

Abstract
Metabolic alterations are a hallmark of cancer controlling tumor progression and metastasis. Among the various metabolic phenotypes encountered in tumors, this review focuses on the contributions of mitochondria, lipid and amino acid metabolism to the metastatic process. Tumor cells require functional mitochondria to grow, proliferate and metastasize, but shifts in mitochondrial activities confer pro-metastatic traits encompassing increased production of mitochondrial reactive oxygen species (mtROS), enhanced resistance to apoptosis and the increased or de novo production of metabolic intermediates of the TCA cycle behaving as oncometabolites, including succinate, fumarate, and d-2-hydroxyglutarate that control energy production, biosynthesis and the redox state. Lipid metabolism and the metabolism of amino acids, such as glutamine, glutamate and proline are also currently emerging as focal control points of cancer metastasis.
Keywords: Tumor metastasis, Oxidative phosphorylation (OXPHOS), Electron transport chain (ETC), Reactive oxygen species (ROS), Tricarboxylic acid cycle (TCA cycle) , Lipogenesis, Glutaminolysis, Proline metabolism
Introduction
While intensive research aims to characterize primary tumor biology for the sake of new therapeutic and diagnostic tools, less attention has been paid until recently to the biology of metastases. Metastasis, however, is estimated to be responsible for ~90 % of cancer-associated deaths [1], representing a yearly toll of ~8,200,000 patients worldwide (Globoscan 2012).1 Growing evidence points to a metabolic control of tumor progression affecting many phenotypic traits of malignancy, including metastasis. While the specific contributions of tumor pH, glycolysis and the pentose phosphate pathway to the metastatic process are addressed in a companion paper, this review focuses on mitochondrial, lipid and amino acid metabolism.
Defects of mitochondrial function have long been suspected to contribute to the development and progression of cancer. Almost a century ago, Otto Warburg [2] initiated research on mitochondrial alterations in cancer and proposed a mechanism to explain the differences in energy metabolism between normal and cancer cells. He suggested that mitochondrial alterations could provide unique therapeutic targets in various cancer types [3]. The concept promoted by Warburg that mitochondrial damage is the cause of cancer is no longer tenable as a general concept [4]. However, specific cancer-associated mutations have been reported in nuclear-encoded mitochondrial enzymes of the tricarboxylic acid (TCA) cycle, including fumarate hydratase (FH) [5], succinate dehydrogenase (SDH) [5] and isocitrate dehydrogenase (IDH) [6]. SDH deficiency has been linked to hereditary paraganglioma and pheochromocytoma, and FH inactivation promotes leiomyoma, leiomyosarcoma and renal cell carcinoma [7, 8]. Besides these mutations, several other cancer-related mitochondrial alterations have been identified, and mitochondria are now emerging as key players in tumor transformation and progression. This is not surprising since mitochondria are not passive bystanders involved in bioenergetics. They rather act as metabolic and signaling hubs, interconnecting metabolism and cell signaling.
From a metabolic standpoint, one of the main activities of mitochondria is to perform the TCA cycle where the acetyl group of acetyl-coenzyme A (acetyl-CoA), mostly derived from pyruvate, is progressively oxidized to CO2 to provide reducing equivalents for oxidative phosphorylation (OXPHOS) (Fig. 1). In addition to pyruvate, the TCA cycle can be fueled by products of anaplerotic reactions (from Greek  [up] and πληρόω [fill in]), among which α-ketoglutarate (αKG) from glutaminolysis is a major substrate [9]. OXPHOS occurs at the mitochondrial electron transport chain (ETC) and represents a highly efficient pathway for ATP generation. Importantly, the TCA cycle also generates intermediates connecting the cycle to other metabolic pathways by means of the so-called cataplerotic reactions (from Greek κατα [down] and πληρόω [deplete]) that drain the TCA cycle from its metabolites. These reactions provide substrates for biosynthesis, thus supporting normal cellular functions, cell growth and proliferation [10].
[up] and πληρόω [fill in]), among which α-ketoglutarate (αKG) from glutaminolysis is a major substrate [9]. OXPHOS occurs at the mitochondrial electron transport chain (ETC) and represents a highly efficient pathway for ATP generation. Importantly, the TCA cycle also generates intermediates connecting the cycle to other metabolic pathways by means of the so-called cataplerotic reactions (from Greek κατα [down] and πληρόω [deplete]) that drain the TCA cycle from its metabolites. These reactions provide substrates for biosynthesis, thus supporting normal cellular functions, cell growth and proliferation [10].
Fig. 1.
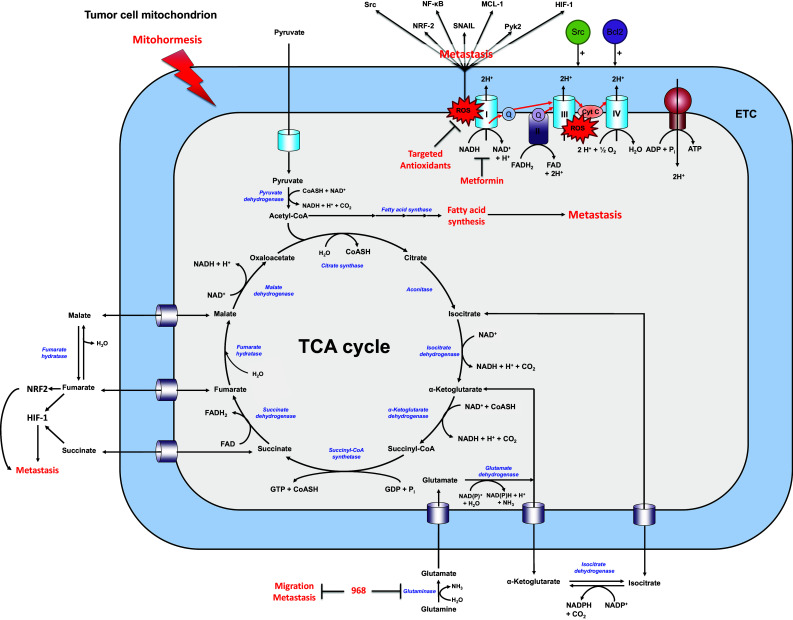
Simplified scheme highlighting the contribution of mitochondria, lipid and amino acid metabolism to tumor metastasis. The scheme depicts a mitochondrion where enzymes are represented in italicized blue font and their substrates in bold black. Upon entering into the mitochondria, pyruvate is broken down during the catabolic part of the tricarboxylic acid (TCA) cycle. This produces reducing agents that fuel the electron transport chain (ETC) to generate the proton motive force needed for the production of ATP and, as a byproduct, mitochondrial reactive oxygen species (mtROS). Increased mtROS levels have been proposed to increase resistance to stress (mitohormesis). Furthermore, several anaplerotic reactions replenish the TCA cycle. Many of these reactions promote tumor metastasis, as indicated in red. Other abbreviations: HIF-1 hypoxia-inducible factor-1, MCL-1 myeloid cell leukemia-1, NF-kB nuclear factor-kB, NRF2 nuclear factor-like 2
Besides metabolism sensu stricto, mitochondria are also critical regulators of compartmentalized cellular signaling. One typical example is the modulation of apoptosis, which mitochondria achieve by regulating the release of cytochrome c and calcium. Mitochondria further host several signaling pathways involved in tumor transformation and growth. For example, the mitochondrial fraction of signal transducer and activator of transcription 3 (STAT3) promotes OXPHOS and pancreatic cancer initiation [11, 12], ERK activation promotes tumor cell resistance to apoptosis by inhibiting the opening of the permeability transition pore [13], and c-Src regulates OXPHOS by phosphorylating several ETC subunits [14]. These signaling pathways are directly related to mitochondrial metabolism. It is therefore not surprising that mitochondria exert critical roles not only in tumorigenesis but also in cancer metastasis, which is the topic of this review.
Oxidative phosphorylation and mitochondrial reactive oxygen species
Mitochondria are involved in OXPHOS, which couples redox reactions of the ETC to ATP synthesis (Fig. 1). During this process, electrons derived from NADH and FADH2 are transferred to molecular oxygen through sequential redox reactions that generate a proton gradient across the inner mitochondrial membrane. Four macromolecular protein complexes located at the inner mitochondrial membrane are required for driving the generation of the gradient, which triggers the activity of the ATP-synthase complex at the inner mitochondrial membrane. Thus, protons transferred from the intermembrane space to the mitochondrial matrix through ATP-synthase along their concentration gradient provide the energy for ATP generation in mitochondria.
While O2 is fully reduced to water to foster ATP synthesis in mitochondria, a small proportion is converted into superoxide [15, 16] (Fig. 1). Superoxide is a radical anion and a highly reactive oxygen species (ROS) acting as a strong oxidant. In normal circumstances, superoxide is detoxified by superoxide dismutases (SODs) that rapidly convert it to H2O2. SODs are excellent catalysts that are rate-limited primarily by the speed of diffusion of the substrate in solution [17]. When superoxide is produced at a supraphysiological rate and ROS rise to a critical level, they can oxidize cytochrome c and impair its binding to the inner mitochondrial membrane [18]. Cytochrome c is normally bound to the phospholipid cardiolipin at the inner mitochondrial membrane where it transfers electrons from complex III to IV of the ETC. Following oxidation, this interaction is repressed and cytochromec is released from mitochondria, thus triggering apoptosis [19]. However, mitochondrial ROS (mtROS) are not only triggering apoptosis. At non-lethal levels, they are essential for cellular adaptation to stress [20] and to hypoxia through activation of transcription factor hypoxia-inducible factor-1 (HIF-1) [21], as well as for the induction of autophagy [22] and mitochondrial biogenesis by activation of the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator-1 (PGC-1) [23].
The initial observation by Warburg that some tumor cells perform glycolysis at a high rate even in presence of oxygen prompted him to formulate the hypothesis according to which mitochondria are not functional in tumors [3]. Except for some specific cases, this hypothesis has not been confirmed [24]. Still, the perception of the role of mitochondria in tumor cells has often been reduced to the provision of biosynthetic intermediates for cellular proliferation [25]. But mitochondria are also emerging as essential mediators of tumorigenesis. In fact, complete abolition of ETC activity was shown to prevent tumorigenesis in a Kirsten Rat Sarcoma (KRAS)-driven mouse model of lung cancer [26], in murine breast cancer and melanoma models [27], and it inhibited the in vivo growth of human breast cancer cells [28]. In models of pancreatic cancer, mitochondrial targeting of STAT3 stimulated ETC activity and tumor transformation [11, 12], further emphasizing the critical role of mitochondria in cancer. Along the same lines, cancer cells responsible for tumor relapse following oncogene ablation specifically rely on OXPHOS for survival [29]. Consistently, the antidiabetic drug metformin, a dual AMPK activator and ETC Complex I inhibitor [30], acts synergistically with chemotherapy to eradicate resistant tumor cells and promote cancer remission [31]. This observation has been recently linked to the activity of metformin as an inhibitor of ETC Complex I [30, 32].
With regard to the specific contribution of OXPHOS to tumor metastasis, several groups challenged the view that ETC is dispensable for tumor migration and metastasis [27, 33–37]. Disseminating cancer cells actually display increased levels of mitochondrial respiration, at least in breast and melanoma models [35]. This is driven by the upregulation of PGC-1α, a master gene regulating mitochondrial biogenesis and metabolism, making PGC-1α expression a marker of poor prognosis in invasive ductal breast carcinoma [35]. Another piece of evidence arguing for an important contribution of mitochondria to tumor metastasis derives from the work of Tan et al. [27] who demonstrated that active mitochondria are required for tumor growth in vivo and that upon experimentally induced loss of mitochondrial DNA (ρ0 cells), cancer cells are capable of restoring mitochondrial respiration by “stealing” mitochondria from host cells. Interestingly, this work further showed that tumor cells display an increased mitochondrial activity (elevated O2 consumption and increased presence of cristae) along with the progressive acquisition of metastatic traits, with circulating tumor cells characterized by an intermediate metabolic phenotype compared to primary and secondary tumors (see Figure 1 in companion paper). One of the advantages conferred by an elevation of OXPHOS activity could be a stronger resistance to apoptosis through the induction of a pro-oxidant state [38]. Altered mitochondrial metabolism has also been proposed to promote metastasis by inducing stress tolerance during nutrient deprivation [39].
In strong contrast to studies advocating for a contribution of OXPHOS to cancer metastasis, a number of independent authors reported that interfering with mitochondrial activity can promote metastasis. In particular, overexpression of oncogenes such as BAX inhibitor-1 [40, 41] or loss of metastasis suppressor genes such as KISS1 [42] induces metastasis together with a switch from an oxidative to a glycolytic metabolism. Mitochondrial DNA (mtDNA) mutations can also promote metastatic dissemination from prostate to bones and the invasive growth of melanoma cells in vivo [43, 44]. Accordingly, specific mtDNA mutations are a marker of poor prognosis in breast cancer and melanoma patients [45, 46]. As a final example, mitochondrial stress resulting from mtDNA depletion or treatment with ionophores that uncouple ETC activity from ATP generation promoted an invasive behavior in human lung cancer cells and C2C12 myoblasts [47, 48].
It has been reported that either increased or reduced OXPHOS activity promotes metastatic dissemination. A potential explanation of this conundrum is that two opposite metabolic phenotypes might contribute to metastasis by regulating different pathways. Indeed, on one hand, increased mitochondrial metabolism has been shown to promote resistance to apoptosis [35, 38, 49–51], whereas, on the other hand, mitochondrial dysfunction can favor a pro-metastatic behavior either by promoting glycolytic compensation [52] or by impacting the NAD+/NADH redox ratio that regulates sirtuin activity, thereby directly promoting tumor metastasis [53]. We recently proposed a parallel but not mutually exclusive interpretation. In different tumor cell models, we indeed observed that either an increased or a dysfunctional mitochondrial activity are equally capable of promoting an invasive tumor phenotype [33]. We found that metabolic states of tumor cells corresponding to increased TCA cycle activity or experimentally induced ETC bottlenecking were associated with increased mtROS generation, and mtROS were a common mediator of metastasis for these two different metabolic phenotypes: targeting mtROS with mitochondria-targeted antioxidants mitoTEMPO or mitoQ inhibited metastatic dissemination in a mouse melanoma B16F10 model and abolished spontaneous metastatic dissemination in a model of MDA-MB-231 human breast cancer in mice [33]. Despite the fact that mtROS can promote apoptosis, depending on their production level, they also behave as second messengers for retrograde mitochondrial signaling to the nucleus [54] (Fig. 1). Because mtROS are short-lived and compartmentalized in mitochondria [20], they can indeed spatially and temporally coordinate a localized signaling cascade by oxidizing specific amino acid residues [55]. One recent example comes from studies in C. elegans where mtROS promoted cellular adaptation to stress by stimulating, e.g., the p38-nuclear factor-like 2 (NRF2) redox-sensitive pathway [56]. This in turn primed the antioxidant machinery, resulting in increased lifespan of the nematodes, a phenomenon that has been defined as mitohormesis [56, 57]. In tumor cells, we found that mtROS can cause metastasis by activating the proto-oncogene Src and the focal adhesion kinase Pyk2, collectively resulting in resistance to anoikis and increased migration, invasion, metastasis take and spontaneous metastasis [33, 58]. Consequently, targeting mtROS generation by complete inhibition of Complex I activity or using specific mitochondria-targeted antioxidants was sufficient to abolish metastasis formation in vivo. The fact that mtROS are relevant for metastasis has been broadly demonstrated, from the initial reports indicating that metastatic cells accumulate more ROS than untransformed cells [59] to the seminal work of Ishikawa et al. [60] demonstrating that specific ROS-inducing mtDNA mutations are sufficient to promote metastasis. In the latter study, increased levels of mtROS activated the anti-apoptotic protein myeloid cell leukemia-1 (MCL-1), a member of the Bcl-2 family promoting tumor cell aggressiveness. To date, several specific mtDNA mutations have been reported to trigger tumor progression through the promotion of ROS production [61–63]. From a molecular standpoint, several downstream targets of mtROS have been identified, including FAK [64], PYK2 [58], SNAIL [65], p38 and NRF-2 [56, 66], nuclear factor-κB (NF-κB) activation mediated by c-Src oxidation [67] and HIF-1 stabilization through mtROS-mediated prolylhydroxylase (PHD) inactivation [21]. These mediators, in turn promote increased cell resistance to stress [68, 69]. Due to the particular nature of ROS, it is important to consider mtROS in a spatial context. Consequently, there are important caveats to consider when targeting mtROS with general antioxidants that can alter the total cellular ROS pool in tumor and host cells and interfere with therapy, explaining the disappointing results of several clinical trials having treated cancer patients with general antioxidants [58, 70].
Although, glycolytic compensation following mtDNA mutation certainly contributes to the pro-metastatic phenotype [42], it is not sufficient by itself [71]. In oncocytoma, for example, total loss of Complex I activity was reported to correlate with the emergence of tumors that rarely progress [72]. Oncocytomas normally occur in endocrine and exocrine tissues (e.g., thyroid, parathyroid, kidneys, salivary and pituitary glands), and are characterized by mitochondrial hyperplasia seen as a compensatory mechanism caused by complete inactivation of Complex I activity [72–75]. Further evidence is required to define the overall levels of mtROS in these tumors and their glycolytic rate. Overall, metabolic analyses of oncocytomas point at the necessity to maintain residual ETC activity for tumor invasiveness.
TCA cycle
The TCA cycle comprises eight consecutive reaction steps, starting from the condensation of acetyl-CoA with a molecule of oxaloacetate by citrate synthase (CS) to form citrate (Fig. 1). Interestingly, CS expression was found to be increased in pancreatic ductal adenocarcinoma [76] and in metastatic versus benign ovarian carcinoma [77]. Evidence is controversial for its role in metastasis, as it has been shown that CS downregulation either decreases (as seen after transient siRNA knock-down) [77] or increases (following stable shRNA expression) [78] invasion. One possible explanation for these opposite observations might be an adaptive phenotype following long-term knock-down of CS.
The product of the CS reaction, citrate, is then converted to isocitrate by aconitase (Aco). This reaction requires the conversion of citrate into the enzyme-bound intermediate cis-aconitate. Aco reactivation is particularly relevant for tumorigenesis in prostate cancer, where Aco is normally inactivated by the high concentration of zinc ions present in prostate cells [79]. In vitro, Aco activation has also been associated with the metastatic behavior of PC-3 M human prostate cancer cells [80].
Isocitrate is converted to αKG by IDHs, with the concomitant generation of NADH by IDH3 localized in mitochondria and NADPH by IDH1 and IDH2 that traffic between the cytoplasm and mitochondria. αKG is not only a metabolic intermediate of the TCA cycle but also an important co-substrate of several enzymes, especially the vast family of oxygenases that performs functions ranging from collagen synthesis to histone demethylation [81]. It is therefore not surprising that αKG controls complex biologic functions, with, for example, high levels of αKG promoting the maintenance of the pluripotency of embryonic stem cells (eSCs) by regulating whole genome methylation [82]. One of the classes of oxygenases requiring αKG as a co-substrate is PHDs, i.e., prolylhydroxylases that behave as oxygen sensors. PHD2 in particular tags HIF-1 subunit α for proteasome-mediated degradation under normoxia [83], and low levels of αKG would therefore theoretically promote normoxic HIF-1 activation and tumor progression. αKG also mediates the activation of mammalian target of rapamycin complex 1 (mTORC1) in synergy with leucine [84]. It increases the lifespan of C. elegans by promoting a moderate mitochondrial inhibition through binding to ATP-synthase [85]. Biological activities of αKG are thus numerous. Yet, blocking the anaplerotic reactions supplementing αKG (such as glutaminolysis and alanine transamination) inhibits cell transformation and tumor invasion [26, 86], while supplementation with cell-permeable αKG is sufficient to rescue the clonogenic potential of tumor cells [86]. High intracellular levels of αKG are likely pro-metastastic, but because αKG also promotes transformation, specific research is still required to discriminate between its impact on tumor transformation and metastasis. Beyond the fact that it catalyzes αKG production, the IDH reaction is of particular interest because heterozygous mutations of IDH1 and IDH2 are quite common in gliomas [87]. These mutations can result in a neomorphic enzymatic activity that allows heterodimeric IDH complexes to catalyze the conversion of αKG into the oncometabolite d-2-hydroxyglutarate (d-2HG) [6]. Mutations of IDH1 and IDH2 genes have been found in colon cancer [88], acute myeloid leukemia [89] and enchondroma [90]. Despite the fact that d-2HG promotes tumorigenesis by altering DNA-methylation genome-wide (primarily by antagonizing αKG-dependent dioxygenases) [91], limited evidence links the IDH1 and IDH2 mutations to an increased metastatic potential [92].
In the TCA cycle, αKG is then sequentially converted to succinyl-CoA (with the generation of NADH), succinate (with the concurrent generation of GTP) and fumarate (by SDH) (Fig. 1). FH then converts fumarate into malate, which is used to generate oxaloacetate by malate dehydrogenase. This final reaction produces one additional molecule of NADH from NAD+. SDH is a macromolecular complex composed of four different subunits located on the internal face of the inner mitochondrial membrane. A major characteristic of this enzymatic complex is that it is shared between the TCA cycle and the ETC where it composes Complex II. Indeed, following succinate oxidation, SDH covalently binds a molecule of FAD that is reduced to FADH2, which is followed by the transfer of two electrons from FADH2 to ubiquinone (also known as coenzyme Q [CoQ]), yielding ubiquinol [CoQH2] in the ETC. Mutations of SDH5, the protein responsible for covalently attaching FAD to SDH, are causally linked to tumorigenesis [7, 8, 93–96] as they lead to accumulation of succinate, which has been identified as an oncometabolite promoting transformation [97]. On the other hand, fumarate accumulation linked to FH deficiency promotes the formation of hereditary uterine fibroids, skin leiomyomas, papillary renal cell cancers, sarcomas, pheochromocytomas and paragangliomas [98–100]. Mechanistically, succinate and fumarate are competitive inhibitors of αKG-dependent dioxygenases (i.e., enzymes that catalyze the incorporation of the two atoms of oxygen of O2 into a substrate). They promote HIF-1 stabilization by inhibiting PHDs [97] and induce a whole genome epigenetic dysregulation by inhibiting both histone demethylases and the ten-eleven translocation (TET) family of 5-methlycytosine hydroxylases [5, 101, 102]. Both metabolites thus share a common way of inducing transformation [103]. In addition, fumarate accumulation results in a spontaneous reaction leading to posttranslational modification of cysteines in proteins known as ‘succination’ (i.e., a chemical modification of proteins formed by a Michael addition reaction between fumarate and thiol groups) [104]. Succination notably impairs the function of Kelch-like ECH-associated protein 1 (KEAP1), thus relieving transcription factor NRF2 from KEAP1-mediated inhibition and promoting an antioxidant response [105, 106]. In contrast, succination can also affect glutathione, promoting oxidative stress [107]. Despite the vast influence of succinate and fumarate on epigenetic regulation and signaling pathways, only a limited amount of evidence links these oncometabolites to metastatic progression. While the experimental re-expression of FH in a FH-deficient renal cell carcinoma line impaired tumor cell migration and invasion in vitro [108], treatment with dimethylfumarate, a cell-permeable form of fumarate, strongly reduced invasion and metastasis formation in melanoma [109–111]. To date, only the loss of SDH5 has been clearly shown to drive the acquisition of a mesenchymal and prometastatic phenotype in lung cancer cells, further correlating with reduced levels of SDH5 in a small group of metastatic versus non-metastatic lung patients [112]. However, no evidence links this effect to the enzymatic activity of SDH. Rather, SDH5 was found to form a complex with glycogen synthase kinase (GSK-3β, a mediator of β-catenin degradation) and protein phosphatase A (PP2A): when present, SDH5 activates GSK-3β and prevents the epithelial-to-mesenchymal transition (EMT). When SDH5 is lost, β-catenin accumulates, translocates to the cell nucleus and promotes EMT [112].
The TCA cycle is amphibolic: it not only mediates catabolic reactions aimed at energy production but also produces precursors for cell growth. Cancer cells are capable of reducing αKG to isocitrate, which has been termed reductive or reverse TCA cycle [113], ultimately increasing citrate and acetyl-CoA levels to promote lipogenesis. Glutamine directly fuels this pathway, especially when oxygen is limiting or when mitochondria are dysfunctional [114, 115]. The trigger for such “anti-clockwise” TCA cycle has been suggested to be an increased αKG/citrate ratio [116]. Although this phenotype has been strongly linked to tumor growth [114, 115, 117–119], little is known about the specific contribution of reductive carboxylation to tumor metastasis. It has nevertheless been reported that steroid receptor coactivator 2 (SRC2) promotes lipogenesis by stimulating glutamine utilization and reductive carboxylation in prostate cancer, and SRC2 is particularly enriched in metastatic lesions in patients with prostate cancer [120, 121]. In animal models, SRC2 depletion strongly reduced tumor cell viability, tumor growth and spontaneous metastasis. Although, these data suggest that the metabolic phenotype characterized by a pronounced reductive carboxylation might promote the emergence of aggressive tumor cell clones prone to metastasis, further experiments are required to test this possibility.
Lipid metabolism
Lipid accumulation is so common in tumors that it can by itself be considered as a hallmark of cancer [122]. The transcription factors, sterol regulatory element-binding proteins -1 and -2 (SREBP-1/-2) are the main transcriptional regulators of the lipogenic program, inducing cholesterol and fatty acid biosynthesis [123, 124]. SREBPs are downstream targets of the phosphoinositide 3-kinase (PI3K)–protein kinase B (PKB/Akt)–mTORC1 signaling pathway. Their inhibition represses tumor growth by depleting lipid rafts at the plasma membrane, thereby impairing Akt activation [125]. Conversely, upregulation of SREBP expression renders tumor cells more resistant to apoptosis and makes them more aggressive, especially in conditions where nutrient and oxygen availability are limited [126]. As a matter of fact, the SREBP signature is a marker of poor prognosis in glioblastoma [126]. Notably, the mucin 1 oncoprotein (which is overexpressed in breast cancer and is important for metastatic progression [127, 128]) and SRC2 (which is overexpressed in prostate metastatic lesions [120], see above) are upstream regulators of SREBPs and, thus, of lipid metabolism. Despite, further studies are required to establish a cause–consequence relationship, regulation of lipid metabolism and cancer metastasis may thus be intertwined.
Acetyl-CoA is at the crossroad between the TCA cycle and lipid synthesis, with its production being critical for both metabolic pathways. Essential gatekeepers regulating de novo lipid synthesis are ATP-citrate lyase (ACLY) that converts citrate to oxaloacetate and acetyl-CoA, and fatty acid synthase (FASN), a macromolecular complex that catalyzes the condensation of carbon skeletons into fatty acids. With a few exceptions, e.g., in liver and mammary glands, FASN expression is low in normal tissues, but it increases during transformation [129]. In the prostate, FASN demonstrated pro-oncogenic properties, as forced overexpression was sufficient to induce resistance to apoptosis and the transformation of normal prostate cells in mice [130]. Similarly, ACLY is required for tumor growth and its inhibition is a promising strategy for tumor therapy [131, 132].
Lipid synthesis is essential for the production of membranes necessary for cell proliferation. In addition to promoting cell proliferation, data indicate that lipid synthesis further contributes to tumor progression and metastasis, with a strong increase in FASN and ACLY expression in breast cancer [132, 133], retinoblastoma [134], lung cancer [132] and colon cancer [135]. Inhibition of FASN and ACLY was found to impair the metastatic progression of colon cancer cells by reducing CD44- and hepatocyte growth factor receptor (HGFR)-mediated signaling, resulting in reduced tumor cell migration and clonogenicity on soft agar [136]. Because lipid synthesis endows tumor cells with an increased resistance to apoptosis [126], tumor cells can be expected to rewire their energy metabolism towards increased lipid synthesis to overcome therapy, an adaptation that further confers a more aggressive phenotype. In contrast, blockade of lipid synthesis can inhibit metastatic dissemination following anti-angiogenic therapy [137]. This observation holds great promise for future combination therapies since one of the major drawbacks reducing the efficacy of anti-angiogenic therapy is the induction of metastases [138]. One of the major future challenges will be the identification of the molecular mechanisms linking lipid synthesis to the acquisition of invasive traits.
Besides the role of lipid synthesis in promoting invasion and metastasis, it is becoming clear that fatty acid oxidation by itself can also promote metastasis. Indeed, simple depletion of exogenous lipids is sufficient to impair breast cancer cell migration in vitro, even in the presence of alternative oxidative fuels glucose and glutamine [139]. Similarly, in the non-small cell lung cancer cell line A549, the addition of transforming growth factor β promotes tumor invasiveness by increasing mitochondrial lipid oxidation [140]. In this model, forced lipid oxidation alone was sufficient to induce EMT. In agreement, cancer-associated adipocytes are emerging as key regulators of metastatic formation in both breast and ovarian cancers where they fuel metastases by providing tumor cells with energy substrates [141, 142].
Amino acid metabolism
Amino acid metabolism is a central part of cellular metabolic homeostasis. Among all natural amino acids, glutamine is one of the most characterized for its role in tumor formation and metastasis [9]. This non-essential amino acid is also one of the most abundant amino acids present in bodily fluids and one of the most heavily depleted amino acid in tumor tissue, indicating the high avidity of tumors for glutamine [143]. Glutaminolysis has been proposed to be as important as glucose metabolism in tumors [144] and is primarily induced by the Myc oncogene [145]. The first step of glutamine metabolism is mediated by glutaminases GLS1 and GLS2 that catalyze the conversion of glutamine to glutamate and ammonia (NH3). Glutamate can further be deaminated to αKG by glutamate dehydrogenase (GDH), with the concurrent production of one molecule of NADH by GDH1 or NADPH by GDH2. αKG can then be used in oxidative TCA cycling, reductive TCA cycling or as a co-factor for biochemical reactions, as detailed above. Alternatively, glutamate can also be transaminated by the glutamic-pyruvic transaminase or by glutamic-oxaloacetic transaminases (GOT1 or GOT2). GOT2 generates aspartate in the mitochondria, which may then generate oxaloacetate in the cytosol after a second transamination reaction catalyzed by GOT1. Oxaloacetate is channeled to malate dehydrogenase, resulting in the production of pyruvate and NADPH. This alternative pathway of glutamate has emerged as a critical regulator of the redox balance in pancreatic ductal adenocarcinoma [146]. Because glutamate can further be used for glutathione synthesis, glutamine metabolism is important not only for energy production but also for redox regulation in cancer. Hence, GLS inhibition impairs tumor cell migration and invasion [86] and promotes apoptosis in cells undergoing EMT as a consequence of decreased resistance to oxidative stress [147].
Interestingly, GDH1, the enzyme controlling NAD+-dependent glutamate deamination, has been reported to be overexpressed in metastases of gallbladder [148] and murine hepatocarcinoma [149]. GDH1 is important for redox homeostasis as it controls αKG production and the subsequent generation of fumarate, which activates the antioxidant enzyme glutathione peroxidase 1 [150]. Thus, GDH1 has been shown to promote tumor progression by increasing tumor cell resistance to oxidative stress. Its specific contribution to the metastatic process remains to be determined.
A more complex story of amino acids and their role in cancer metastasis is that of proline. On one hand, proline oxidase, which catalyzes proline degradation, has been identified as a mitochondrial tumor suppressor due to its pro-apoptotic properties coupled to increased ROS generation [151]. Interestingly, the Myc oncogene inhibits proline degradation [145], by participating in the accumulation of proline in growing tumors [143, 152]. Proline further accumulates in tumors following degradation of the extracellular matrix [153] and elevated proline synthesis in glutamine catabolism [145]. On the other hand, proline degradation can turn into an important source of energy during nutrient deprivation, either by stimulating ATP production or by inducing ROS-mediated autophagy [154]. This pro-oxidant activity of proline has been proposed to promote mitohormesis [56], i.e., adaptation to stress driven by mitochondria (see also above). It most probably explains the observation that increased proline metabolism correlates with invasiveness and resistance to oxidative stress in esophageal squamous cell cancer [155]. Along the same lines, Comes et al. [156] recently reported that proline supplementation on its own was sufficient to trigger a genome-wide methylation remodeling and the acquisition of an EMT-like phenotype by eSC, promoting eSC metastasis in vivo.
Finally, amino acid metabolism could also promote metastasis via a tumor cell-extrinsic action. For example, the seminal work of Uyttenhove et al. [157] revealed that cancer cells can escape immune defenses by stimulating the degradation of tryptophan, an amino acid essential for T-cell replication. Indoleamine 2,3-dioxygenase is the main enzyme involved in tryptophan degradation and constitutes a marker of tumor aggressiveness in ovarian [158], thyroid [159], breast [160] and skin [161] cancers. In another example, enhanced serine biosynthesis has been associated with the induction of osteoclastogenesis and increased formation of bone metastases in breast cancer [162], although the molecular rationale behind this observation still remains to be elucidated.
Concluding remarks
In eukaryotic cells, mitochondria evolved as gatekeepers not only of energy producing metabolism (cataplerosis) but also of biosynthetic metabolism (anaplerosis) and apoptosis. While a vast amount of experimental data point at the key role of mitochondria in promoting tumor cell growth and proliferation in primary tumors, a rather limited number of studies directly addressed their contribution to the metastatic process. The emerging picture is that while mitochondrial metabolism is hijacked by tumor cells to promote cell growth, proliferation, redox homeostasis and survival, mitochondria could also act as bioenergetic sensors conferring migratory, invasive and metastatic phenotypes to cancer cells exposed to harsh microenvironmental conditions. Together with de novo produced d-2-hydroxyglutarate following specific IDH mutations, mtROS and TCA cycle intermediates increasingly produced in (pre)metastatic tumor cells, as well as glutamate originating from glutaminolysis, could be viewed as molecules involved in retrograde signaling from mitochondria to the cell, suggesting that in certain circumstances, mitochondria could drive cancer metastasis.
The origins of the changes affecting mitochondrial metabolism in premetastatic cells, in metastatic progenitor cells and tumor cells populating the metastatic lesion and whether they can/must be reversed at specific steps of the metastatic process remain to be determined. They could be dependent or independent from mutations affecting genomic or mitochondrial DNA. The observation that a metabolic switch from OXPHOS to glycolysis can also promote the metastatic phenotype (see companion paper) argues for the existence of temporally well-defined metabolic adaptations along the metastatic route. Alternatively, different metabolic phenotypes could independently promote tumor metastasis, with the caveat that cause–effect relationships must still be established in most instances. In our opinion, resolving these uncertainties is a task of fundamental importance not only to improve the understanding of the metabolism of metastatic progenitor cells, but also to establish a strong rationale for the development of antimetastatic treatment strategies.
A further degree of complexity of the metastatic process arises from interactions between cellular populations with different metabolic phenotypes inside a given tumor, resulting, e.g., in metabolic symbiosis [163] and commensalism [164]. For example, cancer-associated fibroblasts can fuel the oxidative metabolism of prostate cancer cells [165], which drives EMT and metastatic progression [166]. In this context, there are strong indications that oxidative lipid metabolism, lipogenesis and amino acid metabolism play critical roles that largely remain to be explored from molecular and clinical standpoints. In particular, despite the identification of specific metabolic reactions that promote metastasis, it will be pivotal to understand the molecular determinants driving the metastatic switch. It goes without saying that a better delineation of specific changes affecting mitochondria, lipid and amino acid metabolism could ultimately translate into new, original therapeutic strategies directed against cancer metastasis.
Conclusively, targeting tumor metastasis for therapy now requires distinguishing metabolic changes that can drive tumor metastasis from those that merely result from the acquisition of the metastatic phenotype, and determining whether sequential metabolic changes in a given metastatic progenitor cell along its metastatic route and/or independent metabolic changes in several different metastatic progenitor cell populations account for the metastatic process.
Acknowledgments
Work at the authors’ lab is supported by a Starting Grant from the European Research Council (ERC No. 243188 TUMETABO), Interuniversity Attraction Pole (IAP) grant #UP7-03 from the Belgian Science Policy Office (Belspo), an Action de Recherche Concertée from the Communauté Française de Belgique (ARC 14/19-058), the Belgian Fonds National de la Recherche Scientifique (F.R.S.-FNRS), the Télévie, the Belgian Fondation contre le Cancer (2012-186), the Belgian Federal Agency for Nuclear Control (FANC-AFCN), the Louvain Foundation and the UCL Fonds Spéciaux de la Recherche (FSR). Pierre Sonveaux is a F.R.S.-FNRS Research Associate, Paolo E. Porporato a F.R.S.-FNRS Postdoctoral Fellow and Valéry L. Payen a F.R.S.-FNRS PhD Fellow. Bjorn Baselet is a grantee of the Belgian Nuclear Research Center (SCK·CEN).
Abbreviations
- αKG
α-Ketoglutarate
- Aco
Aconitase
- ACLY
ATP-citrate lyase
- CoA
Coenzyme A
- CS
Citrate synthase
- d-2HG
d-2-Hydroxyglutarate
- EMT
Epithelial-to-mesenchymal transition
- ETC
Electron transport chain
- eSC
Embryonic stem cell
- FASN
Fatty acid synthase
- FH
Fumarate hydratase
- GDH
Glutamate dehydrogenase
- GLS
Glutaminase
- HGFR
Hepatocyte growth factor receptor
- HIF-1
Hypoxia-inducible factor-1
- IDH
Isocitrate dehydrogenase
- KEAP1
Kelch-like ECH-associated protein 1
- KRAS
Kirsten Rat Sarcoma
- MCL-1
Myeloid cell leukemia-1
- mtROS
Mitochondrial reactive oxygen species
- mTORC1
Mammalian target of rapamycin complex 1
- NF-κB
Nuclear factor-κB
- NRF2
Nuclear factor-like 2
- OXPHOS
Oxidative phosphorylation
- PGC-1
Peroxisome proliferator-activated receptor γ coactivator-1
- PHD
Prolylhydroxylase
- PI3K
Phosphoinositide 3-kinase
- PKB/Akt
Protein kinase B
- ROS
Reactive oxygen species
- SDH
Succinate dehydrogenase
- SOD
Superoxide dismutase
- SRC2
Steroid receptor coactivator 2
- SREBP
Sterol regulatory element-binding protein
- STAT3
Signal transducer and activator of transcription 3
- TCA (cycle)
Tricarboxylic acid (cycle)
- TET (enzyme)
Ten-eleven translocation (enzyme)
Footnotes
P. E. Porporato and V. L. Payen equally contributed to this manuscript.
Submitted as a companion paper to “Payen VL, Porporato PE, Baselet B, Sonveaux P. Metabolic changes associated with tumor metastasis, part 1: Tumor pH, glycolysis and the pentose phosphate pathway.”
References
- 1.Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Warburg O. On the origin of cancer cell. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 4.Carew JS, Huang P. Mitochondrial defects in cancer. Mol Cancer. 2002;1:9. doi: 10.1186/1476-4598-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, Guan KL. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaelin WG., Jr SDH5 mutations and familial paraganglioma: somewhere Warburg is smiling. Cancer Cell. 2009;16:180–182. doi: 10.1016/j.ccr.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 8.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW, III, Cornelisse CJ, Devilee P, Devlin B. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 9.DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Payen VL, Brisson L, Dewhirst MW, Sonveaux P. Common responses of tumors and wounds to hypoxia. Cancer J. 2015;21:75–87. doi: 10.1097/PPO.0000000000000098. [DOI] [PubMed] [Google Scholar]
- 11.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang R, Loux T, Tang D, Schapiro NE, Vernon P, Livesey KM, Krasinskas A, Lotze MT, Zeh HJ., III The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc Natl Acad Sci U S A. 2012;109:7031–7036. doi: 10.1073/pnas.1113865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rasola A, Sciacovelli M, Chiara F, Pantic B, Brusilow WS, Bernardi P. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc Natl Acad Sci U S A. 2010;107:726–731. doi: 10.1073/pnas.0912742107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogura M, Yamaki J, Homma MK, Homma Y. Mitochondrial c-Src regulates cell survival through phosphorylation of respiratory chain components. Biochem J. 2012;447:281–289. doi: 10.1042/BJ20120509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muller FL, Liu Y, Van RH. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 2004;279:49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 16.Wellen KE, Thompson CB. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell. 2010;40:323–332. doi: 10.1016/j.molcel.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu JL, Hsieh Y, Tu C, O’Connor D, Nick HS, Silverman DN. Catalytic properties of human manganese superoxide dismutase. J Biol Chem. 1996;271:17687–17691. doi: 10.1074/jbc.271.30.17687. [DOI] [PubMed] [Google Scholar]
- 18.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B, Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–414. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 22.Dewaele M, Maes H, Agostinis P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy. 2010;6:838–854. doi: 10.4161/auto.6.7.12113. [DOI] [PubMed] [Google Scholar]
- 23.St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 24.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B, Pesdar EA, Sobol M, Filimonenko A, Stuart S, Vondrusova M, Kluckova K, Sachaphibulkij K, Rohlena J, Hozak P, Truksa J, Eccles D, Haupt LM, Griffiths LR, Neuzil J, Berridge MV. Mitochondrial Genome Acquisition Restores Respiratory Function and Tumorigenic Potential of Cancer Cells without Mitochondrial DNA. Cell Metab. 2015;21:81–94. doi: 10.1016/j.cmet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 28.Fogal V, Richardson AD, Karmali PP, Scheffler IE, Smith JW, Ruoslahti E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol Cell Biol. 2010;30:1303–1318. doi: 10.1128/MCB.01101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, Kost-Alimova M, Muller F, Colla S, Nezi L, Genovese G, Deem AK, Kapoor A, Yao W, Brunetto E, Kang Y, Yuan M, Asara JM, Wang YA, Heffernan TP, Kimmelman AC, Wang H, Fleming JB, Cantley LC, DePinho RA, Draetta GF. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014;3:e02242. doi: 10.7554/eLife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–7511. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janzer A, German NJ, Gonzalez-Herrera KN, Asara JM, Haigis MC, Struhl K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc Natl Acad Sci U S A. 2014;111:10574–10579. doi: 10.1073/pnas.1409844111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, Feron O, Michiels C, Gallez B, Sonveaux P. A Mitochondrial Switch Promotes Tumor Metastasis. Cell Rep. 2014;8:754–766. doi: 10.1016/j.celrep.2014.06.043. [DOI] [PubMed] [Google Scholar]
- 34.Comito G, Calvani M, Giannoni E, Bianchini F, Calorini L, Torre E, Migliore C, Giordano S, Chiarugi P. HIF-1alpha stabilization by mitochondrial ROS promotes Met-dependent invasive growth and vasculogenic mimicry in melanoma cells. Free Radic Biol Med. 2011;51:893–904. doi: 10.1016/j.freeradbiomed.2011.05.042. [DOI] [PubMed] [Google Scholar]
- 35.LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, de Carvalho FM, Damascena A, Domingos Chinen LT, Rocha RM, Asara JM, Kalluri R. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014;16:992–1003. doi: 10.1038/ncb3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen EI, Hewel J, Krueger JS, Tiraby C, Weber MR, Kralli A, Becker K, Yates JR, III, Felding-Habermann B. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007;67:1472–1486. doi: 10.1158/0008-5472.CAN-06-3137. [DOI] [PubMed] [Google Scholar]
- 37.Caneba CA, Bellance N, Yang L, Pabst L, Nagrath D. Pyruvate uptake is increased in highly invasive ovarian cancer cells under anoikis conditions for anaplerosis, mitochondrial function, and migration. Am J Physiol Endocrinol Metab. 2012;303:E1036–E1052. doi: 10.1152/ajpendo.00151.2012. [DOI] [PubMed] [Google Scholar]
- 38.Chen ZX, Pervaiz S. Bcl-2 induces pro-oxidant state by engaging mitochondrial respiration in tumor cells. Cell Death Differ. 2007;14:1617–1627. doi: 10.1038/sj.cdd.4402165. [DOI] [PubMed] [Google Scholar]
- 39.Caino MC, Chae YC, Vaira V, Ferrero S, Nosotti M, Martin NM, Weeraratna A, O’Connell M, Jernigan D, Fatatis A, Languino LR, Bosari S, Altieri DC. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J Clin Invest. 2013;123:2907–2920. doi: 10.1172/JCI67841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee GH, Yan C, Shin SJ, Hong SC, Ahn T, Moon A, Park SJ, Lee YC, Yoo WH, Kim HT, Kim DS, Chae SW, Kim HR, Chae HJ. BAX inhibitor-1 enhances cancer metastasis by altering glucose metabolism and activating the sodium-hydrogen exchanger: the alteration of mitochondrial function. Oncogene. 2010;29:2130–2141. doi: 10.1038/onc.2009.491. [DOI] [PubMed] [Google Scholar]
- 41.Park J, Kusminski CM, Chua SC, Scherer PE. Leptin receptor signaling supports cancer cell metabolism through suppression of mitochondrial respiration in vivo. Am J Pathol. 2010;177:3133–3144. doi: 10.2353/ajpath.2010.100595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu W, Beck BH, Vaidya KS, Nash KT, Feeley KP, Ballinger SW, Pounds KM, Denning WL, Diers AR, Landar A, Dhar A, Iwakuma T, Welch DR. Metastasis suppressor KISS1 seems to reverse the Warburg effect by enhancing mitochondrial biogenesis. Cancer Res. 2014;74:954–963. doi: 10.1158/0008-5472.CAN-13-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Govindarajan B, Sligh JE, Vincent BJ, Li M, Canter JA, Nickoloff BJ, Rodenburg RJ, Smeitink JA, Oberley L, Zhang Y, Slingerland J, Arnold RS, Lambeth JD, Cohen C, Hilenski L, Griendling K, Martinez-Diez M, Cuezva JM, Arbiser JL. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J Clin Invest. 2007;117:719–729. doi: 10.1172/JCI30102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arnold RS, Sun CQ, Richards JC, Grigoriev G, Coleman IM, Nelson PS, Hsieh CL, Lee JK, Xu Z, Rogatko A, Osunkoya AO, Zayzafoon M, Chung L, Petros JA. Mitochondrial DNA mutation stimulates prostate cancer growth in bone stromal environment. Prostate. 2009;69:1–11. doi: 10.1002/pros.20854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuo SJ, Chen M, Ma GC, Chen ST, Chang SP, Lin WY, Chen YC, Lee TH, Lin TT, Liu CS. Number of somatic mutations in the mitochondrial D-loop region indicates poor prognosis in breast cancer, independent of TP53 mutation. Cancer Genet Cytogenet. 2010;201:94–101. doi: 10.1016/j.cancergencyto.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 46.Ebner S, Lang R, Mueller EE, Eder W, Oeller M, Moser A, Koller J, Paulweber B, Mayr JA, Sperl W, Kofler B. Mitochondrial haplogroups, control region polymorphisms and malignant melanoma: a study in middle European Caucasians. PLoS ONE. 2011;6:e27192. doi: 10.1371/journal.pone.0027192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amuthan G, Biswas G, Ananadatheerthavarada HK, Vijayasarathy C, Shephard HM, Avadhani NG. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene. 2002;21:7839–7849. doi: 10.1038/sj.onc.1205983. [DOI] [PubMed] [Google Scholar]
- 48.Amuthan G, Biswas G, Zhang SY, Klein-Szanto A, Vijayasarathy C, Avadhani NG. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J. 2001;20:1910–1920. doi: 10.1093/emboj/20.8.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, Kaluarachchi K, Bornmann W, Duvvuri S, Taegtmeyer H, Andreeff M. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120:142–156. doi: 10.1172/JCI38942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Girnun GD. The diverse role of the PPARgamma coactivator 1 family of transcriptional coactivators in cancer. Semin Cell Dev Biol. 2012;23:381–388. doi: 10.1016/j.semcdb.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghosh JC, Siegelin MD, Vaira V, Faversani A, Tavecchio M, Chae YC, Lisanti S, Rampini P, Giroda M, Caino MC, Seo JH, Kossenkov AV, Michalek RD, Schultz DC, Bosari S, Languino LR, Altieri DC. Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J Natl Cancer Inst. 2015 doi: 10.1093/jnci/dju502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smallbone K, Gatenby RA, Gillies RJ, Maini PK, Gavaghan DJ. Metabolic changes during carcinogenesis: potential impact on invasiveness. J Theor Biol. 2007;244:703–713. doi: 10.1016/j.jtbi.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 53.Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, LeBoeuf SE, Gay LJ, Yagi T, Felding-Habermann B. Mitochondrial complex I activity and NAD +/NADH balance regulate breast cancer progression. J Clin Invest. 2013;123:1068–1081. doi: 10.1172/JCI64264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Singh RK, Srivastava A, Kalaiarasan P, Manvati S, Chopra R, Bamezai RN. mtDNA germ line variation mediated ROS generates retrograde signaling and induces pro-cancerous metabolic features. Sci Rep. 2014;4:6571. doi: 10.1038/srep06571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chiarugi P. PTPs versus PTKs: the redox side of the coin. Free Radic Res. 2005;39:353–364. doi: 10.1080/10715760400027987. [DOI] [PubMed] [Google Scholar]
- 56.Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, Guthke R, Platzer M, Kahn CR, Ristow M. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial l-proline catabolism to induce a transient ROS signal. Cell Metab. 2012;15:451–465. doi: 10.1016/j.cmet.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ristow M. Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat Med. 2014;20:709–711. doi: 10.1038/nm.3624. [DOI] [PubMed] [Google Scholar]
- 58.Porporato PE, Sonveaux P. Paving the way for a therapeutic prevention of tumor metastasis with agents targeting mitochondrial superoxide. Mol Cell Oncol. 2015 doi: 10.4161/23723548.2014.968043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 60.Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–664. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 61.Chang J, Jung HJ, Jeong SH, Kim HK, Han J, Kwon HJ. A mutation in the mitochondrial protein UQCRB promotes angiogenesis through the generation of mitochondrial reactive oxygen species. Biochem Biophys Res Commun. 2014;455:290–297. doi: 10.1016/j.bbrc.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 62.Kaipparettu BA, Ma Y, Park JH, Lee TL, Zhang Y, Yotnda P, Creighton CJ, Chan WY, Wong LJ. Crosstalk from non-cancerous mitochondria can inhibit tumor properties of metastatic cells by suppressing oncogenic pathways. PLoS One. 2013;8:e61747. doi: 10.1371/journal.pone.0061747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dasgupta S, Hoque MO, Upadhyay S, Sidransky D. Mitochondrial cytochrome B gene mutation promotes tumor growth in bladder cancer. Cancer Res. 2008;68:700–706. doi: 10.1158/0008-5472.CAN-07-5532. [DOI] [PubMed] [Google Scholar]
- 64.Ali MH, Mungai PT, Schumacker PT. Stretch-induced phosphorylation of focal adhesion kinase in endothelial cells: role of mitochondrial oxidants. Am J Physiol Lung Cell Mol Physiol. 2006;291:L38–L45. doi: 10.1152/ajplung.00287.2004. [DOI] [PubMed] [Google Scholar]
- 65.Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, Bissell MJ. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Imhoff BR, Hansen JM. Extracellular redox status regulates Nrf2 activation through mitochondrial reactive oxygen species. Biochem J. 2009;424:491–500. doi: 10.1042/BJ20091286. [DOI] [PubMed] [Google Scholar]
- 67.Lluis JM, Buricchi F, Chiarugi P, Morales A, Fernandez-Checa JC. Dual role of mitochondrial reactive oxygen species in hypoxia signaling: activation of nuclear factor-{kappa}B via c-SRC and oxidant-dependent cell death. Cancer Res. 2007;67:7368–7377. doi: 10.1158/0008-5472.CAN-07-0515. [DOI] [PubMed] [Google Scholar]
- 68.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA, Tuveson DA. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Assefa Z, Vantieghem A, Declercq W, Vandenabeele P, Vandenheede JR, Merlevede W, de WP WP, Agostinis P. The activation of the c-Jun N-terminal kinase and p38 mitogen-activated protein kinase signaling pathways protects HeLa cells from apoptosis following photodynamic therapy with hypericin. J Biol Chem. 1999;274:8788–8796. doi: 10.1074/jbc.274.13.8788. [DOI] [PubMed] [Google Scholar]
- 70.Chandel NS, Tuveson DA. The promise and perils of antioxidants for cancer patients. N Engl J Med. 2014;371:177–178. doi: 10.1056/NEJMcibr1405701. [DOI] [PubMed] [Google Scholar]
- 71.Ishikawa K, Hashizume O, Koshikawa N, Fukuda S, Nakada K, Takenaga K, Hayashi J. Enhanced glycolysis induced by mtDNA mutations does not regulate metastasis. FEBS Lett. 2008;582:3525–3530. doi: 10.1016/j.febslet.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 72.Gasparre G, Romeo G, Rugolo M, Porcelli AM. Learning from oncocytic tumors: why choose inefficient mitochondria? Biochim Biophys Acta. 2011;1807:633–642. doi: 10.1016/j.bbabio.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 73.Tallini G. Oncocytic tumours. Virchows Arch. 1998;433:5–12. doi: 10.1007/s004280050209. [DOI] [PubMed] [Google Scholar]
- 74.Gasparre G, Hervouet E, De LE, Demont J, Pennisi LF, Colombel M, Mege-Lechevallier F, Scoazec JY, Bonora E, Smeets R, Smeitink J, Lazar V, Lespinasse J, Giraud S, Godinot C, Romeo G, Simonnet H. Clonal expansion of mutated mitochondrial DNA is associated with tumor formation and complex I deficiency in the benign renal oncocytoma. Hum Mol Genet. 2008;17:986–995. doi: 10.1093/hmg/ddm371. [DOI] [PubMed] [Google Scholar]
- 75.Mayr JA, Meierhofer D, Zimmermann F, Feichtinger R, Kogler C, Ratschek M, Schmeller N, Sperl W, Kofler B. Loss of complex I due to mitochondrial DNA mutations in renal oncocytoma. Clin Cancer Res. 2008;14:2270–2275. doi: 10.1158/1078-0432.CCR-07-4131. [DOI] [PubMed] [Google Scholar]
- 76.Schlichtholz B, Turyn J, Goyke E, Biernacki M, Jaskiewicz K, Sledzinski Z, Swierczynski J. Enhanced citrate synthase activity in human pancreatic cancer. Pancreas. 2005;30:99–104. doi: 10.1097/01.mpa.0000153326.69816.7d. [DOI] [PubMed] [Google Scholar]
- 77.Chen L, Liu T, Zhou J, Wang Y, Wang X, Di W, Zhang S. Citrate synthase expression affects tumor phenotype and drug resistance in human ovarian carcinoma. PLoS ONE. 2014;9:e115708. doi: 10.1371/journal.pone.0115708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin CC, Cheng TL, Tsai WH, Tsai HJ, Hu KH, Chang HC, Yeh CW, Chen YC, Liao CC, Chang WT (2012) Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci Rep 2:785 [DOI] [PMC free article] [PubMed]
- 79.Singh KK, Desouki MM, Franklin RB, Costello LC. Mitochondrial aconitase and citrate metabolism in malignant and nonmalignant human prostate tissues. Mol Cancer. 2006;5:14. doi: 10.1186/1476-4598-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mycielska ME, Broke-Smith TP, Palmer CP, Beckerman R, Nastos T, Erguler K, Djamgoz MB. Citrate enhances in vitro metastatic behaviours of PC-3M human prostate cancer cells: status of endogenous citrate and dependence on aconitase and fatty acid synthase. Int J Biochem Cell Biol. 2006;38:1766–1777. doi: 10.1016/j.biocel.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 81.Loenarz C, Schofield CJ. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat Chem Biol. 2008;4:152–156. doi: 10.1038/nchembio0308-152. [DOI] [PubMed] [Google Scholar]
- 82.Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518:413–416. doi: 10.1038/nature13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Berra E, Richard DE, Gothie E, Pouyssegur J. HIF-1-dependent transcriptional activity is required for oxygen-mediated HIF-1alpha degradation. FEBS Lett. 2001;491:85–90. doi: 10.1016/S0014-5793(01)02159-7. [DOI] [PubMed] [Google Scholar]
- 84.Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, Hall MN. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell. 2012;47:349–358. doi: 10.1016/j.molcel.2012.05.043. [DOI] [PubMed] [Google Scholar]
- 85.Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, Hu E, Whelan SA, Wang JX, Jung G, Solis GM, Fazlollahi F, Kaweeteerawat C, Quach A, Nili M, Krall AS, Godwin HA, Chang HR, Faull KF, Guo F, Jiang M, Trauger SA, Saghatelian A, Braas D, Christofk HR, Clarke CF, Teitell MA, Petrascheck M, Reue K, Jung ME, Frand AR, Huang J. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature. 2014;510:397–401. doi: 10.1038/nature13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, Cerione RA. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18:207–219. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ward PS, Cross JR, Lu C, Weigert O, Abel-Wahab O, Levine RL, Weinstock DM, Sharp KA, Thompson CB. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene. 2012;31:2491–2498. doi: 10.1038/onc.2011.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, Abbott RM, Vickery TL, Reed JS, Robinson JS, Wylie T, Smith SM, Carmichael L, Eldred JM, Harris CC, Walker J, Peck JB, Du F, Dukes AF, Sanderson GE, Brummett AM, Clark E, McMichael JF, Meyer RJ, Schindler JK, Pohl CS, Wallis JW, Shi X, Lin L, Schmidt H, Tang Y, Haipek C, Wiechert ME, Ivy JV, Kalicki J, Elliott G, Ries RE, Payton JE, Westervelt P, Tomasson MH, Watson MA, Baty J, Heath S, Shannon WD, Nagarajan R, Link DC, Walter MJ, Graubert TA, DiPersio JF, Wilson RK, Ley TJ. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hirata M, Sasaki M, Cairns RA, Inoue S, Puviindran V, Li WY, Snow BE, Jones LD, Wei Q, Sato S, Tang YJ, Nadesan P, Rockel J, Whetstone H, Poon R, Weng A, Gross S, Straley K, Gliser C, Xu Y, Wunder J, Mak TW, Alman BA. Mutant IDH is sufficient to initiate enchondromatosis in mice. Proc Natl Acad Sci USA. 2015;112:2829–2834. doi: 10.1073/pnas.1424400112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fu Y, Zheng Y, Li K, Huang R, Zheng S, An N, Liang A. Mutations in isocitrate dehydrogenase 2 accelerate glioma cell migration via matrix metalloproteinase-2 and 9. Biotechnol Lett. 2012;34:441–446. doi: 10.1007/s10529-011-0800-8. [DOI] [PubMed] [Google Scholar]
- 93.Astuti D, Douglas F, Lennard TW, Aligianis IA, Woodward ER, Evans DG, Eng C, Latif F, Maher ER. Germline SDHD mutation in familial phaeochromocytoma. Lancet. 2001;357:1181–1182. doi: 10.1016/S0140-6736(00)04378-6. [DOI] [PubMed] [Google Scholar]
- 94.Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reineke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peczkowska M, Szmigielski C, Eng C. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 95.Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, Latif F, Maher ER. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst. 2008;100:1260–1262. doi: 10.1093/jnci/djn254. [DOI] [PubMed] [Google Scholar]
- 96.Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, Dahia PL, Liegl B, Ball ER, Raygada M, Lai AH, Kelly L, Hornick JL, O’Sullivan M, de Krijger RR, Dinjens WN, Demetri GD, Antonescu CR, Fletcher JA, Helman L, Stratakis CA. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011;108:314–318. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 98.Kiuru M, Lehtonen R, Arola J, Salovaara R, Jarvinen H, Aittomaki K, Sjoberg J, Visakorpi T, Knuutila S, Isola J, Delahunt B, Herva R, Launonen V, Karhu A, Aaltonen LA. Few FH mutations in sporadic counterparts of tumor types observed in hereditary leiomyomatosis and renal cell cancer families. Cancer Res. 2002;62:4554–4557. [PubMed] [Google Scholar]
- 99.Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance RR, Olpin S, Bevan S, Barker K, Hearle N, Houlston RS, Kiuru M, Lehtonen R, Karhu A, Vilkki S, Laiho P, Eklund C, Vierimaa O, Aittomaki K, Hietala M, Sistonen P, Paetau A, Salovaara R, Herva R, Launonen V, Aaltonen LA. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 100.Castro-Vega LJ, Buffet A, De Cubas AA, Cascon A, Menara M, Khalifa E, Amar L, Azriel S, Bourdeau I, Chabre O, Curras-Freixes M, Franco-Vidal V, Guillaud-Bataille M, Simian C, Morin A, Leton R, Gomez-Grana A, Pollard PJ, Rustin P, Robledo M, Favier J, Gimenez-Roqueplo AP. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet. 2014;23:2440–2446. doi: 10.1093/hmg/ddt639. [DOI] [PubMed] [Google Scholar]
- 101.Kaelin WG., Jr Cancer and altered metabolism: potential importance of hypoxia-inducible factor and 2-oxoglutarate-dependent dioxygenases. Cold Spring Harb Symp Quant Biol. 2011;76:335–345. doi: 10.1101/sqb.2011.76.010975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, Buffet A, Marcaillou C, Bertherat J, Amar L, Rustin P, De RA, Gimenez-Roqueplo AP, Favier J. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 103.King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25:4675–4682. doi: 10.1038/sj.onc.1209594. [DOI] [PubMed] [Google Scholar]
- 104.Nagai R, Brock JW, Blatnik M, Baatz JE, Bethard J, Walla MD, Thorpe SR, Baynes JW, Frizzell N. Succination of protein thiols during adipocyte maturation: a biomarker of mitochondrial stress. J Biol Chem. 2007;282:34219–34228. doi: 10.1074/jbc.M703551200. [DOI] [PubMed] [Google Scholar]
- 105.Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K, Stevens M, Fischer R, Carmeliet P, Maxwell PH, Pugh CW, Frizzell N, Soga T, Kessler BM, El-Bahrawy M, Ratcliffe PJ, Pollard PJ. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524–537. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, Min BW, Tan MH, Zhang Z, Yang XJ, Zhou M, Gardie B, Molinie V, Richard S, Tan PH, Teh BT, Furge KA. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20:511–523. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 107.Zheng L, Cardaci S, Jerby L, MacKenzie ED, Sciacovelli M, Johnson TI, Gaude E, King A, Leach JD, Edrada-Ebel R, Hedley A, Morrice NA, Kalna G, Blyth K, Ruppin E, Frezza C, Gottlieb E. Fumarate induces redox-dependent senescence by modifying glutathione metabolism. Nat Commun. 2015;6:6001. doi: 10.1038/ncomms7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sudarshan S, Shanmugasundaram K, Naylor SL, Lin S, Livi CB, O’Neill CF, Parekh DJ, Yeh IT, Sun LZ, Block K. Reduced expression of fumarate hydratase in clear cell renal cancer mediates HIF-2alpha accumulation and promotes migration and invasion. PLoS One. 2011;6:e21037. doi: 10.1371/journal.pone.0021037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Loewe R, Valero T, Kremling S, Pratscher B, Kunstfeld R, Pehamberger H, Petzelbauer P. Dimethylfumarate impairs melanoma growth and metastasis. Cancer Res. 2006;66:11888–11896. doi: 10.1158/0008-5472.CAN-06-2397. [DOI] [PubMed] [Google Scholar]
- 110.Yamazoe Y, Tsubaki M, Matsuoka H, Satou T, Itoh T, Kusunoki T, Kidera Y, Tanimori Y, Shoji K, Nakamura H, Ogaki M, Nishiura S, Nishida S. Dimethylfumarate inhibits tumor cell invasion and metastasis by suppressing the expression and activities of matrix metalloproteinases in melanoma cells. Cell Biol Int. 2009;33:1087–1094. doi: 10.1016/j.cellbi.2009.06.027. [DOI] [PubMed] [Google Scholar]
- 111.Valero T, Steele S, Neumuller K, Bracher A, Niederleithner H, Pehamberger H, Petzelbauer P, Loewe R. Combination of dacarbazine and dimethylfumarate efficiently reduces melanoma lymph node metastasis. J Invest Dermatol. 2010;130:1087–1094. doi: 10.1038/jid.2009.368. [DOI] [PubMed] [Google Scholar]
- 112.Liu J, Gao L, Zhang H, Wang D, Wang M, Zhu J, Pang C, Wang C. Succinate dehydrogenase 5 (SDH5) regulates glycogen synthase kinase 3beta-beta-catenin-mediated lung cancer metastasis. J Biol Chem. 2013;288:29965–29973. doi: 10.1074/jbc.M113.450106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Holleran AL, Briscoe DA, Fiskum G, Kelleher JK. Glutamine metabolism in AS-30D hepatoma cells. Evidence for its conversion into lipids via reductive carboxylation. Mol Cell Biochem. 1995;152:95–101. doi: 10.1007/BF01076071. [DOI] [PubMed] [Google Scholar]
- 114.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fendt SM, Bell EL, Keibler MA, Olenchock BA, Mayers JR, Wasylenko TM, Vokes NI, Guarente L, Vander Heiden MG, Stephanopoulos G. Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells. Nat Commun. 2013;4:2236. doi: 10.1038/ncomms3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Filipp FV, Scott DA, Ronai ZA, Osterman AL, Smith JW. Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell Melanoma Res. 2012;25:375–383. doi: 10.1111/j.1755-148X.2012.00989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, Boriack R, Rakheja D, Sullivan LB, Linehan WM, Chandel NS, DeBerardinis RJ. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014;7:1679–1690. doi: 10.1016/j.celrep.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sun RC, Denko NC. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014;19:285–292. doi: 10.1016/j.cmet.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dasgupta S, Putluri N, Long W, Zhang B, Wang J, Kaushik AK, Arnold JM, Bhowmik SK, Stashi E, Brennan CA, Rajapakshe K, Coarfa C, Mitsiades N, Ittmann MM, Chinnaiyan AM, Sreekumar A, O’Malley BW. Coactivator SRC-2-dependent metabolic reprogramming mediates prostate cancer survival and metastasis. J Clin Invest. 2015;125:1174–1188. doi: 10.1172/JCI76029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Qin J, Lee HJ, Wu SP, Lin SC, Lanz RB, Creighton CJ, DeMayo FJ, Tsai SY, Tsai MJ. Androgen deprivation-induced NCoA2 promotes metastatic and castration-resistant prostate cancer. J Clin Invest. 2014;124:5013–5026. doi: 10.1172/JCI76412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hsu PP, Sabatini DM. Cancer cell metabolism: warburg and beyond. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 123.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI0215593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yamauchi Y, Furukawa K, Hamamura K, Furukawa K. Positive feedback loop between PI3 K-Akt-mTORC1 signaling and the lipogenic pathway boosts Akt signaling: induction of the lipogenic pathway by a melanoma antigen. Cancer Res. 2011;71:4989–4997. doi: 10.1158/0008-5472.CAN-10-4108. [DOI] [PubMed] [Google Scholar]
- 125.Griffiths B, Lewis CA, Bensaad K, Ros S, Zhang Q, Ferber EC, Konisti S, Peck B, Miess H, East P, Wakelam M, Harris AL, Schulze A. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer Metab. 2013;1:3. doi: 10.1186/2049-3002-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lewis CA, Brault C, Peck B, Bensaad K, Griffiths B, Mitter R, Chakravarty P, East P, Dankworth B, Alibhai D, Harris AL, Schulze A. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene. 2015 doi: 10.1038/onc.2014.439. [DOI] [PubMed] [Google Scholar]
- 127.Pitroda SP, Khodarev NN, Beckett MA, Kufe DW, Weichselbaum RR. MUC1-induced alterations in a lipid metabolic gene network predict response of human breast cancers to tamoxifen treatment. Proc Natl Acad Sci U S A. 2009;106:5837–5841. doi: 10.1073/pnas.0812029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhao Q, Barclay M, Hilkens J, Guo X, Barrow H, Rhodes JM, Yu LG. Interaction between circulating galectin-3 and cancer-associated MUC1 enhances tumour cell homotypic aggregation and prevents anoikis. Mol Cancer. 2010;9:154. doi: 10.1186/1476-4598-9-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Flavin R, Peluso S, Nguyen PL, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010;6:551–562. doi: 10.2217/fon.10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, Inazuka F, Grisanzio C, Palescandolo E, Shin E, Fiore C, Xie W, Kung AL, Febbo PG, Subramanian A, Mucci L, Ma J, Signoretti S, Stampfer M, Hahn WC, Finn S, Loda M. Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer. J Natl Cancer Inst. 2009;101:519–532. doi: 10.1093/jnci/djp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311–321. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 132.Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, Ushijima M, Mashima T, Seimiya H, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008;68:8547–8554. doi: 10.1158/0008-5472.CAN-08-1235. [DOI] [PubMed] [Google Scholar]
- 133.Alo’ PL, Visca P, Marci A, Mangoni A, Botti C, Di TU. Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer. 1996;77:474–482. doi: 10.1002/(SICI)1097-0142(19960201)77:3<474::AID-CNCR8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 134.Camassei FD, Cozza R, Acquaviva A, Jenkner A, Rava L, Gareri R, Donfrancesco A, Bosman C, Vadala P, Hadjistilianou T, Boldrini R. Expression of the lipogenic enzyme fatty acid synthase (FAS) in retinoblastoma and its correlation with tumor aggressiveness. Invest Ophthalmol Vis Sci. 2003;44:2399–2403. doi: 10.1167/iovs.02-0934. [DOI] [PubMed] [Google Scholar]
- 135.Luque-Garcia JL, Martinez-Torrecuadrada JL, Epifano C, Canamero M, Babel I, Casal JI. Differential protein expression on the cell surface of colorectal cancer cells associated to tumor metastasis. Proteomics. 2010;10:940–952. doi: 10.1002/pmic.200900441. [DOI] [PubMed] [Google Scholar]
- 136.Zaytseva YY, Rychahou PG, Gulhati P, Elliott VA, Mustain WC, O’Connor K, Morris AJ, Sunkara M, Weiss HL, Lee EY, Evers BM. Inhibition of fatty acid synthase attenuates CD44-associated signaling and reduces metastasis in colorectal cancer. Cancer Res. 2012;72:1504–1517. doi: 10.1158/0008-5472.CAN-11-4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sounni NE, Cimino J, Blacher S, Primac I, Truong A, Mazzucchelli G, Paye A, Calligaris D, Debois D, De TP, Mari B, De PE, Noel A. Blocking lipid synthesis overcomes tumor regrowth and metastasis after antiangiogenic therapy withdrawal. Cell Metab. 2014;20:280–294. doi: 10.1016/j.cmet.2014.05.022. [DOI] [PubMed] [Google Scholar]
- 138.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Antalis CJ, Uchida A, Buhman KK, Siddiqui RA. Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clin Exp Metastasis. 2011;28:733–741. doi: 10.1007/s10585-011-9405-9. [DOI] [PubMed] [Google Scholar]
- 140.Jiang L, Xiao L, Sugiura H, Huang X, Ali A, Kuro O, DeBerardinis RJ, Boothman DA. Metabolic reprogramming during TGFbeta1-induced epithelial-to-mesenchymal transition. Oncogene. 2015;34:3908–3916. doi: 10.1038/onc.2014.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, Wang YY, Meulle A, Salles B, Le GS, Garrido I, Escourrou G, Valet P, Muller C. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011;71:2455–2465. doi: 10.1158/0008-5472.CAN-10-3323. [DOI] [PubMed] [Google Scholar]
- 142.Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, Yamada SD, Peter ME, Gwin K, Lengyel E. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17:1498–1503. doi: 10.1038/nm.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Medina MA, Marquez J, Nunez DCI. Interchange of amino acids between tumor and host. Biochem Med Metab Biol. 1992;48:1–7. doi: 10.1016/0885-4505(92)90041-V. [DOI] [PubMed] [Google Scholar]
- 144.Board M, Humm S, Newsholme EA. Maximum activities of key enzymes of glycolysis, glutaminolysis, pentose phosphate pathway and tricarboxylic acid cycle in normal, neoplastic and suppressed cells. Biochem J. 1990;265:503–509. doi: 10.1042/bj2650503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW, Phang JM. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc Natl Acad Sci U S A. 2012;109:8983–8988. doi: 10.1073/pnas.1203244109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, Cantley LC, Kimmelman AC. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ulanet DB, Couto K, Jha A, Choe S, Wang A, Woo HK, Steadman M, DeLaBarre B, Gross S, Driggers E, Dorsch M, Hurov JB. Mesenchymal phenotype predisposes lung cancer cells to impaired proliferation and redox stress in response to glutaminase inhibition. PLoS ONE. 2014;9:e115144. doi: 10.1371/journal.pone.0115144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wang JW, Peng SY, Li JT, Wang Y, Zhang ZP, Cheng Y, Cheng DQ, Weng WH, Wu XS, Fei XZ, Quan ZW, Li JY, Li SG, Liu YB. Identification of metastasis-associated proteins involved in gallbladder carcinoma metastasis by proteomic analysis and functional exploration of chloride intracellular channel 1. Cancer Lett. 2009;281:71–81. doi: 10.1016/j.canlet.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 149.Liu S, Sun MZ, Tang JW, Wang Z, Sun C, Greenaway FT. High-performance liquid chromatography/nano-electrospray ionization tandem mass spectrometry, two-dimensional difference in-gel electrophoresis and gene microarray identification of lymphatic metastasis-associated biomarkers. Rapid Commun Mass Spectrom. 2008;22:3172–3178. doi: 10.1002/rcm.3725. [DOI] [PubMed] [Google Scholar]
- 150.Jin L, Li D, Alesi GN, Fan J, Kang HB, Lu Z, Boggon TJ, Jin P, Yi H, Wright ER, Duong D, Seyfried NT, Egnatchik R, DeBerardinis RJ, Magliocca KR, He C, Arellano ML, Khoury HJ, Shin DM, Khuri FR, Kang S. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell. 2015;27:257–270. doi: 10.1016/j.ccell.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Liu Y, Borchert GL, Surazynski A, Hu CA, Phang JM. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: the role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene. 2006;25:5640–5647. doi: 10.1038/sj.onc.1209564. [DOI] [PubMed] [Google Scholar]
- 152.Miyagi Y, Higashiyama M, Gochi A, Akaike M, Ishikawa T, Miura T, Saruki N, Bando E, Kimura H, Imamura F, Moriyama M, Ikeda I, Chiba A, Oshita F, Imaizumi A, Yamamoto H, Miyano H, Horimoto K, Tochikubo O, Mitsushima T, Yamakado M, Okamoto N. Plasma free amino acid profiling of five types of cancer patients and its application for early detection. PLoS ONE. 2011;6:e24143. doi: 10.1371/journal.pone.0024143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Steven FS, Griffin MM, Itzhaki S, Al-Habib A. A trypsin-like neutral protease on Ehrlich ascites cell surfaces: its role in the activation of tumour-cell zymogen of collagenase. Br J Cancer. 1980;42:712–721. doi: 10.1038/bjc.1980.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu W, Glunde K, Bhujwalla ZM, Raman V, Sharma A, Phang JM. Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. Cancer Res. 2012;72:3677–3686. doi: 10.1158/0008-5472.CAN-12-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Togashi Y, Arao T, Kato H, Matsumoto K, Terashima M, Hayashi H, de Velasco MA, Fujita Y, Kimura H, Yasuda T, Shiozaki H, Nishio K. Frequent amplification of ORAOV1 gene in esophageal squamous cell cancer promotes an aggressive phenotype via proline metabolism and ROS production. Oncotarget. 2014;5:2962–2973. doi: 10.18632/oncotarget.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Comes S, Gagliardi M, Laprano N, Fico A, Cimmino A, Palamidessi A, De CD, De FS, Angelini C, Scita G, Patriarca EJ, Matarazzo MR, Minchiotti G. L-Proline induces a mesenchymal-like invasive program in embryonic stem cells by remodeling H3K9 and H3K36 methylation. Stem Cell Reports. 2013;1:307–321. doi: 10.1016/j.stemcr.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van Den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 158.Tanizaki Y, Kobayashi A, Toujima S, Shiro M, Mizoguchi M, Mabuchi Y, Yagi S, Minami S, Takikawa O, Ino K. Indoleamine 2,3-dioxygenase promotes peritoneal metastasis of ovarian cancer by inducing an immunosuppressive environment. Cancer Sci. 2014;105:966–973. doi: 10.1111/cas.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ryu HS, Park YS, Park HJ, Chung YR, Yom CK, Ahn SH, Park YJ, Park SH, Park SY. Expression of indoleamine 2,3-dioxygenase and infiltration of FOXP3 + regulatory T cells are associated with aggressive features of papillary thyroid microcarcinoma. Thyroid. 2014;24:1232–1240. doi: 10.1089/thy.2013.0423. [DOI] [PubMed] [Google Scholar]
- 160.Chen JY, Li CF, Kuo CC, Tsai KK, Hou MF, Hung WC. Cancer/stroma interplay via cyclooxygenase-2 and indoleamine 2,3-dioxygenase promotes breast cancer progression. Breast Cancer Res. 2014;16:410. doi: 10.1186/s13058-014-0410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Speeckaert R, Vermaelen K, van GN, Autier P, Lambert J, Haspeslagh M, van GM, Thielemans K, Neyns B, Roche N, Verbeke N, Deron P, Speeckaert M, Brochez L. Indoleamine 2,3-dioxygenase, a new prognostic marker in sentinel lymph nodes of melanoma patients. Eur J Cancer. 2012;48:2004–2011. doi: 10.1016/j.ejca.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 162.Pollari S, Kakonen SM, Edgren H, Wolf M, Kohonen P, Sara H, Guise T, Nees M, Kallioniemi O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res Treat. 2011;125:421–430. doi: 10.1007/s10549-010-0848-5. [DOI] [PubMed] [Google Scholar]
- 163.Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, Feron O, Dewhirst MW. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008;118:3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 165.Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De DA, Lanciotti M, Serni S, Cirri P, Chiarugi P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012;72:5130–5140. doi: 10.1158/0008-5472.CAN-12-1949. [DOI] [PubMed] [Google Scholar]
- 166.Giannoni E, Taddei ML, Morandi A, Comito G, Calvani M, Bianchini F, Richichi B, Raugei G, Wong N, Tang D, Chiarugi P (2015) Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs oxphos and prostate cancer metastatic spread. Oncotarget. PMID:26183399 [DOI] [PMC free article] [PubMed]