

Abstract
Skeletal muscles of patients with Duchenne muscular dystrophy (DMD) show numerous alterations including inflammation, apoptosis, and necrosis of myofibers. However, the molecular mechanism that explains these changes remains largely unknown. Here, the involvement of hemichannels formed by connexins (Cx HCs) was evaluated in skeletal muscle of mdx mouse model of DMD. Fast myofibers of mdx mice were found to express three connexins (39, 43 and 45) and high sarcolemma permeability, which was absent in myofibers of mdx Cx43fl/flCx45fl/fl:Myo-Cre mice (deficient in skeletal muscle Cx43/Cx45 expression). These myofibers did not show elevated basal intracellular free Ca2+ levels, immunoreactivity to phosphorylated p65 (active NF-κB), eNOS and annexin V/active Caspase 3 (marker of apoptosis) but presented dystrophin immunoreactivity. Moreover, muscles of mdx Cx43fl/flCx45fl/fl:Myo-Cre mice exhibited partial decrease of necrotic features (big cells and high creatine kinase levels). Accordingly, these muscles showed similar macrophage infiltration as control mdx muscles. Nonetheless, the hanging test performance of mdx Cx43fl/flCx45fl/fl:Myo-Cre mice was significantly better than that of control mdx Cx43fl/flCx45fl/fl mice. All three Cxs found in skeletal muscles of mdx mice were also detected in fast myofibers of biopsy specimens from patients with muscular dystrophy. Thus, reduction of Cx expression and/or function of Cx HCs may be potential therapeutic approaches to abrogate myofiber apoptosis in DMD.
Keywords: Connexons, Evans blue uptake, P2X7 receptors, Pannexin1, NF-κB, Cell death
Introduction
Duchenne muscular dystrophy (DMD, MIM#30037), the most frequent human muscular genetic disease, affects 1 of 3500 males born [1]. DMD is caused by mutation in dystrophin (427 kDa; Xp21.2-p21.1), which links the cytoskeleton to the molecular aggregate termed dystroglycan complex [2]. This complex is present on the surface of the sarcolemma where it interacts with the extracellular matrix. The condition is currently being detected at an early age but survival has not changed significantly since there are no effective treatments for this disease [3].
Since the mdx mouse shares several characteristics with the human muscular dystrophy illness, it is frequently used to further knowledge on this disease [4]. In these animals, the absence of dystrophin in myofibers is associated with an early increase in intracellular free Ca2+ [5] and Na+ [6] levels. In addition, membrane permeability to dyes such as procion orange (FW 631 g mol−1) and Evans blue (EB4−, FW 960.82 g mol−1) also increases and has been interpreted as skeletal muscle damage in vivo [7, 8]. Nonetheless, this finding might need reinterpretation since the sarcolemma of denervated myofibers shows similar changes including permeability to EB4− and has been shown to result from permeabilization of the sarcolemma by connexin (Cx) hemichannels [9].
Hemichannels are non-selective cationic channels permeable to ions (e.g. Ca2+ and Na+) and small compounds, including signaling molecules such as ATP and NAD+ and dyes including ethidium (Etd+) and EB4− [9, 10]. Ionic gadolinium (Gd3+), a connexin hemichannel (Cx HC) blocker [11], has been used to prevent muscular damage in mdx mice [6] and to block stretch-activated sarcolemma channels reducing Ca2+ influx in mdx mice myofibers [12]. However, the molecular identity of these channels remains unknown and a putative identity could be Cx HCs since they are mechano-sensitive [13], permeable to Ca2+ [14, 15] and blocked by lanthanides including Gd3+ and La3+ [9]. Moreover, TRPV2 channels have been proposed to be a relevant membrane pathway through which Ca2+ influx increases and consequently intracellular free Ca2+ levels increases in myofibers of mdx mice [16]. Accordingly, a dominant-negative TRPV2 channel in skeletal myofibers renders the sarcolemma of dystrophic myofibers less permeable to Ca2+ and improves muscle performance [16]. Muscles of mdx mice also show upregulation of pannexin1 (Panx1) channels [17] and purinergic ionotropic P2X7 receptors (P2X7Rs) [18, 19]. The possible relevance of at least of Panx1 channels in the muscle pathogenesis has been ruled out [19]. However, it remains unknown whether skeletal muscles of mdx mice express Cx HCs, of which at least Cx43 HCs have been shown to be permeable to Na+ and Ca2+ [15, 20].
In the present study, we observed the presence of Cxs 39, 43 and 45 in dystrophic mouse mdx skeletal muscle. These proteins were found to form functional hemichannels in myofibers of mdx mice, which are absent in normal fibers. We also observed that P2X7Rs and Panx1 channels participate in controlling the sarcolemma permeability of dystrophic myofibers and apoptosis was not detected in myofibers deficient in Cx43/Cx45 expression. However, the latter still show significant necrotic cell death. Since Cxs 40.1, 43 and 45 were also detected in biopsy specimen of DMD patients but not in those of health individuals, it is suggested that de novo expression of functional Cx HCs plays a similar role in the pathogenesis of DMD.
Results
Skeletal myofibers of mdx mice express connexins 39, 43 and 45 and show enhanced hemichannel activity controlled by P2X7Rs
Since sarcolemma permeability to Ca2+ of mdx myofibers is known to be high [12], we decided to evaluate the presence and distribution of proteins known to form non-selective membrane channels. To this end, we studied the presence of Cxs 39, 43 and 45, Panx1 and protein subunits P2X7R and the transient receptor potential type V2 (TRPV2) channel [21–23]. All these proteins, with the exception of Panx1, are not expressed by myofibers after innervation and throughout adulthood [24, 25]. For that we evaluated the levels of each protein aforementioned through western blot analysis in tibialis anterior (TA) muscle homogenates from control (C57) and mdx mice, detecting a significant increase in levels of all protein evaluated in mdx muscle, which did not occur in mdx muscles deficient in Cx43/Cx45 expression (mdx M-C) (Fig. 1). However, levels of Cx43 and Cx45 in control and mdx M-C muscles were comparable probably due to the contamination with other cell types present in tissue homogenates, including vascular and satellite cells that express pannexin1 and connexins [26, 27]. The absence of Cx43 and Cx45 did not affect the increase in Cx39 levels and only partially prevented the increase in P2X7Rs, but levels of Cx43, Cx45, Panx1 and TRPV2 channels were as in control muscles (Fig. 1). Then, the presence of these proteins specifically in myofibers was evaluated by immunofluorescence analysis of TA muscle cross sections. Accordingly, myofibers of TA muscles of control mice only presented Panx1 reactivity (Fig. 2). However, the sarcolemma of mdx myofibers showed high reactivity for all three Cxs studied as well as for P2X7R and TRPV2 channel (Fig. 2). The reactivity of Panx1 in mdx muscles was notably higher than in muscles of control animals (Fig. 2). Despite the lack of Cx43/Cx45 expression TA muscles of mdx Cx43fl/flCx45fl/fl:Myo-Cre mice still presented reactivity for Cx39, Panx1 and P2X7R (Fig. 2). However, it was evident that these muscles were devoid of TRPV2 reactivity (Fig. 2). Throughout this paper, we use the designation C57 as an abbreviation for the control mouse strain C57Bl/6 that harbored the corresponding Cx but not the mdx mutation.
Fig. 1.

Levels of Cx39, Cx43, Cx45, Panx1, P2X7R are increased in skeletal muscles of mdx mice. Tibialis anterior (TA) muscles were extracted from control (C57), mdx and mdx with myofibers deficient in Cx43 and Cx45 expression mice (mdx M-C). Then, muscle tissue was homogenized and processed for western blot analysis. The relative levels of Cx39, Cx43, Cx45, Panx1, P2X7R and TRPV2 channels were analyzed. Relative β-Tubulin levels were measured as sample loading control. a Representative blot. b Quantification of three independent experiments as shown in a
Fig. 2.

The presence and distribution of Cx39, Cx43, Cx45, Panx1, P2X7R and TRPV2 channel in tibialis anterior muscle from C57 and mdx mice. Tibialis anterior (TA) muscles were extracted from control (Cx43fl/flCx45fl/fl) and mdx (Cx43fl/flCx45fl/fl) mice, and cross sections (20 μm) were obtained. Nuclei were stained with DAPI (blue) and antigen–antibody complexes were detected with anti-IgGs conjugated to Cy3 (red). Then, the presence and distribution of Cx39, Cx43, Cx45, Panx1, P2X7R and TRPV2 were analyzed by immunofluorescence. In control (C57) sections only Panx1 showed a positive signal mostly surrounded the myofibers, while in mdx muscles all proteins evaluated were positive and mostly in the sarcolemma (upper panel). Cx39, Panx1, P2X7R and TRPV2 reactivity was evaluated in cross sections of TA from mdx mice deficient in Cx43 and Cx45 (Cx43fl/flCx45fl/fl:Myo-Cre) (bottom panel). Scale bar 50 μm (4 mice and 6 sections analyzed of each animal)
Skeletal myofibers from mdx mice show enhanced membrane permeability to Evans blue that is inhibited by hemichannel and P2XR blockers
Since skeletal myofibers of mdx mice show Evans blue (EB4−) uptake in vivo [7, 8] and the sarcolemma of denervated myofibers is permeable to EB4− through a pathway inhibited by blockers of poorly selective membrane channels [9], we explored whether carbenoxolone (CBX), which is a hemichannel and P2X7R inhibitor [28] and brilliant blue G (BBG) inhibitor of Panx1 channels and P2X7R [28] would affect EB4− uptake in mdx myofibers in vivo. To this end, control and mdx mice were treated with 80 mg/kg CBX or 45 mg/kg BBG 20 min before EB4− injection. Intense intracellular EB4− staining was found in cross sections of TA muscles obtained from mdx mice (Fig. 3). However, muscle samples of control mice or mdx mice treated with CBX or BBG did not show intracellular EB4− staining (Fig. 3). The possible role of membrane damage was then tested in mdx mice injected with Rhodamine Dextran (RD) (10 kDa), which is a molecular probe with a size above the exclusion limit of non-selective channels permeable to small molecules (between 1 and 2 kDa). RD staining was detected only in the interstitial spaces of TA muscles of these animals (Fig. 3), thus indicating the absence of cell membrane damage.
Fig. 3.

Skeletal myofibers from mdx mice show enhanced Evans blue staining inhibited by HC and P2XR blockers. Control (Ctrl) C57, Ctrl Cx43fl/fl Cx45fl/fl, Ctrl Cx43fl/fl Cx45fl/fl:Myo-Cre (M-C), mdx, mdx Cx43fl/flCx45fl/fl and mdx Cx43fl/fl Cx45fl/fl:Myo-Cre (M-C) mice were injected with Evans blue (65 mg/kg EB4−), and only mice that can express Cxs (Ctrl and mdx) were evaluated in the absence or presence of carbenoxolone (CBX, 80 mg/kg), which is a connexin hemichannel and pannexin1 channel inhibitor, brilliant blue G (BBG, 45 mg/kg), a P2X7R and Panx1 channels inhibitor or Rhodamine Dextran (RD). They were euthanized after 6 h. Then, tibialis anterior muscles were dissected and cross sections were obtained, mounted and excited at λ = 530 nm, and fluorescent emission was photographed at λ = 595 nm (20 μm) (n = 4). Scale bar 50 μm (4 mice and 6 sections were analyzed from each animal)
To further test if Cx HCs mediated the EB4− uptake observed in TA muscles of mdx mice, we repeated the assay in mdx Cx43fl/flCx45fl/fl:Myo-Cre (myofibers deficient in Cx43/Cx45 expression) and mdx Cx43fl/flCx45fl/fl mice (control mice). EB4− fluorescence was clearly seen in numerous myofibers in cross sections of TA muscles of mdx Cx43fl/flCx45fl/fl mice (Fig. 3). However, intracellular EB4− staining was absent in all myofibers of TA muscles of mdx Cx43fl/flCx45fl/fl:Myo-Cre mice (Fig. 3), which is consistent with the notion that EB4− crosses the sarcolemma through Cx HCs in myofibers of mdx animals.
The sarcolemma of isolated myofibers presents functional hemichannels
Since the above results suggest the involvement of Cx HCs increasing the sarcolemma permeability of mdx myofibers, we decided to further test the presence of functional hemichannels in freshly isolated myofibers. Ethidium (Etd+) and different channel blockers were used as described previously for denervated or inflamed myofibers [9, 24].
While C57 myofibers presented only Panx1 reactivity, mdx myofibers were positive for Cx39, Cx43 and Cx45 in addition to Panx1 (Fig. 4a). In agreement with the content of HC forming proteins, myofibers of mdx mice presented higher Etd+ uptake (~2.3-fold) than myofibers of control animals (Fig. 4b, d). In both cases, the Etd+ uptake rate was inhibited by 200 µM La3+ (~70 %) (Fig. 4d), which is a blocker of Cx HCs and P2XRs [10, 29]. The Etd+ uptake was also drastically reduced by 10Panx1 peptide, suggesting the involvement of Panx1 channels (Fig. 4d).
Fig. 4.

Myofibers of mdx mice show enhanced Panx1 channels and Cx hemichannels activity controlled by P2X7R. Freshly isolated myofibers from flexor digitorum brevis muscles were used for immunodetection or dye uptake assays. a Distribution of Cx39, Cx43, Cx45, Panx1 and P2X7 (green) was analyzed by confocal microscopy in freshly isolated skeletal muscle fibers from control and mdx mice. Only Panx1 was present in control myofibers (upper panels, white dash line defines the myofiber border) but all proteins appeared in mdx myofibers (bottom panel). b Representative curve of ethidium (Etd+) uptake of isolated myofibers from the same cultures as in a. Each value represents the fluorescence intensity over time of five myofibers (mean ± SD) under basal conditions. c Representative curve of Etd+ uptake in basal conditions as in b, followed by the application of 100 µM oxidized ATP (oATP), a P2XR inhibitor, as indicated with a black trace. d Etd uptake rate (slopes obtained from curves as in b or c) after treatment with 200 µM La3+, 200 µM 10Panx1 peptide, 100 µM oxidized ATP (oATP) or 10 µM A740003. **P < 0.01 (n = 4). Scale bar 20 μm
To unravel the role of different non-selective channels in the mechanism that could underlie the Etd+ uptake difference between control and mdx myofibers, cells were acutely treated (5 min) with oxidized ATP (oATP), which blocks several P2XRs, or A740003, a selective P2X7R blocker [30]. Etd+ uptake was drastically inhibited by oATP and A740003 in mdx myofibers (~80 and ~90 %, respectively), but in normal myofibers these compounds were without effect (Fig. 4c, d), and the lack of Cx43/Cx45 expression in mdx myofibers (mdx:M-C) totally prevented the increase in Etd+ uptake, confirming the results obtained with hemichannel inhibitors (Fig. 4d).
We next decided to evaluate the intracellular free Ca2+ signal in myofibers, because previous works have shown that these channels are permeable to Ca2+ [15] and that mdx myofibers show discrete elevated basal intracellular free Ca2+ levels [31]. Thus, in concordance with the elevated hemichannels activity found in mdx myofibers, we also detected an elevated basal intracellular free Ca2+ signal in these myofibers compared with control (C57) myofibers (Fig. 5), which was not detected in myofibers deficient in Cx43 and Cx45 expression (mdx:M-C myofibers) (Fig. 5).
Fig. 5.

The absence of Cx43/Cx45 in mdx skeletal myofibers prevents the elevation of basal intracellular free Ca2+ levels. Skeletal myofibers were isolated from C57, mdx and mdx:M-C (which do not express Cx43 and Cx45) mice and the basal intracellular Ca2+ signal was measured with FURA-2AM. The graph shows the 340 nm:380 nm fluorescent ratio. ***P < 0.001
The absence of Cx43 and Cx45 abrogates NF-κB activation and iNOS expression in mdx myofibers
Since the absence of Cx HCs abrogates the denervation-induced activation of nuclear factor kappa-light-chain-enhancer in activated B cells (NF-κB) [9], the presence and distribution of activated [phosphorylated p65 subunit (P-p65)] NF-κB was analyzed in mdx Cx43fl/flCx45fl/fl:Myo-Cre mice. While numerous myofibers of control mdx Cx43fl/flCx45fl/fl mice showed P-p65 reactivity in the cytoplasm and nuclei (Fig. 6a), myofibers of mdx Cx43fl/flCx45fl/fl:Myo-Cre mice did not show P-p65 reactivity, indicating that NF-κB was not activated in the absence of Cx43/Cx45 expression (Fig. 6a).
Fig. 6.

The absence of Cx43/Cx45 in skeletal muscles protects from activation of NF-κB and upregulation of iNOS in mdx mice. Cross sections of tibialis anterior (TA) muscles from control Cx43fl/flCx45fl/fl, mdx Cx43fl/flCx45fl/fl and mdx Cx43fl/flCx45fl/fl:Myo-Cre mice were used for immunofluorescence analysis. a Reactivity of phosphorylated p65 subunit of NF-κB (P-p65) (red). Central nuclei positive for P-p65 (white arrow) were detected only in mdx mice that express Cx43 and Cx45 (mdx Cx43fl/flCx45fl/fl). b iNOS reactivity (green) was evident in mdx muscles that express Cxs (upper panels) and absent in mdx muscles that cannot express Cxs (bottom panels). Scale bar 50 μm (4 mice and 6 sections were analyzed per animal)
Since mdx myofibers are known to express iNOS and generate more nitric oxide than control myofibers [32, 33], we tested if Cxs detected in mdx mice are related to the expression of iNOS. iNOS reactivity was mainly located in the periphery of myofibers in TA muscles of mdx Cx43fl/flCx45fl/fl mice whereas it was absent in TA muscles of mdx Cx43fl/flCx45fl/fl:Myo-Cre mice (Fig. 6b).
Lack of Cx43/Cx45 expression completely prevents programed cell death in mdx skeletal muscles
Since apoptotic cells are frequent in mdx muscles [7, 34], we determined whether Cx expression is related to this form of cell death. To study this possibility, myofibers undergoing apoptosis were detected through the analysis of annexin V signal by using an anti-annexin V antibody [35] or by immunodetection of active caspase 3. Cross sections of TA muscles from control C57 mice did not show positive annexin V or active caspase 3 reactivity, whereas ~30 % small cells showed strong annexin V or active caspase 3 reactivity in TA muscles of mdx mice (Fig. 7a, b). In contrast, the absence of Cx43/Cx45 in myofibers of mdx mice completely prevented the appearance of the annexin V and active caspase 3 signal (Fig. 7a, b), suggesting a direct correlation between Cx43/Cx45 expression and apoptosis.
Fig. 7.

The absence of Cx43/Cx45 expression drastically reduces apoptosis and dying fast myofibers are permeabilized to EB4− in mdx mice. Cross sections of tibialis anterior muscles of control Cx43fl/fl Cx45fl/fl, control Cx43fl/fl Cx45fl/fl: Myo-Cre, mdx Cx43fl/fl Cx45fl/fl and mdx Cx43fl/fl Cx45fl/fl:Myo-Cre mice were used to evaluate the reactivity of annexin V (Anx V) or Evan blue (EB4−) staining. a Annexin V reactivity (red). b active Caspase 3 staining (green). c Evans blue (EB4−) staining (red) and annexin V reactivity (green). d EB4− staining (red) and myosin heavy chain I reactivity (MHCI positive, green) to denote slow myofibers. Scale bar 50 μm (4 mice and 6 sections were analyzed per animal)
Since several mdx myofibers presented EB4− uptake (Fig. 3) or annexin V reactivity (Fig. 7a), we evaluated whether apoptotic cells also incorporated EB4−. To study this possibility, muscle sections of mdx mice treated with EB4− were processed for annexin V detection. We found that all myofibers with intracellular EB4− staining were also positive for annexin V (Fig. 7c). In contrast, myofibers deficient in Cx43/Cx45 expression did not show either EB4− staining or annexin V reactivity (Fig. 7c). We then asked the following question: Are EB4− stained fibers fast or slow? To answer this question, muscle sections of mdx mice previously injected with EB4− were processed to detect myosin heavy chain type I (MHC-I), used as a marker of slow twitch fibers. We found that cells labeled with EB4− were not positive for MHC-I (Fig. 7d), indicating that EB4− stained fibers correspond to the fast subtype.
The absence of Cx43/Cx45 expression partially prevents cell death by necrosis
In agreement with the occurrence of muscle regeneration promoted by cell death in skeletal muscle of mdx mice, the percentage of nuclei located centrally (Internal nuclei index) in cross sections of TA muscles was clearly higher in mdx than in C57 mice. However, TA muscles comprised of myofibers deficient in Cx43/Cx45 expression showed a significant reduction in internal nuclei index (Fig. 8). Moreover, the cross-sectional area (CSA) of myofibers analyzed in TA muscle sections stained with hematoxylin–eosin revealed that the absence of Cx43/Cx45 expression confers partial protection. We found that mdx TA muscles presented ~7 % myofibers with a CSA bigger than the largest one found in TA muscles of C57 mice. This was defined as a CSA value higher than 5700 μm2 of the total number of myofibers analyzed (301 myofibers) while in control myofibers only ~0.6 % myofibers from the total number of myofibers analyzed (623 myofibers) were over 5700 μm2, and TA muscles of mdx Cx43fl/flCx45fl/fl: Myo-Cre mice showed a significantly low number of large myofibers (~2.1 %) from the total number of myofibers analyzed (561 myofibers) (Fig. 8). In this way, we measured the total CSA because mdx fibers present very different size, so the average of the CSA is not a good indicator of variations in CSA, as it is shown in Fig. 8c. Then, we arbitrarily defined a threshold size for big myofibers, which was 5700 μm2 and we estimated the percentage of total myofibers analyzed that were above this threshold size. For mdx myofibers it was ~7 % and for control myofibers it was only ~0.6 %. However, in mdx fibers deficient in Cx43/Cx45 expression it was ~2.1 %, conferring a partial protection respect to mdx fibers.
Fig. 8.

Mdx muscles present a higher presence of big myofibers. Cross sections of tibialis anterior muscles obtained from Control (Cx43fl/fl Cx45fl/fl, white circles), mdx (gray circles) and mdx Cx43fl/fl Cx45fl/fl:Myo-Cre (Cx43fl/fl Cx45fl/fl:M-C, black circles) mice. a Distribution of DAPI stained nuclei indicated in the middle panel with white arrowheads. Calibration bar 50 μm. b Quantification of internal nuclei index obtained from DAPI stained muscle cross sections. c Quantification of the cross-sectional area (CSA) in hematoxylin–eosin stained muscle cross sections. The horizontal black line represents the average value of CSA (4 mice and 6 sections were analyzed per animal)
In addition to apoptotic cell death, skeletal muscles of mdx mice and patients with DMD present necrosis reflected by high circulating levels of serum creatine kinase (CK) [36, 37]. As described previously [38], CK activity in serum samples of mdx mice was ~30 times higher than in serum of control animals (Fig. 9a). Moreover, the circulating CK activity of mdx Cx43fl/flCx45fl/fl:Myo-Cre mice was ~30 % lower compared to mdx Cx43fl/flCx45fl/fl mice but remained significantly high compared to control C57 mice (Fig. 9a). This suggests that lack of Cx43/Cx45 expression confers only partial protection from necrotic cell death.
Fig. 9.

The absence of Cx43/Cx45 expression reduces the activity of circulating creatine kinase activity but does not affect the infiltration of inflammatory cells. a Graph showing creatine kinase (CK) activity of control, mdx Cx43fl/fl Cx45fl/fl and mdx Cx43fl/fl Cx45fl/fl:Myo-Cre (mdx M-C) mice. n = 4, **P < 0.01, ***P < 0.005. b Panels showing TA muscles cross sections (20 μm thick) from (top) control (middle) mdx and (bottom) mdx Cx43fl/fl Cx45fl/fl (mdx M-C) mice with DAPI stained nuclei and infiltrated macrophages detected by their ED2 (red) reactivity. Calibration bar 50 µm
Since cell death by necrosis, but not apoptosis, retrieves inflammatory cells, and since muscles of mdx mice are known to present necrosis and macrophage infiltration [34, 39], we tested whether the absence of Cx43/Cx45 expression affects this phenomenon. Toward this aim, we evaluated the presence of inflammatory cells infiltrating muscular tissue through the evaluation of ED2 reactivity, which is a marker of mature macrophages [40]. As expected, mdx muscles presented several (30 % of myofibers presented at least 1 ED2 positive cell) ED2 positive cells, while control muscles were negative (Fig. 9b). Moreover, muscles of mdx Cx43fl/flCx45fl/fl:Myo-Cre mice showed a similar ED2 reactivity as that of muscle of mdx Cx43fl/flCx45fl/fl mice (Fig. 9b), suggesting that cell necrosis persists in the absence of Cx43/Cx45 expression.
The absence of Cx43/Cx45 expression in mdx skeletal muscles induces reappearance of dystrophin and partial muscle performance recovery
Mdx myofibers as well as human myofibers in patients with DMD show a drastic reduction or complete disappearance of dystrophin reactivity [41]. Accordingly, we found that TA muscles of 3-month-old mdx Cx43fl/flCx45fl/fl mice showed very low dystrophin reactivity compared to muscles of control C57 mice (Fig. 10). In contrast, myofibers in mdx Cx43fl/flCx45fl/fl:Myo-Cre mice of the same age showed dystrophin reactivity with intensity and distribution comparable to that in control C57 mice (Fig. 10).
Fig. 10.

The absence of Cx43/Cx45 upregulates the dystrophin immunoreactivity and improves muscle performance. The presence of dystrophin (green) was evaluated by immunofluorescence, in cross sections of control (C57) (top left panel) tibialis anterior muscle, mdx Cx43fl/flCx45fl/fl (top middle panel) and mdx Cx43fl/flCx45fl/fl:Myo-Cre (top right panel) mice. Scale bar 50 μm (4 mice and 6 sections analyzed from each animal). Muscle performance was evaluated with the two (bottom left graph) and four (bottom right graph) leg hanging tests. n = 4, *P < 0.05, **P < 0.01, ***P < 0.005
Since loss of muscle function of mdx mice and DMD patients is attributed to loss of functional dystrophin [42] and since we observed the reappearance of dystrophin and the absence of apoptosis in mdx Cx43fl/flCx45fl/fl:Myo-Cre mice, it was of interest to study whether the absence of Cx43/Cx45 affects muscle performance. This was evaluated by using the two or four limb “wire hanging” test. Muscle performance of 3-month-old mdx mice was drastically reduced in both the two and the four limb test compared to control C57 mice (Fig. 10). Muscle performance of mdx Cx43fl/flCx45fl/fl:Myo-Cre mice in both tests was significantly lower than that of control C57 mice and also significantly higher than that of mdx Cx43fl/flCx45fl/fl mice (Fig. 10).
Skeletal muscles of Duchenne muscular dystrophy patients present Cx40.1, Cx43 and Cx45
Biopsy specimens from two patients with DMD and from one patient with Becker’s muscular dystrophy (BMD), showing the absence and reduction of reactivity to dystrophin, respectively, and whose diagnosis were confirmed through the genetic analysis (Table 1). Assuming that the anti-Cx39 antibodies used cross react with Cx40.1, the Cx39 ortholog found in human [43], all human samples from dystrophic patients (2 DMD and 1 BMD) exhibited positive reactivity to Cx40.1, Cx43 and Cx45 that was not detected in two human control biopsies; one example of control and DMD is shown in Fig. 11.
Table 1.
Data of patients with Duchenne or Becker muscular dystrophy included in this study
| Patient | Gender/age of diagnosis (years) | Clinical features | Serum creatine kinase (U/l) | Skeletal muscles biopsy | Mutation in dystrophin gene | Diagnosis |
|---|---|---|---|---|---|---|
| DMD1105 | M/2 |
Hypotonia Developmental delay |
11,000–16,000 |
Large variability of myofiber size and increased centralization of nuclei Fatty infiltration and endomysial fibrosis Negative reactivity to dystrophin 1, 2 and 3 |
Frameshift deletion of exon 50 | Duchenne’s muscular dystrophy |
| DMD1073 | M/3 |
Pseudohypertrophy of lower limbs Symmetrical hyporeflexia Gower (+) |
4000–14,000 |
Large variability of myofiber size with frequent centered nuclei Intense necrosis-regeneration, fatty infiltration and endomysial fibrosis Negative reactivity to dystrophin 1, 2 and 3 Variable and weak reactivity to β-sarcoglycan |
Point mutation c. 7585<T (GAA>TAA) Glu2529stop | Duchenne’s muscular dystrophy |
| DMD1022 | M/44 | Muscular weakness of upper and lower limbs | 1400 |
Large variability of myofiber size with frequent centered nuclei Scattered necrosis-regeneration with mild fatty infiltration Positive but irregular reactivity to dystrophin 1 and 3, and δ-sarcoglycan |
In-frame deletion of exons 45–48 | Becker’s muscular dystrophy |
Fig. 11.
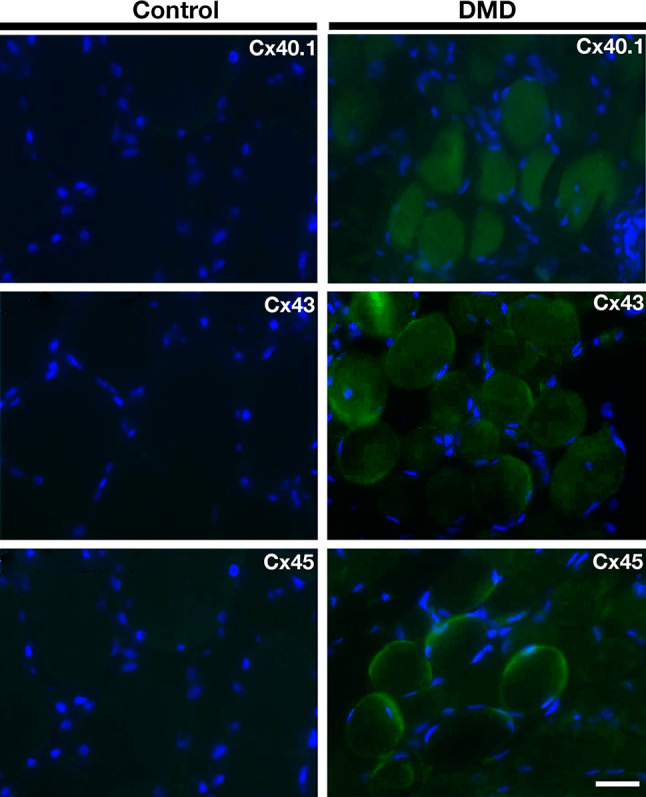
Connexins 40.1, 43 and 45 are present in human biopsies of Duchenne muscular dystrophy patients but not of normal subjects. Sections of biopsies from quadriceps muscles were obtained from a Spanish Duchenne muscular dystrophy patient (DMD, right panels) and control person (control, left panels). The presence of connexins 40.1, 43 or 45 (Cx40.1, Cx43 and Cx45) was analyzed by immunofluorescence assays in three muscle sections. Scale bar 50 µm (2 DMD patients)
Discussion
The results of the present study indicate that skeletal muscles from patients diagnosed with DMD or BMD and mdx mice (animal model of DMD) express Cxs (39, 43 and 45). Accordingly, fast myofibers of mdx mice also express Cxs that form functional hemichannels co-expressed with other non-selective membrane channels. The presence of Cxs in myofibers was associated with activation of NF-κB, expression of iNOS and apoptotic cell death. The absence of Cx43/Cx45 expression in mdx myofibers abrogated all the above changes and partially reduced necrotic cell death and improved muscle performance. These findings suggest that Cx HCs play a critical role in muscle degeneration associated with muscular dystrophy.
In addition to de novo expression of Cxs 39, 43 and 45, mdx myofibers showed upregulation of Panx1 channels and de novo expression of two other non-selective membrane channels, P2X7R and TRPV2, as described previously [16, 17, 19]. Notably, the absence of Cx43/Cx45 expression by mdx fibers abrogated the expression of TRPV2 as observed by western blots and immunofluorescence assays, suggesting that TRPV2 expression is controlled downstream of Cx43/Cx45 expression. In contrast P2X7R and Cx39 expression in fibers of mdx Cx43fl/flCx45fl/fl:Myo-Cre mice was comparable to that of control mdx Cx43fl/flCx45fl/fl fibers, suggesting that it is controlled independently or upstream of Cx43/Cx45 expression. The possible link between dystrophin and the expression of these non-selective membrane channels remains to be elucidated.
Clearly, all the non-selective channels mentioned above could contribute to the increase in sarcolemma permeability to Ca2+ in mdx myofibers, because they are all permeable to this divalent ion [15, 16, 19]. Consequently, each could partially contribute to reaching a critical intracellular free Ca2+ concentration that leads to cell degeneration. In support of this possibility, it has been shown that mdx myofibers express TRPV2 channels and P2X7R, and that TRPV2 knockout or P2X7R inhibition reduces the intracellular Ca2+ signal [16, 19]. Moreover, we found that myofibers deficient in Cx43/Cx45 expression present similar free Ca2+ signal as control myofibers, whereas mdx myofibers presented a discrete by significant increase in basal free Ca2+ signal. These findings suggest that under basal condition, the Ca2+ handling mechanisms of mdx myofibers are slightly overfilled and the elimination of at least one Ca2+ permeable channel is enough to help the cells handle the Ca2+ influx mediated by the remaining Ca2+ permeable channels. These non-selective membrane channels could also increase Na+ influx and high intracellular Na+ concentration would interfere with Ca2+ extrusion via the Na+/Ca2+ exchanger, thus reducing the Ca2+ handling capacity of myofibers as previously proposed [44]. In addition to the increase in permeability to Ca2+, we found an increase in sarcolemma permeability to small molecules, including EB4− (in vivo) and Etd+ (in vitro), which is reminiscent of what was described for denervated skeletal muscles [9]. These findings also indicate that cell staining with EB4− results from dye influx via Cx HCs and not from cell membrane damage as interpreted elsewhere [45]. In agreement with this interpretation, EB4− uptake of mdx myofibers was completely prevented by the acute administration of CBX or BBG, which are inhibitors of Cx HCs and Panx1 channels (CBX) and Panx1 channels and P2X7R [28]. This suggests that these channels play a critical role in mdx sarcolemma permeabilization to small molecules. In support of the role of Cx HCs, we have previously shown that Cx39 HCs, Cx43 HCs and Cx45 HCs are permeable to EB4− [9]. In addition, we observed that myofibers of mdx mice were not stained in vivo with RD (molecular mass 10 kDa), which is a dye with a size well above the exclusion limit of any of the non-selective channels described herein. This finding also indicates that cells positive to extracellular proteins (e.g., Albumin, IgG and IgM; with molecular sizes well above the exclusion limit of Cx HCs) in DMD and mdx muscles [46] should be different from the EB4−-stained cells and might be cells undergoing necrosis (see below).
The absence of Cx43/Cx45 in mdx myofibers was also sufficient to completely prevent Etd+ uptake of isolated mdx myofibers. In support of the role of non-selective channels, Etd+ uptake was completely inhibited by Cx HC (La3+) or P2XR (oATP or A740003) blockers and partially blocked by the 10Panx1 peptide, a Panx1 channel blocker. This point to the orchestration of these three channel types as it occurs in denervated myofibers [9]. Young et al. [19] observed the absence of propidium iodide uptake in mdx myoblasts and myotubes upon prolonged P2X7R activation. This finding might be explained by the low permeability of Cx HCs to propidium iodide [47]. However, the lack of Lucifer yellow uptake under these conditions [19] might be related to cellular differences since EB4− is larger and more negatively charged (molecular mass 960.8 Da and −4 net charge) than Lucifer yellow (MW: 443.4 and −2) and both dyes (EB4− and LY2−) have been shown to permeate Cx HCs [9, 48].
Dystrophic muscles show upregulation of NF-κB and iNOS activity and these responses can be ameliorated by reducing either the Ca2+ influx or Ca2+ release from intracellular stores [49]. Accordingly, we found that myofibers of mdx Cx43fl/flCx45fl/fl:Myo-Cre mdx mice, which are deficient in two Cx HCs, two membrane pathways for Ca2+ influx, present basal intracellular free Ca2+ signal comparable to that of control C57 myofibers, suggesting that the elevated basal Ca2+ signal found in mdx myofibers is the result of functional Cx HC expression. In addition, mdx Cx43fl/flCx45fl/fl:Myo-Cre mdx myofibers did not show NF-κB activation and iNOS expression. These findings suggest that selective inhibition of Cx HCs might be useful to reduce or completely inhibit the inflammasome activation and subsequent generation of proinflammatory cytokines as well as nitric oxide generation, which are elevated in mdx muscles [50, 51].
The observation of apoptosis in skeletal muscles of mdx mice was first reported by two groups [7, 34]. Consistent with our finding of colocalization of annexin V reactivity and EB4− staining in fast myofibers of mdx mice, Matsuda and collaborators had shown that apoptotic myofibers are also stained with EB4− [7]. We now demonstrated that myofibers deficient in Cx43/Cx45 do not exhibit evident apoptosis, which again is consistent with the combined action of different non-selective membrane channels. In line with the latter, the dominant negative inhibition of TRPV2 channels drastically reduced apoptosis and EB4− stained myofibers of mdx mice [16]. Since TRPV2 channels are impermeable to EB4− [9] and Cx HC activity is promoted by elevated intracellular Ca2+ concentrations [10], the reduction in EB4− staining described by Iwata and collaborators [16] might be explained by the resulting reduction in intracellular Ca2+ concentration that would prevent activation of Cx HCs [10].
Mdx mice also exhibit myofiber necrosis, which as a result of the cell lysis induces macrophage infiltration and elevates serum levels of cytosolic proteins including CK [16, 34]. Here, we found that the absence of Cx43/Cx45 expression does not significantly affect the muscle infiltration of ED2 positive cells and only partially prevented (~30 %) the increase in serum CK levels of mdx mice. Similarly, mdx mice under dominant negative inhibition of TRPV2 channels have been shown to present lower serum CK levels than untreated mdx mice [16]. Inhibition of TRPV2 channels should result in systemic inhibition whereas Cx43/Cx45 KO was restricted to myofibers. It should be noted that necrosis is frequently triggered by ischemia–reperfusion or hypoxia–reoxygenation, and the absence of dystrophin in other cell types might contribute to these processes affecting myofiber survival. In fact, dystrophin is expressed in platelets [45] and tunica media of blood vessels [52], and its absence in blood vessel of patients with DMD has been reported [52]. Consequently, dystrophin deficiency in these cells might explain the multiple episodes of thrombosis found in patients with BMD, which is a less aggressive muscle dystrophy than DMD [53]. Treatment with a vasodilator could, thus, alleviate muscle ischemia in patients with BMD [54]. In support of the orchestration of several non-selective channels treatment with Coomassie brilliant blue, which is a blocker of P2X7Rs, reduces the number of degeneration–regeneration cycles in mdx mice [19].
Regeneration, internal nuclei, hypertrophic cells are other common changes found in muscles of mdx mice or DMD patients [46]. Similarly, we found numerous intracellularly nucleated and hypertrophic myofibers in TA muscles of mdx mice. However, both changes were significantly reduced in muscles of mice deficient in Cx43/Cx45 expression, thus suggesting that necrosis was reduced but not completely eliminated.
Muscles of mdx mice deficient in Cx43/Cx45 remarkably showed dystrophin expression comparable to that of control C57BL/6 mice. This might result from less Ca2+ influx via Cx HCs and TRPV2 channels, hence reducing the activation of Ca2+-dependent proteases and reducing proteolysis of numerous proteins, including dystrophin. Despite the presence of high levels of dystrophin in Cx43/Cx45 deficient muscles the muscle function evaluated in the whole animals showed only partial protection compared to control mice. This is probably because the rescued dystrophin is not functional or is just partially functional. It remains to be determined whether selective inhibition of Cx HC in other tissue including the heart, vascular cells and platelets would confer more significant protection in all parameters described here as partially protected. Alternatively, the simultaneous inhibition of all non-selective channels mentioned here might be more effective in protecting mdx mice.
Because we detected the presence of Cxs in skeletal muscle from patients diagnosed with DMD and BMD, based on the observation that these proteins form functional hemichannels and that the absence of Cx43/Cx45 prevented the apoptosis in mdx myofibers, we suggest that these channels could constitute a potential therapeutic target to reduce muscle deterioration in DMD or BMD.
Materials and methods
Reagents
Western lightning chemiluminescence (ECL) detection reagents were obtained from PerkinElmer (Boston, MA, USA) was purchased from Pierce (Pierce biotechnology, Rockford, IL, USA), and anti-rabbit or anti-mouse IgG antibodies-conjugated to Cy2 (green) or Cy3 (red) were purchased from Jackson immunoresearch laboratories (West Grove, PA, USA). Ethidium (Etd+) bromide was from GIBCO/BRL (Grand Island, NY, USA), fluoromount-G was from Electron Microscopy Science (Hatfield, PA, USA), brilliant blue G (BBG), N-benzyl-p-toluene sulphonamide (BTS) and collagenase type I were obtained from Sigma (St. Louis, USA), and A740003 from Tocris Bioscience (Bristol, UK). Phycoerithrin (PE) or FITC conjugated monoclonal anti-annexin V antibody were from BD Biosciences (San Jose, CA, USA) and polyclonal anti-TRPV2 or anti-P2X7R antibodies were from Abcam (Cambridge, MA, USA). Previously described polyclonal anti-Cx39, -43, -Cx45, and -Panx1 antibodies were used [9]. Rabbit polyclonal anti-phosphorylated-p65 NF-kB subunit antibodies were from Cell Signaling, and rabbit polyclonal anti-dystrophin antibodies were from the Developmental Studies Hybridoma Bank.
The dystrophin monoclonal antibody (MANDRA1) developed by Glenn E. Morris was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242, USA.
Animals
All protocols were approved by the Bioethics Committee of the Pontificia Universidad Católica de Chile (protocol no. 176) in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All efforts were made to minimize animal suffering, reduce the number of animals used, and alternatives to in vivo techniques, if available. Two- to three-month-old male C57BL/6 (C57) and mdx mice (C57BL/6 mdx 4cv) were obtained from the animal facility of the Faculty of Biological Sciences at the Pontificia Universidad Católica de Chile. Previously described Cx43fl/flCx45fl/fl and Cx43fl/flCx45fl/fl:Myo-Cre mice were used [9]. The latter were skeletal muscle deficient for Cx43 and Cx45 generated from breeding Cx43fl/fl mice [55] and Cx45fl/fl mice [56] with Myo-Cre mice, which express Cre recombinase under the control of a myogenin promoter and the MEF2C [57] enhancer. Mdx mice lacking Cx43 and Cx45 expression in skeletal muscle were obtained by a four-factor cross starting with mating a Cx43- and Cx45-deficient (in skeletal muscle) male mouse to a homozygous mdx female (the mdx mutation is located on the X chromosome). This led to offspring, all being heterozygously floxed for Cx43 and Cx45, of which 50 % showed Myo-Cre expression but all carried the mutated X chromosome (F1 generation). For generating the F2 generation a Cx43fl/+ Cx45fl/+, mdx/x female was bred with a Cx43fl/fl Cx45fl/fl:Myo-Cre male to generate the desired genotype Cx43fl/fl Cx45fl/fl:Myo-Cre, mdx/y male. Breeding this male (F2) with Cx43fl/fl Cx45fl/fl, mdx/+ females finally yielded the desired mutants in the F3 generation Cx43fl/fl Cx45fl/fl:Myo-Cre, mdx/mdx females and Cx43fl/fl Cx45fl/fl:Myo-Cre, mdx/y males which were interbred.
Genotyping of mdx mice
Genomic DNA from a tail piece (2 mm) was isolated using the proteinase K digestion method [58] and http://jackslab.mit.edu/protocols/dna_isolation_proteinase_k_method, with some modifications. Briefly, samples were incubated overnight in 400 μl of lysis buffer (0.5 % SDS, 100 mM NaCl, 10 mM EDTA and 10 mM Tris–HCl pH 8) containing 300 μg of proteinase K at 55 °C. Then, 300 μl of 6 M NaCl were added, mixed and samples were spun down at 13,000g for 10 min at room temperature (RT). The supernatant was transferred to a new microcentrifuge tube and 700 μl of isopropanol were added, mixed and incubated at RT for 15 min. Samples were spun down at 13,000g for 10 min at RT. Finally, DNA pellet was rinse with 70 % ethanol, air dried and resuspended in 50 μl of nuclease-free water. An aliquot of genomic DNA was diluted 1:10 and 1 μl was used for PCR. Genotyping was performed by primer competition PCR method described previously [59]. This method allows identifying the mdx and wild type genotype mice by different sizes of PCR products. A product of 123 pb confirms the mdx 4cv genotype and for WT genotype the PCR product is 141 pb.
Western blots
Tibialis anterior muscles were washed with ice-cold saline. Tendon-free muscles were minced in small pieces by using a razor blade and then homogenized (homogenizer; Brinkmann) and sonicated (Heat Systems Microson). Tissue homogenates were centrifuged for 15 min at 13,000×g and pellets were discarded. Then, samples were processed for western blot analyses of proteins of interest as described previously [25]. Blots were incubated overnight with appropriate dilutions of primary antibodies diluted in 5 % fat free milk–PBS solution. Then, blots were rinsed with 1 % PBS solution–Tween 20 and incubated for 40 min at room temperature with HRP-conjugated goat anti-rabbit or anti-mouse IgGs (Santa Cruz Biotechnology). After five rinses, immunoreactive proteins were detected using ECL reagents according to the manufacturer’s instructions (PerkinElmer).
Skeletal myofibers isolation
Myofibers of mouse flexor digitorum brevis (FDB) muscles were isolated from anesthetized mice as described previously [9]. Muscles were immersed in cultured medium (DMEM/F12 supplemented with 10 % fetal bovine serum) containing 0.2% collagenase type I and incubated for 2.5 h at 37 °C. Then, they were transferred to a 15 ml Falcon test tube containing 3 ml of Krebs buffer (in mM: 145 NaCl, 5 KCl, 3 CaCl2, 1 MgCl2, 5.6 glucose, 10 HEPES-Na, pH 7.4) plus 10 µM BTS (to reduce muscle damage due to activation of myofiber contraction during the isolation procedure). Muscle tissue was incubated for 5 min with 200 µM Suramin, which is a P2 receptor inhibitor, to prevent activation of P2 receptors by ATP released during the isolation procedure. Then, tissue was gently triturated by passing it 15 times through a wide-tip Pasteur pipette so as to dissociate single myofibers. Dissociated myofibers were centrifuged at 1000 rpm for 10 s (Kubota 8700 centrifuge, Tokyo, Japan) and the sediment was washed two times with HEPES buffered Krebs saline solution containing 10 µM BTS. Finally, myofibers were resuspended in 5 ml HEPES buffered Krebs solution plus 10 µM BTS, plated in plastic culture dishes or placed in 1.5 ml Eppendorf tubes.
Membrane permeability to dyes
The uptake of fluorescent molecules was evaluated using time lapse measurements or snap shot photographs. Freshly isolated myofibers plated onto plastic culture dishes were washed twice with Krebs-buffer solution. For time lapse measurements myofibers were incubated in Krebs buffer solution (plus 10 µM BTS) containing 5 μM ethidium (Etd+) bromide. Etd+ fluorescence was recorded in regions of interest located in nuclei of the myofibers. A water immersion Olympus 51W1I upright microscope (Japan) was used for recording. Images were captured with a Q Imaging model Retiga 13001 fast cooled monochromatic digital camera (12-bit) (Qimaging, Burnaby, BC, Canada) every 30 s. Image processing was performed off-line with ImageJ software (NIH, Bethesda, USA).
For snap shot pictures frozen sections of tibialis anterior (TA) muscles from mice injected with EB4− were analyzed by epifluorescence microscopy (excitation 495 nm, emission 590 nm).
Ca2+ signal measurements
The basal intracellular free Ca2+ signal was measured in isolated myofibers using the Ca2+ indicator FURA-2AM and following the manufacturer’s instructions. Briefly, isolated myofibers were incubated with FURA-2AM (5 μM) in Krebs solution for 50 min at room temperature. Once finished the incubation period, the myofibers were washed with Krebs saline solution without dye, and placed on coverslips to measure the basal free Ca2+ signal in fluorescence microscope Nikon eclipse Ti (Tokyo, Japan) as the 340 nm:380 nm fluorescence ratio.
Immunofluorescence analysis
To detect different proteins in myofibers or cross sections (20 μm) of muscles (fast frozen with iso-methyl-butane cooled in liquid nitrogen) samples were obtained and processed as described previously [9]. Samples were incubated at 4 °C for 12 h with diluted primary anti-Cx39 (1:100), anti-Cx43 (1:300), anti-Cx45 (1:300), anti-Panx1 (1:300), anti-TRPV2 (1:100), anti-P2X7 (1:300) or anti-phosphorylated P65 (1:200) antibodies followed by four washes with HSS and then, incubated with an appropriate dilution of Cy2- or Cy3-conjugated goat anti-rabbit IgG antibodies. Samples were rinsed with HSS, mounted with fluoromount G on glass slides and representative images were acquired in a confocal microscope, Olympus fluoview 1000 (Tokyo, Japan).
For annexin and active caspase 3 detection cross sections of tibialis anterior (TA) muscle were mounted on slides, fixed for 5 min with formaldehyde 4 % w/v and then incubated with blocking solution (NH4Cl 50 mM, BSA 1 %, PBS1X) for 3 h at room temperature. Then, the slices were incubated for 12 h at 4 °C with anti-annexin V antibody conjugated to phycoerythrin or anti-active caspase 3 antibody followed by incubation with a secondary antibody conjugated to FIT-C and washed three times with PBS. Samples were subsequently covered with fluoromount G and glass coverslips. The slices were analyzed with a fluorescence microscope Nikon Eclipse Ti.
The absence of dystrophin in mdx mice was confirmed by immunological detection with anti-dystrophin antibodies.
Cross-sectional area (CSA) and centered nuclei
The CSA of skeletal muscle fibers was measured as described previously [9]. In brief, cryosections of muscles were fixed with 4 % (wt/vol) paraformaldehyde, stained with hematoxylin and eosin and mounted with fluoromount G with DAPI on glass slide for nuclei detection. The CSA of each myofiber was evaluated by using offline analyses with ImageJ software (National Institutes of Health).
Internal nuclei index
Skeletal muscle sections with hematoxylin/eosin and DAPI stained were used. In brief, the internal nucleus in each myofiber and total myofibers in each image were counted. The index was calculated as the ratio between the total internal nuclei and total myofibers in each image.
Creatine kinase activity
Blood was obtained from adult mice (8–9 weeks, ~20 g). The collected blood was incubated (45–60 min) at 37 °C to allow blood to clot. Then, samples were centrifuged (1000 rpm for 5 m) to remove the clot, and the clear supernatant was the serum. The catalytic activity of serum creation kinase was determined at 37 °C on a spectrophotometer at 340 nm in accordance with manufacture’s instructions (Valtek S.A., Santiago, Chile).
Wire hanging test
A metal cloth hanger was hanged around 37 cm above a layer of bedding. Then, a mouse was brought near the wire via the tail. For two limb hanging test, the mouse was left to grasp the wire with two forepaws only, and the hind limbs were lowered in such a way that the mouse only hanged by the two forepaws from the wire. The maximum hanging time was recorded. For four limb hanging test, the mouse was led to grasp in the wire with four limbs, for which the maximum hanging time was recorded.
Selection of DMD patients and mutation and biopsy studies
Following ethical guidelines, the healthy volunteers and DMD patients who participated in this study signed an informed consent approved by the local Ethics Committee in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Diagnosis of Duchenne/Becker muscular dystrophy was achieved based on clinical examination, muscular biopsy and mutation analysis (Table 1). Genomic DNA extracted from peripheral blood leucocytes by standardized procedures (kit Purogene, Qiagen GmbH, Hilden, Germany) were screened for DMD gene duplications/deletions using the MLPA technique (SALSA MLPA probemixes P034/P035 DMD, MRC-Holland, Amsterdam, The Netherlands). In those cases without duplications/deletions, the 79 exons of the DMD gene and part of the intronic regions were sequenced by NGS technology by the Illumina MiSeq team. The amplified fragments were performed using the Multiplicom MASTR DMD kit. All variants detected were confirmed by Sanger sequencing (amplified DNA products were sequenced by the dideoxy termination method, BigDye Sequencing Kit, Applied Biosystems, Foster City, CA, USA). Human biopsies from controls and DMD patients consisted of small fragments of quadriceps muscle and were quickly frozen with isopentane in a container immersed in liquid nitrogen. Cryosections of 10 µm fixed in 4 % paraformaldehyde were processed for histological (H–E and trichrome), histochemical stains [oxidative (NADH, SDH and COX), ATPasa (pH 4.2; 4.6 and 9.4), Oil-red-O and PAS] and immunohistochemistry (dystrophin 1, 2 and 3, alpha, beta, delta and gamma sarcoglycans, merosin and beta-spectrin and Cxs).
Statistical analysis
Results are presented as mean ± standard error (SE). Two populations were compared using the logarithm of ratio and posterior Student’s t test. For multiple comparisons with a single control, a non-parametric one-way ANOVA followed by the Tukey’s multiple comparison test was used. Analyses were carried out using GRAPHPAD software. P < 0.05 was considered statistically significant.
Acknowledgments
We thank Ms. Teresa Vergara and Ms. Paola Fernández for their technical support. We also thank members of the Spanish families, whose participation made this study possible. This work was partially supported by CONICYT/PAI Proyecto de Inserción en la Academia 79140023 (to LAC); Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT): Grant 3130662 (to CP); 1150291 (to JCS), ICM-Economía P09-022-F Centro Interdisciplinario de Neurociencias de Valparaíso (to JCS), grants from the Spanish Ministry of Economy and Competitiveness (Consolider CSD2008-00005 and BFU2013-33821) and the Community of Madrid (Neurotec-P2010/BMD-2460) (to LCB). The research stay of LAC and CP in the Bonn laboratory was supported by a grant of CONICYT and the German Academic Exchange Service (to JCS and KW). Additional work in the Bonn laboratory was funded by the German Research Foundation (Wi 270/33.1 and SFB 645, B2) to KW.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
The authors certify that the experiments comply with the current laws of Chile, where the experiments were performed. All protocols were approved by the Bioethics Committee of the Pontificia Universidad Católica de Chile (Protocol No. 176) in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All efforts were made to minimize animal suffering, reduce the number of animals used, and alternatives to in vivo techniques, if available.
Contributor Information
Luis A. Cea, Email: luiscea@med.uchile.cl
Juan C. Sáez, Email: jsaez@bio.puc.cl
References
- 1.Emery AE. Muscular dystrophy into the new millennium. Neuromuscul Disord. 2002;12:343–349. doi: 10.1016/S0960-8966(01)00303-0. [DOI] [PubMed] [Google Scholar]
- 2.Ervasti JM, Campbell KP. A role for the dystrophin–glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pillers D. A new day for Duchenne’s? The time has come for newborn screening. Mol Genet Metab. 2014;113:11–13. doi: 10.1016/j.ymgme.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 4.Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balnave CD, Allen DG. Intracellular calcium and force in single mouse muscle fibres following repeated contractions with stretch. J Physiol. 1995;488:25–36. doi: 10.1113/jphysiol.1995.sp020943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeung EW, Head SI, Allen DG. Gadolinium reduces short-term stretch-induced muscle damage in isolated mdx mouse muscle fibres. J Physiol. 2003;552:449–458. doi: 10.1113/jphysiol.2003.047373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsuda R, Nishikawa A, Tanaka H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: evidence of apoptosis in dystrophin-deficient muscle. J Biochem. 1995;118:959–964. doi: 10.1093/jb/118.5.959. [DOI] [PubMed] [Google Scholar]
- 8.Hamer PW, McGeachie JM, Davies MJ, Grounds MD. Evans Blue Dye as an in vivo marker of myofibre damage: optimising parameters for detecting initial myofibre membrane permeability. J Anat. 2002;200:69–79. doi: 10.1046/j.0021-8782.2001.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cea LA, Cisterna BA, Puebla C, Frank M, Figueroa XF, Cardozo C, et al. De novo expression of connexin hemichannels in denervated fast skeletal muscles leads to atrophy. Proc Natl Acad Sci USA. 2013;110:16229–16234. doi: 10.1073/pnas.1312331110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sáez JC, Leybaert L. Hunting for connexin hemichannels. FEBS Lett. 2014;588:1205–1211. doi: 10.1016/j.febslet.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Eskandari S, Zampighi GA, Leung DW, Wright EM, Loo DD. Inhibition of gap junction hemichannels by chloride channel blockers. J Membr Biol. 2002;185:93–102. doi: 10.1007/s00232-001-0115-0. [DOI] [PubMed] [Google Scholar]
- 12.Franco A, Jr, Lansman JB. Calcium entry through stretch-inactivated ion channels in mdx myotubes. Nature. 1990;344:670–673. doi: 10.1038/344670a0. [DOI] [PubMed] [Google Scholar]
- 13.Bao L, Sachs F, Dahl G. Connexins are mechanosensitive. Am J Physiol Cell Physiol. 2004;287:C1389–C1395. doi: 10.1152/ajpcell.00220.2004. [DOI] [PubMed] [Google Scholar]
- 14.Fiori MC, Figueroa V, Zoghbi ME, Saéz JC, Reuss L, Altenberg GA. Permeation of calcium through purified connexin 26 hemichannels. J Biol Chem. 2012;287:40826–40834. doi: 10.1074/jbc.M112.383281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schalper KA, Sánchez HA, Lee SC, Altenberg GA, Nathanson MH, Sáez JC. Connexin 43 hemichannels mediate the Ca2+ influx induced by extracellular alkalinization. Am J Physiol Cell Physiol. 2010;299:C1504–C1515. doi: 10.1152/ajpcell.00015.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iwata Y, Katanosaka Y, Arai Y, Shigekawa M, Wakabayashi S. Dominant-negative inhibition of Ca2+ influx via TRPV2 ameliorates muscular dystrophy in animal models. Hum Mol Genet. 2009;18:824–834. doi: 10.1093/hmg/ddn408. [DOI] [PubMed] [Google Scholar]
- 17.Valladares D, Almarza G, Contreras A, Pavez M, Buvinic S, Jaimovich E, et al. Electrical stimuli are anti-apoptotic in skeletal muscle via extracellular ATP. Alteration of this signal in mdx mice is a likely cause of dystrophy. PLoS One. 2013;8:e75340. doi: 10.1371/journal.pone.0075340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeung D, Zablocki K, Lien CF, Jiang T, Arkle S, Brutkowski W, et al. Increased susceptibility to ATP via alteration of P2X receptor function in dystrophic mdx mouse muscle cells. FASEB J. 2006;20:610–620. doi: 10.1096/fj.05-4022com. [DOI] [PubMed] [Google Scholar]
- 19.Young CN, Brutkowski W, Lien CF, Arkle S, Lochmüller H, Zabłocki K, et al. P2X7 purinoceptor alterations in dystrophic mdx mouse muscles: relationship to pathology and potential target for treatment. J Cell Mol Med. 2012;16:1026–1037. doi: 10.1111/j.1582-4934.2011.01397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kondo RP, Wang SY, John SA, Weiss JN, Goldhaber JI. Metabolic inhibition activates a non-selective current through connexin hemichannels in isolated ventricular myocytes. J Mol Cell Cardiol. 2000;32:1859–1872. doi: 10.1006/jmcc.2000.1220. [DOI] [PubMed] [Google Scholar]
- 21.Sáez JC, Schalper KA, Retamal MA, Orellana JA, Shoji KF, Bennett MV. Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp Cell Res. 2010;316:2377–2389. doi: 10.1016/j.yexcr.2010.05.026. [DOI] [PubMed] [Google Scholar]
- 22.North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–1067. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- 23.Perálvarez-Marín A, Doñate-Macian P, Gaudet R. What do we know about the transient receptor potential vanilloid 2 (TRPV2) ion channel? FEBS J. 2013;280:5471–5487. doi: 10.1111/febs.12302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cea LA, Riquelme MA, Cisterna BA, Puebla C, Vega JL, Rovegno M, et al. Connexin- and pannexin-based channels in normal skeletal muscles and their possible role in muscle atrophy. J Membr Biol. 2012;245:423–436. doi: 10.1007/s00232-012-9485-8. [DOI] [PubMed] [Google Scholar]
- 25.Riquelme MA, Cea LA, Vega JL, Boric MP, Monyer H, Bennett MV, et al. The ATP required for potentiation of skeletal muscle contraction is released via pannexin hemichannels. Neuropharmacology. 2013;75:594–603. doi: 10.1016/j.neuropharm.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 26.Riquelme MA, Cea LA, Vega JL, Puebla C, Vargas AA, Shoji KF, et al. Pannexin channels mediate the acquisition of myogenic commitment in C2C12 reserve cells promoted by P2 receptor activation. Front Cell Dev Biol. 2015;3:25. doi: 10.3389/fcell.2015.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Araya R, Eckardt D, Maxeiner S, Krüger O, Theis M, Willecke K, Sáez JC. Expression of connexins during differentiation and regeneration of skeletal muscle: functional relevance of connexin43. J Cell Sci. 2005;118:27–37. doi: 10.1242/jcs.01553. [DOI] [PubMed] [Google Scholar]
- 28.Dahl G, Qiu F, Wang J. The bizarre pharmacology of the ATP release channel pannexin1. Neuropharmacology. 2013;75:583–593. doi: 10.1016/j.neuropharm.2013.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakazawa K, Liu M, Inoue K, Ohno Y. Potent inhibition by trivalent cations of ATP-gated channels. Eur J Pharmacol. 1997;325:237–243. doi: 10.1016/S0014-2999(97)00120-9. [DOI] [PubMed] [Google Scholar]
- 30.Honore P, Donnelly-Roberts D, Namovic MT, Hsieh G, Zhu CZ, Mikusa JP, et al. A-740003 [N-(1-{[(cyanoimino)(5-quinolinylamino) methyl]amino}-2,2-dimethylpropyl)-2-(3,4-dimethoxyphenyl)acetamide], a novel and selective P2X7 receptor antagonist, dose-dependently reduces neuropathic pain in the rat. J Pharmacol Exp Ther. 2006;319:1376–1385. doi: 10.1124/jpet.106.111559. [DOI] [PubMed] [Google Scholar]
- 31.Burr AR, Molkentin JD. Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy. Cell Death Differ. 2015;22:1402–1412. doi: 10.1038/cdd.2015.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bia BL, Cassidy PJ, Young ME, Rafael JA, Leighton B, Davies KE, et al. Decreased myocardial nNOS, increased iNOS and abnormal ECGs in mouse models of Duchenne muscular dystrophy. J Mol Cell Cardiol. 1999;31:1857–1862. doi: 10.1006/jmcc.1999.1018. [DOI] [PubMed] [Google Scholar]
- 33.Vannucchi MG, Corsani L, Azzena GB, Faussone-Pellegrini MS, Mancinelli R. Functional activity and expression of inducible nitric oxide synthase (iNOS) in muscle of the isolated distal colon of mdx mice. Muscle Nerve. 2004;29:795–803. doi: 10.1002/mus.20010. [DOI] [PubMed] [Google Scholar]
- 34.Tidball JG, Albrecht DE, Lokensgard BE, Spencer MJ. Apoptosis precedes necrosis of dystrophin-deficient muscle. J Cell Sci. 1995;108:2197–2204. doi: 10.1242/jcs.108.6.2197. [DOI] [PubMed] [Google Scholar]
- 35.Turpin SM, Lancaster GI, Darby I, Febbraio MA, Watt MJ. Apoptosis in skeletal muscle myotubes is induced by ceramides and is positively related to insulin resistance. Am J Physiol Endocrinol Metab. 2006;291:E1341–E1350. doi: 10.1152/ajpendo.00095.2006. [DOI] [PubMed] [Google Scholar]
- 36.Percy ME, Andrews DF, Thompson MW. Serum creatine kinase in the detection of Duchenne muscular dystrophy carriers: effects of season and multiple testing. Muscle Nerve. 1982;5:58–64. doi: 10.1002/mus.880050111. [DOI] [PubMed] [Google Scholar]
- 37.Glesby MJ, Rosenmann E, Nylen EG, Wrogemann K. Serum CK, calcium, magnesium, and oxidative phosphorylation in mdx mouse muscular dystrophy. Muscle Nerve. 1988;11:852–856. doi: 10.1002/mus.880110809. [DOI] [PubMed] [Google Scholar]
- 38.Manning J, O’Malley D. What has the mdx mouse model of duchenne muscular dystrophy contributed to our understanding of this disease? J Muscle Res Cell Motil. 2015;36:155–167. doi: 10.1007/s10974-015-9406-4. [DOI] [PubMed] [Google Scholar]
- 39.Lagrota-Candido J, Vasconcellos R, Cavalcanti M, Bozza M, Savino W, Quirico-Santos T. Resolution of skeletal muscle inflammation in mdx dystrophic mouse is accompanied by increased immunoglobulin and interferon-gamma production. Int J Exp Pathol. 2002;83:121–132. doi: 10.1046/j.1365-2613.2002.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polfliet MM, Fabriek BO, Daniëls WP, Dijkstra CD, van den Berg TK. The rat macrophage scavenger receptor CD163: expression, regulation and role in inflammatory mediator production. Immunobiology. 2006;211:419–425. doi: 10.1016/j.imbio.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 41.Pigozzo SR, Da Re L, Romualdi C, Mazzara PG, Galletta E, Fletcher S, et al. Revertant fibers in the mdx murine model of Duchenne muscular dystrophy: an age- and muscle-related reappraisal. PLoS One. 2013;8:e72147. doi: 10.1371/journal.pone.0072147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 43.von Maltzahn J, Euwens C, Willecke K, Söhl G. The novel mouse connexin39 gene is expressed in developing striated muscle. J Cell Sci. 2004;117:5381–5392. doi: 10.1242/jcs.01413. [DOI] [PubMed] [Google Scholar]
- 44.Iwata Y, Katanosaka Y, Hisamitsu T, Wakabayashi S. Enhanced Na+/H+ exchange activity contributes to the pathogenesis of muscular dystrophy via involvement of P2 receptors. Am J Pathol. 2007;171:1576–1587. doi: 10.2353/ajpath.2007.070452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cerecedo D, Mondragón R, Cisneros B, Martínez-Pérez F, Martínez-Rojas D, Rendón A. Role of dystrophins and utrophins in platelet adhesion process. Br J Haematol. 2006;134:83–91. doi: 10.1111/j.1365-2141.2006.06120.x. [DOI] [PubMed] [Google Scholar]
- 46.Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 47.Orellana JA, Díaz E, Schalper KA, Vargas AA, Bennett MV, Sáez JC. Cation permeation through connexin 43 hemichannels is cooperative, competitive and saturable with parameters depending on the permeant species. Biochem Biophys Res Commun. 2011;409:603–609. doi: 10.1016/j.bbrc.2011.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garré JM, Retamal MA, Cassina P, Barbeito L, Bukauskas FF, Sáez JC, et al. FGF-1 induces ATP release from spinal astrocytes in culture and opens pannexin and connexin hemichannels. Proc Natl Acad Sci USA. 2010;107:22659–22664. doi: 10.1073/pnas.1013793107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Altamirano F, López JR, Henríquez C, Molinski T, Allen PD, Jaimovich E. Increased resting intracellular calcium modulates NF-κB-dependent inducible nitric-oxide synthase gene expression in dystrophic mdx skeletal myotubes. J Biol Chem. 2012;287:20876–20887. doi: 10.1074/jbc.M112.344929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whitehead NP, Pham C, Gervasio OL, Allen DG. N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J Physiol. 2008;586:2003–2014. doi: 10.1113/jphysiol.2007.148338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lawler JM. Exacerbation of pathology by oxidative stress in respiratory and locomotor muscles with Duchenne muscular dystrophy. J Physiol. 2011;589:2161–2170. doi: 10.1113/jphysiol.2011.207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyatake M, Miike T, Zhao J, Yoshioka K, Uchino M, Usuku G. Possible systemic smooth muscle layer dysfunction due to a deficiency of dystrophin in Duchenne muscular dystrophy. J Neurol Sci. 1989;93:11–17. doi: 10.1016/0022-510X(89)90157-3. [DOI] [PubMed] [Google Scholar]
- 53.Higuchi I, Niiyama T, Uchida Y, Inose M, Nakagawa M, Arimura K, et al. Multiple episodes of thrombosis in a patient with Becker muscular dystrophy with marked expression of utrophin on the muscle cell membrane. Acta Neuropathol. 1999;98:313–316. doi: 10.1007/s004010051086. [DOI] [PubMed] [Google Scholar]
- 54.Martin EA, Barresi R, Byrne BJ, Tsimerinov EI, Scott BL, Walker AE, et al. Tadalafil alleviates muscle ischemia in patients with Becker muscular dystrophy. Sci Transl Med. 2012;4:162ra155. doi: 10.1126/scitranslmed.3004327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Theis M, de Wit C, Schlaeger TM, Eckardt D, Krüger O, Döring B, et al. Endothelium-specific replacement of the connexin43 coding region by a lacZ reporter gene. Genesis. 2001;29:1–13. doi: 10.1002/1526-968X(200101)29:1<1::AID-GENE1000>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 56.Maxeiner S, Dedek K, Janssen-Bienhold U, Ammermüller J, Brune H, Kirsch T, et al. Deletion of connexin45 in mouse retinal neurons disrupts the rod/cone signaling pathway between AII amacrine and ON cone bipolar cells and leads to impaired visual transmission. J Neurosci. 2005;25:566–576. doi: 10.1523/JNEUROSCI.3232-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li S, Czubryt MP, McAnally J, Bassel-Duby R, Richardson JA, Wiebel FF, et al. Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specificgene deletion in mice. Proc Natl Acad Sci USA. 2005;102:1082–1087. doi: 10.1073/pnas.0409103102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trebbin AL, Hoey AJ. A novel and simple method for genotyping the mdx mouse using high-resolution melt polymerase chain reaction. Muscle Nerve. 2009;39:603–608. doi: 10.1002/mus.21215. [DOI] [PubMed] [Google Scholar]
- 59.Shin JH, Hakim CH, Zhang K, Duan D. Genotyping mdx, mdx3cv, and mdx4cv mice by primer competition polymerase chain reaction. Muscle Nerve. 2011;43:283–286. doi: 10.1002/mus.21873. [DOI] [PMC free article] [PubMed] [Google Scholar]