

Abstract
Loss of intestinal barrier functions is a hallmark of inflammatory bowel disease like ulcerative colitis. The molecular mechanisms are not well understood, but likely involve dysregulation of membrane composition, fluidity, and permeability, which are all essentially regulated by sphingolipids, including ceramides of different chain length and saturation. Here, we used a loss-of-function model (CerS2+/+ and CerS2−/− mice) to investigate the impact of ceramide synthase 2, a key enzyme in the generation of very long-chain ceramides, in the dextran sodium salt (DSS) evoked model of UC. CerS2−/− mice developed more severe disease than CerS2+/+ mice in acute DSS and chronic AOM/DSS colitis. Deletion of CerS2 strongly reduced very long-chain ceramides (Cer24:0, 24:1) but concomitantly increased long-chain ceramides and sphinganine in plasma and colon tissue. In naive CerS2−/− mice, the expression of tight junction proteins including ZO-1 was almost completely lost in the colon epithelium, leading to increased membrane permeability. This could also be observed in vitro in CerS2 depleted Caco-2 cells. The increase in membrane permeability in CerS2−/− mice did not manifest with apparent clinical symptoms in naive mice, but with slight inflammatory signs such as an increase in monocytes and IL-10. AOM/DSS and DSS treatment alone led to a further deterioration of membrane integrity and to severe clinical symptoms of the disease. This was associated with stronger upregulation of cytokines in CerS2−/− mice and increased infiltration of the colon wall by immune cells, particularly monocytes, CD4+ and Th17+ T-cells, and an increase in tumor burden. In conclusion, CerS2 is crucial for the maintenance of colon barrier function and epithelial integrity. CerS2 knockdown, and associated changes in several sphingolipids such as a drop in very long-chain ceramides/(dh)-ceramides, an increase in long-chain ceramides/(dh)-ceramides, and sphinganine in the colon, may weaken endogenous defense against the endogenous microbiome.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-017-2518-9) contains supplementary material, which is available to authorized users.
Keywords: Sphingosine, Occludin, Cytokines, FITC dextran, Colon
Introduction
Inflammatory bowel diseases (IBD) affect young people between 15 and 29 years of age. There is no cure for the disease and the chronic inflammation of the colon increases the relative risk for colorectal cancer by about two to threefold [1]. IBD encompasses Crohn’s disease (CD) and ulcerative colitis (UC). While CD is a Th1-dominant immune response and affects any part of the intestine, UC is an atypical Th2-mediated disease, which is restricted to the colonic mucosa [2]. The cause of UC is still unknown, but destruction of the colonic barrier function and subsequent activation and deregulation of various immune cells are the hallmarks of UC [3, 4], which may open novel therapeutic avenues.
The colonic epithelial barrier function is mainly controlled by tight junctions and their molecular components including claudins, occludin, and the junctional adhesion molecules (Jam), which are all transmembrane proteins [5]. Zonula occludens-1 (ZO-1) is a membrane-associated protein which is essential for tight junction assembly. It connects the junctions to the actin cytoskeleton by interacting with the transmembrane proteins occludin, claudins, Jam-A, and actin [6]. Disturbance of tight junctions in UC patients has been associated with changes in expression and location of ZO-1 and other candidates [7], giving rise to the invasion of the colon wall with bacteria and other pathogens, which are normally eliminated by intestinal macrophages expressing markers like CD103, CCR9, and CD14 [8]. Together with dendritic cells (DC), the macrophages constitute one of the first innate defense lines in the gut, and are responsible for the recruitment of other immune cells to the site of infiltration, and maintenance of immune tolerance, which essentially requires control of pro-inflammatory Th17 cells by regulatory T-cells (Treg). Imbalance between Th17 and Treg cells promotes UC development [9].
Sphingolipids are essential for physiological properties of cell membranes and play a distinct role as second messengers in signaling pathways [10, 11]. Consequently, deregulation of sphingolipid metabolism is associated with a number of diseases including cancer, inflammation, demyelination disorders, or sclerotic diseases of the skin and kidney [12, 13]. Ceramide synthases (CerS) catalyze a key step in sphingolipid de novo synthesis involving the attachment of various acyl-CoA side chains to a sphingoid base. Six mammalian CerS (CerS1-6) have been described. They have chain length preferences and introduce side chains form C14-C26. CerS1 mainly synthesizes C18-Cer, CerS4 synthesizes C18-/C20-Cer; CerS5 and CerS6 mainly synthesize C14- and C16-Cer, CerS2 synthesizes the very long-chain C22/C24-Cer, and CerS3 synthesizes very long ceramides with chain length up to C34 [14, 15]. Hence, the tissue distribution and regulation of the different CerS [16] determine the ceramide profiles in specific tissues or cells. This impacts on physiological and pathophysiological processes, because their functions differ depending on the chain length [13].
Recent findings indicate that ceramides play an important role in inflammatory processes. For example, in cystic fibrosis patients, ceramides induce the upregulation of pro-inflammatory mediators [17]. CerS2 null mice, which have low levels of very long-chain ceramides (C24:0 and C24:1) but increased levels of C16-Cer [18], develop severe symptoms of LPS-induced septic shock [19], they are predisposed to inflammatory airway diseases [20] and also to liver carcinoma that spontaneously develops at about 10 months of age in these mice [21]. On the other hand, CerS2−/− mice are partially protected from experimental autoimmune encephalomyelitis, a model of multiple sclerosis, because CerS2-deficient neutrophils lack normal migratory properties, thus, limiting their infiltration into the CNS [22]. Hence, the ‘boon or bane’ of CerS2 crucially depends on the inflammatory model and the site of inflammation or autoimmune attack. Therefore, barrier functions, immune cell migratory properties, and cancer defense all depend on ceramides, and this implies that CerS2 is of particular importance for intestinal inflammatory diseases, in which all three mechanisms determine the outcome.
Therefore, we investigated here AOM/DSS-evoked colitis and colon cancer development in CerS2−/− mice in comparison to CerS2+/+ mice, in terms of the clinical symptoms and of pathophysiological correlates of the disease, including immune cell infiltration, cytokine production, sphingolipid profiles, and histopathological signs of barrier functions.
Methods
Animal models
In all experiments, the ethics guidelines for investigations in conscious animals were followed and the experiments were approved by the local Ethics Committee for Animal Research. The CerS2+/− mice were backcrossed more than ten times onto BL/6 and 129S4/SvJae (each separately). For the experiments, we used the F1 generation of 129S4/SvJae × C57BL/6, which we generated from CerS2+/− BL/6 and heterozygous 129S4/SvJae to get CerS2+/+ (wt) and CerS2−/− CerS2−(ko) mice as described previously [18]. For the chronic inflammation model, mice were treated with AOM and DSS according to [23]. In brief, one injection of 10 mg/kg azoxymethane (Sigma-Aldrich) was given intraperitoneally at day 0, followed by 2% dextran sodium sulfate (DSS) in the drinking water for 7 days. After 14 day recovery, DSS treatment was repeated twice to induce a chronic inflammation. Animals were killed after 8 or 12 weeks by carbon dioxide inhalation. For the acute inflammation model, mice were treated once for 7 days with DSS and killed on day 8.
Imaging of colon inflammation
Bioluminescence imaging was done under 1.5–2% isoflurane anesthesia with an IVIS Lumina Spectrum that employs XENOGEN technology (Caliper LifeSciences). Images of 3–5 mice per group were captured and analyzed with Living Image® software (Perkin Elmer). The inflammation of the gastrointestinal tract was assessed with the bioluminescent XenoLight RediJect Inflammation Probe® (Perkin Elmer), which allows in vivo assessment of myeloperoxidase (MPO) activity. The abdominal region was shaved to reduce the absorption of light. Inflammation Probe® (200 mg/kg, ca. 150 µl) was injected intraperitoneally 4–10 min before imaging. The IVIS settings were Epi-BLI, Em filter open, Ex filter block, fstop 1, binning 8, exposure 120 s and focus B 6.5 cm.
Preparation of tissue for molecular and histological studies
The colon was dissected and then cleared of faeces by holding the colon with forceps and flushing with a syringe filled with ice-cold HBSS without calcium and magnesium. Residual mesenteric fat tissue was resected completely. The colon was opened longitudinally and cut into 1 cm pieces. Ten mg each were quickly frozen on dry ice and stored at −80 °C for mRNA analysis and LC-MSMS. For FACS (fluorescence-activated cell sorting) and MACS (magnetic-activated cell sorting), blood and colon were used immediately. The tissue for immunohistochemistry was embedded in Tissue-Tek (SAKURA), then quickly frozen on dry ice and stored at −80 °C.
Immunohistochemistry, immunocytochemistry, and DIFF-quick staining
To assess morphologic changes in the tissue sections of the mice colon, either haematoxylin and eosin staining was used or a “Diff-Quick” kit (Labor + Technik) according to the manufacturer’s recommendations.
For immunohistochemistry, 4–10 µm tissue sections were kept in 100% ice-cold methanol for 10 min and 100% acetone for 1 min for fixation. For immunocytochemistry, Caco-2 cells were seeded on a 8-well-chamber slide, incubated for 2 days at 37 °C, 5% CO2, and fixed with 4% paraformaldehyde for 10 min. Slices were washed and permeabilized in PBS containing 0.025% Triton X-100 for 2 × 5 min, then blocked in PBS containing 3% bovine serum albumin and 10% normal goat serum for 90 min at room temperature. The sections were incubated with the primary antibody at 4 °C overnight, followed by fluorescence-labeled secondary antibodies diluted 1:800 for 2 h in PBS containing 1% bovine serum albumin and 1% normal goat serum. The following primary antibodies were used at the following dilutions: anti-CerS2, SIGMA (1:200), anti-ZO-1, Thermo-Fischer (1:200), anti-occludin SIGMA (1:200), and Cy3 anti-Rabbit IgG, SIGMA (1:800) as secondary antibody. DAPI (4′,6-Diamidine-2′-phenylindole dihydrochloride) (1:1000) (Applichem) staining was performed for 10 min. Fluorescent measurements were done with the Keyence BZ-9000 microscope at 20-fold magnification.
Click-iT plus TUNEL assay
To study the level of apoptosis in mice treated with 2% DSS, either in the acute model for 7 days or in the chronic model after treatment with AOM and three cycles of 7 days DSS, the terminal deoxynucleotidyl transferase (TdT)-dUTP nick end labeling kit was used. Cryo-embedded colon tissue was sliced into 4 µm sections and fixed with 4% paraformaldehyde. For staining of 3′-OH ends of fragmented DNA, the Click-iT Plus TUNEL Assay (life technologies) was used. In short, the TUNEL assay utilizes EdUTP, which is incorporated at the 3′-OH ends of fragmented DNA by the TdT enzyme. Detection is based on a copper catalyzed covalent reaction between Alexa Flour 488 picoyl azide dye and alkyne. The assay was performed according to the manufacturer’s instructions.
Ex vivo measurement of intestinal permeability
Intestinal permeability was determined as described previously [24]. Briefly, mice were killed by cervical dislocation and intestines from CerS2+/+ and CerS2−/− mice were removed. The intestine was cut into sections of about 5 cm and washed with 37 °C pre-heated PBS. Afterwards, one end of the intestine section was securely closed with a suture loop. From the other end, 300 µl FITC Dextran (MW ~4000) solution (1 mg/ml) was injected with a syringe and directly thereafter also securely closed with a suture loop. The intestinal section was placed into a 50 ml tube containing 20 ml pre-heated Dulbecco’s Modified Eagle Medium (DMEM) and incubated up to 2 h at 37 °C in a water bath. Every 30 min, 200 µl (100 µl twice) aliquots were taken from each tube and replaced by 200 µl fresh medium. At the end of the incubation time, samples and standards were measured for FITC with a TECAN fluorescence reader using excitation/emission at 495 nm/519 nm.
The standard curve was calculated with the R program and the FITC concentration was calculated for each sample. Afterwards, the FITC-Dex permeability of each individual intestinal sac was calculated according to the following formula:
where Q t is the cumulative concentration at time t; C t is the concentration at time t; V r is the volume at receiver side; Q tsum is the sum of all previous Q t, and V s is volume sampled. Q values were blotted versus time (t) and slope (δQ/δt) was calculated by graph Pad Prism 6. The FITC-Dex permeability (Papp) was then calculated as:
where A is the area of tissue and C 0 is the initial concentration.
Cells and reagents
The human colon cancer cell line Caco-2 (ACC-169) was purchased from Deutsche Sammlung für Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany). Caco-2 cells were cultured in Dulbecco’s MEM (modified Eagle’s medium) with Glutamax-I medium containing 10% FCS (fetal calf serum), at 37 °C in an atmosphere containing 5% CO2.
Knockdown of CerS2 in Caco-2 cells by RNA interference
The knockdown of CerS2 was performed by viral transduction of CerS2-specific shRNA (GIPZ-system, Thermo Scientific, Germany) into Caco-2 cells, as described previously [25]. A non-specific, scrambled shRNA (GIPZ-system, Thermo Scientific, Germany) was used as negative control. The constructs were transfected into HEK293T-cells with lentiviral packaging vectors using calcium-phosphate. Viruses were collected 48 h after transfection and used to infect Caco-2 cells with MOI10. Stable cell lines, Caco-CerS2−/− (knockdown of CerS2) or Caco-NC (scrambled shRNA), were generated by puromycin (3 µg/ml) selection. Transduction efficacy and permanent shRNA expression were monitored by analyzing the GFP-Tag in the GIPZ-system under fluorescence microscopy.
Detection of proteins by western blot
Membrane fractions of Caco-CerS2−/− and Caco-NC cells were prepared as described previously [26]. The protein content was determined using the Bradford method. Forty micrograms of membrane extracts were separated electrophoretically on 8–12% SDS-PAGE (sodiumdodecylsulfate-polyacrylamide gel electrophoresis) and proteins electro-blotted onto a nitrocellulose membrane (Hybond-C, Amersham, Freiburg, Germany). Membranes were stained with 0.5% ponceau in 1% acetic acid to confirm equal loading. After 2 h incubation of the membranes in blocking buffer (Odyssey blocking reagent (LI-COR Biosciences, Bad Homburg, Germany), diluted 1:1 in PBS), they were incubated overnight at 4 °C with the respective primary antibody directed against either occludin (1:500, SIGMA, mouse monoclonal), ZO-1 (1:1000, Thermo-Fischer, rabbit polyclonal), CerS2 (1:200, SIGMA, rabbit polyclonal), Actin (1:1000, SIGMA, mouse monoclonal), and GAPDH (1:1000, Ambion, mouse monoclonal) diluted in blocking buffer in the presence of 0.1% Tween 20. Membranes were then washed three times with 0.1% Tween 20 in PBS and incubated with an IRDye800 or IRDye700 conjugated secondary antibody (BIOTREND Chemikalien GmbH) in blocking buffer for 1 h. After extensive rinsing in 0.1% Tween 20 in PBS, protein-antibody complexes conjugated with IRDye800/700 were visualised on the Odyssey Infrared Imaging System (LI-COR, Biosciences).
Isolation of IEL and LP immune cells
Lamina propria cells (LPs) and intraepithelial-lymphocytes (IELs) from the colon were isolated using the Octo-MACS dissociator with heaters and the lamina propria dissociation Kit (Miltenyi) according to the manufacturer’s instructions. Briefly, large intestines were removed and carefully cleaned of their mesentery. Tumors were excised and the colon was opened longitudinally and cleared of fecal contents. The large intestine was then cut into pieces 0.5 cm in length, which were transferred into 50-ml conical tubes and shaken at 250 rpm for 20 min at 37 °C in Hanks’ balanced-salt solution without Ca2+ and Mg2+ (GIBCO) supplemented with 5% (vol/vol) FBS containing 2-mM EDTA. This process was repeated once more and followed by an additional washing step with HBSS. Cell suspension was passed through a 100-µm strainer and the remaining colon tissue was washed and then minced, transferred to a gentle MACS C tube, and dissociated using the gentle MACS Octo-Dissociator with heaters. Cell suspensions were collected, passed through a strainer, and pelleted by 10-min centrifugation at 300g.
Real-time qPCR
The mRNA from colon and Caco-2 cells was extracted by phenol–chloroform and precipitated with ethanol [27]. Briefly, about 5 mg of colon was homogenized in TRI- Reagent® (SIGMA) and chloroform (80 µl per 350 µl phenol) was added. After centrifugation, the upper aqueous phase was washed with isopropanol and ethanol, dried, and resuspended in water. The cDNA synthesis was performed using the Verso™ cDNA Synthesis kit (Thermo Scientific, Schwerte, Germany) including random hexamers. The expression levels of CerS1, CerS2, CerS3, CerS4, CerS5, CerS6, GAPDH, and PPIA (peptidyl prolyl isomerase) were determined using Maxima SYBR Green (Thermo Scientific, Schwerte, Germany) with an ABI Prism 7500 Sequence Detection System (Applied Biosystems, Austin, USA). Relative mRNA expression was determined using the comparative CT (cycle threshold) method, normalizing relative values to the expression level of murine PPIA. The sequences for the primer sets are shown in Supplement Table 1. Linearity of the assays was determined by serial dilutions of the templates for each primer set separately.
FACS analysis of cell surface immune cell markers
Single cell suspensions were prepared from WBCs (after lysis of erythrocytes), spleen (by mechanical disruption through a 70-μm cell strainer), lamina propria cells, and IELs. Non-specific antibody binding to Fc-γ receptors was blocked with mouse FcR Blocking Reagent (Miltenyi Biotec, Bergisch Gladbach, Germany) for 15 min at room temperature, followed by incubation with an antibody cocktail consisting of CD3-PE-CF594, CD4-BV711, CD11b-BV510, CD11c-AlexaFlour700, CD14-PE, CD19-APC-H7, CD25-PE-Cy7, CD80-FITC, GITR-FITC, Ly6G-APC-Cy7, MHC-II-BV605, NK1.1-PE, 7-AAD-PE-Cy5 (BD, Heidelberg, Germany), CD45-Vioblue (Miltenyi Biotec, Bergisch Gladbach, Germany), CD8-eFluor655, CD36-APC, Ly6C-PerCP-Cy5.5 (eBioscience, Frankfurt, Germany), and F4/80-PE-Cy7 (BioLegend, Fell, Germany) for 15 min at room temperature. Flow cytometric absolute count standard (Polyscience, Eppelheim, Germany) was used to determine absolute cell counts. Samples were acquired with an LSRII/Fortessa flow cytometer (BD, Heidelberg, Germany) and analyzed using FlowJo software v10 (Treestar, Ashland, USA). All antibodies were previously titrated to determine optimal concentrations. Antibody-capturing CompBeads (BD, Heidelberg, Germany) were used for single-color compensation to create multi-color compensation matrices. For gating, fluorescence minus one (FMO) controls were used. The instrument calibration was controlled daily using cytometer setup and tracking beads (BD, Heidelberg, Germany). Cell populations described in this manuscript were identified as follows: monocytes (CD45hi, CD11b+, Ly6G−, Ly6C+, and CD11c−), Tregs (CD45hi, CD3+, CD8−, CD4+, CD25+, and GITRhi), and IL-17 positive T-cells (CD8+ T-cells : CD3+, CD8+, NK1.1−, and IL-17+; immature T-cells: CD3+, CD4−, CD8−, γ/δ−, and Nk1.1− IL-17+; CD4/IL-17+: CD3+, CD4+, NK1.1−, and IL-17+).
FACS analysis of intracellular and plasma cytokines
1 × 106 isolated WBCs were cultured in RPMI + Glutamax + 10% FBS overnight and then harvested. Harvested cells were resuspended in PBS + 10% FCS and stimulated with 50-ng/ml phorbol 12-myristate 13-acetate (PMA) (Adipo-Gen) and 500-ng/ml ionomycin (SIGMA) for 3 h at 37 °C. For blocking the intracellular protein transport processes, Golgi-Stop (BD) + 5 µg Brefeldin A (Adipo-Gen) were mixed and incubated for 4 h. After washing with PBS, cells were blocked with FcR Blocking (Miltenyi) for 20 min on ice. Cells were stained extracellularly with CD45-AlexaFlour700, CD3-PE-CF594, CD4-BV711, CD8-BV650, γδ-TCR-APC, and NK1.1-BV510 (BD) for 30 min on ice. After washing with PBS, cells were fixed and permeabilized using the fixation/permeabilization solution kit (BD) according to the manufacturer’s instructions. A second blocking step for 15 min on ice was performed before intracellular staining with IL-17-PE (eBioscience) for 30 min on ice. Samples were acquired with an LSRII/Fortessa flow cytometer (BD, Heidelberg, Germany) and analyzed using FlowJo software v10 (Treestar, Ashland, USA).
For analysis of plasma cytokines (IL-10 and IL-6), we used the Cytometric Bead Array (CBA) Mouse Th1/Th2/Th17 Cytokine Kit (BD) according to the manufacturer’s instructions. In short, the assay provides several beads (of known size) conjugated with a specific antibody directed against distinct cytokines. By incubating with a fluorescence coupled antibody, the bead–analyte complex could be measured by flow cytometry, whereby the fluorescent signal is proportional to the amount of bound analyte.
Liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis of sphingolipids
Quantification of sphingolipids was performed by high-performance liquid chromatography–tandem mass spectrometry. For quantification of sphingolipids, the tissue samples were first mixed with water and homogenized to a suspension of 0.05-ng/µl tissue using a swing mill (Retsch, Haan, Germany) with 4 zirconium oxide grinding balls for each sample (25 Hz for 2.5 min).
Afterwards, 40 µl of the tissue suspension (in total 2 mg tissue) were mixed with 160-µl water, 200-µl extraction buffer (citric acid 30 mM, and disodium hydrogen phosphate 40 mM) and 20 µl of the internal standard solution containing sphingosine-d7, sphinganine-d7, sphingosine-1-phosphate-d7, C17:0 Cer, C16:0 Cer-d31, C18:0 Cer-d3, C24:0 Cer-d4, C17:0 LacCer, C18:0 DHC-d3, C16:0 LacCer-d3, and C18:0 GluCer-d5. The mixture was extracted twice with 600-µl methanol/chloroform/hydrochloric acid (15:83:2, v/v/v). The collected lower organic phases were evaporated at 45 °C under a gentle stream of nitrogen and reconstituted in 100 µl of tetrahydrofuran/water (9:1, v/v) with 0.2% formic acid and 10-mM ammonium formate. Afterwards, amounts of sphingosine, sphingosine-1-phosphate, sphinganine, sphinganine-1-phosphate, C16:0-Cer, C18:0-Cer, C18:1-Cer, C20:0-Cer, C24:0-Cer, C24:1-Cer, GluCer-C16:0, C16:0-dhCer, C18:0-dhCer, and C24:1-dhCer were analyzed by LC–MS/MS. A Luna C8 column (150 mm × 2 mm ID, 3-μm particle size, 100 Å pore size; Phenomenex, Aschaffenburg, Germany) was used for chromatographic separation. The HPLC mobile phases consisted of water with 0.2% formic acid and 2-mM ammonium formate (A) and acetonitrile/isopropanol/acetone (50:30:20, v/v/v) with 0.2% formic acid (B). For separation, a gradient program was used at a flow rate of 0.3 ml/min. The initial buffer composition 55% (A)/45% (B) was held for 0.7 min and then within 4.0 min linearly changed to 0% (A)/100% (B) and held for 13.3 min. Subsequently, the composition was linearly changed within 1.0 min to 75% (A)/25% (B) and then held for another 2.0 min. The running time was 21 min and the injection volume was 15 μl. After every sample, the column was washed by injecting the sample solvent for a 12-min run. The MS/MS analyses were performed using a triple quadrupole mass spectrometer API4000 (Sciex, Darmstadt, Germany) equipped with an ESI (Electrospray Ionization) ion source. The analysis was done in multiple reaction monitoring (MRM) mode with a dwell time of 25 ms.
Data acquisition was done using Analyst Software V 1.6 and quantification was performed with MultiQuant Software V 3.0 (both Sciex, Darmstadt, Germany), employing the internal standard method (isotope dilution mass spectrometry). Variations in accuracy of the calibration standards were less than 15% over the whole range of calibration, except for the lower limit of quantification, where a variation in accuracy of 20% was accepted.
Multi-epitope-ligand cartography (MELC)
The MELC technology is an immunohistochemical imaging method that allows the visualization of 20–40 proteins on the same slide and has been described previously [28, 29]. Briefly, colon tissue was embedded in tissue freezing medium (Leica Microsystems, Nussloch, Germany), and cryosections of 10-µm thickness were applied on silane-coated coverslips, fixed in 4% paraformaldehyde in PBS for 15 min, permeabilized with 0.1% Triton X100 in PBS for 15 min, and blocked with 3% BSA in PBS for 1 h. The sample was placed on the stage of a Leica DM IRE2 and a picture was taken. Then, by a robotic process, the sample was incubated for 15 min with bleachable fluorescence-labeled antibodies (see Table 2) and rinsed with PBS. Afterwards, the phase contrast and fluorescence signals were imaged by a cooled, charge-coupled device camera (Apogee KX4; Apogee Instruments, Roseville, CA, USA, 2× binning results in images of 1024 × 1024 pixels). To delete fluorescence signals, a bleaching step was performed. A post-bleaching image was recorded and the next antibody was applied. The post-bleaching image was subtracted from the following fluorescence image during the data analysis. Using the corresponding phase contrast images, fluorescence images produced by each antibody were aligned pixel-wise. Images were corrected for illumination faults using flat-field correction. After the MELC run, the tissue slices were stained with Diff-Quick (Dade Behring).
Table 2.
Sphingolipid levels determined by LC–MS/MS in colon and blood
| ng/mg tissue | CerS2+/+ control | CerS2+/+ DSS | CerS2−/− Control | CerS2−/− DSS | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | n | Mean | SD | n | Mean | SD | n | Mean | SD | n | |
| Sphingolipids colon | ||||||||||||
| Sphingosine | 0.22 | 0.07 | 7 | 0.25 | 0.22 | 24 | 0.33 | 0.10 | 5 | 0.23 | 0.10 | 11 |
| Sphinganine | 0.11 | 0.06 | 7 | 0.13 | 0.11 | 24 | 0.42 | 0.11 | 5 | 0.35 | 0.29 | 11 |
| S1P | 0.03 | 0.01 | 3 | 0.04 | 0.02 | 19 | 0.02 | 0.01 | 2 | 0.04 | 0.02 | 5 |
| GluCer 16:0 | 1.62 | 0.95 | 7 | 2.45 | 4.53 | 24 | 1.56 | 0.57 | 5 | 3.67 | 3.24 | 11 |
| C16:0-Cer | 10.43 | 5.53 | 7 | 7.84 | 6.01 | 22 | 13.43 | 5.56 | 5 | 6.44 | 2.46 | 11 |
| C14:0-Cer | 0.12 | 0.08 | 7 | 0.06 | 0.06 | 24 | 0.09 | 0.03 | 5 | 0.04 | 0.03 | 11 |
| C18:0-Cer | 1.92 | 1.05 | 7 | 1.75 | 0.93 | 24 | 2.58 | 1.11 | 5 | 1.21 | 0.39 | 11 |
| C20:0-Cer | 2.39 | 1.06 | 7 | 2.24 | 1.33 | 24 | 2.59 | 0.74 | 5 | 1.49 | 0.46 | 11 |
| C24:0-Cer | 1.12 | 0.69 | 6 | 1.41 | 1.19 | 24 | 0.45 | 0.10 | 3 | 0.53 | 0.40 | 6 |
| C24:1-Cer | 3.38 | 2.66 | 6 | 3.43 | 2.17 | 24 | 0.45 | 0.10 | 3 | 1.53 | 0.40 | 6 |
| C16:0-sphinganine | 0.91 | 0.39 | 7 | 0.97 | 0.96 | 24 | 1.98 | 0.85 | 5 | 1.14 | 0.39 | 11 |
| C18:0-sphinganine | 0.09 | 0.06 | 7 | 0.09 | 0.07 | 24 | 0.12 | 0.06 | 5 | 0.16 | 0.10 | 11 |
| C24:1-sphinganine | 0.16 | 0.06 | 6 | 0.22 | 0.19 | 24 | ND | ND | 5 | ND | ND | 6 |
| ng/ml plasma | CerS2+/+ control | CerS2+/+ DSS | CerS2−/− Control | CerS2−/− DSS | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | n | Mean | SD | n | Mean | SD | n | Mean | SD | n | |
| Sphingolipids plasma | ||||||||||||
| Sphingosine | 12.10 | 3.67 | 8 | 12.54 | 2.91 | 7 | 25.63 | 10.45 | 9 | 24.20 | 17.43 | 8 |
| Sphinganine | 10.19 | 5.81 | 8 | 11.81 | 2.62 | 7 | 80.03 | 47.06 | 9 | 42.94 | 31.52 | 8 |
| S1P | 479.75 | 152.60 | 8 | 312.46 | 63.99 | 7 | 1055.86 | 470.28 | 9 | 389.65 | 58.27 | 8 |
| SA1P | 157.5 | 72.92 | 8 | 74.31 | 9.20 | 7 | 468.90 | 66.18 | 4 | 370.56 | 184.35 | 8 |
| GluCer 16:0 | 262.76 | 410.64 | 8 | 670.03 | 557.45 | 7 | 13545 | 8543.9 | 9 | 2334.36 | 1931.64 | 8 |
| C16:0-Cer | 40.83 | 18.16 | 3 | 60.70 | 27.84 | 6 | 264.60 | 171.81 | 4 | 167.47 | 93.69 | 3 |
| C20:0-Cer | 49.96 | 26.12 | 8 | 99.39 | 48.64 | 7 | 26.37 | 18.46 | 9 | 42.08 | 45.79 | 8 |
| C24:0-Cer | 735.79 | 338.85 | 8 | 600.81 | 156.69 | 7 | 65.01 | 72.46 | 8 | 9.9 | 17.73 | 8 |
| C24:1-Cer | 470.75 | 295.36 | 8 | 625.90 | 151.99 | 7 | 39.66 | 72.56 | 9 | ND | ND | 8 |
Data are mean ± SD. Significance of means was examined by one-way ANOVA with post hoc Turkey’s correction. p < 0.05. Significant changes of sphingolipid levels in untreated CerS2−/− mice in comparison to untreated CerS2+/+ mice are shown in bold font
Statistics
Sphingolipid levels, mRNA levels and FACS data are presented as mean ± SEM (standard error of the mean). Statistical analyses were done with GraphPad Prism 6 software. The heatmap of Sphingolipid levels was done by “R”, standardized by z-transformation. Significant differences between groups were assessed using one-way ANOVA for three or more groups or two-tailed, two-sided Student’s t tests for two groups. In case of significant ANOVAs, groups were mutually compared with t tests employing a correction of alpha according to Tukey. Time courses, for example of body weight and colitis scores, were analyzed by rm-ANOVA for repeated measurements with the within factor “time” and between factor “treatment”. Individual outliers were identified with GraphPad Prism and were excluded. Groups comprised of 8–10 animals for clinical courses of the disease, and 3–4 animals for molecular, biochemical, and immunologic experiments. The exact numbers are given in the respective figure legends.
Results
CerS2 deficiency aggravates chronic AOM/DSS and acute DSS-induced colitis
The AOM/DSS-induced chronic and the acute DSS colitis models were used to assess whether CerS2 deficiency impacted on the development of colon inflammation and cancer development. The CerS2+/+ wild-type mice have a mixed genetic background involving Sv129, which is resistant to DSS colitis. Therefore, CerS2+/+ mice exhibited only very mild symptoms in the chronic AOM/DSS evoked disease. In contrast, CerS2−/− mice developed severe clinical symptoms (Fig. 1) including aggravated weight loss and clinical disease scores (Fig. 1a, b) and morphologic manifestations of inflammatory destruction of the colon epithelium, including ulcers, erythema due to stronger blood flow, and colon shrinkage (Fig. 1d). In vivo imaging of living mice confirmed the stronger bowel inflammation in CerS2−/− mice (Fig. 1c). After 12 weeks, we also observed an enhanced tumor burden in the large intestine in CerS2−/− mice compared with control mice. Figure 1e shows examples of different CerS2+/+ and CerS2−/− colon swiss rolls after 12-week treatment. In the acute DSS evoked disease, CerS2−/− mice also developed severe inflammation which could be detected by imaging in vivo as well as inflammatory destruction of the colon (Fig. 2a, b). Hence, CerS2−/− mice were highly susceptible to AOM/DSS and acute DSS induced colon inflammation and the associated cancer development.
Fig. 1.
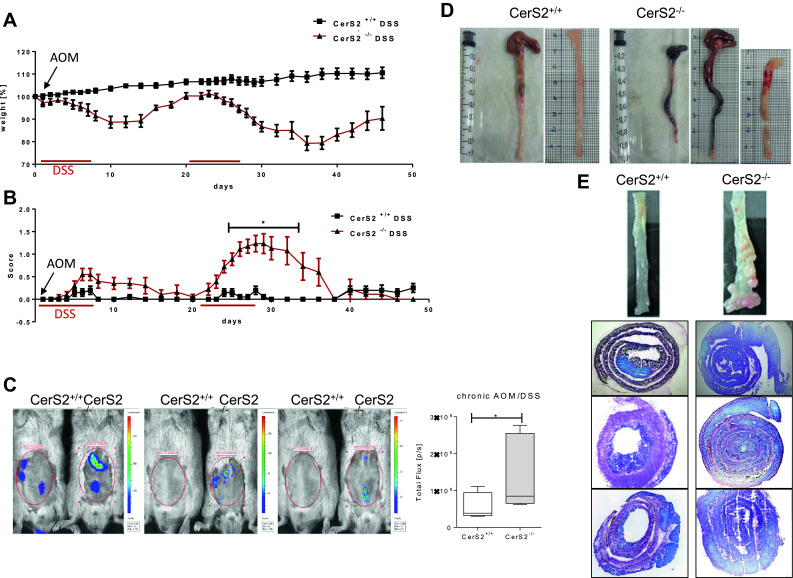
CerS2 knockout increases the clinical course and pathology in AOM/DSS induced colitis. 129S4/SvJae × C57BL/6 mice were treated once with AOM and additionally 2.0% DSS in the drinking water for seven consecutive days every 3 weeks to induce ulcerative colitis. a Time courses of body weight from CerS2−/− and CerS2+/+ mice (n = 20 in each group) show a strong loss of body weight after DSS phases. b Severity of colitis was monitored by a representative score (0 = no symptoms, 1 = diarrhea, 2 = bloody faeces, 3 = abort). Asterisks indicate statistically significant differences between groups [repeated-measures (rm) analysis of variance (ANOVA), *p < 0.05. c Bioluminescence imaging of inflammation with an IVIS Lumina Spectrum. After two cycles of DSS treatment, colitis was detected in living animals by i.p injection of 200 mg/kg inflammation probe® and subsequent detection of bioluminescence in the abdomen of CerS2+/+ and CerS2−/− mice. The total luminescent flux (p/s) in the abdominal regions of interest was quantified. Data are mean ± standard deviation (SD) of n = 9; statistical analysis was done by unpaired t test, *p < 0.05. d Representative examples of colons from CerS2+/+ and CerS2−/− mice after two cycles of DSS treatment. Colons from CerS2−/− mice are bloody and shortened in comparison to CerS2+/+ mice. e Pictures of whole colon and swiss rolls of CerS2+/+ and CerS2−/− mice after 12 weeks and three cycles of DSS
Fig. 2.

Bioluminescence imaging of inflammation and representative examples of colons from CerS2+/+ and CerS2−/− mice after acute DSS treatment. Mice were treated with 2.0% DSS in the drinking water. a Bioluminescence imaging of inflammation with an IVIS Lumina Spectrum. After 7 days of DSS treatment, colitis was detected in living animals by i.p injection of 200 mg/kg inflammation probe® and subsequent detection of bioluminescence in the abdomen of CerS2+/+ and CerS2−/− mice. The total luminescent flux (p/s) in the abdominal regions of interest was quantified. Data are mean ± standard deviation (SD) of n = 4, statistical analysis was done by unpaired t test, and *p < 0.05. b Representative example of colons from CerS2+/+ and CerS2−/− mice after 7 days of DSS treatment. Colons from CerS2−/− mice are bloody in comparison to CerS2+/+ mice
Regulation of CerS expression and shift of sphingolipid profiles after AOM/DSS treatment
We next assessed the expression of the CerS isoforms and sphingolipid profiles in plasma and colon tissue to confirm the expected profile changes from CerS2-knockout and to investigate whether AOM/DSS treatment leads to alterations in other ceramide synthase isoforms. CerS2 mRNA expression was undetectable in colon tissue from CerS2−/− mice (Table 1). AOM/DSS treatment tended to increase the expression of CerS1, CerS2, CerS3, CerS5, and glycosylceramide synthase (GCS) mRNA in the colon in wild-type CerS2+/+ mice, which did not occur or was reduced in CerS2−/− mice. Immunohistochemistry of CerS2 in colon tissue revealed that CerS2 is expressed mainly in colon epithelial cells in CerS2+/+ mice, but undetectable in CerS2−/− (Supplement 1). The localization agrees with the human tissue atlas (http://www.proteinatlas.org/ENSG00000143418-CERS2/tissue/colon#imid_6864894).
Table 1.
RNA levels of CerS1-6 and glycosylceramide synthase (GCS) in colons of CerS2+/+ and CerS2−/− mice with and without treatment of AOM and two cycles of DSS [data are the mean ± SD and mice in each group (n)]
| mRNA | CerS2+/+ control | CerS2+/+ DSS | CerS2−/− control | CerS2−/− DSS | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | n | Mean | SD | n | Mean | SD | n | Mean | SD | n | |
| CerS1 | 1.01 | 0.35 | 5 | 249.1 | 362.0 | 12 | 4.55 | 5.12 | 6 | 16.24 | 16.21 | 4 |
| CerS2 | 13.16 | 24.31 | 5 | 60.95 | 72.33 | 12 | 0.84 | 0.57 | 6 | 4.11 | 2.68 | 4 |
| CerS3 | 2.21 | 1.95 | 6 | 91.82 | 147.8 | 12 | 4.39 | 3.14 | 6 | 18.19 | 28.27 | 4 |
| CerS4 | 12.29 | 14.22 | 6 | 12.31 | 17.19 | 13 | 11.98 | 12.45 | 6 | 9.89 | 10.80 | 4 |
| CerS5 | 3.44 | 3.90 | 6 | 9.24 | 7.77 | 13 | 5.48 | 4.35 | 6 | 8.12 | 9.59 | 4 |
| CerS6 | 7.64 | 9.70 | 6 | 10.52 | 15.92 | 15 | 5.31 | 7.51 | 6 | 3.84 | 6.67 | 4 |
| GCS | 1.15 | 0.70 | 3 | 10.55 | 12.33 | 10 | 0.51 | 0.23 | 3 | 1.17 | 1.55 | 2 |
Significance of means was examined by one-way ANOVA with post hoc Turkey’s correction and revealed no significant changes; therefore, trends are indicated in different fonts (“bold” indicates that the level is upregulated in comparison to CerS2+/+ control mice, “italics” indicates that the level is downregulated in comparison to CerS2+/+ control mice)
As expected, the levels of C24:0- and C24:1-Cer, which are mainly produced by CerS2, were strongly reduced in colon and plasma of CerS2−/− mice in comparison to CerS2+/+ mice (Fig. 3; Table 2). In contrast, long-chain ceramides and dihydroceramides (C16:0 and C18:0), as well as sphinganine tended to be increased in the colon of CerS2−/− control mice as compared to CerS2+/+. In general, CerS2+/+ mice did not show any significant lipid changes after AOM/DSS treatment, which is in line with the relative resistance of this mouse line to AOM/DSS treatment due to their genetic background [30]. A non-significant increase in C24:1-Cer was detected in the colon of CerS2−/− mice after DSS treatment, likely due to the strong inflammation in the mice and in line with our previous results in the DSS model [31]. This may be due to the reverse activity of neutral ceramidase, which is able to synthesize ceramides using sphingosine and fatty acids as substrates at the plasma membrane [32, 33]. We cannot exclude the possibility that CerS3 may also contribute to this effect. The concentration of complex sphingolipids, such as GluCer-C16:0, did not change after AOM/DSS treatment in either genotype (Fig. 3; Table 2), but GluCer-C16:0 was increased in plasma of naive CerS2−/− mice, and decreased after DSS treatment (Fig. 3; Table 2). Further alterations were observed for sphingosine (S), sphingosine-1-phosphate (S1), sphinganine (SA), and sphinganine-1-phosphate (SA1P) (Fig. 3; Table 2).
Fig. 3.

Heat map of sphingolipid levels in colon (a) and plasma (b) of CerS2+/+ and CerS2−/− mice with and without treatment with AOM and two cycles of DSS. Sphingolipids were determined by LC–MS/MS. The absolute sphingolipid levels were standardized by Z-transformation (x-mean/SD) for each analyte. High values with a relative number of one are defined as dark red, middle range values are yellow (0), and low values (−1) are shown in dark green. Black squares are values which lie under the detection limit. Calculation and graphics were carried out with the R program
Disruption of membrane barrier integrity in CerS2−/− mice and human colon cells
The observed lipid profile changes pointed to alterations in membrane properties which are closely linked to barrier function. To test this hypothesis, we assessed tight junction protein expression and membrane permeability. CerS2−/− mice showed a loss of expression of zonula occludens-1 (ZO-1) and occludin expression in the colon tissue after chronic AOM/DSS and acute DSS treatment (Fig. 4a, b). While ZO-1 had a clear apical membranous localization in epithelial cells in naive CerS2+/+ mice, it showed patchy staining in the whole cytoplasm after AOM/DSS, but its expression was not lost. In contrast, in CerS2−/− mice, ZO-1 was only weakly expressed in colon epithelial cells in naive mice and almost undetectable after AOM/DSS treatment (Fig. 4a). The decrease of tight junction proteins is an early event in the DSS evoked disease, since ZO-1 and occludin diminished also in the acute DSS colitis model already after 7 days DSS in CerS2−/− mice (Fig. 4b). This is not related to apoptosis, as the results of the TUNEL assay revealed. We detected only weak TUNEL staining in the colon of CerS2−/− mice after acute DSS treatment. This was much more obvious after chronic AOM/DSS treatment. In contrast, in CerS2+/+ mice, only a few apoptotic cells could be detected in both disease models (Fig. 4c, d).
Fig. 4.
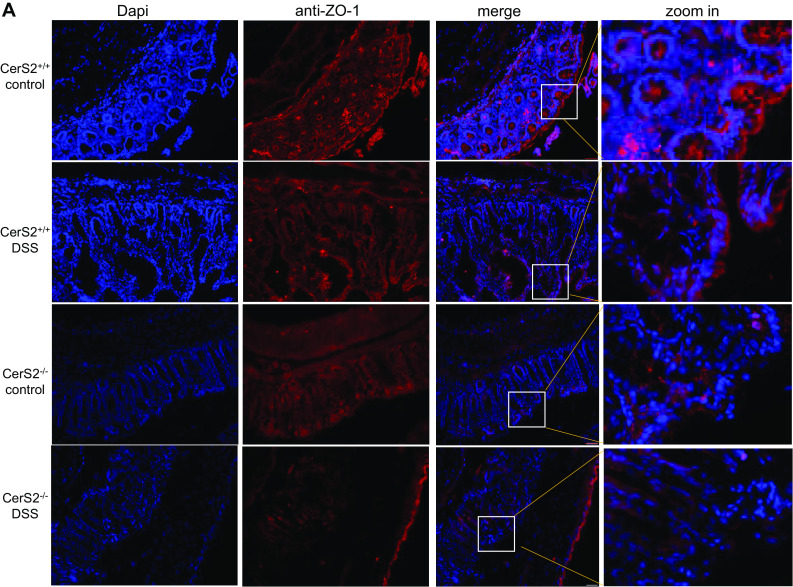
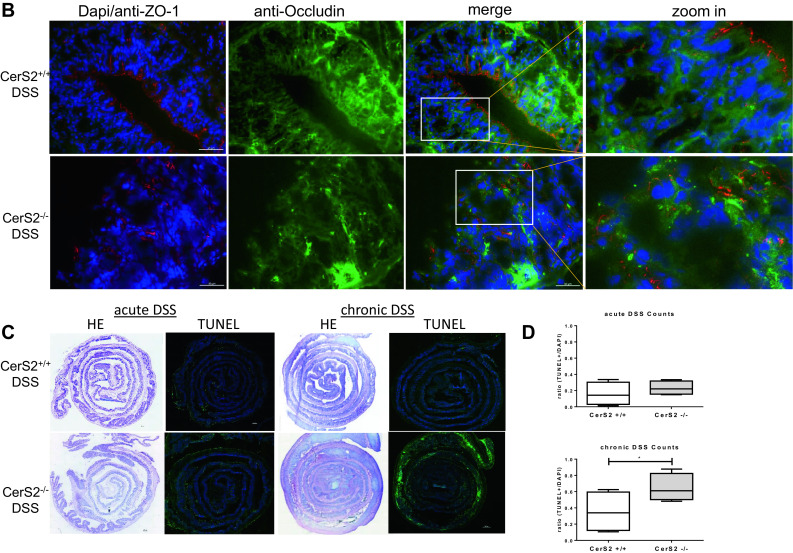
a Immuno-fluorescence staining of ZO-1 in the chronic DSS model. Colon sections were fixed with ice cold aceton and 100% ice-cold methanol and permeabilized in PBS containing 0.025% Triton X-100. After blocking, the tissue sections were incubated with anti-ZO-1 (1:200) at 4 °C overnight, followed by a Cy3 labeled anti-rabbit antibody diluted 1:800 for 2 h. Subsequently, tissue sections were stained with DAPI (1:1000). Fluorescent measurements were done with the Keyence BZ-9000 microscope with 20-fold magnification. Representative pictures are shown from CerS2+/+ and CerS2−/− mice with or without AOM and two cycles of DSS treatment. Red bars indicate 50 µm. b Immuno-fluorescence staining of ZO-1 and occludin on 4-µm colon sections of CerS2+/+ and CerS2−/− treated with DSS for 7 days. Fluorescent measurements were done with the Keyence BZ-9000 microscope at 40-fold magnification. White bars indicate 100 µm. c H&E staining and TUNEL assay of 4-µm colon sections from mice treated with DSS for 7 days (acute DSS model) as well as after treatment with AOM and 3× 7 days DSS (chronic DSS model). TUNEL positive cells are stained by a Click-iT reaction with AF-488 and subsequently stained with DAPI. Single pictures were taken with the Keyence BZ-9000 microscope with tenfold magnification and recomposed by the software. d AF-488 positive cells and Dapi positive cells were counted by the Keyence software. The total count of TUNEL positive cells in each picture (as shown in c) was normalized to Dapi. Data are mean ± standard deviation (SD) of n = 5 each group. Statistical analysis was done by unpaired t test, *p ≤ 0.05
To extend these data to the human system, we depleted CerS2 in Caco-2 cells. In shRNACerS2 transduced Caco-2 cells, the very long-chain sphingolipids were reduced, whereas long-chain sphingolipids increased (Supplement 2). Western-blot analysis of ZO-1 and occludin demonstrated a clear reduction of both proteins in CerS2-depleted Caco2 cells (Caco-shCerS2) in comparison to Caco-2 cells transfected with a control shRNA (Caco-NC) (Fig. 5a).
Fig. 5.
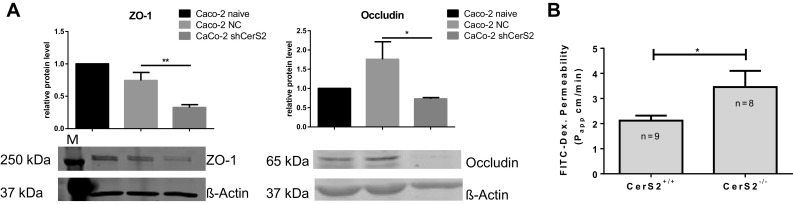
a ZO-1 and occludin expression in human Caco-2 cells after lentiviral depletion of CerS2. Western blot analysis of ZO-1 (1:1000) and occludin (1:500) in membrane fractions isolated from confluent grown CaCo-2 cells. Values are presented as mean ± SD, *p < 0.05 of three independent replicates. b Ex vivo FITC dextran permeability assay of CerS2−/− and CerS2+/+ mice colons. Permeability of intestinal sacs was calculated by measurement of FITC dextran diffusion from the sac lumen into the outer medium. FITC-Dex. permeability of each sac is shown for untreated CerS2+/+ and CerS2−/− mice. Significance of means was examined with unpaired t test. Data are mean ± SEM
Our immunohistochemical data indicated that even in untreated CerS−/− mice, the barrier function is already impaired in comparison to CerS2+/+ mice. To test this idea, we measured the permeability of the intestine ex vivo using FITC dextran. The classical in vivo permeability assay using oral FITC dextran was not applicable to our mice, because the motility of cilia depends on C24:1-Cer [34], which was reduced in CerS2−/− mice, hence resulting in differences in intestinal transport of FITC dextran. To circumvent this problem, we used the ex vivo permeability assay [24] (Fig. 5b). The permeability of colon sacs in CerS2−/− control mice was significantly increased compared to CerS2+/+ control mice (Fig. 5b).
Deregulation of immune cell balance in CerS2−/− mice
The observed disruption of barrier function in the colonic epithelial layer in CerS2−/− mice may affect the immunologic defense of the colon wall. To address this question, we analyzed immune cell subtypes in blood and colon epithelium (intestinal epithelial lymphocytes, IEL) and lamina propria lymphocytes (LPL) by FACS and MELC (muli-epitope ligand cartography) analysis. Blood monocytes (CD11b+, Ly6C+, and Ly6G−) were increased in naive CerS2−/− (Fig. 6a), accompanied by a higher IL-10 level in plasma (Fig. 6c), suggesting that the immune system was activated even without AOM/DSS. After DSS treatment, there was a strong increase in monocytes, Tregs, and IL-17+ T-cells in blood and colon of CerS2−/− mice (Fig. 6a, b), associated with an increase in IL-6 in plasma (Fig. 6c), none of which occured in CerS2+/+ mice, again showing that the controls were fairly resistant to the disease. MELC analysis confirmed the changes in the immune cell distribution in the colon, which is exemplified by the monocyte infiltration. CD11b+, Ly6C+, and F480 positive cells were more abundant in CerS2−/− mice compared with CerS2+/+ mice (Supplement 3 A–D).
Fig. 6.
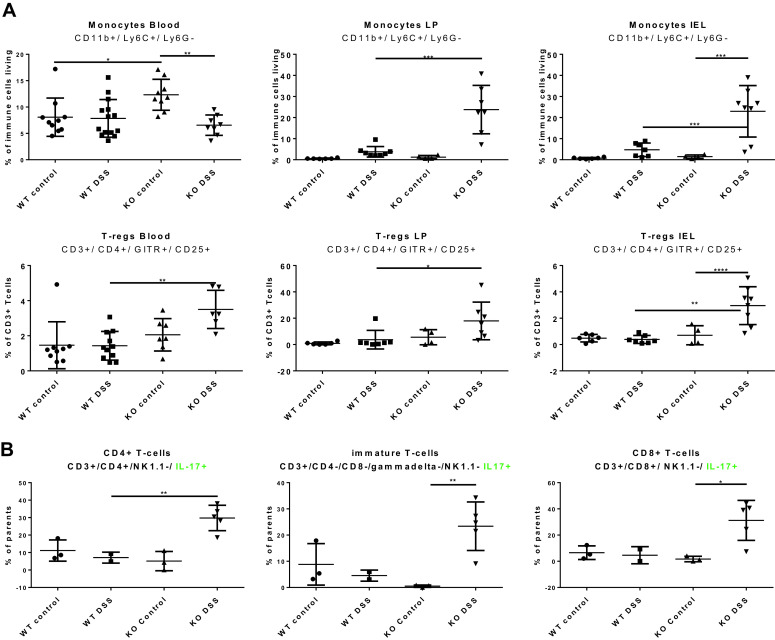
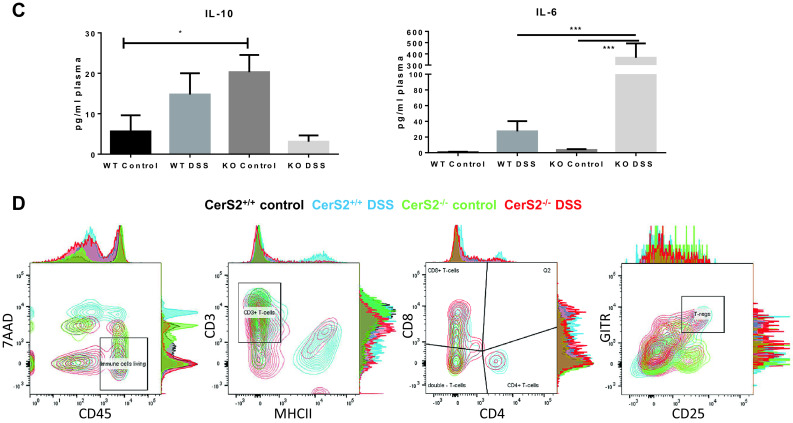
a FACS analysis of immune cells in blood and colon (lamina propria and intraepithelial leukocytes) of CerS2+/+ and CerS2−/− mice with and without AOM and two cycles of DSS treatment. Monocytes were identified as CD3−, CD11b+, Ly6C+, and Ly6G−. Tregs are CD3+, CD4+ GITR+, and CD25+. b FACS analysis of IL-17 positive T-cells in blood of CerS2+/+ and CerS2−/− mice with and without AOM and after two cycles of DSS treatment. The following markers were used for the identification of the different subtypes: CD8+ T-cells: CD3+, CD8+, NK1.1−, IL-17+; immature T-cells: CD3+, CD4−, CD8−, γ/δ−, and Nk1.1− IL-17+; CD4/IL-17+: CD3+, CD4+, NK1.1−, and IL-17+. c Plasma cytokine concentration in CerS2+/+ and CerS2−/− mice with and without AOM and two cycles of DSS treatment. Data are the mean ± SEM, n = 6–13 in each group. Statistical analyses were done with GraphPad Prism 6 software. Significance of means was examined with one-way ANOVA with post hoc Tukey’s correction p < 0.05. Representative FACS gates are presented in d. Gating strategy to determine Tregs is shown. Blood cells from mice were first selected for CD45 positive cells. CD45 positive cells were further selected for CD3-positive/MHC-II-negative cells. CD3-positive cells were selected for CD8 or CD4 expression and CD4-positive cells additionally were selected for CD25 and GITR expression. CD25/GITR positive cells were Tregs
Discussion
The present study shows that CerS2−/− mice are much more sensitive towards AOM/DSS evoked colitis than CerS2+/+ mice, owing to a disruption of intestinal epithelial barrier functions, resulting in stronger infiltration of pro-inflammatory lymphocytes and myeloid cells. Long-standing colonic inflammation is associated with an enhanced risk of colorectal cancer, as we confirmed in our CerS2−/− mice. The molecular mechanisms by which inflammation promotes clonal expansion are not fully understood, but Choi et al. proposed that IBD is related to clonal evolution based on three major mechanisms: IBD-associated inflammation (I) generates a mutagenic pressure, (II) provides a selective advantage to those cells able to survive an inflammatory insult, and (III) provides a selective advantage to cells able to repopulate the healing mucosa more rapidly [35]. The way in which CerS2 deficiency affects these mechanisms is not known and will be investigated in future experiments.
Previously, it was shown that the membrane structure of CerS2−/− mice is altered in comparison to CerS2+/+ mice, particularly that of detergent-resistant membranes (DRMs) [36], which contribute to the organization of tight junctions and the regulation of paracellular permeability in epithelial cells [37]. In particular, claudins and occludin accumulate in these DRMs and displacement of these tight junction proteins impairs membrane barrier functions [38]. The loss of zonula occludens-1 and occludin in the colon of CerS2−/− mice points to disturbances of tight junction assembly. The direct impact of CerS2 on tight junction proteins could be confirmed by western blots of CerS-2 depleted Caco-2 cells which also displayed reduced ZO-1 and occludin expression in comparison to control Caco-2 cells. Further membrane barrier proteins, such as the gap junction protein, connexin 32, also depend on their localization to DRMs. Consequently, connexin 32 was dislocated and increasingly degraded in CerS2−/− mice [39], likely contributing to the loss of intestinal barrier function.
CerS and GCS in the colon were upregulated in CerS2+/+ but not in CerS−/− mice after AOM/DSS treatment. Clearly, the CerS2 response is deficient in CerS2−/− mice, but whether this is related to cytokine effects, cell–cell contact, or intracellular signaling pathway cannot be concluded on the basis of our current data. It has been shown that membrane microdomains enriched with very long-chain LacCer are signaling platforms for membrane receptors that bind to microorganism [40]. The glycosylated very long-chain ceramides are reduced in CerS2−/− mice as well as in CerS2-depleted Caco-2 cells. Thus, alterations in microdomains might be responsible for the reduced effect on CerS and GCS expression in the CerS2−/− mice. Very little is known about the stimuli regulating CerS expression, but we know that CerS expression is regulated at the transcriptional, posttranscriptional as well as posttranslational level [16, 41], so that multiple factors may play a role. Changes in CerS expression did not result in changes in the respective ceramides, which could be attributed to the fact that ceramides act as substrates for several other enzymes (like glucosylceramide synthase, sphingomyelin synthase, and ceramidase) and are rapidly metabolized to more complex sphingolipids or degraded. Therefore, CerS2 deficiency not only affected ceramide profiles, but also sphingosine-1-phosphate (S1P), which is another major player in DSS-induced colitis. Knockout or inhibition of sphingosine kinase 1 (SK1) or blocking of sphingosine-1-phosphate receptors (S1PRs) attenuated DSS induced inflammation in mice [42–45]. In contrast, deletion of intestinal neutral ceramidase [46], leading to a decrease in S1P in colon epithelium but not in plasma, worsens inflammation after DSS treatment [47] and inversely, deletion of S1P phosphatase 2 (SGPP2) leading to an increase in intestinal S1P, attenuated DSS induced colitis [48]. Hence, although somewhat complex, it appears that S1P derived from colon epithelium protects against DSS-induced inflammation, whereas high plasma S1P promotes DSS-evoked colitis, likely by affecting immune cell homing and egress to and from lymphoid organs [49]. In line with this interpretation, CerS2−/− mice had a high plasma S1P level in naive mice without DSS, which might result in enhanced immune cell traffic to the colon, resulting in high monocyte infiltration and increased IL-17 expressing lymphocytes. Thus, S1P may be involved, but its inverse regulation and function in plasma and colon remains unresolved.
The loss of colon epithelial barrier function is likely a major reason for the aggravation of the disease in CerS2−/− mice. Beside the differences in membrane physiology discussed above, direct epithelial damage may contribute to this effect. Sphinganine and sphingosine as well as the imbalances between long-chain and very long-chain ceramides/(dh)ceramides are toxic for colon cancer cells [50–53]. Furthermore, it has been shown that medium-chain-length fatty acids as well as Western-style diet, which contains high amounts of medium- to long-chain-length fatty acids, augment DSS induced colitis in mice [54, 55]. Instead, long-chain polyunsaturated fatty acids (PUFA) with n-3 have been shown to be protective in various colitis models [56]. Until now, the possibility that the inclusion of very long-chain sphingolipids to diets impacts the outcome of colitis has not been tested. However, due to the fact that these lipids are not cell permeable, the benefit is likely to be questionable.
In conclusion, we show that CerS2 deficiency increases the susceptibility towards AOM/DSS induced chronic colitis as well as acute colitis, resulting from imbalances in ceramide profiles, in particular a loss of very long-chain ceramides in favour of the shorter variants, which leads to disruption of intestinal epithelial barrier functions with enhanced immune cell infiltration (Fig. 7). The barrier disruption was evident in naive CerS2−/− mice and only intensified with AOM/DSS, suggesting that CerS2-deficiency causes a life-long defect of pathogen defense, possibly with microbiome changes and an increased risk of autoimmune-mediated inflammatory bowel disease. Translated to humans, it may be useful to diagnose ceramide profile changes in colon biopsies and eventually reset the balance by alimentary treatments.
Fig. 7.
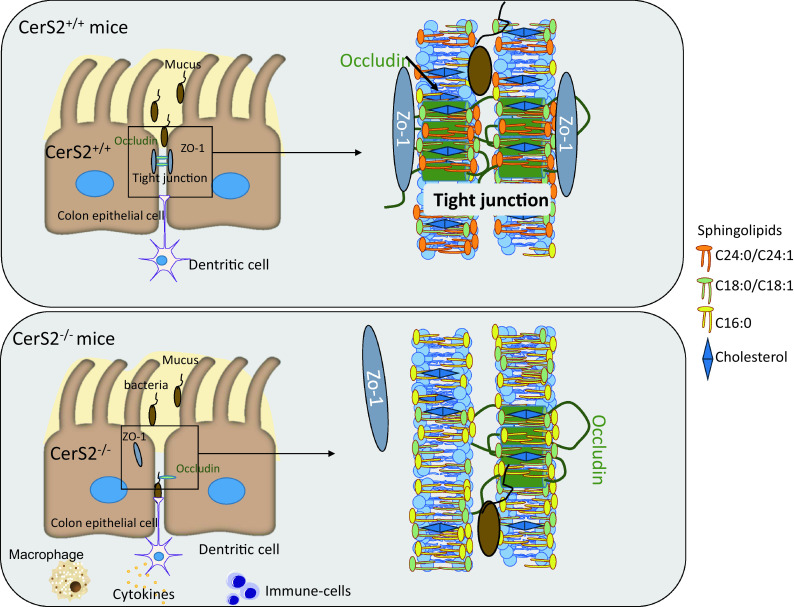
Scheme of the molecular mechanisms contributing to severe colitis in CerS2−/− mice. In CerS2−/− mice, the formation of tight junctions is disturbed by the loss of very long-chain sphingolipids in the membrane of colon epithelial cells. This leads to an invasion of bacteria from the colon lumen and activation of the immune system. DSS treatment enhances this effect as it leads to a further destruction of tight junctions and enhanced inflammation resulting in colitis
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplement 1 Immuno-fluorescence staining of CerS2. Colon sections were fixed with 100% ice-cold methanol and permeabilized in PBS containing 0.025% Triton X-100. Subsequent tissue sections were either stained immunohistochemically by DIFF-Quick staining or used for immuno-fluorescence staining. This procedure determines whether the DIFF-Quick sections are comparable to the fluorescence stained section. After blocking, the tissue sections were incubated with anti-CerS2 at 4 °C overnight, followed by a Cy3 labeled anti-rabbit antibody diluted 1:800 for 2 h. Subsequently, tissue sections were stained with DAPI (1:1000). Fluorescent measurements were done with the Keyence BZ-9000 microscope with 20-fold magnification. Representative pictures are shown from CerS2+/+ and CerS2−/− mice with or without AOM and two cycles of DSS treatment. (PPTX 2860 kb)
Supplement 2 Depletion of CerS2 in human Caco-2 cells. CerS2 was depleted in Caco-2 cells by lentiviral transduction of a CerS2-shRNA. As a control, we transduced a non-specific (NC) shRNA. A) mRNA for CerS expressed in Caco-2 cells (CerS1 and Cer3 are only very weakly expressed in Caco-2 cells). CerS2 mRNA was reduced by 80% in CerS2-shRNA treated Caco-2 cells. B) CerS2-protein expression was also significantly reduced in these cells. We show one representative Western-blot of three and the calculation of the relative protein expression (CerS2/GAPDH, CaCo-2 untreated cells were set to 100%) based on findings from three blots. C) Down-regulation of CerS2 in Caco-2 cells led to alterations in various sphingolipid levels. Sphingolipid levels were determined by LC-MS/MS. Changes in sphingolipids are comparable to those observed in the colon of CerS2−/− mice in comparison to CerS2+/+ mice (see Table 2). Statistical analysis was done by unpaired t test, p* < 0.05; p** < 0.01; p*** < 0.001 (PPTX 413 kb)
Supplement 3 MELC analysis of colon sections from CerS2+/+ and CerS2−/− mice with and without AOM and two cycles of DSS treatment. Ten micrometer cryosections of the colon were applied to silane-coated coverslips, fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X100, and blocked with 3% BSA. The sample was placed on the stage of an Leica DM IRE2 and a phase contrast picture was taken. Then, the sample was automatically incubated for 15 min with fluorescence-labeled antibodies [FITC-Ly-6C (Gr1) antibody, FITC-CD11b and FITC-F4/80 antibody] and rinsed with PBS. To delete fluorescence signals, a bleaching step was performed after each antibody incubation step and at least 7-amino actinomycin D (7-AAD) was used to stain nuclei. After the MELC run, the tissue slices were stained with Diff-Quick (Dade Behring) (PPTX 10272 kb)
Supplement Table 1 RT-PCR primer used in this study. (PPTX 41 kb)
Acknowledgements
We thank Prof. Anthony Futerman and Dr. Yael Pewzner-Jung for providing us CerS2-wt and -ko breeding mice. Many thanks to Kerstin Birod and Annett Häussler for technical assistance. Furthermore, the authors would like to thank Prof. Simone Fulda for kindly providing viral packaging plasmids. The authors thank Prof. Dr. Michael Parnham for the linguistic revision of the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) [SFB 1039/1, A03, A08, B03, B04, B05, B06, Z01]; DFG project [GR2011/3-2], and the LOEWE Lipid Signaling Forschungszentrum Frankfurt (LiFF).
Author contributions
SO: data collection, data analysis, and writing; KSch: Melk-analysis; AW: FACS analysis, writing; DT and ST: LC–MSMS analysis; JSch and NF: data analysis and calculation; M-SW: data interpretation; HHR: AOM/DSS mouse model; BB, GG, and IT: writing and figures, SG: study design, writing, data interpretation, data collection, and data analysis.
References
- 1.Carter MJ, Lobo AJ, Travis SP, Ibd Section BSoG Guidelines for the management of inflammatory bowel disease in adults. Gut. 2004;53(Suppl 5):V1–16. doi: 10.1136/gut.2004.043372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474(7351):298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 4.Lopez-Posadas R, Neurath MF, Atreya I. Molecular pathways driving disease-specific alterations of intestinal epithelial cells. Cell Mol Life Sci. 2017;74(5):803–826. doi: 10.1007/s00018-016-2363-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suzuki T. Regulation of intestinal epithelial permeability by tight junctions. Cell Mol Life Sci. 2013;70(4):631–659. doi: 10.1007/s00018-012-1070-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wroblewski LE, Peek RM., Jr Targeted disruption of the epithelial-barrier by Helicobacter pylori. Cell Commun Signal. 2011;9(1):29. doi: 10.1186/1478-811X-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kucharzik T, Walsh SV, Chen J, Parkos CA, Nusrat A. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol. 2001;159(6):2001–2009. doi: 10.1016/S0002-9440(10)63051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gren ST, Grip O. Role of monocytes and intestinal macrophages in Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2016;22(8):1992–1998. doi: 10.1097/MIB.0000000000000824. [DOI] [PubMed] [Google Scholar]
- 9.Gong Y, Lin Y, Zhao N, He X, Lu A, Wei W, Jiang M. The Th17/Treg immune imbalance in ulcerative colitis disease in a Chinese Han population. Mediators Inflamm. 2016;2016:7089137. doi: 10.1155/2016/7089137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol. 2008;8(10):753–763. doi: 10.1038/nri2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castro BM, Prieto M, Silva LC. Ceramide: a simple sphingolipid with unique biophysical properties. Prog Lipid Res. 2014;54:53–67. doi: 10.1016/j.plipres.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 12.Aguilera-Romero A, Gehin C, Riezman H. Sphingolipid homeostasis in the web of metabolic routes. Biochim Biophys Acta. 2014;1841(5):647–656. doi: 10.1016/j.bbalip.2013.10.014. [DOI] [PubMed] [Google Scholar]
- 13.Grosch S, Schiffmann S, Geisslinger G. Chain length-specific properties of ceramides. Prog Lipid Res. 2012;51(1):50–62. doi: 10.1016/j.plipres.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 14.Ben-David O, Futerman AH. The role of the ceramide acyl chain length in neurodegeneration: involvement of ceramide synthases. Neuromol Med. 2010;12(4):341–350. doi: 10.1007/s12017-010-8114-x. [DOI] [PubMed] [Google Scholar]
- 15.Eckl KM, Tidhar R, Thiele H, Oji V, Hausser I, Brodesser S, Preil ML, Onal-Akan A, Stock F, Muller D, Becker K, Casper R, Nurnberg G, Altmuller J, Nurnberg P, Traupe H, Futerman AH, Hennies HC. Impaired epidermal ceramide synthesis causes autosomal recessive congenital ichthyosis and reveals the importance of ceramide acyl chain length. J Invest Dermatol. 2013;133(9):2202–2211. doi: 10.1038/jid.2013.153. [DOI] [PubMed] [Google Scholar]
- 16.Wegner MS, Schiffmann S, Parnham MJ, Geisslinger G, Grosch S. The enigma of ceramide synthase regulation in mammalian cells. Prog Lipid Res. 2016;63:93–119. doi: 10.1016/j.plipres.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Becker KA, Riethmuller J, Zhang Y, Gulbins E. The role of sphingolipids and ceramide in pulmonary inflammation in cystic fibrosis. Open Respir Med J. 2010;4:39–47. doi: 10.2174/1874306401004010039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pewzner-Jung Y, Park H, Laviad EL, Silva LC, Lahiri S, Stiban J, Erez-Roman R, Brugger B, Sachsenheimer T, Wieland F, Prieto M, Merrill AH, Jr, Futerman AH. A critical role for ceramide synthase 2 in liver homeostasis: I. alterations in lipid metabolic pathways. J Biol Chem. 2010;285(14):10902–10910. doi: 10.1074/jbc.M109.077594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ali M, Saroha A, Pewzner-Jung Y, Futerman AH. LPS-mediated septic shock is augmented in ceramide synthase 2 null mice due to elevated activity of TNFalpha-converting enzyme. FEBS Lett. 2015;589(17):2213–2217. doi: 10.1016/j.febslet.2015.06.045. [DOI] [PubMed] [Google Scholar]
- 20.Petrache I, Kamocki K, Poirier C, Pewzner-Jung Y, Laviad EL, Schweitzer KS, Van Demark M, Justice MJ, Hubbard WC, Futerman AH. Ceramide synthases expression and role of ceramide synthase-2 in the lung: insight from human lung cells and mouse models. PLoS One. 2013;8(5):e62968. doi: 10.1371/journal.pone.0062968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pewzner-Jung Y, Brenner O, Braun S, Laviad EL, Ben-Dor S, Feldmesser E, Horn-Saban S, Amann-Zalcenstein D, Raanan C, Berkutzki T, Erez-Roman R, Ben-David O, Levy M, Holzman D, Park H, Nyska A, Merrill AH, Jr, Futerman AH. A critical role for ceramide synthase 2 in liver homeostasis: II. Insights into molecular changes leading to hepatopathy. J Biol Chem. 2010;285(14):10911–10923. doi: 10.1074/jbc.M109.077610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barthelmes J, de Bazo AM, Pewzner-Jung Y, Schmitz K, Mayer CA, Foerch C, Eberle M, Tafferner N, Ferreiros N, Henke M, Geisslinger G, Futerman AH, Grosch S, Schiffmann S. Lack of ceramide synthase 2 suppresses the development of experimental autoimmune encephalomyelitis by impairing the migratory capacity of neutrophils. Brain Behav Immun. 2015;46:280–292. doi: 10.1016/j.bbi.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 23.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2(3):541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 24.Mateer SW, Cardona J, Marks E, Goggin BJ, Hua S, Keely S. Ex vivo intestinal sacs to assess mucosal permeability in models of gastrointestinal disease. J Vis Exp. 2016;108:e53250. doi: 10.3791/53250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moser CV, Meissner M, Laarmann K, Olbrich K, King-Himmelreich TS, Wolters MC, Geisslinger G, Niederberger E. The protein kinase IKKepsilon contributes to tumour growth and tumour pain in a melanoma model. Biochem Pharmacol. 2016;103:64–73. doi: 10.1016/j.bcp.2015.12.016. [DOI] [PubMed] [Google Scholar]
- 26.Maier TJ, Janssen A, Schmidt R, Geisslinger G, Grosch S. Targeting the beta-catenin/APC pathway: a novel mechanism to explain the cyclooxygenase-2-independent anticarcinogenic effects of celecoxib in human colon carcinoma cells. FASEB J. 2005;19(10):1353–1355. doi: 10.1096/fj.04-3274fje. [DOI] [PubMed] [Google Scholar]
- 27.Chomczynski P. An reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques. 1993;15:532–537. [PubMed] [Google Scholar]
- 28.Pierre S, Scholich K. Toponomics: studying protein–protein interactions and protein networks in intact tissue. Mol BioSyst. 2010;6(4):641–647. doi: 10.1039/b910653g. [DOI] [PubMed] [Google Scholar]
- 29.Pierre S, Maeurer C, Coste O, Becker W, Schmidtko A, Holland S, Wittpoth C, Geisslinger G, Scholich K. Toponomics analysis of functional interactions of the ubiquitin ligase PAM (Protein Associated with Myc) during spinal nociceptive processing. Mol Cell Proteomics. 2008;7(12):2475–2485. doi: 10.1074/mcp.M800201-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Perse M, Cerar A. Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol. 2012;2012:718617. doi: 10.1155/2012/718617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zschiebsch K, Fischer C, Pickert G, Haeussler A, Radeke H, Grosch S, Ferreiros N, Geisslinger G, Werner ER, Tegeder I. Tetrahydrobiopterin attenuates DSS-evoked colitis in mice by rebalancing redox and lipid signaling. J Crohns Colitis. 2016;10(8):965–978. doi: 10.1093/ecco-jcc/jjw056. [DOI] [PubMed] [Google Scholar]
- 32.Hwang YH, Tani M, Nakagawa T, Okino N, Ito M. Subcellular localization of human neutral ceramidase expressed in HEK293 cells. Biochem Biophys Res Commun. 2005;331(1):37–42. doi: 10.1016/j.bbrc.2005.03.134. [DOI] [PubMed] [Google Scholar]
- 33.El Bawab S, Birbes H, Roddy P, Szulc ZM, Bielawska A, Hannun YA. Biochemical characterization of the reverse activity of rat brain ceramidase. A CoA-independent and fumonisin B1-insensitive ceramide synthase. J Biol Chem. 2001;276(20):16758–16766. doi: 10.1074/jbc.M009331200. [DOI] [PubMed] [Google Scholar]
- 34.Kong JN, Hardin K, Dinkins M, Wang G, He Q, Mujadzic T, Zhu G, Bielawski J, Spassieva S, Bieberich E. Regulation of Chlamydomonas flagella and ependymal cell motile cilia by ceramide-mediated translocation of GSK3. Mol Biol Cell. 2015;26(24):4451–4465. doi: 10.1091/mbc.E15-06-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi CR, Bakir IA, Hart AL, Graham TA. Clonal evolution of colorectal cancer in IBD. Nat Rev Gastroenterol Hepatol. 2017;14(4):218–229. doi: 10.1038/nrgastro.2017.1. [DOI] [PubMed] [Google Scholar]
- 36.Park JW, Park WJ, Kuperman Y, Boura-Halfon S, Pewzner-Jung Y, Futerman AH. Ablation of very long acyl chain sphingolipids causes hepatic insulin resistance in mice due to altered detergent-resistant membranes. Hepatology. 2013;57(2):525–532. doi: 10.1002/hep.26015. [DOI] [PubMed] [Google Scholar]
- 37.Nusrat A, Parkos CA, Verkade P, Foley CS, Liang TW, Innis-Whitehouse W, Eastburn KK, Madara JL. Tight junctions are membrane microdomains. J Cell Sci. 2000;113(Pt 10):1771–1781. doi: 10.1242/jcs.113.10.1771. [DOI] [PubMed] [Google Scholar]
- 38.Sugibayashi K, Onuki Y, Takayama K. Displacement of tight junction proteins from detergent-resistant membrane domains by treatment with sodium caprate. Eur J Pharm Sci. 2009;36(2–3):246–253. doi: 10.1016/j.ejps.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 39.Park WJ, Park JW, Erez-Roman R, Kogot-Levin A, Bame JR, Tirosh B, Saada A, Merrill AH, Jr, Pewzner-Jung Y, Futerman AH. Protection of a ceramide synthase 2 null mouse from drug-induced liver injury: role of gap junction dysfunction and connexin 32 mislocalization. J Biol Chem. 2013;288(43):30904–30916. doi: 10.1074/jbc.M112.448852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iwabuchi K, Nakayama H, Iwahara C, Takamori K. Significance of glycosphingolipid fatty acid chain length on membrane microdomain-mediated signal transduction. FEBS Lett. 2010;584(9):1642–1652. doi: 10.1016/j.febslet.2009.10.043. [DOI] [PubMed] [Google Scholar]
- 41.Wegner MS, Wanger RA, Oertel S, Brachtendorf S, Hartmann D, Schiffmann S, Marschalek R, Schreiber Y, Ferreiros N, Geisslinger G, Grosch S. Ceramide synthases CerS4 and CerS5 are upregulated by 17beta-estradiol and GPER1 via AP-1 in human breast cancer cells. Biochem Pharmacol. 2014;92(4):577–589. doi: 10.1016/j.bcp.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 42.Snider AJ, Kawamori T, Bradshaw SG, Orr KA, Gilkeson GS, Hannun YA, Obeid LM. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J. 2009;23(1):143–152. doi: 10.1096/fj.08-118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schulze T, Golfier S, Tabeling C, Rabel K, Graler MH, Witzenrath M, Lipp M. Sphingosine-1-phospate receptor 4 (S1P(4)) deficiency profoundly affects dendritic cell function and TH17-cell differentiation in a murine model. FASEB J. 2011;25(11):4024–4036. doi: 10.1096/fj.10-179028. [DOI] [PubMed] [Google Scholar]
- 44.Maines LW, Fitzpatrick LR, French KJ, Zhuang Y, Xia Z, Keller SN, Upson JJ, Smith CD. Suppression of ulcerative colitis in mice by orally available inhibitors of sphingosine kinase. Dig Dis Sci. 2008;53(4):997–1012. doi: 10.1007/s10620-007-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deguchi Y, Andoh A, Yagi Y, Bamba S, Inatomi O, Tsujikawa T, Fujiyama Y. The S1P receptor modulator FTY720 prevents the development of experimental colitis in mice. Oncol Rep. 2006;16(4):699–703. [PubMed] [Google Scholar]
- 46.Kono M, Dreier JL, Ellis JM, Allende ML, Kalkofen DN, Sanders KM, Bielawski J, Bielawska A, Hannun YA, Proia RL. Neutral ceramidase encoded by the Asah2 gene is essential for the intestinal degradation of sphingolipids. J Biol Chem. 2006;281(11):7324–7331. doi: 10.1074/jbc.M508382200. [DOI] [PubMed] [Google Scholar]
- 47.Snider AJ, Wu BX, Jenkins RW, Sticca JA, Kawamori T, Hannun YA, Obeid LM. Loss of neutral ceramidase increases inflammation in a mouse model of inflammatory bowel disease. Prostaglandins Other Lipid Mediat. 2012;99(3–4):124–130. doi: 10.1016/j.prostaglandins.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang WC, Liang J, Nagahashi M, Avni D, Yamada A, Maceyka M, Wolen AR, Kordula T, Milstien S, Takabe K, Oravecz T, Spiegel S. Sphingosine-1-phosphate phosphatase 2 promotes disruption of mucosal integrity, and contributes to ulcerative colitis in mice and humans. FASEB J. 2016;30(8):2945–2958. doi: 10.1096/fj.201600394R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maeda Y, Seki N, Kataoka H, Takemoto K, Utsumi H, Fukunari A, Sugahara K, Chiba K. IL-17-producing Vgamma4 + gammadelta T cells require sphingosine 1-phosphate receptor 1 for their egress from the lymph nodes under homeostatic and inflammatory conditions. J Immunol. 2015;195(4):1408–1416. doi: 10.4049/jimmunol.1500599. [DOI] [PubMed] [Google Scholar]
- 50.Ahn EH, Schroeder JJ. Sphinganine causes early activation of JNK and p38 MAPK and inhibition of AKT activation in HT-29 human colon cancer cells. Anticancer Res. 2006;26(1A):121–127. [PubMed] [Google Scholar]
- 51.Ahn EH, Schroeder JJ. Induction of apoptosis by sphingosine, sphinganine, and C(2)-ceramide in human colon cancer cells, but not by C(2)-dihydroceramide. Anticancer Res. 2010;30(7):2881–2884. [PubMed] [Google Scholar]
- 52.Hartmann D, Lucks J, Fuchs S, Schiffmann S, Schreiber Y, Ferreiros N, Merkens J, Marschalek R, Geisslinger G, Grosch S. Long chain ceramides and very long chain ceramides have opposite effects on human breast and colon cancer cell growth. Int J Biochem Cell Biol. 2012;44(4):620–628. doi: 10.1016/j.biocel.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 53.Hartmann D, Wegner MS, Wanger RA, Ferreiros N, Schreiber Y, Lucks J, Schiffmann S, Geisslinger G, Grosch S. The equilibrium between long and very long chain ceramides is important for the fate of the cell and can be influenced by co-expression of CerS. Int J Biochem Cell Biol. 2013;45(7):1195–1203. doi: 10.1016/j.biocel.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 54.Laroui H, Ingersoll SA, Liu HC, Baker MT, Ayyadurai S, Charania MA, Laroui F, Yan Y, Sitaraman SV, Merlin D. Dextran sodium sulfate (DSS) induces colitis in mice by forming nano-lipocomplexes with medium-chain-length fatty acids in the colon. PLoS One. 2012;7(3):e32084. doi: 10.1371/journal.pone.0032084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim IW, Myung SJ, Do MY, Ryu YM, Kim MJ, Do EJ, Park S, Yoon SM, Ye BD, Byeon JS, Yang SK, Kim JH. Western-style diets induce macrophage infiltration and contribute to colitis-associated carcinogenesis. J Gastroenterol Hepatol. 2010;25(11):1785–1794. doi: 10.1111/j.1440-1746.2010.06332.x. [DOI] [PubMed] [Google Scholar]
- 56.Lewis JD, Abreu MT. Diet as a trigger or therapy for inflammatory bowel diseases. Gastroenterology. 2017;152(2):398–414 e396. doi: 10.1053/j.gastro.2016.10.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement 1 Immuno-fluorescence staining of CerS2. Colon sections were fixed with 100% ice-cold methanol and permeabilized in PBS containing 0.025% Triton X-100. Subsequent tissue sections were either stained immunohistochemically by DIFF-Quick staining or used for immuno-fluorescence staining. This procedure determines whether the DIFF-Quick sections are comparable to the fluorescence stained section. After blocking, the tissue sections were incubated with anti-CerS2 at 4 °C overnight, followed by a Cy3 labeled anti-rabbit antibody diluted 1:800 for 2 h. Subsequently, tissue sections were stained with DAPI (1:1000). Fluorescent measurements were done with the Keyence BZ-9000 microscope with 20-fold magnification. Representative pictures are shown from CerS2+/+ and CerS2−/− mice with or without AOM and two cycles of DSS treatment. (PPTX 2860 kb)
Supplement 2 Depletion of CerS2 in human Caco-2 cells. CerS2 was depleted in Caco-2 cells by lentiviral transduction of a CerS2-shRNA. As a control, we transduced a non-specific (NC) shRNA. A) mRNA for CerS expressed in Caco-2 cells (CerS1 and Cer3 are only very weakly expressed in Caco-2 cells). CerS2 mRNA was reduced by 80% in CerS2-shRNA treated Caco-2 cells. B) CerS2-protein expression was also significantly reduced in these cells. We show one representative Western-blot of three and the calculation of the relative protein expression (CerS2/GAPDH, CaCo-2 untreated cells were set to 100%) based on findings from three blots. C) Down-regulation of CerS2 in Caco-2 cells led to alterations in various sphingolipid levels. Sphingolipid levels were determined by LC-MS/MS. Changes in sphingolipids are comparable to those observed in the colon of CerS2−/− mice in comparison to CerS2+/+ mice (see Table 2). Statistical analysis was done by unpaired t test, p* < 0.05; p** < 0.01; p*** < 0.001 (PPTX 413 kb)
Supplement 3 MELC analysis of colon sections from CerS2+/+ and CerS2−/− mice with and without AOM and two cycles of DSS treatment. Ten micrometer cryosections of the colon were applied to silane-coated coverslips, fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X100, and blocked with 3% BSA. The sample was placed on the stage of an Leica DM IRE2 and a phase contrast picture was taken. Then, the sample was automatically incubated for 15 min with fluorescence-labeled antibodies [FITC-Ly-6C (Gr1) antibody, FITC-CD11b and FITC-F4/80 antibody] and rinsed with PBS. To delete fluorescence signals, a bleaching step was performed after each antibody incubation step and at least 7-amino actinomycin D (7-AAD) was used to stain nuclei. After the MELC run, the tissue slices were stained with Diff-Quick (Dade Behring) (PPTX 10272 kb)
Supplement Table 1 RT-PCR primer used in this study. (PPTX 41 kb)