

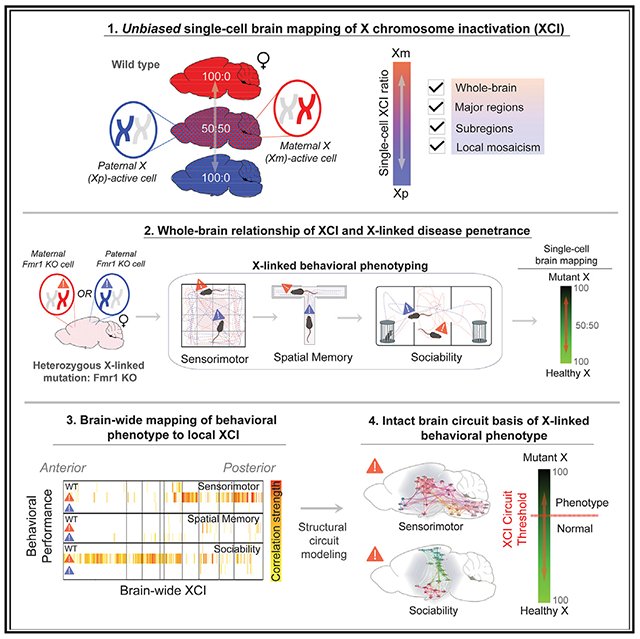
SUMMARY
The precise anatomical degree of brain X chromosome inactivation (XCI) that is sufficient to alter X-linked disorders in females is unclear. Here, we quantify whole-brain XCI at single-cell resolution to discover a prevalent activation ratio of maternal to paternal X at 60:40 across all divisions of the adult brain. This modest, non-random XCI influences X-linked disease penetrance: maternal transmission of the fragile X mental retardation 1 (Fmr1)-knockout (KO) allele confers 55% of total brain cells with mutant X-active, which is sufficient for behavioral penetrance, while 40% produced from paternal transmission is tolerated. Local XCI mosaicism within affected maternal Fmr1-KO mice further specifies sensorimotor versus social anxiety phenotypes depending on which distinct brain circuitry is most affected, with only a 50%–55% mutant X-active threshold determining penetrance. Thus, our results define a model of X-linked disease penetrance in females whereby distributed XCI among single cells populating brain circuitries can regulate the behavioral penetrance of an X-linked mutation.
Graphical Abstract
In brief
Szelenyi et al. demonstrate that adult brain XCI is systematically biased toward maternal X-active cells, which is sufficient for disease penetrance of the X-linked Fmr1-KO allele. Furthermore, local XCI mosaicism distinguishes phenotypic outcomes of individuals based on mutant X-active cells populating distinct brain circuits.
INTRODUCTION
The X chromosome expresses more brain-specific genes than any other chromosome,1 and X-linked mutations give rise to a number of neurodevelopmental disorders, such as Rett syndrome (RTT), fragile X syndrome (FXS), and other forms of X-linked mental retardation.2,3 In female eutherian mammals, X chromosome inactivation (XCI) is thought to be a random process ensuring X dosage compensation compared to males.1,4 Non-random (skewed or biased) XCI favoring the maternal Xm or paternal Xp chromosome can occur as a stochastic fluctuation or a developmental selection against an X chromosome carrying a deleterious mutation. Such selection bias favoring the healthy X chromosome has been proposed to occur in unaffected or only mildly affected female carriers of X-linked brain disorders, with the degree of skewing needed to reduce phenotypic penetrance typically defined as ≥80:20 ratio favoring the healthy X.5–12
Human evidence for the role of XCI in disease remains inconclusive, as some studies report correlation between disease manifestations and ≥80:20 XCI skewing, for example, in RTT and FXS,13–20 while others fail to identify consistent evidence to support this model,6,11,21–26 including three studies that directly examined XCI in post-mortem brains of patients with RTT instead of relying on the indirect measure of XCI in blood.6,11,26 In RTT model mice, correlations have been observed between XCI selection against the mutant allele and severity of the disease phenotype27,28 suggesting that XCI skewing in a single brain region (e.g., cerebellum) can directly influence disease state. In healthy mice, the existence of non-random XCI partially favoring maternal X-active cells has been documented separately by two groups,29,30 which could theoretically offset or worsen the final display of X-linked disease states depending upon the parent of origin of the mutant X.
Despite these findings, the ultimate degrees by which XCI skewing varies in healthy brain, and its sufficiency to regulate X-linked disorders, remains unclear.
RESULTS
Brain XCI is systematically biased
In the current study, we obtained an unbiased and complete survey of brain XCI by applying our serial two-photon tomography (STPT)-based imaging and computational pipeline31 to quantify maternal versus paternal active X chromosome distribution using knockin Mecp2-GFP reporter mice32 in which X-linked methyl-CpG-binding protein is tagged with GFP (Mecp2-GFP) and acts as a cellular reporter of the selection of the active X chromosome (Figures S1 and S2).
We first compared the total number of brain cells with maternal X active in Mecp2-GFP(m/+) mice that inherited the X-linked Mecp2-GFP allele maternally (Xm; n = 18) and the total number of brain cells with paternal X active in Mecp2-GFP(p/+) mice that inherited the Mecp2-GFP reporter allele paternally (Xp; n = 19; Figures 1A–1C). This comparison revealed significantly more GFP+ cells in the brains of the maternal Mecp2-GFP(m/+) mice, demonstrating an average 58:42 bias toward higher paternal XCI and, consequently, an average 58:42 ratio of cells with maternal Xm active to paternal Xp active in wild-type (WT) brain (Figures 1C and 1D; Table S1). Notably, though, this average ~60:40 paternal XCI bias comprised a considerable individual variability, including extreme examples of 84:16 Xm selection bias and 25:75 Xp selection bias (Figure 1D). Therefore, both stochastic variability in XCI and the systematic paternal inactivation bias play roles in determining the overall Xm:Xp ratio in each brain.
Figure 1. Whole-brain quantification of maternal and paternal active X chromosome distribution.
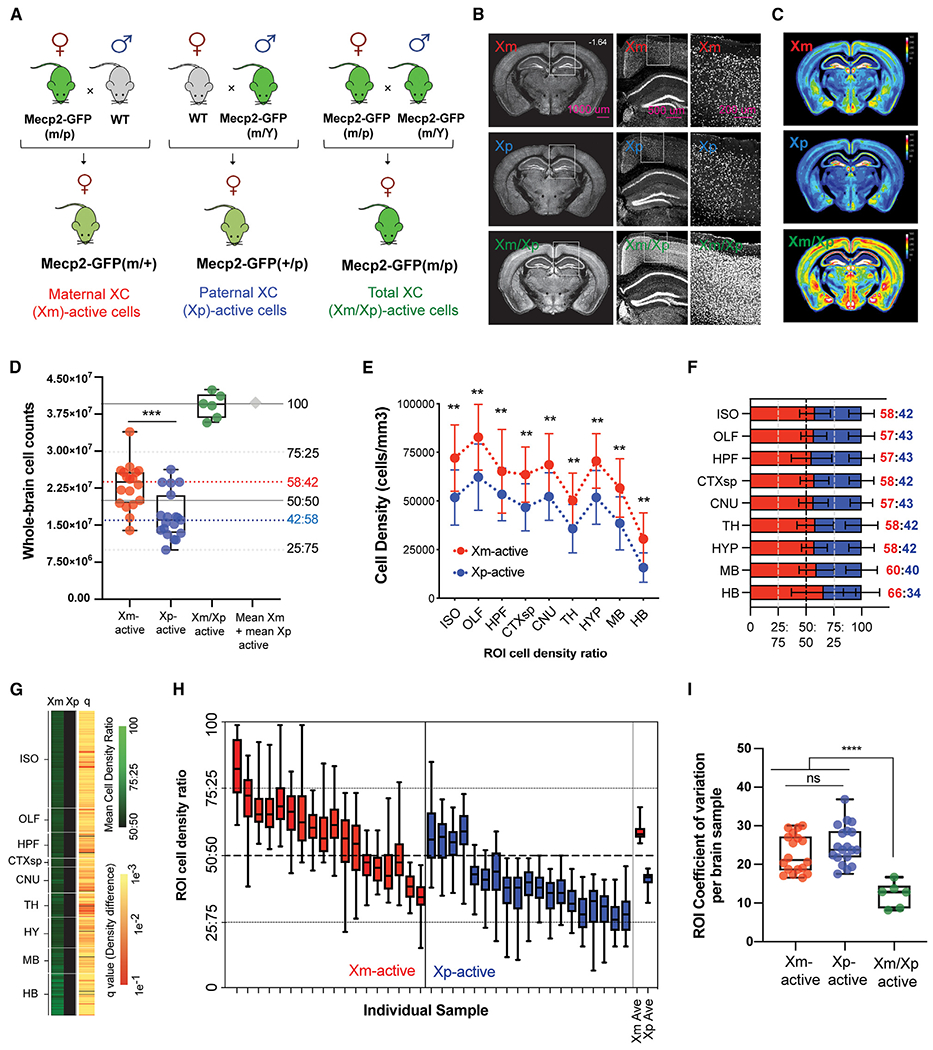
(A) Breeding strategy for generating maternal, paternal, and homozygous Mecp2-GFP reporter mice.
(B) Representative STPT images from brains with maternal (Xm; top), paternal (Xp; middle), and homozygous (Xm/Xp; bottom) Mecp2-GFP+ cells.
(C) Mean Mecp2-GFP+ cell density across genotypes represented as voxelized heatmaps on a 16-color gradient scale from black (0 cells/voxel) and yellow (150 cells/voxel) to white (300 cells/100 μm sphere voxel).
(D) Whole-brain cell counts for Xm-GFP+ (n = 18), Xp-GFP+ (n = 19), and homozygous Xm-GFP+/Xp-GFP+ cells (n = 6). ***p < 0.0005, Welch’s t test. The sum of the heterozygous Xm-GFP+ and Xp-GFP+ cell counts is shown as a gray diamond. Dashed red and blue lines show the data-derived Xm-active and Xp-active median ratios, respectively.
(E) Mecp2-GFP+-labeled cell densities (cells/mm3) across all major ontological brain divisions. **p < 0.01, two-way mixed-effects ANOVA with Holm-Sidak-corrected post hoc test.
(F) Data from (E) converted into stacked cell density ratios. Data from (E)–(F) represent mean ± SD.
(G) XCI selection across all atlased brain regions. Left two columns: heatmap visualization of normalized mean Xm-active and Xp-active region of interest (ROI) cell density on a color gradient of black (50%) to green (100%). Right column: statistical significance across all ROIs (false discovery rate [FDR]-corrected Student’s t tests), with each q value indicated by a color gradient from red (0.1) to yellow (0.001).
(H) Stochastic variability of XCI selection across all brain regions in each brain analyzed. The average data for Xm-GFP and Xp-GFP brains are plotted on the far right. Box-and-whisker plots display median, interquartile range, and 95th percentiles of the data.
(I) Quantification of brain-wide ROI stochastic variability by coefficient of variation (CV) analysis. ***p < 0.005 from one-way ANOVA with Dunnett’s post hoc test. Box-and-whisker plot data from (D), (H), and (I) display median, interquartile range, and 95th percentiles of the data.
Next, we asked whether the paternal XCI bias seen at the whole-brain level exists similarly across all brain areas or whether there may be regional differences in XCI patterns in the brain. This analysis revealed that the average ~60:40 bias for maternal X selection is seen across all major brain divisions, including the isocortex (58:42), cortical subplate (58:42), olfactory areas (57:43), hippocampal formation (57:43), cerebral nuclei (57:43), thalamus (58:42), hypothalamus (58:42), midbrain (60:40), and hindbrain (66:34) (Figures 1E and 1F; Tables S1 and S2), as well as across all local subregions in these areas (Figure 1G; Tables S1 and S2). Cortical fluorescence-activated cell sorting (FACS) experiments in genetically defined Fezf2+ excitatory or parvalbumin+ inhibitory cell types confirmed STPT-derived cell count measurements in which a composite 61:39 bias for maternal selection was observed (Figure S3).
Local mosaicism further distinguishes XCI patterning of individual brains
All anatomical segmentations of the whole-brain data allowed us to measure regional XCI variability—or local mosaicism—in each individual brain (Figures 1H and 1I; Table S1) and determine whether brains with highly skewed Xm:Xp ratios comprise different (or the same) regional variability compared to brains with the overall Xm:Xp ratio being close to equal. To this end, we generated brain-wide XCI heatmaps of each individual brain sample to visualize regional variability in Xm and Xp selection (Figure S4) as well as collapsed box-and-whisker plots (Figure 1H). This revealed a similar variability from the mean for all imaged brains independent of their overall Xm:Xp ratio, with the mean coefficient of variation (CV) for both Mecp2-GFP(m/+) and Mecp2-GFP(p/+) groups being ~20% compared to a CV of ~10% in homozygous Mecp2-GFP(m/p) mice (Figure 1I).
Maternal, but not paternal, transmission of an X-linked FMR1-KO allele is sufficient for behavioral penetrance due to biased brain XCI
Having quantified the XCI patterns in WT mice, we next asked whether the identified biases persist and affect disease penetrance in a female heterozygous mouse model of FXS, an X-linked disorder caused by the loss of expression of the fragile X mental retardation 1 (Fmr1) protein.33,34 To this end, we crossed the Fmr1-knockout (KO) mouse model of FXS35 with the Mecp2-GFP X reporter line, generating heterozygous Fmr1-KO/+ female mice with the KO allele inherited either maternally in Fmr1-KO(m)/Mecp2-GFP(p) mice or paternally in Fmr1-KO(p)/Mecp2-GFP(m) mice (Figure 2A). We note that while this FXS mouse model has been studied extensively as a complete KO in male hemizygous Fmr1-KO/Y mice, only three studies reported modest phenotypes (synaptic and social) in female heterozygous Fmr1-KO/+ mice, with the KO allele always transmitted maternally.36–38
Figure 2. Maternal transmission of the Fmr1-KO allele is sufficient for behavioral penetrance due to persistent brain XCI bias.
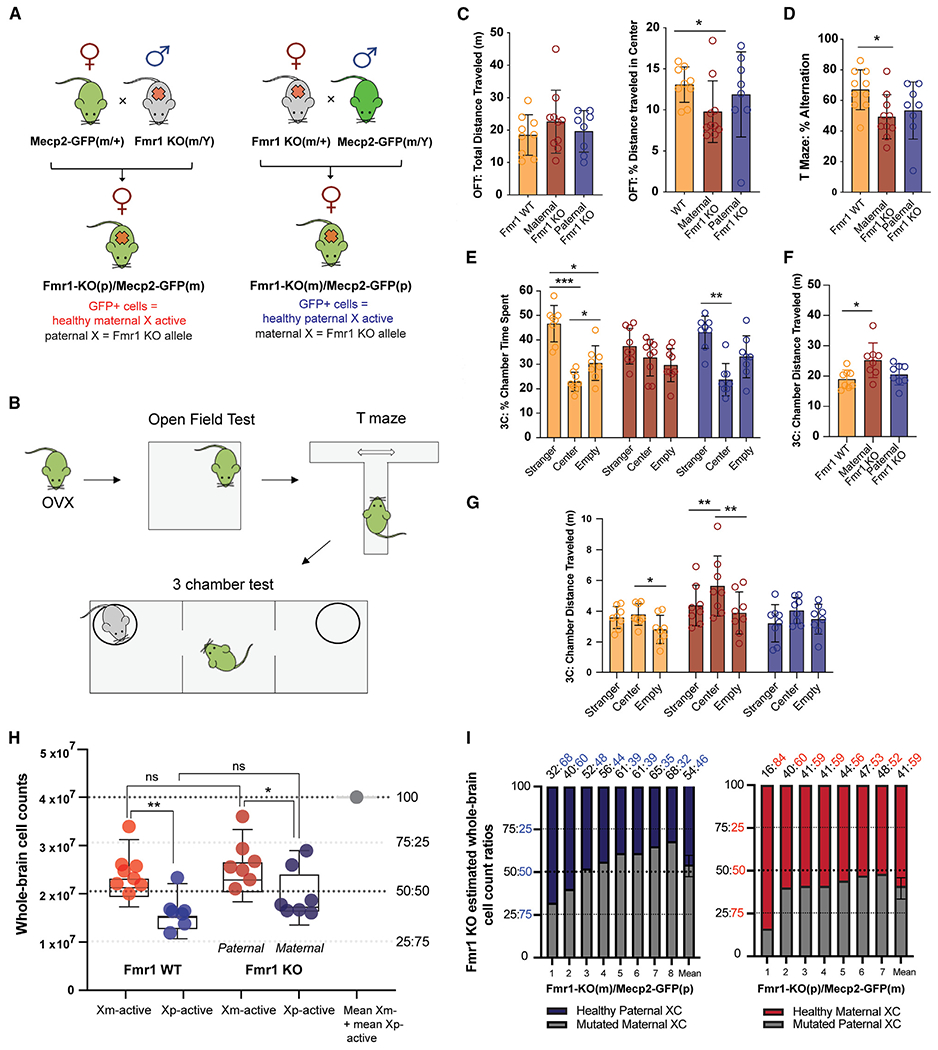
(A) Reciprocal Fmr1-KO and Mecp2-GFP crosses created to derive female heterozygous Fmr1-KO mice with healthy X-active cells reported with the reciprocally inherited Mecp2-GFP allele.
(B) Behavioral testing: mice from all groups were first ovariectomized (OVX) and then sequentially tested in the open field test (OFT), T maze, and 3-chamber test (n = 8 for each group). Data for each individual test are derived from sequential tests of the same animals.
(C) OFT results: left, total distance traveled; right, percentage of distance traveled in center arena. *p < 0.05, Welch’s ANOVA and Dunnett’s T3 post hoc test.
(D) Spatial alternation results from T maze test. *p < 0.05, Welch’s ANOVA, Dunnett’s T3 post hoc test.
(E–G) 3-chamber test results. (E) Percentage of time spent in each chamber. *p < 0.05, **p < 0.01, and ***p < 0.001, two-way mixed-effects ANOVA, Holm-Sidak post hoc test. (F) Total distance traveled. *p < 0.05, Welch’s ANOVA and Dunnett’s T3 post hoc tests. (G) Chamber distance traveled. *p < 0.05 and **p < 0.01, two-way mixed-effects ANOVA, Holm-Sidak post hoc test.
(H) Quantification of whole-brain Xm versus Xp selection in (left) WT mice and (right) heterozygous Fmr1-KO mice. *p < 0.05 and **p < 0.01, ANOVA with Holm-Sidak post hoc test. Box-and-whisker plot data display mean, interquartile range, and 95th percentiles of the data.
(I) Stacked bar graphs of each maternal Fmr1-KO(m/+) (left) and paternal Fmr1-KO(+/p) (right) whole-brain X selection from (H): healthy Xp-active cells in Fmr1-KO(m)/+ brains are highlighted in dark blue (left), and healthy Xm-active cells in Fmr1-KO(p/+) are highlighted in red (right). Fmr1-KO:WT cellular ratios of each sample are listed on top.
Data from (C)–(G) represent mean ± SD.
We first assayed the impact of the maternal versus paternal Fmr1-KO allele transmission across three behavioral tests that were used previously to identify disease-related phenotypes in FXS mice: the open field test (OFT) to assess sensorimotor functions, the T-maze spontaneous alternation test to assess working memory, and finally, the 3-chamber test to assay sociability and social preference (Figure 2B). These experiments revealed that the Fmr1-KO(m)/Mecp2-GFP(p) heterozygous female mice with maternal KO allele transmission showed deficits in all three tests, while in contrast, the paternal Fmr1-KO(p)/Mecp2-GFP(m) heterozygous mice did not differ from control Mecp2-GFP sibling mice in any measurement (Figures 2C–2G). The behavioral deficits of the maternal Fmr1-KO(m)/Mecp2-GFP(p) mice included (1) reduced travel distance across the center arena in the OFT (Figure 2C), (2) reduced frequency of spontaneous alterations in the T-maze (Figure 2D), and (3) a complete lack of social preference with increased hyperactivity in the 3-chamber social interaction test (Figures 2E–2G).
We next imaged the brains of all mice used in the above behavioral tests by STPT as done for WT brains in Figure 1 and determined the distribution of healthy X-active cells marked by Mecp2-GFP expression from the same X chromosome. These measurements revealed that the whole-brain Xm:Xp ratio in the maternal Fmr1-KO(m)/Mecp2-GFP(p) mice was 54:46, representing an average 54% of mutant Fmr1-KO X-active brain cells compared to 46% healthy X-active cells, while that of paternal Fmr1-KO(p)/Mecp2-GFP(m) mice was 41:59, conversely reflecting an average 41% of mutant Fmr1-KO X-active cells compared to 59% healthy X-active cells (Figures 2H and 2I; Tables S3 and S4). Additionally, the regional Xm:Xp ratio differences were more pronounced for cortical versus subcortical areas (Figure S5), suggesting a modest compensation favoring the selection of the healthy paternal Xp chromosome subcortically compared to WT brains. And third, the whole-brain Xm:Xp ratio in the paternal Fmr1-KO(p)/Mecp2-GFP(m) mice was 41:59, reflecting an average 41% of Fmr1-KO mutant X-active compared to 59% healthy X-active cells (Figures 2H and 2I).
Local XCI status maps FXS behavioral phenotypes across distinct assay-specific sets of brain regions
The brain-wide cellular distribution measurements of Fmr1 WT versus KO alleles also allowed us to test how local mosaicism of regional XCI may further influence the observed phenotypes in the affected maternal Fmr1-KO(m)/Mecp2-GFP(p) mice. To test this hypothesis, we correlated the Fmr1 WT:KO cell ratios across all brain regions to behavioral scores from all three behaviors for each mouse tested. This correlation analysis identified two distinct sets of anatomical regions in the maternal Fmr1-KO(m)/Mecp2-GFP(p) brains, in which the Fmr1 WT:KO cell ratios were correlated to behavioral performance in either the OFT or the 3-chamber test (Figures 3A and S6; Table S5).
Figure 3. Individual FXS behavioral phenotypes map distinctly to mosaic brain-wide XCI patterns.
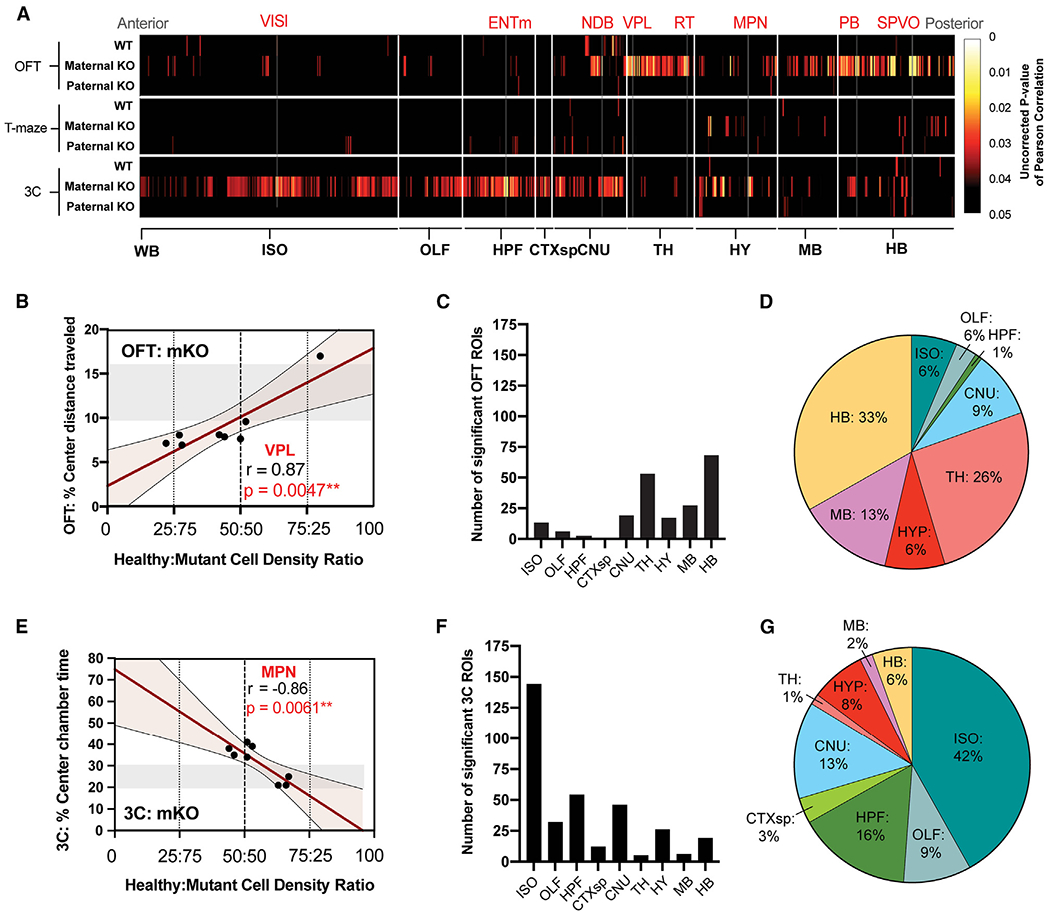
(A) Pearson’s correlational analysis of healthy:mutant cell density ratios correlated to behavioral performance across all behaviors. Data are displayed as 2D heatmaps of statistically significant correlations across brain ROIs grouped by major ontological structures. Uncorrected p values of Pearson correlations are represented on a color gradient scale from 0.05 (black) to 0.005 (red) to 0.0005 (yellow). Results from Fmr1 WT (“WT”), maternal Fmr1-KO(m)/Mecp2-GFP(p) (“Maternal KO”), and paternal Fmr1-KO(p)/Mecp2-GFP(m) (“Paternal KO”) mice are grouped by each behavioral test: top, OFT; middle, T maze; and bottom, 3 chamber (3C).
(B and E) Representative scatterplot displays of correlated ROI density from (A) shown for (B) ventral posterolateral nucleus (VPL) of thalamus in OFT and (E) hypothalamic medial preoptic nucleus (MPN) of hypothalamus in 3-chamber tests.
(C and F) Number of significantly correlated ROIs from maternal Fmr1-KO(m)/Mecp2-GFP(p) mice listed by major brain structure for (C) OFT and (F) 3-chamber tests.
(D and G) 100% pie charts of significantly correlated ROIs from maternal Fmr1-KO(m)/Mecp2-GFP(p) mice grouped by major brain structure and represented as a percentage of total correlated ROIs for (D) OFT and (G) 3-chamber test.
See Table S1 for all acronyms of brain regions.
The first set of brain regions comprised areas in which the Fmr1 WT:KO cell ratio was positively correlated to OFT behavioral performance, specifically the distance traveled in the center of the OFT arena. These regions included primarily sensory structures of the thalamus, midbrain, and hindbrain (Figures 3A–3D; Table S5), such as the sensory ventral posterolateral nucleus of the thalamus39 (Figure 3B). The second set of brain regions comprised areas in which the Fmr1 WT:KO cell ratio was inversely correlated to the time spent in the center of the 3-chamber apparatus. These regions, in contrast to the first set, contained primarily cortical, hippocampal, and hypothalamic brain areas (Figures 3A and 3E–3G), including the hypothalamic medial preoptic nucleus that is well known for regulating social behavior40 (Figure 3E). The same correlation analysis failed to identify a distinct set of brain regions with Fmr1 WT:KO ratios related to behavioral performance in the T-maze test, in which the Fmr1-KO(m)/Mecp2-GFP(p) mice showed only a modest level of impairment (Figure 3A).
In addition, whole-brain Fmr1 WT:KO ratios, in contrast to the regional ratios described above, showed only a trend toward a positive correlation in the OFT and a negative correlation in the 3-chamber task (Figure S7). Finally, as expected, we also did not observe any significant correlations for brain regions in the paternal Fmr1-KO(p)/Mecp2-GFP(m) mice, which did not show any behavioral phenotypes (Figures 3A and S6; Table S5).
Intact brain circuitry represents anatomical correlates of FXS behavioral penetrance
The identification of the two sets of brain regions with Fmr1-KO allele density linked to behavioral phenotypes suggests a representation of two distinct anatomically connected brain circuits underlying sensorimotor- and exploratory-related versus social- and anxiety-related behaviors. To test this hypothesis further, we next applied an established structural connectivity matrix analysis derived from a whole-brain connectivity model of the mouse brain41 (Figures 4B, 4C, 4G, and 4H). This analysis indeed revealed much higher connection densities for brain regions within each putative behavioral circuit than for matching randomly sampled brain structures: specifically, the OFT circuit density (Figures 4B and 4C) and 3-chamber circuit density (Figures 4G and 4H) represented the 93rd and 100th percentiles of each sample circuit’s distribution, respectively (Table S6).
Figure 4. Distinct intact brain circuitries represent anatomical correlates of FXS behavioral outcomes.
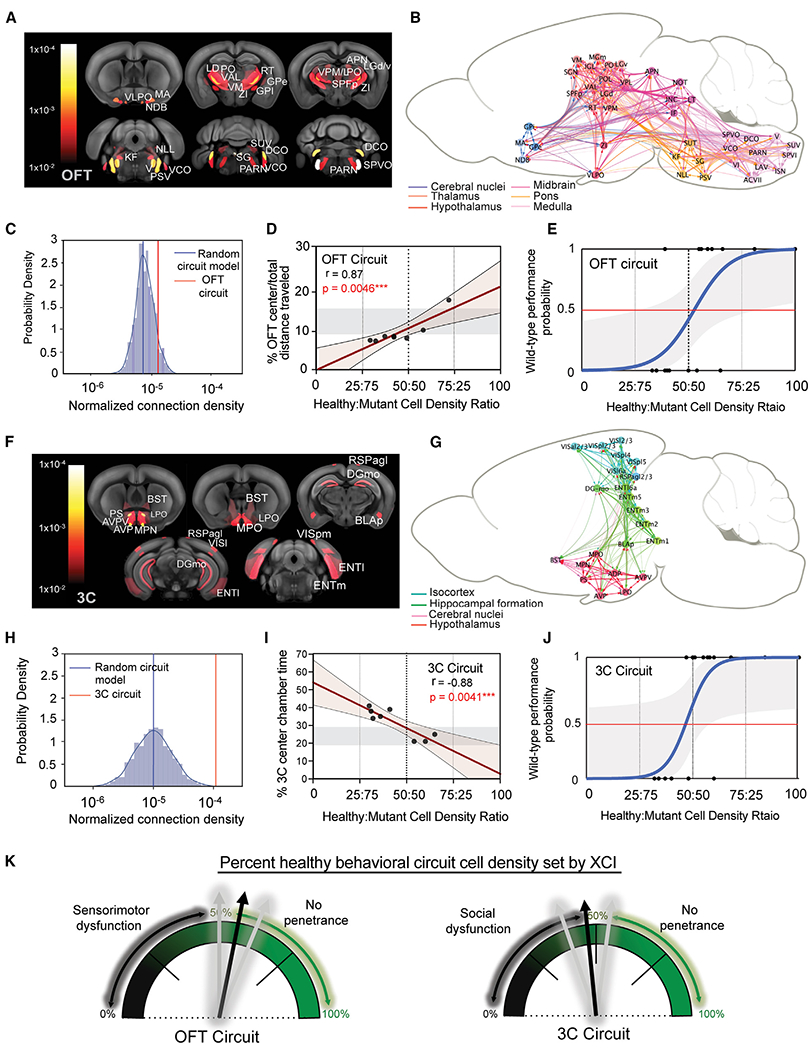
(A and F) Select ROIs from maternal Fmr1-KO(m)/Mecp2-GFP(p) mice with significant correlation (cutoff at p = 0.01)to performance in (A) OFT and (F) 3-chamber tests are heat mapped and overlaid on a reference mouse brain template.
(B and G) Visualization of structural connectivity weights within behaviorally correlated brain networks of ROIs in the (B) OFT and (G) 3-chamber tests.
(C and H) Log-scaled and normalized median connection densities of the (C) OFT and (H) 3-chamber ROI networks (red lines) overlaid on the probability density of 1,000 in-silico-generated random ROI networks of the same inter-regional distance and total ROI number (blue line = median). One-sample t test: OFT, p = 6.36 × 20−247; 3 chamber: p = 0.0.
(D and I) Linear regression models of behavioral scores and brain circuit cell density ratios. Regression and statistical test values are listed for each image. Shadowed rectangle in each group represents control Fmr1 WT range of behavioral scores for comparison.
(E and J) Logistic regression modeling of WT behavioral performance predicted by percentage of healthy cell density in behavioral circuits from all mice (n = 22) within (E) OFT (x2(1) = 14.88; log-likelihood ratio = −14.42; equal-odds ratio = 54%; p = 0.00011) and (J) 3 chamber (x2(1) = 13.88; log-likelihood ratio = −12.89; equal-odds ratio = 49%; p = 0.00019) behaviors. Grayed area represents ±95% confidence intervals.
(K) Cartoon depiction model of OFT (left) and 3-chamber (right) logistic modeling results, which portrays how female FXS phenotypes are determined by XCI-distributed healthy cell densities occupying the behavioral circuits identified in this study.
See Table S1 for all ROI acronyms.
The identified correlations of local Fmr1-KO cell densities to behavioral phenotypes also suggest that the distribution of the Fmr1-KO allele across the two behavioral circuits can in fact predict disease penetrance in each animal. To test this hypothesis, we next calculated the Fmr1 WT:KO allele ratios selectively across the two behavioral circuits and regressed these values against the behavioral performance in the OFT and 3-chamber test (Table S7). As shown in Figure 4, Fmr1 WT:KO allelic ratios of cell density in behavioral circuits highly predicted individual behavioral performance for only maternal Fmr1-KO(m)/Mecp2-GFP(p) heterozygous mice in both the OFT (Figure 4D) and 3-chamber (Figure 4I) assays and not for control or paternal Frm1-KO(p)/Mecp2-GFP(m) mice (Figure S8).
Finally, we probed the local Fmr1-KO cell density amount sufficient for phenotypic manifestations of disease-related behaviors. Likelihood-ratio tests performed on binary logistic regression models revealed that the Fmr1-KO allele distributions across each brain circuit indeed significantly distinguishes normal from disease-related performance in both the OFT (Figure 4E) and 3-chamber test (Figure 4J). The equal-odds ratio of normal versus disease-related behavioral outcome was calculated to be 55.20% ± 5.95% healthy cell density percentage in the OFT brain circuit and 49.18% ± 5.19% healthy cell density percentage in the 3-chamber brain circuit (Figure 4K).
DISCUSSION
We have applied our whole-brain microscopy pipeline with adult XCI reporter mice to directly measure XCI across all cells of the intact mouse brain. This approach allowed for an anatomically unbiased and highly quantitative mapping of brain XCI which both complements and extends previous reports of XCI measurements derived from singular brain regions, bulk tissue, or peripheral cells as a proxy for the brain.
We first discovered a 60:40 whole-brain bias in maternal versus paternal X activation in the adult brain. This result is consistent with neonatal whole-brain measurements from hybrid mice strains,30 which, taken together, developmentally suggest that whole-brain XCI is set prior to the neonatal period and is maintained into adulthood. During early development, progressive and complete inactivation of the Xp occurs in mouse development at the 2-cell stage up until embryo implantation.42 This effect appears to be guided by repetitive element Xp epigenetic marks remnant from post-meiotic silencing in spermatocytes.42 After embryo implantation, all cells of the epiblast erase these imprints and transition into “random” XCI 1 day post-implantation.43 Considering our results from adult brain show preferential inactivation of paternal X, incomplete erasure of Xp marks found during pre-implantation could contribute toward the small but favorable inactivation during somatic cell XCI onset. Alternatively, pre-implantation Xp marks could be completely erased, but subsequent proliferation or differentiation effects caused by cis- or trans-acting factors from Xm or Xp, respectively, could lead to the bias in the brain. The existence of Xm-only brain-expressed genes (paternally imprinted XC genes)44 supports this possibility; however, our functional understanding of these genes is very limited. Future efforts are warranted to understand the postnatal progression and somatic intricacies of XCI more broadly.
Through atlas-based segmentation of all brain regions, we further demonstrate that the whole-brain maternal bias is spatially distributed in an anatomically even fashion and not in particular brain systems. Our orthogonal FACS dataset importantly reproduced the STPT-derived 60:40 maternal X-active bias in the cortex, which also agrees with previous bulk XCI RNA sequencing (RNA-seq) measurements made from the frontal cortex of hybrid strain mice.29 Importantly, this indicates that our unbiased single-cell brain mapping XCI dataset accurately captures XCI ratios consistent with other counting methods, including FACS and RNA-seq. Furthermore, we also identified a remarkable ~20% stochastic variability beyond the overall whole-brain XCI status, which introduces a layer of mosaic patterning across local brain areas within individuals—a result that quantitatively captures regional XCI variability first observed from a dual XCI reporter mice line approach.45
Our measurements in heterozygous Fmr1-KO/+ mice revealed that the modest maternal whole-brain bias and regional stochastic variability can provide a clear and explainable effect on phenotypic penetrance of behavior. At the whole-brain level, >55% occupancy of mutant cells—pervasively seen in maternal Fmr1-KO mice—is sufficient to cause behavioral phenotypes, while >60% of healthy brain cells—only observed in paternal Fmr1-KO mice—is, conversely, sufficient to prevent phenotypic manifestations. Whole-brain XCI ratios failed to correlate with behavioral performance across assays. Instead, by employing a paired brain-wide correlational analysis and structural connectivity modeling approach, we revealed a direct correlation between behavioral-assay-specific performance with intact brain-circuit-specific XCI ratios. A ~50%–55% mutant cell density threshold distributed within distinct brain circuits determined the appearance of behavioral phenotypes.
Specifically, the OFT-correlated brain circuit consisted of subcortical regions collectively involved in sensorimotor (e.g., sensory thalamic and hindbrain nuclei) and arousal (e.g., magnocellular nucleus,46 ventrolateral preoptic area,47 reticular nucleus48) function, an anatomical basis consistent with sensorimotor gating abnormalities reliably documented in patients with FXS49 and model mice.50 The 3-chamber behavioral effects phenocopied the socially avoidant disposition of maternal FMR1-deficient females,51,52 a patient population representing 20% of total autism spectrum disorder cases,53 by displaying reduced time spent in the social chamber and increased hyperreactivity and time spent in the center chamber. Alternatively, the 3-chamber-correlated brain circuit consisted of key regions known to control spatial navigation and object recognition (entorhinal cortex,54 lateral visual area55) or social behavior and anxiety (medial preoptic area,40 cortical amygdala,56 basolateral amygdala,57 and bed nucleus of stria terminalis58), indicating an integrated spatio-social anxiety putative function of the circuit.
In conclusion, the current study establishes a model of X-linked penetrance that connects intact behavioral-circuit-specific XCI ratios with the behavioral penetrance of individual females. The results provide insight into the highly variable nature of phenotypes displayed by heterozygous female patients of X-linked syndromes.
Limitations of the study
Our model’s framework of X-linked behavioral penetrance is currently limited to a single X-linked example of FXS in C57 mice. Therefore, further testing in other inherited X-linked disorders and mouse strain backgrounds is critical to further understand the model’s generalizability. XCI-defined thresholds of X-linked penetrance in brain circuits are currently hypothesized to vary from our current study due to factors such as the specific nature of X-linked mutation59 and strain- or species-specific variation of Xce alleles.60
Additionally, our model was derived from screening three standard behavioral assays disrupted by FXS and, although limited in scope, suggests the potential for identifying a broader range of affected behavioral-circuit-linked phenotypes through high-throughput behavioral screening and circuit mapping assays. Alternatively, future work employing more complex behavioral procedures can offer targeted insights into dysregulated behavioral circuitries influenced by X-linked mutations.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests regarding this Report should be directed to and will be fulfilled by the lead contact, Eric R. Szelenyi (szelenyi@uw.edu).
Materials availability
This study did not generate new reagents.
Data and code availability
Raw cell count and calculated cell density data from samples of all whole-brain imaging experiments are published with the paper as Supplemental tables. All behavioral data is published with the paper as supplemental tables. All structural connectivity raw data and circuit cell density calculations are published with the paper as supplemental tables. Raw FACS sorting data have been deposited at FlowRepository.org and are publicly available as of the date of publication. All microscopy images are available from lead contact upon request.
Custom code for structural connectivity modeling analysis is hosted on Github and Zenodo (https://doi.org/10.5281/zenodo.10815678) and is publicly available as of date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Adult mice (8–10 weeks old) were used for whole-brain imaging experiments. Animals were housed under a 12-h light/dark cycle (0600 ON, 1800 OFF), had access to food and water ad libitum, and were housed with littermates. All experimental procedures were performed in accordance with CSHL Animal Care and Use Committee Guidelines. The Mecp2-GFP mouse line was obtained from the Jackson laboratory (stock # 014610), and was backcrossed and maintained on a C57Bl6/J background for over 6 generations since its donation to Jackson laboratory, and at least 2 generations in our laboratory. Mecp2 is a gene located at chromosomal position X A7.3 and is subject to XCI, thus is expressed only from the active X.61 Developed in the laboratory of Adrian Bird, this mouse line contains an in-frame knock-in cassette at the 3′ UTR of the Mecp2 locus.32 Driven and regulated by the endogenous Mecp2 promoter/enhancers, Mecp2-GFP expression leads to normal Mecp2 levels and subcellular localization of Mecp2 protein that is fused at the C terminus with EGFP. Expression of the fusion allele does not alter neuronal physiology32 and mice are successfully bred to homozygosity without behavioral or reproductive complications (data not shown). In addition, strong expression of Mecp2-GFP favors neurons of many types32 thereby circumventing biased effects of XCI determinations based on expression profile. Mecp2-GFP(m/+) or Mecp2-GFP(p/+) mice were obtained in separate heterozygotes by crossing homozygous females or hemizygous males with wild-type C57Bl6/J (JAX stock # 000664) mice. A subset of these wild type reporter mice was derived from Fmr1 KO or WT crosses that generated Mecp2-GFP(m/+)/Fmr1 KO(+/+) (n = 7) and Mecp2-GFP(+/p)/Fmr1 KO(+/+) (n = 8) mice that are congenic to the C57Bl6/J crosses. Homozygous reporter mice were obtained by crossing homozygous Mecp2-GFP(m/p) females with hemizygous Mecp2-GFP(m/Y) males. Fmr1 KO mice were obtained from the Jackson laboratory (#003025). These mice were originally developed in the Oostra laboratory and contain a gene-disrupting neomycin resistance cassette in exon 5 of the FMR1 locus.35 Mecp2-GFP(m/+)/Fmr1 KO(+/p) female mice were generated by breeding Mecp2-GFP(m/p) females with hemizygous Fmr1 KO(m/Y) males. For imaging only, Mecp2-GFP(m/+)/Fmr1 KO(+/+) female mice were generated by separately breeding homozygous Mecp2-GFP(m/p) females with hemizygous Fmr1 KO(+/Y) males. Conversely, Mecp2-GFP(+/p)/Fmr1 KO(m/+) or Mecp2-GFP(+/p)/Fmr1 KO(+/+) wild type littermates were generated by breeding Fmr1 KO(m/+) females with hemizygous Mecp2-GFP(m/Y) males. Using this genetic strategy, double heterozygous mice used for behavior and imaging experiments contained the Mecp2-GFP and Fmr1 KO alleles on opposing X chromosomes. All transgenic mice were maintained on a C57Bl6/J background. For FACS experiments, the Fezf2-2A-CreER Cre driver line62 (Jax stock# 036296) was used to inducibly label layer V-VI pyramidal neurons, a subset of excitatory neurons. Parvalbumin (PV)- 2A-CreER (JAX stock # 028580; kindly provided by Huang Laboratory, Duke University) was used to inducibly label a subset of inhibitory cortical neurons. Ai14 mouse line (JAX stock #007914) was used as the Cre reporter mouse, which expresses CAG-driven tdTomato upon Cre expression and recombination at the Cre reporter allele located in the Rosa26 locus. Single-cell XCI was FACS-counted within Fezf2 and PV + cortical neurons in triple transgenic mice containing Mecp2-GFP, cell-type specific Cre driver, and Ai14 Cre reporter alleles. The triple transgenic mice were generated by first crossing homozygous female or hemizygous Mecp2-GFP mice with Ai14 homozygous mice. Resulting double transgenics were inbred to generate double homo- or hemizygous Mecp2/Ai14 mice. Males were crossed into female Cre-driver lines to label active Xp and females were crossed into males to label active Xm in Fez2 or PV + neurons. Inductions of CreER to allow tdTomato labeling of Fezf2 (n = 13; 4 maternal Mecp2-GFP+; 9 paternal) and PV-expressing cells (n = 14; 11 maternal Mecp2-GFP+; 3 paternal) were performed by administering two intraperitoneal (I.P.) injections of tamoxifen (2mg dissolved in corn oil) (Sigma, T5648) at P21 and P28.
METHOD DETAILS
Brain sample preparation
Animals were euthanized via transcardial perfusion under ketamine/dexmedetomidine anesthesia. Dissected brains were post-fixed overnight in 4% paraformaldehyde at 4 C, incubated for 48 h in 0.1 M glycine/0.1 M PB for auto fluorescent quenching, and then stored in 0.05 M PB at 4 C until confocal or serial two-photon tomography imaging (STPT; see below). Prior to STPT imaging, brains were embedded 4% oxidized agarose in 0.05 M PB using custom molds and holders to maintain consistent embedding position. Embedded brains were crosslinked in 0.2% sodium borohydrate solution for 3h at room temperature or overnight at 4 C prior to STPT processing (below).
Immunohistochemistry and confocal imaging
Neuronal expression of the Mecp2-GFP allele was studied though immunostaining and confocal imaging. 50 μm vibratome-processed, free-floating coronal sections of homozygous Mecp2-GFP mice brains (n = 2) were processed. Sections were washed 3 times in PBS followed by blocking for 1 h at room temperature in PBS- T (PBS, 0.2% Triton X-100) containing 5% donkey serum. Sections were then incubated overnight at 4 C in blocking solution containing rabbit anti-NeuN (Millipore, ABN78) primary antibody at 1:1000. After washing, NeuN-stained sections were incubated with anti-rabbit AlexaFluor-568-conjugated secondary antibody (Thermo-Scientific, A10042) diluted 1:500 for 1 h at room temperature. After washing excess secondary antibody, sections were mounted, DAPI- counterstained (Prolong Gold Antifade Mountant, Thermo Fisher), and coverslipped for imaging. Confocal images were acquired with a Zeiss LSM780 confocal microscope using a 561 laser and corresponding dichroic and filter sets. Single plane images were captured with a 40x oil immersion objective. Total colocalized populations for each marker of every FOV (212.55 μm Xx 212.55 μm Y) were manually quantified using Fiji image processing package.
Serial two-photon tomography whole-brain imaging
The Tissuecyte1000 instrument was used for all imaging experiments (TissueVision). This system combines a high-speed multi-photon microscope with a fully integrated vibratome for automated z-sectioning and image acquisition throughout the entire whole-mount sample. Embedded sample brains were imaged with a 20x objective at 50 μM below the sample surface. 270 total serial sections were acquired at 50 μm z-resolution (~13.5 mm total z-length), with each section being comprised of a 12 (x axis, 700 μm) x 16 (y axis, 700 μm) field of view (FOV) mosaic. Images were acquired with laser scan settings of 1 μm/pixel at an integration time of 1 us. A laser wavelength of 910 nm with ~322 mW power at the end of the objective was used for optimal excitation/emission of Mecp2-GFP fluorescence. Constant laser settings and PMT detector settings were used for all samples.
Automated Mecp2-GFP+ cell detection and counting
Raw image tiles for each brain were illumination corrected, stitched in 2D with MATLAB and aligned in 3D using Fiji software. For reliable automated Mecp2-GFP detection from full brain datasets, we implemented convolutional networks (CNs). CN training for detection of Mecp2-GFP+ cells in the STPT datasets was accomplished with CN training performed on human marked-up ground truth data (biological expert identified Mecp2-GFP+ nuclei) of MeCP2-GFP brains. CN performance was determined based on F-score calculations (F-score = the harmonic mean of the precision and recall, where precision is the ratio of correctly predicted cells divided by all predicted cells and recall is ratio of correctly predicted cells divided by ground truth positive cells; ~1800 Mecp2-GFP+ cells were marked/expert/brain). Composite F-scores for Mecp2-GFP CN was obtained by determining F-scores in 8 FOVs (400 (X) um by 400 (Y) um) representing different cellular density and imaging content in 3 separate heterozygous Mecp2-GFP+ brains (24 FOVs total). Stable precision and recall was seen for all regions analyzed, delivering a composite F-score of 0.84 (Figure S2). In the CN output images, signal smaller than 10 μm2 was removed as noise. In order to normalize the performance of CN for each brain, the brightness of Mecp2-GFP+ signal for each sample was normalized by the mean and standard deviation of tissue autofluorescence signal from a coronal section corresponding to bregma position of +0.20 mm. We did not analyze Mecp2-GFP+ cells in the cerebellum due to faulty brain-to-brain warping of this region (data not shown).
3D brain registration and anatomical segmentation
Registration of individual brains to a standardized reference space was computationally achieved as published previously.31 In short, affine transform was calculated using 4 resolution levels and B-spline with 3. Advanced Mattes mutual information was the metric used to measure similarity between moving and fixed images. Image similarity function is estimated and minimized for a set of randomly chosen samples with each 23 images in a multi-resolution and iterative fashion. Entire warping of whole-brain images is done using Elastix. Anatomical segmentation of Allen Brain Atlas (ABA) labels onto sample brains was made possible also as previously published.31 Version 2.2 ABA labels (836 total) were transformed onto individually registered samples. Quality control of ROI segmentation found and excluded 95 ROIs total from analysis due to erroneous counting most likely caused by small ROI size and/or warping location (Full ROI list found in Table 1). In addition, cell counts from layer 6 a and b were combined into one layer, layer a.
2D-3D cell count correction and density measurements
Detected 2D cell count values obtained at 50 μm Z resolution were transformed by a stereological 3D conversion factor obtained by the following way (Figure S2). First, counting boxes of 200 μm x 200 μm x 50 μm (xyz) were acquired at 2.5 μm Z resolution via optical imaging within 6 brain regions comprising major anatomical divisions of a female heterozygous Mecp2-GFP mouse brain. 20 optical images were acquired at a depth range that spanned 50 μm around the normal 50 μm focal depth (i.e., 25–75 μm below the tissue surface). Second, Mecp2-GFP CN was run on the middle optical section corresponding to the 50 μm depth. Third, manual markup of Mecp2-GFP+ nuclei was performed in each counting box using the stereological counting rules of Williams and Rakic.63 Lastly, a conversion factor for each region was calculated by dividing manual 3D counts by 2D CN count of the middle section. This factor was averaged over the 6 regions reaching a final conversion factor of 2.6. (Figure S2). ROI cellular density was obtained by 1) transforming ABA labels onto individual brains, 2) converting ROI assigned pixel space to mm3, 3) dividing 2.5 μm Z-corrected absolute cell counts by mm3 values by to arrive at cells/mm3.
Cortical dissociation and fluorescence-activated cell sorting
Unilateral cortical hemispheres were used for cell-type specific XCI studies. Cortical dissections were performed from freshly decapitated mice brain in Hibernate-A (Gibco; A11473DJ) media supplemented with glutamax (Gibco; 35050) and B27 supplement (Thermo Scientific; 17504001). Single hemispheres were minced briefly with a razorblade, transferred to a new tube and incubated in pre-activated papain (10 U/ml; Worthington-biochem; LK003716) diluted in 10 mL Hibernate-A supplemented with glutamax for 15 min at 37 C. At 5 min of incubation, 2 μg of DNase I (Roche; 10104159001) was added to solution to prevent cell clumping. 3 triturations were performed over the 15 min to facilitate single-cell suspensions. Suspensions were then carefully transferred to an Opti-prep (Sigma; D1556) density gradient column diluted in hibernate-A (with B27) and spun for 15 min at 800 rcf. Optiprep media was removed and neuronal pellet was resuspended in fresh 5 mL hibernate A (with B27) and respun at 200 rcf for 5 min. Cell pellets were resuspended in 1 mL PBS (20mM HEPES, pH 7.0; 1% FBS) and mesh filtered to remove debris. Unstained controls were independently stained and samples were co-stained with 1 μM DAPI (to mark damaged cells) and DRAQ5 (Cell Signaling Technology; 4084) (to label viable cells) for at least 10 min prior to sorting. Cell analysis and sorting was performed using a FACSAria II SORP (BD Biosciences, San Jose, CA) at 25psi with the 100 μm nozzle. Fluorescent parameters included DAPI, DRAQ5, GFP and tdTomato. DAPI was excited by the 355nm UV laser and its emission was collected with a 450/50 filter. The 633nm red laser was used to excite the DRAQ5 and its emission collected with the 780/60 filter. tdTomato was excited by the 561nm yellow/green laser and emission collected with the 582/15 filter. Lastly, GFP was excited by the 488nm blue laser and emission was collected with the 530/30 filter. Unstained and single-color controls were used to set PMT voltages and eliminate spectral overlap between fluorescent channels. Experimental samples were first gated on DAPI−/DRAQ5+ populations. This gate was then applied to a scatterplot for elimination of debris and then doublet discrimination. Single cells were viewed in a dot plot of GFP-A (x axis) and TdTomato-A (y axis). Both the tdTomato+/GFP+ and tdTomato+/GFP− populations were then sorted until ≥100,000 total events were reached. We restricted our FACS XCI determinations to precent tdTomato+/GFP+ detected events from total single cells detected in order to limit potential confounds of the total tdTomato+ gated counts due to, 1) tamoxifen induction timing and post-induction dissection timing variability, which was experimentally minimized but cannot be ruled out as a cause in variable total recombined cells across animals, and 2) the gating strategy set to discriminate tdTomato+/GFP− from tdTomato+/GFP+ cells in which many tdTomato+ cells were left undetected between the two gates.
Behavioral testing
6–8-month-old ovariectomized female mice were behaviorally phenotyped in a sequential series of tests. All mice were ovariectomized at least 2 weeks prior to testing in order to remove estrous cycle influences from behavior. Each behavioral test was separated by 2–7 days to avoid acute post-testing and handling effects. Mecp2-GFP(+/p)/Fmr1 KO(+/+) mice served as behavioral controls for all behaviors studied. The following tests were sequentially performed on each mouse:
Open field test (OFT)
To measure activity and anxiety in an open field, unhabituated mice were placed in a 40 × 40 × 40 cm2 open plexiglass box containing a layer of fresh bedding. The open field arena was located in a non-sound-proof, enclosed environment under dim lighting. All mice were housed in the same facility room behavioral testing was performed. An overhead camera visually captured all tests and ANY-maze (Stoelting) automated behavior tracking software was used for real-time activity/location recording and analysis. A 20 × 20 cm center square designated within the tracking settings defined the center and perimeter boundaries of the arena. The software measured total and center distance traveled. For center-specific activity, center distance was normalized to total distance traveled and presented as percent total distance traveled. Adequate cleaning of the maze with bleach, water and drying was performed between each mouse. Fresh bedding was added to the arena for each subject.
T-maze
We studied mouse spatial memory by measuring spontaneous spatial alternations in the T-maze. Spontaneous alternation is an innate exploratory behavior possessed by rodents which is hippocampus-dependent and serves as an index of spatial and working memory. The dimensions of the T-maze used was 35 cm stem length, 28 cm arm length, 10 cm arm height, and 5 cm lane width (Stoelting). For testing, the T-maze was located in a non-sound-proof, enclosed environment under dim lighting. All mice were housed in the same facility room behavioral testing was performed in. To begin the test, each mouse was carefully placed at the stem start position of the maze and was freely allowed to enter either arm. To prevent the mouse from entering the other arm after its initial choice, a metal block was placed at the entrance of the empty arm once the subject committed exploration to an arm. The subjects were allowed to freely explore the chosen arm and stem until it explored back to start of the stem. Once the beginning position was reached, the mouse was held in-between the start position and a metal block placed proximally to the start position for 5 s. The metal block was then removed and the mouse was allowed again to enter an arm of its choice. Manual scoring of each arm choice and time to experimental completion was made after 14 trials. No more than 3 min/trial was allowed for each subject and encouragement was given to each subject at 3 min (in the form of hand movement behind the mouse) to return to start position. Mice that did not complete more than 9 trials were excluded from analysis. Adequate cleaning of the maze with bleach, water and drying was performed between each mouse. The number of trial-to-trial arm entry alternations (e.g., left-to-right or right-to-left) was calculated and expressed as a percent of total trials.
3-chamber test
Sociability was measured using the 3-chamber test based on the protocol developed in the Crawley laboratory.64 The 3-chamber apparatus used consisted of a plexiglass box (60 × 40 × 22(h) cm) partitioned into 3 chambers (20 cm/each) (Stoelting). Doors (4 × 8 cm) connecting chambers allowed the mice to freely explore all areas of the box. The apparatus was located in a non-sound-proof, enclosed environment under dim lighting. All mice were housed in the same facility room that behavioral testing was performed in. An overhead camera visually captured all test sessions and ANY-maze (Stoelting) automated behavior tracking software was used for real-time activity/location recording and analysis. Chamber designations in tracking software were user-defined and used for chamber-specific activity measurements. Two metal-barred cylindrical cages (7 cm (diameter) x 15 cm (height); 3 mm bar diameter and 7 mm spacing) were used for stranger mouse containment in one chamber and for an empty enclosure in the opposite-sided chamber. The cage bars are spaced such that close sniffing is the only interaction type possible. Ovariectomized adult female Fmr1 WT mice were used as stranger mice and were habituated to an enclosure cage for 10 min at least 1 day prior to any experiments. Each stranger mouse (n = 8) was used 4 times only and were rotated every 4 experiments for use. Test mice were habituated to an empty 3 chamber apparatus for 10 min prior to actual experiments. For testing, mice were allowed to freely explore all chambers for 10 min. For each experiment the enclosed stranger mouse was placed in the left chamber and the empty enclosure on the right. Chamber time spent and distance traveled was quantified for each chamber. Percent time spent or distance traveled was calculated as total value/individual chamber value.
Quantification of structural connectivity within behaviorally correlated brain regions
We determined if OFT and 3-chamber significantly correlated ROI groups (herein referred to as “circuits”) represented structural connected brain circuitry by comparing the median ROI circuit connection weight to a distribution of randomly sampled circuits of the same size for both tasks. ROIs having significant correlation of p < 0.01 were included in the analysis. We used the normalized connection density, a measure of connection strength normalized by both source and target region sizes, from the regional structural connectivity matrix.41 We restricted the population of structures for each circuit to ROIs from which we could draw to an intermediate level of the ontology represented by 292 ‘summary structures’. The intersection of these summary structures with the sets of ROIs for each task resulted in sets of 39 and 13 ROIs for the open field and 3-chamber tasks, respectively (Table S6). These ROIs included: OFT: ACVII, APN, DCO, GPe, GPi, IF, IGL, ISN, LAV, LGd, LGv, LT, MA, MG, NDB, NLL, NOT, PARN, PB, PO, POL, PSV, RT, SG, SGN, SPFp, SPVI, SPVO, SUT, SUV, V, VAL, VCO, VI, VLPO, VM, VPL, VPM, ZI; 3 chamber: ADP, AVP, AVPV, BST, ENTl, ENTm, LC, LPO, MPN, MPO, PS, VISl, VISpl. Additionally, since mesoscale connectivity is distance dependent and the model in Knox et al. is spatially dependent,41 we restricted the selection of random ROI circuits to have similar inter-regional distance dependence as that of their respective cell density correlated ROI circuits.
The procedure for this selection is as follows: Given a set of N cell density correlated summary structure level ROIs.
Randomly draw a set of N regions from the set of summary structures.
Compute the pairwise inter-regional distances for the set of sampled regions.
- Compute the Kolmogorov-Smirnov statistic to measure the difference in distributions of distances for the sampled and cell-count correlated networks
- If the KS statistic shows a significant difference in distributions (having a p value <0.01), reject the sample and return to (1).
- Else, return the sample.
The above procedure is repeated 1000 times, after which the median normalized connection density of each experimental circuit is compared to the distribution of the sample medians. Since these connectivity measures are log-normally distributed, the t statistic is computed in log-transformed space to test the significance of the difference. Visualization of ROI-ROI connectivity for each behavioral circuit of ROIs was created with Cytoscape network visualization and analytic program (Version 3.7.1). Significantly correlated ROIs of the deepest ontological distance from root structures were chosen for visualization, except for the ROIs not annotated at the summary structure level and hence not found in the structural connectivity matrix. Those ROIs included: BSTmg, BSTpr, BSTif, BSTpr, PVHap, TTv3, isl, islm, MPNc, MPNl, PAA3, and COApl3. Log-transformed normalized connection densities were used for edge sizes and scaled for presentation. Edges with sizes <0 were excluded from the visualization.
QUANTIFICATION AND STATISTICAL ANALYSIS
Whole-brain absolute cell counts were compared amongst Xm-active and Xp-active reporter brains using a Welch’s t test. A 2-way mixed effects ANOVA ([2] X-active/KO or WT allele parent-of-origin x [9] major ROI) was used to compare XC-active (Figure 1) or WT:KO allele (Figure 2) parent-of-origin across cell densities from major ROIs. Holm-Sidak post-hoc tests were used to assess simple between-subjects effects. Brain-wide screens for skewed XCI were statistically performed using Benjamini-Hochberg FDR-corrected student’s t tests on X-active cell density groups. Welch’s ANOVAs with Dunnet’s T3 post-hoc testing was used to compare group performances in the OFT, T maze, and 3 chamber total distance traveled. 3 chamber results were analyzed with 2-way mixed effects ANOVAs with Holm-Sidak post-hoc tests of between- and/or within-subjects simple effects. Whole-brain absolute healthy cell counts in Fmr1 WT and KO mice were analyzed via ANOVA with Holm-Sidak post-hoc comparisons. Correlational screens were used to localize the physical source of behavioral penetrance using Pearson’s correlation amongst cell density and behavioral score across 736 ROIs. In this analysis, we did not correct the p values against Type I error risk in favor of revealing ROI networks or patterns that share behavioral dependencies. Additionally, noise correlations (Table S5) in the control WT groups did not surpass 5% of ROIs in both OFT (20/736 ROIs = 2.7%) and 3 chamber (5/736 ROIs = 0.6%) screens, further supporting the use of uncorrected p values in this dataset. ROI circuits of behavioral penetrance were defined by p < 0.01 bins of significance and each bin’s distributed cell count, volume, and density across each OFT and 3 chamber circuit were calculated (Table S7) and used for linear and logistic regression modeling. Logistic regression was performed on healthy cell density percent in behavioral circuits as the continuous, independent variable and WT performance as the categorical, dependent variable. Mice (n = 22) from all genotypes were categorized as WT or mutant performers for each test based on the performance range of Fmr1 WT mice. Fmr1 WT mice were coded as containing 100% healthy cell density. A likelihood ratio test was performed on each logistic model to determine statistical significance. All statistical testing was performed with Graphpad Prism software version 7.0 and R (R Core Team). Alpha level was set at 0.05 in all analyses except where otherwise noted above.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-NeuN (rabbit) polyclonal | Millipore | Cat# ABN78; AB_10807945 |
| DRAQ5 | Cell Signaling Technology | Cat#4084 |
| Hibernate A | Gibco | Cat#A11473DJ |
| Glutamax | Gibco | Cat#35050 |
| Pre-activated papain | Worthington-biochem | Cat#LK003716 |
| DNase I | Roche | Cat#10104159001 |
| B27 supplement | Thermo Scientific | Cat#17504001 |
| DAPI | Thermo Scientific | Cat#D1306 |
| Donkey anti-rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 |
Thermo Fisher Scientific | Cat#A10042 |
| Tamoxifen | Sigma | Cat#T5648 |
| Deposited data | ||
| Raw and analyzed data | This paper | Tables S1–S7 |
| Experimental models: Organisms/strains | ||
| Mouse: Mecp2tm3.1Bird/J (Mecp2-GFP) | The Jackson Laboratory | IMSR_JAX:014610 |
| Mouse: C57BL/6J | The Jackson Laboratory | IMSR_JAX:000664 |
| Mouse: B6.129P2-Fmr1tm1Cgr/J (Fmr1-KO) | The Jackson Laboratory | IMSR_JAX:003025 |
| Mouse: B6; 129S4-Fezf2tm1.1(cre/ERT2)Zjh/J (Fezf2-P2A-CreER) | The Jackson Laboratory | IMSR_JAX:036296 |
| Mouse: B6; 129S4-Pvalbtm2(cre/ERT2)Zjh/J (PV-P2A-CreER) | The Jackson Laboratory | IMSR_JAX:028580 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (Ai14) | The Jackson Laboratory | IMSR_JAX:007914 |
| Software and algorithms | ||
| STPT image stitching algorithm | Kim Laboratory | https://doi.org/10.5281/zenodo.6517742 |
| Convolutional neural network for cell counting | Osten Laboratory | http://brainarchitecture.org/ost |
| Elastix for image registration | https://elastix.lumc.nl/ | https://elastix.lumc.nl/ |
| ImageJ | NIH | RRID:SCR_003070 |
| Graphpad Prism | Graphpad Software | https://www.graphpad.com/features |
| R for statistical computing | The R Foundation | https://www.r-project.org/ |
| MATLAB | Mathworks | https://www.mathworks.com/products/matlab.html |
| ANY-maze | Stoelting | https://www.any-maze.com/ |
| Cytoscape | Cytoscape Consortium | https://cytoscape.org/ |
| X chromosome structural connectivity modeling analysis code and data | Allen Institute | https://github.com/AllenInstitute/chromosome-network-modeling; https://doi.org/10.5281/zenodo.10815678 |
| Other | ||
| Tissuecyte 1000: Serial 2-photon microscope | TissueVision | https://www.tissuevision.com/tissuecyte |
| LSM780 confocal microscope | Zeiss | https://www.zeiss.com/microscopy/en/products/light-microscopes/confocal-microscopes.html |
| FACSAria II SORP | BD Biosciences | https://www.bdbiosciences.com/en-us/products/instruments/flow-cytometers/research-cell-sorters/ |
Highlights.
Adult brain XCI favors maternal X-active cells at 60:40 ratios across all regions
Local XCI mosaicism shapes individual variability beyond whole-brain status by 20%
X-linked Fmr1-KO allele is penetrant in maternal carriers with biased brain XCI
XCI within intact brain circuitry distinguishes individual phenotypic outcomes
ACKNOWLEDGMENTS
We would like to thank CSHL Hillside animal husbandry services for their support and efforts and members of the Osten lab and Dr. Kirstin Baldwin for their inputs on the study. We thank Dr. Sam A. Golden for his guidance in the resubmission of the manuscript. This work was supported by grants R01 MH096946 and U01 MH105971 to P.O. and funds from The Gertrude and Louis Feil Family Trust to P.O.; R01MH116176 and RF1MH124605 to Y.K.; and the Washington Research Foundation Postdoctoral Fellowship, the CoMotion Innovation Gap Fund, and a Washington Research Foundation Phase I Technology Commercialization grant to E.R.S.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114068.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Nguyen DK, and Disteche CM (2006). Dosage compensation of the active X chromosome in mammals. Nat. Genet 38, 47–53. 10.1038/ng1705. [DOI] [PubMed] [Google Scholar]
- 2.Ropers H-H, and Hamel BCJ (2005). X-linked mental retardation. Nat. Rev. Genet 6, 46–57. 10.1038/nrg1501. [DOI] [PubMed] [Google Scholar]
- 3.Skuse DH (2005). X-linked genes and mental functioning14 Spec No 1. Hum. Mol. Genet, R27–R32. 10.1093/hmg/ddi112. [DOI] [PubMed] [Google Scholar]
- 4.Avner P, and Heard E (2001). X-chromosome inactivation: counting, choice and initiation. Nat. Rev. Genet 2, 59–67. 10.1038/35047580. [DOI] [PubMed] [Google Scholar]
- 5.Bittel DC, Theodoro MF, Kibiryeva N, Fischer W, Talebizadeh Z, and Butler MG (2008). Comparison of X-chromosome inactivation patterns in multiple tissues from human females. J. Med. Genet 45, 309–313. 10.1136/jmg.2007.055244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gibson JH, Williamson SL, Arbuckle S, and Christodoulou J (2005). X chromosome inactivation patterns in brain in Rett syndrome: implications for the disease phenotype. Brain Dev. 27, 266–270. 10.1016/j.braindev.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Minks J, Robinson WP, and Brown CJ (2008). A skewed view of X chromosome inactivation. J. Clin. Invest 118, 20–23. 10.1172/JCI34470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orstavik KH (2009).X chromosome inactivation in clinical practice. Hum. Genet 126, 363–373. 10.1007/s00439-009-0670-5. [DOI] [PubMed] [Google Scholar]
- 9.Plenge RM, Stevenson RA, Lubs HA, Schwartz CE, and Willard HF (2002). Skewed X-chromosome inactivation is a common feature of X-linked mental retardation disorders. Am. J. Hum. Genet 71, 168–173. 10.1086/341123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renault NKE, Pritchett SM, Howell RE, Greer WL, Sapienza C, Ørstavik KH, and Hamilton DC (2013). Human X-chromosome inactivation pattern distributions fit a model of genetically influenced choice better than models of completely random choice. Eur. J. Hum. Genet 21, 1396–1402. 10.1038/ejhg.2013.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shahbazian MD, Sun Y, and Zoghbi HY (2002). Balanced X chromosome inactivation patterns in the Rett syndrome brain. Am. J. Med. Genet 111, 164–168. 10.1002/ajmg.10557. [DOI] [PubMed] [Google Scholar]
- 12.Vianna EQ, Piergiorge RM, Gonçalves AP, Dos Santos JM, Calassara V, Rosenberg C, Krepischi ACV, Boy da Silva RT, Dos Santos SR, Ribeiro MG, et al. (2020). Understanding the Landscape of X-linked Variants Causing Intellectual Disability in Females Through Extreme X Chromosome Inactivation Skewing. Mol. Neurobiol 57, 3671–3684. 10.1007/s12035-020-01981-8. [DOI] [PubMed] [Google Scholar]
- 13.Amir RE, Van den Veyver IB, Schultz R, Malicki DM, Tran CQ, Dahle EJ, Philippi A, Timar L, Percy AK, Motil KJ, et al. (2000). Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Ann. Neurol 47, 670–679. [PubMed] [Google Scholar]
- 14.Archer H, Evans J, Leonard H, Colvin L, Ravine D, Christodoulou J, Williamson S, Charman T, Bailey MES, Sampson J, et al. (2007). Correlation between clinical severity in patients with Rett syndrome with a p.R168X or p.T158M MECP2 mutation, and the direction and degree of skewing of X-chromosome inactivation. J. Med. Genet 44, 148–152. 10.1136/jmg.2006.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bienvenu T, Carrié A, de Roux N, Vinet MC, Jonveaux P, Couvert P, Villard L, Arzimanoglou A, Beldjord C, Fontes M, et al. (2000). MECP2 mutations account for most cases of typical forms of Rett syndrome. Hum. Mol. Genet 9, 1377–1384. 10.1093/hmgZ9.9.1377. [DOI] [PubMed] [Google Scholar]
- 16.Heine-Surñer D, Torres-Juan L, Morlà M, Busquets X, Barceló F, Picó G, Bonilla L, Govea N, Bernués M, and Rosell J (2003). Fragile-X syndrome and skewed X-chromosome inactivation within a family: a female member with complete inactivation of the functional X chromosome. Am. J. Med. Genet 122A, 108–114. 10.1002/ajmg.a.20160. [DOI] [PubMed] [Google Scholar]
- 17.Villard L, Kpebe A, Cardoso C, Chelly PJ, Tardieu PM, and Fontes M (2000). Two affected boys in a Rett syndrome family: clinical and molecular findings. Neurology 55, 1188–1193. 10.1212/wnl.55.8.1188. [DOI] [PubMed] [Google Scholar]
- 18.Wan M, Lee SS, Zhang X, Houwink-Manville I, Song HR, Amir RE, Budden S, Naidu S, Pereira JL, Lo IF, et al. (1999). Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotspots. Am. J. Hum. Genet 65, 1520–1529. 10.1086/302690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zappella M, Meloni I, Longo I, Hayek G, and Renieri A (2001). Preserved speech variants of the Rett syndrome: molecular and clinical analysis. Am. J. Med. Genet 104, 14–22. 10.1002/ajmg.10005. [DOI] [PubMed] [Google Scholar]
- 20.Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, et al. (2005). Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet 77, 442–453. 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Auranen M, Vanhala R, Vosman M, Levander M, Varilo T, Hietala M, Riikonen R, Peltonen L, and Järvelä I (2001). MECP2 gene analysis in classical Rett syndrome and in patients with Rett-like features. Neurology 56, 611–617. 10.1212/wnl.56.5.611. [DOI] [PubMed] [Google Scholar]
- 22.Nielsen JB, Henriksen KF, Hansen C, Silahtaroglu A, Schwartz M, and Tommerup N (2001). MECP2 mutations in Danish patients with Rett syndrome: high frequency of mutations but no consistent correlations with clinical severity or with the X chromosome inactivation pattern. Eur. J. Hum. Genet 9, 178–184. 10.1038/sj.ejhg.5200600. [DOI] [PubMed] [Google Scholar]
- 23.Sirianni N, Naidu S, Pereira J, Pillotto RF, and Hoffman EP (1998). Rett syndrome: confirmation of X-linked dominant inheritance, and localization of the gene to Xq28. Am. J. Hum. Genet 63, 1552–1558. 10.1086/302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smeets E, Schollen E, Moog U, Matthijs G, Herbergs J, Smeets H, Curfs L, Schrander-Stumpel C, and Fryns JP (2003). Rett syndrome in adolescent and adult females: clinical and molecular genetic findings. Am. J. Med. Genet 122A, 227–233. 10.1002/ajmg.a.20321. [DOI] [PubMed] [Google Scholar]
- 25.Taylor AK, Safanda JF, Fall MZ, Quince C, Lang KA, Hull CE, Carpenter I, Staley LW, and Hagerman RJ (1994). Molecular predictors of cognitive involvement in female carriers of fragile X syndrome. JAMA 271, 507–514. [PubMed] [Google Scholar]
- 26.Zoghbi HY, Percy AK, Schultz RJ, and Fill C (1990). Patterns of X chromosome inactivation in the Rett syndrome. Brain Dev. 12, 131–135. 10.1016/s0387-7604(12)80194-x. [DOI] [PubMed] [Google Scholar]
- 27.Young JI, and Zoghbi HY (2004). X-Chromosome Inactivation Patterns Are Unbalanced and Affect the Phenotypic Outcome in a Mouse Model of Rett Syndrome. Am. J. Hum. Genet 74, 511–520. 10.1086/382228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watson CM, Pelka GJ, Radziewic T, Shahbazian MD, Christodoulou J, Williamson SL, and Tam PPL (2005). Reduced proportion of Purkinje cells expressing paternally derived mutant Mecp2308 allele in female mouse cerebellum is not due to a skewed primary pattern of X-chromosome inactivation. Hum. Mol. Genet 14, 1851–1861. 10.1093/hmg/ddi191. [DOI] [PubMed] [Google Scholar]
- 29.Gregg C, Zhang J, Butler JE, Haig D, and Dulac C (2010). Sex-specific parent-of-origin allelic expression in the mouse brain. Science 329, 682–685. 10.1126/science.1190831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X, Soloway PD, and Clark AG (2010). Paternally biased X inactivation in mouse neonatal brain. Genome Biol. 11, R79. 10.1186/gb-2010-11-7-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim Y, Venkataraju KU, Pradhan K, Mende C, Taranda J, Turaga SC, Arganda-Carreras I, Ng L, Hawrylycz MJ, Rockland KS, et al. (2015). Mapping social behavior-induced brain activation at cellular resolution in the mouse. Cell Rep. 10, 292–305. 10.1016/j.celrep.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, Guy J, Kastan NR, Robinson ND, de Lima Alves F, et al. (2013). Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci 16, 898–902. 10.1038/nn.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, and Nelson DL (1991). Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822. 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 34.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, and Zhang FP (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 35.(1994). Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 78, 23–33. [PubMed] [Google Scholar]
- 36.Gauducheau M, Lemaire-Mayo V, D’Amato FR, Oddi D, Crusio WE, and Pietropaolo S (2017). Age-specific autistic-like behaviors in heterozygous Fmr1-KO female mice. Autism Res. 10, 1067–1078. 10.1002/aur.1743. [DOI] [PubMed] [Google Scholar]
- 37.Nolan SO, Hodges SL, and Lugo JN (2020). High-throughput analysis of vocalizations reveals sex-specific changes in Fmr1 mutant pups. Gene Brain Behav. 19, e12611. 10.1111/gbb.12611. [DOI] [PubMed] [Google Scholar]
- 38.Yau SY, Bostrom CA, Chiu J, Fontaine CJ, Sawchuk S, Meconi A, Wortman RC, Truesdell E, Truesdell A, Chiu C, et al. (2016). Impaired bidirectional NMDA receptor dependent synaptic plasticity in the dentate gyrus of adult female Fmr1 heterozygous knockout mice. Neurobiol. Dis 96, 261–270. 10.1016/j.nbd.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 39.O’Reilly C, Iavarone E, Yi J, and Hill SL (2021). Rodent somatosensory thalamocortical circuitry: Neurons, synapses, and connectivity. Neurosci. Biobehav. Rev 126, 213–235. 10.1016/j.neubiorev.2021.03.015. [DOI] [PubMed] [Google Scholar]
- 40.McHenry JA, Otis JM, Rossi MA, Robinson JE, Kosyk O, Miller NW, McElligott ZA, Budygin EA, Rubinow DR, and Stuber GD (2017). Hormonal gain control of a medial preoptic area social reward circuit. Nat. Neurosci 20, 449–458. 10.1038/nn.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knox JE, Harris KD, Graddis N, Whitesell JD, Zeng H, Harris JA, Shea-Brown E, and Mihalas S (2019). High-resolution data-driven model of the mouse connectome. Netw. Neurosci 3, 217–236. 10.1162/netn_a_00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee JT, and Bartolomei MS (2013). X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell 152, 1308–1323. 10.1016/j.cell.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 43.Kojima Y, Tam OH, and Tam PPL (2014). Timing of developmental events in the early mouse embryo. Semin. Cell Dev. Biol 34, 65–75. 10.1016/j.semcdb.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 44.Raefski AS, and O’Neill MJ (2005). Identification of a cluster of X-linked imprinted genes in mice. Nat. Genet 37, 620–624. 10.1038/ng1567. [DOI] [PubMed] [Google Scholar]
- 45.Wu H, Luo J, Yu H, Rattner A, Mo A, Wang Y, Smallwood PM, Erlanger B, Wheelan SJ, and Nathans J (2014). Cellular resolution maps of X chromosome inactivation: implications for neural development, function, and disease. Neuron 81, 103–119. 10.1016/j.neuron.2013.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szymusiak R. (1995). Magnocellular nuclei of the basal forebrain: substrates of sleep and arousal regulation. Sleep 18, 478–500. 10.1093/sleep/18.6.478. [DOI] [PubMed] [Google Scholar]
- 47.Kroeger D, Absi G, Gagliardi C, Bandaru SS, Madara JC, Ferrari LL, Arrigoni E, Münzberg H, Scammell TE, Saper CB, and Vetrivelan R (2018). Galanin neurons in the ventrolateral preoptic area promote sleep and heat loss in mice. Nat. Commun 9, 4129. 10.1038/s41467-018-06590-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lewis LD, Voigts J, Flores FJ, Schmitt LI, Wilson MA, Halassa MM, and Brown EN (2015). Thalamic reticular nucleus induces fast and local modulation of arousal state. Elife 4, e08760. 10.7554/eLife.08760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frankland PW, Wang Y, Rosner B, Shimizu T, Balleine BW, Dykens EM, Ornitz EM, and Silva AJ (2004). Sensorimotor gating abnormalities in young males with fragile X syndrome and Fmr1-knockout mice. Mol. Psychiatr 9, 417–425. 10.1038/sj.mp.4001432. [DOI] [PubMed] [Google Scholar]
- 50.Nielsen DM, Derber WJ, McClellan DA, and Crnic LS (2002). Alterations in the auditory startle response in Fmr1 targeted mutant mouse models of fragile X syndrome. Brain Res. 927, 8–17. 10.1016/s0006-8993(01)03309-1. [DOI] [PubMed] [Google Scholar]
- 51.Williams TA, Porter MA, and Langdon R (2014). Social approach and emotion recognition in fragile X syndrome. Am. J. Intellect. Dev. Disabil 119, 133–150. 10.1352/1944-7558-119.2.133. [DOI] [PubMed] [Google Scholar]
- 52.Williams TA, Langdon R, and Porter MA (2013). Hyper-reactivity in fragile X syndrome females: generalised or specific to socially-salient stimuli? A skin conductance study. Int. J. Psychophysiol 88, 26–34. 10.1016/j.ijpsycho.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 53.Clifford S, Dissanayake C, Bui QM, Huggins R, Taylor AK, and Loesch DZ (2007). Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J. Autism Dev. Disord 37, 738–747. 10.1007/s10803-006-0205-z. [DOI] [PubMed] [Google Scholar]
- 54.Fyhn M, Molden S, Witter MP, Moser EI, and Moser M-B (2004). Spatial representation in the entorhinal cortex. Science 305, 1258–1264. 10.1126/science.1099901. [DOI] [PubMed] [Google Scholar]
- 55.Wang Q, Sporns O, and Burkhalter A (2012). Network analysis of corticocortical connections reveals ventral and dorsal processing streams in mouse visual cortex. J. Neurosci 32, 4386–4399. 10.1523/JNEUROSCI.6063-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scott R, Aubry A, Cuttoli R.D.d., Rachel F-F, Lyonna P, Cathomas F, Burnett C, Yang Y, yuan chongzhen, Lablanca A, et al. (2023). A critical role for cortical amygdala circuitry in shaping social encounters. Res. Sq 10.21203/rs.3.rs-3015820/v1. [DOI] [Google Scholar]
- 57.Tovote P, Fadok JP, and Lüthi A (2015). Neuronal circuits for fear and anxiety. Nat. Rev. Neurosci 16, 317–331. 10.1038/nrn3945. [DOI] [PubMed] [Google Scholar]
- 58.Bayless DW, and Shah NM (2016). Genetic dissection of neural circuits underlying sexually dimorphic social behaviours. Philos. Trans. R. Soc. Lond. B Biol. Sci 371, 20150109. 10.1098/rstb.2015.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun Z, Fan J, and Wang Y (2022). X-Chromosome Inactivation and Related Diseases. Genet. Res 2022, 1391807. 10.1155/2022/1391807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Calaway JD, Lenarcic AB, Didion JP, Wang JR, Searle JB, McMillan L, Valdar W, and Pardo-Manuel de Villena F (2013). Genetic architecture of skewed X inactivation in the laboratory mouse. PLoS Genet. 9, e1003853. 10.1371/journal.pgen.1003853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun Y, Yang Y, Luo Y, Chen M, Wang L, Huang Y, Yang Y, and Dong M (2021). Lack of MECP2 gene transcription on the duplicated alleles of two related asymptomatic females with Xq28 duplications and opposite X-chromosome inactivation skewing. Hum. Mutat 42, 1429–1442. 10.1002/humu.24262. [DOI] [PubMed] [Google Scholar]
- 62.Matho KS, Huilgol D, Galbavy W, He M, Kim G, An X, Lu J, Wu P, Di Bella DJ, Shetty AS, et al. (2021). Genetic dissection of the glutamatergic neuron system in cerebral cortex. Nature 598, 182–187. 10.1038/s41586-021-03955-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Williams RW, and Rakic P (1988). Three-dimensional counting: an accurate and direct method to estimate numbers of cells in sectioned material. J. Comp. Neurol 278, 344–352. 10.1002/cne.902780305. [DOI] [PubMed] [Google Scholar]
- 64.Yang M, Silverman JL, and Crawley JN (2011). Automated three-chambered social approach task for mice. Curr Protoc Neurosci Chapter. Curr. Protoc. Neurosci 8 (8.26). 10.1002/0471142301.ns0826s56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw cell count and calculated cell density data from samples of all whole-brain imaging experiments are published with the paper as Supplemental tables. All behavioral data is published with the paper as supplemental tables. All structural connectivity raw data and circuit cell density calculations are published with the paper as supplemental tables. Raw FACS sorting data have been deposited at FlowRepository.org and are publicly available as of the date of publication. All microscopy images are available from lead contact upon request.
Custom code for structural connectivity modeling analysis is hosted on Github and Zenodo (https://doi.org/10.5281/zenodo.10815678) and is publicly available as of date of publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.