

Abstract
Cystic fibrosis transmembrane conductance regulator (CFTR) channel gating is predominantly regulated by protein kinase A (PKA)-dependent phosphorylation. In addition to regulating CFTR channel activity, PKA phosphorylation is also involved in enhancing CFTR trafficking and mediating conformational changes at the interdomain interfaces of the protein. The major cystic fibrosis (CF)-causing mutation is the deletion of phenylalanine at position 508 (F508del); it causes many defects that affect CFTR trafficking, stability, and gating at the cell surface. Due to the multiple roles of PKA phosphorylation, there is growing interest in targeting PKA-dependent signaling for rescuing the trafficking and functional defects of F508del-CFTR. This review will discuss the effects of PKA phosphorylation on wild-type CFTR, the consequences of CF mutations on PKA phosphorylation, and the development of therapies that target PKA-mediated signaling.
Keywords: Phosphorylation, Channel gating, Interdomain interactions, Trafficking, Cytoskeleton, Modulators
Introduction
Cystic fibrosis (CF) is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The gene encodes for CFTR, an anion channel that consists of two membrane-spanning domains (MSDs), two nucleotide-binding domains (NBDs), and a unique regulatory (R) domain. The interface between the MSDs and the NBDs is conferred by intracellular loops (ICLs) that extend from the MSDs and connect with the NBDs via coupling helices (CHs). Channel gating of CFTR is regulated primarily by protein kinase A (PKA) phosphorylation, although several other serine/threonine kinases and tyrosine kinases have also been implicated in its regulation [1–3].
The major CF-causing mutation is the deletion of phenylalanine at position 508 (F508del). The mutation disrupts the thermostability of NBD1 and the interface between ICL4 and NBD1, leading to defects in CFTR assembly, processing and channel gating [4, 5]. Drug discovery efforts have resulted in the first Food and Drug Administration (FDA)-approved drug known as KALYDECO® (VX-770), a potentiator that enhances CFTR channel gating at the cell surface. VX-770 was initially approved for CFTR gating mutations such as G551D (the mutation of glycine to aspartic acid at position 551). A few years later, it was approved for patients with the F508del mutation in combination with VX-809, a corrector compound that partially rescues the misfolded mutant protein and promotes its trafficking to the cell surface. Together, the potentiator VX-770 and corrector VX-809 drug cocktail is marketed as ORKAMBITM. Our intention for this review is to highlight recent insights on the PKA-dependent regulation of CFTR functional expression, the effects of CF-causing mutations and the activity of recently approved modulators.
CFTR synthesis, trafficking and stability are modified by PKA-mediated phosphorylation
PKA phosphorylation regulates CFTR expression and trafficking via the biosynthetic pathway
The promoter region of the CFTR gene contains a cyclic adenosine monophosphate (cAMP)-response element (CRE) [6, 7]. McDonald and colleagues hypothesized that PKA phosphorylation may regulate CFTR gene expression by activating this regulatory region and/or promote CFTR synthesis by increasing the mRNA stability of the CFTR gene (Fig. 1) [6, 7]. After its synthesis, CFTR possesses multiple PKA consensus sites which have low basal levels of phosphorylation. Previous radioactivity and mass spectrometry-based studies have shown that nine dibasic PKA consensus sites are located in the regulatory insertion (RI) of NBD1 (Serine 422 or S422), the regulatory extension (RE) in the amino-terminus of the R domain (S660) and the remainder of the R domain [8–17]. These sites exhibit low levels of phosphorylation in the resting (i.e., inactive) state of the protein and increased levels of phosphorylation after the addition of cAMP agonists (particularly at S660, S700, S737, S768, S795 and S813) [8–17].
Fig. 1.
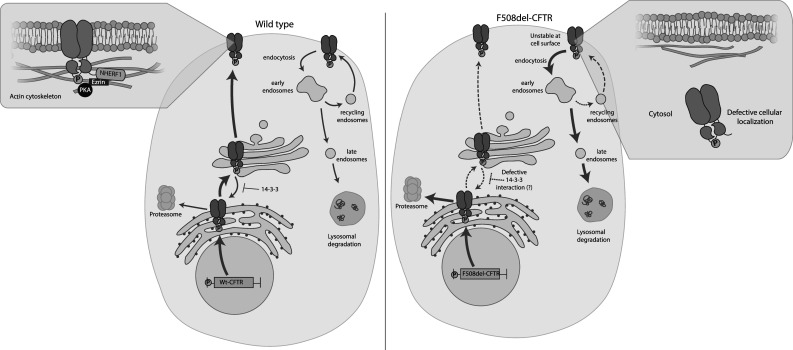
The deletion of phenylalanine at position 508 (F508del) alters phosphoregulation of cystic fibrosis transmembrane conductance regulator (CFTR) trafficking. Left panel: protein kinase A (PKA) phosphorylation promotes CFTR gene expression via the cyclic adenosine monophosphate (cAMP) response element. Phosphorylation also enhances interactions of wild-type CFTR (Wt-CFTR) protein with 14-3-3, which facilitates CFTR exit from the endoplasmic reticulum (ER) and promotes its forward trafficking to the cell surface. Zoom in of the cell surface: phosphorylation enhances the interaction between CFTR and the Na+/H+ exchanger regulatory factor (NHERF1)–ezrin–actin complex which stabilizes the protein at the cell surface. Ezrin also brings PKA closer to CFTR which is essential for regulating channel gating. At the cell surface, CFTR can be internalized by endocytosis into early endosomes which can then recycle back to the cell surface or undergo lysosomal degradation. PKA phosphorylation can also enhance cell surface expression of CFTR by promoting recycling. Right panel: the major population of F508del-CFTR is retained in the ER and undergoes ER-associated degradation via the proteasome. A limited number of F508del-CFTR protein can escape the ER and reach the Golgi apparatus; however, aberrant exposure of “retention motifs” redirects the protein back to the ER. F508del-CFTR may also have impaired interactions with 14-3-3 due to defective phosphorylation of the mutant, resulting in decreased forward trafficking. Zoom in of the cell surface: the small population of F508del-CFTR that reaches the cell surface is unstable and is targeted for degradation via the peripheral protein quality control, possibly due to defective interactions with the NHERF1–ezrin–actin complex. The phosphorylation defect also results in reduced recycling leading to decreased cell surface expression of F508del-CFTR. Not depicted for clarity: CFTR trafficks within vesicles in the cell
One of the roles of constitutive phosphorylation relates to CFTR protein trafficking from the endoplasmic reticulum (ER) to the Golgi apparatus. After exiting the ER, partially assembled CFTR protein interacts with β-coat protein (COP), a component of the COPI complex that mediates ER retention [18, 19]. Co-immunoprecipitation studies show that phosphorylation decreases the interaction of CFTR with β-COP by enhancing its affinity to 14-3-3 (the β and ε isoforms), a protein involved in the forward trafficking of CFTR from the ER to the Golgi and cell surface (Fig. 1) [18].
Nuclear magnetic resonance (NMR) studies have shown that PKA phosphorylation of S768, S795 and S813 in the R domain led to binding of CFTR to the 14-3-3β isoform in the ER [18]. A recent study by the Ottmann group shows that the R domain cooperatively binds to 14-3-3 using fluorescence polarization-binding assays with R domain constructs containing individually phosphorylated PKA sites [20]. Their results suggest that phosphorylation of S768 in the R domain created the strongest binding site of CFTR to 14-3-3, and this “anchor” facilitated binding of additional sites (S712, S753, S795, S813) to 14-3-3 [20].
Role of PKA phosphorylation in cell surface stability of CFTR
CFTR residence at the cell surface is determined by multiple factors, including its relative affinity to cytoskeletal and scaffolding proteins as well as endocytic proteins (Fig. 1). Phosphorylation has been found to play a role in increasing the abundance of CFTR at the cell surface by enhancing its interaction with cytoskeletal scaffolding proteins such as the Na+/H+ exchanger regulatory factor (NHERF1) and the ezrin/radixin/moesin (ERM) complex. NHERF1 and ezrin interacts with the actin cytoskeleton (NHERF1-ezrin-actin complex); together the complex binds to CFTR via its PDZ motif [21–24]. Ezrin has also been shown to tether PKA to the CFTR-containing complex, thereby amplifying its functional expression as a channel [25–27]. Taken together, these studies suggest that phosphorylation promotes interactions between CFTR and the cytoskeleton, increasing the abundance and stability of CFTR at the cell surface [23, 24, 28–30].
PKA phosphorylation also enhances cell surface residence of CFTR by promoting recycling of CFTR via endosomes to the cell surface as assessed in recent fluorescence detection technology studies applying fluorogen-activating proteins (FAPs) [31]. This technology enables efficient labeling of CFTR at the plasma membrane in live epithelial cells [31]. The Wt-CFTR protein residing in Rab11-positive recycling endosomes was actively delivered to the cell surface following activation of PKA [31]. Hence, the steady-state residence time for Wt-CFTR at the cell surface is enhanced by PKA through two mechanisms: decreased internalization via endocytosis and increased exocytosis via recycling endosomes.
Regulation of CFTR channel gating by PKA phosphorylation
ATP-dependent (liganded) gating is regulated by PKA-mediated phosphorylation
There have been many informative studies of phosphorylation and nucleotide-dependent gating of the chloride channel activity of CFTR [12, 32–35] that are discussed in recent reviews [35–38]. Our current understanding of the molecular basis for ATP regulated channel gating of CFTR has been guided by biophysical studies of CFTR channel gating [35, 36, 39, 40], biochemical and ATPase activity studies by our laboratory [41–45] and mechanistic insights provided by similar ATP-binding cassette (ABC) prokaryotic proteins [46–49].
PKA-mediated phosphorylation of the R domain is absolutely required for nucleotide-dependent gating (Fig. 2) [12, 13, 50]. Following this obligatory modification, ATP binds to two sites at the NBD1:NBD2 interface [51]. Site 1, which is comprised of the Walker A and B motifs of NBD1 and the non-canonical Walker C motif of NBD2 (LSHGH), binds but does not hydrolyze the nucleotide [41]. Site 2, which is comprised of the Walker A and B motifs of NBD2 and the canonical Walker C motif of NBD1 (LSGGQ), binds and hydrolyzes ATP (i.e., confers ATPase activity) [41].
Fig. 2.

F508del-CFTR exhibits defective phosphorylation-dependent conformational changes essential for channel gating. Top panel: In Wt-CFTR, PKA increases phosphorylation at multiple sites which include S422 in the regulatory insertion (RI) of NBD1, S660 in the regulatory extension (RE) located at the amino-terminus of the R domain and other sites in the R domain. CFTR has two adenosine triphosphate (ATP)-binding sites at the NBD dimers: site 1 is non-canonical and does not hydrolyze ATP, whereas site 2 is canonical and exhibits ATPase activity. PKA phosphorylation decreases the alpha helical content of the RE and RI which removes their steric hindrances on nucleotide-binding domain 1 (NBD1). This enhances interactions at the NBD1:NBD2 interface (necessary for ATP binding) as well as the intracellular loop 1 (ICL1):NBD1 and ICL4:NBD1 interfaces (necessary for conveying conformational changes from the NBDs to the membrane spanning domains for channel gating). Bottom panel: F508del-CFTR exhibits defective phosphorylation at S660 in the RE. F508del-CFTR may also exhibit defective phosphorylation at S422 in the RI and/or other phosphorylation sites in the R domain, but this could not be confirmed with our mass spectrometry methods (sites depicted as question marks). F508del-NBD1 retains aberrant interactions with the RE and RI upon PKA phosphorylation; this prevents NBD1 from interacting with NBD2, ICL1 and ICL4 which results in impaired ATP binding/hydrolysis and defective conformational changes necessary for channel gating
Recent studies by Csanády and colleagues support a model wherein ATP binding to both nucleotide-binding sites enhances dimerization of NBD1 and NBD2 which is obligatory for channel opening [52]. ATPase activity at site 2 subsequently promotes destabilization of the NBD dimer, leading to channel closure [52]. This scheme is supported by mutagenesis studies which show that disruption of key residues in the putative catalytic base (e.g., mutation of glutamic acid to glutamine at position 1371 or E1371Q) inhibited ATPase activity of purified full-length CFTR and decreased the rate of channel closure [35, 36, 41, 53]. This model mostly conforms to the generally accepted mechanism underlying the transport activity of ABC proteins [46].
The mechanisms underlying PKA-mediated regulation of ATP-dependent gating remain unclear. Patch clamp studies conducted by Szellas and Nagel show that PKA activation decreased the Michaelis–Menten constant (K M) of the Mg–ATP-dependent channel opening by two-fold [54], suggesting that it enhances nucleotide affinity to one or both sites. Our group showed that PKA phosphorylation increases the apparent affinity of Mg–ATP in ATPase activity measurements using purified and reconstituted CFTR protein [55], suggesting that phosphorylation enhances nucleotide affinity to the catalytic site (Site 2). A recent study suggests that PKA phosphorylation stabilizes the post-catalytic transition state rather than modifying a pre-catalytic state [56]. Hence, fundamental questions regarding the regulation of ATPase activity and channel gating of CFTR by PKA phosphorylation remain unanswered.
ATP-independent (unliganded) gating is regulated by PKA phosphorylation
Primarily through studies of the ATP-binding mutant, G551D-CFTR, and the NBD2 deletion mutant (deletion of residues 1172–1480), it became clear that CFTR could mediate ATP-independent (or unliganded) channel openings. These unliganded channel openings appear to be regulated by phosphorylation, potentially via allosteric interactions between the R domain and the membrane domains and/or intracellular loops [57]. Future studies are required to understand the molecular basis for such ATP-independent gating and its regulation.
Conformational changes underlying phosphorylation-regulated channel activation
As a result of elegant biochemical, structural and computational studies, the R domain has been modeled as a flexible, disordered region of the protein that undergoes dynamic interactions with other CFTR domains [58, 59]. Although the R domain is disordered, it possesses regions with the propensity to form alpha helices in the non-phosphorylated state [59–62]. PKA phosphorylation was found to reduce this propensity: results from biophysical and computational studies suggest that the isolated R domain becomes less compact with phosphorylation [59–62]. There is a growing consensus that these phosphorylation-dependent dynamic changes in the R domain allosterically modulates intramolecular interactions in CFTR that are critical for its activity as an anion channel [59–64]. In the next paragraphs, we will discuss studies of phosphorylation-mediated changes in domain:domain interactions in the context of isolated domains and in the context of the full-length protein.
Phosphorylation-regulated changes in NBD1 interactions
Although the majority of the canonical, dibasic PKA sites are located within the R domain of CFTR, there is one phosphorylation site in the RI of NBD1, S422. S660 is located in the amino-terminus of the R domain, but was modeled in X-ray crystallography studies of an NBD1 fragment described as the RE region [65]. Previous NMR studies on isolated NBD1 and regions of the R domain reported that in the closed CFTR state, the RE and RI regions create steric hindrances on NBD1 which prevent it from interacting with NBD2, ICL1 and ICL4 (Fig. 2) [59–62]. These NMR studies found that upon phosphorylation of S422 and S660, the alpha helical content of the RI and RE decreases respectively, removing their steric hindrances on NBD1 and increasing interactions at the NBD1:NBD2, ICL1:NBD1 and ICL4:NBD1 interfaces [59]. Interactions at the NBD1:NBD2 interface are essential for the formation of the two ATP-binding pockets; interactions at the ICL:NBD1 interfaces may be important for relaying conformational changes from the NBDs to the MSDs upon ATP binding and hydrolysis [59, 60, 62]. Taken together, these interactions are necessary for channel gating and may also contribute to the stability of full-length CFTR [59, 60, 62].
Phosphorylation-regulated changes in intramolecular interactions
Changes in the affinity of domain:domain interactions in the context of the full-length protein have been assessed by chemical cross-linking, peptide-mediated competition of gating transitions and electron microscopy studies of the purified protein. Consistent with NMR studies discussed in the previous section, results from cysteine cross-linking studies suggest that phosphorylation enhances interactions at the NBD1:NBD2 interface [66, 67] and promotes direct interactions between specific regions of the R domain and structure(s) in the membrane domains (the amino- and/or the carboxy- terminal[s]) [68–70]. Electron microscopy studies also show that full-length CFTR protein becomes more compact after phosphorylation, supporting the idea that this modification regulates intramolecular interactions [70].
Cysteine cross-linking studies have found that NBD1 interacts with ICL1 (MSD1) and ICL4 (MSD2), while NBD2 interacts with ICL2 (MSD1) and ICL3 (MSD2) [48, 67]. However, these studies were unable to detect any changes at those interfaces upon PKA phosphorylation [67]. The failure to detect changes in cysteine cross-linking suggests that the ICL:NBD1 or ICL:NBD2 interfaces may interact regardless of phosphorylation state; alternatively, phosphorylation may cause subtle conformational changes at those interfaces during channel gating, too subtle for cysteine cross-linking methods to detect [67]. Interestingly, single channel studies have found that cysteine cross-linking at the domain-swapping interfaces (ICL4:NBD1 and ICL2:NBD2) inhibited phosphorylation-dependent channel gating, whereas cross-linking at the interfaces of the same half of the protein (ICL1:NBD1 and ICL3:NBD2) did not [48, 67]. Furthermore, Ehrhardt and colleagues reported that competition with an ICL1 peptide for the native ICL1:NBD1 interaction abolished channel gating and ATPase activity [71], suggesting that this interaction exerts a positive effect on ATP-dependent gating in the native protein. Together, these studies suggest that dynamic interactions at the ICL:NBD interfaces are essential for phosphorylation-dependent channel gating, yet uncertainty remains regarding the relative role of these interactions in channel opening and closing.
The ICLs of CFTR may also interact with each other as there were reports of an ICL tetrahelix bundle based on homology modeling and cysteine cross-linking studies [48, 72]. Recent cysteine cross-linking studies have found that E267 (ICL2) and K1060 (ICL4) interact with each other; both residues are also in close proximity to the NBDs and CHs and thus may be involved in conveying conformational changes to the MSDs for channel gating [72]. Switching the charge of E267 and K1060 or cross-linking the cysteine pair (E267C/K1060C) abolished PKA sensitivity and CFTR gating, suggesting that helix bundle interactions may be important for mediating phosphorylation-dependent effects [72].
Molecular consequences of disease-causing mutations and impact on regulation by PKA-dependent phosphorylation
G551D-CFTR
The G551D mutation at NBD1 results in defective channel gating but does not affect trafficking of the mutant protein to the cell surface [73]. Our iodide efflux studies with purified and reconstituted full-length G551D-CFTR protein show that the mutant displays defective phosphorylation- and ATP-dependent gating [74, 75]. The dysfunction of G551D-CFTR may be due to the mutation disrupting ATP binding at site 2, as ATPase activity assays from our laboratory show that the G551D mutation significantly decreases the apparent affinity of CFTR for ATP [75]. However, it was reported that mutation of the G551 residue in NBD1 of the human multidrug resistance protein 1 (a similar ABC transporter) does not affect ATP binding; an alternative explanation is that the mutation may prevent conformational changes upon ATP binding at the NBD1:NBD2 interface [40, 76]. Furthermore, G551D may prevent phosphorylation from enhancing interactions at the NBD1:NBD2 interface which may explain the defective channel gating and decreased apparent ATP affinity of CFTR. However, additional studies must be conducted to test this hypothesis.
F508del-CFTR
The major CF-causing mutation, F508del, causes destabilization of NBD1 and misassembly of ICL:NBD interactions [4, 5]. These intrinsic molecular defects disrupt protein assembly and trafficking, resulting in ER-associated degradation of the mutant protein [77–81]. The limited number of mutant proteins that reach the cell surface are intrinsically unstable at 37 degrees Celsius (°C), exhibit altered channel function and are subject to retrieval from the cell surface and disposal by lysosomal degradation (Fig. 1) [82, 83]. In the following paragraphs, we will discuss how phosphorylation-dependent regulation of trafficking, surface stability and function is disrupted in F508del-CFTR.
F508del alters the intrinsic propensity of CFTR for phosphorylation
Our group has shown that F508del-CFTR can be phosphorylated at the ER as detected by immunoblotting and immunofluorescence microscopy [8]. Selected reaction monitoring-mass spectrometry (SRM–MS) detected that phosphorylation of S660 at the RE was significantly reduced in immuno-purified F508del-CFTR when compared to immuno-purified Wt-CFTR (Fig. 2) [8]. We interpreted the reduced propensity for phosphorylation at this site (and possibly other PKA consensus sites) to reflect the multiple conformational defects induced by this deletion mutation.
The defective propensity for phosphorylation at S660 may be related to aberrant intramolecular interactions between NBD1 and NBD2 or between NBD1 and CHs conferred by ICL1 and/or ICL4 (Fig. 2). Such aberrant interactions were previously determined in NMR studies of protein fragments and chemical cross-linking studies [60, 84, 85]. NMR studies of isolated F508del-NBD1 bearing stabilizing mutations revealed an increased affinity between the phospho-regulated-RI and the core of NBD1 [60]. Deletion of the RI partially restored intramolecular interactions at the ICL4:NBD1 and ICL2:NBD2 interfaces and improved F508del-CFTR channel activation [86]. Hence, the effect of the F508del mutation on these interactions that lead to reduced rates of phosphorylation may contribute to defective trafficking and slower rates of phosphorylation-dependent channel activation [87, 88]. Unfortunately, we could not monitor the phosphorylation propensity of S422 in the RI with our SRM-MS studies due to its weak signal. It would have been particularly interesting to determine if phosphorylation at S422 is also reduced in the F508del-CFTR protein given the proposed relevance of enhanced RI:NBD1 affinity in preventing normal assembly of this mutant [86].
F508del-CFTR protein targeted to ER from the Golgi via retrograde trafficking: a potential consequence of aberrant phosphorylation
A small population of F508del-CFTR can escape ER quality control and reach the cell surface; however, the conformational defects of the mutant protein can lead to the aberrant exposure of dibasic “retention motifs” (RXR) which promote re-direction of F508del-CFTR from the Golgi back to the ER (Fig. 1) [89–91]. As previously discussed, COPI-mediated trafficking is competed by phosphorylation-regulated interactions between CFTR and 14-3-3 [18]. Due to the limitations of the amount of F508del-CFTR that could be studied by SRM-MS in our previous work, we could not monitor the effect of this mutation on the sites that interact with 14-3-3 (e.g., pS768 and pS753). Hence, it remains unclear whether a defective interaction between F508del-CFTR and 14-3-3 is a result of defective phosphorylation at these sites.
Cell surface F508del-CFTR protein is targeted to the lysosomes and exhibits defective phosphorylation-regulated endosomal recycling
A small population of F508del-CFTR may escape ER retention (this amount is increased at low temperature culture conditions) to reach the cell surface; however, the mutant protein is rapidly removed by peripheral protein quality control, primarily via endocytosis and delivery to lysosomes (Fig. 1). The loss of F508del-CFTR may be due to its inherent thermal instability and reduced interactions with the NHERF1–ezrin–actin complex [82]. As previously mentioned, the NHERF1–ezrin–actin complex is important for the localization, stability and PKA-regulated channel activity of CFTR at the cell surface [28, 92]. In addition, the fraction of F508del-CFTR that is delivered to the recycling endosomal compartment from the cell surface exhibits a defect in PKA-regulated exocytosis [31]. Taken together, defective endosomal trafficking by F508del-CFTR partially reflects its altered regulation by PKA phosphorylation [31].
Impact of mutation-targeted therapies on phosphorylation-dependent channel gating
Correctors
Low temperature culture conditions (i.e., 27 °C) partially rescue the misassembly and mistrafficking of F508del-CFTR [93]. Similarly, small molecule corrector compounds have been identified to partially recapitulate this rescue effect [4, 5, 94–96]. The mutant protein that is “corrected” by low temperature incubation at the plasma membrane exhibits a decline in surface expression and channel open probability with increasing temperatures as measured in single channel studies [5]. Interestingly, treatment with the corrector VX-809 or its analog, C18, maintained the open probability of F508del-CFTR with increasing temperatures [5]. Previous studies have suggested that correctors may rescue phosphorylation-dependent channel gating of F508del-CFTR by either stabilizing intramolecular interactions at the ICL:NBD interfaces [4, 5] or by enhancing interactions with the NHERF1–ezrin–actin complex [97] that stabilize cell surface expression. Interestingly, SRM-MS studies from our laboratory showed that the corrector C18 does not significantly rescue the intrinsic phosphorylation defect exhibited by F508del-CFTR, supporting the idea that protein abundance rather than the specific, phosphorylation-regulated activity of each mutant protein is enhanced by C18 and related correctors [8].
Potentiators
The potentiator, VX-770, was shown to enhance channel activity of the gating mutant, G551D-CFTR, and F508del-CFTR (after rescue of its trafficking defect) [98]. Interestingly, and implicit in its potentiation of the ATP-binding mutant, G551D, VX-770 can potentiate CFTR in an ATP-independent manner [74, 99]. Hwang and colleagues proposed that VX-770 may bind to the transmembrane regions of CFTR and shift the equilibrium from the post-hydrolytic state towards the pre-hydrolytic stage, increasing the opening time of CFTR by allowing ATP molecules to rebind to site 2 [99]. Our iodide efflux studies of purified and reconstituted CFTR protein show that VX-770 directly binds to full-length CFTR; the addition of VX-770 led to a two-fold increase in the iodide efflux rate in proteoliposomes containing PKA-phosphorylated CFTR, but had no effect on non-phosphorylated CFTR. We interpreted these findings as suggesting that VX-770 potentiation is phosphorylation dependent [74]. On the contrary, another group reported that VX-770 can enhance the activity of CFTR with the R domain removed, suggesting that VX-770 does not act on the R domain and its activity is phosphorylation independent [99]. Alternatively, we suggest that the R domain deletion mutant may mimic the effects of R domain phosphorylation, a hypothesis that we will be testing in the future.
Targeting PKA-dependent signaling as a therapeutic strategy
Targeting PKA-dependent phosphorylation to rescue mistrafficking of F508del-CFTR
Studies have shown that targeting cAMP/PKA phosphorylation can promote the interaction of F508del-CFTR with 14-3-3 (β and ε isoforms), enhancing the forward trafficking of the mutant protein and stabilizing it at the cell surface [18]. Recent fluorescence polarization studies have found that the drug fusicoccin-A, a phytotoxin produced by the fungus Phomopsis amygdali, significantly increased binding of the pS753 epitope in a R domain peptide of CFTR to 14-3-3 [20]. Hence, modification of the PKA-dependent interaction between F508del-CFTR and 14-3-3 may constitute a potential therapeutic target for rescuing F508del-CFTR trafficking.
Targeting PKA-dependent phosphorylation to rescue defects in stability and dysfunction of F508del-CFTR after correction of its mistrafficking defect
The groups of Loureiro and Abbattiscianni have assessed the impact of modulating the phosphorylation-regulated interaction between F508del-CFTR and the NHERF1–ezrin–actin complex in enhancing the functional expression of the mutant after correction of its trafficking defect [97, 100]. These groups showed that phosphorylation of ezrin enhanced the interaction of F508del-CFTR with NHERF1 and actin [97]. Further, ezrin phosphorylation was found to increase actin polymerization and stabilize F508del-CFTR at the cell surface of CFBE cells, possibly by preventing its recognition by the peripheral protein quality control [97, 100].
The concentration of intracellular cAMP within CFTR-containing microdomains is regulated by proximal phosphodiesterases (PDEs). A recent study introduced the application of cyclic nucleotide PDE inhibitors to rescue the dysfunction of F508del-CFTR [101]. The PDE inhibitor, RPL554, was shown to increase CFTR activity in CFBE cells [101]. RPL554 and other PDE inhibitors, milrinone and rolipram, were also found to significantly enhance VX-770 potentiation in CFBE cells [101]. Interestingly, this study has found that RPL554 may be therapeutically beneficial for CF as it significantly increased cilia beating for mucociliary clearance, possibly by enhancing PKA phosphorylation of the dynein light chains of cilia [101].
In addition, the cAMP concentrations proximal to CFTR are regulated by the activity of the multidrug resistance protein 4 (MRP4), another ABC protein which has been shown to directly mediate the efflux of cAMP [102–106]. Inhibition of MRP4 using MK-571 led to a localized increase in cAMP evoked by addition of the beta-adrenergic agonist, adenosine, resulting in an increase in Wt-CFTR channel function in the apical membrane [104]. Future studies are required to evaluate the potential for MRP4 inhibition to enhance the rescue activity of modulators targeting mutant forms of CFTR after their delivery to the cell surface.
Concluding statements
In summary, PKA phosphorylation modifies multiple steps in the life cycle of the CFTR protein, including its biosynthesis, trafficking, cell surface stability and function. Disease-causing mutations exhibit defects in one or more of these steps, and the therapeutic potential of targeting PKA phosphorylation to ameliorate these defects is being explored. Given the importance of the location of CFTR in apical macromolecular complexes for robust regulation by PKA, therapies targeting F508del-CFTR modification by PKA will need to be added in concert with other interventions aimed at first promoting proper assembly and forward trafficking of CFTR out of the ER. Finally, phosphorylation by PKA and possibly other kinases offer a tremendous potential to augment the therapeutic efficacy of CFTR modulators.
Acknowledgments
S. Chin was supported by a NSERC scholarship (PGS-D) and the research activities in the Bear Lab supported in part by the Canadian Institutes of Health Research and Cystic Fibrosis Canada.
References
- 1.Billet A, et al. Potential sites of CFTR activation by tyrosine kinases. Channels (Austin) 2016;10(3):247–251. doi: 10.1080/19336950.2015.1126010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Billet A, et al. Regulation of the cystic fibrosis transmembrane conductance regulator anion channel by tyrosine phosphorylation. Faseb J. 2015;29(9):3945–3953. doi: 10.1096/fj.15-273151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Billet A, Hanrahan JW. The secret life of CFTR as a calcium-activated chloride channel. J Physiol. 2013;591(21):5273–5278. doi: 10.1113/jphysiol.2013.261909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabeh WM, et al. Correction of both NBD1 energetics and domain interface is required to restore DeltaF508 CFTR folding and function. Cell. 2012;148(1–2):150–163. doi: 10.1016/j.cell.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okiyoneda T, et al. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat Chem Biol. 2013;9(7):444–454. doi: 10.1038/nchembio.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDonald RA, et al. Basal expression of the cystic fibrosis transmembrane conductance regulator gene is dependent on protein kinase A activity. Proc Natl Acad Sci U S A. 1995;92(16):7560–7564. doi: 10.1073/pnas.92.16.7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matthews RP, McKnight GS. Characterization of the cAMP response element of the cystic fibrosis transmembrane conductance regulator gene promoter. J Biol Chem. 1996;271(50):31869–31877. doi: 10.1074/jbc.271.50.31869. [DOI] [PubMed] [Google Scholar]
- 8.Pasyk S, et al. The major cystic fibrosis causing mutation exhibits defective propensity for phosphorylation. Proteomics. 2015;15(2–3):447–461. doi: 10.1002/pmic.201400218. [DOI] [PubMed] [Google Scholar]
- 9.Gadsby DC, Nairn AC. Regulation of CFTR channel gating. Trends Biochem Sci. 1994;19(11):513–518. doi: 10.1016/0968-0004(94)90141-4. [DOI] [PubMed] [Google Scholar]
- 10.Csanady L, et al. Preferential phosphorylation of R-domain Serine 768 dampens activation of CFTR channels by PKA. J Gen Physiol. 2005;125(2):171–186. doi: 10.1085/jgp.200409076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng SH, et al. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell. 1991;66(5):1027–1036. doi: 10.1016/0092-8674(91)90446-6. [DOI] [PubMed] [Google Scholar]
- 12.Chang XB, et al. Protein kinase A (PKA) still activates CFTR chloride channel after mutagenesis of all 10 PKA consensus phosphorylation sites. J Biol Chem. 1993;268(15):11304–11311. [PubMed] [Google Scholar]
- 13.Seibert FS, et al. Influence of phosphorylation by protein kinase A on CFTR at the cell surface and endoplasmic reticulum. Biochim Biophys Acta. 1999;1461(2):275–283. doi: 10.1016/S0005-2736(99)00163-7. [DOI] [PubMed] [Google Scholar]
- 14.Wilkinson DJ, et al. CFTR activation: additive effects of stimulatory and inhibitory phosphorylation sites in the R domain. Am J Physiol. 1997;273(1 Pt 1):L127–L133. doi: 10.1152/ajplung.1997.273.1.L127. [DOI] [PubMed] [Google Scholar]
- 15.Hegedus T, et al. Role of individual R domain phosphorylation sites in CFTR regulation by protein kinase A. Biochim Biophys Acta. 2009;1788(6):1341–1349. doi: 10.1016/j.bbamem.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 16.Neville DC, et al. Evidence for phosphorylation of serine 753 in CFTR using a novel metal-ion affinity resin and matrix-assisted laser desorption mass spectrometry. Protein Sci. 1997;6(11):2436–2445. doi: 10.1002/pro.5560061117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McClure M, et al. Purification of CFTR for mass spectrometry analysis: identification of palmitoylation and other post-translational modifications. Protein Eng Des Sel. 2012;25(1):7–14. doi: 10.1093/protein/gzr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang X, et al. Phosphorylation-dependent 14-3-3 protein interactions regulate CFTR biogenesis. Mol Biol Cell. 2012;23(6):996–1009. doi: 10.1091/mbc.E11-08-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rennolds J, et al. Cystic fibrosis transmembrane conductance regulator trafficking is mediated by the COPI coat in epithelial cells. J Biol Chem. 2008;283(2):833–839. doi: 10.1074/jbc.M706504200. [DOI] [PubMed] [Google Scholar]
- 20.Stevers LM, et al. Characterization and small-molecule stabilization of the multisite tandem binding between 14-3-3 and the R domain of CFTR. Proc Natl Acad Sci U S A. 2016;113(9):E1152–E1161. doi: 10.1073/pnas.1516631113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cantiello HF. Role of the actin cytoskeleton in the regulation of the cystic fibrosis transmembrane conductance regulator. Exp Physiol. 1996;81(3):505–514. doi: 10.1113/expphysiol.1996.sp003953. [DOI] [PubMed] [Google Scholar]
- 22.Louvet-Vallee S. ERM proteins: from cellular architecture to cell signaling. Biol Cell. 2000;92(5):305–316. doi: 10.1016/S0248-4900(00)01078-9. [DOI] [PubMed] [Google Scholar]
- 23.Naren AP, et al. A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc Natl Acad Sci U S A. 2003;100(1):342–346. doi: 10.1073/pnas.0135434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li C, et al. Molecular assembly of cystic fibrosis transmembrane conductance regulator in plasma membrane. J Biol Chem. 2004;279(23):24673–24684. doi: 10.1074/jbc.M400688200. [DOI] [PubMed] [Google Scholar]
- 25.Moyer BD, et al. The PDZ-interacting domain of cystic fibrosis transmembrane conductance regulator is required for functional expression in the apical plasma membrane. J Biol Chem. 2000;275(35):27069–27074. doi: 10.1074/jbc.M004951200. [DOI] [PubMed] [Google Scholar]
- 26.Short DB, et al. An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton. J Biol Chem. 1998;273(31):19797–19801. doi: 10.1074/jbc.273.31.19797. [DOI] [PubMed] [Google Scholar]
- 27.Sun F, et al. E3KARP mediates the association of ezrin and protein kinase A with the cystic fibrosis transmembrane conductance regulator in airway cells. J Biol Chem. 2000;275(38):29539–29546. doi: 10.1074/jbc.M004961200. [DOI] [PubMed] [Google Scholar]
- 28.Monterisi S, et al. CFTR regulation in human airway epithelial cells requires integrity of the actin cytoskeleton and compartmentalized cAMP and PKA activity. J Cell Sci. 2012;125(Pt 5):1106–1117. doi: 10.1242/jcs.089086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alshafie W, et al. VIP regulates CFTR membrane expression and function in Calu-3 cells by increasing its interaction with NHERF1 and P-ERM in a VPAC1- and PKCepsilon-dependent manner. Am J Physiol Cell Physiol. 2014;307(1):C107–C119. doi: 10.1152/ajpcell.00296.2013. [DOI] [PubMed] [Google Scholar]
- 30.Lobo MJ, et al. EPAC1 activation by cAMP stabilizes CFTR at the membrane by promoting its interaction with NHERF1. J Cell Sci. 2016;129(13):2599–2612. doi: 10.1242/jcs.185629. [DOI] [PubMed] [Google Scholar]
- 31.Holleran JP, et al. Regulated recycling of mutant CFTR is partially restored by pharmacological treatment. J Cell Sci. 2013;126(Pt 12):2692–2703. doi: 10.1242/jcs.120196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bear CE, et al. Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR) Cell. 1992;68(4):809–818. doi: 10.1016/0092-8674(92)90155-6. [DOI] [PubMed] [Google Scholar]
- 33.Aleksandrov AA, Cui L, Riordan JR. Relationship between nucleotide binding and ion channel gating in cystic fibrosis transmembrane conductance regulator. J Physiol. 2009;587(Pt 12):2875–2886. doi: 10.1113/jphysiol.2009.170258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou Z, et al. The two ATP binding sites of cystic fibrosis transmembrane conductance regulator (CFTR) play distinct roles in gating kinetics and energetics. J Gen Physiol. 2006;128(4):413–422. doi: 10.1085/jgp.200609622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vergani P, et al. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature. 2005;433(7028):876–880. doi: 10.1038/nature03313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aleksandrov AA, Aleksandrov LA, Riordan JR. CFTR (ABCC7) is a hydrolyzable-ligand-gated channel. Pflugers Arch. 2007;453(5):693–702. doi: 10.1007/s00424-006-0140-z. [DOI] [PubMed] [Google Scholar]
- 37.Gadsby DC, Vergani P, Csanady L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440(7083):477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheung JC, et al. Molecular basis for the ATPase activity of CFTR. Arch Biochem Biophys. 2008;476(1):95–100. doi: 10.1016/j.abb.2008.03.033. [DOI] [PubMed] [Google Scholar]
- 39.Scott-Ward TS, et al. Chimeric constructs endow the human CFTR Cl- channel with the gating behavior of murine CFTR. Proc Natl Acad Sci U S A. 2007;104(41):16365–16370. doi: 10.1073/pnas.0701562104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bompadre SG, et al. G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects. J Gen Physiol. 2007;129(4):285–298. doi: 10.1085/jgp.200609667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramjeesingh M, et al. The intact CFTR protein mediates ATPase rather than adenylate kinase activity. Biochem J. 2008;412(2):315–321. doi: 10.1042/BJ20071719. [DOI] [PubMed] [Google Scholar]
- 42.Stratford FL, et al. The Walker B motif of the second nucleotide-binding domain (NBD2) of CFTR plays a key role in ATPase activity by the NBD1-NBD2 heterodimer. Biochem J. 2007;401(2):581–586. doi: 10.1042/BJ20060968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kogan I, et al. Studies of the molecular basis for cystic fibrosis using purified reconstituted CFTR protein. Methods Mol Med. 2002;70:143–157. doi: 10.1385/1-59259-187-6:143. [DOI] [PubMed] [Google Scholar]
- 44.Kogan I, et al. Perturbation of the pore of the cystic fibrosis transmembrane conductance regulator (CFTR) inhibits its atpase activity. J Biol Chem. 2001;276(15):11575–11581. doi: 10.1074/jbc.M010403200. [DOI] [PubMed] [Google Scholar]
- 45.Kidd JF, et al. A heteromeric complex of the two nucleotide binding domains of cystic fibrosis transmembrane conductance regulator (CFTR) mediates ATPase activity. J Biol Chem. 2004;279(40):41664–41669. doi: 10.1074/jbc.M407666200. [DOI] [PubMed] [Google Scholar]
- 46.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443(7108):180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 47.Locher KP. Review. Structure and mechanism of ATP-binding cassette transporters. Philos Trans R Soc Lond B Biol Sci. 2009;364(1514):239–245. doi: 10.1098/rstb.2008.0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Serohijos AW, et al. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci U S A. 2008;105(9):3256–3261. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mornon JP, Lehn P, Callebaut I. Atomic model of human cystic fibrosis transmembrane conductance regulator: membrane-spanning domains and coupling interfaces. Cell Mol Life Sci. 2008;65(16):2594–2612. doi: 10.1007/s00018-008-8249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winter MC, Welsh MJ. Stimulation of CFTR activity by its phosphorylated R domain. Nature. 1997;389(6648):294–296. doi: 10.1038/38514. [DOI] [PubMed] [Google Scholar]
- 51.Hwang TC, Sheppard DN. Gating of the CFTR Cl- channel by ATP-driven nucleotide-binding domain dimerisation. J Physiol. 2009;587(Pt 10):2151–2161. doi: 10.1113/jphysiol.2009.171595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mihalyi C, Torocsik B, Csanady L. Obligate coupling of CFTR pore opening to tight nucleotide-binding domain dimerization. Elife. 2016;5:e18164. doi: 10.7554/eLife.18164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li C, Ramjeesingh M, Bear CE. Purified cystic fibrosis transmembrane conductance regulator (CFTR) does not function as an ATP channel. J Biol Chem. 1996;271(20):11623–11626. doi: 10.1074/jbc.271.20.11623. [DOI] [PubMed] [Google Scholar]
- 54.Szellas T, Nagel G. Apparent affinity of CFTR for ATP is increased by continuous kinase activity. FEBS Lett. 2003;535(1–3):141–146. doi: 10.1016/S0014-5793(02)03892-9. [DOI] [PubMed] [Google Scholar]
- 55.Li C, et al. ATPase activity of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1996;271(45):28463–28468. doi: 10.1074/jbc.271.45.28463. [DOI] [PubMed] [Google Scholar]
- 56.Zwick M, et al. How phosphorylation and ATPase activity regulate anion flux though the cystic fibrosis transmembrane conductance regulator (CFTR) J Biol Chem. 2016;291(28):14483–14498. doi: 10.1074/jbc.M116.721415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang W, et al. ATP-independent CFTR channel gating and allosteric modulation by phosphorylation. Proc Natl Acad Sci U S A. 2010;107(8):3888–3893. doi: 10.1073/pnas.0913001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ostedgaard LS, Baldursson O, Welsh MJ. Regulation of the cystic fibrosis transmembrane conductance regulator Cl- channel by its R domain. J Biol Chem. 2001;276(11):7689–7692. doi: 10.1074/jbc.R100001200. [DOI] [PubMed] [Google Scholar]
- 59.Baker JM, et al. CFTR regulatory region interacts with NBD1 predominantly via multiple transient helices. Nat Struct Mol Biol. 2007;14(8):738–745. doi: 10.1038/nsmb1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kanelis V, et al. NMR evidence for differential phosphorylation-dependent interactions in WT and DeltaF508 CFTR. EMBO J. 2010;29(1):263–277. doi: 10.1038/emboj.2009.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hudson RP, et al. Conformational changes relevant to channel activity and folding within the first nucleotide binding domain of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2012;287(34):28480–28494. doi: 10.1074/jbc.M112.371138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dawson JE, Farber PJ, Forman-Kay JD. Allosteric coupling between the intracellular coupling helix 4 and regulatory sites of the first nucleotide-binding domain of CFTR. PLoS One. 2013;8(9):e74347. doi: 10.1371/journal.pone.0074347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grimard V, et al. Phosphorylation-induced conformational changes of cystic fibrosis transmembrane conductance regulator monitored by attenuated total reflection-Fourier transform IR spectroscopy and fluorescence spectroscopy. J Biol Chem. 2004;279(7):5528–5536. doi: 10.1074/jbc.M311014200. [DOI] [PubMed] [Google Scholar]
- 64.Chappe V, et al. Phosphorylation of CFTR by PKA promotes binding of the regulatory domain. EMBO J. 2005;24(15):2730–2740. doi: 10.1038/sj.emboj.7600747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lewis HA, et al. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 2004;23(2):282–293. doi: 10.1038/sj.emboj.7600040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mense M, et al. In vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain heterodimer. EMBO J. 2006;25(20):4728–4739. doi: 10.1038/sj.emboj.7601373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He L, et al. Multiple membrane-cytoplasmic domain contacts in the cystic fibrosis transmembrane conductance regulator (CFTR) mediate regulation of channel gating. J Biol Chem. 2008;283(39):26383–26390. doi: 10.1074/jbc.M803894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fu J, Kirk KL. Cysteine substitutions reveal dual functions of the amino-terminal tail in cystic fibrosis transmembrane conductance regulator channel gating. J Biol Chem. 2001;276(38):35660–35668. doi: 10.1074/jbc.M105079200. [DOI] [PubMed] [Google Scholar]
- 69.Naren AP, et al. CFTR chloride channel regulation by an interdomain interaction. Science. 1999;286(5439):544–548. doi: 10.1126/science.286.5439.544. [DOI] [PubMed] [Google Scholar]
- 70.Bertrand CA, et al. SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J Gen Physiol. 2009;133(4):421–438. doi: 10.1085/jgp.200810097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ehrhardt A, et al. Channel gating regulation by the cystic fibrosis transmembrane conductance regulator (CFTR) first cytosolic loop. J Biol Chem. 2016;291(4):1854–1865. doi: 10.1074/jbc.M115.704809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang W, Roessler BC, Kirk KL. An electrostatic interaction at the tetrahelix bundle promotes phosphorylation-dependent cystic fibrosis transmembrane conductance regulator (CFTR) channel opening. J Biol Chem. 2014;289(44):30364–30378. doi: 10.1074/jbc.M114.595710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gregory RJ, et al. Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide-binding domains 1 and 2. Mol Cell Biol. 1991;11(8):3886–3893. doi: 10.1128/MCB.11.8.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Eckford PD, et al. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiator VX-770 (ivacaftor) opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner. J Biol Chem. 2012;287(44):36639–36649. doi: 10.1074/jbc.M112.393637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pasyk S, et al. Direct interaction of a small-molecule modulator with G551D-CFTR, a cystic fibrosis-causing mutation associated with severe disease. Biochem J. 2009;418(1):185–190. doi: 10.1042/BJ20081424. [DOI] [PubMed] [Google Scholar]
- 76.Ren XQ, et al. Function of the ABC signature sequences in the human multidrug resistance protein 1. Mol Pharmacol. 2004;65(6):1536–1542. doi: 10.1124/mol.65.6.1536. [DOI] [PubMed] [Google Scholar]
- 77.Riordan JR. Assembly of functional CFTR chloride channels. Annu Rev Physiol. 2005;67:701–718. doi: 10.1146/annurev.physiol.67.032003.154107. [DOI] [PubMed] [Google Scholar]
- 78.Younger JM, et al. A foldable CFTR{Delta}F508 biogenic intermediate accumulates upon inhibition of the Hsc70-CHIP E3 ubiquitin ligase. J Cell Biol. 2004;167(6):1075–1085. doi: 10.1083/jcb.200410065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Younger JM, et al. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126(3):571–582. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 80.Farinha CM, Amaral MD. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol Cell Biol. 2005;25(12):5242–5252. doi: 10.1128/MCB.25.12.5242-5252.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Okiyoneda T, et al. Delta F508 CFTR pool in the endoplasmic reticulum is increased by calnexin overexpression. Mol Biol Cell. 2004;15(2):563–574. doi: 10.1091/mbc.E03-06-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Okiyoneda T, et al. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science. 2010;329(5993):805–810. doi: 10.1126/science.1191542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gentzsch M, et al. Endocytic trafficking routes of wild type and DeltaF508 cystic fibrosis transmembrane conductance regulator. Mol Biol Cell. 2004;15(6):2684–2696. doi: 10.1091/mbc.E04-03-0176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Loo TW, Bartlett MC, Clarke DM. Processing mutations disrupt interactions between the nucleotide binding and transmembrane domains of P-glycoprotein and the cystic fibrosis transmembrane conductance regulator (CFTR) J Biol Chem. 2008;283(42):28190–28197. doi: 10.1074/jbc.M805834200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.He L, et al. Correctors of DeltaF508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. 2013;27(2):536–545. doi: 10.1096/fj.12-216119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aleksandrov AA, et al. Regulatory insertion removal restores maturation, stability and function of DeltaF508 CFTR. J Mol Biol. 2010;401(2):194–210. doi: 10.1016/j.jmb.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang F, et al. Deletion of phenylalanine 508 causes attenuated phosphorylation-dependent activation of CFTR chloride channels. J Physiol. 2000;524(Pt 3):637–648. doi: 10.1111/j.1469-7793.2000.00637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Drumm ML, et al. Chloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytes . Science. 1991;254(5039):1797–1799. doi: 10.1126/science.1722350. [DOI] [PubMed] [Google Scholar]
- 89.Hegedus T, et al. F508del CFTR with two altered RXR motifs escapes from ER quality control but its channel activity is thermally sensitive. Biochim Biophys Acta. 2006;1758(5):565–572. doi: 10.1016/j.bbamem.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 90.Kim Chiaw P, et al. Functional rescue of DeltaF508-CFTR by peptides designed to mimic sorting motifs. Chem Biol. 2009;16(5):520–530. doi: 10.1016/j.chembiol.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 91.Chang XB, et al. Removal of multiple arginine-framed trafficking signals overcomes misprocessing of delta F508 CFTR present in most patients with cystic fibrosis. Mol Cell. 1999;4(1):137–142. doi: 10.1016/S1097-2765(00)80196-3. [DOI] [PubMed] [Google Scholar]
- 92.Favia M, et al. Na +/H + exchanger regulatory factor 1 overexpression-dependent increase of cytoskeleton organization is fundamental in the rescue of F508del cystic fibrosis transmembrane conductance regulator in human airway CFBE41o- cells. Mol Biol Cell. 2010;21(1):73–86. doi: 10.1091/mbc.E09-03-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Denning GM, et al. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358(6389):761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 94.Farinha CM, et al. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem Biol. 2013;20(7):943–955. doi: 10.1016/j.chembiol.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 95.Pedemonte N, et al. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest. 2005;115(9):2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Van Goor F, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108(46):18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Abbattiscianni AC, et al. Correctors of mutant CFTR enhance subcortical cAMP-PKA signaling through modulating ezrin phosphorylation and cytoskeleton organization. J Cell Sci. 2016;129(6):1128–1140. doi: 10.1242/jcs.177907. [DOI] [PubMed] [Google Scholar]
- 98.Van Goor F, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009;106(44):18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jih KY, Hwang TC. Vx-770 potentiates CFTR function by promoting decoupling between the gating cycle and ATP hydrolysis cycle. Proc Natl Acad Sci U S A. 2013;110(11):4404–4409. doi: 10.1073/pnas.1215982110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Loureiro CA, et al. A molecular switch in the scaffold NHERF1 enables misfolded CFTR to evade the peripheral quality control checkpoint. Sci Signal. 2015;8(377):ra48. doi: 10.1126/scisignal.aaa1580. [DOI] [PubMed] [Google Scholar]
- 101.Turner MJ, et al. The dual phosphodiesterase 3 and 4 inhibitor RPL554 stimulates CFTR and ciliary beating in primary cultures of bronchial epithelia. Am J Physiol Lung Cell Mol Physiol. 2016;310(1):L59–L70. doi: 10.1152/ajplung.00324.2015. [DOI] [PubMed] [Google Scholar]
- 102.Li C, Naren AP. Analysis of CFTR interactome in the macromolecular complexes. Methods Mol Biol. 2011;741:255–270. doi: 10.1007/978-1-61779-117-8_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li C, Naren AP. CFTR chloride channel in the apical compartments: spatiotemporal coupling to its interacting partners. Integr Biol (Camb) 2010;2(4):161–177. doi: 10.1039/b924455g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Moon C, et al. Compartmentalized accumulation of cAMP near complexes of multidrug resistance protein 4 (MRP4) and cystic fibrosis transmembrane conductance regulator (CFTR) contributes to drug-induced diarrhea. J Biol Chem. 2015;290(18):11246–11257. doi: 10.1074/jbc.M114.605410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li C, et al. Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell. 2007;131(5):940–951. doi: 10.1016/j.cell.2007.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Arora K, Naren AP. Pharmacological correction of cystic fibrosis: molecular mechanisms at the plasma membrane to augment mutant CFTR function. Curr Drug Targets. 2016;17(11):1275–1281. doi: 10.2174/1389450117666151209114343. [DOI] [PubMed] [Google Scholar]