

Abstract
The critical role of the placenta in supporting a healthy pregnancy is mostly ensured by the extraembryonic trophoblast lineage that acts as the interface between the maternal and the foetal compartments. The diverse trophoblast cell subtypes that form the placenta originate from a single layer of stem cells that emerge from the embryo when the earliest cell fate decisions are occurring. Recent studies show that these trophoblast stem cells exhibit extensive plasticity as they are capable of differentiating down multiple pathways and are easily converted into embryonic stem cells in vitro. In this review, we discuss current knowledge of the mechanisms and control of the epigenesis of mouse trophoblast stem cells through a comparison with the corresponding mechanisms in pluripotent embryonic stem cells. To illustrate some of the more striking manifestations of the epigenetic plasticity of mouse trophoblast stem cells, we discuss them within the context of two paradigms of epigenetic regulation of gene expression: the imprinted gene expression of specific loci and the process of X-chromosome inactivation.
Keywords: Trophoblast stem cells, Placenta, Epigenetic plasticity, Multipotency, Mouse extraembryonic development
Introduction
Plasticity is defined as the ability of a cell to change fate in response to extrinsic factors. It notably describes the ability of neurons to dynamically modify the neural network in response to environmental stimuli (thereby allowing the brain to ensure functions such as learning and memory). The term “plasticity” is also used in the stem cell field to refer to the ability of a cell to commit to various lineage fates. Developmental plasticity is tightly linked to epigenetic mechanisms, since a permissive chromatin state allows flexible and dynamic reprogramming of the epigenome that underlies gene expression profile. Conrad Waddington’s epigenetic landscape represents the developmental process of cell fate decision, generating a wide diversity of cell types through a cascade of heritable changes in gene expression and cell phenotype. In this review, we discuss current knowledge of the “epigenesis” of trophoblast stem cells, in a broad sense, including both the cell potency and the permissiveness of the cell epigenome.
The zygote is the only unequivocally totipotent cell since it gives rise to the whole conceptus including extraembryonic tissues. Zygotic divisions are accompanied by a progressive restriction in cell potency and the appearance of distinct cell lineages. At the morula stage, a first dichotomy is created between outer, polarised cells and inner, apolar cells. External epithelial cells constitute the multipotent, extraembryonic, trophectoderm from which all trophoblast cell types originate [1, 2]. Internal cells, on the other hand, form the inner cell mass of the blastocyst, which then segregate into the inner epiblast and the overlying primitive endoderm. The latter is a multipotent, extraembryonic lineage that contributes to the yolk sac, while the epiblast is pluripotent since it gives rise to all adult tissues including the germ cells.
Trophoblast cells together with maternal and foetal cells form the complex structure of the placenta. Trophoblast cells exert a variety of functions during development. Initially, they ensure that the embryo implants into the uterus and promote the invasion of the maternal vasculature into the conceptus. In the mature placenta, specialised trophoblast cells enable the blood-mediated exchanges of nutrients and gas between the mother, the foetus and the disposal of wastes. Other trophoblast subtypes secrete hormones and cytokines into the maternal blood flow that direct the perpetual remodelling of blood vessels, optimise the nutrient exchanges to the benefit of the foetus and protect the foetus from rejection by the maternal immune system [3–5]. Consequently, although trophoblast cells do not contribute to the embryo per se, their role is essential for the normal development of the foetus and for a healthy pregnancy. In humans, defects in placental development are associated with multiple pregnancy complications, including pre-eclampsia, foetal growth restriction, pre-term birth and miscarriage [5]. A better understanding of trophoblast cell biology and trophoblast stem cell in particular is therefore necessary to gain insight into placentation and associated misregulations.
Studies of early developmental stages are hampered by the access to embryos and by the low number of cells. In the mouse, stem cell lines derived from each blastocyst lineages have been invaluable cellular models allowing for ex vivo characterisation of the corresponding lineage. In particular, mouse embryonic stem (ES) cells and mouse trophoblast stem (TS) cells are representative of the pluripotent epiblast and of the multipotent trophectoderm, respectively [6–8]. Both cell types show extensive self-renewal ability, can be differentiated ex vivo into lineage-specific specialised subtypes and, upon aggregation into chimaera embryos, contribute specifically to epiblast and trophoblast tissues, respectively. The remarkable plasticity of ES cells has raised much scientific interest given their potential therapeutic applications in regenerative medicine. Notably, human ES cells have been instrumental to investigate the determinants of pluripotency/plasticity and develop ex vivo models of oriented differentiation into various cell types [9]. In contrast, mouse TS cell plasticity has been much less thoroughly studied and no real human equivalent of mouse TS cells has yet been established. Please note that, for the purposes of this review we will use the term TS cells to refer to stem cell lines in culture while trophoblast stem cells or trophectoderm cells will be used to designate stem cells in vivo. We will compare the regulation of cell plasticity at work in the trophoblast lineage with the mechanisms controlling the plasticity of embryonic stem cells and we will focus on two paradigms of long-range epigenetic regulation of gene expression: the imprinted gene expression of specific loci and the process of X-chromosome inactivation, to illustrate some of the most striking manifestations of the epigenetic plasticity of trophoblast stem cells.
Plasticity of trophoblast stem cell fate
Multipotency of trophoblast stem cells
Specialised trophoblast cell types, while showing a remarkable diversity of functions, arise from a single layer of cells surrounding the blastocyst called the trophectoderm (TE). This multipotent, exclusively extraembryonic, stem compartment is subdivided into two regions. The mural TE lies in direct contact with the blastocoel cavity while the polar TE borders on the inner cell mass (ICM) (Fig. 1a). Soon after the beginning of implantation, cells of the mural TE differentiate into primary trophoblast giant cells (TGCs I). During this very specific differentiation process, although the cells stop dividing, DNA fibres keep replicating. These cycles of genomic DNA endo-replication eventually lead to polyploid, terminally differentiated and highly specialised cells. In contrast, cells of the polar TE continue to proliferate to give rise, early after implantation, to the extraembryonic ectoderm (ExE) of the egg-cylinder embryo [10] (Fig. 1a). ExE cells constitute the precursors of the various placental lineages. They further differentiate to form the ectoplacental cone (EPC) and, at later stages, the chorionic plate. The EPC generates the spongiotrophoblasts, which serve as a structural support of the placenta, the glycogen cells (of yet unknown function), which invade the decidua and, finally, the secondary TGCs (TGCs II), which lie at the interface between the decidua and the spongiotrophoblasts [5, 11]. The TGCs II are produced either directly by the EPC or via an intermediate progenitor population of spongiotrophoblastic cells. The chorionic plate attaches to the epiblast-derived allantois. Then this dual structure undergoes villous branching to form the innermost part of the placenta called labyrinth, the site where exchanges between mother and foetus take place [3, 12–14] (Fig. 1a).
Fig. 1.

Phenotypic plasticity of the mouse trophoblast lineage. a The development of the trophoblast lineage highlights the diversity of cell phenotypes among specialised subtypes. TE trophectoderm, Epi epiblast, PrE primitive endoderm, EPC ectoplacental cone, ExE extraembryonic ectoderm, TGC trophoblast giant cell, SpT spongiotrophoblast, VE visceral endoderm, PE parietal endoderm. b Progressive restriction of CDX2 and OCT4 expression, respectively, in outer and inner cells at the morula–blastocyst transition (left panel) involves a cascade of regulation mediated by the Hippo pathway, FGF signalling and OCT4/CDX2 mutual repression (right panel). c ES and TS cells derived from the blastocyst lineages and maintained in culture can be differentiated or transconverted into the other lineage
Because of this intricate development, TGCs have been subdivided into several subtypes that exert slightly different functions depending on their location in the mature placenta (Fig. 1a). The parietal TGCs include both TGCs I and II, and lie at the border of the decidua where they are responsible for the anchoring of the placenta into the maternal endometrium [3, 5]. In contrast, while the TGCs lining the maternal blood canals in the spongiotrophoblast zone and in the labyrinth and the TGCs lining the spiral arteries in the decidua can be distinguished from each other and from parietal TGCs based on their gene expression profiles, their exact functions remain unclear [15]. The sinusoidal TGCs lining the maternal sinusoids in the labyrinth together with two distinct layers of multinucleated syncytiotrophoblast cells form a trilaminar structure [15]. Within this structure, the TGCs and the spongiotrophoblasts produce the cytokines and the hormones that promote the remodelling of the maternal vasculature, optimise the transport of nutrients from the mother to the foetus and dampen the maternal immune system [3, 5].
Another striking example of trophoblast stem cell plasticity is their capacity to give rise to a variety of cell morphologies including mononuclear diploid cells (spongiotrophoblast, glycogen cells), multinucleated cells (syncytiotrophoblast) and mononuclear highly polyploid cells (TGCs) of variable size and DNA content [15]. Trophoblast stem cells are also able to switch, along the differentiation process, from polarised epithelial cells to invasive cells and are able to infiltrate into the uterine tissues. In the mouse, two major steps of trophoblast invasion have been identified: the early invasion of TGCs along maternal blood arteries, which initiates soon after the blastocyst implantation and the delayed invasion (from E14,5) of glycogen cells into the decidual stroma [11]. The acquisition of this migratory ability represents the first event of epithelial–mesenchymal transition (EMT) in development [16, 17]. EMT is a highly regulated key process during embryo morphogenesis, while, in adult tissues, the aberrant reactivation of the EMT program promotes the transformation of tumour cells into metastatic cells [18].
Multipotency constitutes the most crucial aspect of trophoblast stem cell plasticity. The specificity of each trophoblast subtype is progressively acquired during development and is often associated with drastic changes in cellular characteristics. Further investigations will be required to clearly define the roles of some trophoblast subtypes and it is likely that additional subtypes will be identified.
Regulatory mechanisms controlling the identity of trophoblast stem cells
The TE is the first cell lineage to segregate during mammalian pre-implantation development. At the morula stage, two successive waves of both symmetric and asymmetric cell divisions create a morphological distinction between external epithelial cells and internal apolar cells [1, 2]. These changes in embryo architecture are accompanied by a progressive restriction of the expression of the CDX2 (caudal-type homeobox 2) and of the OCT4 (octamer-binding protein 4, also known as POU5F1) transcription factors in the TE and in the ICM, respectively [19, 20] (Fig. 1b). CDX2-mediated activation of a positive feedback loop together with the induction of CDX2 target genes, stabilises Cdx2 expression in TE cells and mediates the establishment of the TE lineage [1, 2, 20–23]. The transcription factor TEAD4 (transcriptional enhancer activator domain 4) acts upstream of Cdx2 to regulate the specification of the TE. Although TEAD4 is expressed in all the blastomeres of the pre-implantation embryo, the subcellular localisation of TEAD4 co-activators, YAP (Yes-associated protein) and WWTR1 (WW domain containing transcriptional regulator 1), regulates TEAD4 activity in inner and outer cells [24]. In the inner cells, the Hippo pathway components LATS1/2 trigger the phosphorylation and subsequent cytoplasmic localisation of YAP and WWTR1. Conversely, in the outer cells, the Hippo pathway is inactive, which allows the nuclear accumulation of YAP and WWTR1 and the subsequent activation of Cdx2 and of other TE-specific genes by TEAD4 [25] (Fig. 1b).
How do the Hippo pathway and TE specification relate to cell polarity and the blastomere position in the embryo? During pre-implantation development CDX2 expression is detected after the first signs of cell polarisation [26]. In addition, after surgical removal of the TE, the most external cells of the isolated ICM re-polarise to generate, de novo, a Cdx2-expressing TE layer [25, 27]. Both observations concur to show that blastomere positioning/polarisation precedes lineage specification. This conclusion is supported by analyses of the regulation of the Hippo pathway in inner and outer cells of ES cell aggregates and in morula-like structures in which the blastomeres have been dissociated and randomly re-aggregated mechanically. In these studies, the cell position within the embryo-like structure and/or the polarisation of the cell appeared to regulate the Hippo pathway [25]. Refined experiments involving embryo fragmentation to dissociate polar from apolar cells further suggested that cell polarity and not the outer position of the cells within the embryo structure is the major determinant for Cdx2 induction [28]. Although it has been postulated that heterogeneous levels of Cdx2 expression detected from the eight-cell stage onwards may also influence the outer or inner positioning of cell progenies, cell positioning and polarisation directly promote the TE fate, thus highlighting the importance of environmental cues for the establishment of the TE lineage [26, 29].
In the embryo, undifferentiated trophoblasts are systematically localised near the embryonic compartment since both the polar TE and the ExE lie near the ICM and of the epiblast, respectively. This suggests that early cell-to-cell exchanges between the two lineages may regulate their specification. Accordingly, the transfer of TE vesicles deprived of ICM into recipient uteri mostly fails to drive the formation of a decidua while another striking example of trophoblast stem cell plasticity the insertion of an ectopic ICM into empty TE vesicles prior to uterine transfer rescues the implantation capacity [30]. In addition, sparse ExE cells grown in the absence of epiblast cells stop dividing and differentiate into TGCs. In contrast, enclosing trophoblastic explants inside embryonic pockets promotes a continued proliferation of undifferentiated diploid cells [31]. Cell contacts between undifferentiated trophoblasts also seem to inhibit—to some extent—spontaneous trophoblast differentiation in vitro since compact ExE or EPC explants exhibit a reduced number of TGCs compared to low-density cell cultures upon long-term culture [32]. These experiments show that cell communication amongst undifferentiated trophoblasts and, more significantly, between the extraembryonic layer and the embryonic compartment is critical to maintain the pool of proliferative trophoblast stem cells.
Several lines of evidence suggest that FGF signalling mediates the crosstalk between extraembryonic and embryonic compartments. First, from the mouse blastocyst stage onwards, the expression of FGF4 (fibroblast growth factor 4) is restricted to the embryonic lineage while FGFR2 (fibroblast growth factor receptor 2) expression appears to be specific to the TE lineage [33–35]. Second, homozygous mutations of either Fgf4 or Fgfr2 lead to embryonic lethality, associated with defects in ICM development [33, 36]. Third, exogenous treatment of ExE explants with FGF4 allows the maintenance of a proliferative and undifferentiated pool of trophoblast cells and is required for the derivation of TS cell lines [8, 19]. Taken together, these observations indicate that the FGF4–FGFR2 pathway participates, at least in part, in the bidirectional interaction between embryonic and trophoblastic compartments (for further details on the downstream cascades involved in this inter-relation see [37–41]) (Fig. 1b).
This crosstalk with the epiblast, critical for the maintenance of trophoblast stem cells in vivo, raises questions about the identity of TS cells in culture. TS cells are isolated and grown ex vivo on a feeder layer of primary mouse embryonic fibroblasts, in a medium containing, in particular, FGF4 and serum [8, 42]. Under these conditions, TS cells exhibit the morphological and transcriptional features of undifferentiated trophoblast cells in vivo and contribute specifically to the TE lineage in mouse chimaeras. Intriguingly, however, there is a decrease in spontaneous TGC differentiation and an increase in cell proliferation rate along TS cell passages, which suggest that TS cells undergo some changes upon derivation and/or prolonged culture (personal observations and [42]). Developing mixed cell culture systems containing TS and ES cells might be a way to mimic the physiological context and could provide an alternative model to identify new actors involved in the communication between the two lineages. Alternatively, a standardisation of culture conditions could limit the variability of TS cell culture quality that is linked to fluctuations of serum composition and of embryonic-fibroblast-conditioned-media [8, 42]. Along this line, Kubaczka et al. developed a simple, serum-free, chemically defined medium able to support derivation and prolonged culture of TS cells on feeder-cell-free Matrigel substrates [43]. TS cells cultured under these conditions contribute exclusively to TE derivatives in mouse chimaeras, thus showing the maintenance of the trophoblast stem cell identity. Using this new medium as a starting point, future research will help in defining which ingredients/factors are essential for TS cell growth and could provide new insights into the paracrine signals involved in the maintenance and self-renewal of trophoblast stem cells.
Lineage conversion from or towards the TS fate
Another marker of cellular plasticity is the ability of a cell type to de-differentiate, i.e. to climb back up the differentiation slope of the Waddington landscape to be reprogrammed towards a less specialised cell type within the same lineage. The most extreme example of de-differentiation is the resetting of an adult somatic cell into a totipotent cell through the transfer of an adult nucleus into an enucleated oocyte (somatic cell nuclear transfer or SCNT) [44]. Somatic cells can also be reprogrammed to a pluripotent state through fusion with ES cells and the formation of heterokaryons [45]. More recently, somatic cells have been directly reprogrammed into induced pluripotent stem (iPS) cells via forced expression of four pluripotency transcription factors [46]. A way to measure the extent of the plasticity of the trophoblast lineage would therefore be to determine if differentiated trophoblast subtypes can be reprogrammed into TS-like cells or if TS cells can be reprogrammed to become totipotent. To date, few experiments have addressed these questions. A single recent study reports that SCNT using TS cells as donor cells results in low cloning efficiency compared to SCNT using adult somatic cells suggesting that TS cells are probably less prone to de-differentiation than some adult cell types or than embryonic fibroblasts [47].
Cellular plasticity may also be assessed using the trans-differentiation ability of the cell type, i.e. the capacity to change lineage identity. The generation of heterokaryons between lymphocytes and TS cells induces the expression of TE markers and the loss of lymphocyte marker expression in the somatic nucleus [48]. TS cells thus show similarities with ES cells in their capacity to dominantly reprogram somatic nuclei [45]. More significantly, in two independent studies, the induction of the four pluripotency-reprogramming factors led to the conversion of TS cells into pluripotent cells, characterised by a loss of TE markers and the activation of endogenous pluripotency genes [49, 50] (Fig. 1c). These TS-derived iPS cells show a global gene expression profile similar to ES cells and to adult-derived iPS cells and they contribute to the soma and to the germ line in mouse chimaeras [49, 50]. The efficiency of the TS cell reprogramming into iPS is, however, debated due to contradictory observations. Wu et al. found that induction of Oct4 alone was sufficient to convert TS cells into iPS cells and that the reprogramming efficiencies were similar to those obtained with mouse embryonic fibroblasts [50]. In contrast, Kuckenberg et al. were unable to reprogram TS cells using Oct4 only and reported that mouse embryonic fibroblasts were more easily reprogrammed by the four pluripotency-reprogramming factors than TS cells [49]. The use of distinct TS cell lines may well account for these differences. Nevertheless, both studies clearly establish the capacity of TS cells to be converted into an ES-like state and therefore confirm their extensive plasticity. Interestingly, inducing the four pluripotency-reprogramming factors in TGCs failed to convert these cells into iPS cells thereby suggesting that either the reprogramming ability characterises only the undifferentiated TS state or that the high degree of ploidy of TGCs prevents efficient genome reprogramming [49].
The close relationship between ES and TS cell lineages is also exemplified by the ease of converting ES into TS-like cells (Fig. 1c). Merely transferring ES cells onto a collagen IV substrate in TS culture conditions is sufficient to trigger the formation of a fraction of TS-like cells [51]. Alternatively, ES cell reprogramming towards TS-like state is robustly achieved through the repression of Oct4, through an ectopic induction of TS determinants CDX2 or EOMES or by the ectopic expression of an active form of Ras, thereby pointing out the importance of the MAPK pathway for the establishment and/or the maintenance of the TE fate [22, 39, 52]. A recent, in-depth analysis of ES-derived TS-like cells, however, questioned the real identity of these cells [53]. Indeed although these cells acquired the morphology of TS cells, they express TE-specific genes at lower levels compared to bona fide TS cells, they show a reduced proliferation rate and a DNA methylation profiles intermediary between ES and TS cells. In addition, TS-like cells colonise the placenta of chimeric mice with a low efficiency. Collectively, these observations suggest that ES cells can be reprogrammed into TS-like cells so that they partially lose their identity and acquire an intermediate state similar, but not identical, to the induced fate. When injected in chimeric embryos, it is likely that the in vivo environment allows these partially reprogrammed cells to complete their reprogramming and to contribute to the developing tissues.
Epigenetic regulation of trophoblast stem cell plasticity
Characteristics of trophoblast stem cell chromatin
In ES cell, the chromatin appears dramatically decondensed, markedly enriched in active histone marks and exhibits a loose and dynamic binding of chromatin architectural proteins such as HP1 (heterochromatin protein 1), the linker histone H1 or core histones H2B and H3 [54, 55]. This atypical, very open, chromatin configuration is associated with widespread, low-level, transcriptional activities at both coding and noncoding regions of the genome [56]. Upon differentiation, the loss of pluripotency is accompanied by the formation of large condensed domains of heterochromatin, by the accumulation of repressive histone marks and by a tissue-specific definition of the transcriptional landscape [54, 56]. These observations led to the idea that the open chromatin architecture enables low-level, genome-wide, transcription and thereby contributes to the plasticity of pluripotent ES cells. Direct visualization of the chromatin structures through electron spectroscopic imaging further indicated that early epiblast cells share the open chromatin features of ES cells ex vivo while undifferentiated cells of the polar TE display significantly larger domains of compact chromatin [57]. Cells of the mural TE undergoing TGC differentiation, however, show a very specific condensation pattern characterised by a rim of compact chromatin along the nuclear envelop and very few dispersed fibres towards the centre of the nucleus while ExE cells exhibit large blocks of condensed chromatin (Fig. 2a). Notably, the ExE chromatin structure is hardly distinguishable from that of the committed epiblast cells of post-implantation embryos [57]. This widespread, decondensed, chromatin thus specifically defines pluripotent stem cells both in vivo and ex vivo while multipotent trophoblast progenitor cells show more compact chromatin structures and undergo additional, lineage-specific, levels of condensation upon differentiation.
Fig. 2.

Characteristics of the epigenome of mouse TS and ES cells. a Global chromatin architecture of the embryonic and of the trophoblast lineages in vivo. Pluripotent cells of the early epiblast are characterised by a widely dispersed chromatin whereas committed epiblast cells and cells of the extraembryonic ectoderm (ExE) display large domains of condensed chromatin. Cells of the polar trophectoderm (TE) exhibit an intermediate chromatin architecture, which is markedly distinct from the chromatin structure of committed cells of the mural TE. b Characteristics of bivalent promoters in ES and TS cells. Bivalent promoters in ES cells carry both the repressive H3K27me3 histone mark and the permissive H3K4me2/3 histone marks. Upon differentiation, these marks resolve into H3K27me3 at repressed trophoblast-specific gene promoters and into H3K4me2/3 at the promoter of genes specific of each differentiated lineage. In the trophoblast lineage an additional repressive histone mark, H3K9me2/3, comes into play. It participates in the initial repression of developmental genes at the TS cell state and, upon TS cell differentiation, in the repression of specific genes in each trophoblast subtype. c DNA methylation profiles in ES and in TS cells. Repetitive elements are hypomethylated in TS cells and hypermethylated in ES cells. The majority of gene promoters are undermethylated in ES cells specifically. However, differential methylation is observed at a number of lineage-specific genes
Chromatin states of specific cell types are also defined by the combination of histone marks found at the genome scale. Very few histone modifications have, however, been analysed genome-wide in TS cells. Among those that have been, the levels of H3K27me3 appear to be globally lower in TS cells and in the TE than in ES cells and in the ICM. This is probably linked to the low expression levels of EED, a component of the PRC2 (polycomb repressive complex 2) responsible for H3K27 trimethylation [58, 59]. In addition, ChIP analyses have revealed that the few genomic regions that are enriched in H3K27me3 in TS cells are only rarely localised near transcription start sites (TSS) [58, 60]. These observations suggest that, in TS/TE cells, the H3K27me3 repressive mark plays a role that is different to the direct modulation of gene expression observed in ES/ICM cells.
A specificity of ES cell chromatin structure that is considered as a biomarker of ES cell plasticity is the co-enrichment in antagonist histone modifications H3K27me3 and H3K4me2/3 at the promoter of developmental genes [48, 58, 61, 62]. Upon differentiation, these “bivalent domains” tend to resolve toward either H3K27me3 or H3K4me2/3 enrichment at repressed or induced genes, respectively. These particular chromatin signatures have been proposed to maintain silencing of developmental genes in pluripotent cells while keeping them poised for activation during embryogenesis. Importantly, these bivalent domains have also been observed in vivo in blastomeres of eight-cell embryos, in the morula and in the ICM/epiblast of the early/late blastocyst, which further supports the biological relevance of such combination of histone marks in pluripotent cells [58, 60, 61]. TS and TE cells also harbour H3K27me3–H3K4me2/3 bivalent promoters, although at a smaller number of genes compared to ES cells [48, 58, 60, 61]. Similarly, some promoters that are activated in specialised trophoblast cell types exhibit such bivalent histone marks in the ExE, indicating that these bivalent domains may also be used in trophoblast progenitors to prime genes that are differentially regulated upon placental development [58] (Fig. 2b).
Several differences in the chromatin composition are observed between in vivo tissues and TS cell cultures. First, some of the developmentally regulated promoters exhibit higher levels of H3K4me3 and lower levels of H3K27me3, in TS cells than in vivo [58]. This suggests that the technique of chromatin profiling of the whole genome that is only possible using large populations of cultured cells, may underestimate the occurrence of these bivalent domains in the trophoblast lineage in vivo. Second, as opposed to observations made in vivo, TS cells show bivalent marks at a subset of gene promoters that are not regulated during placental development. Furthermore, some of these promoters tend to retain the H3K4me2/3 mark upon differentiation into spongiotrophoblasts or into TGCs despite the lack of gene activation [61]. These observations question the functionality and, thereby, the biological significance of H3K27me3–H3K4me2/3 bivalent domains in the TS cell model (Fig. 2b).
More interestingly, changes in gene expression during TS cell differentiation also correlate with changes in the level of the repressive mark H3K9me3 at gene promoters. Among such promoters, several appear to be strongly co-enriched in H3K9me3 and H3K4me3 in undifferentiated TS cells as compared to ES cells [58]. Sequential ChIP assay on TS cells confirms that both modifications are present on the same alleles [58, 61]. Importantly, these H3K9me3/H3K4me2/3 trophoblast-specific bivalent domains have also been observed in vivo in the ExE and evolved into H3K9me3-enriched or into H3K4me3-enriched status upon EPC formation at repressed and induced genes, respectively [58]. H3K9me3 also accumulates during the differentiation process at promoters that are “inappropriately” co-enriched in H3K27me3 and in H3K4me2/3 suggesting that H3K9me3 ensures the definitive silencing of these genes in differentiated TS cells [49, 50, 61].
Other histone modifications may play specific roles in the regulation of trophoblast stem cell plasticity. Notably histone H4 lysine 20 trimethylation (H4K20me3), a mark usually associated with pericentric heterochromatin, appears enriched at many promoters in TS cells as compared to ES cells suggesting that gene repression is relatively stable in trophoblast cells [48]. How this repressive mark interacts with H3K9me3 and with H3K27me3 has yet to be analysed in detail.
In conclusion, H3K27me3–H3K4me2/3 bivalent domains, that sustain epigenetic plasticity in pluripotent cells, are rather rare in cells of the TE lineage. In addition, their efficiency at creating a poised state at ad hoc genes remains to be unambiguously established. In contrast, the repressive H3K9me3 modification appears to play several roles in the trophoblast lineage, not only in locking the silent state of “inappropriately” H3K27me3–H3K4me2/3 co-enriched TS promoters, but also in establishing bivalent domains through co-enrichment with H3K4me3 at developmentally regulated trophoblast-specific genes. Therefore, bivalent domains that combine trophoblast-specific repressive histone marks with the permissive H3K4me2/3 modification may be one of the driving forces underlying trophoblast stem cell plasticity.
Characteristics of trophoblast DNA methylation
The first analyses of DNA methylation status using Southern-blot to compare extraembryonic and epiblast derivatives revealed a significant undermethylation of repetitive elements and of some protein coding genes in extraembryonic tissues [63, 64]. This initial observation was later confirmed using immunostaining of 5-methylcytosines (5mC) at the blastocyst and egg-cylinder stages and by mass spectrometry quantification of 5mC in the trophoblast and in the epiblast of the early post-implantation embryos [65, 66]. In TS cell cultures, major satellite repeats remain hypomethylated compared to ES or to epiblast cells. However, the overall methylation levels are slightly increased compared to TE derivatives in vivo, indicating that the distribution of DNA methylation may be slightly modified by the TS cell derivation procedure [66]. MeDIP (methylated DNA immuno-precipitation) approaches, that allow for gene-by-gene definition of DNA methylation profiles, have revealed a lineage-specific hypermethylation of CpG islands and/or of promoters of the Oct4 and Nanog pluripotency genes and of their targets in TS cells as compared to ES cells (Fig. 2c). This suggests that DNA methylation plays a role in the repression of the pluripotency network in TS cells [53, 66–71]. In contrast, TE markers such as Cdx2, Eomes and Fgfr2 appear to be hypomethylated in both cell types. More recent MeDIP studies have however identified several loci that are specifically methylated in ES cells, including Elf5, Tead4 and Hand1, for which the role in trophoblast development is well established [53, 67, 72, 73] (Fig. 2c). Analyses of DNA methylation profiles in ExE and in epiblast cells of E6,5 embryos validated, in vivo, the differentially methylated regions identified during the TS vs ES cell comparisons [66, 70]. At earlier developmental time points, however, these loci appear hypomethylated in both the TE and the ICM, irrespective of the expression profile of the associated genes [70, 71]. This suggests that alternative repression mechanisms must be at work in the early blastocyst to ensure lineage-specific gene expression and that DNA methylation may act as a secondary lock to maintain lineage-specific gene silencing after implantation.
Collectively, these observations reveal a distinct usage of DNA methylation for the maintenance of trophoblast and epiblast cell identities whereby this repressive mark is specifically localised at gene regulatory sequences in TS or ExE cells while it is rather used to repress the activity of repeat sequences in ES or epiblast cells. Although the global hypomethylation of the TS/ExE genome may appear, at first glance, as a mark of loose epigenetic repression, the methylation of specific promoters and/or CpG islands underlines a tight regulation of key genes and, thereby, a tight control of trophoblast stem cell identity by DNA methylation marks (Fig. 2c).
Effect of lineage conversion on epigenetic profiles of trophoblast stem cells
Conversion of TS cells into pluripotent cells has been accomplished either via co-induction of pluripotency-reprogramming factors or via forced expression of Oct4 alone. In both cases, this reprogramming is accompanied by DNA methylation at the Elf5 promoter and by demethylation of the Nanog and Oct4 promoters, further supporting the critical role of differential DNA methylation of lineage-specific genes in the regulation of TS and ES cell identities [49, 50]. Reciprocally, TS-like cells generated by Cdx2 induction in ES cells become progressively methylated at the Nanog and Oct4 promoters during the conversion process [74]. The DNA methylation profile of reprogrammed cells represents, however, an intermediate state between ES and TS cell epigenotypes. Notably, the loci that are hypermethylated in ES cells, including Elf5 and Tead4, appear particularly refractory to demethylation during identity change. Thus, despite their high degree of plasticity, specific hypermethylation of a small number of gatekeeper genes limits the efficiency of reprogramming of ES into TS cells [53]. Similar TS-specific DNA methylation barriers preventing the full reprogramming of TS into ES cells are likely to be at work in the TE lineage.
Beyond changes in DNA methylation states, the Cdx2-induced conversion of ES cells into TS-like cells is accompanied by a loss of acetylated histone H3 at the Nanog and Oct4 promoters and by the recruitment of CDX2 and BRG1 (a component of the chromatin remodelling complex SWI/SNF) at the Oct4 promoter [71, 74]. This conversion is also characterised by a rapid decrease in EED expression, reaching the levels measured in endogenous TS cells [61]. In contrast, the expression of the H3K9 methyltransferase, SUV39H1, increases and the enzyme is recruited at ES-specific bivalent promoters that acquire H3K9 trimethylation [61]. Simultaneously, another H3K9 methyltransferase, ESET/SETDB1, unbinds from several TS-specific promoters including Cdx2, which become de-repressed [75]. Thus, distinct H3K9 methyltransferase are involved in the regulation of ES/TS identity switch. Suv39h1 preferentially acts on ES-specific bivalent promoters to trigger their H3K9 di- and/or trimethylation while Eset-dependent H3K9 di- and/or trimethylation is released from trophoblast-specific promoters to allow gene activation.
Taken together, these observations indicate that the plasticity of epigenetic control participates greatly in the modulation of gene expression that underlies the ES/TS identity switch. However, stable epigenetic silencing of certain loci may constitute a real roadblock that limits the completion of these lineage conversions ex vivo.
Effects of experimental perturbation of epigenetic marks on the trophoblast stem cell identity
Upon treatment with the DNA methylation inhibitor 5-aza-dC (5-aza-2-deoxycytidine), Oct4 expression becomes detectable in TS cells while Nanog expression levels remain low [68, 69]. In vivo, aberrant Oct4 expression is detected in the placenta of embryos that are deficient for the maintenance DNA-methyltransferase DNMT1 [69]. DNA methylation thus appears to play an essential role in the repression of Oct4 expression in trophoblast cells in vivo while Nanog silencing seems more resistant to demethylation in TS cells, probably because of the presence of additional repressive modifications such as dimethylated H3K27 and H3K9 at the Nanog promoter [68]. In contrast, when Dnmt1 −/− ES cells are grown under TS cell culture conditions, a significant fraction of the cells differentiate into TGCs, transiently inducing some TS cell markers and ultimately expressing genes specific to specialised trophoblast subtypes [73]. The significant contribution of these cells to the TE in chimeric blastocysts supports the hypothesis that Dnmt1 −/− TS-like cells go through a genuine trophoblast stem state during their spontaneous differentiation into TGCs [73]. Importantly, both TS and ES cells appear tolerant to a complete absence of DNA methylation since both Dnmt1/3a/3b deficient-TS and -ES cells that completely lack DNA-methyltransferase activity may be maintained in culture [76]. Upon LIF (leukaemia inhibitory factor) removal, however, triple KO ES cells differentiate into cells exhibiting a TS-like morphology [76]. Similarly, in chimaeras generated by aggregating triple KO ES cells with wild type blastomeres, methylation-deficient cells are mostly found in the placenta and rarely in the embryo proper [76]. These observations indicate that hypomethylation is more compatible with the trophoblast than with the epiblast fate, supporting the link between hypomethylation and trophoblast identity.
The involvement of histone acetylation in the dichotomy between ES and TS cells has been assessed using trichostatin A (TSA), a histone deacetylase inhibitor. TSA-treated TS cells appear to express Oct4 but not Nanog, despite the TS-specific hypoacetylation of histones H3 and H4 observed at the regulatory regions of both pluripotency genes [68, 69]. As observed when inhibiting DNA methylation, the plasticity of gene expression in TSA-treated TS cells varies among genes, with Nanog exhibiting a more robust silencing than Oct4, consistent with the presence of additional repressive histone marks at the Nanog promoter [68]. Extending the analysis to a larger number of genes, however, reveals that TSA-mediated increases in histone acetylation trigger the conversion of ES cells towards a TS-like fate while TSA-treated TS cells tend to differentiate into specialised trophoblast cell types [77]. Thus, increased global acetylation levels correlate with a reduction of the potency of the cells.
Similarly, knocking-down the ESET H3K9 methyltransferase in ES cells results in cell conversion into a TS-like phenotype—although no prolonged culture of TS-like cells could be obtained—while Eset-KD TS cells show enhanced spontaneous differentiation [75]. In addition, two-cell embryos injected with Eset shRNA give rise to morula showing increased expression of trophoblast markers associated with defects in ICM outgrowth. Later on during development, Eset depleted blastomeres contribute preferentially to the TE of chimeric embryos [75]. These observations are consistent with Eset being mostly involved in the silencing of TS-specific genes in ES cells (see above).
LSD1 (lysine specific demethylase 1 also known as KDM1A) catalyses the demethylation of the permissive histone mark H3K4me1/2 and/or of the repressive mark H3K9me1/2 and has been identified as an epigenetic regulator of the entry of TS cells into differentiation. Interestingly, the epiblast-restricted deletion of Lsd1 results in delayed embryonic defects and delayed lethality compared to the ubiquitous deletion showing that this demethylase plays a specific and early role in the extraembryonic compartment prior to its role in epiblast-derived tissues. Indeed, the Lsd1 depletion induces a TE-autonomous decrease in the number of Eomes-expressing cells in the ExE of mutant embryos [78]. Lsd1 depletion in cultured TS cells is accompanied by an enlargement of the cells and by an increase of cell migration and invasion, while induced Lsd1 depletion during TS cell differentiation favours the formation of syncytiotrophoblasts over spongiotrophoblasts or TGCs [78]. The histone demethylase Lsd1 is therefore an epigenetic actor in TS cell plasticity by regulating both the onset and the path of differentiation. Intriguingly, in Lsd1 mutant TS cells the early onset of differentiation mainly results from an increase of H3K4me1/2 at the Ovol2 promoter associated with an over-expression of the Ovol2 gene [78]. Ovol2 encodes a zinc-finger transcription factor of yet undetermined target genes.
De-repression of gene expression through a perturbation of dedicated epigenetic marks appears to induce the loss of TS/TE cell potency as exemplified by the spontaneous differentiation of trophoblast stem cells observed upon such perturbations. Intriguingly, instead of being the consequence of a global epigenetic deregulation, this effect seems to be mostly caused by a deregulation of a few specific genes. In ES cells, similar perturbations predominantly induce the conversion of ES cells towards a TS-like fate suggesting that one function of repressive marks in this cell type is to prevent the expression of TS-specific genes. In the near future, it will be important to address the effect of a loss of permissive histone marks on both stem cell types.
Plasticity of the regulation of imprinted loci and of X-chromosome inactivation in the trophoblast lineage
During pre-implantation development, the genome undergoes successive waves of epigenetic reprogramming. These steps are necessary to reset the epigenomes inherited from the gametes, to establish a totipotent state and, later on, for the cells to commit into distinct blastocyst lineages. Parent-of-origin-specific silencing of imprinted genes and the chromosome-wide silencing of one X-chromosome in females are paradigms of such developmentally regulated epigenetic phenomena.
Trophoblast-specific regulation of imprinted gene expression
Imprinted genes show a strict mono-allelic expression of either the paternal or the maternal allele. In mice, genomic imprinting concerns more than 100 genes [79–81]. These genes tend to cluster into large genomic regions within which gene expression is coordinated over several hundred kbps. Genomic imprinting employs highly specialised regulatory mechanisms to ensure large-scale, parent-of-origin-specific gene expression. First, the allele specificity is controlled by a cis-acting Imprint Control Region (ICR) that carries a germline DNA methylation imprint on one chromosome. Second, the methylation status of the ICR dictates, in most cases, the mono-allelic expression of lncRNAs, which coat in cis, one of the two parental alleles, to create a repressive nuclear domain through the recruitment of chromatin remodelers [82, 83] (Fig. 3a). In addition, these lncRNAs are generally transcribed in the antisense orientation and partially overlap at least one of the coding genes that they repress. Whether they function through interference with transcription due to their antisense orientation and/or if the transcript itself ensures regulatory functions remains to be elucidated. One possible mechanism is that antisense lncRNAs may serve as matrices producing microRNAs, which target the degradation of complementary protein coding transcripts [84].
Fig. 3.
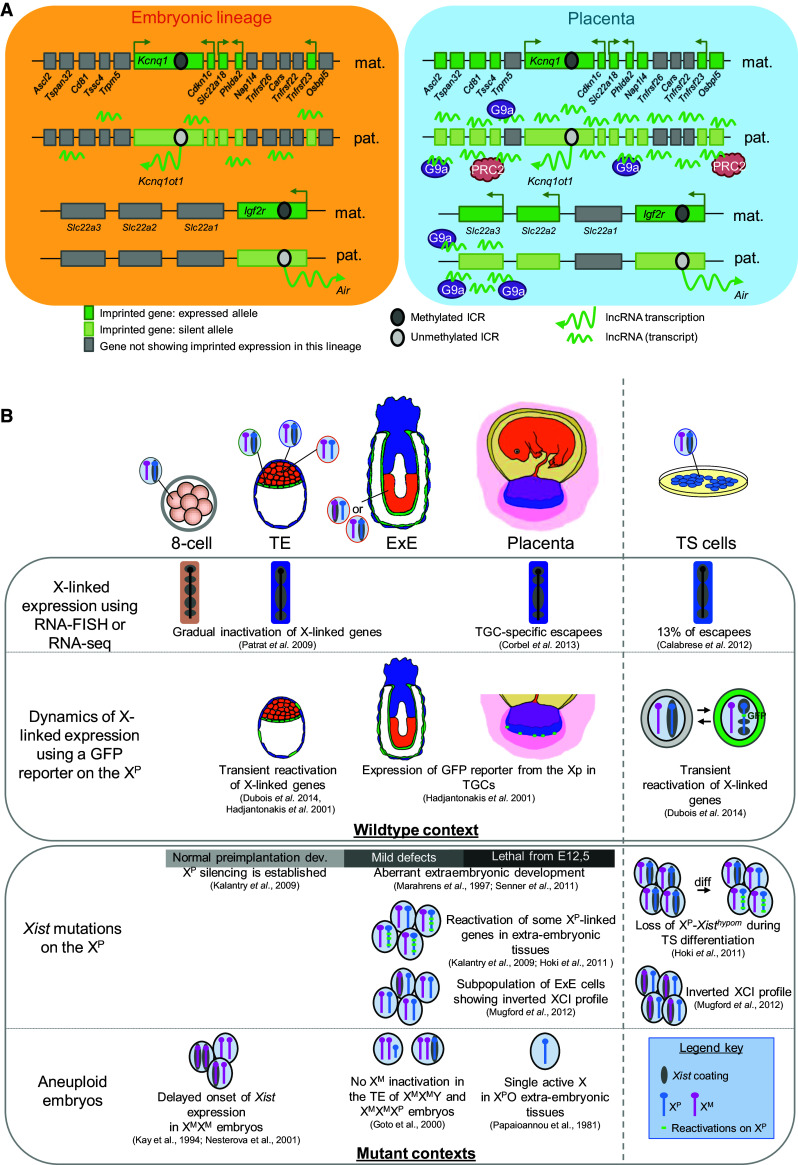
Trophoblast-specific features of imprinted gene expression and X-chromosome inactivation in the mouse. a Distinct regulations of the Kcnq1 and of the Igf2r imprinted gene clusters in the placenta compared to the embryonic lineages. At both clusters, differential methylations of the imprinted control regions (ICR) control the mono-allelic expression of a lncRNA which, in turn, regulates the allelic expression of protein coding genes. The association of the lcnRNAs Kncnq1ot1 and Air extends to additional placenta-specific imprinted genes of the Kcnq1 and of the Igf2r loci, respectively in the placenta compared to embryonic lineages. This association is accompanied by extraembryonic-specific recruitment of histone modifiers G9a and PRC2 (polycomb repressive complex 2). b Main features characterising the plasticity of X-chromosome inactivation in the mouse trophoblast lineage and in mouse TS cells. The upper panel summarises observations made in wildtype mouse embryos and the lower panel summarises observations made in various mutant contexts. References to the ad hoc studies are indicated. X P paternal X chromosome, X M maternal X-chromosome, TE trophectoderm, ExE extraembryonic ectoderm, TGC trophoblast giant cells, XCI X-chromosome inactivation
A major difference in the regulation of the various imprinted loci is apparent in the developmental time point at which imprinted expression initiates. For example, at the Kcnq1 locus, mono-allelic expression starts around the 2–4 cell stage whilst gene expression at the Igf2r locus remains bi-allelic until implantation [82, 83]. In addition, within most clusters, certain genes show parent-of-origin-specific expression only in given tissues while others are ubiquitously imprinted. Interestingly, this tissue-specific imprinting appears to occur preferentially in the extraembryonic compartment, especially in the placenta, and mostly concerns genes located at the extremities of the clusters [85–87] (Fig. 3a). These observations suggest that, although the parental imprint per se is present and maintained from the fertilised oocyte onwards, its activity is regulated both by developmental factors and by lineage-specific factors.
This specificity raises questions about the plasticity of imprinted gene regulation in the extraembryonic compartment: does the extension of imprinted expression to additional genes in the placenta reflect a looser regulation of gene expression or, on the contrary, does it reflect a specific control of gene dosage that is required for the proper functioning of extraembryonic lineages? Since extraembryonic derivatives are short-lived organs that are lost at the end of pregnancy, epigenetic regulations at play in these tissues may be subject to a reduced evolutionary pressure compared to embryonic tissues. Many imprinted genes, however, play crucial roles in the development of the foetus and, more particularly, of the placenta [86–88]. For example, mutations in imprinted genes such as Peg3, Igf2 or Peg1/Mest result in foetal growth retardation and/or in impaired growth of the placenta, associated with reduced postnatal survival rates [89–91]. This suggests that these genes need, on the other hand, to be tightly regulated during extraembryonic development. Both hypotheses may be reconciled if one considers that extraembryonic tissues take advantage of a relaxed control of the genomic range of silencing to modulate the dosage of genes at the extremity of the clusters. Pushing this hypothesis further, the organisation of the genes within each cluster may have been favoured throughout evolution—genes subject to strict, ubiquitous, mono-allelic expression being localised at the vicinity of the ICRs and those expressed in single dose, specifically in extraembryonic tissues being positioned at the border of the cluster.
The regulation of extraembryonic-specific imprinted genes seems to involve histone modifications rather than DNA methylation [81, 87, 92]. For example, at the Kcnq1 imprinted cluster, placenta-specific imprinted genes carry repressive histone marks on the silent allele and permissive histone marks on the active alleles, which are established concomitantly with the specification of the trophoblast lineage [93] (Fig. 3a). In the absence of DNA methylation, placenta-specific mono-allelic expression is maintained, indicating that DNA methylation is not required for the control of imprinted expression of these genes.
LncRNAs may also play a role in the regulation of the extraembryonic-specific regulation of imprinted clusters. The lncRNA Kcnq1ot1 acts as a repressor of gene expression at the Kcnq1 cluster, although it remains unclear how the RNA molecule exerts this repressive function [94]. Both the human KCNQ1OT1 and mouse Kcnq1ot1 transcripts have been shown to accumulate at their respective clusters [95, 96] (Fig. 3a). Furthermore, biochemical analyses indicated that the mouse Kcnq1ot1 RNA associates more strongly with the chromatin of the Kcnq1 region in the placenta than in foetal tissues [94]. Interestingly, this association is accompanied by a placenta-specific interaction with PRC2 and G9a which correlates with an enrichment in H3K27me3 and in H3K9me3 at Kcnq1 extraembryonic-specific imprinted genes in this organ [94]. The regulation of placenta-specific imprinted genes may also include a specific nuclear organisation since the Kcnq1 locus appears co-localised with the nucleolar compartment preferentially in this tissue compared to others [94]. Similarly, at the Igf2r cluster, the lncRNA Air and the H3K9 methyltransferase G9a are associated, in placental tissues, with the paternal Slc22a3 promoter that is repressed in these cells but not with the Igf2r promoter that shows an ubiquitous imprinted profile [87, 97] (Fig. 3a). The truncation of Air leads to reduced recruitment of G9a and to bi-allelic expression of Slc22a3 in the placenta, strongly suggesting that this lncRNA represses transcription through an accumulation of repressive histone marks specifically at genes imprinted in the extraembryonic compartment [97].
Our current knowledge of imprinted gene clusters thus suggests that lncRNAs regulate ubiquitous and placenta-specific imprinted genes in a distinct manner. In particular, lncRNAs seem to act through the recruitment of histone modifiers at the promoter of placenta-imprinted genes specifically in extraembryonic tissues. Few imprinted clusters have been extensively studied yet. Consequently, these examples might not reflect the full variety of mechanisms employed in the regulation of such imprinted regions. Further investigation will provide useful insight into the trophoblast-specific regulation of imprinting, and notably the specific roles of lncRNAs and histone marks in the regulation of genes that are specifically imprinted in the placenta.
Plasticity of X-chromosome inactivation during development
In mammals, the X-chromosome inactivation (XCI) process ensures the dosage compensation of X-linked genes between the sexes through the transcriptional silencing of one of the two Xs in females. This chromosome-wide silencing is achieved by the cis-coating of the targeted chromosome with the Xist lncRNA, followed by the exclusion of RNA polymerase II from this chromosome and through a series of epigenetic modifications of the X chromatin including the gain of repressive histone marks, the loss of permissive histone marks, the accumulation of the histone variant macroH2A and of the chromatin remodelling protein ATRX. Later on, the inactive state is locked by DNA methylation of CpGs at X-linked promoters, by a shift of replication timing to the late S-phase and by the association of the inactive X with the nuclear matrix (for review see [98, 99] and references therein).
This epigenetic reprogramming of the X-chromosome occurs during pre-implantation development and tightly correlates with the specification of the blastocyst lineages [100, 101]. At the four-cell stage in the mouse, a first wave of X-inactivation specifically targeting the paternal X-chromosome (XP) initiates. The parent-of-origin specificity of this XCI is thought to be controlled by a germinal mark, laid on the XM during oogenesis, which protects this chromosome from inactivation during early embryogenesis. At the blastocyst stage, this imprinted XCI is reversed in cells of the inner cell mass while the XP remains silent in the TE and in the primitive endoderm. Around implantation, a second wave of random XCI, characterised by the silencing of one of the two X-chromosomes irrespective of its parental origin is established in the epiblast [102–104]. The inactive state is then inherited during subsequent cell divisions throughout both extraembryonic and embryonic development. Thus, the placenta exhibits a homogeneous profile of XCI whereas the foetus and, later on, the adult, are mosaic for the expression of paternal and maternal X-linked alleles (Fig. 3b). Differences in the XCI processes at work in the epiblast and in the trophoblast lineages further evidence the epigenetic plasticity of trophoblast stem cells. These differences can be investigated ex vivo in ES and TS cells. ES cells carry two active Xs and undergo random XCI upon differentiation in vitro. In contrast, TS cells carry a silent XP and an active maternal X (XM) and allow the study of the changes in imprinted XCI characteristics that take place during the specification of the various trophoblast subtypes.
Trophoblast-specific features of XCI
While random XCI is maintained throughout adult life, imprinted XCI undergoes reversal shortly after being initiated implying an inherent plasticity of this process. Moreover, during pre-implantation development, XP-linked genes are not only asynchronously inactivated but some paternal alleles are still active in the TE at the blastocyst stage [104] (Fig. 3b). This incomplete silencing is associated with low-levels of methylation of CpG islands associated with promoter regions in trophoblast tissues compared to the late epiblast [66]. Consistent with this, Dnmt1 −/− mutant embryos display some reactivation of a reporter transgene on the XP in the embryonic but not in the extraembryonic compartment, showing that DNA methylation is not significantly implicated in the maintenance of imprinted XCI in vivo [105]. In female TS cells, however, X-linked CpG islands appear methylated at levels similar to cells showing random XCI suggesting that some CpG islands have gained methylation during the TS cell derivation procedure, which may affect the epigenetic status of the inactive XP, likely reinforcing the stability of imprinted XCI in cultured cells compared to the in vivo context [66].
The Xist lncRNA is necessary for imprinted XCI since the paternal transmission of a null mutation of the Xist gene induces the death of the embryos while, upon maternal transmission, the embryos develop normally. Surprisingly, however, embryos mutated on the paternal Xist allele show only mild defects at the egg-cylinder stage and lethality of mutant embryos does not occur before E12,5 [106, 107] (Fig. 3b). Monitoring the expression of X-linked genes in such Xist mutant embryos, Kalantry et al. showed that, while the silencing of the XP is properly established at the 8- and 16-cell stages, a XP-linked GFP transgene appears to be re-activated in a growing number of extraembryonic cells around implantation [108]. Similarly, Hoki et al., reported frequent events of reactivation of XP-linked genes in a subset of extraembryonic tissues carrying a paternal hypomorphic Xist mutation and in differentiated TS cells derived from the same genetic background [109] (Fig. 3b). Thus, Xist appears dispensable for the initiation of imprinted XCI but necessary for its maintenance in trophoblast tissues.
Other observations underlining the plasticity of imprinted XCI in the extraembryonic lineage have come from the study of aneuploid embryos (Fig. 3b). In XPO embryos, the single X-chromosome remains active in a subset of extraembryonic cells revealing a degree of flexibility of the imprinted XCI process [110]. XMXM parthenogenetic embryos show a delay in the establishment of XCI during pre-implantation development and heterogeneous populations of cells displaying 0, 1 or 2 Xist-coated chromosomes at the morula stage and in the TE of early blastocysts indicating that some cells, at these stages, are able to overcome the repressive maternal imprint to trigger XCI in the absence of a paternal contribution [111, 112]. Paradoxically, inactivation of a single X-chromosome and a lack of XCI have been reported to characterise the trophoblast lineage of disomic XMXMXP and XMXMY embryos, respectively [113] (Fig. 3b). Moreover, in these mutant embryos, the persistence of two active X-chromosomes in extraembryonic tissues has been associated with placental defects [111, 113–117]. The discrepancies in XCI patterns between gynogenetic and disomic embryos suggest that the mechanisms of imprinted XCI change during development and/or that the genetic background (and, notably, the presence of a supernumerary sex chromosome) may modify the ability of embryos to cope with two XM.
Plasticity of the inactive X in trophoblast stem cells and their derivatives
Although a proper dosage compensation of X-linked gene expression is necessary for female extraembryonic development to occur normally, placental progenitor cells are markedly enriched in genes escaping from X-inactivation as compared to epiblast-derived cells [118]. RNA-seq analyses of X-linked gene expression in female TS cells have revealed that some 13 % of X-linked genes are significantly expressed from the inactive chromosome, a percentage dramatically higher than the proportion of genes escaping from random XCI in various adult tissues [119–121] (Fig. 3b). Escapees identified in TS cells and verified in vivo have all been found to escape from imprinted XCI in the extraembryonic ectoderm and in the ectoplacental cone, suggesting that the escape is not merely acquired during derivation and/or culture of TS cells [120]. Similar analyses led on reciprocal mouse crosses intriguingly suggested that the probability to be expressed from the inactive X depends on the genetic background of the XP [120]. Additionally, few genes found to be efficiently inactivated in TS cells by RNA-seq were shown to escape from imprinted XCI using RNA-FISH, an approach detecting the transcriptional activity of the loci as opposed to RNA-seq, which measures, under the conditions used, the levels of mature X-linked transcripts [118, 120]. Collectively these results suggest that a degree of relaxation of XP silencing may be tolerated at some specific X-linked loci in the TE lineage. This relaxation is highly variable from mouse strain to mouse strain and/or amongst TS cell lines and may be regulated at the transcriptional and/or post-transcriptional levels.
This flexibility of silencing of specific X-linked genes is further supported by pioneering observations of spontaneous reactivations of GFP or LacZ transgenes located on the XP as well as of reactivations of several endogenous XP-linked genes in TGCs [12, 122–125] (Fig. 3b). This instability of imprinted XCI compared to random XCI also characterises undifferentiated TS and TE cells since transient reactivations of several X-linked genes occur in a significant fraction of cells both ex vivo and in vivo [126] (Fig. 3b). In addition, few trophoblast cells show a complete switch of X-inactivation profiles in embryos carrying a paternal mutation of Xist [127] (Fig. 3b). Most strikingly, using a heterozygous mutation in the X-linked Hprt1 gene as a reporter for XCI relaxation events, we could isolate TS cells showing a complete reversal of XCI profiles. This reversal is mediated by a loss of Xist coating and of H3K27me3 accumulation on the XP leading to its global reactivation (Prudhomme et al., to be published). The resulting two-active-X state appears highly unstable since either cells rapidly evolve towards a re-establishment of XP inactivation or, more rarely, towards a switch of XCI profile. Interestingly, this secondary inactivation is established homogeneously, within clonal populations of cells indicating that the two-active-X state in TS cells is epigenetically distinct from that characterising ES cells (Prudhomme et al., to be published). These studies in living cells reveal the dynamics of XP silencing and suggest that genes expressed from the XP undergo cycles of ON and OFF states. Indeed, these observations suggest that the entire process of XCI may be reversed and fully re-established de novo in undifferentiated TS cells.
Compared to a randomly inactivated X chromosome in somatic tissues, the silencing of the XP in the trophoblast lineage appears highly unstable. Although this relaxation of silencing is especially pronounced in specialised trophoblast subtypes, studies in trophoblast stem cells ex vivo and in vivo revealed an accrued and highly dynamic plasticity of XCI that may predispose the trophoblast lineage for the reactivation events observed at later developmental time points. Importantly, the X chromosome is enriched in genes involved in placental development and functions [128]. One may therefore speculate that the epigenetic metastable states characterising the plasticity of X-inactivation in the trophoblast lineage have been selected during evolution as a labile regulatory mechanism able to adjust the level of X-linked placental gene expression in response to environmental pressures and/or to compensate for deleterious mutations that may affect the XM.
Conclusions and perspectives
Multipotent trophoblast stem cells clearly exhibit a remarkable phenotypic plasticity characterised by a broad spectrum of differentiation potentials, including atypical cell morphologies such as highly polyploid and multinucleated cells, and by the ability to be experimentally converted into pluripotent cells. Looking at the epigenome, repetitive elements appear extensively hypomethylated in placental precursors cells as compared to pluripotent epiblast cells. In contrast, lineage-specific genes are predominantly repressed by DNA methylation in trophoblast stem cells. Furthermore, accumulating evidences suggests that bivalent chromatin domains combining repressive and permissive histone modifications, usually considered as a hallmark of pluripotency, also exist and are functional in trophoblast stem cells but rely on a distinct combination of histone marks. The acquisition of pluripotency has recently been correlated with the expression of a specific set of lncRNAs (long non-coding RNA) that may act at the interface between chromatin regulation and gene expression patterning [129, 130]. Along the same idea it will be interesting to address whether a specific lncRNA landscape also characterises trophoblast stem cells. The role of lncRNAs in the regulation of epigenetic plasticity of trophoblast stem cells is most apparent in the control of epigenetic processes such as the regulation of imprinted genes or of X-chromosome inactivation. The molecular mechanisms at play in these two phenomena are correlated with a looser control of the boundaries of the domains subject to mono-allelic expression in the trophoblast lineage. This leads to an expansion of mono-allelic silencing to additional genes at parentally imprinted gene cluster, to spontaneous and local relaxations of X-linked silencing or, more dramatically, to a reversal of XCI profiles.
The placental lineage constitutes a relatively recent evolutionary innovation that, however, shows striking differences across modern Eutherians. The anatomy of the mature placenta exhibits tremendous interspecies diversification and proteins secreted by this organ are poorly conserved between species [13, 131, 132]. Intriguingly, global hypomethylation of repetitive elements in the trophoblast lineage leads to elevated transcriptional activity of endogenous retrovirus sequences (ERVs) [11, 63]. Some of these ERVs have been co-opted in some specialised trophoblasts to serve specific placental functions and Long Terminal Repeats (LTRs), the regulatory elements flanking ERVs, may be used as enhancers of placenta-specific genes (for review see [16, 133, 134]). Therefore, ERVs may represent a predominant driving force underlying the rapid evolution of placental functions. The recent observation that a significant fraction of trophoblast core enhancers in mouse TS cells is derived from a mouse-specific ERV family further supports this hypothesis [135]. Thus, hypomethylation of repetitive elements in the trophoblast lineage may have been selected during evolution since elevated ERV activity facilitates the rapid species-specific adaptation of the placenta. Consequently, this rapid evolution may have contributed to the diversification of the reproductive strategies (gestation length, litter sizes) across placental Mammals. The epigenetic plasticity of the trophoblast lineage may have facilitated the lineage-specific activity of co-opted ERV sequences and, probably, more generally, other changes in gene expression thereby participating in a crucial manner to the evolutionary history of the placenta.
Acknowledgments
We apologise to the authors who have contributed to related studies or aspects of trophoblast stem cell plasticity that could not be addressed here due to the format restrictions of the review. We thank Dr. Graham Hayhurst for critical reading of the manuscript. J.P. was supported by a doctoral fellowship from the Région Ile-de-France (DIM-StemPôle), and by a grant from the REVIVE Labex. CM is supported on a permanent basis by the French National Institute for Scientific and Medical Research (INSERM).
References
- 1.Rossant J, Tam PP. Blastocyst lineage formation, early embryonic asymmetries and axis patterning in the mouse. Development. 2009;136(5):701–713. doi: 10.1242/dev.017178. [DOI] [PubMed] [Google Scholar]
- 2.Saiz N, Plusa B. Early cell fate decisions in the mouse embryo. Reproduction. 2013;145(3):R65–R80. doi: 10.1530/REP-12-0381. [DOI] [PubMed] [Google Scholar]
- 3.Cross JC, et al. Trophoblast functions, angiogenesis and remodeling of the maternal vasculature in the placenta. Mol Cell Endocrinol. 2002;187(1–2):207–212. doi: 10.1016/S0303-7207(01)00703-1. [DOI] [PubMed] [Google Scholar]
- 4.Georgiades P, Ferguson-Smith AC, Burton GJ. Comparative developmental anatomy of the murine and human definitive placentae. Placenta. 2002;23(1):3–19. doi: 10.1053/plac.2001.0738. [DOI] [PubMed] [Google Scholar]
- 5.John R, Hemberger M. A placenta for life. Reprod Biomed Online. 2012;25(1):5–11. doi: 10.1016/j.rbmo.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 6.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292(5819):154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 7.Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci USA. 1981;78(12):7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka S, et al. Promotion of trophoblast stem cell proliferation by FGF4. Science. 1998;282(5396):2072–2075. doi: 10.1126/science.282.5396.2072. [DOI] [PubMed] [Google Scholar]
- 9.Efthymiou AG, et al. Self-renewal and cell lineage differentiation strategies in human embryonic stem cells and induced pluripotent stem cells. Expert Opin Biol Ther. 2014;14(9):1333–1344. doi: 10.1517/14712598.2014.922533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Copp AJ. Interaction between inner cell mass and trophectoderm of the mouse blastocyst. II. The fate of the polar trophectoderm. J Embryol Exp Morphol. 1979;51:109–120. [PubMed] [Google Scholar]
- 11.Hemberger M, Hughes M, Cross JC. Trophoblast stem cells differentiate in vitro into invasive trophoblast giant cells. Dev Biol. 2004;271(2):362–371. doi: 10.1016/j.ydbio.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 12.Hemberger M, et al. Genetic and developmental analysis of X-inactivation in interspecific hybrid mice suggests a role for the Y chromosome in placental dysplasia. Genetics. 2001;157(1):341–348. doi: 10.1093/genetics/157.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maltepe E, Bakardjiev AI, Fisher SJ. The placenta: transcriptional, epigenetic, and physiological integration during development. J Clin Invest. 2010;120(4):1016–1025. doi: 10.1172/JCI41211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rugg-Gunn PJ, et al. Cell-surface proteomics identifies lineage-specific markers of embryo-derived stem cells. Dev Cell. 2012;22(4):887–901. doi: 10.1016/j.devcel.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simmons DG, Fortier AL, Cross JC. Diverse subtypes and developmental origins of trophoblast giant cells in the mouse placenta. Dev Biol. 2007;304(2):567–578. doi: 10.1016/j.ydbio.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 16.Logan PC, Mitchell MD, Lobie PE. DNA methyltransferases and TETs in the regulation of differentiation and invasiveness of extra-villous trophoblasts. Front Genet. 2013;4:265. doi: 10.3389/fgene.2013.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perry JK, et al. Regulation of invasive growth: similar epigenetic mechanisms underpin tumour progression and implantation in human pregnancy. Clin Sci. 2010;118:451–457. doi: 10.1042/CS20090503. [DOI] [PubMed] [Google Scholar]
- 18.Richard G, Puisieux A, Caramel J. Antagonistic functions of EMT-inducers in melanoma development: implications for cancer cell plasticity. Cancer Cell Microenviron. 2014;1:e61. [Google Scholar]
- 19.Nichols J, et al. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95(3):379–391. doi: 10.1016/S0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- 20.Strumpf D, et al. Cdx2 is required for correct cell fate specification and differentiation of trophectoderm in the mouse blastocyst. Development. 2005;132(9):2093–2102. doi: 10.1242/dev.01801. [DOI] [PubMed] [Google Scholar]
- 21.Albert M, Peters AH. Genetic and epigenetic control of early mouse development. Curr Opin Genet Dev. 2009;19(2):113–121. doi: 10.1016/j.gde.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 22.Niwa H, et al. Interaction between Oct3/4 and Cdx2 determines trophectoderm differentiation. Cell. 2005;123(5):917–929. doi: 10.1016/j.cell.2005.08.040. [DOI] [PubMed] [Google Scholar]
- 23.Zernicka-Goetz M, Morris SA, Bruce AW. Making a firm decision: multifaceted regulation of cell fate in the early mouse embryo. Nat Rev Genet. 2009;10(7):467–477. doi: 10.1038/nrg2564. [DOI] [PubMed] [Google Scholar]
- 24.Nishioka N, et al. Tead4 is required for specification of trophectoderm in pre-implantation mouse embryos. Mech Dev. 2008;125(3–4):270–283. doi: 10.1016/j.mod.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 25.Nishioka N, et al. The Hippo signaling pathway components Lats and Yap pattern Tead4 activity to distinguish mouse trophectoderm from inner cell mass. Dev Cell. 2009;16(3):398–410. doi: 10.1016/j.devcel.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 26.Ralston A, Rossant J. Cdx2 acts downstream of cell polarization to cell-autonomously promote trophectoderm fate in the early mouse embryo. Dev Biol. 2008;313(2):614–629. doi: 10.1016/j.ydbio.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 27.Stephenson RO, Yamanaka Y, Rossant J. Disorganized epithelial polarity and excess trophectoderm cell fate in preimplantation embryos lacking E-cadherin. Development. 2010;137(20):3383–3391. doi: 10.1242/dev.050195. [DOI] [PubMed] [Google Scholar]
- 28.Kondratiuk I, et al. Delay of polarization event increases the number of Cdx2-positive blastomeres in mouse embryo. Dev Biol. 2012;368(1):54–62. doi: 10.1016/j.ydbio.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 29.Jedrusik A, et al. Role of Cdx2 and cell polarity in cell allocation and specification of trophectoderm and inner cell mass in the mouse embryo. Genes Dev. 2008;22(19):2692–2706. doi: 10.1101/gad.486108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gardner RL, Papaioannou VE, Barton SC. Origin of the ectoplacental cone and secondary giant cells in mouse blastocysts reconstituted from isolated trophoblast and inner cell mass. J Embryol Exp Morphol. 1973;30(3):561–572. [PubMed] [Google Scholar]
- 31.Rossant J, Tamura-Lis W. Effect of culture conditions on diploid to giant-cell transformation in postimplantation mouse trophoblast. J Embryol Exp Morphol. 1981;62:217–227. [PubMed] [Google Scholar]
- 32.Ilgren EB. On the control of the trophoblastic giant-cell transformation in the mouse: homotypic cellular interactions and polyploidy. J Embryol Exp Morphol. 1981;62:183–202. [PubMed] [Google Scholar]
- 33.Arman E, et al. Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc Natl Acad Sci USA. 1998;95(9):5082–5087. doi: 10.1073/pnas.95.9.5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haffner-Krausz R, et al. Expression of Fgfr2 in the early mouse embryo indicates its involvement in preimplantation development. Mech Dev. 1999;85(1–2):167–172. doi: 10.1016/S0925-4773(99)00082-9. [DOI] [PubMed] [Google Scholar]
- 35.Niswander L, Martin GR. Fgf-4 expression during gastrulation, myogenesis, limb and tooth development in the mouse. Development. 1992;114(3):755–768. doi: 10.1242/dev.114.3.755. [DOI] [PubMed] [Google Scholar]
- 36.Feldman B, et al. Requirement of FGF-4 for postimplantation mouse development. Science. 1995;267(5195):246–249. doi: 10.1126/science.7809630. [DOI] [PubMed] [Google Scholar]
- 37.Bedzhov I, et al. Developmental plasticity, cell fate specification and morphogenesis in the early mouse embryo. Phil Trans R Soc London B Biol Sci. 2014;369:20130538. doi: 10.1098/rstb.2013.0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guzman-Ayala M, et al. Nodal protein processing and fibroblast growth factor 4 synergize to maintain a trophoblast stem cell microenvironment. Proc Natl Acad Sci USA. 2004;101(44):15656–15660. doi: 10.1073/pnas.0405429101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu CW, et al. Ras-MAPK signaling promotes trophectoderm formation from embryonic stem cells and mouse embryos . Nat Genet. 2008;40(7):921–926. doi: 10.1038/ng.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rossant J. Stem cells from the mammalian blastocyst. Stem Cells. 2001;19(6):477–482. doi: 10.1634/stemcells.19-6-477. [DOI] [PubMed] [Google Scholar]
- 41.Saba-El-Leil MK, et al. An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep. 2003;4(10):964–968. doi: 10.1038/sj.embor.embor939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Himeno E, Tanaka S, Kunath T. Isolation and manipulation of mouse trophoblast stem cells. Curr Protoc Stem Cell Biol. 2008;Chapter 1:Unit 1E.4. doi: 10.1002/9780470151808.sc01e04s7. [DOI] [PubMed] [Google Scholar]
- 43.Kubaczka C, et al. Derivation and maintenance of murine trophoblast stem cells under defined conditions. Stem Cell Rep. 2014;2(2):232–242. doi: 10.1016/j.stemcr.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krueger WH, et al. Natural and artificial routes to pluripotency. Int J Dev Biol. 2010;54(11–12):1545–1564. doi: 10.1387/ijdb.103199wk. [DOI] [PubMed] [Google Scholar]
- 45.Soza-Ried J, Fisher AG. Reprogramming somatic cells towards pluripotency by cellular fusion. Curr Opin Genet Dev. 2012;22(5):459–465. doi: 10.1016/j.gde.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 46.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 47.Ogawa H, et al. Deficiency of genomic reprogramming in trophoblast stem cells following nuclear transfer. Cell Reprogram. 2015;17(2):115–123. doi: 10.1089/cell.2014.0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santos J, et al. Differences in the epigenetic and reprogramming properties of pluripotent and extra-embryonic stem cells implicate chromatin remodelling as an important early event in the developing mouse embryo. Epigenetics Chromatin. 2010;3:1. doi: 10.1186/1756-8935-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuckenberg P, et al. Lineage conversion of murine extraembryonic trophoblast stem cells to pluripotent stem cells. Mol Cell Biol. 2011;31(8):1748–1756. doi: 10.1128/MCB.01047-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu T, et al. Reprogramming of trophoblast stem cells into pluripotent stem cells by Oct4. Stem Cells. 2011;29(5):755–763. doi: 10.1002/stem.617. [DOI] [PubMed] [Google Scholar]
- 51.Schenke-Layland K, et al. Collagen IV induces trophoectoderm differentiation of mouse embryonic stem cells. Stem Cells. 2007;25(6):1529–1538. doi: 10.1634/stemcells.2006-0729. [DOI] [PubMed] [Google Scholar]
- 52.Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000;24(4):372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- 53.Cambuli F, et al. Epigenetic memory of the first cell fate decision prevents complete ES cell reprogramming into trophoblast. Nat Commun. 2014;5:5538. doi: 10.1038/ncomms6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meshorer E, Misteli T. Chromatin in pluripotent embryonic stem cells and differentiation. Nat Rev Mol Cell Biol. 2006;7(7):540–546. doi: 10.1038/nrm1938. [DOI] [PubMed] [Google Scholar]
- 55.Meshorer E, et al. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev Cell. 2006;10(1):105–116. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Efroni S, et al. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell. 2008;2(5):437–447. doi: 10.1016/j.stem.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ahmed K, et al. Global chromatin architecture reflects pluripotency and lineage commitment in the early mouse embryo. PLoS One. 2010;5(5):e10531. doi: 10.1371/journal.pone.0010531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rugg-Gunn PJ, et al. Distinct histone modifications in stem cell lines and tissue lineages from the early mouse embryo. Proc Natl Acad Sci USA. 2010;107(24):10783–10790. doi: 10.1073/pnas.0914507107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saha B, et al. EED and KDM6B coordinate the first mammalian cell lineage commitment to ensure embryo implantation. Mol Cell Biol. 2013;33(14):2691–2705. doi: 10.1128/MCB.00069-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dahl JA, et al. Histone H3 lysine 27 methylation asymmetry on developmentally-regulated promoters distinguish the first two lineages in mouse preimplantation embryos. PLoS One. 2010;5(2):e9150. doi: 10.1371/journal.pone.0009150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alder O, et al. Ring1B and Suv39h1 delineate distinct chromatin states at bivalent genes during early mouse lineage commitment. Development. 2010;137(15):2483–2492. doi: 10.1242/dev.048363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 63.Chapman V, et al. Cell lineage-specific undermethylation of mouse repetitive DNA. Nature. 1984;307(5948):284–286. doi: 10.1038/307284a0. [DOI] [PubMed] [Google Scholar]
- 64.Rossant J, et al. Undermethylation of structural gene sequences in extraembryonic lineages of the mouse. Dev Biol. 1986;117(2):567–573. doi: 10.1016/0012-1606(86)90325-8. [DOI] [PubMed] [Google Scholar]
- 65.Santos F, et al. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol. 2002;241(1):172–182. doi: 10.1006/dbio.2001.0501. [DOI] [PubMed] [Google Scholar]
- 66.Senner CE, et al. DNA methylation profiles define stem cell identity and reveal a tight embryonic-extraembryonic lineage boundary. Stem Cells. 2012;30(12):2732–2745. doi: 10.1002/stem.1249. [DOI] [PubMed] [Google Scholar]
- 67.Farthing CR, et al. Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS Genet. 2008;4(6):e1000116. doi: 10.1371/journal.pgen.1000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hattori N, et al. Epigenetic regulation of Nanog gene in embryonic stem and trophoblast stem cells. Genes Cells. 2007;12(3):387–396. doi: 10.1111/j.1365-2443.2007.01058.x. [DOI] [PubMed] [Google Scholar]
- 69.Hattori N, et al. Epigenetic control of mouse Oct-4 gene expression in embryonic stem cells and trophoblast stem cells. J Biol Chem. 2004;279(17):17063–17069. doi: 10.1074/jbc.M309002200. [DOI] [PubMed] [Google Scholar]
- 70.Nakanishi MO, et al. Trophoblast-specific DNA methylation occurs after the segregation of the trophectoderm and inner cell mass in the mouse periimplantation embryo. Epigenetics. 2012;7(2):173–182. doi: 10.4161/epi.7.2.18962. [DOI] [PubMed] [Google Scholar]
- 71.Wang K, et al. Brg1 is required for Cdx2-mediated repression of Oct4 expression in mouse blastocysts. PLoS One. 2010;5(5):e10622. doi: 10.1371/journal.pone.0010622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Donnison M, et al. Loss of the extraembryonic ectoderm in Elf5 mutants leads to defects in embryonic patterning. Development. 2005;132(10):2299–2308. doi: 10.1242/dev.01819. [DOI] [PubMed] [Google Scholar]
- 73.Ng RK, et al. Epigenetic restriction of embryonic cell lineage fate by methylation of Elf5. Nat Cell Biol. 2008;10(11):1280–1290. doi: 10.1038/ncb1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carey TS, et al. Transcriptional reprogramming and chromatin remodeling accompanies Oct4 and Nanog silencing in mouse trophoblast lineage. Stem Cells Dev. 2014;23(3):219–229. doi: 10.1089/scd.2013.0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yuan P, et al. Eset partners with Oct4 to restrict extraembryonic trophoblast lineage potential in embryonic stem cells. Genes Dev. 2009;23(21):2507–2520. doi: 10.1101/gad.1831909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sakaue M, et al. DNA methylation is dispensable for the growth and survival of the extraembryonic lineages. Curr Biol. 2010;20(16):1452–1457. doi: 10.1016/j.cub.2010.06.050. [DOI] [PubMed] [Google Scholar]
- 77.Kidder BL, Palmer S. HDAC1 regulates pluripotency and lineage specific transcriptional networks in embryonic and trophoblast stem cells. Nucleic Acids Res. 2012;40(7):2925–2939. doi: 10.1093/nar/gkr1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu D, et al. Lysine-specific demethylase 1 regulates differentiation onset and migration of trophoblast stem cells. Nat Commun. 2014;5:3174. doi: 10.1038/ncomms4174. [DOI] [PubMed] [Google Scholar]
- 79.Barlow DP. Genomic imprinting: a mammalian epigenetic discovery model. Annu Rev Genet. 2011;45:379–403. doi: 10.1146/annurev-genet-110410-132459. [DOI] [PubMed] [Google Scholar]
- 80.Bartolomei MS1, Ferguson-Smith AC. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol. 2011;3(7):a002592. doi: 10.1101/cshperspect.a002592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Duffie R, Bourc’his D. Parental epigenetic asymmetry in mammals. Curr Top Dev Biol. 2013;104:293–328. doi: 10.1016/B978-0-12-416027-9.00009-7. [DOI] [PubMed] [Google Scholar]
- 82.Mohammad F, Mondal T, Kanduri C. Epigenetics of imprinted long noncoding RNAs. Epigenetics. 2009;4(5):277–286. doi: 10.4161/epi.4.5.9242. [DOI] [PubMed] [Google Scholar]
- 83.Santoro F, Barlow DP. Developmental control of imprinted expression by macro non-coding RNAs. Semin Cell Dev Biol. 2011;22(4):328–335. doi: 10.1016/j.semcdb.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 84.Ito M, et al. A trans-homologue interaction between reciprocally imprinted miR-127 and Rtl1 regulates placenta development. Development. 2015;142(14):2425–2430. doi: 10.1242/dev.121996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hanna JH, Saha K, Jaenisch R. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell. 2010;143(4):508–525. doi: 10.1016/j.cell.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bressan FF, et al. Unearthing the roles of imprinted genes in the placenta. Placenta. 2009;30(10):823–834. doi: 10.1016/j.placenta.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 87.Hudson QJ, et al. Genomic imprinting mechanisms in embryonic and extraembryonic mouse tissues. Heredity (Edinb) 2010;105(1):45–56. doi: 10.1038/hdy.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Radford EJ, Ferron SR, Ferguson-Smith AC. Genomic imprinting as an adaptative model of developmental plasticity. FEBS Lett. 2011;585(13):2059–2066. doi: 10.1016/j.febslet.2011.05.063. [DOI] [PubMed] [Google Scholar]
- 89.Li L, et al. Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science. 1999;284(5412):330–333. doi: 10.1126/science.284.5412.330. [DOI] [PubMed] [Google Scholar]
- 90.Constancia M, et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature. 2002;417(6892):945–948. doi: 10.1038/nature00819. [DOI] [PubMed] [Google Scholar]
- 91.Lefebvre L, et al. Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat Genet. 1998;20(2):163–169. doi: 10.1038/2464. [DOI] [PubMed] [Google Scholar]
- 92.Weaver JR, Susiarjo M, Bartolomei MS. Imprinting and epigenetic changes in the early embryo. Mamm Genome. 2009;20(9–10):532–543. doi: 10.1007/s00335-009-9225-2. [DOI] [PubMed] [Google Scholar]
- 93.Lewis A, et al. Epigenetic dynamics of the Kcnq1 imprinted domain in the early embryo. Development. 2006;133(21):4203–4210. doi: 10.1242/dev.02612. [DOI] [PubMed] [Google Scholar]
- 94.Pandey RR, et al. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. 2008;32(2):232–246. doi: 10.1016/j.molcel.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 95.Murakami K, Oshimura M, Kugoh H. Suggestive evidence for chromosomal localization of non-coding RNA from imprinted LIT1. J Hum Genet. 2007;52(11):926–933. doi: 10.1007/s10038-007-0196-4. [DOI] [PubMed] [Google Scholar]
- 96.Terranova R, et al. Polycomb group proteins Ezh2 and Rnf2 direct genomic contraction and imprinted repression in early mouse embryos. Dev Cell. 2008;15(5):668–679. doi: 10.1016/j.devcel.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 97.Nagano T, et al. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science. 2008;322(5908):1717–1720. doi: 10.1126/science.1163802. [DOI] [PubMed] [Google Scholar]
- 98.Gendrel AV, Heard E. Noncoding RNAs and epigenetic mechanisms during X-chromosome inactivation. Annu Rev Cell Dev Biol. 2014;30:561–580. doi: 10.1146/annurev-cellbio-101512-122415. [DOI] [PubMed] [Google Scholar]
- 99.Morey C, Avner P. The demoiselle of X-inactivation: 50 years old and as trendy and mesmerising as ever. PLoS Genet. 2011;7(7):e1002212. doi: 10.1371/journal.pgen.1002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Barakat TS, Gribnau J. X chromosome inactivation in the cycle of life. Development. 2012;139(12):2085–2089. doi: 10.1242/dev.069328. [DOI] [PubMed] [Google Scholar]
- 101.Deuve JL, Avner P. The coupling of X-chromosome inactivation to pluripotency. Annu Rev Cell Dev Biol. 2011;27:611–629. doi: 10.1146/annurev-cellbio-092910-154020. [DOI] [PubMed] [Google Scholar]
- 102.Mak W, et al. Reactivation of the paternal X chromosome in early mouse embryos. Science. 2004;303(5658):666–669. doi: 10.1126/science.1092674. [DOI] [PubMed] [Google Scholar]
- 103.Okamoto I, et al. Epigenetic dynamics of imprinted X inactivation during early mouse development. Science. 2004;303(5658):644–649. doi: 10.1126/science.1092727. [DOI] [PubMed] [Google Scholar]
- 104.Patrat C, et al. Dynamic changes in paternal X-chromosome activity during imprinted X-chromosome inactivation in mice. Proc Natl Acad Sci USA. 2009;106(13):5198–5203. doi: 10.1073/pnas.0810683106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sado T, et al. X inactivation in the mouse embryo deficient for Dnmt1: distinct effect of hypomethylation on imprinted and random X inactivation. Dev Biol. 2000;225(2):294–303. doi: 10.1006/dbio.2000.9823. [DOI] [PubMed] [Google Scholar]
- 106.Marahrens Y, et al. Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev. 1997;11(2):156–166. doi: 10.1101/gad.11.2.156. [DOI] [PubMed] [Google Scholar]
- 107.Senner CE, et al. Disruption of a conserved region of Xist exon 1 impairs Xist RNA localisation and X-linked gene silencing during random and imprinted X chromosome inactivation. Development. 2011;138(8):1541–1550. doi: 10.1242/dev.056812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kalantry S, et al. Evidence of Xist RNA-independent initiation of mouse imprinted X-chromosome inactivation. Nature. 2009;460(7255):647–651. doi: 10.1038/nature08161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hoki Y, et al. Incomplete X-inactivation initiated by a hypomorphic Xist allele in the mouse. Development. 2011;138(13):2649–2659. doi: 10.1242/dev.061226. [DOI] [PubMed] [Google Scholar]
- 110.Papaioannou VE, West JD. Relationship between the parental origin of the X chromosomes, embryonic cell lineage and X chromosome expression in mice. Genet Res. 1981;37(2):183–197. doi: 10.1017/S0016672300020152. [DOI] [PubMed] [Google Scholar]
- 111.Kay GF, et al. Imprinting and X chromosome counting mechanisms determine Xist expression in early mouse development. Cell. 1994;77(5):639–650. doi: 10.1016/0092-8674(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 112.Nesterova TB, et al. Loss of Xist imprinting in diploid parthenogenetic preimplantation embryos. Dev Biol. 2001;235(2):343–350. doi: 10.1006/dbio.2001.0295. [DOI] [PubMed] [Google Scholar]
- 113.Goto Y, Takagi N. Maternally inherited X chromosome is not inactivated in mouse blastocysts due to parental imprinting. Chromosome Res. 2000;8(2):101–109. doi: 10.1023/A:1009234217981. [DOI] [PubMed] [Google Scholar]
- 114.Goto Y, Takagi N. Tetraploid embryos rescue embryonic lethality caused by an additional maternally inherited X chromosome in the mouse. Development. 1998;125(17):3353–3363. doi: 10.1242/dev.125.17.3353. [DOI] [PubMed] [Google Scholar]
- 115.Okamoto I, Tan S, Takagi N. X-chromosome inactivation in XX androgenetic mouse embryos surviving implantation. Development. 2000;127(19):4137–4145. doi: 10.1242/dev.127.19.4137. [DOI] [PubMed] [Google Scholar]
- 116.Shao C, Takagi N. An extra maternally derived X chromosome is deleterious to early mouse development. Development. 1990;110(3):969–975. doi: 10.1242/dev.110.3.969. [DOI] [PubMed] [Google Scholar]
- 117.Tada T, Takagi N, Adler ID. Parental imprinting on the mouse X chromosome: effects on the early development of X0, XXY and XXX embryos. Genet Res. 1993;62(2):139–148. doi: 10.1017/S0016672300031736. [DOI] [PubMed] [Google Scholar]
- 118.Merzouk S, et al. Lineage-specific regulation of imprinted X inactivation in extraembryonic endoderm stem cells. Epigenetics Chromatin. 2014;7:11. doi: 10.1186/1756-8935-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Berletch JB, et al. Escape from X inactivation varies in mouse tissues. PLoS Genet. 2015;11(3):e1005079. doi: 10.1371/journal.pgen.1005079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Calabrese JM, et al. Site-specific silencing of regulatory elements as a mechanism of X inactivation. Cell. 2012;151(5):951–963. doi: 10.1016/j.cell.2012.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yang F, et al. Global survey of escape from X inactivation by RNA-sequencing in mouse. Genome Res. 2010;20(5):614–622. doi: 10.1101/gr.103200.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Corbel C, et al. Unusual chromatin status and organization of the inactive X chromosome in murine trophoblast giant cells. Development. 2013;140(4):861–872. doi: 10.1242/dev.087429. [DOI] [PubMed] [Google Scholar]
- 123.Hadjantonakis AK, et al. An X-linked GFP transgene reveals unexpected paternal X-chromosome activity in trophoblastic giant cells of the mouse placenta. Genesis. 2001;29(3):133–140. doi: 10.1002/gene.1016. [DOI] [PubMed] [Google Scholar]
- 124.Tam PP, Williams EA, Tan SS. Expression of an X-linked HMG-lacZ transgene in mouse embryos: implication of chromosomal imprinting and lineage-specific X-chromosome activity. Dev Genet. 1994;15(6):491–503. doi: 10.1002/dvg.1020150608. [DOI] [PubMed] [Google Scholar]
- 125.Tan SS, Williams EA, Tam PP. X-chromosome inactivation occurs at different times in different tissues of the post-implantation mouse embryo. Nat Genet. 1993;3(2):170–174. doi: 10.1038/ng0293-170. [DOI] [PubMed] [Google Scholar]
- 126.Dubois A, et al. Spontaneous reactivation of clusters of X-linked genes is associated with the plasticity of X-inactivation in mouse trophoblast stem cells. Stem Cells. 2014;32(2):377–390. doi: 10.1002/stem.1557. [DOI] [PubMed] [Google Scholar]
- 127.Mugford JW, Yee D, Magnuson T. Failure of extra-embryonic progenitor maintenance in the absence of dosage compensation. Development. 2012;139(12):2130–2138. doi: 10.1242/dev.076497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hemberger M. The role of the X chromosome in mammalian extra embryonic development. Cytogenet Genome Res. 2002;99(1–4):210–217. doi: 10.1159/000071595. [DOI] [PubMed] [Google Scholar]
- 129.Guttman M, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477(7364):295–300. doi: 10.1038/nature10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Flynn RA, Chang HY. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell. 2014;14(6):752–761. doi: 10.1016/j.stem.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Furukawa S, Kuroda Y, Sugiyama A. A comparison of the histological structure of the placenta in experimental animals. J Toxicol Pathol. 2014;27(1):11–18. doi: 10.1293/tox.2013-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Knox K, Baker JC. Genomic evolution of the placenta using co-option and duplication and divergence. Genome Res. 2008;18(5):695–705. doi: 10.1101/gr.071407.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chuong EB. Retroviruses facilitate the rapid evolution of the mammalian placenta. BioEssays. 2013;35(10):853–861. doi: 10.1002/bies.201300059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hemberger M. Genetic-epigenetic intersection in trophoblast differentiation: implications for extraembryonic tissue function. Epigenetics. 2010;5(1):24–29. doi: 10.4161/epi.5.1.10589. [DOI] [PubMed] [Google Scholar]
- 135.Chuong EB, et al. Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat Genet. 2013;45(3):325–329. doi: 10.1038/ng.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]