

Abstract
In the study of regulated cell death, the rapidly expanding field of regulated necrosis, in particular necroptosis, has been drawing much attention. The signaling of necroptosis represents a sophisticated form of a death pathway. Anti-caspase mechanisms (e.g., using inhibitors of caspases, or genetic ablation of caspase-8) switch cell fate from apoptosis to necroptosis. The initial extracellular death signals regulate RIP1 and RIP3 kinase activation. The RIP3-associated death complex assembly is necessary and sufficient to initiate necroptosis. MLKL was initially identified as an essential mediator of RIP1/RIP3 kinase-initiated necroptosis. Recent studies on the signal transduction using chemical tools and biomarkers support the idea that MLKL is able to make more functional sense for the core machinery of the necroptosis death complex, called the necrosome, to connect to the necroptosis execution. The experimental data available now have pointed that the activated MLKL forms membrane-disrupting pores causing membrane leakage, which extends the prototypical concept of morphological and biochemical events following necroptosis happening in vivo. The key role of MLKL in necroptosis signaling thus sheds light on the logic underlying this unique “membrane-explosive” cell death pathway. In this review, we provide the general concepts and strategies that underlie signal transduction of this form of cell death, and then focus specifically on the role of MLKL in necroptosis.
Keywords: Regulated cell death, Necroptosis, Necrosome, MLKL, Pore-forming protein
Introduction
Regulated cell death has long been appreciated as the built-in brake valve system for organisms to balance with cell survival during development, maintenance of tissue homeostasis and elimination of infected cells in pathological conditions. Building on the classic work in C. elegans and various animal models, the apoptosis pathway was the first evolutionarily conserved regulated cell death signaling pathway that was discovered. A large body of investigation has firmly established a central role of caspase activation and the following regulated proteolysis that leads to apoptosis execution. In contrast to apoptosis, which manifests with specific morphological and biochemical markers, necrotic cell death was initially regarded as a passive form of cell death that results from acute cellular injury or overwhelming stresses. The impressive body of knowledge about the molecular apparatus of apoptosis sets up expectations to getting a comparable clarity of understanding about the regulated necrosis signaling, which was observed independent of caspase activity [1]. Since the late 1980s, researchers have observed that, under certain conditions, caspase inhibition does not block cell death induced by members of the tumor necrosis factor (TNF) family of cytokines, but rather shifts cell fate to necrotic death [1–6]. These observations highlight a long-standing ambiguity about whether necrosis was also a form of regulated cell death like apoptosis.
The necrosome machinery closes the gap between the death signal and membrane disruption
Necrosome assembly
The complexity of necrosis is revealed by a string of defined biochemical signals that occur under tight regulation when the necrotic death complex (necrosome) is assembled. Receptor-interacting kinase 1 (RIP1, also known as RIPK1) was the first molecule identified as a necrosome component required for Fas ligand-induced caspase-independent necrotic cell death [7]. RIP1 plays pleiotropic roles in multiplying signaling processes, such as NF-κB activation and apoptosis [8–11]. Its kinase activity was found to be specifically required in death receptor activation-induced regulated necrosis, also termed necroptosis. Upon death ligand ligation, RIP1 binds with death receptors via their respective death domains (DD). Once FADD or caspase activity is inhibited by genetic or chemical methods, RIP1 forms the death complex with receptor-interacting kinase 3 (RIP3, also known as RIPK3) through their homotypic interaction motif (RHIM) domains, and activates their kinase activities [12–14]. Thereafter, the activated RIP kinases respond to and link the death signals to a downstream substrate, the pseudokinase mixed-lineage kinase domain-like protein (MLKL) [15].
Determinants of necrosome complex assembly
TNF receptor 1 (TNFR1) ligation leads to the recruitment of TRADD, RIP1, TRAF2 and cIAP1/2, which is known as complex I. The E3 ubiquitin ligases, cIAP1/2, block RIP1 transition to the necrosome by ubiquitination of RIP1 that mediates NF-κB activation [16]. Loss of cIAP1/2 function, caused by treatment with Smac mimetics, promotes RIP1 kinase activation and necrosome complex formation [17]. A20, an inhibitor of NF-κB signaling, was recently reported to restrict ubiquitination of RIP3 at Lys5 (K5) and protect from the formation of the necrosome [18].
Death receptor-activated apoptosis and necroptosis share a common regulatory signal complex (complex IIB), which includes RIP1, FADD, caspase-8 and cFLIP. Mice with deletions of any of the complex-IIB genes display severe embryonic developmental defects that lead to either embryonic (Casp8 −/− ; Fadd −/− ; cFlip −/−) or postnatal lethality (Rip1 −/−) [19, 20]. RIP3- or MLKL-deficient mice do not show developmental or homeostasis defects [13, 21–23]. The lethality caused by deficiency in caspase-8 or FADD can be rescued by loss of either RIP3 or MLKL [20, 24–27]. In addition, the neonatal lethality of Rip1 −/− mice was rescued by concurrent loss of caspase-8 and RIP3 [28–30]. Therefore, RIP1 and caspase-8 work synergistically as regulatory components to prevent both forms of cell death during development. It is worth noting that kinase-dead RIP1 (K45A; D138N) knock-in mice develop normally into adulthood [29, 31–33]. These data implicate that the kinase activity of RIP1 is dispensable for instruction of these two death pathways in regulating multiorgan development.
Necrosome assembly provides a structural basis for RIP3 oligomerization
Necroptosis induced by the TNF family of cytokines (TNF, CD95/FasL, TRAIL) requires RIP1 for assembling the necrosome components. However, exceptions have been found. For instance, RIP3 and MLKL are required in Toll-like receptor signaling (TLR3/TLR4), but in an RIP1-dispensable way. Instead, another RHIM domain-containing protein, TRIF, is associated with RIP3 via its RHIM domain, which is analogous to the RIP1–RIP3 necrosome complex [30, 34–36]. Likewise, murine cytomegalovirus (MCMV)-induced necroptosis employs a DAI–RIP3–MLKL axis. In addition, the virus DNA sensor, DAI, was shown to be associated with RIP3 also through its RHIM domain [34, 37, 38]. Additionally, the herpes simplex virus (HSV)-1 effector protein, ICP6, binds with RIP1/3 through their virus–host RHIM interactions, which leads to either pro-necroptotic or anti-necroptotic effects in mice and humans, respectively [39–42]. Given this shared mode of necroptosis induction across physiological and disease processes in varying species, a simple working definition for necroptosis initiation includes the RHIM domain protein-mediated activation of RIP3 functionality (Fig. 1).
Fig. 1.
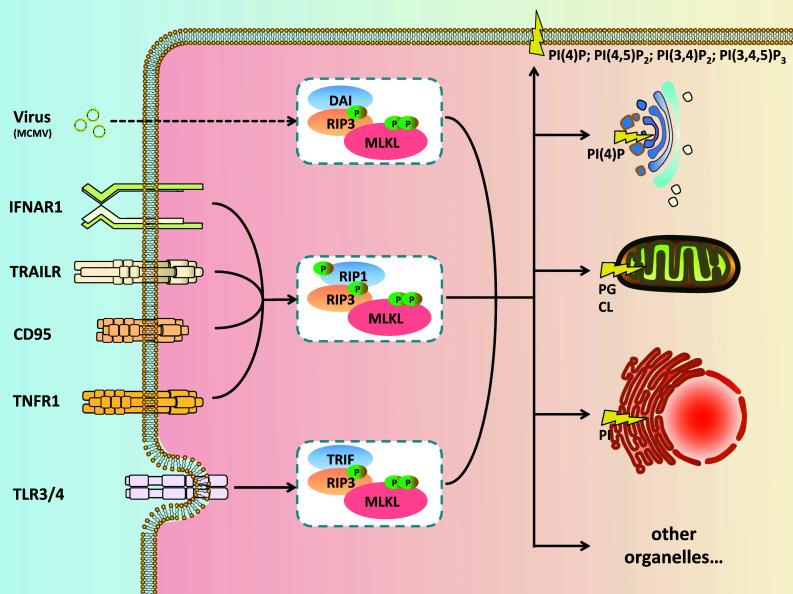
Necrosome assembly and signal propagation. Ligation of members of the tumor necrosis factor receptor superfamily (TNFRSF, including TNFR1, CD95, TRAILR) leads to the recruitment of receptor-interacting protein kinase 1 (RIP1). RIP1 and RIP3 form a pro-necrotic death complex via their RHIM domains. RIP3 is then activated by auto-phosphorylation at Ser227 (Ser232 for mouse RIP3). The activated RIP3 recruits and phosphorylates its substrate MLKL at Thr357/Ser358 (Ser345 for mouse MLKL), a step which defines the formation of the functional necrosome. Besides TNFRSF, activation of type I interferon receptor (IFNAR1) also triggers the formation of a RIP1–RIP3–MLKL-containing necrosome. Moreover, pattern recognition receptors (PRRs) drive necrosis in an RIP1-independent manner. Toll-like receptor-induced TRIF–RIP3–MLKL necrosome formation (for TLR3, e.g., by sense dsRNA such as poly I:C; for TLR4 in response to LPS) also depends on RHIM interaction between TRIF and RIP3. Likewise, infection with M45 mutant murine cytomegalovirus (MCMV) leads to RHIM-mediated interaction between DAI and RIP3. Of all necrosome protein complexes reported to date, MLKL binds to activated RIP3 to propagate the necrotic death signal, regardless of what RHIM protein RIP3 utilizes for its self-activation. Activated MLKL binds phosphatidylinositol phosphates (PIPs), cardiolipin (CL) and phosphatidylglycerol (PG), which navigate necrosomes to different phospholipid-rich cellular compartments. Once targeted to membranes, MLKL disrupts membrane integrity and finally causes necroptotic cell death
A complementary study to assess the mutual dependency of RIP1 and RIP3 has been to generate a death receptor-free environment by building up an artificial dimerization system for RIP1 and RIP3. Cook et al. found that necroptosis can be induced either by dimerization of RIP1 or RIP3 in immortalized mouse embryonic fibroblasts (MEFs) [43]. Wu et al. expressed combinations of wild-type and RHIM mutant RIP1 and RIP3 in 293T cells [44]. They emphasized that the necroptotic signal is propagated from the RIP1–RIP3 amyloid scaffold by recruiting additional free RIP3 molecules. Orozco et al. added a point that RIP1 might negatively regulate the spontaneous oligomerization of RIP3 when RIP3 is maintained at a relatively low level, although they faintly induced necroptosis by expressing RIP3 dimer in their system [45]. These discrepant data of dimerized RIP3 were explained by side effects of the different positions that the protein was tagged with the dimerization domains.
Nonetheless, we assemble here a scenario where the death signal flows from RIP1 to RIP3 through their RHIM domains, and additional molecules of RIP3 would be recruited afterward for auto-phosphorylation-driven signal propagation. Meanwhile, an emerging theme is the highly conditional influence of these artificial systems, in that RIP1 is needed under some circumstances but not in others. Altogether, these data lend further support to the notion that RIP3 oligomerization is the minimal functional unit that is required to drive necrosome assembly.
How RIP3 activates MLKL
Mixed-lineage kinase domain-like protein was found to be specifically required for the RIP3-dependent necroptosis pathway. It was initially identified as a RIP3-binding protein via its C-terminal kinase-like domain. A series of kinase assays combined with proteomics studies revealed that MLKL is the natural target of RIP3 kinase. Recruitment of MLKL depends on auto-phosphorylation of RIP3 at S227 (S232 for mouse RIP3) [15]. In a following study, Xie et al. presented the co-crystal structure of the mouse RIP3 kinase domain (residues 1–318) with the MLKL kinase-like domain (residues 182–464) at 2.5 Å resolution [46]. The binding interface between RIP3 and MLKL was depicted aligning in a parallel fashion. The phosphate group of the phosphorylated Ser232 in RIP3 was clearly visible, which accepts an H-bond from the hydroxyl group of Ser404 in MLKL. In addition, other observed phosphorylated residues, Ser184 and Thr231, of RIP3 were found not to be involved in the interface with MLKL, and had little impact on RIP3–MLKL complex formation. At the center of their interface, Phe27 of RIP3 and Phe234 of MLKL stack against each other through π–π interactions, providing the most prominent bond strength for RIP3 and MLKL [46]. A cochaperone complex, HSP90–CDC37, was discovered as RIP3-binding partner [47]. Knocking down CDC37 or using HSP90 inhibitors efficiently blocks necroptosis by preventing auto-phosphorylation on RIP3–Ser227 and RIP3 punctae formation. Besides, one of the negative regulators of this auto-phosphorylation on RIP3, protein phosphatase 1B (Ppm1b), was recently found to suppress necroptosis by dephosphorylation of RIP3 [48]. Taken together, the auto-phosphorylation of RIP3 at S227 (S232 for mouse RIP3) shapes up the interface of RIP3 in recognizing its substrate MLKL.
Evidence suggests that p-MLKL is the key for necroptotic signal transfer. When activated RIP3 binds to MLKL, it subsequently phosphorylates MLKL at T357/S358 (S345 for mouse MLKL) [15]. A study of doxycycline (DOX)-inducible MLKL mutants expressed in mlkl −/–MEFs also confirmed that phosphorylation of Ser345 is critical for RIP3-mediated necroptosis, while other reported sites (Ser347 and Thr349) either play minor roles or seem to be irrelevant [49].
MLKL phosphorylation as a biomarker of necroptosis activation
Based on the knowledge that MLKL is phosphorylated by RIP3, a rabbit monoclonal antibody was developed to specifically recognize phosphorylated MLKL (p-MLKL) and serve as a marker for necroptosis [50]. This work advances the field by allowing to detect necroptosis in a more accurate way. Given the specificity of this phospho-antibody, an emerging picture of the properties of necrotic diseases has helped us gain a broader understanding of under what conditions this form of cell death occurs. For example, drug-induced liver injury (DILI) was the first reported clinical disorder that exhibits necrotic damage by showing strong p-MLKL signals in the diseased compartments [50]. After that, researchers also observed p-MLKL signals in human non-alcoholic steatohepatitis (NASH) samples. Recent studies implicated that MLKL-dependent necroptosis is highly prevalent in isolating and removing pathogens [39]. In addition, necroptosis is implicated in regulating chemical-induced cell injuries, such as cerulein-induced pancreatitis [21]. Furthermore, excessive necrotic cell death is associated with ischemia reperfusion-induced damage in the brain as well as in patients with neurodegenerative disorders [51]. In a study of human multiple sclerosis (MS) pathological specimens, robust p-MLKL signals were detected [52]. Apart from these clinical conditions, p-MLKL signals also occur in primate ovarian tissues (human and rhesus monkey), which may contribute to follicular atresia and luteolysis in females, eventually leading to menopause [53]. Application of this phospho-antibody as a biomarker to reflect necroptotic cell death in patients will provide us sufficient proof-of-principle support for the development of pharmaceutical agents that interfere with these necroptotic diseases.
Necrosome inhibitors
A range of chemical screens for necroptosis inhibitors has significantly increased our understanding of necrotic signaling; however, each has had its drawbacks. The first well-defined necroptosis inhibitor is necrostatin-1 (Nec-1), which blocks necroptosis by targeting RIP1 kinase activity [54, 55]. More in vivo tests of Nec-1 derivatives were explored in animal models, such as 7N-1 which blocks demyelination induced by cuprizone and encephalomyelitis EAE [34, 52]. However, since RIP1 also contributes to other processes beyond necroptosis, such as the regulation of inflammatory cytokine release and apoptosis, an independent focus has also been placed on RIP3 and MLKL inhibitors. But, the challenge for RIP3 inhibitors resides in the potential gain of function of triggering apoptosis. A collection of RIP3 inhibitors induced apoptosis while simultaneously blocking necroptosis, which could be explained by the discovery that overexpression of kinase-dead RIP3 (K51A), causes the cells to die from apoptosis [56]. In line with this, recently, RIP3 kinase-dead knock-in mice (D161N) were found to be embryonic lethal due to vast apoptosis [31]. A larger pool of RIP3 inhibitors is in the process of being further characterized for their apoptotic toxicity.
All of these data above exemplify the urgent need for finding less-toxic necroptosis inhibitors. So far, the MLKL inhibitor, NSA, shows the least amount of toxicity to cells [15]. NSA blocks necroptosis by targeting Cys86 on MLKL, thus interfering with MLKL oligomerization. In the studies of necroptosis signaling, NSA was proven to be a unique tool for dissecting the downstream process of necroptosis without disturbing necrosome death complex assembly. However, because NSA has an exclusive recognition of human MLKL, this limits its application in mouse models. In 2014, another mouse MLKL inhibitor, referred to as compound 1 by Hildebrand et al., was identified to inhibit the necroptosis pathway by delaying MLKL translocation to the membrane, but its toxicity and multiple targets on the other components of necrosome preclude the application of this MLKL inhibitor [57]. New necroptosis inhibitors that target mouse MLKL will expand our knowledge for the in vivo significance of MLKL-dependent necroptosis. In addition, a series of chemical tools also anchor our interpretation of the underlying logic of MLKL activation.
Emerging concepts in the study of necroptosis execution
Membrane pore-forming machinery by MLKL
Phosphorylation drives MLKL oligomerization and membrane translocation
Within the past few years, it has become clear that the phosphorylation of MLKL builds on the most formidable engine of necrosome machinery, which ignite and propel the death signal toward membranes. As we review here, there is now much clearer understanding of the crucial role of MLKL in necroptosis execution.
Downstream necroptosis signals work through MLKL. Accordingly, phosphorylated MLKL would be expected to transduce death signals. Both in vivo and in vitro biochemical analyses characterized the oligomerization nature of MLKL. Necroptosis induction causes a molecular weight shift of MLKL, which appears on non-reducing PAGE gels. Wang et al. demonstrated that phosphorylation of MLKL turns on its oligomerization [50]. However, researchers have not reached a consensus on the minimal units of the MLKL oligomer [50, 58–60]. Interpretations of these data, using crosslinkers or gel filtration, should be regarded with caution, given the present technical limitations. Only alternative approaches with higher resolution, such as NMR and crystallography technology, could provide a conclusive physical picture of MLKL oligomerization.
Biochemical fractionation revealed that MLKL translocates to the plasma membrane after necroptosis is induced. By following the phospho-MLKL signals, it was found that this translocation was not limited to the plasma membrane; organelle membranes were also targeted by p-MLKL. This targeting to membranes is facilitated by the N-terminal coil–coil domain of MLKL, which possesses a patch of positively charged amino acid that enables MLKL to interact with phospholipids. Pleckstrin homology (PH) domains are well known for their binding to phosphatidylinositol lipids (PIPs)-containing lipids within biological membranes. Different PH domains possess specificities for different lipids [61–64]. It is noteworthy that MLKL has a broader affinity for PIPs than PH domains. Both Wang and Dondelinger found that the N-terminal MLKL directly binds with phosphatidylinositol phosphates (PIPs) [50, 65]. In addition, Wang et al. found that cardiolipin (CL) could also bind to MLKL.
Different PIPs may direct MLKL to different cellular compartments. The lipid composition of these membranes varies from organelle to organelle even though these distinct membrane systems are in communication through intracellular trafficking by vesicles. Most membranes are rich in phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylinositol (PI). PC and PI are enriched in the ER; phosphatidylglycerol (PG) and CL are synthesized in and confined to mitochondria [66, 67]. For instance, plasma membranes have the most abundant PI(4)P and PI(4,5)P2. This could explain the vast damage on cell membranes after necroptosis induction. Cardiolipin (CL) is mostly distributed in the mitochondrial inner membrane. During necroptosis, interference with mitochondrial fusion has been described. This was well timed for CL cytosolic exposure, and the CL-enriched microdomain provides a signaling platform for MLKL-mediated death signals on mitochondria [68–70], which would strengthen the intrinsic amplification of death signals. In the same spirit, other PIPs have their distinctive organelle distribution, which would give MLKL the proper guidance to the designated membrane compartments.
Evidence that MLKL punches membranes
Membrane rupture and organelle swelling have remained cornerstone features of necrosis. Investigators have generally regarded membrane damage as a basic morphological criterion for necrosis. The released cellular content of necrotic cells will trigger an immune response. Thus, the exposure of DAMPs (damage-associated molecular pattern) [71] has been considered as a signature indicator for necrosis.
Using the classic liposome leakage assay, MLKL was found to translocate to PIPs- or CL-containing membranes and to disrupt the membrane integrity in a dose-dependent manner [50] (Fig. 2a, adapted from [72]). NSA can block these membrane disruptions. Since NSA is targeted to the N-terminus of MLKL, this discovery is consistent with the finding that the N-terminal domain mediates liposome damage and cell death. Moreover, when the intracellular levels of MLKL-binding PIPs were downregulated by interfering with their production processes, MLKL-dependent necroptosis was largely attenuated [65]. These experiments extensively characterized the pore-forming property of MLKL on membrane structures. It puts forth the possibility that MLKL is the executioner of necroptosis by punching holes in cellular membranes. It has become increasingly clear that the pore-forming properties of MLKL are further influenced by the interplay between the N-terminus of MLKL and phospholipids.
Fig. 2.
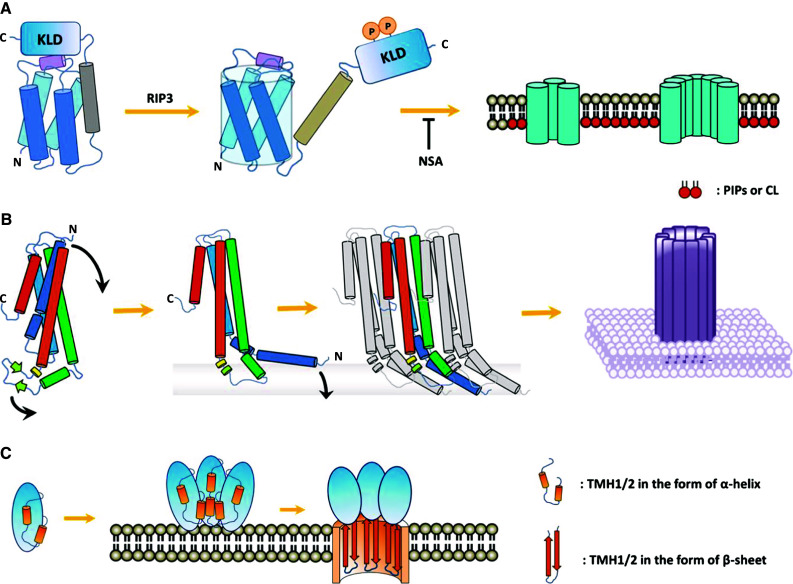
Overview of the membrane-punching mechanisms of pore-forming proteins. a Oligomerized MLKL punches membranes. The MLKL monomer is sequestered in an inactivated state by its C-terminal kinase-like domain (KLD). Phosphorylation of MLKL releases the auto-inhibition on the amino-terminal MLKL and enables MLKL to bind with PIPs or CL. NSA blocks MLKL from oligomerization and membrane translocation. b A representative α-PFT protein, cytolysin A (ClyA, also known as HlyE), forms a pore on a target membrane. The β-tongue, consisting of two β-strands between the third and fourth helices, locks ClyA in a compacted soluble state. Once ClyA binds to the membrane, the C-terminal helix is released from the bundle, touches the membrane and further recruits other molecules. After assembled into oligomers, the aggregated C-terminal helices insert into the membrane to form pores. c Structural transitions of CDC pore-forming proteins. Beta-PFTs are assembled into oligomeric pre-pores and bind to the membrane. The tandem transmembrane helical (TMH) units that are initially buried in the core of the pre-pore intermediate will be exposed and inserted into the membrane as a β-barrel. Perforin and the complement share similar primary structures of both monomer and oligomer, whether there is a transition station remains unclear
The structure of the MLKL N-terminal region has been determined by nuclear magnetic resonance spectroscopy, which reveals that the four-helix bundle (4HBD) with an additional helix at the top is likely to be the key for MLKL function. Further, fluorescence spectroscopy measurements indicate that much of the 4HBD inserts into membranes, but not the intermediate helix [72]. Moreover, 4HBD is sufficient to induce liposome leakage, while the C-terminal helix inhibits this activity [50, 72]. It has also been reported that expressing truncated forms of MLKL lacking the pseudokinase domain can lead to constitutive cell death in both Mlkl −/− and WT MEF cells or HEK293T cells [57, 65], indicating an essential role of the MLKL N-terminal domain in necroptosis signaling and implicating the C-terminal pseudokinase domain as a suppressor to restrain the N-terminal 4HBD function. A recent work by Quarato et al. has further confirmed that the interaction between the brace and N-terminal 4HBD exerts the “inhibitory plug” regulation. Moreover, they also showed once MLKL integrates with the membrane, its N-terminal helix bundle utilizes a “rolling over” mechanism to expose additional high-affinity PIP-binding sites, which added another layer of distinct PIP-binding sites responsible for robust association to the membrane [73].
Apart from the aforementioned MLKL activation pathways, artificial systems can also lead to the activation of MLKL and to execution of necroptosis. Taking advantage of the HBD*-4-OHT dimerization system, Chen and colleagues demonstrated that activation of MLKL by forcing the protein or its N-terminus together (tetramerization here) directly triggers necroptosis, which can bypass RIP3 signals [58]. In accordance with the artificial system, they also observed MLKL tetramers in TNF-induced necroptosis. Using the FKBPv-AP20187 dimerization system, Wang and colleagues reported that polymerization of phosphomimic-MLKL leads to more necroptosis than WT-MLKL in RIP3-deficient cells [50].
Controversies: channel theories
A range of experimental criteria has been employed to monitor ion disturbances, which potentially contribute to necroptosis. In a cell-based experiment, Cai et al. found that TRPM7-mediated Ca2+ influx is required for necroptosis execution [59]. This finding suggested an activation process of non-voltage-sensitive Ca2+ channels downstream of MLKL. Adding to this, Chen et al. provided an experimental indication of a potential Na+ channel downstream of MLKL, suggesting that the membrane translocation of MLKL is truly associated with membrane disruption [58]. These perspectives, however, conflict with the phenomenon that Ca2+/Na+ depletion blocks necroptosis, which largely depends on the experimental settings and the specific backgrounds of cell lines. Nevertheless, more exploration is required on how such channels mesh with membrane rupture happening, which involves many biological processes. The main doubt about these data hinges on the presumption that either Ca2+/Na+ influx is the cause or one of the consequences that the osmotic pressure posed by the MLKL perforator elicits.
Other pore-forming proteins
Apart from MLKL, Mother Nature makes judicious use of finite pore-forming mechanisms to control cell death, of which there are a certain number of pore-forming toxins (PFTs) produced typically by bacteria, complement proteins or perforins released by cytotoxic T lymphocytes or natural killer cells. All these proteins share the ability to form pores in the plasma membrane. According to the secondary structure involved in pore formation, PFTs can be classified into α-PFTs and β-PFTs [74, 75]. α-PFTs form pores using helices (Fig. 2b, adapted from [76]) while β-PFTs, such as cholesterol-dependent cytolysins (CDCs), insert into membranes by their β-sheet structure [77–79]. Their membrane-damaging processes can be divided into three steps: oligomerization, membrane insertion, and pore formation. The membrane attack complex (MAC) of complement is a part of the mammalian innate immune system, which is formed by sequential assembly of C5b with C6, C7 and C8, and polymerization of C9, resulting in transmembrane pore formation and loss of membrane integrity [80, 81]. Recent findings provided some insights into the similarities between PFTs and MAC in the process of pore formation [82, 83]. Aside from the humoral cytotoxicity that is caused by the complement system, the lesion caused by cytotoxic T lymphocytes (CTLs) is also formed by polymerization of a pore-forming protein, called perforin, to form cylindrical pores on the target-cell membrane. The pores formed by perforin are similar to those observed in complement-mediated lysis, which mechanistically form pores in a CDC-like manner [84–87] (Fig. 2c adapted from [88]).
The pore-forming proteins mentioned above share general characteristic principles. Thus, the impact of the pore-forming mechanism spans many fields of study, such as the conversion from water-soluble monomers to intramembranous oligomers, membrane insertion by specific secondary structure motifs, selective recognition of membrane lipids, and induction of cell lysis. The finding that MLKL does not fit into any of these known pore-forming protein categories also, in turn, sheds light on new paradigms for defining pore-forming proteins.
In addition, gasdermin D (GSDMD) was recently identified as a critical component in pyroptosis, another kind of necrosis driven by inflammatory caspases, for which the membrane-damaging mechanism has remained a mystery for years. GSDMD can be functionally divided into gasdermin-N and gasdermin-C domains. Once inflammatory caspases (caspase-1, -4, -5 and -11) are activated, they specifically cleave GSDMD behind D257 (D276 in mouse) and enable the gasdermin-N domains to sufficiently drive pyroptosis [89, 90]. However, the biophysical properties by which the N-terminal fragment elicits pyroptotic cell death remains unknown, and it remains to be explored whether it shares a similar membrane-damaging mechanism with the known pore-forming proteins or whether it has a requirement for a receptor on the target membrane.
Concluding remarks and perspectives
Necroptosis can be triggered by various death signals. We have deliberately focused on mechanisms that convey the signal from RIP kinases to MLKL. We believe that wide variations in the RIP1/RIP3 kinases interplay/hierarchy reflect the differences in experimental settings, which will ultimately be traced to variations in the potential modulatory factors for these death kinases. Recent progress on necroptosis signaling advances our knowledge on the core machinery, most importantly on MLKL activation and its highly ordered organization on liposome structures leading to membrane disruption. More biophysical analysis about the membrane-binding characteristics of MLKL is needed, which will further our understanding of the executioner role of MLKL during necroptosis.
Apart from the notion that MLKL receives death signals from RIP3 kinase by direct phosphorylation, it was not at all obvious how MLKL transduces signals to its downstream effectors. The phenomenon of MLKL nuclear translocation before cell death was found by immunofluorescence and biochemical fractionation [91]. Although the functional relevance of this population of p-MLKL for necroptosis execution is still unclear, the consequences of different subcellular translocations of p-MLKL remain to be systemetically clarified. We do not claim that this summary provides a complete explanation of how necrotic damage appears on membranes. Even if the pore forming of MLKL is not the whole story of membrane rupture, it will be fruitful to pursue the regulators of RIP1/RIP3, and the potential co-factors of MLKL. Though many aspects of necrosome signaling are still shrouded in mystery, it is clear that new techniques such as a combination of cellular analysis and biophysical investigation can illuminate the unique complexity of the necrosome machinery.
Acknowledgments
We thank Ms. Sasha Sa for critical reading and comments on the manuscript. The work of L.S. is sponsored by Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences and The One Hundred Talents Program of Chinese Academy of Sciences.
References
- 1.Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S. Caspase-independent cell killing by Fas-associated protein with death domain. J Cell Biol. 1998;143(5):1353–1360. doi: 10.1083/jcb.143.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, Vandenabeele P. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. 1998;188(5):919–930. doi: 10.1084/jem.188.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187(9):1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vercammen D, Vandenabeele P, Beyaert R, Declercq W, Fiers W. Tumour necrosis factor-induced necrosis versus anti-Fas-induced apoptosis in L929 cells. Cytokine. 1997;9(11):801–808. doi: 10.1006/cyto.1997.0252. [DOI] [PubMed] [Google Scholar]
- 5.Grooten J, Goossens V, Vanhaesebroeck B, Fiers W. Cell membrane permeabilization and cellular collapse, followed by loss of dehydrogenase activity: early events in tumour necrosis factor-induced cytotoxicity. Cytokine. 1993;5(6):546–555. doi: 10.1016/S1043-4666(05)80003-1. [DOI] [PubMed] [Google Scholar]
- 6.Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol. 1988;141(8):2629–2634. [PubMed] [Google Scholar]
- 7.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1(6):489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 8.Lee TH, Shank J, Cusson N, Kelliher MA. The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem. 2004;279(32):33185–33191. doi: 10.1074/jbc.M404206200. [DOI] [PubMed] [Google Scholar]
- 9.Ting AT, Pimentel-Muinos FX, Seed B. RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J. 1996;15(22):6189–6196. [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4(4):387–396. doi: 10.1016/S1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 11.Grimm S, Stanger BZ, Leder P. RIP and FADD: two “death domain”-containing proteins can induce apoptosis by convergent, but dissociable, pathways. Proc Natl Acad Sci USA. 1996;93(20):10923–10927. doi: 10.1073/pnas.93.20.10923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 13.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 14.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1–2):213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 16.Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30(6):689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 17.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Hacker G, Leverkus M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43(3):449–463. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Onizawa M, Oshima S, Schulze-Topphoff U, Oses-Prieto JA, Lu T, Tavares R, Prodhomme T, Duong B, Whang MI, Advincula R, Agelidis A, Barrera J, Wu H, Burlingame A, Malynn BA, Zamvil SS, Ma A. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol. 2015 doi: 10.1038/ni.3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, Mak TW, Wallach D, Green DR. Survival function of the FADD–CASPASE-8–cFLIP(L) complex. Cell Rep. 2012;1(5):401–407. doi: 10.1016/j.celrep.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. Catalytic activity of the caspase-8–FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471(7338):363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, Yang Z, Wu SQ, Chen L, Han J. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23(8):994–1006. doi: 10.1038/cr.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, Young SN, Varghese LN, Tannahill GM, Hatchell EC, Majewski IJ, Okamoto T, Dobson RC, Hilton DJ, Babon JJ, Nicola NA, Strasser A, Silke J, Alexander WS. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39(3):443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 23.Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004;24(4):1464–1469. doi: 10.1128/MCB.24.4.1464-1469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471(7338):373–376. doi: 10.1038/nature09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van Loo G, Pasparakis M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477(7364):330–334. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- 26.Gunther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, Becker C. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477(7364):335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, Bloch W, Haase I, Pasparakis M. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity. 2011;35(4):572–582. doi: 10.1016/j.immuni.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 28.Rickard JA, O’Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, Vince JE, Lawlor KE, Ninnis RL, Anderton H, Hall C, Spall SK, Phesse TJ, Abud HE, Cengia LH, Corbin J, Mifsud S, Di Rago L, Metcalf D, Ernst M, Dewson G, Roberts AW, Alexander WS, Murphy JM, Ekert PG, Masters SL, Vaux DL, Croker BA, Gerlic M, Silke J. RIPK1 regulates RIPK3–MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157(5):1175–1188. doi: 10.1016/j.cell.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 29.Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, Sundararajan A, Guo H, Roback L, Speck SH, Bertin J, Gough PJ, Balachandran S, Mocarski ES. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci USA. 2014;111(21):7753–7758. doi: 10.1073/pnas.1401857111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, Janke LJ, Kelliher MA, Kanneganti TD, Green DR. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157(5):1189–1202. doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose-Girma M, Warming S, Dixit VM. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343(6177):1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 32.Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, Lee TH, Chan FK, Pasparakis M, Kelliher MA. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol. 2014;193(4):1539–1543. doi: 10.4049/jimmunol.1400590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Cook M, Finger J, Hughes-Earle A, Harris PA, Kaiser WJ, Mocarski ES, Bertin J, Gough PJ. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J Immunol. 2014;192(12):5476–5480. doi: 10.4049/jimmunol.1400499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288(43):31268–31279. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA. 2011;108(50):20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma Y, Temkin V, Liu H, Pope RM. NF-kappaB protects macrophages from lipopolysaccharide-induced cell death: the role of caspase 8 and receptor-interacting protein. J Biol Chem. 2005;280(51):41827–41834. doi: 10.1074/jbc.M510849200. [DOI] [PubMed] [Google Scholar]
- 37.Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11(3):290–297. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, Hofmann K, Vazquez J, Benedict CA, Tschopp J. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009;10(8):916–922. doi: 10.1038/embor.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mocarski ES, Guo H, Kaiser WJ. Necroptosis: the Trojan horse in cell autonomous antiviral host defense. Virology. 2015;479–480:160–166. doi: 10.1016/j.virol.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang Z, Wu SQ, Liang Y, Zhou X, Chen W, Li L, Wu J, Zhuang Q, Chen C, Li J, Zhong CQ, Xia W, Zhou R, Zheng C, Han J. RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe. 2015;17(2):229–242. doi: 10.1016/j.chom.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 41.Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, Kaiser WJ, Mocarski ES. Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe. 2015;17(2):243–251. doi: 10.1016/j.chom.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Li Y, Liu S, Yu X, Li L, Shi C, He W, Li J, Xu L, Hu Z, Yu L, Yang Z, Chen Q, Ge L, Zhang Z, Zhou B, Jiang X, Chen S, He S. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci USA. 2014;111(43):15438–15443. doi: 10.1073/pnas.1412767111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cook WD, Moujalled DM, Ralph TJ, Lock P, Young SN, Murphy JM, Vaux DL. RIPK1- and RIPK3-induced cell death mode is determined by target availability. Cell Death Differ. 2014;21(10):1600–1612. doi: 10.1038/cdd.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu XN, Yang ZH, Wang XK, Zhang Y, Wan H, Song Y, Chen X, Shao J, Han J. Distinct roles of RIP1–RIP3 hetero- and RIP3–RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014 doi: 10.1007/978-1-4614-9302-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Orozco S, Yatim N, Werner MR, Tran H, Gunja SY, Tait SW, Albert ML, Green DR, Oberst A. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014;21(10):1511–1521. doi: 10.1038/cdd.2014.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie T, Peng W, Yan C, Wu J, Gong X, Shi Y. Structural insights into RIP3-mediated necroptotic signaling. Cell reports. 2013;5(1):70–78. doi: 10.1016/j.celrep.2013.08.044. [DOI] [PubMed] [Google Scholar]
- 47.Li D, Xu T, Cao Y, Wang H, Li L, Chen S, Wang X, Shen Z. A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc Natl Acad Sci USA. 2015;112(16):5017–5022. doi: 10.1073/pnas.1505244112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen W, Wu J, Li L, Zhang Z, Ren J, Liang Y, Chen F, Yang C, Zhou Z, Sean SuS, Zheng X, Zhang Z, Zhong CQ, Wan H, Xiao M, Lin X, Feng XH, Han J. Ppm1b negatively regulates necroptosis through dephosphorylating Rip3. Nat Cell Biol. 2015;17(4):434–444. doi: 10.1038/ncb3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, Oberst A, Quarato G, Low J, Cripps JG, Chen T, Green DR. Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 2015 doi: 10.1038/cdd.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54(1):133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 51.Liu S, Wang X, Li Y, Xu L, Yu X, Ge L, Li J, Zhu Y, He S. Necroptosis mediates TNF-induced toxicity of hippocampal neurons. Biomed Res Int. 2014;2014:290182. doi: 10.1155/2014/290182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, Ye J, Zhang X, Chang A, Vakifahmetoglu-Norberg H, Geng J, Py B, Zhou W, Amin P, Berlink Lima J, Qi C, Yu Q, Trapp B, Yuan J. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015;10(11):1836–1849. doi: 10.1016/j.celrep.2015.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blohberger J, Kunz L, Einwang D, Berg U, Berg D, Ojeda SR, Dissen GA, Frohlich T, Arnold GJ, Soreq H, Lara H, Mayerhofer A. Readthrough acetylcholinesterase (AChE-R) and regulated necrosis: pharmacological targets for the regulation of ovarian functions? Cell Death Dis. 2015;6:e1685. doi: 10.1038/cddis.2015.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 56.Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, Ouellette M, King BW, Wisnoski D, Lakdawala AS, DeMartino MP, Casillas LN, Haile PA, Sehon CA, Marquis RW, Upton J, Daley-Bauer LP, Roback L, Ramia N, Dovey CM, Carette JE, Chan FK, Bertin J, Gough PJ, Mocarski ES, Kaiser WJ. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56(4):481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, Pierotti C, Garnier JM, Dobson RC, Webb AI, Tripaydonis A, Babon JJ, Mulcair MD, Scanlon MJ, Alexander WS, Wilks AF, Czabotar PE, Lessene G, Murphy JM, Silke J. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA. 2014 doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, Han J. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24(1):105–121. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16(1):55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murphy JM, Lucet IS, Hildebrand JM, Tanzer MC, Young SN, Sharma P, Lessene G, Alexander WS, Babon JJ, Silke J, Czabotar PE. Insights into the evolution of divergent nucleotide-binding mechanisms among pseudokinases revealed by crystal structures of human and mouse MLKL. Biochem J. 2014;457(3):369–377. doi: 10.1042/BJ20131270. [DOI] [PubMed] [Google Scholar]
- 61.Rameh LE, Arvidsson A, Carraway KL, 3rd, Couvillon AD, Rathbun G, Crompton A, VanRenterghem B, Czech MP, Ravichandran KS, Burakoff SJ, Wang DS, Chen CS, Cantley LC. A comparative analysis of the phosphoinositide binding specificity of pleckstrin homology domains. J Biol Chem. 1997;272(35):22059–22066. doi: 10.1074/jbc.272.35.22059. [DOI] [PubMed] [Google Scholar]
- 62.Marte BM, Downward J. PKB/Akt: connecting phosphoinositide 3-kinase to cell survival and beyond. Trends Biochem Sci. 1997;22(9):355–358. doi: 10.1016/S0968-0004(97)01097-9. [DOI] [PubMed] [Google Scholar]
- 63.Salim K, Bottomley MJ, Querfurth E, Zvelebil MJ, Gout I, Scaife R, Margolis RL, Gigg R, Smith CI, Driscoll PC, Waterfield MD, Panayotou G. Distinct specificity in the recognition of phosphoinositides by the pleckstrin homology domains of dynamin and Bruton’s tyrosine kinase. EMBO J. 1996;15(22):6241–6250. [PMC free article] [PubMed] [Google Scholar]
- 64.Garcia P, Gupta R, Shah S, Morris AJ, Rudge SA, Scarlata S, Petrova V, McLaughlin S, Rebecchi MJ. The pleckstrin homology domain of phospholipase C-delta 1 binds with high affinity to phosphatidylinositol 4,5-bisphosphate in bilayer membranes. Biochemistry. 1995;34(49):16228–16234. doi: 10.1021/bi00049a039. [DOI] [PubMed] [Google Scholar]
- 65.Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, Bertin J, Gough PJ, Savvides S, Martinou JC, Bertrand MJ, Vandenabeele P. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7(4):971–981. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 66.van Meer G, de Kroon AI. Lipid map of the mammalian cell. J Cell Sci. 2011;124(Pt 1):5–8. doi: 10.1242/jcs.071233. [DOI] [PubMed] [Google Scholar]
- 67.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008;9(2):112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sorice M, Manganelli V, Matarrese P, Tinari A, Misasi R, Malorni W, Garofalo T. Cardiolipin-enriched raft-like microdomains are essential activating platforms for apoptotic signals on mitochondria. FEBS Lett. 2009;583(15):2447–2450. doi: 10.1016/j.febslet.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 69.Sorice M, Circella A, Cristea IM, Garofalo T, Di Renzo L, Alessandri C, Valesini G, Esposti MD. Cardiolipin and its metabolites move from mitochondria to other cellular membranes during death receptor-mediated apoptosis. Cell Death Differ. 2004;11(10):1133–1145. doi: 10.1038/sj.cdd.4401457. [DOI] [PubMed] [Google Scholar]
- 70.Stepanyants N, Macdonald PJ, Francy CA, Mears JA, Qi X, Ramachandran R. Cardiolipin’s propensity for phase transition and its reorganization by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Mol Biol Cell. 2015;26(17):3104–3116. doi: 10.1091/mbc.E15-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38(2):209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 72.Su L, Quade B, Wang H, Sun L, Wang X, Rizo J. A plug release mechanism for membrane permeation by MLKL. Structure. 2014 doi: 10.1016/j.str.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Quarato G, Guy CS, Grace CR, Llambi F, Nourse A, Rodriguez DA, Wakefield R, Frase S, Moldoveanu T, Green DR. Sequential engagement of distinct MLKL phosphatidylinositol-binding sites executes necroptosis. Mol Cell. 2016 doi: 10.1016/j.molcel.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gouaux E. Channel-forming toxins: tales of transformation. Curr Opin Struct Biol. 1997;7(4):566–573. doi: 10.1016/S0959-440X(97)80123-6. [DOI] [PubMed] [Google Scholar]
- 75.Lesieur C, Vecsey-Semjen B, Abrami L, Fivaz M, Gisou van der Goot F. Membrane insertion: the strategies of toxins (review) Mol Membr Biol. 1997;14(2):45–64. doi: 10.3109/09687689709068435. [DOI] [PubMed] [Google Scholar]
- 76.Mueller M, Grauschopf U, Maier T, Glockshuber R, Ban N. The structure of a cytolytic alpha-helical toxin pore reveals its assembly mechanism. Nature. 2009;459(7247):726–730. doi: 10.1038/nature08026. [DOI] [PubMed] [Google Scholar]
- 77.Gilbert RJ, Mikelj M, Dalla Serra M, Froelich CJ, Anderluh G. Effects of MACPF/CDC proteins on lipid membranes. Cell Mol Life Sci. 2013;70(12):2083–2098. doi: 10.1007/s00018-012-1153-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shatursky O, Heuck AP, Shepard LA, Rossjohn J, Parker MW, Johnson AE, Tweten RK. The mechanism of membrane insertion for a cholesterol-dependent cytolysin: a novel paradigm for pore-forming toxins. Cell. 1999;99(3):293–299. doi: 10.1016/S0092-8674(00)81660-8. [DOI] [PubMed] [Google Scholar]
- 79.Tweten RK, Hotze EM, Wade KR. The unique molecular choreography of giant pore formation by the cholesterol-dependent cytolysins of Gram-positive bacteria. Annu Rev Microbiol. 2015;69:323–340. doi: 10.1146/annurev-micro-091014-104233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I—molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262. doi: 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. 2015;6:257. doi: 10.3389/fimmu.2015.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aleshin AE, Schraufstatter IU, Stec B, Bankston LA, Liddington RC, DiScipio RG. Structure of complement C6 suggests a mechanism for initiation and unidirectional, sequential assembly of membrane attack complex (MAC) J Biol Chem. 2012;287(13):10210–10222. doi: 10.1074/jbc.M111.327809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aleshin AE, DiScipio RG, Stec B, Liddington RC. Crystal structure of C5b-6 suggests structural basis for priming assembly of the membrane attack complex. J Biol Chem. 2012;287(23):19642–19652. doi: 10.1074/jbc.M112.361121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Voskoboinik I, Dunstone MA, Baran K, Whisstock JC, Trapani JA. Perforin: structure, function, and role in human immunopathology. Immunol Rev. 2010;235(1):35–54. doi: 10.1111/j.0105-2896.2010.00896.x. [DOI] [PubMed] [Google Scholar]
- 85.Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. 2015 doi: 10.1038/nri3839. [DOI] [PubMed] [Google Scholar]
- 86.Tschopp J, Masson D, Stanley KK. Structural/functional similarity between proteins involved in complement- and cytotoxic T-lymphocyte-mediated cytolysis. Nature. 1986;322(6082):831–834. doi: 10.1038/322831a0. [DOI] [PubMed] [Google Scholar]
- 87.Law RH, Lukoyanova N, Voskoboinik I, Caradoc-Davies TT, Baran K, Dunstone MA, D’Angelo ME, Orlova EV, Coulibaly F, Verschoor S, Browne KA, Ciccone A, Kuiper MJ, Bird PI, Trapani JA, Saibil HR, Whisstock JC. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature. 2010;468(7322):447–451. doi: 10.1038/nature09518. [DOI] [PubMed] [Google Scholar]
- 88.Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. 2015;15(6):388–400. doi: 10.1038/nri3839. [DOI] [PubMed] [Google Scholar]
- 89.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 90.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 91.Yoon S, Bogdanov K, Kovalenko A, Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2015 doi: 10.1038/cdd.2015.92. [DOI] [PMC free article] [PubMed] [Google Scholar]