

Abstract
In the process of leukocyte migration from the circulation across the vascular wall, the crosstalk with endothelial cells that line the blood vessels is essential. It is now firmly established that in endothelial cells important signaling events are initiated upon leukocyte adhesion that impinge on the regulation of cell–cell contact and control the efficiency of transendothelial migration. In addition, several external factors such as shear force and vascular stiffness were recently identified as important regulators of endothelial signaling and, consequently, leukocyte transmigration. Here, I review recent insights into endothelial signaling events that are linked to leukocyte migration across the vessel wall. In this field, protein phosphorylation and Rho-mediated cytoskeletal dynamics are still widely studied using increasingly sophisticated mouse models. In addition, activation of tyrosine phosphatases, changes in endothelial cell stiffness as well as different vascular beds have all been established as important factors in endothelial signaling and leukocyte transmigration. Finally, I address less-well-studied but interesting components in the endothelium that also control transendothelial migration, such as the ephrins and their Eph receptors, that provide novel insights in the complexity associated with this process.
Keywords: VE-cadherin, Tyrosine phosphorylation, Mechanosignaling, Rho GTPases
Introduction
For experienced cell biologists as well as laymen young and old, moving cells are among the most fascinating biological phenomena to watch. Particularly intriguing is the migration of leukocytes across the endothelial layer which lines the inside of blood vessels, a hallmark of inflammation. This transendothelial migration (TEM, also known as diapedesis) occurs constitutively but its frequency increases in response to various stimuli and under different conditions, with acute and (chronic) inflammation as the best-known examples. TEM is accompanied by dramatic shape changes of the leukocyte that occur rapidly in a sometimes turbulent environment, culminating in the breaching of what intuitively appears to be an essential, not-to-be-breached barrier: the vascular endothelium [1, 2] The now firmly established multi-step process of TEM has been introduced many times in ever so many reviews and research papers over the past ~25 years [3–5]. I therefore choose to omit such an introduction from the main text and refer readers, less familiar with the TEM paradigm, to Box 1.
Many of the steps that together comprise leukocyte TEM have been extensively studied over past decades, in particular at the level of the leukocyte [6]. This sequence of cellular behavior, defined originally by the groundbreaking work of Eugene Butcher and Timothy Springer [7, 8], is key to inflammatory responses and immune surveillance. The field has studied in detail myriad chemokines and their G-protein coupled receptors as well as a growing number of adhesion molecules and their ligands (i.e. integrins, cadherins, etc.), both on leukocytes and on the endothelium. It would appear from most literature that in this ‘pas de deux’, only the leukocytes are dancing, but this is not the case. Over the past 15–20 years we have seen a marked increase in research on the endothelial cells and their contribution to efficient leukocyte TEM (see Box 1) [3, 9–11]. Note that although TEM is studied mainly in in vitro and in vivo settings that mimic acute inflammation, it is generally assumed that chronic inflammatory disorders are also based on an analogous multi-step TEM process. Similarly, it is accepted that analogous events that entail the passage of cells across vascular barriers, such as tumor cell metastasis and stem cell homing, are driven by such multi-step mechanisms [12–14].
It is important to underscore that TEM can occur under quite different conditions (absence or presence of shear force, resting or inflamed vessels, etc.) or different tissues (soft or stiff), which will affect one or more aspects of the classical multistep paradigm. Also the endothelium itself is far from homogeneous. This is true at the level of tissues and organs [15, 16], at the level of individual cells and at the subcellular level, where different domains in the apical membrane and intercellular junctions regulate TEM [11, 17, 18]. Consequently, the mechanisms that control TEM may not be quite as generic as we may assume them to be. Moreover, to complicate matters further, variants of transmigratory behavior such as ‘hesitant’ and ‘reverse’ TEM have been identified in recent years [19–21]. Finally, the majority of mechanistic conclusions that form the basis of current models are derived from in vitro assays where endothelial cells are cultured on artificial substrates such as plastic or glass. More and more however, relevant data is generated in vivo using intravital imaging in mice [19, 21–25]. Although intravital imaging is lagging behind in structural and molecular resolution as compared to high resolution in vitro imaging., this technique has become invaluable for our appreciation of tissue- and leukocyte-specific aspects and behavior, as well as of the overall kinetics of TEM in real vessels in live animals. In parallel, more sophisticated ex vivo and in vitro models for human vessels are being developed [26–28] which will allow the field to bridge the gap between current in vitro approaches with human cells and in vivo work in mice. This is important not only because molecular pathways and responses can differ substantially between mice and men (see also [29, 30]), but also to answer to calls from society and government agencies to reduce as much as possible the use of laboratory animals.
Leukocyte TEM is the subject of active investigation already for many years and covers ~2000 publications which address many aspects that cannot all be discussed here. In fact, I choose to limit the topic (i.e. endothelial signaling) and time-frame covered to the most recent ~5 years, taking off where several earlier key reviews ended [1, 6, 31]. I will start with a brief summary for those readers less familiar with endothelial signaling in TEM to set the stage of the field some 5 years ago. Next I will focus on a series of molecular mechanisms that control TEM and that have either been studied in more detail in recent years (such as new insights on the role of protein phosphorylation) or represent emerging concepts (i.e. mechano-signaling).
Previously on: endothelial signaling in leukocyte TEM
Textbook images tend to depict the endothelium as a rather inert layer of bulky cells, a representation which is not very close to our current view of this cell type. Firstly, vascular endothelial cells are usually quite flat and as such very different from epithelial cells. Importantly, however, like epithelial cells they do show apical–basal polarity [32] and asymmetric distribution of proteins, such as caveolin, during migration [33]. Most recently, EC polarity was implicated in lumen formation during angiogenesis [34]. Secondly, endothelial cells are not inert at all; not in resting conditions, and certainly not during inflammatory responses and leukocyte TEM. The first study that introduced the concept of endothelial signaling contributing to leukocyte TEM was based on the finding that adhesion of neutrophils, mimicked by stimulation with soluble receptor ligands, raises intracellular calcium levels [35] and promotes permeability in the endothelium. Moreover, clamping of intracellular calcium in the endothelium was found to limit neutrophil-induced permeability as well as TEM across monolayers of HUVEC. Thus, adherent leukocytes trigger signaling events in the endothelium that appear to promote efficient TEM.
Following up on this seminal paper by the Silverstein lab, many groups identified endothelial signaling events that were suggested to be required for efficient TEM. Most notably, the activation of a rho-rhokinase (ROCK)–myosin light chain kinase (MLCK)-pathway which stimulates endothelial, actomyosin-based contractility has gained a lot of interest [36–40]. This is because this pathway is central to agonist-induced loss of endothelial cell–cell contact and increased permeability as induced by thrombin or histamine, for example [41, 42]. And indeed, many studies showed that inhibition of any of these components reduced leukocyte TEM in vitro. These studies focused largely on intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1)-mediated signaling (reviewed in [9, 31, 43, 44]). In addition to these, many other cell surface molecules have been implicated over the past two decades in leukocyte adhesion and TEM (e.g. platelet endothelial cell adhesion molecule-1 (PECAM-1), the junctional adhesion molecules (JAMs), CD99, endothelial cell-selective adhesion molecule (ESAM), etc. [19, 45–50]). Although it is likely that any clustered transmembrane molecule will initiate some form of intracellular signaling, the pathways downstream of adhesion molecules, other than ICAM-1 or VCAM-1, have not or only partially been described.
In addition to leukocyte adhesion-induced contractility in EC, it was recognized early on that protein phosphorylation was an important aspect of EC signaling in TEM as well. Many studies have looked at protein tyrosine phosphorylation and investigated the role of the non-receptor tyrosine kinase p60Src, in addition to the Pyk2 tyrosine kinase and various Ser/Thr kinases [37, 51–58]. In parallel, several groups established a role for Rac1-dependent, NADPH oxidase (NOX)-generated reactive oxygen species (ROS), e.g. downstream from cross-linked VCAM-1 [59–61]. These ROS, known to inhibit intracellular protein tyrosine phosphatases thus promoting increased tyrosine kinase activity, were suggested to be central to the kinase-based regulation of VE-cadherin [59, 62, 63] or were proposed to activate metallo-proteases in the extracellular environment, which, in turn, were shown to activate the intracellular tyrosine phosphatase PTP1B [64]. The VCAM-1-mediated Rac1–NOX–ROS pathway was recently linked to Pyk2-mediated regulation of VE-PTP [62], initiating its dissociation from VE-cadherin, a requirement for efficient leukocyte TEM [65]. Importantly, recent studies using elegant mouse models expressing phosphorylation-deficient mutants of VE-cadherin have provided new insights in the relevance and regulation of this pathway (see below).
A confounding issue in biochemical studies on leukocyte-induced endothelial signaling has always been that simultaneous lysis of the two types of cell makes it hard to discriminate which protein, activated or modified by posttranslational modification, is derived from which cell type. To circumvent this problem, may labs, including ours, resorted to the use of anti-ICAM-1 or anti-VCAM-1 antibodies [59]. Initially, these were used in solution, in combination with a secondary cross-linking antibody. This approach results in a global activation of available ICAM-1/VCAM-1 molecules on the cell surface, leading to what we now consider as exaggerated cellular responses, accompanied by dramatic EC contraction and formation of large intercellular gaps [59]. Later on, many laboratories have used small antibody-coated beads to induce more local signaling events that could be imaged using increasingly sophisticated tools, such as GFP-fusion proteins [66–68], or biosensors. Yet, this approach also has important drawbacks, because such beads, although resembling leukocytes in size, tend to be immobile. This leads to prolonged local signaling which may simplify its detection, but should be interpreted with caution. The use of such beads, however, has stimulated work on so-called ‘docking structures’ or ‘transmigratory cups’; endothelial apical membrane protrusions, initially described by Barreiro et al. [69, 70], that also form around adherent leukocytes. The identification of these structures sparked a series of studies addressing the connection between clustered ICAM-1/VCAM-1 and the endothelial cortical actin cytoskeleton. Like any other transmembrane adhesion molecule, ICAM1/VCAM-1 require cytoskeletal anchoring for proper adhesive function. A number of actin-binding and -regulating proteins were found by us and other labs to bind to the (very short) intracellular domains of ICAM-1/VCAM-1 and to regulate their clustering and adhesive function as part of a complicated feed-back loop [51, 53, 66, 71–76]. The finding that actin dynamics in EC is a key feature driving local clustering of integrin ligands and concomitant leukocyte behavior and TEM was extended later to the notion that endothelial cell stiffness, a function of actin dynamics and crosslinking, also regulates leukocyte adhesion, spreading and TEM [17, 74] (discussed in more detail below).
Finally, a topic that has been very much present in the field over the past 10–15 years is the phenomenon of transcellular migration, where leukocytes cross the endothelial barrier by migrating through a EC body, rather than through a cell–cell junction (i.e. the paracellular pathway) [18, 77–79]. It is now well established that the transcellular pathway exists in vivo and in vitro, but its relative importance and/or tissue- or cell type specificity has been a topic of debate. The in vivo results by the Vestweber lab [65] that showed that a F-actin-locked VE-cadherin mutant, restricting junctional opening and permeability, efficiently prevents leukocyte TEM, left little room for a (compensatory) transcellular pathway, at least in the inflamed skin, lung and cremaster muscle models used in this work. Whereas we think that we understand in part which intracellular signaling in EC may control junctional integrity during TEM, such signaling has remained largely obscure for transcellular diapedesis, with the exception of experiments implicating membrane traffic-associated proteins such as caveolin or VASP [18, 78, 80]. The more recent insight [81] that cellular/vascular stiffness may also be a factor in the choice of which transmigration route is used is interesting, as it may explain some earlier findings in the field (see below).
In the current overview, I make an attempt to sketch where the TEM-field is in 2016 focusing on endothelial signaling. New imaging and cellular analysis tools, i.e. hardware, software and biosensors, have entered this area of research, in addition to the use of very sophisticated in vivo models. Many of the topics discussed below represent extensions of earlier findings, but I have also included a brief discussion of some recently identified, potentially interesting, EC components at the end.
Protein kinases and transendothelial migration
It is well established that leukocyte TEM is regulated by protein phosphorylation in EC. Both Ser/Thr kinases as well as tyrosine kinases and -phosphatases have been implicated, sometimes based on the use of pharmacological inhibitors, but also using elegant mouse models. Here we will address some recent studies on the contribution of these two classes of protein kinases to leukocyte TEM.
Ser/Thr kinases
A role for endothelial protein kinase C (PKC) activity in leukocyte TEM was proposed already close to 20 years ago in the context of hypoxia-induced phosphorylation of PECAM-1 in HUVEC, promoting monocyte TEM [82]. Later, PKC activity was linked to clustered ICAM-1 induced endothelial actin stress fiber formation in rat brain EC and its inhibition was found to reduce T lymphocyte TEM [36, 37]. The group of Cook-Mills identified PKCα, downstream from NADPH oxidase, as a VCAM-1-regulated component in mouse microvascular cells, which controls phosphorylation and activation of the cytosolic tyrosine phosphatase PTP1B [83]. This was found to mediate TEM as inhibition of the pathway by blocking endothelial PTP1B reduced TEM of splenic T lymphocytes [64, 83]. Intriguingly, a more recent study in PTP1B−/− mice showed that PTP1B acts as a negative regulator of allergic responses and that loss of PTP1B promotes leukocyte adhesion in vivo in OVA-challenged mice and TEM in vitro across WT EC [84]. A subsequent study using chimeric mice (PTP1B deficiency in non-hematopoietic cells vs. WT leukocytes) and mice with induced deletion of PTP1B in the endothelium showed that endothelial PTP1B is required for leukocyte influx in the lung [85]. Thus, these studies identified markedly different contributions to TEM for PTP1B in leukocytes vs. PTP1B in the vascular endothelium.
A recent study, [86] focusing on the role of endothelial PKCβ in the regulation of T cell influx into the brain, showed that the PKCβ inhibitor and oral drug LY-317615 was effective in reducing T cell influx. The mechanism involved was partially resolved and shown to rely on inhibition of PKCβ-mediated upregulation of mRNAs encoding junctional proteins, in particular claudin-5, in brain endothelial cells. This is intriguing as claudin-5 expression is suppressed in brain inflammation. The concomitant LY-317615-induced stabilization of the blood–brain barrier was proposed to be responsible for retention of CD4+ T-cells in the vasculature, although additional PKCβ-dependent mechanisms that reduce effective TEM of T cells could not be excluded, such as those involving the small GTPase Rac1 or the TGF-β pathway [86]. Importantly, LY-317615 did reduce experimental autoimmune encephalomyelitis (EAE) in vivo in a mouse model for multiple sclerosis, most likely by suppressing influx of encephalitogenic T cells. This data highlights the potential clinical application of targeting the vascular (i.e. brain) endothelium to limit inflammatory disease.
In a recent paper implicating soluble adenylate cyclase (sAC), Watson et al. [87] showed that the homophilic adhesion molecule CD99 also employs intracellular signaling in EC to regulate leukocyte TEM. It was shown previously that inhibition of CD99 blocks TEM by stalling the transmigration leukocyte halfway through the endothelial cell–cell junctions. The analysis by Watson et al. indicates that CD99 engagement leads to recruitment and activation of sAC, resulting in the generation of cAMP and activation of PKA. In line with this model, pharmacological inhibition of sAC reduces leukocyte TEM in vivo to a similar level as blocking CD99 using antibodies. An interesting finding in this work was that clustering of CD99 using an adhesion-blocking antibody restored neutrophil TEM, indicating that it is the clustering-induced intracellular signaling, rather than the adhesive function of CD99 which is required to promote TEM. What remains unknown is how endothelial PKA signaling controls TEM and why inhibition of this pathway blocks TEM by preventing full passage of the leukocyte across inter-endothelial junctions.
Several recent studies addressed the role of endothelial PI3-kinases (PI3K) in the control of endothelial barrier function and leukocyte TEM. Nakhaei-Nejad et al. [88] showed that pretreatment of HUVEC monolayers with the PI3K inhibitors wortmannin or LY-294002, or transfection with siRNA to reduce expression of the regulatory p85 subunit of Class 1A PI3K impaired transmigration (20–70 % reduction) of human peripheral blood lymphocytes under flow. This was linked to defects not in lymphocyte adhesion and locomotion on the EC surface, but rather in the ability to cross an endothelial cell–cell junction [88]. Similar findings were reported by Cain et al. [89] using various PI3K inhibitors as well as wortmannin. Focusing more on the role of individual p110 catalytic subunits of PI3K, the group of Anne Ridley further showed, using siRNA approaches in HUVECs, that loss of p110α induced the most prominent inhibition of TNFα-induced permeability and paracellular transmigration of monocytic THP-1 cells. Also here, there was no effect of the loss of endothelial p110α on the adhesion of the leukocytes [90].
The signaling pathway which is relevant downstream of this PI3K-mediated regulation of TEM was suggested to involve the Pyk2 tyrosine kinase, which was previously implicated in the regulation of VE-cadherin function [91]. SiRNA-mediated loss of p110α was found to reduce the association of tyrosine phosphorylated Pyk2 with VE-cadherin and to reduce the tyrosine phosphorylation of VE-cadherin itself. Moreover, siRNA-mediated loss of Pyk2 reduced TNFα-induced permeability and, in conjunction with loss of Rac1, reduced TEM of THP1 cells [90]. PI3K (p110α) was also found to be required for basal RhoA (but not RhoB or RhoC) and Rac1 activity in TNFα-treated EC. This is unusual as RhoA and Rac1 each have distinct effects on actin-based junctional phenotypes: RhoA, in conjunction with myosin, drives the generation of mechanical pulling forces and formation of focal adherens junctions [92], whereas Rac1 promotes formation of cell–cell contacts due to its stimulation of actin polymerization and membrane protrusions [93].
The Ser/Thr kinase Akt (Protein kinase B) is a major target of PI3K and Akt has been firmly established as an important regulator of many cellular responses, including inflammation [94]. Yet, a role for endothelial Akt in leukocyte TEM has not been studied extensively yet. Using isoform-specific knock out mice, Di Lorenzo et al. [95] have shown that endothelial Akt1, but not Akt2, is required for the induction of paw edema and for the concomitant influx of monocytes and EC. Mechanistically, this was linked to Akt1-mediated regulation of permeability as siRNA-mediated loss of Akt1 markedly protected against histamine-induced loss of transendothelial electrical resistance. Downstream from Akt1 in EC, there appears to be an important role for eNOS (endothelial Nitric Oxide Synthase). ENOS is central to the regulation of endothelial integrity through its main product, Nitric Oxide, which protects against vascular constriction and as such stabilizes blood vessels and limits increases in blood pressure. Several studies have implicated eNOS in ICAM-1-dependent leukocyte TEM. Martinelli et al. [96] found that eNOS activity, but not Akt or PI3K activity, is required for lymphocyte TEM across various types of primary and immortalized EC. A more recent study in eNOS-deficient mice showed that leukocyte influx in a model for lung inflammation was reduced by 50 % as compared to the influx in control mice [97]. In this study, ICAM-1 mediated activation of eNOS was found to be downstream of a p60Src-Akt-dependent signaling pathway. This pathway was linked to phosphorylation of PECAM-1, albeit that adhesive activity of PECAM-1 was not altered.
Thus, several studies have provided evidence for components of an endothelial PI3K/Akt-eNOS signaling unit to regulate TEM. However, the relative contribution of the various components and the mechanistic connection to junctional proteins and TEM remain to be established further.
Tyrosine phosphorylation
An intriguing and complicated issue in the field of TEM and vascular permeability has been (and still is) the regulation by protein tyrosine kinases, particularly of endothelial junctional proteins and actin-binding adapter proteins. Regarding the latter, the phosphorylation of the adapter protein cortactin by the cytosolic tyrosine kinase p60src has been most intensely studied. Several studies identified Src as a key regulator of ICAM-1 clustering and function, through its phosphorylation of cortactin, [71]. Consequently, Src-mediated cortactin phosphorylation was shown to be required for efficient leukocyte TEM in vitro and cortactin was later shown to be essential for endothelial barrier function and neutrophil TEM in vivo [73].
Regarding junctional proteins, the tyrosine phosphorylation of VE-cadherin has gained the most interest over the years. This topic has recently also been studied in detail in elegant mouse models. The first papers on this topic showed that not only cell density and agonists such as VEGF and histamine, but also adhesion of activated neutrophils could induce tyrosine phosphorylation of VE-cadherin and associated proteins [98–101]. The consensus has been that tyrosine phosphorylation of junctional proteins reduces their adhesive function and leads to a (transient) loss of cell–cell contact. Over the past 15 years, this area of research was seemingly accelerated by the development of commercially available, assumed phospho-specific antibodies to VE-cadherin. These reagents have been used in search for a functional link of phosphorylation with VE-cadherin-mediated vascular permeability and the regulation of leukocyte TEM. However, recent studies have shown that most, if not all, of these commercial antibodies were not specific. To resolve this, the Vestweber and Dejana labs have gone to considerable lengths to develop and use well-characterized site-specific anti-phospho-VE-cadherin antibodies [102, 103]. Use of these antibodies, in combination with site-specific Y-F mutants of VE-cadherin for specificity conformation, have now both clarified and complicated the overall picture of regulation of junctional proteins by tyrosine phosphorylation (Fig. 1).
Fig. 1.
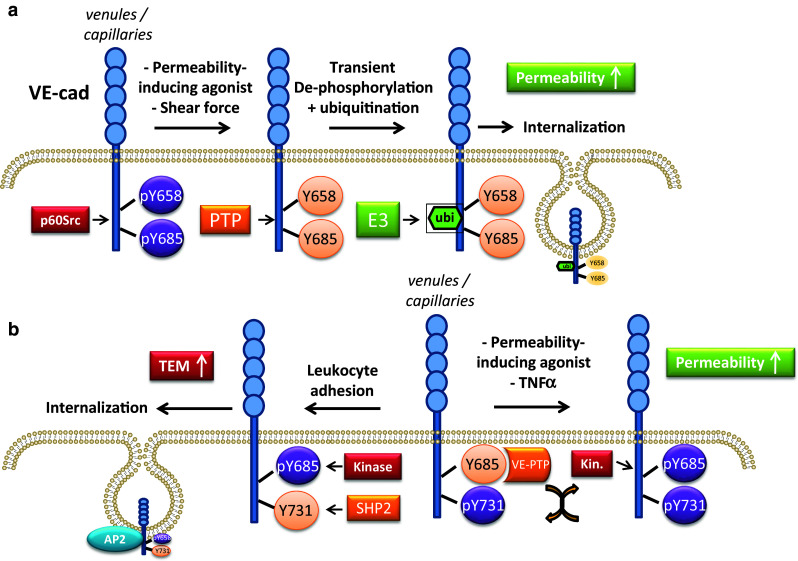
Differential phosphorylation of VE-cadherin in control of TEM and permeability. a Schematic overview of (de)phosphorylation events as described in Orsenigo et al. [102]. Here, VE-cadherin is phosphorylated constitutively on Y658 and Y685 in venules and capillaries. Permeability-inducing stimuli transiently as well as shear forces constitutively downregulate this phosphorylation. Subsequent ubiquitination by an unknown ubiquitin E3-ligase initiates the internalization of de-phosphorylated VE-cadherin. b In the model derived from data by Wessel et al., Y731 is constitutively phosphorylated in venules and capillaries and Y685 is bound t the VE-PTP phosphatase, keeping phosphorylation of Y685 low. Permeability-inducing stimuli induced dissociation of VE-PTP allowing phosphorylation of Y685. Y731 does not appear to be involved in control of permeability, In contrast, leukocyte adhesion induced de-phosphorylation of Y731 by SHP2, concomitant with increased phosphorylation of Y685. The latter even may involve a p60src-FAK pathway that was implicated in VEGF-stimulated tumor cell extravasation. These events initiate binding of the AP2 component α-adaptin, triggering internalization of Y731 dephosphorylated VE-cadherin. Note that the indicated events are supposed to take place at EC junctions
Several residues in the cytoplasmic domain of VE-cadherin (Y645; Y658; Y685 Y731; Y733) were analyzed in the initial studies for their requirement for lymphocyte or neutrophil TEM [57, 104]. These studies relied on ectopic expression of Y-F VE-cadherin mutants in human or murine endothelial cells followed by analysis of neutrophil [57] or lymphocyte [104] TEM under static conditions. In these studies Y645, Y731 and Y733 were identified as essential for efficient TEM. Using a combination of pharmacological and dominant negative inhibitors, the Src and Pyk2 tyrosine kinases were implicated in the phosphorylation of VE-cadherin in human EC [57] but not in those of the mouse [104].
The Dejana lab developed antibodies specific for Y658 and Y685 in the intracellular domain of VE-cadherin [103]. These antibodies detect phosphorylated VE-cadherin in resting venules and capillaries but, intriguingly, not in arterioles. This difference was proposed to be caused by differences in hemodynamics: moderate shear stress promoted Src-mediated phosphorylation of Y658 on VE-cadherin, but higher stress, such as in arteries, inhibited phosphorylation. This was shown both in vitro as well as in vivo as deduced from elegant in vivo shunt experiments in rats, connecting the jugular vein to the carotid artery. Importantly, active Src (pY418-Src) was found at cell–cell contacts in venules but was absent from arterioles. This mechanism to regulate cell–cell junctions was also implicated in the induction of vascular permeability induced by agonists such as bradykinin [103]. Regarding regulation of permeability, the Orsenigo study showed that permeability-inducing stimuli led to a dephosphorylation of both Y658 and Y685, initiating ubiquitination and internalization of VE-cadherin (Fig. 1).
What is relevant to consider is that TEM of leukocytes occurs primarily via the paracellular pathway and thus requires local and transient opening of cell–cell contacts. Consequently, in many studies, the regulation of permeability has been considered particularly relevant for TEM. Although one cannot deny the functional connection between vascular permeability and leukocyte diapedesis, this relationship is more complex than originally thought. This was most strikingly shown by a study from the Vestweber lab, in which it was demonstrated that regulation of vascular permeability in mice, induced by VEGF or histamine, requires the phosphorylation of Y685 in the intracellular domain of VE-cadherin, but not the phosphorylation of Y731 [102]. This was analyzed using novel phospho-specific antibodies and using transgenic mice in which point-mutants of VE-cadherin were targeted to the endogenous VE-cadherin locus. Intriguingly, induction of phosphorylation of Y685, which is also a target of Src [105], occurred selectively in venules and a subset of capillaries, but not in arterioles. Also, these authors reported that phosphorylation of Y685 is absent in unstimulated blood vessel endothelium or human EC cultured in vitro but is rapidly induced by VEGF and histamine as well as TNFα and adherent T-cells. The reason for the lack of baseline pY685 signal in the study from the Vestweber lab, in marked contrast with the findings by Orsenigo et al. [103], is unknown but could be due to differences in antibody sensitivity or the fact that the Vestweber lab developed antibodies that were specific for either the mouse and human pY685 peptides. In contrast to pY685, pY731 is detectable in unstimulated endothelium and its phosphorylation is not affected by VEGF or TNFα.
In marked contrast to VE-cadherin Y685, the phosphorylation of which regulates permeability, Y731 was found to be relevant for TEM in an acute inflammation model (Il-1β-stimulated cremaster muscle). In fact, the dephosphorylation of Y731 was induced by T-cell adhesion to bEnd.5 mouse cells in vitro and was shown to be required for efficient TEM in vivo and in vitro. This dephosphorylation at Y731 required the tyrosine phosphatase SHP2, but not the VE-cadherin associated phosphatases VE-PTP or DEP1, and was suggested to control α-adaptin-mediated internalization of VE-cadherin [102]. The model that emerges from this work is that kinase driven phosphorylation (of Y685) controls permeability in venules whereas SHP2-driven dephosphorylation (of Y731) and dissociation of the associated VE-PTP controls leukocyte TEM by regulating VE-cadherin internalization.
In addition to the above-described tyrosine phosphorylation at endothelial cell–cell junctions, there is some information on the role of focal adhesion-related tyrosine kinases, in particular the Focal Adhesion Kinase FAK, in TEM. Transmigration of breast cancer cells across lung microvascular EC was blocked by siRNA-mediated reduction of FAK or the expression of a dominant negative protein (coined FRNK) [106]; similar findings were reported later for neutrophils migrating across TNFα-treated HUVEC [107]. Jean et al. [108] recently provided evidence that FAK phosphorylates Y658 in tumor samples, as well as, in response to VEGF-A stimulation, in lung tissue from mice and in murine and human ECs in vitro. This effect was accompanied by VEGF-induced Src and FAK translocation to junctions. Further experiments showed that FAK-mediated Y658 phosphorylation regulates EC permeability and tumor cell TEM in vitro. Moreover, genetically engineered mice, expressing a kinase-dead FAK in the endothelium, showed reduced lung infiltration by tumor cells concomitant with reduced tumor growth in the lungs as well as reduced lung metastasis [108]. A related study also identified FAK as an important mediator of VEGF-induced TEM of B16 melanoma cells [109].
Clearly, protein tyrosine phosphorylation is a central regulatory event in the endothelial control of TEM [110]. It is interesting to conclude that such tyrosine phosphorylation is associated with proteins that control cell–cell contact and with proteins that control cell–matrix adhesion. There is limited insight into the crosstalk between these two adhesive systems in EC in the context of TEM, but it is likely that the endothelial environment, provided by the matrix and associated cells such as pericytes and smooth muscle cells, is yet another important regulatory component in driving TEM efficiency. Sensing this environment is a key function of integrins and analysis of the associated signal transducers such as FAK and their functional connection to the regulation of VE-cadherin function and TEM, e.g. through force sensing molecules such as vinculin [92], represents a challenge for future research.
Endothelial guanine nucleotide exchange factors (GEFs)
In marked contrast to the large number of studies on the role of endothelial Rho GTPases in leukocyte transmigration [1, 11], there is only limited information on which of the Rho-or Rac1-activating GEFs is relevant. The Rac1GEF P-Rex1 (phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchange factor 1) was identified as a key regulator of TNFα-induced endothelial permeability and neutrophil TEM [111]. Importantly, loss of P-REX1 was shown to reduce TNFα-induced lung permeability in vivo, concomitant with a reduction in influx of neutrophils and macrophages in the BAL fluid. Mechanistically, P-Rex1-mediated and PI3-K-dependent activation of Rac1 was found to result in production of ROS which are known to induce a transient loss of cell–cell contact. Moreover, siRNA-mediated loss of P-Rex1 impaired TNFα-induced upregulation of ICAM-1 expression, which may well explain the concomitant reduction in TEM [111].
A small number of RhoGEFs has been identified to be more directly, i.e. downstream of activated, i.e. clustered ICAM-1, involved in regulating neutrophil TEM. In TNFα-activated, primary HUVEC, the RhoA homologue RhoG is activated as a consequence of ICAM-1 clustering, an effect that is mediated by SGEF (SH3-containing GEF) [68]. SGEF binds, via its SH3 domain, directly to a proline-rich sequence in the intracellular domain of ICAM-1. This also makes SGEF specific for ICAM-1-induced signaling, since the intracellular domain of VCAM-1 does not encode such a proline-rich region. Importantly, SGEF activity was not required for its binding to ICAM-1 [68], which indicates that this association is a direct consequence of ICAM-1 clustering, rather than of downstream signaling. Analogous to the induction of dorsal membrane ruffles in fibroblasts [112], SGEF and RhoG mediate formation of apical membrane protrusions that form in response to beads-induced ICAM-1 clustering as well as leukocyte adhesion. These ‘docking structures’ or ‘transmigratory cups’ are now a well-established endothelial feature that accompanies leukocyte TEM, be it though the para- and the transcellular pathway [68, 69, 80, 113].
The relevance of SGEF for leukocyte TEM in vivo was more recently further underscored using mice genetically deficient for functional SGEF. These otherwise viable mice were crossed with ApoE−/− deficient mice, and the progeny was fed Western Diet to induce atherosclerosis. Subsequent analysis showed that, at various locations along the aorta, the lack of SGEF resulted in a 40–60 % reduction in size of atherosclerotic lesions [114]. This effect was accompanied by (1) lack of RhoG activation in the endothelium, (2) reduced formation of apical membrane protrusions around adherent anti-ICAM-1 coated beads and (3) a reduction in beads adhesion to the aortic wall. This data suggests that also in vivo, the ICAM-1–SGEF–RhoG signaling axis mediates efficient leukocyte TEM driving atherosclerosis and likely also other inflammatory disorders.
Another GEF implicated in formation of apical membrane protrusions is Trio. Trio encodes two GEF domains; the N-terminal domain activates Rac1/RhoG and the C-terminal domain activates RhoA [115]. In addition, Trio encodes a Ser/Thr protein kinase domain in its C-terminus. In primary HUVEC, Trio expression is strongly upregulated by TNFα [116]. Trio and Trio-mediated Rac1 activation are in turn required for TNFα-induced upregulation of ICAM-1 and VCAM-1 as well as, consequently, for neutrophil TEM. Trio is also more directly involved in neutrophil TEM, since ICAM-1 clustering rapidly activates Trio and, consequently, Rac1 and RhoG in TNFα-treated primary HUVEC [117]. Interestingly, ICAM-1 induced Trio activation depends on filamin. Filamin is a dimeric actin-binding and -regulating protein that was previously shown to bind to clustered ICAM-1 [66]. Moreover, the first GEF domain of Trio (known as TrioD1) binds, via its SH3 domain, to filamin [118]. In primary HUVEC, filamin acts as an organizing scaffold required for Trio activation, and is not strictly required for the ICAM-1–Trio interaction [113]. This scaffolding role is further underscored by the notion that filamin can bind to several small GTPases, including Rac1 and RhoA [119]. The functional relationship between SGEF- and Trio-mediated, ICAM-1-induced docking structures is as yet unresolved. Trio function requires filamin, a protein that in vitro associates to beads-clustered ICAM-1 with a significant delay [66, 74]. Thus, an ICAM-1–SGEF complex may form early after ICAM-1 clustering, later followed by an ICAM-1–filamin–Trio complex. All this was studied in human EC; it is relevant to mention that the C-terminal domain of murine ICAM-1 does not bind to filamin [74]. This suggests that in the murine vasculature, the ICAM-1–SGEF signaling pathway may be dominant in the control of leukocyte TEM.
Most recently, the GEF LARG (Leukemia-Associated Rho GEF, ARHGEF12) was identified as a RhoGEF that is activated downstream from clustered ICAM-1. This activation was further potentiated by force, induced by pulling using magnetic tweezers on adherent, anti-ICAM-1-coated beads [120]. Activation by force was specific for LARG and not observed for other RhoGEFs, i.e. p190RhoGEF, p115, PDZ-RhoGEF or GEFH1. Furthermore, LARG was identified as being involved in force-induced and ICAM-1 mediated activation of RhoA, phosphorylation of myosin light chain and TEM of neutrophils across TNFα-treated microvascular EC. The magnetic tweezer experiments also showed that exertion of force on ICAM-1 induced EC stiffening, at least at the level of the clustered ICAM-1. This suggests that adherent, crawling leukocytes will also induce local EC stiffening which is in line with various studies that showed that cell spreading as well as TEM is potentiated by EC stiffening [17, 74, 121–123]. Whether LARG-mediated EC stiffening is primarily relevant for leukocyte adhesion and spreading on ICAM-1 or also for intercellular gap formation is currently unknown.
Mechanosignaling and TEM
There is increasing interest in the relevance of endothelial and vascular stiffness for inflammation and, more specifically, leukocyte-vessel wall interactions and TEM. Endothelial cells in culture are large (up 100 μm in diameter), flat cells in which the F-actin cytoskeleton, as visualized by phalloidin, presents itself mainly in the form of stress fibers and cortical bundles. These stress fibers are in part linked to integrins in focal adhesions (FAs) and in part to cell–cell contacts, where they co-localize with VE-cadherin. Although differently organized, also small actin fibers extending radially from cortical bundles link to VE-cadherin at cell–cell contacts [124]. VE-cadherin in less permeable, stable junctions is organized in a linear fashion, whereas VE-cadherin in dynamic, remodeling, junctions is jagged and co-localizes with F-actin as well as with actin-regulating proteins such as vinculin [92, 124]. It is primarily at these remodeling Focal Adherens Junctions that the cytoskeleton exerts tension on the VE-cadherin complex. This tension is an important regulatory factor in vascular permeability and consequently also in TEM, albeit that this relationship is not as direct as originally anticipated. Although increased permeability will promote TEM, the process does require firm adhesion of leukocytes to the endothelium. Conversely, although crosslinking of cell-surface ICAM-1 using antibodies in solution has been found to activate RhoA [36] and induce intercellular gap formation, it remains to be demonstrated whether very local ICAM-1 clustering, as induced by adherent and migrating leukocytes, induces similar signals. In addition, such signaling may show spatial differences, as a result of which ICAM-1 engagement on the cell’s center induces other events as compared to ICAM-1 engagement at cell–cell contacts. It is fair to assume that new methodologies based on high resolution imaging of endothelial signaling events and cytoskeletal dynamics during leukocyte TEM will soon provide additional insights in the mechanistic links between adhesion-induced signaling, junctional integrity and TEM.
Regarding mechanical events and TEM, it is important to consider two related factors that impinge on the process. The first is the presence or absence of flow-mediated shear forces. It is now well accepted in the field that the presence of flow significantly promotes TEM of leukocytes in in vitro assays, a phenomenon coined chemorheotaxis [125], and that in the presence of flow, chemokines do not need to be presented in a gradient to induce TEM. The stimulating effects of flow directly relate to the second important mechanical factor, which is the stiffness of the endothelium. This is a relevant factor, since adherent cells, including leukocytes, require a certain optimal substrate stiffness for firm adhesion, spreading and crawling. In EC, as well as many other cell types, Rho-regulated cytoskeletal reorganization leads to increased cellular stiffness [126]. Exposure of EC to flow activates RhoA and also increases cellular stiffness, which may well explain the shear force-mediated stimulation of TEM. It is important to underscore that most information on flow-induced leukocyte TEM has been obtained using EC cultured on glass, i.e. very stiff, flow chambers. It is very likely that this will influence both the EC as well as the leukocytes, as it might promote intercellular gap formation as well as durotaxis, respectively.
The relationship between endothelial and vascular stiffness and inflammation, as well as the relevance of stiffness sensing and durotaxis (i.e. migration in the direction of optimal stiffness) and tenertaxis (migration to sites of low stiffness [81]) in TEM have recently been extensively reviewed elsewhere [17, 126]. However, there is an interesting analogy between the relevance of endothelial stiffness for leukocyte TEM and other, seemingly distantly related findings regarding behavior of transmigrating leukocytes.
Two recent studies addressed directly the mechanistic relation between endothelial cell stiffness and leukocyte TEM [74, 81]. In the study from our lab by Schaefer et al. [74], endothelial cell stiffness, regulated by the actin crosslinking protein α-actinin4, was found to positively correlate with the adhesion, spreading and TEM under flow across TNFα-stimulated HUVEC, of primary human neutrophils. We detected a cell-autonomous, central-to-peripheral gradient of increasing stiffness within TNFα-stimulated primary human EC. This drives crawling neutrophils towards cell–cell contacts, thus promoting TEM. In contrast, the study of Martinelli et al. [81], showed that TEM of human T cells migrating across rat or human microvascular EC from various sources, occurred at sites of reduced stiffness, as deduced from relatively low levels of F-actin. In this paper, increased junctional integrity associated with increased junctional stiffness, was found to enhance transcellular diapedesis at the expense of paracellular TEM, whereas increased permeability was found to promote leukocytes to use the paracellular pathway.
We recently proposed Schaefer and Hordijk [17] that the apparent differences between these findings could in part be due to experimental set-up: the probe for AFM measurements used in the Schaefer study was larger (i.e. 10 μm) as the one used in the Martinelli study (<1 μm). This could explain the detection of small, relatively soft areas in the apical cell surface. Importantly, even when transmigrating at junctions, cells round off prior to diapedesis. This could be the result of relatively low F-actin levels within a 3–5 μm area in the vicinity of VE-cadherin Schaefer and Hordijk [17]. Thus, whereas a stiffness gradient may drive spread leukocytes towards the cellular periphery, a very local reduction in stiffness, at or around cell–cell contacts, may promote cell rounding and the crossing of the endothelial barrier.
The mechanisms that drive transcellular migration have been studied at various levels and EC. Martinelli et al. [80] recently showed that long-term flow stabilizes cell–cell contacts and promotes transcellular vs. paracellular diapedesis, without altering TEM quantitatively. Moreover, in addition to local differences in endothelial F-actin [81], local recruitment and fusion of endothelial vesicles and local differences in surface levels of ICAM-1 have all been postulated to control a leukocytes’ choice to use the transcellular pathway [77, 127]. Regarding the latter, elevated levels of surface ICAM-1, either induced by transfection or by treatment with variable doses of TNFα, were found to reduce crawling distances [127] and promote transcellular migration. In this study, cells that were on ICAM1/2-deficient endothelium showed a lack of polarization and crawling. Surprisingly, the limited number of cells that did transmigrate, almost exclusively used the transcellular route. This result appears reminiscent of two earlier reports on transcellular migration. In the first, T-cells deficient for the Rac1 GEF Tiam1 were found not to be deficient in adhesion to activated bEnd.3 endothelial cells, but to show much reduced crawling on immobilized ICAM-1 or on activated endothelial cells [128]. This was associated with an increase in transcellular diapedesis. Tiam1-deficient lymphocytes also showed reduced polarization under these conditions, which may well relate to their lack of efficient Rac1 activation. More recently, Kumar et al. [129] showed that neutrophils, deficient in the small GTPase Rap1b (but not Rap1a), show a marked spreading defect on CD11b-coated plates although adhesion as such was not different from WT cells. This phenotype was accompanied by increased podosome formation by Rap1b−/− neutrophils and a >3-fold increase in the use of the transcellular route during TEM across bEnd.3 cells or HUVECs.
Together, these studies indicate that leukocytes that are impaired in cell spreading and/or crawling (i.e. due to locally reduced substrate stiffness, too much or too little ICAM1/2; lack of Tiam1–Rac1 signaling or lack of Rap1b) show a shift towards the use of the transcellular pathway for TEM. This poses the question whether leukocytes, in close interaction with the endothelium, actively ‘choose’ for transcellular TEM or whether this pathway represents a default route for relatively immobile cells that do not adhere in the vicinity of an intercellular junction. The formation of podosomes [79] is likely directly linked to limited migration, since crawling cells may not reside at any position long enough for full podosome formation to occur. However, this supposition requires further experimental analysis.
New players
There are several signaling proteins/components that were recently proposed to be important for leukocyte TEM. Although some of these observations may appear phenomenological and not very well developed yet, they are possibly of interest to the field which is why I will briefly discuss some of these ‘new players in TEM’ here.
LSP1
The F-actin binding protein LSP1 (leukocyte-specific protein 1) has not been studied extensively, despite its apparent in vivo relevance for neutrophil TEM. Already 10 years ago, the group of Paul Kubes showed, using bone marrow transplantation experiments, that it is the LSP1 in the endothelium, rather than in the neutrophils, which controls leukocyte TEM in the inflamed cremaster muscle model [130]. Recipient LSP1−/− mice showed a ~70 % reduction in emigration of wild type leukocytes whereas LSP1−/− leukocytes in WT mice did not show inhibition of transmigration. Moreover, lack of LSP1 protected significantly from histamine-induced microvascular permeability in cremasteric venules. Intriguingly, in a subsequent study it was shown that loss of endothelial LSP1 results in increased permeability which accompanies leukocyte transmigration [24]. Since lack of endothelial LSP1 did not impair leukocyte adhesion, this protein could be more relevant for preserving endothelial integrity in the context of TEM. LSP1 is a substrate for several protein kinases, and some of its effects may relate to its regulation by phosphorylation. Hossain et al. [131] showed that ICAM-1 clustering induces LSP1 phosphorylation both in vitro and in vivo, downstream from p38 MAPK. The mechanistic connection between LSP1 phosphorylation and its regulation of neutrophil TEM remain to be further established.
EphrinB1/2
The cell-surface associated and transmembrane ephrins (5 GPI-linked ephrin A ligands; 3 transmembrane ephring B ligands) and their Eph receptors (9 EphA receptors; 5 EphB receptors) [132, 133] are exquisitely fit to play a role in the interactions between different types of endothelium with different types of leukocyte. Upon contact, these receptors and their ligands show bidirectional (‘forward’ and ‘reverse’) signaling in both contacting cells. Moreover, these receptors and their ligands show differential expression in tissues, circulating cells as well as in different types of cancer [134, 135]. Isoforms of both EphB and EphA receptors and their ligands have been implicated in endothelial cell activation and TEM of both leukocytes and cancer cells.
Ectopic expression in EC of Ephrin-B2, but not a mutant lacking the cytoplasmic tail, promoted TEM under static conditions of monocytic J774 cells [135] (Fig. 2a). More recently, Liu et al. [136] showed that endogenous ephrin-B1/2 on EC in the skin are upregulated in response to inflammation, and that siRNA-mediated downregulation of these ephrins inhibits TEM of monocytic THP-1 cells under static conditions. These authors propose a model in which ephrin-B1 engagement by EphB2 receptors on monocytes would signal towards E-selectin upregulation and loss of VE-cadherin localization and increased permeability. Conversely, inflammation-stimulated expression of ephrin-B2, which is known to associate with CD31 and to accumulate at cell–cell junctions upon engagement [137], was suggested to promote monocyte TEM. Another recent study by van Gils et al. [138] identified ephrin-B2 as a guidance cue which is upregulated in atherosclerotic vascular tissue and promotes monocyte recruitment. Funk et al. [139] found increased expression of EphA4 and ephrin-A1 in atherosclerosis-prone endothelium in vivo and showed that activating EphA4 with Fc-ephrinA1 induces EC activation, leading to VCAM-1 expression and monocyte adhesion.
Fig. 2.

Schematic representation of some ‘new players’ in leukocyte TEM. a Summary of ephrin-Eph interactions implicated in TEM. As detailed in the body text, several ephrin-Eph receptor interactions have been implicated in leukocyte TEM. These involve Eph receptors and ephrin ligands on endothelial cells, as well as on transmigration leukocytes or cancer cells. Some of these interactions result in increased permeability, and effects on ICAM-1 and VCAM-1 recruitment or expression which drives TEM and atherosclerosis. However, the underlying signaling pathways remain to be further elucidated. PC prostate cancer. b The ReticulonA/B proteins have been found to negatively regulate p60Src/Pyk2-mediated tyrosine phosphorylation of VE-cadherin, downstream from ICAM-1 clustering. ReticulonA/B proteins are largely residing in the endoplasmic reticulum, but are also found at the plasma membrane. The TRPC6 calcium channel as well as the Pannexin 1 ATP transporter have both been identified as positive regulators of leukocyte TEM. TRPC6 regulates, downstream, from clustered PECAM-1, the LBRC which mediated TEM. Pannexin 1-driven ATP export can activate leukocytes, thus stimulating TEM in the extracellular milieu. LBRC later border recycling compartment
The functional diversity of Eph receptors and their ligands is further underscored by the findings that in HUVEC as well as EC in vivo, high levels of EphA2 were detected, which were found to regulate TEM of ephrin–A4 positive B-cells [140]. Intriguingly, these authors showed that clustering of EphA2 on HUVEC recruited ICAM-1 and VCAM-1, but not vice versa, suggestive for a close interaction between these leukocyte receptors in the context of TEM. On the leukocyte-side of things, chronic lymphocytic leukemia cells were found to express increased levels of ephrin-A4, which impairs chemotaxis and TEM [140]. Along similar lines, the EphA2 receptor on prostate cancer cell lines and its ligand ephrin-A1 on the endothelium were shown to be required for cancer cell TEM [141].
There is substantial information on bidirectional signaling by ephrins and their Eph receptors, as well as on the crosstalk with well-established intracellular signaling pathways driving cell motility and invasion, as well as proliferation and inflammation (reviewed also in [142]). Most of this research has been performed in the context of cancer and angiogenesis, but there is already sufficient evidence that these regulators of heterotypic cell–cell contact bear relevance for leukocyte TEM. It remains to be seen whether the ephrin and/or Eph-receptor induced endothelial signaling in TEM employs known or novel pathways. However, given the therapeutic potential of this class of receptors/ligands [142], further detailed analysis of their contribution from within the vessel wall to cancer metastasis as well as cardiovascular disease [143] will be of much interest to the field.
Ion channels
As alluded to in the beginning of this overview, the transient increase of intracellular calcium was one of the first signaling events in EC identified to regulate leukocyte TEM [35].
Only very recently there has been additional mechanistic insight into this finding. Weber et al. [144] reported that in HUVEC, the transient receptor potential canonical 6 (TRPC6) calcium channel controls leukocyte TEM (Fig. 2b). Their data suggest that TRPC6 associated with clustered PECAM-1 upon leukocyte binding, regulated lateral border recycling compartment (LBRC) trafficking and thus controlled TEM. The LBRC is tubulo-vesicular structure, localized near endothelial cell–cell contacts that is required for efficient leukocyte TEM (see [3] for a recent review). TRPC6 represents the first endothelial calcium channel that has been directly linked to leukocyte transmigration.
However, apart from the established relevance of calcium in orchestrating endothelial signaling in TEM, not many additional ion fluxes in EC have directly been linked to TEM. Indirectly, the control of endothelial or vessel wall stiffness by e.g. sodium influx (through the epithelial sodium channel ENaC) [145] will affect TEM, since EC stiffness controls adhesion and TEM [74, 81, 126]. A recent study identified a potassium channel (TREK1, TWIK-related potassium channel-1) as a negative regulator of immune cell traffic across the blood–brain barrier [146]. Mice, deficient for TREK1, which is also down-regulated in inflamed EC, showed increased expression of ICAM-1, VCAM-1 and PECAM-1, reduced phosphorylation of ERK1/2, increased lymphocyte influx and -disease scores following induction of EAE. These effects on brain pathology could be reduced by treatment of the mice with the TREK-1-activating fatty acid α-linolenic acid, a component of linseed oil. However, more detailed insights in the endothelial mechanisms controlling TEM downstream of this potassium channel are currently lacking and will require additional research.
An intriguing variant of this mode of regulation was recently described by Lohman et al. [147]. These authors showed that ATP-release from EC by the Pannexin 1 channels is induced by TNFα, through a Src-mediated signaling pathway, and that this ATP in the extracellular milieu promotes leukocyte adhesion and TEM, as a consequence of purinergic receptor signaling (Fig. 2b). This was shown in vivo using intravital imaging (cremaster muscle model) in mice which inducibly lacked Pannexin 1 in their endothelial cells. The effects of released ATP appears in part to be autocrine, since TNFα-induced VCAM-1 upregulation is Pannexin 1 dependent, the lack of which could be bypassed by exogenous ATP. This recent finding adds to existing literature on adenosine nucleotides promoting leukocyte adhesion and migration [148, 149] and add a new mode of regulation to the field of leukocyte TEM.
Reticulon-4B
Reticulon-4A/B proteins (also known as Nogo-A/B) are proteins with two membrane-spanning domains that reside in the endoplasmic reticulum as well as the plasma membrane [150]. This family of proteins and their receptors are pleiotropic, with well characterized functions in neuronal development, including a negative regulation of neuronal growth [150]. More recently, the Nogo-B receptor was implicated in lipid biosynthesis and protein glycosylation [151]. The alternative Nogo receptor, PirB, was found to limit macrophage recruitment to vein grafts, and lack of PirB promoted recovery in hindlimb ischemia models in mice. Endothelial Nogo-B has been directly implicated in leukocyte TEM [152]. Mice lacking NogoA/B showed reduced leukocyte influx in air-pouch models for acute inflammation and bone marrow transplantation experiments showed that it is the host (i.e. endothelial) Nogo proteins that were responsible for these effects. In vitro studies in human dermal microvascular EC showed that downregulation of Nogo-B was sufficient to reduce TEM of neutrophils and monocytes under flow. Mechanistically, Nogo-B was found to control the activation of p60Src and Pyk2 downstream of clustered ICAM-1 and to inhibit ICAM-1 induced phosphorylation of VE-cadherin (Fig. 2b). These authors [152] suggest that these effects of loss of Nogo-B may reflect a role for ER-plasma membrane coupling in controlling EC signaling downstream of ICAM-1. This phenomenon may also relate to the leukocyte-adhesion induced release of intracellular calcium from the endoplasmic reticulum stores and its relevance for TEM.
Concluding remarks
As can be deduced from the above, the past 5 years or so have seen both an extension of our insights in previously identified phenomena driving leukocyte TEM as well as the introduction of new ‘players’ and concepts. Clearly, the field has profited from the broadly growing interest in the role of external force on cellular behavior. The increasing use of sophisticated technologies such as atomic force microscopy has opened up new ways to link cellular stiffness to EC signaling and responses. Also, the use of physiological flow as an essential requirement for in vitro TEM studies, as opposed to the classical, static Transwell assays, has become more broadly established. Additional approaches include the use of FRET-based biosensors not only in vitro but increasingly also in mouse models [153, 154]. Although performing reliable, quantitative imaging in a pulsating blood vessel is challenging, there clearly is progress in this area which will undoubtedly be continued in the coming years, spurred by improved methods of detection and image analysis software. Another important aspect that deserves more attention is the tissue specificity of leukocyte TEM, also against the backdrop of the relevance of substrate stiffness for EC behavior and integrity [155, 156]. This has been addressed in the past already but will require more elaborate in vivo and ex vivo approaches. The notion that primary venous cells in cell culture adopt an arterial phenotype [28] suggests that many in vitro data are likely more representative of what occurs in arteries rather than in veins. In this context, it is also of interest that the first studies linking specific microRNAs to vascular inflammation and leukocyte traffic have been published recently [157, 158]. This directly relates to the signaling capacity of circulating microRNA-loaded microvesicles and their potential to control leukocyte TEM and inflammation at distant sites [159], a topic which will very likely be addressed in more detail the coming years.
Finally, more elaborate models for human vessels (i.e. comprising more complex matrices and microfluidics) that can also take into account interactions with pericytes and smooth muscle cells as well as with platelets, are required to do more translational research which is relevant to address clinical questions on microvascular function and inflammation. The isolation and use of circulating EC provides another very interesting approach to complement already existing and more recent technologies to address patient-specific behavior of EC and to allow a next step towards personalized medicine at the level of the vascular wall.
Box 1: The classical transmigration paradigm for acute inflammation
The swift response of our bodies’ defense mechanism to foreign invaders, such as bacteria on a sharp kitchen knife, requires that circulating leukocytes such as neutrophils deploy to the area of infestation a.s.a.p. While so doing, however, these cells (our ‘first line of defense’) encounter a number of problems to which an adequate response is mandatory. This response requires the leukocytes, following their recruitment to the inflamed site, to initiate a now well-established standard operating protocol for their approach (see schematic and text below).
The first phase of this multi-step process is the rolling along and adhesion to the inner wall of the blood vessel. This wall is paved with a monolayer of endothelial cells (EC) which can be locally activated by inflammatory cytokines. Upregulated by these cytokines, carbohydrate-conjugated endothelial cell surface proteins known as selectins (mainly E-selectin) orchestrate the ‘first contact’; their low-affinity interactions with cognate receptors such as PSGL-1 (P-selectin glycoprotein ligand-1) are key to the rolling behavior of leukocytes under flow. The flow in itself can induce leukocyte to initiate the TEM program, a response known as chemorheotaxis. Despite the continuous contact with large numbers of red blood cells, many rolling leukocytes remain associated with the vessel wall. Locally accumulated chemokines instruct the leukocytes to transform their circular shape into a spread-out, polarized and motile form, imitating directional motility (chemotaxis). This transformation is not possible without the rapid activation of special adhesion molecules, the β1 and β2 integrins, that allow the cells to further resist the external forces imposed by the blood plasma and its constituents. Activated integrins will lock on to their endothelial ligands ICAM-1 and VCAM-1, securing firm leukocyte adhesion. These ligands may also present as a gradient on the endothelial cells’ surface, driving haptotaxis, migration along a gradient of adhesion molecules.
The crawling along the vessel wall is next, directing the cells to optimal sites for TEM that are marked by largely unknown markers. Some of these may involve local differences in EC stiffness, leading to durotaxis. Next is the key process of TEM: the cells’ reversion to a round shape, possibly driven by regions of low stiffness, a phenomenon recently coined ‘tenertaxis’. Subsequently, the cells will protrude and cross the endothelial barrier. Once on the other side, the turbulence of the blood is no more but now the cells need to crawl in a novel way through dense, 3-dimensional networks of extracellular matrix (ECM) proteins and along other types of cell, such as pericytes and smooth muscle cells, in search for the inflammatory culprit.
The inflamed vascular endothelium, in the meantime, has been anything but oblivious to the spectacle and has welcomed the leukocytes not only by presenting sufficient adhesion sites in the shape of upregulated ICAM-1 and VCAM-1, but also by forming ‘docking structures’ or ‘transmigratory cups’ that promote the entrance into the inflamed tissue. These endothelial responses are driven by complicated intracellular signaling machinery, of which new components and interactions are still being discovered.
To conclude, be it by crossing the endothelial barrier through the seemingly obvious way of passage, i.e. through the intercellular junction, or by penetration of an endothelial cell’s body while aiming for an as yet unknown target, this incursion of inflamed tissue results in a rapid resolution of the original problem through the tracking down and killing of invader pathogens.
References
- 1.Nourshargh S, Hordijk PL, Sixt M. Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol. 2010;11:366–378. doi: 10.1038/nrm2889. [DOI] [PubMed] [Google Scholar]
- 2.Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity. 2014;41:694–707. doi: 10.1016/j.immuni.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Muller WA. The regulation of transendothelial migration: new knowledge and new questions. Cardiovasc Res. 2015;107:310–320. doi: 10.1093/cvr/cvv145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol. 2015;15:692–704. doi: 10.1038/nri3908. [DOI] [PubMed] [Google Scholar]
- 5.Scheiermann C, Frenette PS, Hidalgo A. Regulation of leucocyte homeostasis in the circulation. Cardiovasc Res. 2015;107:340–351. doi: 10.1093/cvr/cvv099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 7.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 8.Butcher EC. Leukocyte–endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez-Borja M, van Buul JD, Hordijk PL. The regulation of leucocyte transendothelial migration by endothelial signalling events. Cardiovasc Res. 2010;86:202–210. doi: 10.1093/cvr/cvq003. [DOI] [PubMed] [Google Scholar]
- 10.Muller WA. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol. 2011;6:323–344. doi: 10.1146/annurev-pathol-011110-130224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heemskerk N, van Rijssel J, van Buul JD. Rho-GTPase signaling in leukocyte extravasation: an endothelial point of view. Cell Adh Migr. 2014;8:67–75. doi: 10.4161/cam.28244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reymond N, d’Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13:858–870. doi: 10.1038/nrc3628. [DOI] [PubMed] [Google Scholar]
- 13.Chavakis E, Dimmeler S. Homing of progenitor cells to ischemic tissues. Antioxid Redox Signal. 2011;15:967–980. doi: 10.1089/ars.2010.3582. [DOI] [PubMed] [Google Scholar]
- 14.Voermans C, van Hennik PB, van der Schoot CE. Homing of human hematopoietic stem and progenitor cells: new insights, new challenges? J Hematother Stem Cell Res. 2001;10:725–738. doi: 10.1089/152581601317210827. [DOI] [PubMed] [Google Scholar]
- 15.Aird WC. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ. Res. 2007;100:174–190. doi: 10.1161/01.RES.0000255690.03436.ae. [DOI] [PubMed] [Google Scholar]
- 16.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100:158–173. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- 17.Schaefer A, Hordijk PL. Cell-stiffness-induced mechanosignaling—a key driver of leukocyte transendothelial migration. J Cell Sci. 2015;128:2221–2230. doi: 10.1242/jcs.163055. [DOI] [PubMed] [Google Scholar]
- 18.Carman CV. Mechanisms for transcellular diapedesis: probing and pathfinding by ‘invadosome-like protrusions’. J Cell Sci. 2009;122:3025–3035. doi: 10.1242/jcs.047522. [DOI] [PubMed] [Google Scholar]
- 19.Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, Nash GB, Chavakis T, Albelda SM, Rainger GE, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol. 2011;12:761–769. doi: 10.1038/ni.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Voisin MB, Nourshargh S. Neutrophil transmigration: emergence of an adhesive cascade within venular walls. J Innate Immun. 2013;5:336–347. doi: 10.1159/000346659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, Chavakis T, Brohi K, Imhof BA, Nourshargh S. Leukotriene B4-neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo. Immunity. 2015;42:1075–1086. doi: 10.1016/j.immuni.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Proebstl D, Voisin MB, Woodfin A, Whiteford J, D’Acquisto F, Jones GE, Rowe D, Nourshargh S. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med. 2012;209:1219–1234. doi: 10.1084/jem.20111622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillipson M, Kaur J, Colarusso P, Ballantyne CM, Kubes P. Endothelial domes encapsulate adherent neutrophils and minimize increases in vascular permeability in paracellular and transcellular emigration. PLoS One. 2008;3:e1649. doi: 10.1371/journal.pone.0001649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petri B, Kaur J, Long EM, Li H, Parsons SA, Butz S, Phillipson M, Vestweber D, Patel KD, Robbins SM, et al. Endothelial LSP1 is involved in endothelial dome formation, minimizing vascular permeability changes during neutrophil transmigration in vivo. Blood. 2011;117:942–952. doi: 10.1182/blood-2010-02-270561. [DOI] [PubMed] [Google Scholar]
- 25.Sanz MJ, Kubes P. Neutrophil-active chemokines in in vivo imaging of neutrophil trafficking. Eur J Immunol. 2012;42:278–283. doi: 10.1002/eji.201142231. [DOI] [PubMed] [Google Scholar]
- 26.Zheng Y, Chen J, Craven M, Choi NW, Totorica S, Diaz-Santana A, Kermani P, Hempstead B, Fischbach-Teschl C, Lopez JA, et al. In vitro microvessels for the study of angiogenesis and thrombosis. Proc Natl Acad Sci USA. 2012;109:9342–9347. doi: 10.1073/pnas.1201240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng Y, Chen J, Lopez JA. Flow-driven assembly of VWF fibres and webs in in vitro microvessels. Nat Commun. 2015;6:7858. doi: 10.1038/ncomms8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Geemen D, Smeets MW, van Stalborch AM, Woerdeman LA, Daemen MJ, Hordijk PL, Huveneers S. F-actin-anchored focal adhesions distinguish endothelial phenotypes of human arteries and veins. Arterioscler Thromb Vasc Biol. 2014;34:2059–2067. doi: 10.1161/ATVBAHA.114.304180. [DOI] [PubMed] [Google Scholar]
- 29.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takao K, Miyakawa T. Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2015;112:1167–1172. doi: 10.1073/pnas.1401965111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wittchen ES. Endothelial signaling in paracellular and transcellular leukocyte transmigration. Front Biosci (Landmark Ed) 2009;14:2522–2545. doi: 10.2741/3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muller WA, Gimbrone MA., Jr Plasmalemmal proteins of cultured vascular endothelial cells exhibit apical–basal polarity: analysis by surface-selective iodination. J Cell Biol. 1986;103:2389–2402. doi: 10.1083/jcb.103.6.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parat MO, Anand-Apte B, Fox PL. Differential caveolin-1 polarization in endothelial cells during migration in two and three dimensions. Mol Biol Cell. 2003;14:3156–3168. doi: 10.1091/mbc.E02-11-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giampietro C, Deflorian G, Gallo S, Di MA, Pradella D, Bonomi S, Belloni E, Nyqvist D, Quaranta V, Confalonieri S, et al. The alternative splicing factor Nova2 regulates vascular development and lumen formation. Nat Commun. 2015;6:8479. doi: 10.1038/ncomms9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR, Silverstein SC. Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells. J Cell Biol. 1993;120:1371–1380. doi: 10.1083/jcb.120.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Etienne S, Adamson P, Greenwood J, Strosberg AD, Cazaubon S, Couraud PO. ICAM-1 signaling pathways associated with Rho activation in microvascular brain endothelial cells. J Immunol. 1998;161:5755–5761. [PubMed] [Google Scholar]
- 37.Etienne-Manneville S, Manneville JB, Adamson P, Wilbourn B, Greenwood J, Couraud PO. ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J Immunol. 2000;165:3375–3383. doi: 10.4049/jimmunol.165.6.3375. [DOI] [PubMed] [Google Scholar]
- 38.Walters CE, Pryce G, Hankey DJ, Sebti SM, Hamilton AD, Baker D, Greenwood J, Adamson P. Inhibition of Rho GTPases with protein prenyltransferase inhibitors prevents leukocyte recruitment to the central nervous system and attenuates clinical signs of disease in an animal model of multiple sclerosis. J Immunol. 2002;168:4087–4094. doi: 10.4049/jimmunol.168.8.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wojciak-Stothard B, Williams L, Ridley AJ. Monocyte adhesion and spreading on human endothelial cells is dependent on Rho-regulated receptor clustering. J Cell Biol. 1999;145:1293–1307. doi: 10.1083/jcb.145.6.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson PW, Randi AM, Ridley AJ. Intercellular adhesion molecule (ICAM)-1, but not ICAM-2, activates RhoA and stimulates c-fos and rhoA transcription in endothelial cells. J Immunol. 2002;169:1007–1013. doi: 10.4049/jimmunol.169.2.1007. [DOI] [PubMed] [Google Scholar]
- 41.Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res. 2000;87:335–340. doi: 10.1161/01.RES.87.4.335. [DOI] [PubMed] [Google Scholar]
- 42.Nieuw Amerongen GP, Beckers CM, Achekar ID, Zeeman S, Musters RJ, van Hinsbergh VW. Involvement of Rho kinase in endothelial barrier maintenance. Arterioscler Thromb Vasc Biol. 2007;27:2332–2339. doi: 10.1161/ATVBAHA.107.152322. [DOI] [PubMed] [Google Scholar]
- 43.van Buul JD, Hordijk PL. Signaling in leukocyte transendothelial migration. Arterioscler Thromb Vasc Biol. 2004;24:824–833. doi: 10.1161/01.ATV.0000122854.76267.5c. [DOI] [PubMed] [Google Scholar]
- 44.van Buul JD, Kanters E, Hordijk PL. Endothelial signaling by Ig-like cell adhesion molecules. Arterioscler Thromb Vasc Biol. 2007;27:1870–1876. doi: 10.1161/ATVBAHA.107.145821. [DOI] [PubMed] [Google Scholar]
- 45.Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;178:449–460. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schenkel AR, Chew TW, Muller WA. Platelet endothelial cell adhesion molecule deficiency or blockade significantly reduces leukocyte emigration in a majority of mouse strains. J Immunol. 2004;173:6403–6408. doi: 10.4049/jimmunol.173.10.6403. [DOI] [PubMed] [Google Scholar]
- 47.Lou O, Alcaide P, Luscinskas FW, Muller WA. CD99 is a key mediator of the transendothelial migration of neutrophils. J Immunol. 2007;178:1136–1143. doi: 10.4049/jimmunol.178.2.1136. [DOI] [PubMed] [Google Scholar]
- 48.Wegmann F, Petri B, Khandoga AG, Moser C, Khandoga A, Volkery S, Li H, Nasdala I, Brandau O, Fassler R, et al. ESAM supports neutrophil extravasation, activation of Rho, and VEGF-induced vascular permeability. J Exp Med. 2006;203:1671–1677. doi: 10.1084/jem.20060565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Del Maschio A, De Luigi A, Martin-Padura I, Brockhaus M, Bartfai T, Fruscella P, Adorini L, Martino G, Furlan R, De Simoni MG, et al. Leukocyte recruitment in the cerebrospinal fluid of mice with experimental meningitis is inhibited by an antibody to junctional adhesion molecule (JAM) J Exp Med. 1999;190:1351–1356. doi: 10.1084/jem.190.9.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ostermann G, Weber KS, Zernecke A, Schroder A, Weber C. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol. 2002;3:151–158. doi: 10.1038/ni755. [DOI] [PubMed] [Google Scholar]
- 51.Durieu-Trautmann O, Chaverot N, Cazaubon S, Strosberg AD, Couraud PO. Intercellular adhesion molecule 1 activation induces tyrosine phosphorylation of the cytoskeleton-associated protein cortactin in brain microvessel endothelial cells. J Biol Chem. 1994;269:12536–12540. [PubMed] [Google Scholar]
- 52.Minshall RD, Tiruppathi C, Vogel SM, Niles WD, Gilchrist A, Hamm HE, Malik AB. Endothelial cell-surface gp60 activates vesicle formation and trafficking via G(i)-coupled Src kinase signaling pathway. J Cell Biol. 2000;150:1057–1070. doi: 10.1083/jcb.150.5.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tilghman RW, Hoover RL. The Src-cortactin pathway is required for clustering of E-selectin and ICAM-1 in endothelial cells. FASEB J. 2002;16:1257–1259. doi: 10.1096/fj.01-0969fje. [DOI] [PubMed] [Google Scholar]
- 54.Wang Q, Pfeiffer GR, Gaarde WA. Activation of SRC tyrosine kinases in response to ICAM-1 ligation in pulmonary microvascular endothelial cells. J Biol Chem. 2003;278:47731–47743. doi: 10.1074/jbc.M308466200. [DOI] [PubMed] [Google Scholar]
- 55.Potter MD, Barbero S, Cheresh DA. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem. 2005;280:31906–31912. doi: 10.1074/jbc.M505568200. [DOI] [PubMed] [Google Scholar]
- 56.Yang L, Kowalski JR, Zhan X, Thomas SM, Luscinskas FW. Endothelial cell cortactin phosphorylation by Src contributes to polymorphonuclear leukocyte transmigration in vitro. Circ Res. 2006;98:394–402. doi: 10.1161/01.RES.0000201958.59020.1a. [DOI] [PubMed] [Google Scholar]
- 57.Allingham MJ, van Buul JD, Burridge K. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol. 2007;179:4053–4064. doi: 10.4049/jimmunol.179.6.4053. [DOI] [PubMed] [Google Scholar]
- 58.Dasgupta B, Muller WA. Endothelial Src kinase regulates membrane recycling from the lateral border recycling compartment during leukocyte transendothelial migration. Eur J Immunol. 2008;38:3499–3507. doi: 10.1002/eji.200838605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Wetering S, van den Berk N, van Buul JD, Mul FP, Lommerse I, Mous R, ten Klooster JP, Zwaginga JJ, Hordijk PL. VCAM-1-mediated Rac signaling controls endothelial cell–cell contacts and leukocyte transmigration. Am J Physiol Cell Physiol. 2003;285:C343–C352. doi: 10.1152/ajpcell.00048.2003. [DOI] [PubMed] [Google Scholar]
- 60.Matheny HE, Deem TL, Cook-Mills JM. Lymphocyte migration through monolayers of endothelial cell lines involves VCAM-1 signaling via endothelial cell NADPH oxidase. J Immunol. 2000;164:6550–6559. doi: 10.4049/jimmunol.164.12.6550. [DOI] [PubMed] [Google Scholar]
- 61.Deem TL, Cook-Mills JM. Vascular cell adhesion molecule 1 (VCAM-1) activation of endothelial cell matrix metalloproteinases: role of reactive oxygen species. Blood. 2004;104:2385–2393. doi: 10.1182/blood-2004-02-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vockel M, Vestweber D. How T cells trigger the dissociation of the endothelial receptor phosphatase VE-PTP from VE-cadherin. Blood. 2013;122:2512–2522. doi: 10.1182/blood-2013-04-499228. [DOI] [PubMed] [Google Scholar]
- 63.van Wetering S, van Buul JD, Quik S, Mul FP, Anthony EC, ten Klooster JP, Collard JG, Hordijk PL. Reactive oxygen species mediate Rac-induced loss of cell–cell adhesion in primary human endothelial cells. J Cell Sci. 2002;115:1837–1846. doi: 10.1242/jcs.115.9.1837. [DOI] [PubMed] [Google Scholar]
- 64.Cook-Mills JM, Marchese ME, Abdala-Valencia H. Vascular cell adhesion molecule-1 expression and signaling during disease: regulation by reactive oxygen species and antioxidants. Antioxid Redox Signal. 2011;15:1607–1638. doi: 10.1089/ars.2010.3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schulte D, Kuppers V, Dartsch N, Broermann A, Li H, Zarbock A, Kamenyeva O, Kiefer F, Khandoga A, Massberg S, et al. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J. 2011;30:4157–4170. doi: 10.1038/emboj.2011.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanters E, van Rijssel J, Hensbergen PJ, Hondius D, Mul FP, Deelder AM, Sonnenberg A, van Buul JD, Hordijk PL. Filamin B mediates ICAM-1-driven leukocyte transendothelial migration. J Biol Chem. 2008;283:31830–31839. doi: 10.1074/jbc.M804888200. [DOI] [PubMed] [Google Scholar]
- 67.van Buul JD, van Rijssel J, Van Alphen FP, van Stalborch AM, Mul EP, Hordijk PL. ICAM-1 clustering on endothelial cells recruits VCAM-1. J Biomed Biotechnol. 2010;2010:120328. doi: 10.1155/2010/120328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Buul JD, Allingham MJ, Samson T, Meller J, Boulter E, Garcia-Mata R, Burridge K. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J Cell Biol. 2007;178:1279–1293. doi: 10.1083/jcb.200612053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barreiro O, Yanez-Mo M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, Furthmayr H, Sanchez-Madrid F. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol. 2002;157:1233–1245. doi: 10.1083/jcb.200112126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barreiro O, Yanez-Mo M, Sala-Valdes M, Gutierrez-Lopez MD, Ovalle S, Higginbottom A, Monk PN, Cabanas C, Sanchez-Madrid F. Endothelial tetraspanin microdomains regulate leukocyte firm adhesion during extravasation. Blood. 2005;105:2852–2861. doi: 10.1182/blood-2004-09-3606. [DOI] [PubMed] [Google Scholar]
- 71.Yang L, Kowalski JR, Yacono P, Bajmoczi M, Shaw SK, Froio RM, Golan DE, Thomas SM, Luscinskas FW. Endothelial cell cortactin coordinates intercellular adhesion molecule-1 clustering and actin cytoskeleton remodeling during polymorphonuclear leukocyte adhesion and transmigration. J Immunol. 2006;177:6440–6449. doi: 10.4049/jimmunol.177.9.6440. [DOI] [PubMed] [Google Scholar]
- 72.van Buul JD, Hordijk PL. Endothelial adapter proteins in leukocyte transmigration. Thromb Haemost. 2009;101:649–655. [PubMed] [Google Scholar]
- 73.Schnoor M, Lai FP, Zarbock A, Klaver R, Polaschegg C, Schulte D, Weich HA, Oelkers JM, Rottner K, Vestweber D. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J Exp Med. 2011;208:1721–1735. doi: 10.1084/jem.20101920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schaefer A, Te RJ, Ritz K, Hoogenboezem M, Anthony EC, Mul FP, de Vries CJ, Daemen MJ, Figdor CG, van Buul JD, et al. Actin-binding proteins differentially regulate endothelial cell stiffness, ICAM-1 function and neutrophil transmigration. J Cell Sci. 2014;127:4470–4482. doi: 10.1242/jcs.154708. [DOI] [PubMed] [Google Scholar]
- 75.Celli L, Ryckewaert JJ, Delachanal E, Duperray A. Evidence of a functional role for interaction between ICAM-1 and nonmuscle alpha-actinins in leukocyte diapedesis. J Immunol. 2006;177:4113–4121. doi: 10.4049/jimmunol.177.6.4113. [DOI] [PubMed] [Google Scholar]
- 76.Schnoor M. Endothelial actin-binding proteins and actin dynamics in leukocyte transendothelial migration. J Immunol. 2015;194:3535–3541. doi: 10.4049/jimmunol.1403250. [DOI] [PubMed] [Google Scholar]
- 77.Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood. 2005;106:584–592. doi: 10.1182/blood-2004-12-4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Millan J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol. 2006;8:113–123. doi: 10.1038/ncb1356. [DOI] [PubMed] [Google Scholar]
- 79.Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HD, Dvorak HF, Dvorak AM, Springer TA. Transcellular diapedesis is initiated by invasive podosomes. Immunity. 2007;26:784–797. doi: 10.1016/j.immuni.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. 2004;167:377–388. doi: 10.1083/jcb.200404129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martinelli R, Zeiger AS, Whitfield M, Sciuto TE, Dvorak A, Van Vliet KJ, Greenwood J, Carman CV. Probing the biomechanical contribution of the endothelium to lymphocyte migration: diapedesis by the path of least resistance. J Cell Sci. 2014;127:3720–3734. doi: 10.1242/jcs.148619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kalra VK, Shen Y, Sultana C, Rattan V. Hypoxia induces PECAM-1 phosphorylation and transendothelial migration of monocytes. Am J Physiol. 1996;271:H2025–H2034. doi: 10.1152/ajpheart.1996.271.5.H2025. [DOI] [PubMed] [Google Scholar]
- 83.Deem TL, Abdala-Valencia H, Cook-Mills JM. VCAM-1 activation of endothelial cell protein tyrosine phosphatase 1B. J Immunol. 2007;178:3865–3873. doi: 10.4049/jimmunol.178.6.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Berdnikovs S, Pavlov VI, Abdala-Valencia H, McCary CA, Klumpp DJ, Tremblay ML, Cook-Mills JM. PTP1B deficiency exacerbates inflammation and accelerates leukocyte trafficking in vivo. J Immunol. 2012;188:874–884. doi: 10.4049/jimmunol.1004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Berdnikovs S, Abdala-Valencia H, Cook-Mills JM. Endothelial cell PTP1B regulates leukocyte recruitment during allergic inflammation. Am J Physiol Lung Cell Mol Physiol. 2013;304:L240–L249. doi: 10.1152/ajplung.00375.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lanz TV, Becker S, Osswald M, Bittner S, Schuhmann MK, Opitz CA, Gaikwad S, Wiestler B, Litzenburger UM, Sahm F, et al. Protein kinase Cbeta as a therapeutic target stabilizing blood–brain barrier disruption in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2013;110:14735–14740. doi: 10.1073/pnas.1302569110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Watson RL, Buck J, Levin LR, Winger RC, Wang J, Arase H, Muller WA. Endothelial CD99 signals through soluble adenylyl cyclase and PKA to regulate leukocyte transendothelial migration. J Exp Med. 2015;212:1021–1041. doi: 10.1084/jem.20150354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nakhaei-Nejad M, Hussain AM, Zhang QX, Murray AG. Endothelial PI 3-kinase activity regulates lymphocyte diapedesis. Am J Physiol Heart Circ Physiol. 2007;293:H3608–H3616. doi: 10.1152/ajpheart.00321.2007. [DOI] [PubMed] [Google Scholar]
- 89.Cain RJ, Vanhaesebroeck B, Ridley AJ. Different PI 3-kinase inhibitors have distinct effects on endothelial permeability and leukocyte transmigration. Int J Biochem Cell Biol. 2012;44:1929–1936. doi: 10.1016/j.biocel.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 90.Cain RJ, Vanhaesebroeck B, Ridley AJ. The PI3K p110alpha isoform regulates endothelial adherens junctions via Pyk2 and Rac1. J Cell Biol. 2010;188:863–876. doi: 10.1083/jcb.200907135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.van Buul JD, Anthony EC, Fernandez-Borja M, Burridge K, Hordijk PL. Proline-rich tyrosine kinase 2 (Pyk2) mediates vascular endothelial-cadherin-based cell–cell adhesion by regulating beta-catenin tyrosine phosphorylation. J Biol Chem. 2005;280:21129–21136. doi: 10.1074/jbc.M500898200. [DOI] [PubMed] [Google Scholar]
- 92.Huveneers S, Oldenburg J, Spanjaard E, VanDerKrogt G, Grigoriev I, Akhmanova A, Rehmann H, de Rooij J. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J Cell Biol. 2012;196:641–652. doi: 10.1083/jcb.201108120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Timmerman I, Heemskerk N, Kroon J, Schaefer A, van Rijssel J, Hoogenboezem M, van UJ, Goedhart J, Gadella TW, Jr, Yin T et al (2015) A local VE-cadherin/Trio-based signaling complex stabilizes endothelial junctions through Rac1. J Cell Sci [DOI] [PubMed]
- 94.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Di LA, Fernandez-Hernando C, Cirino G, Sessa WC. Akt1 is critical for acute inflammation and histamine-mediated vascular leakage. Proc Natl Acad Sci USA. 2009;106:14552–14557. doi: 10.1073/pnas.0904073106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Martinelli R, Gegg M, Longbottom R, Adamson P, Turowski P, Greenwood J. ICAM-1-mediated endothelial nitric oxide synthase activation via calcium and AMP-activated protein kinase is required for transendothelial lymphocyte migration. Mol Biol Cell. 2009;20:995–1005. doi: 10.1091/mbc.E08-06-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu G, Place AT, Chen Z, Brovkovych VM, Vogel SM, Muller WA, Skidgel RA, Malik AB, Minshall RD. ICAM-1-activated Src and eNOS signaling increase endothelial cell surface PECAM-1 adhesivity and neutrophil transmigration. Blood. 2012;120:1942–1952. doi: 10.1182/blood-2011-12-397430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lampugnani MG, Corada M, Andriopoulou P, Esser S, Risau W, Dejana E. Cell confluence regulates tyrosine phosphorylation of adherens junction components in endothelial cells. J Cell Sci. 1997;110(Pt 17):2065–2077. doi: 10.1242/jcs.110.17.2065. [DOI] [PubMed] [Google Scholar]
- 99.Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci. 1998;111(Pt 13):1853–1865. doi: 10.1242/jcs.111.13.1853. [DOI] [PubMed] [Google Scholar]
- 100.Andriopoulou P, Navarro P, Zanetti A, Lampugnani MG, Dejana E. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler Thromb Vasc Biol. 1999;19:2286–2297. doi: 10.1161/01.ATV.19.10.2286. [DOI] [PubMed] [Google Scholar]
- 101.Tinsley JH, Wu MH, Ma W, Taulman AC, Yuan SY. Activated neutrophils induce hyperpermeability and phosphorylation of adherens junction proteins in coronary venular endothelial cells. J Biol Chem. 1999;274:24930–24934. doi: 10.1074/jbc.274.35.24930. [DOI] [PubMed] [Google Scholar]
- 102.Wessel F, Winderlich M, Holm M, Frye M, Rivera-Galdos R, Vockel M, Linnepe R, Ipe U, Stadtmann A, Zarbock A, et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat Immunol. 2014;15:223–230. doi: 10.1038/ni.2824. [DOI] [PubMed] [Google Scholar]
- 103.Orsenigo F, Giampietro C, Ferrari A, Corada M, Galaup A, Sigismund S, Ristagno G, Maddaluno L, Koh GY, Franco D, et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat Commun. 2012;3:1208. doi: 10.1038/ncomms2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Turowski P, Martinelli R, Crawford R, Wateridge D, Papageorgiou AP, Lampugnani MG, Gamp AC, Vestweber D, Adamson P, Dejana E, et al. Phosphorylation of vascular endothelial cadherin controls lymphocyte emigration. J Cell Sci. 2008;121:29–37. doi: 10.1242/jcs.022681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wallez Y, Cand F, Cruzalegui F, Wernstedt C, Souchelnytskyi S, Vilgrain I, Huber P. Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: identification of tyrosine 685 as the unique target site. Oncogene. 2007;26:1067–1077. doi: 10.1038/sj.onc.1209855. [DOI] [PubMed] [Google Scholar]
- 106.Earley S, Plopper GE. Disruption of focal adhesion kinase slows transendothelial migration of AU-565 breast cancer cells. Biochem Biophys Res Commun. 2006;350:405–412. doi: 10.1016/j.bbrc.2006.09.056. [DOI] [PubMed] [Google Scholar]
- 107.Parsons SA, Sharma R, Roccamatisi DL, Zhang H, Petri B, Kubes P, Colarusso P, Patel KD. Endothelial paxillin and focal adhesion kinase (FAK) play a critical role in neutrophil transmigration. Eur J Immunol. 2012;42:436–446. doi: 10.1002/eji.201041303. [DOI] [PubMed] [Google Scholar]
- 108.Jean C, Chen XL, Nam JO, Tancioni I, Uryu S, Lawson C, Ward KK, Walsh CT, Miller NL, Ghassemian M, et al. Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J Cell Biol. 2014;204:247–263. doi: 10.1083/jcb.201307067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jouve N, Bachelier R, Despoix N, Blin MG, Matinzadeh MK, Poitevin S, Aurrand-Lions M, Fallague K, Bardin N, Blot-Chabaud M, et al. CD146 mediates VEGF-induced melanoma cell extravasation through FAK activation. Int J Cancer. 2015;137:50–60. doi: 10.1002/ijc.29370. [DOI] [PubMed] [Google Scholar]
- 110.Adam AP. Regulation of endothelial adherens junctions by tyrosine phosphorylation. Mediators Inflamm. 2015;2015:272858. doi: 10.1155/2015/272858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Naikawadi RP, Cheng N, Vogel SM, Qian F, Wu D, Malik AB, Ye RD. A critical role for phosphatidylinositol (3,4,5)-trisphosphate-dependent Rac exchanger 1 in endothelial junction disruption and vascular hyperpermeability. Circ Res. 2012;111:1517–1527. doi: 10.1161/CIRCRESAHA.112.273078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ellerbroek SM, Wennerberg K, Arthur WT, Dunty JM, Bowman DR, Demali KA, Der C, Burridge K. SGEF, a RhoG guanine nucleotide exchange factor that stimulates macropinocytosis. Mol Biol Cell. 2004;15:3309–3319. doi: 10.1091/mbc.E04-02-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.van Rijssel J, Kroon J, Hoogenboezem M, Van Alphen FP, de Jong RJ, Kostadinova E, Geerts D, Hordijk PL, van Buul JD. The Rho-guanine nucleotide exchange factor Trio controls leukocyte transendothelial migration by promoting docking structure formation. Mol Biol Cell. 2012;23:2831–2844. doi: 10.1091/mbc.E11-11-0907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Samson T, van Buul JD, Kroon J, Welch C, Bakker EN, Matlung HL, van den Berg TK, Sharek L, Doerschuk C, Hahn K, et al. The guanine-nucleotide exchange factor SGEF plays a crucial role in the formation of atherosclerosis. PLoS One. 2013;8:e55202. doi: 10.1371/journal.pone.0055202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bellanger JM, Lazaro JB, Diriong S, Fernandez A, Lamb N, Debant A. The two guanine nucleotide exchange factor domains of Trio link the Rac1 and the RhoA pathways in vivo. Oncogene. 1998;16:147–152. doi: 10.1038/sj.onc.1201532. [DOI] [PubMed] [Google Scholar]
- 116.van Rijssel J, Timmerman I, Van Alphen FP, Hoogenboezem M, Korchynskyi O, Geerts D, Geissler J, Reedquist KA, Niessen HW, van Buul JD. The Rho-GEF Trio regulates a novel pro-inflammatory pathway through the transcription factor Ets2. Biol Open. 2013;2:569–579. doi: 10.1242/bio.20134382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van Rijssel J, Hoogenboezem M, Wester L, Hordijk PL, van Buul JD. The N-terminal DH-PH domain of Trio induces cell spreading and migration by regulating lamellipodia dynamics in a Rac1-dependent fashion. PLoS One. 2012;7:e29912. doi: 10.1371/journal.pone.0029912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bellanger JM, Astier C, Sardet C, Ohta Y, Stossel TP, Debant A. The Rac1- and RhoG-specific GEF domain of Trio targets filamin to remodel cytoskeletal actin. Nat Cell Biol. 2000;2:888–892. doi: 10.1038/35046533. [DOI] [PubMed] [Google Scholar]
- 119.Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, Shapiro SS. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001;2:138–145. doi: 10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- 120.Lessey-Morillon EC, Osborne LD, Monaghan-Benson E, Guilluy C, O’Brien ET, Superfine R, Burridge K. The RhoA guanine nucleotide exchange factor, LARG, mediates ICAM-1-dependent mechanotransduction in endothelial cells to stimulate transendothelial migration. J Immunol. 2014;192:3390–3398. doi: 10.4049/jimmunol.1302525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stroka KM, Aranda-Espinoza H. Neutrophils display biphasic relationship between migration and substrate stiffness. Cell Motil Cytoskeleton. 2009;66:328–341. doi: 10.1002/cm.20363. [DOI] [PubMed] [Google Scholar]
- 122.Stroka KM, Aranda-Espinoza H. Endothelial cell substrate stiffness influences neutrophil transmigration via myosin light chain kinase-dependent cell contraction. Blood. 2011;118:1632–1640. doi: 10.1182/blood-2010-11-321125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stroka KM, Hayenga HN, Aranda-Espinoza H. Human neutrophil cytoskeletal dynamics and contractility actively contribute to trans-endothelial migration. PLoS One. 2013;8:e61377. doi: 10.1371/journal.pone.0061377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Huveneers S, de Rooij J. Mechanosensitive systems at the cadherin-F-actin interface. J Cell Sci. 2013;126:403–413. doi: 10.1242/jcs.109447. [DOI] [PubMed] [Google Scholar]
- 125.Cinamon G, Shinder V, Alon R. Shear forces promote lymphocyte migration across vascular endothelium bearing apical chemokines. Nat Immunol. 2001;2:515–522. doi: 10.1038/88710. [DOI] [PubMed] [Google Scholar]
- 126.Huveneers S, Daemen MJ, Hordijk PL. Between Rho(k) and a hard place: the relation between vessel wall stiffness, endothelial contractility, and cardiovascular disease. Circ Res. 2015;116:895–908. doi: 10.1161/CIRCRESAHA.116.305720. [DOI] [PubMed] [Google Scholar]
- 127.Abadier M, Haghayegh JN, Cardoso AL, Boscacci R, Vestweber D, Barnum S, Deutsch U, Engelhardt B, Lyck R. Cell surface levels of endothelial ICAM-1 influence the transcellular or paracellular T-cell diapedesis across the blood–brain barrier. Eur J Immunol. 2015;45:1043–1058. doi: 10.1002/eji.201445125. [DOI] [PubMed] [Google Scholar]
- 128.Gerard A, van der Kammen RA, Janssen H, Ellenbroek SI, Collard JG. The Rac activator Tiam1 controls efficient T-cell trafficking and route of trans-endothelial migration. Blood. 2009;113:6138–6147. doi: 10.1182/blood-2008-07-167668. [DOI] [PubMed] [Google Scholar]
- 129.Kumar S, Xu J, Kumar RS, Lakshmikanthan S, Kapur R, Kofron M, Chrzanowska-Wodnicka M, Filippi MD. The small GTPase Rap1b negatively regulates neutrophil chemotaxis and transcellular diapedesis by inhibiting Akt activation. J Exp Med. 2014;211:1741–1758. doi: 10.1084/jem.20131706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liu L, Cara DC, Kaur J, Raharjo E, Mullaly SC, Jongstra-Bilen J, Jongstra J, Kubes P. LSP1 is an endothelial gatekeeper of leukocyte transendothelial migration. J Exp Med. 2005;201:409–418. doi: 10.1084/jem.20040830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hossain M, Qadri SM, Xu N, Su Y, Cayabyab FS, Heit B, Liu L. Endothelial LSP1 modulates extravascular neutrophil chemotaxis by regulating nonhematopoietic vascular PECAM-1 expression. J Immunol. 2015;195:2408–2416. doi: 10.4049/jimmunol.1402225. [DOI] [PubMed] [Google Scholar]
- 132.Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol. 2002;3:475–486. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- 133.Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–180. doi: 10.1038/nrc2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hafner C, Schmitz G, Meyer S, Bataille F, Hau P, Langmann T, Dietmaier W, Landthaler M, Vogt T. Differential gene expression of Eph receptors and ephrins in benign human tissues and cancers. Clin Chem. 2004;50:490–499. doi: 10.1373/clinchem.2003.026849. [DOI] [PubMed] [Google Scholar]
- 135.Pfaff D, Heroult M, Riedel M, Reiss Y, Kirmse R, Ludwig T, Korff T, Hecker M, Augustin HG. Involvement of endothelial ephrin-B2 in adhesion and transmigration of EphB-receptor-expressing monocytes. J Cell Sci. 2008;121:3842–3850. doi: 10.1242/jcs.030627. [DOI] [PubMed] [Google Scholar]
- 136.Liu H, Devraj K, Moller K, Liebner S, Hecker M, Korff T. EphrinB-mediated reverse signalling controls junctional integrity and pro-inflammatory differentiation of endothelial cells. Thromb Haemost. 2014;112:151–163. doi: 10.1160/TH13-12-1034. [DOI] [PubMed] [Google Scholar]
- 137.Korff T, Dandekar G, Pfaff D, Fuller T, Goettsch W, Morawietz H, Schaffner F, Augustin HG. Endothelial ephrinB2 is controlled by microenvironmental determinants and associates context-dependently with CD31. Arterioscler Thromb Vasc Biol. 2006;26:468–474. doi: 10.1161/01.ATV.0000200081.42064.e7. [DOI] [PubMed] [Google Scholar]
- 138.van Gils JM, Ramkhelawon B, Fernandes L, Stewart MC, Guo L, Seibert T, Menezes GB, Cara DC, Chow C, Kinane TB, et al. Endothelial expression of guidance cues in vessel wall homeostasis dysregulation under proatherosclerotic conditions. Arterioscler Thromb Vasc Biol. 2013;33:911–919. doi: 10.1161/ATVBAHA.112.301155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Funk SD, Yurdagul A, Jr, Albert P, Traylor JG, Jr, Jin L, Chen J, Orr AW. EphA2 activation promotes the endothelial cell inflammatory response: a potential role in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:686–695. doi: 10.1161/ATVBAHA.111.242792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Trinidad EM, Ballesteros M, Zuloaga J, Zapata A, Alonso-Colmenar LM. An impaired transendothelial migration potential of chronic lymphocytic leukemia (CLL) cells can be linked to ephrin-A4 expression. Blood. 2009;114:5081–5090. doi: 10.1182/blood-2009-03-210617. [DOI] [PubMed] [Google Scholar]
- 141.Tawadros T, Brown MD, Hart CA, Clarke NW. Ligand-independent activation of EphA2 by arachidonic acid induces metastasis-like behaviour in prostate cancer cells. Br J Cancer. 2012;107:1737–1744. doi: 10.1038/bjc.2012.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Boyd AW, Bartlett PF, Lackmann M. Therapeutic targeting of EPH receptors and their ligands. Nat Rev Drug Discov. 2014;13:39–62. doi: 10.1038/nrd4175. [DOI] [PubMed] [Google Scholar]
- 143.Funk SD, Orr AW. Ephs and ephrins resurface in inflammation, immunity, and atherosclerosis. Pharmacol Res. 2013;67:42–52. doi: 10.1016/j.phrs.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 144.Weber EW, Han F, Tauseef M, Birnbaumer L, Mehta D, Muller WA. TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. J Exp Med. 2015;212:1883–1899. doi: 10.1084/jem.20150353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kusche-Vihrog K, Jeggle P, Oberleithner H. The role of ENaC in vascular endothelium. Pflugers Arch. 2014;466:851–859. doi: 10.1007/s00424-013-1356-3. [DOI] [PubMed] [Google Scholar]
- 146.Bittner S, Ruck T, Schuhmann MK, Herrmann AM, Moha ou MH, Bobak N, Gobel K, Langhauser F, Stegner D, Ehling P, et al. Endothelial TWIK-related potassium channel-1 (TREK1) regulates immune-cell trafficking into the CNS. Nat Med. 2013;19:1161–1165. doi: 10.1038/nm.3303. [DOI] [PubMed] [Google Scholar]
- 147.Lohman AW, Leskov IL, Butcher JT, Johnstone SR, Stokes TA, Begandt D, DeLalio LJ, Best AK, Penuela S, Leitinger N, et al. Pannexin 1 channels regulate leukocyte emigration through the venous endothelium during acute inflammation. Nat Commun. 2015;6:7965. doi: 10.1038/ncomms8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bao Y, Chen Y, Ledderose C, Li L, Junger WG. Pannexin 1 channels link chemoattractant receptor signaling to local excitation and global inhibition responses at the front and back of polarized neutrophils. J Biol Chem. 2013;288:22650–22657. doi: 10.1074/jbc.M113.476283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Barletta KE, Ley K, Mehrad B. Regulation of neutrophil function by adenosine. Arterioscler Thromb Vasc Biol. 2012;32:856–864. doi: 10.1161/ATVBAHA.111.226845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Schwab ME. Functions of Nogo proteins and their receptors in the nervous system. Nat Rev Neurosci. 2010;11:799–811. doi: 10.1038/nrn2936. [DOI] [PubMed] [Google Scholar]
- 151.Harrison KD, Park EJ, Gao N, Kuo A, Rush JS, Waechter CJ, Lehrman MA, Sessa WC. Nogo-B receptor is necessary for cellular dolichol biosynthesis and protein N-glycosylation. EMBO J. 2011;30:2490–2500. doi: 10.1038/emboj.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Di LA, Manes TD, Davalos A, Wright PL, Sessa WC. Endothelial reticulon-4B (Nogo-B) regulates ICAM-1-mediated leukocyte transmigration and acute inflammation. Blood. 2011;117:2284–2295. doi: 10.1182/blood-2010-04-281956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Johnsson AK, Dai Y, Nobis M, Baker MJ, McGhee EJ, Walker S, Schwarz JP, Kadir S, Morton JP, Myant KB, et al. The Rac-FRET mouse reveals tight spatiotemporal control of Rac activity in primary cells and tissues. Cell Rep. 2014;6:1153–1164. doi: 10.1016/j.celrep.2014.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mizuno R, Kamioka Y, Kabashima K, Imajo M, Sumiyama K, Nakasho E, Ito T, Hamazaki Y, Okuchi Y, Sakai Y, et al. In vivo imaging reveals PKA regulation of ERK activity during neutrophil recruitment to inflamed intestines. J Exp Med. 2014;211:1123–1136. doi: 10.1084/jem.20132112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Krishnan R, Klumpers DD, Park CY, Rajendran K, Trepat X, van Bezu J, van Hinsbergh VW, Carman CV, Brain JD, Fredberg JJ, et al. Substrate stiffening promotes endothelial monolayer disruption through enhanced physical forces. Am J Physiol Cell Physiol. 2011;300:C146–C154. doi: 10.1152/ajpcell.00195.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Huynh J, Nishimura N, Rana K, Peloquin JM, Califano JP, Montague CR, King MR, Schaffer CB, Reinhart-King CA. Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci Transl Med. 2011;3:112ra122. doi: 10.1126/scitranslmed.3002761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Reijerkerk A, Lopez-Ramirez MA, van Het HB, Drexhage JA, Kamphuis WW, Kooij G, Vos JB, van der Pouw Kraan TC, van Zonneveld AJ, Horrevoets AJ, et al. MicroRNAs regulate human brain endothelial cell-barrier function in inflammation: implications for multiple sclerosis. J Neurosci. 2013;33:6857–6863. doi: 10.1523/JNEUROSCI.3965-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Poissonnier L, Villain G, Soncin F, Mattot V. miR126-5p repression of ALCAM and SetD5 in endothelial cells regulates leucocyte adhesion and transmigration. Cardiovasc Res. 2014;102:436–447. doi: 10.1093/cvr/cvu040. [DOI] [PubMed] [Google Scholar]
- 159.Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, Kaluza D, Treguer K, Carmona G, Bonauer A, et al. MicroRNA-34a regulates cardiac ageing and function. Nature. 2013;495:107–110. doi: 10.1038/nature11919. [DOI] [PubMed] [Google Scholar]