

Abstract
Duchenne muscular dystrophy (DMD) is a progressive muscle wasting disease caused by the absence of dystrophin, a membrane-stabilizing protein encoded by the DMD gene. Although mouse models of DMD provide insight into the potential of a corrective therapy, data from genetically homologous large animals, such as the dystrophin-deficient golden retriever muscular dystrophy (GRMD) model, may more readily translate to humans. To evaluate the clinical translatability of an adeno-associated virus serotype 9 vector (AAV-9)-microdystrophin (μDys5) construct, we performed a blinded, placebo-controlled study in which 12 GRMD dogs were divided among four dose groups [control, 1E13 vector genomes per kilogram (vg/kg), 1E14 vg/kg, and 2E14 vg/kg; n = 3 each], treated intravenously at 3 months of age with a canine codon-optimized microdystrophin construct, rAAV9-CK8e-c-μDys5, and followed for 90 days after dosing. All dogs received 1 mg/kg prednisone for a total of 5 weeks from Day −7 through Day 28. We observed dose-dependent increases in tissue vector genome copy numbers, μDys5 protein in multiple appendicular muscles, the diaphragm, and heart, limb and respiratory muscle functional improvement, and reduction of histopathologic lesions. As expected, given that a truncated dystrophin protein was generated, phenotypic test results and histopathologic lesions did not fully normalize. All administrations were well tolerated, and adverse events were not seen. These data suggest that systemically administered AAV-microdystrophin may be dosed safely and could provide therapeutic benefit for patients with DMD.
One Sentence Summary:
AAV9-microdystrophin gene therapy results in molecular, histopathologic, and functional benefits in a canine model of Duchenne muscular dystrophy.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder affecting approximately 1 in 5,000 newborn human males in whom absence of the protein dystrophin causes progressive degeneration of skeletal and cardiac muscle (1). There has been particular interest in potentially corrective genetic therapies, including dystrophin transgene delivery using viral vectors. Gene therapy in DMD has been complicated by two major factors, the limited carrying capacity of conventional viral vectors (2) and the potential for an immune response to the viral construct (3).
The DMD gene transcript is about 14 kb, well exceeding the capacity of first-generation adenovirus (Ad, about 8.2 kb) and adeno-associated virus (AAV, about 4.7 kb) vectors (4). To overcome this size limitation, shortened dystrophin transgenes that retain the key elements necessary for protein function have been developed (5). The therapeutic potential of these miniaturized genes was predicated on the fact that some sizeable in-frame DMD gene deletions cause a much less severe syndrome known as Becker’s muscular dystrophy (BMD) (6). After encouraging results from mdx mice treated intramuscularly with AAV- microdystrophin constructs (7, 8), a small DMD clinical trial was conducted (9, 10). Although evidence of dystrophin production was observed, a T lymphocyte response was noted to the transgene protein and preexisting dystrophin epitopes (9) with interferon-γ enzyme-linked immunosorbent spot (ELISPOT) assays.
Studies had been done on a somewhat delayed basis in a genetically homologous canine disease originally characterized in golden retriever dogs and termed either canine X-linked muscular dystrophy (CXMD) or golden retriever muscular dystrophy (GRMD) (11). Dystrophic dogs treated by intramuscular injection of AAV-microdystrophin constructs showed a marked immune response to either the transgene protein (12) or AAV capsids (13), although ELISPOT assays were not done. In both the canine studies and human trial, a ubiquitous cytomegalovirus (CMV) promoter was used, with the potential for greater gene expression in nonmuscle tissues and an associated immune response. Subsequent studies showed that persistent microdystrophin expression could be achieved after intramuscular injection of AAV constructs with a CMV promoter if dogs were immunosuppressed (14, 15). To reduce the immune response to the microdystrophin transgene, muscle-specific regulatory cassettes (16) that restrict microdystrophin expression to skeletal and cardiac muscle have been increasingly employed (17). Providing proof of concept, a single dystrophic dog treated with a microdystrophin construct that included a muscle-specific promoter showed dystrophin expression for 8 weeks without immunosuppression (18).
Localized gene therapy has been seen as the first step towards systemic administration and associated body wide effects. Intravascular delivery may be less immunogenic, given that side effects of intramuscular injection likely occur partly due to tissue injury and the concentrated antigen load (19). Consistent with this point, systemic administration of AAV-microdystrophin constructs achieved widespread dystrophin expression and, in some cases, improved skeletal muscle and/or cardiac function in mdx mice (20–22) and dystrophic dogs (23, 24), without a substantial immune response.
Here, we report data from a blinded, placebo-controlled study in which GRMD dogs were treated intravenously with a single dose of an AAV9 vector carrying a codon-optimized, canine version of the microdystrophin-5 (μDys5) cDNA. The μDys5 transgene encodes a functionally enhanced microdystrophin that includes the localization domain for neuronal nitric oxide synthase (nNOS) (4, 17). A human version of μDys5 being used in the Solid Biosciences (SGT-001) clinical trial (https://clinicaltrials.gov/ct2/show/NCT03368742) is distinct from earlier generation microdystrophins used in other ongoing trials (4). We observed dose-dependent increases in tissue vector genome copy numbers and improvement in several functional and pathologic outcomes in treated dogs. These results support the potential value of systemic AAV-microdystrophin to treat patients with DMD.
RESULTS
A single infusion of rAAV9-CK8e-c- μDys5 or vehicle was administered IV at increasing doses into four groups of GRMD dogs using a 3-month experimental timeline.
We used a construct containing the canine codon-optimized version of the microdystrophin μDys5 transgene, rAAV9-CK8e-c-μDys5 (25) (Fig. 1A), to lessen the potential immunogenicity of a human transgene being expressed in dogs (26, 27). A dose escalation study was devised, using doses that largely paralleled the 1.5E13 to 6.23E14 vg/kg range previously used in GRMD microdystrophin studies (23, 24, 26). Dogs were randomly assigned to one of four treatment groups (n = 3 for each) that received a single intravenous dose of the rAAV9-CK8e-c-μDys5 construct or vehicle: Group 1: 1E13 vg/kg, Low Dose; Group 2: 1E14 vg/kg, Mid Dose; Group 3: 2E14 vg/kg, High Dose; and Group 4: 0 vg/kg, Vehicle (Control) (Fig. 1B). We expected to see a clear dose response with the ten-fold increase between the low and mid dose groups and a further incremental increase by doubling the middle dose to the high dose. A standard 3-month timeline was employed, performing baseline and terminal studies at 3 and 6 months of age, (11).
Fig. 1. Experimental design and timeline for AAV9-microdystrophin injection in GRMD dogs.

(A) An AAV9 construct containing a microdystrophin (μDys5) consisting of the N-terminal actin binding domain (ABD1), two hinges (H1 and H4), five spectrin-like repeats (R1, R16, R17, R23, and R24), and the cysteine-rich (CR) domain with the muscle-specific CK8e expression cassette was injected. (B) Dogs were randomly assigned to one of four groups (n = 3 for each) that received a single intravenous dose of the AAV9-μDys5 or vehicle based on the dose of AAV-9 received: Group 1: 1E13 vg/kg, Low Dose; Group 2: 1E14 vg/kg, Mid Dose; Group 3: 2E14 vg/kg, High Dose; and Group 4: 0 vg/kg, Vehicle (Control). (C) AAV9-μDys5 was administered on Day 0 (baseline) when dogs were about 90±10 days (3 months) of age. Prednisone (1 mg/kg orally) was begun 7 days prior to AAV9-μDys5 injection and continued without tapering the dose for a total of 35 days. Anti-AAV9 circulating antibodies were measured at baseline and on several subsequent days until termination at about study Day 90. Phenotypic testing was done within 5 days of the AAV9-μDys5 injection and then repeated on Days 45 and 90 (about 4½ and 6 months of age), each ± 5 days to accommodate scheduling the various tests.
To lessen the potential immune response, we gave prednisone (1 mg/kg) orally to all GRMD dogs for 5 weeks, without tapering the dose, starting on Day - 7 and ending on Day 28 (Fig. 1C). Diphenhydramine was administered subcutaneously (2 mg/kg) 30 minutes before dosing to decrease the likelihood of a hypersensitivity response, as we have done previously when administering proteins to GRMD dogs (28).
All GRMD dogs were determined to be seronegative for AAV9 at 90±10 days (~ 3 months) of age (Day 0; baseline). Anti-AAV9 circulating antibodies were measured on several subsequent days until termination at about study Day 90 (table S1). Baseline phenotypic testing was done within 5 days before the AAV9-μDys5 injection and then repeated for all tests but MRI on Days 45 and 90 (about 4½ and 6 months of age), each ± 5 days to accommodate scheduling the various tests. MRI was performed only at baseline and Day 90. In assessing all findings, we were mindful of the well-established tendency for phenotypic variation in GRMD, whereby functional and pathologic findings vary among affected dogs (29). For functional studies, therapeutic efficacy was best shown by calculating the percent difference between baseline and post-treatment values, allowing each dog to essentially serve as its own control.
Dose-dependent AAV9 vector DNA was evident in body fluids and stool by Day 7 and then tapered.
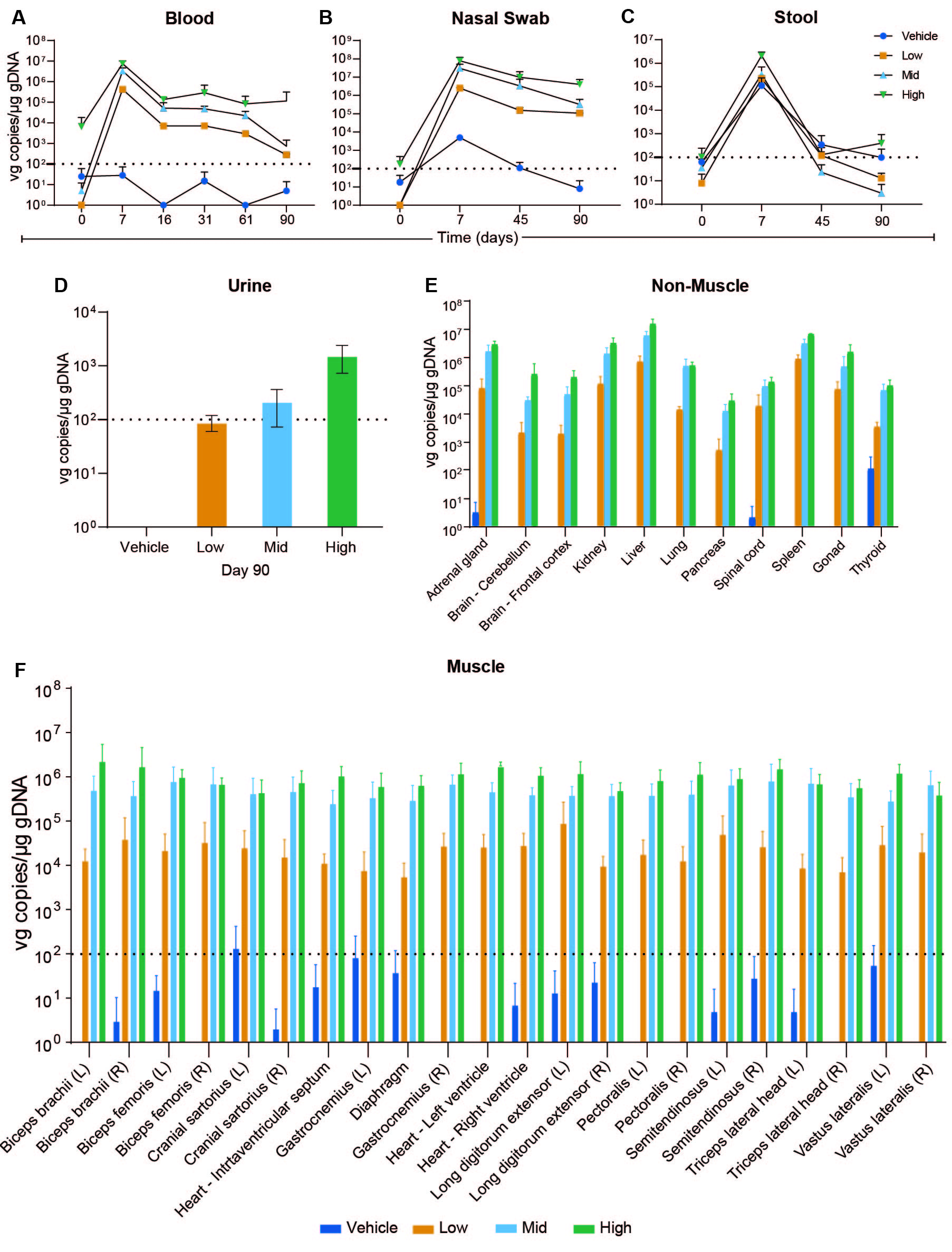
Blood sample vg copies/μg gDNA group means were below the positive cut off (>100 vg copies/μg gDNA) at baseline (fig. S1A), except for the high dose group that had a mean positive value of 6,685 vg/μg gDNA. This reflected a markedly elevated value of 18,250 vg/μg gDNA in a single dog that likely occurred because all dogs were closely housed, potentially allowing body fluids to be shared. Group means in all dose groups were positive at Day 7 after injection and subsequently decreased at Day 16. A small increase occurred from Day 16 to Day 31 in the low and mid-dose groups followed by a continual decrease from Day 31 to Day 90. Mean concentrations from vehicle-injected control dogs remained below the positive cut off at each time point (fig. S1A).
We assessed viral shedding in treated dogs using a positive cut off >100 vg copies/μg gDNA. Nasal swabs showed dose-dependent increases at Day 7 (fig. S1B). A continuous decrease from Day 7 to Day 90 was observed in each of the three dose groups. By Day 90, all groups remained positive in a dose-dependent manner. Nasal swabs from the vehicle-injected control dogs were positive on Days 7, potentially because closely housed dogs shared body fluids as discussed above. Likewise, the baseline sample mean for the high dose group was minimally positive.
Stool sample group means were dose dependent at Day 7 (fig. S1C). By Day 90, only the high dose group remained positive. Like the nasal swabs, stool group means for the controls were positive on Days 7 and 45 but returned to below the limit of detection by Day 90.
Urine group means on Day 90 were positive for the mid and high vg/μg dose groups, whereas those from the vehicle-injected and low dose groups were negative (fig. S1D).
Different AAV serotypes have variable distribution patterns and tissue tropism, with AAV9 demonstrating relative tropism for skeletal muscle and the heart in mice after systemic injection (30). In our study, most tissues showed dose-related positivity (>100 vg copies/μg gDNA) at Day 90, with considerable overlap and similar concentrations in skeletal muscles and the heart (Fig. 2 and fig. S1E and F). The vg group means in tissues collected from vehicle-injected control dogs remained below the positive cut off, except for the left cranial sartorius (133 vg/μg gDNA) and thyroid (107 vg/μg gDNA).
Fig. 2. Biodistribution of vector genomes (vg).

qPCR analysis of vector genome copies per microgram of genomic DNA in muscles (left) and non-muscle tissues (right) at study day 90. Means and standard deviation for all study subjects within a dose group and time point are plotted. LV = left ventricle.
Dose-dependent increases in μDys5 expression were observed in AAV-μDys5-treated GRMD dogs.
We have previously shown widespread skeletal muscle and, to a lesser extent, cardiac expression of AAV9-transgene constructs after IV injection in both wild type (WT) (31) and GRMD (26) dogs using nonspecific Rous sarcoma virus (RSV) and CMV promoters, respectively. In the current study, muscle-specific expression was achieved by using the CK8e regulatory cassette derived from enhancer and promoter regions of the muscle creatine kinase gene (16).
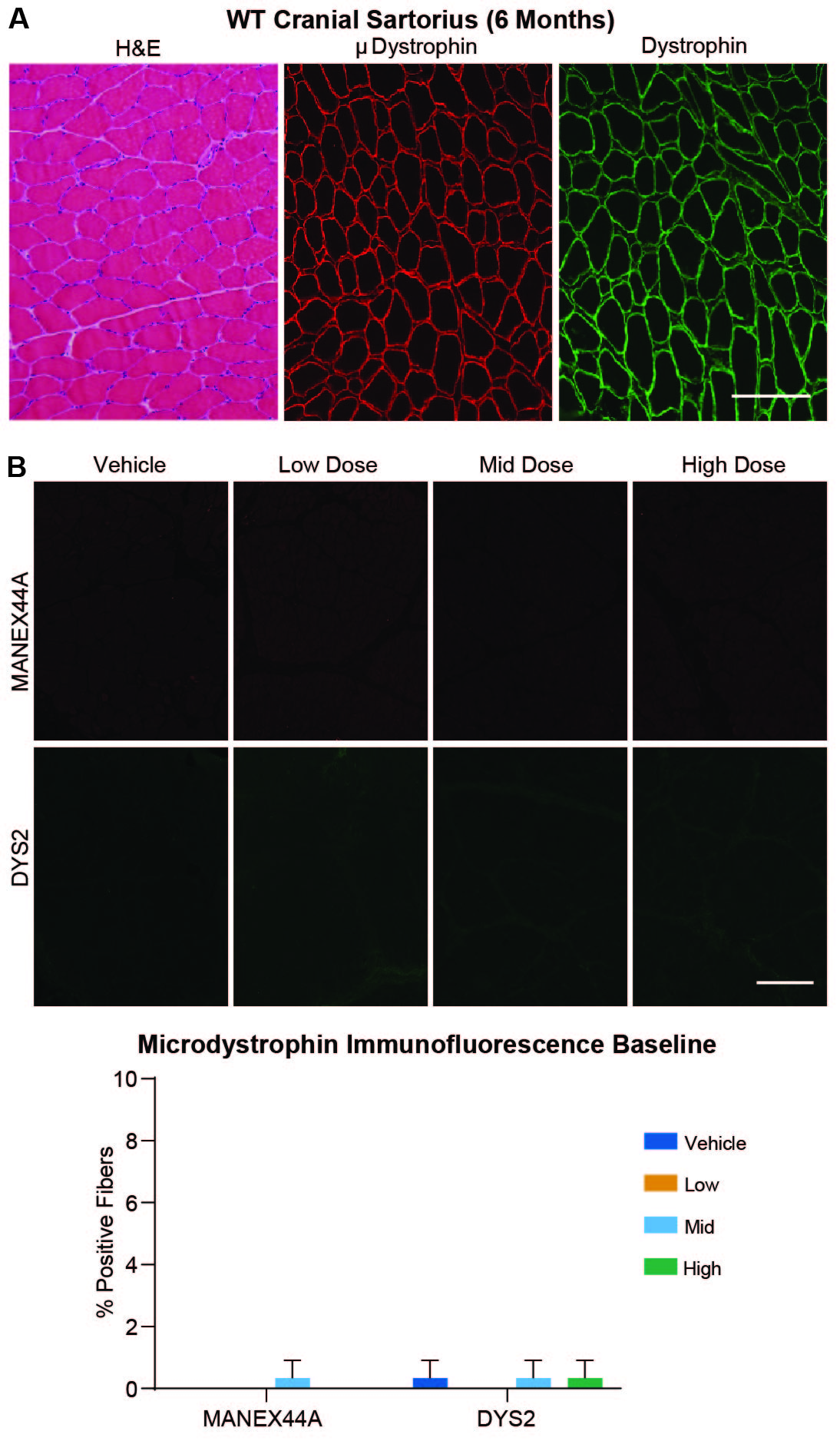
Quantitation of microdystrophin-positive fibers by immunofluorescence IF after AAV-microdystrophin-based therapy in DMD (32) and GRMD (33) is confounded by the presence of dystrophin-positive “revertant fibers,” which presumably result from alternative splicing that reestablishes the mRNA reading frame. To help distinguish revertant and μDys5-positive fibers, we used two dystrophin antibodies, MANEX44A (34) and DYS2 (fig. S2, A and B). The MANEX44A antibody is selective for spectrin-like repeat R17 of human dystrophin and has cross-species compatibility for canine dystrophin and rAAV9-CK8e-c-μDys5 microdystrophin. DYS2 is reactive to a sequence in the C-terminal region of dystrophin not found in μDys5 microdystrophin.
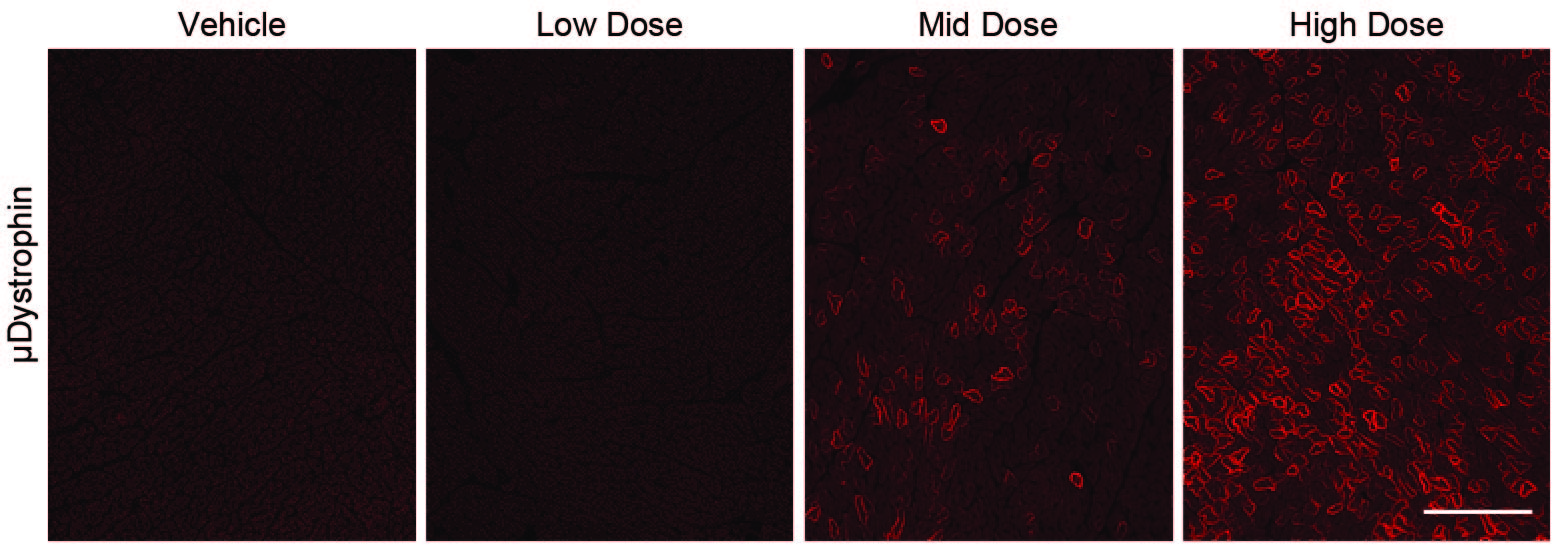
The average percent of the cross-sectional area containing skeletal muscle fibers positive for DYS2 staining across all doses, muscles, dogs, and time points ranged from 0 to 1%, in keeping with the absence of full-length dystrophin other than baseline revertant fibers (fig. S2B). Average expression for μDys5 via MANEX44A staining at baseline in the vastus lateralis and cranial sartorius muscles ranged from 0 to 0.3%, consistent with the values for DYS2 staining (fig. S2B), confirming that no μDys5 was present prior to dosing. At the interim time point (about Day 45), the biceps femoris exhibited a clear dose response, with group means for μDys5 expression ranging from 0.3% for the vehicle-injected control group to 88.3% for the high dose group (fig. S3, A and B).
The tissue distribution of μDys5 protein expression was quantified according to the percentage of tissue section area that included positive myofibers or cardiomyocytes. With respect to this endpoint, a comparable dose response was observed at the terminal (about Day 90) time point, with group means ranging from 5.3 to 93% for the low and high dose groups (Fig. 3A and fig. S4; table S2). Area positivity in the low dose group ranged from 1.3% in the gastrocnemius and vastus lateralis to 20.8% in the semitendinosus (table S2). For the mid dose group, the average was 61.0%, ranging from a minimum of 30% in the diaphragm to 73.3% in the semitendinosus and vastus lateralis. The average for the high dose group was 79.2%, with the maximum seen in the semitendinosus and biceps femoris (93%) and minimum in the diaphragm (58.3%). Across all dose groups, the semitendinosus exhibited the highest percent of cross-sectional area positive for μDys5 expression and the diaphragm exhibited the lowest. All control samples were negative for μDys5 via MANEX44A staining other than baseline skeletal muscle revertant fibers.
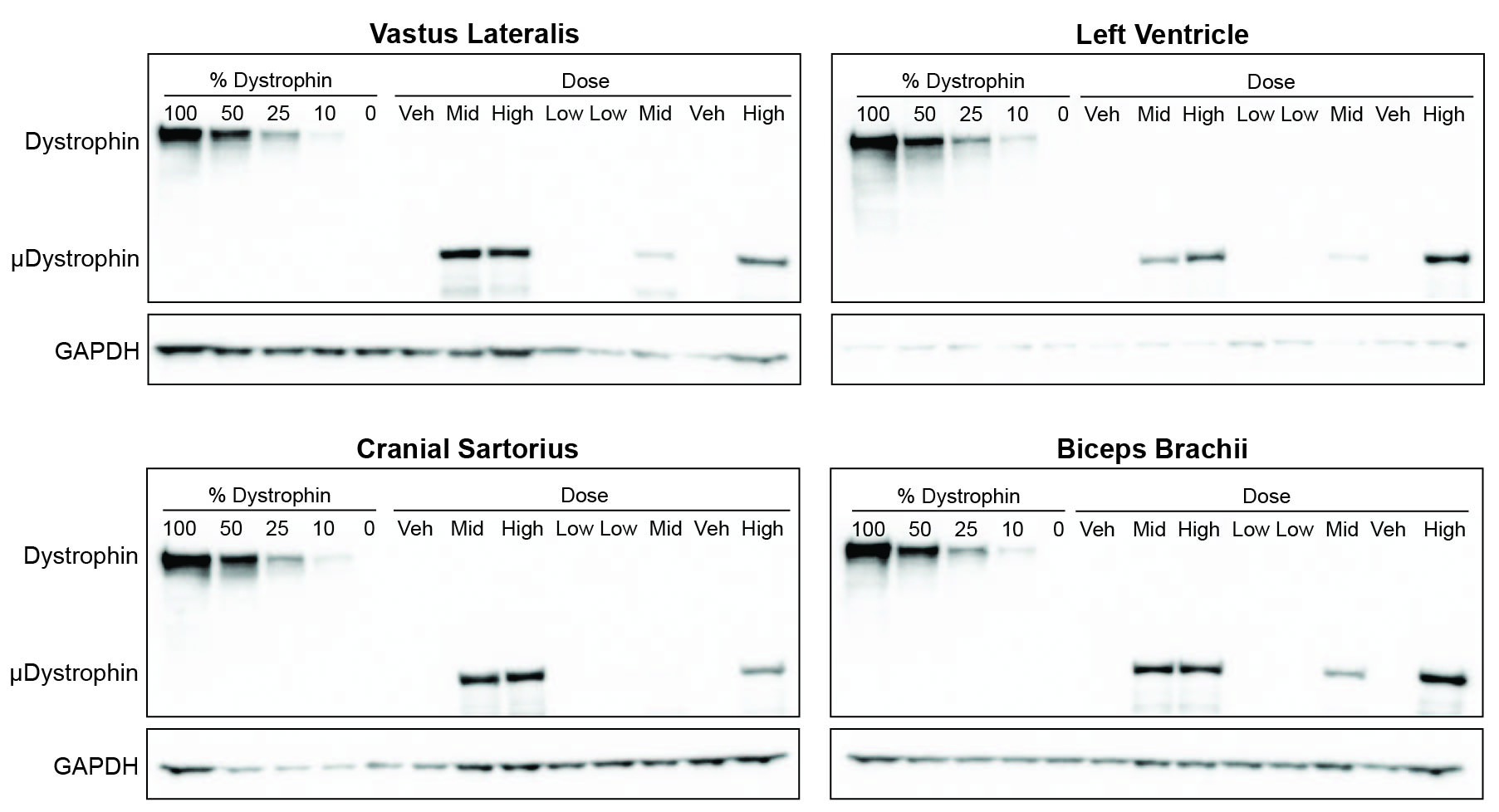
Fig. 3. Protein expression quantified by immunofluorescence (IF) and western blot.
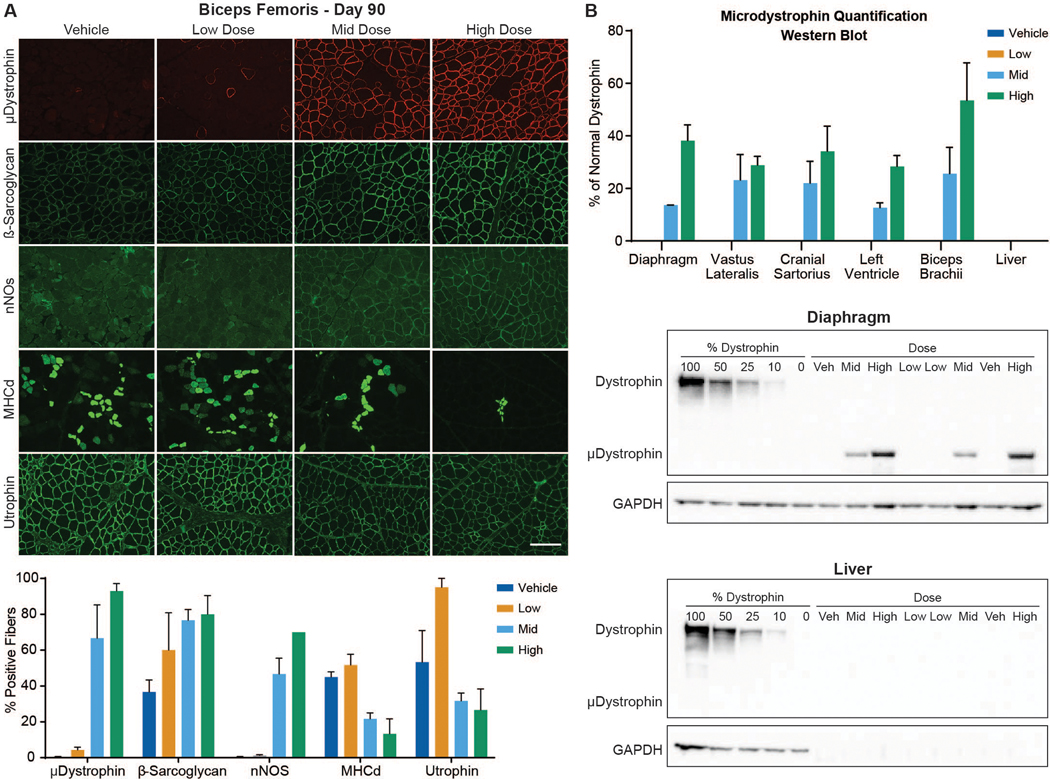
(A) Imaging by IF of biceps femoris muscle sections at Day 90 (top) and a histogram depicting group means of the percent cross sectional area containing positive fibers (bottom). Antibody-mediated IF stains include microdystrophin (top row, red), the dystrophin glycoprotein complex (DGC) members β-sarcoglycan and nNOS (rows 2 and 3; both green), MHCd (row 4, green), and utrophin (row 5; green) (bar = 100μm). (B). Western blot quantification of microdystrophin protein in tissues collected from dogs at necropsy as a % of normal dystrophin (top). Each blot (middle: diaphragm and bottom: liver) contains 13 lanes in the following order: Lanes 1–5 – Dystrophin standard curve composed of 100%, 50%, 25%, 10%, and 0% of normal dystrophin; Lanes 6–13 – Vehicle, Mid, High, Low, Low, Mid, Vehicle, High. GAPDH used to confirm sample loading.
For cardiac muscle, the low dose group had a <1% increase in the average cross-sectional area positive for μDys5 compared to the vehicle-injected controls (figs. S4 and S5). The average overall percent of cardiac sample positive for μDys5 for the mid dose group was 9.7%, with a maximum percentage in the left ventricle (LV) (15%) and minimum in the right ventricle (RV) (6%). For the high dose group, the average overall percent of cardiac sample area positive for μDys5 expression was 26.7%, with a maximum in the interventricular septum (IVS) (33.3%) and minimum in the RV (18.3%).
To better determine the functionality of μDys5, we assessed four additional proteins in terminal biceps femoris samples, β-sarcoglycan (43-DAG), developmental myosin heavy chain (MHCd), utrophin, and nNOS. We expected that β-sarcoglycan would be restored by microdystrophin expression and that the reduced drive for muscle turnover in treated dogs would lessen expression of both MHCd and utrophin. In the absence of the nNOS binding domain, DMD and canine dystrophic muscle is more susceptible to functional ischemia (35, 36). With its inclusion in the μDys5 transgene and restoration of the dystrophin-associated complex in treated dogs, increased nNOS was anticipated.
Expression of these proteins in the Day 90 terminal biceps femoris sections generally followed our projections that β-sarcoglycan and nNOS would increase while utrophin and MHCd decreased, each in a dose-responsive manner (Fig. 3A; table S3). A similar pattern was seen in the interim Day 45 biopsy samples (table S4).
Protein expression was also determined by western blot (WB). Densitometry was recorded in single bands at ~ 427 kDa for dystrophin and ~ 147 kDa for μDys5. μDys5 protein concentrations at Day 90 were below the limit of quantification (BLOQ; < 10%) for each dose in the liver and for all muscle samples from the vehicle-injected control and low dose groups (Fig. 3B and fig. S6; table S5). Other non-targeted tissues were not tested. Group mean protein expression ranged from 12.6% to 30.5% of normal dystrophin for the mid dose group and 28.3% to 53.4% in the high dose group (Fig. 3B). The 2-fold increase in dose resulted in a 2.8-fold increase in expression in the diaphragm, about a 2-fold increase in the left ventricle, and a range of 0.9–2.8-fold increase in skeletal muscles assessed (table S6).
To further quantify μDys5 protein, mass spectrometry (MS) was performed using protocols modified from previously published methods (37, 38) in which values were given as the percent of normal dystrophin protein expression. μDys5 protein concentrations were BLOQ (<10%) for all muscle samples from the vehicle-injected controls. One biceps brachii sample was 10% of normal in the low dose group. Group mean concentrations ranged from 14.6% to 46.7% for the mid dose group and from 26.6% to 61.3% in the high dose group (table S7). The 2-fold increase from 1E14 to 2E14 vg/kg for the mid and high dose groups resulted in a 3.2-fold increase in expression in the diaphragm, about an 1.6 increase in the left ventricle, and from 1.0–3.2 in skeletal muscles assessed (table S8). Values for WB and MS strongly correlated (Spearman r = 0.87; R2 = 0.75; P < 0.0001) (fig. S7).
Anti-AAV9 circulating antibodies were negative at baseline, positive at Day 7, and largely persisted at Day 90.
We saw a dose-related increase in anti-AAV9 circulating antibodies in treated GRMD dogs (fig. S8, and table S9). Control, baseline, and Day 1 and 3 samples were negative (<6,000 mU/mL, positive cut off). Antibodies were detected starting at Day 7 and increased further on Day 16 (fig. S8 and table S9). Most of these differences were not statistically significant when compared across dose groups. Some control values were higher than baseline on Day 7 and subsequent measurements but remained below the 6E3 threshold for positivity. This minor increase could have occurred due to co-housing, as with the AAV DNA.
Antibodies plateaued beyond Day 16, with relatively minor further increases. The high dose group consistently had a higher trajectory except for a dip at day 61 (fig. S1) when a blood sample was inadvertently not collected, potentially causing an artifactually lower value.
By Day 90, all dose groups showed a slight decrease in circulating antibodies. All concentrations remained above baseline, but the difference was only significant between the high dose and vehicle groups (P < 0.05). The average peak for all dose groups was 3.91E7 mU/mL, about a 6.5E3-fold increase over the positive cut off. By Day 90, this had decreased to an average of about a 4.5E3-fold increase, and concentrations did not differ across dose groups.
AAV9-μDys treated dogs had anti-dystrophin antibodies on western blot at Day 90 after administration.
In addition to AAV-directed humoral immunity, there is the potential for a response to micro-dystrophin acting as a neoantigen (3). Studies in the mdx mouse have suggested dystrophin-positive “revertant fibers” may provide tolerance (27). However, GRMD dogs treated systemically with an AAV2/8-microdystrophin did demonstrate anti-dystrophin antibodies (24). Moreover, mdx mice in which dystrophin was restored through morpholino-induced exon skipping developed antibodies to both full-length and truncated exon-skipped dystrophin isoforms (39). To further clarify this issue, we measured IgG antibodies towards the 427 kDa full-length dystrophin and 147 kDa microdystrophin proteins. Extracts from normal dystrophin-positive, GRMD dystrophin-negative, and AAV9-μDys-treated, microdystrophin-positive GRMD muscle samples were probed using an anti-dystrophin positive control antibody or serum from animals in vehicle and AAV9-μDys treated groups as the primary antibody. IgG antibodies towards both proteins were observed, with the response to microdystrophin being dose-dependent (fig. S9).
Dose-related lowering of skeletal muscle histopathologic lesion scores and fibrosis was seen in AAV9-μDys5-treated dogs.
Pathologic studies were done to assess potential toxicity and efficacy. No test article-related toxicologic changes were found upon histopathologic assessment of a range of non-muscle tissues 90 days after dose administration. Microscopic lesions were assessed in skeletal muscles at baseline (Day 0), Day 45, and terminally at Day 90 and the heart at Day 90 to show potential efficacy (Fig. 4, A to C).
Fig. 4. AAV9-μDys5-treated dogs demonstrate reduced histopathologic lesions and fibrosis.
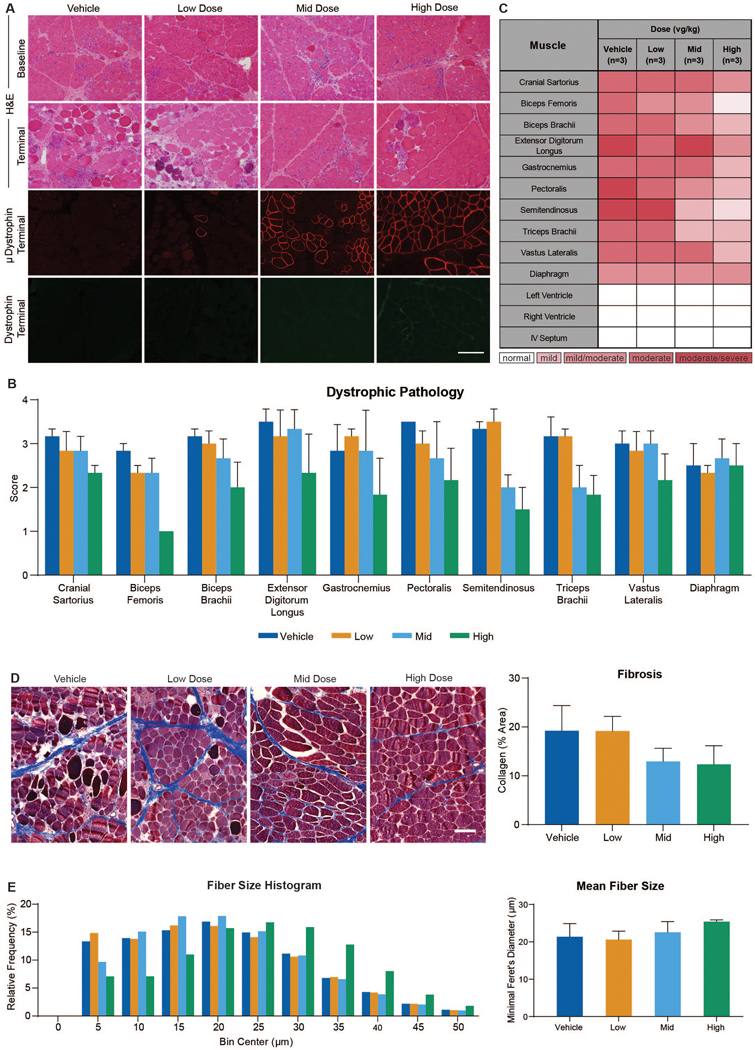
(A) Images of cranial sartorius muscle at baseline and Day 90 from GRMD dog with H&E staining (upper two panels). Immunofluorescence of microdystrophin (red) and dystrophin (green) at Day 90 (lower two panels). (B) Average lesion scores varied across skeletal muscles but were generally lower in the mid and high dose groups. (C) A heat map demonstrating lesion severity in skeletal muscle and cardiac samples from all dogs obtained at Day 90. (D) Representative images from each dose group of muscle collected at necropsy and stained with Masson’s trichrome (left), together with a histogram of mean collagen per group quantified as a percent of area (right). (E) Data presented as a histogram of relative frequency of fiber size using a bin of 5 μm and a maximum bin of 50 μm and mean fiber size for each dose group (left). Fiber size evaluation of biceps femoris muscles collected at necropsy using minimal Feret’s diameter measurements (right). Bar = 100μm in A and D.
We biopsied two muscles with distinctive phenotypes, the cranial sartorius (33) and vastus lateralis (40), at baseline to assess the degree of preexisting disease and treatment effect. Based on a semi-quantitative grading system of dystrophic pathology (see supplementary materials and methods), histopathologic lesions at baseline ranged from mild/moderate to moderate for the vastus lateralis and from mild to mild/moderate for the cranial sartorius (table S10).
Overall tissue histopathology was generally improved following treatments with mid and high vector doses. At the Day 45 interim time point, lesions observed in the biceps femoris were less pronounced in treated dogs, ranging from moderate/severe for vehicle-injected controls to very mild for the high dose group (fig. S3C and table S10). Lesion scores tracked inversely with microdystrophin immunofluorescence at Day 45 (fig. S3, B and C). A similar dose-related effect was seen in the biceps femoris and other muscles at the terminal (Day 90) time point, as mid and high dose-treated dogs had less severe lesions compared to the low dose and vehicle-injected controls (Fig. 4, A to C).
All samples at baseline and from the interim biopsies and necropsy showed inflammation characteristic of primary dystrophic changes. Inflammation was generally restricted to areas of active myofiber degeneration and regeneration and only extended beyond necrotic regions in tissues scored as “severe.” Comparing lesion severity across dose groups, most muscles had moderate/severe lesions in the controls, whereas none of the dogs in the high dose group had severe scores (Fig. 4C). There was no evidence of inflammation in the rAAV9-CK8e-c-μDys5 treated dogs beyond that seen in vehicle-injected controls. Cell infiltrates typical of inflammatory myopathy were not observed.
Because fibrosis occurs by 6 months in GRMD dogs (41), staining for collagen in terminal necropsies at this age could identify a treatment effect. Biceps femoris muscle from dogs in the mid (13.0±2.7) and high (12.4±3.9) dose groups had lower collagen percent positive areas on Masson’s trichrome stains compared to the control (19.2±5.2) and low (19.1±3.0) dose groups (Fig. 4D). Although individual dose groups and the controls did not differ, the combined mid/high groups had significantly lower areas (12.6±2.7) than the combined control/low dose (19.2±3.4) (P < 0.01) dogs.
Calculation of fiber size diameter provides important, albeit indirect, evidence of disease progression and, in turn, response to treatment in both DMD (42) and GRMD (40). Populations of either smaller or larger myofibers can occur, owing to the combined effects of regeneration and compensatory hypertrophy. We hypothesized that less necrosis in treated dogs would reduce the need for regeneration, resulting in fewer smaller fibers and a higher mean fiber diameter. Although the differences between treated and control dogs were not significant, dogs from the mid (22.5±2.4; p = 0.68) and high (25.4±0.39; P = 0.12) dose groups had larger fiber means compared to the control (21.4±2.9) and low dose (20.6±1.8; P = 0.77) groups (Fig. 4E). The high dose dogs had more fibers in the larger fiber bins. Differences between the combined control/low (21.0±2.4) and mid/high (24.0±2.2) dose groups were more pronounced, with a trend towards significance (P=0.07).
All LV, RV, and IVS heart samples were designated as normal for all dose groups.
Skeletal muscle magnetic resonance imaging (MRI) did not show evidence of efficacy in AAV9-μDys5 treated dogs.
Skeletal muscle magnetic resonance imaging (MRI) has been employed as a surrogate biomarker for DMD, showing greater sensitivity than functional tests in natural history studies and clinical trials (43). We have also utilized MRI to define the progression of GRMD (44) and response of affected dogs to therapy (28). Data from MRI and nuclear magnetic spectroscopy (NMR) collected from two GRMD dogs treated with an AAV-microdystrophin construct showed changes reflecting a reduction in lesion severity (24). For the studies reported here, methods developed by our group at the University of North Carolina-Chapel Hill (UNC-CH) were employed at Texas A&M using a similar Siemen’s 3T MRI unit. Biomarkers found to distinguish lesions of GRMD dogs from WT canine muscle at UNC-CH (44) were assessed at baseline and Day 90. Upon analyzing changes in these biomarkers between Days 0 and 90, there were few differences across the dose groups and no overall pattern suggesting efficacy (table S11).
AAV9-μDys5 treated dogs exhibited dose-related percent changes in muscle torque.
Studies in large animal models often are underpowered due to practical considerations relating to cost and availability. In comparing the percent gain in μDys5 cross-sectional immunofluorescence (IF) staining among dog groups, we noted the average percent positive for μDys5 expression for the mid dose (1E14 vg/kg) dogs was 21.5-fold higher than the low dose (1E13 vg/kg), whereas the increase was limited to 1.3-fold between the mid and high (2E14 vg/kg) doses. Given this dose effect, to increase the study’s power, we subsequently compared limb and respiratory functional data from the combined control and low dose groups to that of the combined mid and high dose dogs.
We have sought to develop muscle function outcome methods in GRMD dogs that parallel those used in DMD and mdx mice. Tests to measure extension and flexion tibiotarsal joint (TTJ) torque (45), 6MWT (46), extension contraction decrement (ECD) (47), and joint angles (29) have now been used in several GRMD preclinical trials (11, 28, 48). Dogs were assessed using these tests at baseline and 45- and 90-days post-treatment.
Strength must be considered in the context of potential treatment effects on body size. Body mass (kg) is lower in GRMD versus WT dogs at 3, 4.5, 6, and 12 months (45). Proportionally greater increases could suggest treatment benefit. In tests of TTJ torque, neither baseline nor absolute/percent change values at Day 90 differed across individual or combined groups (Table 1). Extension and flexion torque (Nm/m) were corrected for the length of the TTJ lever arm to arrive at force (Nm). Absolute and body-mass-corrected values were determined. Percent change was calculated from Day 0 to interim and results at Days 45 and 90. Extension torque has best defined a treatment effect in other GRMD studies (28, 48) and was viewed as the primary outcome parameter.
Table 1.
Skeletal muscle limb and respiratory function tests (Mean±SD).
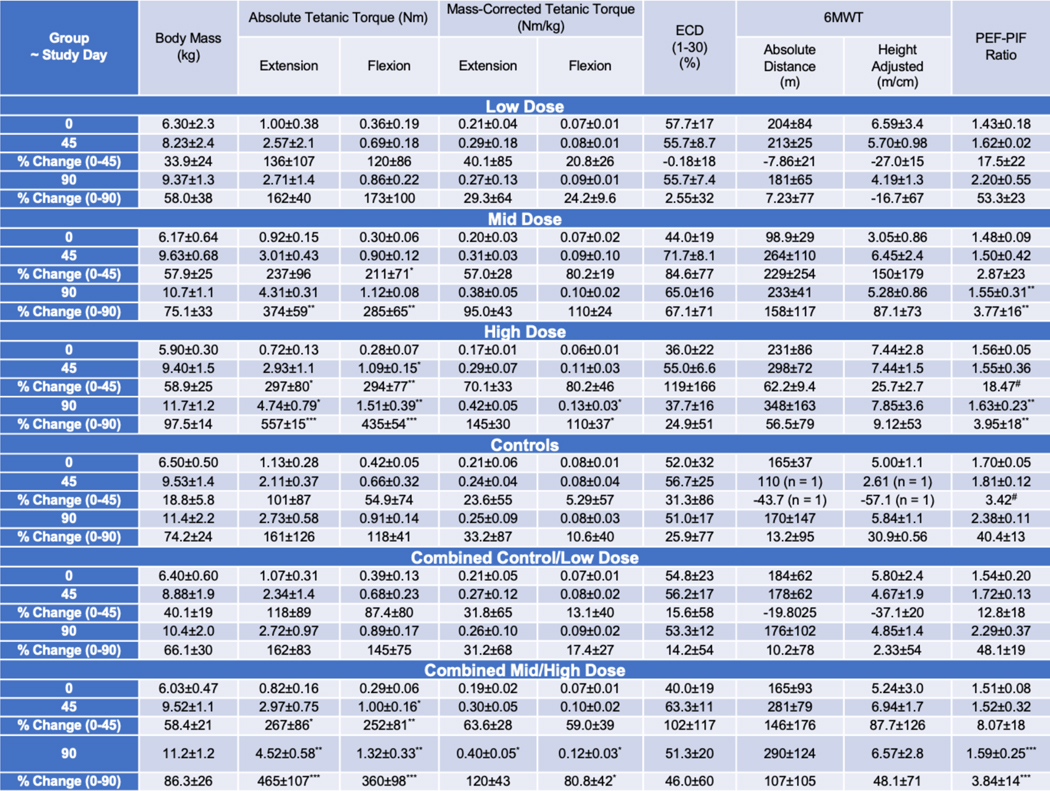
|
Torque values did not differ across the four treatment groups at baseline (Day 0) (p > 0.05) (Table 1). Dose-related treatment effects were seen at Day 45 and became more pronounced at Day 90. Differences were best reflected by percent change from baseline to Day 90. A dose-effect across individual dose groups became more dramatic when combined data were compared (Fig. 5A) and could also be seen when percent change in absolute extension (Fig. 5B) and flexion (Fig. 5C) torque from Day 0 to Days 45 and 90 was plotted.
Fig. 5. Absolute tibiotarsal joint extension and flexion torque percent change for combined dose groups from baseline to Days 45 (interim) and 90 (final).

(A) Dose-related increases in the percent change for both absolute extension and flexion torque from baseline to Day 45 (left) and to Day 90 (right). Means and standard deviation for all study subjects within a dose group and time point are plotted. (B and C) Percent changes for extension (B) and flexion (C) torque plotted for individual dogs show evidence of a dose effect. Each dose group n = 3; combined dose groups n = 6. Combined ‘Control & Low dose’ versus ‘Mid & High dose’ groups were compared using the Mann-Whitney-Wilcoxon test. * P<0.05; ** P<0.01; *** P<0.001.
As an example of the overall pattern, although the percent change for absolute tetanic extension torque from Days 0 to 45 only trended towards being significantly higher in the mid dose group (237±96) versus controls (101±87) (P =0.10), the difference reached significance at Day 90 (374±59 versus 161±126; P < 0.01) (Table 1). Further reflecting the dose effect, differences for the high dose versus controls groups were significant at both time points (P < 0.05 at Day 45 and P < 0.001 at Day 90). Absolute extension torque percent change values were more pronounced when combined data for the mid/high and control/low dose groups were compared (465±107 versus 162±83; P < 0.001) (Fig. 5B). Body-mass corrected values showed less pronounced differences, with percent change from Days 0 to 90 for extension torque only trending towards being significantly higher in the combined mid/high dose (120±43) versus control/low dose (31±68) (P = 0.06) dogs.
The flexion torque and percent change values generally tracked with those for extension, but changes tended to be more pronounced, showing evidence of earlier treatment effects (Fig. 5B; Table 1). For example, even with the mid dose group alone, the percent change for absolute tetanic flexion from Days 0 to 45 (211±71) was significantly higher than for controls (54.9±74) (P < 0.05). The difference was even more pronounced at Day 90 (285±65 versus 118±41; P < 0.01). Percent change for body mass-corrected flexion at Day 90 was also significantly higher for the combined mid-high dose (80.8±42) versus control-low dose (17.4±27) dogs (P < 0.05).
On comparisons of 6MWT results across individual groups, the high dose-treated dogs walked further absolute distances (m) than controls at Day 90 (348±163 versus 170±147), with an associated higher percent change from baseline (56.5±79 versus 13.2±95.36) (Table 1). Comparable differences in absolute distances walked (290±124) and percent change (107±105) were seen in the combined high/mid dose versus control/low dose (176±102, 10.2±78) dogs. The mid dose dogs had decidedly lower baseline values, which could have artificially led to a greater apparent treatment effect. Regardless, with the small group sizes and large SD values, none of these differences reached significance (P = 0.26 and 0.27 for the combined groups). Analogous differences were seen for height-adjusted distance walked.
Values for ECD from 0–30 stimulations in some individual dogs suggested a potential treatment effect at Day 90 but when compared across groups and over particular time periods did not differ (Table 1). For example, the ECD of the high dose-treated dogs at Day 90 was lower than in the controls, 37.7±16 versus 51.0±17, but the P value was only 0.72. Moreover, the 1–30 stimulation ECD values for the combined control/low (53.3±12) and mid/high (51.3±20) dose dogs were essentially identical (P = 0.96). Occasional outlier values made it impossible to interpret percent change for the different time periods across the groups.
There were few differences between joint angles in treated and control dogs. The overall pattern of changes did not suggest a treatment effect (table S12).
Respiratory function was less impaired in AAV9-μDys5-treated dogs.
Cardiopulmonary disease is a major cause of morbidity and mortality in patients with DMD (49). We have previously shown objective evidence of pulmonary dysfunction by assessing respiratory inductance plethysmography (RIP) in adult GRMD versus phenotypically normal carrier dogs (50). Dystrophic dogs had markedly higher tidal breathing peak expiratory flows (PEF), with an associated increase in peak expiratory to inspiratory (PEF:PIF) ratios. Anticipating, as we ultimately showed (51), that similar changes would be seen in younger GRMD dogs, we compared the RIP PEF:PIF ratio across the treatment groups at Day 90 and for percent change from Day 0 to 90 (Fig. 6, A to C; Table 1). The major differences were seen at Day 90, with the PEF:PIF ratios for the mid (1.55±0.31) and high (1.63±0.23) dose-treated dogs being lower than that of the control group (2.38±0.11) (P < 0.01 for both). The PEF:PIF ratios for low dose/control dogs increased over time whereas those for the mid/high dose dogs were relatively stable (Fig. 6B). Differences were more pronounced when data from the combined mid/high (1.59±0.25) and control/low dose (2.29±0.37) groups were compared (P < 0.001) (Fig. 6C, left). The percent change in PEF:PIF ratios for individual and especially combined groups also showed a treatment effect (Fig. 6C, right; Table 1). Values for the combined mid/high dose dogs were relatively stable from baseline to Day 90 (3.84±14; P=0.74) compared to a marked increase (48.1±19; P < 0.001) typical of disease progression in the control/low dose dogs.
Fig. 6. Respiratory peak expiratory flow: peak inspiratory flow (PEF:PIF) ratios from baseline to end of study.

(A) Adult GRMD dog showing the instrumentation for respiratory inductance plethysmography (RIP). The solid black arrows point to the elastic inductance bands that circle the chest and the abdomen. The bands are looped through an inner lycra undershirt. Leads from the bands pass through the loose, outer mesh shirt and directly into the telemetry unit within the outer pouch (open arrow). (B) Graphs showing the absolute (left) PEF:PIF ratios at Day 90 and the percent change from Day 0 to Days 45 and 90 (right). Each dose group n = 3; combined dose groups n = 6. Combined ‘Control & Low dose’ versus ‘Mid & High dose’ groups were compared using the Mann-Whitney-Wilcoxon test. *** P<0.001. (C) RIP peak expiratory flow:peak inspiratory flow (PEF:PIF) ratios for individual dogs show a dose effect, with a disease-associated increase in the control/low dose dogs compared to relative stability of values for the mid/high dose groups.
Cardiac indices did not differ between AAV9-μDys5-treated and control dogs.
We and others have conducted systematic studies of cardiac function in GRMD dogs. Findings have generally pointed towards a delayed onset of cardiac dysfunction, in keeping with DMD cardiomyopathy (52). Accordingly, we did not expect a treatment effect over the 3–6-month age period and were primarily motivated to assess safety.
All electrocardiograms at baseline and study Days 45 and 90 were qualitatively and quantitatively within normal limits (table S13). No test-article-related abnormalities in rhythm, waveform morphology, heart rate, RR interval, PR interval, QRS duration, QT interval, or QTc interval were observed at any dose based on comparison of pre- and post-dose group and control values.
Similarly, baseline echocardiography and cardiac magnetic resonance imaging were within normal limits (table S14). Values after dosing did not differ between treated and control dogs. Thus, from a safety perspective, AAV9-μDys5 treatment was not detrimental to cardiac function.
Serum muscle enzyme values trended lower in AAV9-μDys5-treated dogs.
Hematologic and serum chemistry parameters were assayed to detect evidence of both potential toxicity and efficacy. Hematological changes typical of a stress leukogram (neutrophila with no left shift, lymphopenia, and eosinopenia) were seen in most dogs during prednisone dosing and generally resolved after conclusion of prednisone. No hematologic or chemical changes suggestive of organ injury were seen.
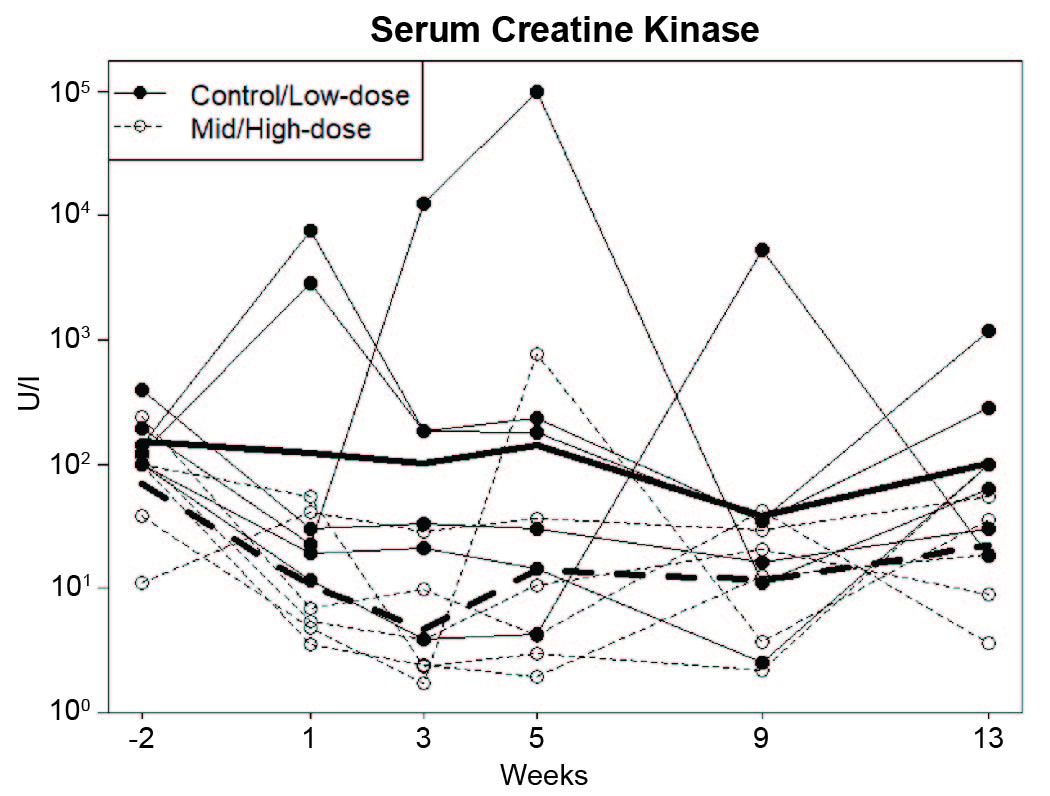
In DMD, certain serum enzymes, including CK as well as the transaminases, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (53), are elevated. Notably, these enzymes are also increased to a lesser degree in BMD patients with in-frame deletions (54), so would not be expected to fully normalize following treatment with AAV-microdystrophin. This is consistent with previous data showing serum CK was reduced but remained elevated in mdx mice treated with μDys5 (55).
In keeping with published findings (56), most GRMD dogs had high CK, ALT, and AST values on Day (–8) prior to beginning the 5-week course of prednisone (fig. S10; tables S15–17). These baseline values did not differ across dose groups. Regarding values after treatment, consideration should be given to a previous study showing that prednisone can lower CK in GRMD (57). With that said, by Day 7 (Week 1), there was a trend towards lower CK values in the mid and high dose-treated dogs (table S15). This was more pronounced on Day 16 (Week 3) and persisted through Day ~ 90 (Week 13). Demonstration of the dose effect was accentuated by combining CK data from the control/low and mid/high dose dogs (fig. S10). Values for serum ALT and AST (tables 16 and 17) generally paralleled CK. There was considerable overlap of values but a trend for a dose effect could be seen, with higher group means in the control and low dose dogs. Overall, none of the serum enzyme differences reached significance.
DISCUSSION
The mdx mouse model of DMD is mildly affected and often underestimates immunologic side effects (11, 58). Outbred dogs more reliably show the immune response to genetic and cell-based therapies (59, 60). An apparent immunologic reaction in dystrophic dogs treated intramuscularly with AAV-microdystrophin (12, 13) was attributed, in part, to interspecies (human versus canine) differences in dystrophin epitopes (27) and use of a CMV promoter with resulting dystrophin expression in nonmuscle tissues (16). Both of these potential sources of immunity were eliminated in the current study. GRMD dogs treated systemically with AAV9-canine-μDys5 under control of a muscle-specific promoter showed dose-dependent increases in tissue vector genome copy numbers, μDys5 protein in multiple appendicular muscles, the diaphragm, and heart, limb and respiratory muscle functional improvement, and a reduction in histopathologic lesions without severe adverse side effects. These results tend to substantiate a role for dystrophin interspecies differences and nonmuscle expression as sources of immunity in microdystrophin therapy. However, direct comparisons in which separate groups of GRMD dogs treated with human transgene and a nonspecific promoter would be required to confirm this observation.
Speaking further to the potential for an immune response, T-cells reacted to microdystrophins on an ELISPOT assay in DMD patients (9, 61). No positive T-cell responses were observed in the limited number of ELISPOTs done in GRMD dogs of our study. Moreover, there was no inflammatory response beyond that typically seen in dystrophic muscle. Tied to the potential for an immune response, questions have been raised about whether sustained dystrophin expression could be achieved with a single treatment (4). Dystrophic dogs treated systemically with an AAV2/8-microdystrophin construct showed persistent dose-dependent dystrophin expression for at least 14 months, even without immunosuppression (24). Clinical stabilization and reduction of histopathologic changes was evident for as long as 24 months. These findings largely paralleled the therapeutic benefit seen in GRMD dogs reported here. Importantly, our study is the first in which a microdystrophin systematically tested in mdx mice and then dystrophic dogs has been assessed in DMD clinical trials.
Beyond concerns about immunity and loss of transgene transduction over time, it has not been clear that < 4.7 kb microdystrophins carried by AAV vectors will hold up to rigorous demands placed on muscles by larger animals. Extending mdx studies to dystrophic dogs, Duan and colleagues demonstrated relative phenotypic rescue, including a reduction of ECD and histopathologic changes, after localized AAV-microdystrophin injection in the small extensor carpi ulnaris (ECU) muscle (15). A follow up study in which this same construct was delivered systemically to immunosuppressed GRMD dogs showed widespread dystrophin expression and reduced histopathologic lesions (23). However, the ECU had minimal dystrophin expression and no improvement in histopathologic changes or function. The results of our current study are promising because TTJ extension torque, which we have used as the primary outcome parameter in other preclinical trials (28, 48), improved in treated versus control dogs. Still greater improvement was seen for TTJ flexion, with changes noted even in the mid dose group at the Day 45 interim time point. This increase in flexion torque contrasts with a paradoxical decrease seen with both prednisone (48, 57) or a Nemo Binding Domain (NBD) peptide (28) treatment, probably reflecting the obvious mechanistic differences. Prednisone and NBD peptide reduce inflammation and enhance muscle regeneration, whereas microdystrophin should stabilize the myofiber membrane and decrease eccentric contraction injury. In this study, ECD did not differ between the treated and control dogs, indicating incomplete myofiber protection. Similarly, CK trended lower in treated dogs but remained elevated. Both of these findings would be expected, given that microdystrophin should ameliorate but not cure DMD, enabling a transition to a Becker phenotype (5).
With more aggressive respiratory support, cardiomyopathy has supplanted pulmonary complications as the major cause of death in DMD (62). Thus, with any treatment, it is important to target the heart. The AAV9 vector was chosen partly because of its high expression in both cardiac and skeletal muscle (63). Similar dual tissue tropism is seen with expression cassettes developed in the Hauschka lab (16). Supportiing the choice of AAV-9 under control of a muscle-specific promoter, dose-related increases in μDys5 expression were observed in all sections of the heart assessed. Although the late onset of cardiomyopathy across DMD, the mdx mouse, and GRMD (52) precluded demonstrating therapeutic efficacy, the absence of deleterious cardiac effects supported a positive safety profile. More encouragingly, treated dogs showed dose-related improved respiratory function, evidenced by relative stabilization of the PEF:PIF ratio.
There are currently four ongoing DMD AAV-microdystrophin clinical programs (4, 5, 64), including Solid Biosciences’ SGT-001 (https://clinicaltrials.gov/ct2/show/NCT03368742), the human codon-optimized variant of the construct used in this GRMD study. Each is unique to itself, with different inclusion criteria, constructs, doses, and outcome parameters. Data are incomplete and many methods of analysis are proprietary, making it difficult and premature to compare results across programs. While no unexpected immune histopathologic or clinical side effects were observed in GRMD dogs treated with SGT-001 reported here, serious adverse events (SAEs) linked to complement activation, including systemic inflammatory response syndrome and thrombocytopenia, have been noted in the ongoing DMD clinical trial (65). SAEs have also been documented in the Sarepta, Pfizer, and Genethon microdystrophin gene therapy trials and in patients receiving AAV genetic constructs for other neuromuscular diseases (66–68).
Several factors could account for the immune response in the DMD trials. Much attention has focused on the established role of AAV in activating complement during the innate immune response (69). Given the absence of severe side effects and immunologic abnormalities throughout our study, we did not assess complement or employ aggressive immunosuppression regimens. Protein transcribed by the R16/17 spectrin-like repeats of the SGT-001 nNOS binding-domain might be a source of immunity. In this context, a recent collaboration among the four programs employing AAV-microdystrophin constructs specifically studied the potential role the transgene played in five DMD patients with SAEs. All had genomic deletions involving N-terminal epitopes, with ELISPOTs reactive to the corresponding N-terminal peptide pool (70). None of those who received SGT001 had reactions to the transgene.
This study had several limitations. Because of practical issues related to cost and availability, canine studies must always be focused. We were not motivated to repeat studies already done by the Chamberlain laboratory in mice to establish benefits provided by the nNOS binding domain. Similarly, no comparisons were made between human versus canine transgenes or specific versus nonspecific promoters that might have shed light on the immune response. Even when canine studies are focused, they are often marginally powered to detect differences with all outcomes that are assessed. The power of our study was increased by comparing results from the combined control/low dose and mid/high dose groups, but numbers were still relatively low. And, despite analyzing combined data, some values showed a wide standard deviation, reflecting variation from dog to dog of μDys5 expression and associated functional and pathologic treatment effect. This likely contributed to the failure of these tests to demonstrate efficacy. Outcomes that are reproducible, with relatively low standard deviations, are more likely to yield meaningful results. We have generally used TTJ torque as our primary outcome and have had considerably less experience and success using 6MWT, ECD, joint angles, and MRI. Regarding MRI, the Siemens 3T unit and analysis methods were like those employed in earlier natural history and treatment studies at UNC-CH (28, 44). Still, minor differences in MRI protocols and analysis methods probably played a role in our inability to distinguish differences at the current sample size. Regardless of these limitations, the results from this blinded, placebo-controlled study in GRMD dogs highlight the promise of AAV-microdystrophin for the treatment of DMD and the utility of large animal models to evaluate alternative therapeutic approaches.
MATERIALS AND METHODS
Study Design
This study explored the clinical translatability of an adeno-associated virus serotype 9 vector (AAV-9)-microdystrophin construct in the GRMD model of DMD. Dogs (12 total; 4 groups of 3 each) received one of three doses (1E13 vg/kg, 1E14 vg/kg, and 2E14 vg/kg) of rAAV9-CK8e-c-μDys5 or vehicle intravenously at 3 months of age and were then monitored for 90 days after dosing using a comprehensive set of analyses that included biodistribution, μDys5 protein expression, limb muscle torque, histopathology, clinical pathology, immune response, MRI, and respiratory and cardiac function.
Based on power analysis (45), we have typically used TTJ extension torque as our primary outcome parameter in GRMD preclinical studies (28, 48). Original natural history studies anticipated that a single measurement at the end of treatment would be compared to a control value. Our preclinical trials have generally assessed 5 or 6 dogs in the treatment arm, comparing them to a like number of untreated dogs (48) or natural history controls (28).
The original study design called for a two-phased study, with an initial cohort of 12 dogs divided among the three doses and a control group (n = 3 for each), as reported here. Based on the outcome of these studies, we anticipated a second cohort of nine GRMD dogs would be assessed, with three each added to the two dose groups having the most promising results and a set of controls (n = 6 each). However, after a treatment effect was seen with the 12 dogs of the first cohort, the second phase was cancelled.
The 12 dogs were randomly assigned to one of the four groups as they were produced in the GRMD colony at Texas A&M University over an 18-month period (see “Animals” below). Study personnel were blinded from dose formulations and animal group assignments throughout the study, including at dose administration and collection of outcome data. Vehicle control dose formulations were identical in visual appearance and viscosity to AAV test article and labeled similarly to all other formulations. The vehicle used for the control group was equivalent to the formulation buffer used for test article manufacturing and was also used to normalize dose formulation volumes for all other groups.
Functional measures were repeated over the 3-month course of the study. Laboratory assessments included an average minimum of 2–3 technical replicates per assay.
Animals
Dogs were used and cared for according to principles outlined in the National Research Council Guide for the Care and Use of Laboratory Animals. Studies were approved by the TAMU Institutional Animal Care and Use Committee (IACUC) through two protocols: TAMU IACUC 2014–0117, Advanced Gene Therapy for Treatment of Cardiomyopathy and Respiratory Insufficiency in Duchenne Muscular Dystrophy and TAMU IACUC 2017–0248, Gene Therapy for Duchenne Muscular Dystrophy. Although phenotypic data have not distinguished a difference between male hemizygous and female homozygous GRMD dogs (71), an effort was made to balance males and females among the different dose and control groups. This was achieved with inclusion of 6 each males and females.
Statistical analysis
Means and standard deviations for body mass, skeletal muscle limb functional, and respiratory data were calculated for the four treatment groups (low, mid, and high dose plus controls). Because of the small group sizes and wide standard deviations for some variables, non-parametric tests were used. For each time point (baseline, interim, final), the four groups were first compared using a Kruskal-Wallis test and, if differences were noted, each pair was further compared using the Dunn post hoc test. Combined ‘Control & Low dose’ versus ‘Mid & High dose’ groups were compared using the Mann-Whitney-Wilcoxon test. In comparing time points across ages, data were combined into a single longitudinal linear mixed model, using the same tests to compare the four groups and each pair. Another linear mixed model was used to compare the combined ‘Control & Low dose’ versus ‘Mid & High dose’ groups. Also, multiple comparison correction was applied. To quantify longitudinal characteristics of the MRI biomarkers, we adopted a linear mixed model, as previously reported (44). The relationship between WB and MS protein values was defined by Spearman’s rank-order correlation. Resulting P values for all analyses were corrected for multiple comparisons and considered significant at P < 0.05.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements:
We thank veterinarians and technical staff of the Texas A&M Comparative Medicine Program for excellent care provided to dogs in this study. Q. Nguyen and X. Chen are acknowledged for their efforts in developing the CK8e muscle-specific expression cassette.
Funding:
This research was supported by a grant from Solid Biosciences, Inc to J.N.K. and a U.S. Department of Defense (DOD), Congressionally Directed Medical Research Programs (CDMRP), Duchenne Muscular Dystrophy Research Program (DMDRP) grant (MD120087) to B.J.B. Additional funding for contributions to the study were provided by the National Institutes of Health to J.S.C. (R01 AR40864) and D.D. (NS-90634). M.J.S. received equity funding from Solid Biosciences.
Footnotes
Data and materials availability: The CK8e muscle-specific promoter used in this study was made available from the University of Washington and J.S.C./S.D.H. under a material agreement with the University of Missouri-Columbia and D.D. The canine version of the CK8e-μDys5 vector was subsequently cloned by the Duan lab. All data associated with this study are presented in the paper or the Supplementary Materials.
Competing interests: D.D. and J.S.C. are members of the scientific advisory board, equity holders, and inventors on patents licensed to Solid Biosciences, Inc. D.D. is an inventor on related intellectual property owned by the University of Missouri: Synthetic mini/micro-dystrophin genes to restore nNOS to the sarcolemma. U.S. Patent Number 7,892,824. J.S.C. and S.D.H. are inventors on related intellectual property owned by the University of Washington, including: a) Compositions and methods for systemic nucleic acid sequence delivery, U.S. Patent # 7,655,467; b) Micro-dystrophins and Related Methods of Use, U.S. Patent Number 10,479,821; c) Regulatory cassettes for muscle-specific gene expression, US Patent Provisional Application 63/173,295. The Duan, Chamberlain, Lawlor, and Kornegay laboratories have received research support from Solid Biosciences, Inc; M.J.S, K.J.B, D.G., J.P.G., C.A.M., and J.S.S. are or were employees of Solid Biosciences, Inc; J.N.K. was a paid consultant for Solid Biosciences, Inc; P.P.N. is a paid consultant for Agada Biosciences.
REFERENCES
- 1.Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A, Duchenne muscular dystrophy. Nat. Rev. Dis. Primers, 2021. Feb 18;7(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts M, Dickson G, The future of Duchenne muscular dystrophy gene therapy: shrinking the dystrophin gene. Curr. Opin. Mol. Ther. 4, 343–348 (2002). [PubMed] [Google Scholar]
- 3.Wells DJ, Ferrer A, Wells KE, Immunological hurdles in the path to gene therapy for Duchenne muscular dystrophy. Expert Rev. Mol. Med. 4, 1–23 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Duan D, Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol. Ther. 26, 2337–2356 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crudele JM, Chamberlain JS, AAV-based gene therapies for the muscular dystrophies. Hum. Mol. Genet. R1, R102–R107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM, An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 2, 90–95 (1988). [DOI] [PubMed] [Google Scholar]
- 7.Wang B, Li J, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc. Natl. Acad. Sci. U.S.A. 97, 13714–13719 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, Harper HA, Robinson AS, Engelhardt JF, Brooks SV, Chamberlain JS, Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 8, 253–261 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, Bowles D, Gray S, Li C, Galloway G, Malik V, Coley B, Clark KR, Li J, Xiao X, Samulski J, McPhee SW, Samulski RJ, Walker CM, Dystrophin immunity in Duchenne’s muscular dystrophy. N. Engl. J. Med. 363, 1429–1437 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowles DE, McPhee SW, Li C, Gray SJ, Samulski JJ, Camp AS, Li J, Wang B, Monahan PE, Rabinowitz JE, Grieger JC, Govindasamy L, Agbandje-McKenna M, Xiao X, Samulski RJ, Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol. Ther. 20, 443–455 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kornegay JN, The golden retriever model of Duchenne muscular dystrophy. Skelet. Muscle 2017. May 19;7(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yuasa K, Yoshimura M, Urasawa N, Ohshima S, Howell JM, Nakamura A, Hijikata T, Miyagoe-Suzuki Y, Takeda S, Injection of a recombinant AAV serotype 2 into canine skeletal muscles evokes strong immune responses against transgene products. Gene Ther. 14, 1249–1260 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Wang Z, Allen JM, Riddell SR, Gregorevic P, Storb R, Tapscott SJ, Chamberlain JS, Kuhr CS, Immunity to adeno-associated virus-mediated gene transfer in a random-bred canine model of Duchenne muscular dystrophy. Hum. Gene Ther. 18, 18–26 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Wang Z, Kuhr CS, Allen JM, Blankinship M, Gregorevic P, Chamberlain JS, Tapscott SJ, Storb R, Sustained AAV-mediated dystrophin expression in a canine model of Duchenne muscular dystrophy with a brief course of immunosuppression. Mol. Ther. 15, 1160–1166 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Shin JH, Pan X, Hakim CH, Yang HT, Yue Y, Zhang K, Terjung RL, Duan D, Microdystrophin ameliorates muscular dystrophy in the canine model of Duchenne muscular dystrophy. Mol. Ther. 21, 750–757 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salva MZ, Himeda CL, Tai PW, Nishiuchi E, Gregorevic P, Allen JM, Finn EE, Nguyen QG, Blankinship MJ, Meuse L, Chamberlain JS, Hauschka SD, Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol. Ther. 15, 320–329 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Ramos JN, Hollinger K, Bengtsson NE, Allen JM, Hauschka SD, Chamberlain JS, Development of novel micro-dystrophins with enhanced functionality. Mol. Ther. 27, 623–635 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koo T, Okada T, Athanasopoulos T, Foster H, Takeda S S, Dickson GJ, Long-term functional adeno-associated virus-microdystrophin expression in the dystrophic CXMDj dog. Gene Med. 13, 497–506 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Guilbaud M, Devaux M, Couzinié C, Le Duff J, Toromanoff A, Vandamme C, Jaulin N, Gernoux G, Larcher T, Moullier P, Le Guiner C, Adjali O, Five years of successful inducible transgene expression following locoregional adeno-associated virus delivery in nonhuman primates with no detectable immunity. Hum. Gene Ther. 30, 802–813 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gregorevic P, Blankinship MJ, Allen JM, Chamberlain JS, Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol. Ther. 16, 657–664 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang B, Li J, Fu FH, Xiao X, Systemic human minidystrophin gene transfer improves functions and life span of dystrophin and dystrophin/utrophin-deficient mice. J. Orthop. Res. 27, 421–426 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Bostick B, Shin JH, Yue Y, Duan D, AAV-microdystrophin therapy improves cardiac performance in aged female mdx mice. Mol. Ther. 19, 1826–1832 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yue Y, Pan X, Hakim CH, Kodippili K, Zhang K, Shin JH, Yang HT, McDonald T, Duan D, Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum. Mol. Genet. 15, 5880–5890 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Le Guiner C, Servais L, Montus M, Larcher T, Fraysse B, Moullec S, Allais M, François V, Dutilleul M, Malerba A, Koo T, Thibaut JL, Matot B, Devaux J. Le Duff, Deschamps JY, Barthelemy I, Blot S, Testault I, Wahbi K, Ederhy S, Martin S, Veron P, Georger C, Athanasopoulos T, Masurier C, Mingozzi F, Carlier P, Gjata B, Hogrel JY, Adjali O, Mavilio F, Voit T, Moullier P, Dickson G, Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun. 2017. Jul 25;8:16105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hakim CH, Wasala NB, Pan X, Kodippili K, Yue Y, Zhang K, Yao G, Haffner B, Duan SX, Ramos J, Schneider JS, Yang NN, Chamberlain JS, Duan D, A five-repeat microdystrophin gene ameliorated dystrophic phenotype in the severe DBA/2J-mdx model of Duchenne muscular dystrophy. Mol. Ther. Methods Clin. Dev. 6, 216–230, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kornegay JN, Li J, Bogan JR, Bogan DJ, Chen C, Zheng H, Wang B, Qiao C, Howard JF Jr, Xiao X, Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs. Mol. Ther. 18, 1501–1508 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrer A, Wells KE, Wells DJ, Immune responses to dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Gene Ther. 7, 1439–1446 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Kornegay JN, Peterson JM, Bogan DJ, Kline W, Bogan JR, Dow JL, Fan Z, Wang J, Ahn M, Zhu H, Styner M, Guttridge DC, NBD delivery improves the disease phenotype of the golden retriever model of Duchenne muscular dystrophy. Skelet Muscle 4:18 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kornegay JN, Bogan JR, Bogan DJ, Childers MK, Li J, Nghiem P, Detwiler DA, Larsen CA, Grange RW, Bhavaraju-Sanka RK, Tou S, Keene BP, Howard JF, Jr J. Wang, Fan Z, Schatzberg SJ, Styner MA, Flanigan KM, Xiao X, Hoffman EP, Canine models of Duchenne muscular dystrophy and their use in therapeutic strategies. Mamm. Genome 23, 85–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE, Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 16, 1073–1080 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Yue Y, Ghosh A, Long C, Bostick B, Smith BF, Kornegay JN, Duan D, A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs. Mol. Ther. 16, 1944–1952 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arechavala-Gomeza V, Kinali M, Feng L, Guglieri M, Edge G, Main M, Hunt D, Lehovsky J, Straub V, Bushby K, Sewry CA, Morgan JE, Muntoni F, Revertant fibres and dystrophin traces in Duchenne muscular dystrophy: implication for clinical trials. Neuromuscul. Disord. 20, 295–301 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Kornegay JN, Cundiff DD, Bogan DJ, Bogan JR, Okamura CS, The cranial sartorius muscle undergoes true hypertrophy in dogs with golden retriever muscular dystrophy. Neuromuscul. Disord. 13, 493–500 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Thanh LT, Nguyen TM, Helliwell TR, Morris GE, Characterization of revertant muscle fibers in Duchenne muscular dystrophy, using exon-specific monoclonal antibodies against dystrophin. Am. J. Hum. Genet. 56, 725–731 (1995). [PMC free article] [PubMed] [Google Scholar]
- 35.Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, Judge L, Bostick B, Chamberlain JS, Terjung RL, Duan D, Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Invest. 119, 624–635 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi Y,M., Rader EP, Crawford RW, Iyengar NK, Thedens DR, Faulkner JA, Parikh SV, Weiss RM, Chamberlain JS, Moore SA, Campbell KP, Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature 456, 511–515 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown KJ, Marathi R, Fiorillo AA, Ciccimaro EF, Sharma S, Rowlands DS, Rayavarapu S, Nagaraju K, Hoffman EP, Hathout Y, Accurate quantitation of dystrophin protein in human skeletal muscle using mass spectrometry. J. Bioanal. Biomed. Dec 18;Suppl 7. pii: 001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farrokhi V, Walsh J, Palandra J, Brodfuehrer J, Caiazzo T, Owens J, Binks S. Neelakantan, Yong F, Dua P, Guiner C. Le, Neubert H, Dystrophin and mini-dystrophin quantification by mass spectrometry in skeletal muscle for gene therapy development in Duchenne muscular dystrophy. Gene Ther. 2021. Nov 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vila MC, Novak JS, Benny Klimek M, Li N, Morales M, Fritz AG, Edwards K, Boehler JF, Hogarth MW, Kinder TB, Zhang A, Mazala D, Fiorillo AA, Douglas B, Chen YW, van den Anker J, Lu QL, Hathout Y, Hoffman EP, Partridge TA, Nagaraju K, Morpholino-induced exon skipping stimulates cell-mediated and humoral responses to dystrophin in mdx mice. J. Pathol. 248, 339–335 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Childers MK, Okamura CS, Bogan DJ, Bogan JR, Sullivan MJ, Kornegay JN, Myofiber injury and regeneration in a canine homologue of Duchenne muscular dystrophy. Am. J. Phys. Med. Rehabil. 80, 175–181 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Valentine BA, Cooper BJ, Cummings JF, de Lahunta A, Canine X-linked muscular dystrophy: morphologic lesions. J. Neurol. Sci. 97, 1–23 (1990). [DOI] [PubMed] [Google Scholar]
- 42.Wang JF, Forst J, Schröder S, Schröder JM JM, Correlation of muscle fiber type measurements with clinical and molecular genetic data in Duchenne muscular dystrophy. Neuromuscul. Disord. 9, 150–158 [DOI] [PubMed] [Google Scholar]
- 43.Barnard AM, Willcocks RJ, Finanger EL, Daniels MJ, Triplett WT, Rooney WD, Lott DJ, Forbes SC, Wang DJ, Senesac CR, Harrington AT, Finkel RS, Russman BS, Byrne BJ, Tennekoon GI, Walter GA, Sweeney HL, Vandenborne K, Skeletal muscle magnetic resonance biomarkers correlate with function and sentinel events in Duchenne muscular dystrophy. PLoS One. Mar 19;13(3):e0194283 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan Z, Wang J, Ahn M, Shiloh-Malawsky Y, Chahin N, Elmore S, Bagnell CR Jr, Wilber K, An H, Lin W, Zhu H, Styner M, Kornegay JN, Characteristics of magnetic resonance imaging biomarkers in a natural history study of golden retriever muscular dystrophy. Neuromuscul. Disord. 24, 178–191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kornegay JN, Bogan DJ, Bogan JR, Childers MK, Cundiff DD, Petroski GF, Schueler RO, Contraction force generated by tarsal joint flexion and extension in dogs with golden retriever muscular dystrophy. J. Neurol. Sci. 166, 115–121 (1999). [DOI] [PubMed] [Google Scholar]
- 46.Acosta AR, Van Wie E, Stoughton WB, Bettis AK, Barnett HH, LaBrie NR, Balog-Alvarez CJ, Nghiem PP, Cummings KJ, Kornegay JN, Use of the six-minute walk test to characterize golden retriever muscular dystrophy. Neuromuscul. Disord, 26, 865–872 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Tegeler CJ, Grange RW, Bogan DJ, Markert CD, Case D, Kornegay JN, Childers MK, Eccentric contractions induce rapid isometric torque drop in dystrophic-deficient dogs. Muscle Nerve 42, 130–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu JMK, Okamura CS, Bogan DJ, Bogan JR, Childers MK, Kornegay JN, Effects of prednisone in canine muscular dystrophy. Muscle Nerve 30, 767–773 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, Case LE, Cripe L, Hadjiyannakis S, Olson AK, Sheehan DW, Bolen J, Weber DR, Ward LM (DMD Care Considerations Working Group), Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 17, 4347–4361 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeVanna JC, Kornegay JN, Bogan DJ, Bogan JR, Dow JL, Hawkins EC, Assessment of respiratory dysfunction in dogs with golden retriever muscular dystrophy: Arterial blood gas analysis and tidal breathing spirometry. Neuromuscul. Disord. 24, 63–73 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hawkins EC, Bettis AK, Kornegay JN, Expiratory dysfunction in young dogs with golden retriever muscular dystrophy. Neuromuscul. Disord. 30, 930–937 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.L-J Guo JH Soslow AK. Bettis KJ. Cummings MW. Lenox MW. Miller JN Kornegay CF. Spurney, Natural history of cardiomyopathy in adult dogs with golden retriever muscular dystrophy. J. Am. Heart Assoc. Aug 20;8(16):e012443 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Veropalumbo C, Del Giudice E, Esposito G, Maddaluno S, Ruggiero L, Vajro P, Aminotransferases and muscular diseases: a disregarded lesson. Case reports and review of the literature. J. Paediatr. Child Health 48, 886–890 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Aston JP, Kingston HM, Ramasamy I, Walters EG, Stansbie D, Plasma pyruvate kinase and creatine kinase activity in Becker muscular dystrophy. J. Neurol. Sci. 65, 307–314 (1984). [DOI] [PubMed] [Google Scholar]
- 55.Rodgers BD, Bishaw Y, Kagel D, Ramos JN, Maricelli JW, Micro-dystrophin gene therapy partially enhances exercise capacity in older adult mdx mice. Mol. Ther. Methods Clin. Dev. 17, 122–132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Valentine BA, Blue JT, Shelley SM, Cooper BJ, Increased serum alanine aminotransferase activity associated with muscle necrosis in the dog. J. Vet. Intern. Med. 4, 140–143 (1990). [DOI] [PubMed] [Google Scholar]
- 57.Barthélémy I I, Uriarte A, Drougard C, Unterfinger Y, Thibaud JL, Blot S, Effects of an immunosuppressive treatment in the GRMD dog model of Duchenne muscular dystrophy. PLoS One. 2012;7(11):e48478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Manning J, O’Malley D, What has the mdx mouse model of Duchenne muscular dystrophy contributed to our understanding of this disease? J. Muscle Res. Cell Motil. 36, 155–167 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Arruda VR, Stedman HH, Haurigot V, Buchlis G, Baila S, Favaro P, Chen Y, Franck HG, Zhou S, Wright JF, Couto LB, Jiang H, Pierce GF, Bellinger DA, Mingozzi F, Nichols TC, High KA, Peripheral transvenular delivery of adeno-associated viral vectors to skeletal muscle as a novel therapy for hemophilia B. Blood 115, 4678–4688 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Storb R, Thomas ED, Graft-versus-host disease in dog and man: the Seattle experience. Immunol. Rev. 88, 215–238 (1985). [DOI] [PubMed] [Google Scholar]
- 61.Flanigan KM, Campbell K, Viollet L, Wang W, Gomez AM, Walker CM, Mendell JR, Anti-dystrophin T cell responses in Duchenne muscular dystrophy: prevalence and a glucocorticoid treatment effect. Hum. Gene Ther. 24, 797–806 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheeran D, Khan S, Khera R, Bhatt A, Garg S, Grodin JL, Morlend R, Araj FG, Amin AA, Thibodeau JT, Das S, Drazner MH, Mammen PPA, Predictors of death in adults with Duchenne muscular dystrophyassociated cardiomyopathy. J. Am. Heart Assoc. 6, e006340 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fang H, Lai NC, Gao MH, Miyanohara A, Roth DM, Tang T, Hammond HK, Comparison of adeno-associated virus serotypes and delivery methods for cardiac gene transfer. Version 2. Hum. Gene Ther. Methods. 23, 234–241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Elangkovan N, Dickson G, Gene therapy for Duchenne muscular dystrophy. J. Neuromuscul. Dis. 8(s2), S303–S316 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Solid Biosciences provides update regarding SGT-001 phase I/II clinical hold on IGNITE DMD. https://www.solidbio.com/about/media/press-releases/solid-biosciences-provides-update-regarding-sgt-001-phase-i-ii-clinical-hold-on-ignite-dmd (2020).
- 66.Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, Goodspeed K, Gray SJ, Kay CN, Boye SL, Boye SE, George LA, Salabarria S, Corti M, Byrne BJ, Tremblay JP, Current clinical applications of in vivo gene therapy with AAVs. Mol. Ther. 29, 464–488 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mendell JR, Shieh PB, Sahenk Z, Lehman KJ, Lowes LP, Reash NF, Iammarino MA, Alfano LN, Powers B, Woods JD, Skura CL, Mao HC, Staudt LA, Potter RA, Griffin DA, Lewis S, Hu L, Upadhyay S, Singh T, Rodino-Klapac LR, A Phase 2 clinical trial evaluating the safety and efficacy of delandistrogene moxeparvovec (SRP-9001) in patients with Duchenne muscular dystrophy. Presented at the 2022 Muscular Dystrophy Association (MDA) Conference, Nashville, TN, USA, March 13–16, 2022 (https://investorrelations.sarepta.com/static-files/56830179-3bb0-4796-8fdc-1b43c989a899). [Google Scholar]
- 68.Butterfield RJ, Shieh PB, Yong F, Binks M, McDonnell TG, Ryan KA, Delnomdedieu M, Belluscio BA, Neelakantan S, Levy DI, Schwartz PF, Smith EC, Results of one year follow-up after treatment with fordadistrogene movaparvovec (PF-06939926) for Duchenne muscular dystrophy (DMD) in a phase 1b, open-label study. Mol Ther 30 (5S1), S4 (2022). [Google Scholar]
- 69.Zaiss AK, Cotter MJ, White LR, Clark SA, Wong NC, Holers VM, Bartlett JS, Muruve DA, Complement is an essential component of the immune response to adeno-associated virus vectors. J. Virol. 82, 2727–2740 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bonnemann CG, Belluscio BA , Braun S. , Morris C, Singh T, Muntoni F, A collaborative analysis by clinical trial sponsors and academic experts of anti-transgene SAEs in studies of gene therapy for DMD. Mol Ther 30 (5S1), S2 (2022). [Google Scholar]
- 71.Kornegay JN, Bogan JR, Bogan DJ, Childers MK, Grange RW, Golden retriever muscular dystrophy (GRMD): Developing and maintaining a colony and physiological functional measurements. In Duan D (ed) Muscle Gene Therapy: Methods and Protocols. Methods in Molecular Biology, vol. 709, Humana Press, New York, pp 105–123 (2011). [DOI] [PubMed] [Google Scholar]
- 72.Bartlett RJ, Winand NJ, Secore SL, Singer JT, Fletcher S, Wilton S, Bogan DJ, Metcalf-Bogan JR, Bartlett WT, Howell JM, Cooper BJ, Kornegay JN, Mutation segregation and rapid carrier detection of X-linked muscular dystrophy in dogs. Am. J. Vet. Res. 57, 650–654 (1996). [PubMed] [Google Scholar]
- 73.Patronek GJ, Waters DJ, Glickman LT, Comparative longevity of pet dogs and humans: implications for gerontology research. J. Gerontol. A. Biol. Sci. Med. Sci. 52, B171–8 (1997). [DOI] [PubMed] [Google Scholar]
- 74.McDonald CM, Abresch RT, Carter GT, Fowler WM, Jr ER Johnson, Kilmer DD, Sigford BJ, Profiles of neuromuscular diseases. Duchenne muscular dystrophy. Am. J. Phys. Med. Rehabil. 74 (Suppl 5), 70–92 (1995). [DOI] [PubMed] [Google Scholar]
- 75.Haurigot V, Mingozzi F, Buchlis G, Hui DJ, Chen Y, Basner-Tschakarjan E, Arruda VR, Radu A, Franck HG, Wright JF, Zhou S, Stedman HH, Bellinger DA, Nichols TC, High KA, Safety of AAV factor IX peripheral transvenular gene delivery to muscle in hemophilia B dogs. Mol. Ther. 18, 1318–1329 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vandenberghe LH, Xiao R, Lock M, Lin J, Korn M, Wilson JM, Efficient serotype-dependent release of functional vector into the culture medium during adeno-associated virus manufacturing. Hum. Gene Ther. 21, 1251–1257 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meng H, Janssen PM, Grange RW, Yang L, Beggs AH, Swanson LC, Cossette SA, Frase A, Childers MK, Granzier H, Gussoni E, Lawlor MW, Tissue triage and freezing for models of skeletal muscle disease. J. Vis. Exp. Jul 15;(89) (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Matsumura K, Tomé FM, Ionasescu V, Ervasti JM, Anderson RD, Romero NB, Simon D, Récan D, Kaplan JC, Fardeau M, Campbell KP, Deficiency of dystrophin-associated proteins in Duchenne muscular dystrophy patients lacking COOH-terminal domains of dystrophin. J. Clin. Invest. 92, 866–871 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.