- 25.Brown CJ, Takayama S, Campen AM, Vise P, Marshall TW, Oldfield CJ, Williams CJ, Keith Dunker A. Evolutionary rate heterogeneity in proteins with long disordered regions. J Mol Evol. 2002;55:104–110. doi: 10.1007/s00239-001-2309-6. [DOI] [PubMed] [Google Scholar]
- 26.Bellay J, Han S, Michaut M, Kim T, Costanzo M, Andrews BJ, Boone C, Bader GD, Myers CL, Kim PM. Bringing order to protein disorder through comparative genomics and genetic interactions. Genome Biol. 2011;12:R14. doi: 10.1186/gb-2011-12-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loewenstein Y, Raimondo D, Redfern OC, Watson J, Frishman D, Linial M, Orengo C, Thornton J, Tramontano A. Protein function annotation by homology-based inference. Genome Biol. 2009;10:207. doi: 10.1186/gb-2009-10-2-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holt C, Sawyer L. Caseins as rheomorphic proteins: interpretation of primary and secondary structures of the alpha(s1)-, beta- and kappa-caseins. J Chem Soc Faraday Trans. 1993;89:2683–2692. doi: 10.1039/FT9938902683. [DOI] [Google Scholar]
- 30.Tompa P, Fuxreiter M, Oldfield CJ, Simon I, Dunker AK, Uversky VN. Close encounters of the third kind: disordered domains and the interactions of proteins. BioEssays. 2009;31:328–335. doi: 10.1002/bies.200800151. [DOI] [PubMed] [Google Scholar]
- 31.Hurst LD. The K a/K s ratio: diagnosing the form of sequence evolution. Trends Genet. 2002;18:486. doi: 10.1016/S0168-9525(02)02722-1. [DOI] [PubMed] [Google Scholar]
- 32.Tucker PK, Lundrigan BL. Rapid evolution of the sex determining locus in Old World mice and rats. Nature. 1993;364:715–717. doi: 10.1038/364715a0. [DOI] [PubMed] [Google Scholar]
- 33.Whitfield LS, Lovell-Badge R, Goodfellow PN. Rapid sequence evolution of the mammalian sex-determining gene SRY. Nature. 1993;364:713–715. doi: 10.1038/364713a0. [DOI] [PubMed] [Google Scholar]
- 34.Csizmok V, Felli IC, Tompa P, Banci L, Bertini I. Structural and dynamic characterization of intrinsically disordered human securin by NMR spectroscopy. J Am Chem Soc. 2008;130:16873–16879. doi: 10.1021/ja805510b. [DOI] [PubMed] [Google Scholar]
- 35.Dunker AK, Lawson JD, Brown CJ, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/S1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 36.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 37.Atkins JD, Boateng SY, Sorensen T, McGuffin LJ. Disorder prediction methods, their applicability to different protein targets and their usefulness for guiding experimental studies. Int J Mol Sci. 2015;16:19040–19054. doi: 10.3390/ijms160819040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ciechanover A. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin–proteasome system and onto human diseases and drug targeting. Biochim Biophys Acta. 2012;1824:3–13. doi: 10.1016/j.bbapap.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 39.Tomko RJ, Jr, Hochstrasser M. Molecular architecture and assembly of the eukaryotic proteasome. Annu Rev Biochem. 2013;82:415–445. doi: 10.1146/annurev-biochem-060410-150257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varshavsky A. Naming a targeting signal. Cell. 1991;64:13–15. doi: 10.1016/0092-8674(91)90202-A. [DOI] [PubMed] [Google Scholar]
- 41.Inobe T, Fishbain S, Prakash S, Matouschek A. Defining the geometry of the two-component proteasome degron. Nat Chem Biol. 2011;7:161–167. doi: 10.1038/nchembio.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin–proteasome system. Nat Rev Mol Cell Biol. 2008;9:679–690. doi: 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prakash S, Tian L, Ratliff KS, Lehotzky RE, Matouschek A. An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat Struct Mol Biol. 2004;11:830–837. doi: 10.1038/nsmb814. [DOI] [PubMed] [Google Scholar]
- 44.Guharoy M, Bhowmick P, Sallam M, Tompa P. Tripartite degrons confer diversity and specificity on regulated protein degradation in the ubiquitin–proteasome system. Nat Commun. 2016;7:10239. doi: 10.1038/ncomms10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein-folding—a synthesis. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 46.Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tompa P, Fuxreiter M. Fuzzy complexes: polymorphism and structural disorder in protein–protein interactions. Trends Biochem Sci. 2008;33:2–8. doi: 10.1016/j.tibs.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 48.Tompa P. On the supertertiary structure of proteins. Nat Chem Biol. 2012;8:597–600. doi: 10.1038/nchembio.1009. [DOI] [PubMed] [Google Scholar]
- 49.Uversky VN. The triple power of D 3: protein intrinsic disorder in degenerative diseases. Front Biosci (Landmark edition) 2014;19:181–258. doi: 10.2741/4204. [DOI] [PubMed] [Google Scholar]
- 50.Guharoy M, Pauwels K, Tompa P. SnapShot: intrinsic structural disorder. Cell. 2015;161(1230–1230):e1. doi: 10.1016/j.cell.2015.05.024. [DOI] [PubMed] [Google Scholar]
- 51.Pauwels K, Tompa P. Editorial: function and flexibility: friend or foe? Front Mol Biosci. 2016;3:31. doi: 10.3389/fmolb.2016.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Babu MM, van der Lee R, de Groot NS, Gsponer J. Intrinsically disordered proteins: regulation and disease. Curr Opin Struct Biol. 2011;21:1–9. doi: 10.1016/j.sbi.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 53.Bodart JF, Wieruszeski JM, Amniai L, Leroy A, Landrieu I, Rousseau-Lescuyer A, Vilain JP, Lippens G. NMR observation of Tau in Xenopus oocytes. J Magn Reson. 2008;192:252–257. doi: 10.1016/j.jmr.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 54.Theillet F-X, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, Verzini S, Lorenz D, van Rossum M, Goldfarb D, Selenko P. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature. 2016;530:45–50. doi: 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- 55.Bernadó P, Svergun DI. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol Biosyst. 2011;8:151–167. doi: 10.1039/C1MB05275F. [DOI] [PubMed] [Google Scholar]
- 56.Receveur-Brechot V, Durand D. How random are intrinsically disordered proteins? A small angle scattering perspective. Curr Protein Pept Sci. 2012;13:55–75. doi: 10.2174/138920312799277901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fontana A, De Laureto PP, Spolaore B, Frare E, Picotti P, Zambonin M. Probing protein structure by limited proteolysis. Acta Biochim Pol. 2004;51:299–321. [PubMed] [Google Scholar]
- 58.Luchinat E, Banci L. A unique tool for cellular structural biology: in-cell NMR. J Biol Chem. 2016;291:3776–3784. doi: 10.1074/jbc.R115.643247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Varadi M, Vranken W, Guharoy M, Tompa P. Computational approaches for inferring the functions of intrinsically disordered proteins. Front Mol Biosci. 2015;2:1–8. doi: 10.3389/fmolb.2015.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uversky VN. Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta Proteins Proteom. 2013;1834:932–951. doi: 10.1016/j.bbapap.2012.12.008. [DOI] [PubMed] [Google Scholar]
- 61.Uversky VN. Biophysical methods to investigate intrinsically disordered proteins: avoiding an “elephant and blind men” situation. Adv Exp Med Biol. 2015;870:215–260. doi: 10.1007/978-3-319-20164-1_7. [DOI] [PubMed] [Google Scholar]
- 62.Anfinsen C (1972) Nobel lecture: studies on the principles that govern the folding of protein chains. Nobelprize.org. Nobel Media AB 2014. http://www.nobelprize.org/nobel_prizes/chemistry/laureates/1972/anfinsen-lecture.html. Accessed 12 Jun 2017
- 63.Gershenson A, Gierasch LM, Pastore A, Radford SE. Energy landscapes of functional proteins are inherently risky. Nat Chem Biol. 2014;10:884–891. doi: 10.1038/nchembio.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ellis RJ. Macromolecular crowding: obvious but under appreciated. Trends Biochem Sci. 2001;26:597–604. doi: 10.1016/S0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- 65.Minton AP. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J Biol Chem. 2001;276:10577–10580. doi: 10.1074/jbc.R100005200. [DOI] [PubMed] [Google Scholar]
- 66.Politou A, Temussi PA. Revisiting a dogma: the effect of volume exclusion in molecular crowding. Curr Opin Struct Biol. 2015;30:1–6. doi: 10.1016/j.sbi.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 67.Rivas G, Minton AP. Macromolecular crowding in vitro, in vivo, and in between. Trends Biochem Sci. 2016;41:970–981. doi: 10.1016/j.tibs.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ellis RJ, Minton AP. Join the crowd. Nature. 2003;425:27–28. doi: 10.1038/425027a. [DOI] [PubMed] [Google Scholar]
- 69.Szasz C, Alexa A, Toth K, Rakacs M, Langowski J, Tompa P. Protein disorder prevails under crowded conditions. Biochemistry. 2011;50:5834–5844. doi: 10.1021/bi200365j. [DOI] [PubMed] [Google Scholar]
- 70.Schuler B, Soranno A, Hofmann H, Nettels D. Single-molecule FRET spectroscopy and the polymer physics of unfolded and intrinsically disordered proteins. Annu Rev Biophys. 2016;45:207–231. doi: 10.1146/annurev-biophys-062215-010915. [DOI] [PubMed] [Google Scholar]
- 71.Soranno A, Koenig I, Borgia MB, Hofmann H, Zosel F, Nettels D, Schuler B. Single-molecule spectroscopy reveals polymer effects of disordered proteins in crowded environments. Proc Natl Acad Sci USA. 2014;111:4874–4879. doi: 10.1073/pnas.1322611111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Flaugh SL, Lumb KJ. Effects of macromolecular crowding on the intrinsically disordered proteins c-Fos and p27(Kip1) Biomacromolecules. 2001;2:538–540. doi: 10.1021/bm015502z. [DOI] [PubMed] [Google Scholar]
- 73.Kuznetsova IM, Turoverov KK, Uversky VN. What macromolecular crowding can do to a protein. Int J Mol Sci. 2014;15:23090–23140. doi: 10.3390/ijms151223090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Theillet FX, Binolfi A, Frembgen-Kesner T, Hingorani K, Sarkar M, Kyne C, Li C, Crowley PB, Gierasch L, Pielak GJ, Elcock AH, Gershenson A, Selenko P. Physicochemical properties of cells and their effects on intrinsically disordered proteins (IDPs) Chem Rev. 2014;114:6661–6714. doi: 10.1021/cr400695p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pastore A, Temussi PA. The two faces of Janus: functional interactions and protein aggregation. Curr Opin Struct Biol. 2012;22:30–37. doi: 10.1016/j.sbi.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 76.Wirth AJ, Gruebele M. Quinary protein structure and the consequences of crowding in living cells: leaving the test-tube behind. BioEssays. 2013;35:984–993. doi: 10.1002/bies.201300080. [DOI] [PubMed] [Google Scholar]
- 77.Serber Z, Doetsch V. In-cell NMR spectroscopy. Biochemistry. 2001;40:14317–14323. doi: 10.1021/bi011751w. [DOI] [PubMed] [Google Scholar]
- 78.Daniels AJ, Williams RJP, Wright PE. The character of the stored molecules in chromaffin granules of the adrenal medulla: a nuclear magnetic resonance study. Neuroscience. 1978;3:573–585. doi: 10.1016/0306-4522(78)90022-2. [DOI] [PubMed] [Google Scholar]
- 79.Gibson TJ, Seiler M, Veitia RA. The transience of transient overexpression. Nat Methods. 2013;10:715–721. doi: 10.1038/nmeth.2534. [DOI] [PubMed] [Google Scholar]
- 80.Banci L, Barbieri L, Bertini I, Luchinat E, Secci E, Zhao Y, Aricescu AR. Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nat Chem Biol. 2013;9:297–299. doi: 10.1038/nchembio.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barbieri L, Luchinat E, Banci L. Characterization of proteins by in-cell NMR spectroscopy in cultured mammalian cells. Nat Protoc. 2016;11:1101–1111. doi: 10.1038/nprot.2016.061. [DOI] [PubMed] [Google Scholar]
- 82.Bekei B, Rose HM, Herzig M, Dose A, Schwarzer D, Selenko P. In-cell NMR in mammalian cells: part 1. Methods Mol Biol. 2012;895:43–54. doi: 10.1007/978-1-61779-927-3_4. [DOI] [PubMed] [Google Scholar]
- 83.Ogino S, Kubo S, Umemoto R, Huang S, Nishida N, Shimada I. Observation of NMR signals from proteins introduced into living mammalian cells by reversible membrane permeabilization using a pore-forming toxin, streptolysin O. J Am Chem Soc. 2009;131:10834–10835. doi: 10.1021/ja904407w. [DOI] [PubMed] [Google Scholar]
- 84.Amata I, Maffei M, Igea A, Gay M, Vilaseca M, Nebreda AR, Pons M. Multi-phosphorylation of the intrinsically disordered unique domain of c-src studied by in-cell and real-time NMR spectroscopy. ChemBioChem. 2013;14:1820–1827. doi: 10.1002/cbic.201300139. [DOI] [PubMed] [Google Scholar]
- 85.Bekei B, Rose HM, Herzig M, Stephanowitz H, Krause E, Selenko P. In cell NMR in mammalian cells: part 3. Methods Mol Biol. 2012;896:107–122. doi: 10.1007/978-1-61779-927-3_6. [DOI] [PubMed] [Google Scholar]
- 86.Bryant JE. Retracted article: In-cell protein dynamics. Mol Biosyst. 2006;2:406–410. doi: 10.1039/b604684c. [DOI] [PubMed] [Google Scholar]
- 87.Bryant JE, Lecomte JTJ, Lee AL, Young GB, Pielak GJ. Protein dynamics in living cells. Biochemistry. 2007;46:8206–8207. doi: 10.1021/bi700744h. [DOI] [PubMed] [Google Scholar]
- 88.Waudby CA, Mantle MD, Cabrita LD, Gladden LF, Dobson CM, Christodoulou J. Rapid distinction of intracellular and extracellular proteins using NMR diffusion measurements. J Am Chem Soc. 2012;134:11312–11315. doi: 10.1021/ja304912c. [DOI] [PubMed] [Google Scholar]
- 89.Barnes CO, Pielak GJ. In-cell protein NMR and protein leakage. Proteins. 2011;79:347–351. doi: 10.1002/prot.22906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Axford D, Ji X, Stuart DI, Sutton G. In cellulo structure determination of a novel cypovirus polyhedrin. Acta Crystallogr D Biol Crystallogr. 2014;70:1435–1441. doi: 10.1107/S1399004714004714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Doerr A. Single-particle cryo-electron microscopy. Nat Methods. 2016;13:23–24. doi: 10.1038/nmeth.3700. [DOI] [PubMed] [Google Scholar]
- 92.Yi P, Wang Z, Feng Q, Pintilie Grigore D, Foulds Charles E, Lanz Rainer B, Ludtke Steven J, Schmid Michael F, Chiu W, O’Malley Bert W. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol Cell. 2015;57:1047–1058. doi: 10.1016/j.molcel.2015.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mahamid J, Pfeffer S, Schaffer M, Villa E, Danev R, Kuhn Cuellar L, Förster F, Hyman AA, Plitzko JM, Baumeister W. Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science. 2016;351:969–972. doi: 10.1126/science.aad8857. [DOI] [PubMed] [Google Scholar]
- 94.Doerr A. Cryo-electron tomography. Nat Methods. 2017;14:34. doi: 10.1038/nmeth.4115. [DOI] [Google Scholar]
- 95.Feng Y, De Franceschi G, Kahraman A, Soste M, Melnik A, Boersema PJ, De Laureto PP, Nikolaev Y, Oliveira AP, Picotti P. Global analysis of protein structural changes in complex proteomes. Nat Biotechnol. 2014;32:1036–1044. doi: 10.1038/nbt.2999. [DOI] [PubMed] [Google Scholar]
- 96.Ebbinghaus S, Dhar A, McDonald JD, Gruebele M. Protein folding stability and dynamics imaged in a living cell. Nat Methods. 2010;7:319–323. doi: 10.1038/nmeth.1435. [DOI] [PubMed] [Google Scholar]
- 97.Frederick KK, Michaelis VK, Corzilius B, Ong TC, Jacavone AC, Griffin RG, Lindquist S. Sensitivity-enhanced NMR reveals alterations in protein structure by cellular milieus. Cell. 2016;163:620–628. doi: 10.1016/j.cell.2015.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R, McKnight SL. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–767. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Aumiller WM, Jr, Keating CD. Phosphorylation-mediated RNA/peptide complex coacervation as a model for intracellular liquid organelles. Nat Chem. 2016;8:129–137. doi: 10.1038/nchem.2414. [DOI] [PubMed] [Google Scholar]
- 100.Brangwynne CP, Tompa P, Pappu RV. Polymer physics of intracellular phase transitions. Nat Phys. 2015;11:899–904. doi: 10.1038/nphys3532. [DOI] [Google Scholar]
- 101.Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154:727–736. doi: 10.1016/j.cell.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, Russo PS, Jiang QX, Nixon BT, Rosen MK. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483:336–340. doi: 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tompa P. Hydrogel formation by multivalent IDPs. A reincarnation of the microtrabecular lattice? Intrinsically Disord Proteins. 2013;1:e24068. doi: 10.4161/idp.24068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Burke KA, Janke AM, Rhine CL, Fawzi NL. Residue-by-residue view of in vitro FUS granules that bind the c-terminal domain of RNA polymerase II. Mol Cell. 2015;60:231–241. doi: 10.1016/j.molcel.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, Forman-Kay JD, Baldwin AJ. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell. 2015;57:936–947. doi: 10.1016/j.molcel.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, Pozniakovski A, Poser I, Maghelli N, Royer LA, Weigert M, Myers EW, Grill S, Drechsel D, Hyman AA, Alberti S. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162:1066–1077. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- 107.Brangwynne CP. Phase transitions and size scaling of membrane-less organelles. J Cell Biol. 2013;203:875–881. doi: 10.1083/jcb.201308087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bertini I, Felli IC, Gonnelli L, Kumar MVV, Pierattelli R. 13C direct-detection biomolecular NMR spectroscopy in living cells. Angew Chem Int Ed Engl. 2011;50:2339–2341. doi: 10.1002/anie.201006636. [DOI] [PubMed] [Google Scholar]
- 109.Bartels T, Choi JG, Selkoe DJ. Alpha-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, Ju S, Cookson MR, Asturias FJ, Agar JN, Webb BN, Kang C, Ringe D, Petsko GA, Pochapsky TC, Hoang QQ. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci USA. 2011;108:17797–17802. doi: 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Iljina M, Tosatto L, Choi ML, Sang JC, Ye Y, Hughes CD, Bryant CE, Gandhi S, Klenerman D. Arachidonic acid mediates the formation of abundant alpha-helical multimers of alpha-synuclein. Sci Rep. 2016;6:33928. doi: 10.1038/srep33928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 115.Uversky VN, Li J, Fink AL. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J Biol Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 116.Eliezer D, Kutluay E, Bussell R, Jr, Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 117.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 118.Trexler AJ, Rhoades E. N-terminal acetylation is critical for forming alpha-helical oligomer of alpha-synuclein. Protein Sci. 2012;21:601–605. doi: 10.1002/pro.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT, Tsika E, Coune P, Prudent M, Lion N, Eliezer D, Moore DJ, Schneider B, Aebischer P, El-Agnaf OM, Masliah E, Lashuel HA. Alpha-synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287:15345–15364. doi: 10.1074/jbc.M111.318949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Binolfi A, Theillet FX, Selenko P. Bacterial in-cell NMR of human alpha-synuclein: a disordered monomer by nature? Biochem Soc Trans. 2012;40:950–954. doi: 10.1042/BST20120096. [DOI] [PubMed] [Google Scholar]
- 121.Dettmer U, Newman AJ, Luth ES, Bartels T, Selkoe D. In vivo cross-linking reveals principally oligomeric forms of alpha-synuclein and beta-synuclein in neurons and non-neural cells. J Biol Chem. 2013;288:6371–6385. doi: 10.1074/jbc.M112.403311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ohrfelt A, Zetterberg H, Andersson K, Persson R, Secic D, Brinkmalm G, Wallin A, Mulugeta E, Francis PT, Vanmechelen E, Aarsland D, Ballard C, Blennow K, Westman-Brinkmalm A. Identification of novel alpha-synuclein isoforms in human brain tissue by using an online nanoLC–ESI–FTICR–MS method. Neurochem Res. 2011;36:2029–2042. doi: 10.1007/s11064-011-0527-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Johnson M, Coulton AT, Geeves MA, Mulvihill DP. Targeted amino-terminal acetylation of recombinant proteins in E. coli . PLoS One. 2010;5:e15801. doi: 10.1371/journal.pone.0015801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Maltsev AS, Ying J, Bax A. Impact of N-terminal acetylation of alpha-synuclein on its random coil and lipid binding properties. Biochemistry. 2012;51:5004–5013. doi: 10.1021/bi300642h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kang L, Moriarty GM, Woods LA, Ashcroft AE, Radford SE, Baum J. N-terminal acetylation of alpha-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 2012;21:911–917. doi: 10.1002/pro.2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gurry T, Ullman O, Fisher CK, Perovic I, Pochapsky T, Stultz CM. The dynamic structure of alpha-synuclein multimers. J Am Chem Soc. 2013;135:3865–3872. doi: 10.1021/ja310518p. [DOI] [PubMed] [Google Scholar]
- 127.Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, von Saucken VE, Sanderson JB, Jaenisch R, Bartels T, Selkoe D. Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun. 2015;6:7314. doi: 10.1038/ncomms8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Daughdrill GW, Hanely LJ, Dahlquist FW. The C-terminal half of the anti-sigma factor FlgM contains a dynamic equilibrium solution structure favoring helical conformations. Biochemistry. 1998;37:1076–1082. doi: 10.1021/bi971952t. [DOI] [PubMed] [Google Scholar]
- 129.Dedmon MM, Patel CN, Young GB, Pielak GJ. FlgM gains structure in living cells. Proc Natl Acad Sci USA. 2002;99:12681–12684. doi: 10.1073/pnas.202331299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Smith AE, Zhou LZ, Pielak GJ. Hydrogen exchange of disordered proteins in Escherichia coli . Protein Sci. 2015;24:706–713. doi: 10.1002/pro.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cleveland DW, Hwo SY, Kirschner MW. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol. 1977;116:227–247. doi: 10.1016/0022-2836(77)90214-5. [DOI] [PubMed] [Google Scholar]
- 132.Smet C, Leroy A, Sillen A, Wieruszeski JM, Landrieu I, Lippens G. Accepting its random coil nature allows a partial NMR assignment of the neuronal Tau protein. ChemBioChem. 2004;5:1639–1646. doi: 10.1002/cbic.200400145. [DOI] [PubMed] [Google Scholar]
- 133.Harbison NW, Bhattacharya S, Eliezer D. Assigning backbone NMR resonances for full length tau isoforms: efficient compromise between manual assignments and reduced dimensionality. PLoS One. 2012;7:e34679. doi: 10.1371/journal.pone.0034679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Luchinat E, Barbieri L, Rubino JT, Kozyreva T, Cantini F, Banci L. In-cell NMR reveals potential precursor of toxic species from SOD1 fALS mutants. Nat Commun. 2014;5:5502. doi: 10.1038/ncomms6502. [DOI] [PubMed] [Google Scholar]
- 135.Bhowmick A, Brookes DH, Yost SR, Dyson HJ, Forman-Kay JD, Gunter D, Head-Gordon M, Hura GL, Pande VS, Wemmer DE, Wright PE, Head-Gordon T. Finding our way in the dark proteome. J Am Chem Soc. 2016;138:9730–9742. doi: 10.1021/jacs.6b06543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Perdigao N, Heinrich J, Stolte C, Sabir KS, Buckley MJ, Tabor B, Signal B, Gloss BS, Hammang CJ, Rost B, Schafferhans A, O’Donoghue SI. Unexpected features of the dark proteome. Proc Natl Acad Sci USA. 2015;112:15898–15903. doi: 10.1073/pnas.1508380112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N, Yeh LS. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32:D115–D119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.O’Donoghue SI, Sabir KS, Kalemanov M, Stolte C, Wellmann B, Ho V, Roos M, Perdigao N, Buske FA, Heinrich J, Rost B, Schafferhans A. Aquaria: simplifying discovery and insight from protein structures. Nat Methods. 2015;12:98–99. doi: 10.1038/nmeth.3258. [DOI] [PubMed] [Google Scholar]
- 139.Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- 140.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::AID-PROT50>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 141.Huntley MA, Golding GB. Simple sequences are rare in the protein data bank. Proteins. 2002;48:134–140. doi: 10.1002/prot.10150. [DOI] [PubMed] [Google Scholar]
- 142.Tompa P, Varadi M. Predicting the predictive power of IDP ensembles. Structure. 2014;22:177–178. doi: 10.1016/j.str.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 143.Aznauryan M, Delgado L, Soranno A, Nettels D, Huang JR, Labhardt AM, Grzesiek S, Schuler B. Comprehensive structural and dynamical view of an unfolded protein from the combination of single-molecule FRET, NMR, and SAXS. Proc Natl Acad Sci USA. 2016;113:E5389–E5398. doi: 10.1073/pnas.1607193113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fisher CK, Stultz CM. Constructing ensembles for intrinsically disordered proteins. Curr Opin Struct Biol. 2011;21:426–431. doi: 10.1016/j.sbi.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ito Y, Selenko P. Cellular structural biology. Curr Opin Struct Biol. 2010;20:640–648. doi: 10.1016/j.sbi.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 146.Borcherds W, Theillet FX, Katzer A, Finzel A, Mishall KM, Powell AT, Wu H, Manieri W, Dieterich C, Selenko P, Loewer A, Daughdrill GW. Disorder and residual helicity alter p53–Mdm2 binding affinity and signaling in cells. Nat Chem Biol. 2014;10:1000–1002. doi: 10.1038/nchembio.1668. [DOI] [PubMed] [Google Scholar]
- 147.Banci L, Barbieri L, Bertini I, Cantini F, Luchinat E. In-cell NMR in E. coli to monitor maturation steps of hSOD1. PLoS One. 2011;6:e23561. doi: 10.1371/journal.pone.0023561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Danielsson J, Inomata K, Murayama S, Tochio H, Lang L, Shirakawa M, Oliveberg M. Pruning the ALS-associated protein SOD1 for in-cell NMR. J Am Chem Soc. 2013;135:10266–10269. doi: 10.1021/ja404425r. [DOI] [PubMed] [Google Scholar]
- 149.König I, Zarrine-Afsar A, Aznauryan M, Soranno A, Wunderlich B, Dingfelder F, Stüber JC, Plückthun A, Nettels D, Schuler B. Single-molecule spectroscopy of protein conformational dynamics in live eukaryotic cells. Nat Methods. 2015;12:773–779. doi: 10.1038/nmeth.3475. [DOI] [PubMed] [Google Scholar]
- 150.Di Tommaso P, Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang JM, Taly JF, Notredame C. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011;39:W13–W17. doi: 10.1093/nar/gkr245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ait-Bara S, Carpousis AJ, Quentin Y. RNase E in the gamma-proteobacteria: conservation of intrinsically disordered noncatalytic region and molecular evolution of microdomains. Mol Genet Genom. 2015;290:847–862. doi: 10.1007/s00438-014-0959-5. [DOI] [PMC free article] [PubMed] [Google Scholar]