

Abstract
The skin forms a vital barrier between an organism’s external environment, providing protection from pathogens and numerous physical and chemical threats. Moreover, the intact barrier is essential to prevent water and electrolyte loss without which terrestrial life could not be maintained. Accordingly, acute disruption of the skin through physical or chemical trauma needs to be repaired timely and efficiently as sustained skin pathologies ranging from mild irritations and inflammation through to malignancy impact considerably on morbidity and mortality. The Nuclear Hormone Receptor Family of transcriptional regulators has proven to be highly valuable targets for addressing a range of pathologies, including metabolic syndrome and cancer. Indeed members of the classic endocrine sub-group, such as the glucocorticoid, retinoid, and Vitamin D receptors, represent mainstay treatment strategies for numerous inflammatory skin disorders, though side effects from prolonged use are common. Emerging evidence has now highlighted important functional roles for nuclear receptors belonging to the adopted and orphan subgroups in skin physiology and patho-physiology. This review will focus on these subgroups and explore the current evidence that suggests these nuclear receptor hold great promise as future stand-alone or complementary drug targets in treating common skin diseases and maintaining skin homeostasis.
Keywords: Nuclear receptor, Wound healing, Orphan receptors, PPAR, LXR, NR4A
Introduction
The skin is the largest organ in the body and performs an essential role in an animal’s health and survival by providing a physical barrier between the organism and the external environment. This physical barrier forms a crucial first line of defence against invading pathogens and must also protect against a range of physical and chemical insults. Furthermore, an effective skin barrier is crucial to prevent uncontrolled loss of water and solutes.
Like all complex organs, the development and differentiation of the skin are co-ordinated by numerous regulatory pathways. Once established the integrity of the skin barrier must be maintained to ensure the ongoing health of the individual. An acute or chronically compromised skin barrier underpins numerous skin pathologies that can range from the mildly irritating through to debilitating or fatal patient outcomes. Moreover, many inflammatory skin disorders, such as psoriasis and atopic dermatitis, can exact a significant social and psychological toll on the individual. Accordingly, understanding the mechanistic basis of these pathologies and identifying novel ‘drug-able’ targets is essential not only for maintaining skin health and preventing or treating skin disease, but will undoubtedly impact the overall heath of the individual.
Skin structure and physiology
Structurally, the skin is made up of several distinct cellular layers from the outer epidermis overlaying the dermis sitting over a layer of sub-cutaneous adipose tissue (Fig. 1). The outermost epidermis is further divided into four distinct layers (inner to outer), such as the stratum basale (SB), stratum spinosum (SS), stratum granulosum (SG), and the stratum corneum (SC). Keratinocytes are the predominant cell type in the skin. These distinctive layers of the epidermis are formed by a vertical differentiation program of keratinocytes from proliferative keratinocyte populations in the SB, which migrate and differentiate through the respective layers to finally form the protein rich, anuclear cells termed corneocytes of the stratum corneum (Fig. 1). The protein rich corneocytes, embedded in a lipid rich matrix, are crucial to the formation of the permeability barrier that affords protection from the external environment and the loss of water and electrolytes. Formation of the SC requires expression of proteins, such as loricrin, involucrin, and filaggrin to form the cornified envelope. In addition, the corneocytes provide a scaffold that is required for the organization of extracellular lipids into lamellar membranes [1, 2]. In addition to providing a physical and chemical barrier, keratinocytes also respond to external insults to propagate immune responses by secreting pro-inflammatory cytokines in response to a range of stimuli [3].
Fig. 1.
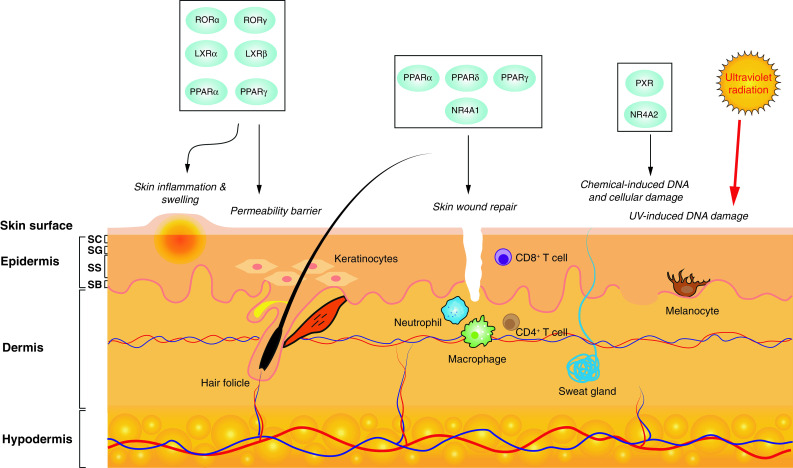
Skin is comprised of the outer epidermis, the dermis, and the hypodermis. The epidermis is made up of four distinct layers, such as stratum basale (SB), stratum spinosum (SS), stratum granulosum (SG), and the stratum corneum (SC), representing keratinocytes at different stages of differentiation. Members of the adopted and orphan nuclear receptors are involved in critical processes, such as wound repair, permeability barrier homeostasis, and repair of UV damage. A number of receptors may be targeted to address skin inflammation that underpins a number of acute and chronic skin disorders
Melanocytes are neural crest derived pigment cells that reside at the dermal–epidermal interface and make direct contact with approximately 20–30 keratinocytes via nerve, such as dendritic projections. Melanocytes protect the keratinocytes form UV radiation via the synthesis of the melanin polymer in specialized organelles termed melanosomes, which are transferred to associated keratinocytes via the dendritic processes [4]. Malignant transformation of melanocytes gives rise to the most aggressive form of skin cancer, melanoma.
Lipid permeability barrier formation and wound healing
Epidermal lipid homeostasis is vital for the formation and maintenance of the skins permeability barrier. In addition, skin lipids play an important anti-microbial role in the defence against external pathogens [5]. The permeability barrier is localised primarily in the stratum corneum, which is comprised of protein rich corneocytes (terminally differentiated and anuclear keratinocytes) embedded in a lipid rich extracellular matrix [6]. The stratum corneum is formed by 10–25 layers of corneocytes arranged in a “bricks and mortar” formation in parallel to the skin surface embedded in the extracellular matrix. The hydrophobic extracellular lipid matrix of the stratum corneum provides a barrier against loss of water and electrolytes, known as trans-epidermal water loss (TEWL). The lipid composition of the epidermis is predominantly ceramides (approximately 50 %) together with cholesterol and free fatty acids [7]. Ageing and a number of common skin pathologies, including atopic dermatitis and psoriasis, are known to disrupt the permeability of the skin. Accordingly, therapeutic avenues that can modulate epidermal lipid homeostasis provide enticing candidates to address a range of skin pathologies [5].
Maintaining the integrity of the skin barrier following physical or chemical trauma is vital. Skin wound healing is a multistep process with three major overlapping phases that proceed sequentially from (1) inflammation, (2) new tissue formation, and (3) finally, tissue remodeling. A number of nuclear receptors have been shown to be involved in the respective stages and hence form ideal therapeutic targets to facilitate this process, which is of particular relevance in disease states, such as diabetes, in which impaired healing processes leads to chronic wounds that form a major contribution to disease morbidity [8].
Skin cancer
Collectively, skin cancers are the most common forms of cancer in humans with lifetime incidence rates approaching one in five and are divided into the non-melanoma skin cancers, basal cell carcinoma (BCC), and squamous cell carcinoma (SCC) and melanoma [9]. BCCs form the majority of non-melanoma skin cancers, while they rarely metastasise pose a significant burden on patient morbidity and consequently health care systems globally [10]. SCCs are more likely to metastasise and accordingly have a significantly higher mortality rate, while melanoma, which arises from transformation from melanocytes in the skin, is the most aggressive form of skin cancer that has a propensity to become highly metastatic and carries the highest rate of mortality rate of all skin cancers [10]. Ultraviolet radiation exposure (UVR) is a major contributing factor for the development of all skin cancer types [11, 12] in concert with the degree of skin pigmentation [13]. Not surprisingly, most skin cancers can be attributed to direct UV signature mutations (C → T and CC → TT), although a number key oncogenic lesions carry non-UV signature mutations presumably from indirect factors, such as reactive oxygen species, which can be produced following UVR exposure [9]. The importance of robust DNA repair mechanisms in the skin for the protection from UVR, arguably the most common external mutagen to which humans are exposed to, is exemplified by the significantly high rates of skin cancer observed in patients with Xeroderma pigmentosum [14].
Targeting nuclear receptors in skin health and disease
To date, nuclear receptors have proven to be valuable targets in treating a range of skin conditions most notably via agents, including glucocorticoids, retinoids (vitamin A derivatives), and deltanoids (vitamin D analogues). Most prominent among these are the glucocorticoids, a steroid class of drugs, which are the most commonly prescribed drugs for topical treatment of inflammatory skin conditions [15]. While glucocorticoids are favoured for their anti-inflammatory activity, side effects that commonly arise from the long-term use include skin atrophy, fragility, and bruising [16–19]. Moreover, undesirable side effects, such as impaired permeability barrier, have been observed after only the short-term glucocorticoid-based treatment regimes [20]. For example, while glucocorticoids are an effective strategy to reduced inflammation in allergic contact dermatitis, the efficacy of these agents can be limited by well-recognised deleterious side effects, including impaired cutaneous permeability barrier homeostasis that ultimately counteracts the positive effects of these drugs [21].
The importance of drugs targeting the classic endocrine sub-class of nuclear hormone receptors, such as the glucocorticoid and retinoid receptors, in the skin is well established. However, the development of non-desirable side effects from both acute and long-term treatment with these agents argues for the development of alternative or complementary strategies to achieve the same clinical outcomes but circumventing less desirable side effects. This desired complementarity can potentially be achieved by targeting additional or alternative members of the nuclear receptor family. Current research has identified important roles for the adopted and orphan sub-classes of the nuclear receptor family that are crucial to maintaining the integrity of the skin barrier and may be targeted to address issues, such as inflammation, wound repair, and cytoprotection against UV irradiation (Fig. 1). Accordingly, this review will focus on emerging evidence that highlights the potential of ‘adopted’ and orphan nuclear receptors in skin physiology and patho-physiology and explore their potential as drug-able therapeutic targets in these contexts.
The peroxisome proliferator-activated receptors
The peroxisome proliferator-activated receptors (PPARs) are a subfamily of nuclear receptors comprising three members, such as PPARα, PPARγ, and PPARβ/δ (henceforth referred to as PPARδ) that are activated by a wide variety of lipid-based ligands [22]. With high levels of expression detected in key metabolic tissues and crucial roles recognised in regulating metabolic homeostasis, these receptors are best known as regulators of carbohydrate and lipid metabolism, and accordingly, identification of synthetic ligands targeting the PPAR family has proven to be highly effective agents in the clinic to address metabolic syndrome [22]. All three family members are expressed at varying levels in human and rodent skin under normal and pathological conditions [23]. Investigating the diverse biological functions of these receptors in normal skin homeostasis and in models of skin disease suggests that the PPAR family may prove to be valuable clinical targets for treating a range of skin pathologies in the future. Indeed, the potent anti-inflammatory effects of activated PPARs further highlight the potential of PPAR modulators in addressing epidermal pathologies in which aberrant inflammation is frequently a key component [24].
Expression and function in skin
Expression of all PPAR family members has been observed to varying levels in rodent skin and human keratinocytes [25–27]. Moreover, the activation of PPAR α, δ, or γ has major effects on epidermal/keratinocyte function and is known to exert beneficial effects in skin homeostasis and integrity [28–32]. Critical keratinocyte differentiation markers, such as involucrin, loricrin, profilaggrin, and transglutaminase 1, are increased in response to the topical application of PPAR ligands to mouse skin or treatment of cultured primary human keratinocytes [33–37]. Furthermore, the activation of PPAR α, δ, or γ inhibits keratinocyte proliferation in vivo and in in vitro models [38–41]. Importantly, the maintenance and restoration of the epidermal permeability barrier function are promoted by PPAR α, δ, or γ ligands resulting in an acceleration of barrier recovery following acute disruption, with anti-inflammatory functions of the receptors making a major contribution to this effect [32, 35, 36, 42].
Mouse knock-out models of both PPARα and PPARγ exhibit only modest disruptions in skin physiology and general morphology and appear to have an intact permeability barrier function, suggesting that these receptors are not required for the normal development and maintenance of the epidermal barrier [35, 42–44]. Similarly, no major disruption in basal permeability barrier was evident in PPARδ knock-out mice; however, these animals show a significant delay in barrier recovery rates following acute barrier disruption [45].
Glucocorticoid treatments commonly administered to ameliorate epidermal inflammation are known to decrease lipid synthesis in the epidermis leading to a disruption to the permeability barrier [20]. PPAR activators administered in conjunction with topical corticosteroids have been shown to normalise keratinocyte differentiation and diminish corticosteroid-mediated atrophy of the epidermal layer, suggesting that PPAR modulators can be used to prevent common side effects of GC monotherapy [29, 46].
Wound healing
To date, PPARδ and PPARα have been found to play the most prominent role in the crucial process of skin healing after injury, although more recent evidence, discussed below, argues that targeting PPARγ function in macrophages may show utility in augmenting the repair process.
Both PPARα and PPARδ expressions have been found to increase dramatically in response to skin wounding and healing, although with different timings, suggesting distinct roles for each receptor in the healing process. PPARα upregulation is transient and restricted to the early inflammatory phase of the repair process [47]. PPARα null mice were found to have a transient delay in initiating wound healing which appears to hinge predominantly on a reduction in neutrophil and monocyte/macrophage recruitment to the wound bed, ultimately resulting in uncontrolled inflammation in the initial phase of the repair process [47]. Conversely, elevated PPARδ expression is maintained throughout all the stages of the healing process and PPARδ knock-out mice exhibit a delay in repair throughout the entire process, delaying repair by 2–3 days [47]. Primary cultures of PPARδ mutant keratinocytes result in a notable defect in migration and adhesion, and given that keratinocyte migration to the wound edge is a pivotal step in the repair process, this defect is likely to be a major contributor to the delayed repair in the PPARδ transgenic mice [47]. The increased expression of PPARα and PPARδ at the wound edge is driven by pro-inflammatory cytokines, such as TNFα and IFNγ, while the progressive PPARδ repression as epithelial repair proceeds is mediated by TGFβ-mediated repression of the PPARδ promoter [47–50].
While the earlier work of Michelik et al. [47] suggested a negligible role for PPARγ in skin wound repair, more recent evidence argues that reduced macrophage PPARγ expression accounts for impaired wound healing in diabetic patients [51]. Central to this activity is the recognised transition of macrophages in the context of normal wound healing from pro-inflammatory to “healing-associated” phenotypes [52, 53]. In the context of diabetes, this transition is frequently impaired, primarily due to sustained IL1β production within the diabetic wound leading to prolonged wound inflammation [54–56]. Using myloid cell specific PPARγ null mice, it was determined that PPARγ was crucial for the macrophage phenotype switch within the wound and that loss of the receptor was sufficient for prolonged wound inflammation and delayed healing. Crucially, topical administration of PPARγ agonists to skin wounds in diabetic mice was capable of facilitating the macrophage phenotype switch with a commensurate improvement in wound healing [51]. Together, these data suggest an additional avenue with which to address poor wound healing in diabetic individuals, a common symptomatic feature of diabetes, which represents a significant burden associated with this disease.
Skin disease
Given the well-established utility of targeting the anti-inflammatory activities of the PPAR family in a range of pathologies [24], it is not surprising that these avenues will likely extend to various diseases of the skin, such as psoriasis and atopic dermatitis, many of which are underpinned by acute or chronic inflammation.
PPARγ activation is known to mediate anti-inflammatory actions in numerous patho-physiological contexts [24, 57]. Mechanistically, these effects are largely driven by a process known as trans-repression, whereby the expression of pro-inflammatory mediators is repressed by ligand-activated PPARγ via the recruitment of co-repressor complexes by PPARγ to the promoter region of these genes [58–60]. The activation of PPARγ by the Thiazolidine (TZD) class of drugs can attenuate epidermal hyper-proliferation, in concert with reduced inflammation in rodent models of inflammatory skin disease [35, 38, 61]. PPARγ expression is reduced in psoriatic plaques, potentially laying a foundation for chronic inflammation of the skin [27]. Importantly, systemic administration of two TZD agents, pioglitazone and troglitazone were found to improve skin lesions in psoriasis patients, while a third agent, rosiglitazone, was found to have a negligible effect [62–65]. These observations may be attributed to off target/non-PPARγ-mediated mechanisms of the different TZDs or alternatively illustrate the complexities of differential activation of these receptors by different chemical agents. In the context of atopic dermatitis, studies in both humans and rodents have shown that systemic delivery of PPARγ modulators can ameliorate the symptoms of this condition [28, 30, 66–68]. Mast cells are central mediators of the pathogenesis of atopic dermatitis and as such represent important cellular targets in treating this disease [69]. Significantly, pharmacological activation of PPARγ has been found to inhibit the maturation of mast cells in the skin, while transgenic knock-down accelerated mast cell differentiation [70]. Accordingly, in light of the pivotal role of mast cells activity in allergen and inflammatory process in dermatitis, it is perhaps not surprising that PPARγ activation was capable of attenuating atopic and contact dermatitis in rodent models [70].
PPARα activators have also been shown to inhibit both irritant and contact dermatitis in mouse skin [32]. Filagrin mutations or decreased filagrin expression is frequently associated with the manifestation of atopic dermatitis [71]. Using filagrin deficient skin reconstructions, Wallmeyer and colleagues have recently demonstrated the ability of the PPARα agonist WY14643 to normalise the disturbed free fatty acid profile of the skin, ultimately improving the skin barrier function [72]. The potential utility of PPARα agonism in the treatment of both atopic dermatitis and the prevention of glucocorticoid induced barrier disruption is further supported by recent work that demonstrated that PPAR activation is crucial for pseudo-ceramide-mediated attenuation of inflammation and barrier improvement in a murine model of atopic dermatitis [73].
Interestingly, PPARδ expression is upregulated in psoriatic plaques compared to non-lesional skin [27]. Moreover, the activation of PPARδ has been found to trigger a psoriasis-like condition in rodent skin [74]. Together, these observations suggest that PPARδ antagonism might prove to be an effective strategy in the context of psoriasis, a concept further supported by observed reduction of PPARδ induced psoriasis-like symptoms in rodent skin with concurrent treatment with PPARδ antagonists [75].
Skin cancer
With UV damage being the leading cause of mutations that underpin skin carcinogenesis, a number of studies have been initiated to determine if PPAR modulation may impact on the development of skin cancer. The activation of PPARα in human skin reduces the UV-induced inflammation, which may afford some protection against UV-mediated skin carcinogenesis [76], while PPARα agonists exhibited a protective effect in mouse skin exposed to chemical carcinogens [77]. Chemically induced skin tumours using DMBA have been found to be more severe in PPARδ knock-out mice, an effect that has been attributed to a role for PPARδ inhibiting keratinocyte proliferation [78, 79]. Similarly, PPARγ activation has been shown to inhibit chemical and UV-mediated skin carcinogenesis [80–82].
Treatment of melanoma cells with PPARγ activators has been reported to inhibit the proliferation and promote apoptosis of melanoma cells [83–85]. Moreover, PPARγ modulates β-catenin signalling in a ligand-dependent manner in melanoma cells [85]. PPARα agonists have also been found to be capable of reducing the metastatic potential of melanoma cells in a rodent model [86]. While some small clinical trials that have been conducted to explore the clinical utility of treating melanoma have shown some promise, more comprehensive studies are needed to determine if PPAR agonist may prove to be useful adjunct therapies in treating this disease.
The liver-X-receptors
Expression and function in the skin
Despite the name expression of both Liver-X receptors, LXRα and LXRβ, has been identified in human and rodent skin [87–89], oxysterol ligands for LXRs have been found to promote the differentiation of cultured normal human keratinocytes as evidenced by elevated expression of proteins, such as involucrin and transglutaminase-1, which correlated with a twofold increase in cornified envelope formation [88]. These oxysterol ligands were also found to accelerate the differentiation of the fetal rat epidermis and barrier formation in vivo when injected into the amniotic fluid during gestation [87]. Topical application of LXR agonists to murine skin similarly promoted epidermal differentiation and studies using LXRα and LXRβ null loss of function transgenic mice revealed that LXRβ but not LXRα was required for this effect. While this may suggest that LXRβ is the primary target of LXR agonists in the skin, expression analysis only detected this isoform in the murine epidermis in contrast to the presence of both isoforms in human keratinocytes and the rat epidermis [90].
Given the importance of lipid homeostasis in the skin, it is not surprising to find that improved permeability barrier function driven by natural and synthetic LXR ligands is mediated by increased expression of components of the SC lipid matrix, including cholesterol, fatty acids, sphingolipids, and importantly barrier specific ceremides [91]. Furthermore, in response to acute barrier disruption, LXR agonists enhanced lamellar body formation, secretion, and post-secretory processing [91].
LXRs have also been proposed as potential therapeutic targets to address UV-mediated photo-ageing of the skin. Intriguingly, the skin of LXRβ−/−null mice was found to share many of the features associated with chronologically aged skin including a similar gene expression profile of aged skin identified through analyses comparing young and aged human skin [92]. Furthermore, the synthetic ligand TO901317 reduced the abnormal thickening of the skin in response to UV irradiation [92, 93].
Skin disease
Oxysterol ligands and synthetic agonists of LXRs have proven to be effective agents in reducing cutaneous inflammation and epidermal thickening in rodent models of irritant and allergic contact dermatitis [93]. Utilizing transgenic knock-out lines in these studies confirmed LXRβ as the key mediator of this effect with the oxysterol 22(R)-hydroxycholesterol unable to reduce inflammation in the absence of LXRβ; however, loss of LXRα in these studies also partially reduced the anti-inflammatory effect or the 22-ROH [93]. Atopic dermatitis (AD) is a chronic inflammatory dermatitis that also presents with prominent deficits in critical skin barrier functions, such as stratum corneum cohesion and hydration and anti-microbial defence [94–97]. Significant reduction in epidermal hyperplasia and inflammation in the context of a hapten induced AD, such as pathology, in hairless mice were observed following the topical application of the synthetic LXR activators TO901317 and GW3965, suggesting that such agents might prove effective in treating AD in humans [98]. Notably, while synthetic LXR agonists and naturally occurring ligands, such as 22-ROH, have shown utility in other inflammatory skin models, 22-ROH administration was not able to recapitulate the anti-inflammatory effect and improved barrier function elicited by their synthetic counterparts [98].
Recent analysis suggests that the targeted activation of LXRs may provide a promising strategy for future treatment of melanoma. Treatment of mouse B16 and human A-357 melanoma cell lines with the synthetic agonist T0901317 elicited a significant inhibition of proliferation in vitro. Moreover, oral administration of T0901317 inhibited the growth of B16 cells injected into C57BL/6 mice largely through induction of apoptosis [99]. While an alternative study did not observe the same growth inhibition in vivo for either T0901317 or GW3965, a robust inhibition of tumour metastasis was observed [100]. This effect was found to be mediated by melanoma cell autonomous and non-cell autonomous (stromal) induction of ApoE driven primarily by LXRβ which blocks multiple facets of the metastasis process, including tumour cell invasion, endothelial recruitment, and distal site colonisation. Moreover, this effect was recapitulated in multiple genetic contexts and using cells with acquired resistance to the conventional and emerging cancer therapies, arguing strongly for the potential of these agents as adjunct therapies or for the treatment of relapsing tumours [100].
The NR4A family
The NR4A subfamily consists of three highly related nuclear receptors: neuron-derived clone 77 [NR4A1; NUR77]; nuclear receptor related 1 [NR4A2; NURR1]; and neuron-derived clone 1 [NR4A3; NOR1]. Current evidence suggests these receptors are true orphan receptors, as no endogenous ligands have been identified so far; hence, the transcriptional response of NR4A receptors has been suggested to operate in a ligand-independent manner [101], predominantly through the activation of multiple cell-signalling pathways. Despite the lack of endogenous ligands, numerous agents capable of specifically modulating the NR4A family of receptors have emerged raising the prospect of future targeting of these receptors to treat a wide range of pathologies [102]. The NR4A receptors influence the activities of a diverse range of genes that control cell survival, proliferation, migration, apoptosis, angiogenesis, and DNA repair and inflammation; all of which have long been recognised to modulate the onset and progression of human diseases and malignancies [103, 104].
Exposure of melanocytes to ultraviolet radiation (UVR) activates the melanocortin-1-receptor (MC1R)-signalling pathway [105], leading to the activation of two independent pathways, melanogenesis (pigment synthesis) and DNA repair. Interestingly, the activation of the MC1R-signalling pathway has been found to induce the expression of all three NR4A members in B16 mouse melanoma cells and primary human melanocytes in a rapid and transient manner [106], suggesting that NR4As are early response genes involved in mediating melanocyte responses to UV irradiation. Within the nucleus, the NR4A receptors translocate into distinct nuclear foci following exposure to genotoxic stress, including UVR, ionising radiation (IR), and oxidative stress [107–109]. Recent studies using in vitro models of UVR or IR-induced DNA damage observed the co-localisation of NR4A2 nuclear foci with DNA repair factors, such as γH2AX, DDB2, and XPC, suggesting that these foci represent sites of DNA damage that are undergoing active DNA repair [108, 109]. Dual loss of functional NR4A1 and NR4A2 in melanocytes following UV irradiation results in impaired clearance of UV-induced DNA lesion, such as cyclobutane pyrimidine (CPD) dimer [106].
The importance of DNA damage response (DDR), which detects and repairs DNA damage, has long been recognised as a vital mechanism to prevent cellular transformation. Unrepaired DNA damage in melanocytes can be mutagenic, ultimately leading to melanoma development. Given that the formation of foci is a hallmark of DDR proteins, emerging evidence points to the possibility that the recruitment of NR4A receptors to foci may contribute to an enhanced DDR in cells by potentially aiding the recruitment of additional repair machineries, opening and stabilising the local chromatin environment, strengthening interaction between proteins, and providing signalling frameworks that may contribute to cross-talk between different DDR cascades. Indeed, supporting evidence revealed that NR4A2 is involved in nucleotide excision repair [108] and DNA double-strand breaks [109] following DNA damage. It is likely that the NR4A nuclear receptors represent central components in an inducible DNA repair pathway that provides an additional level of protection to UV-sensitive cells [110]. Since lack of DNA repair is a precursor to cancer predisposition, establishing NR4A receptors as potential anticancer therapeutic targets in a drug-inducible model may be an emerging theme in the field of melanoma prevention.
The pharmacological application of NR4A receptor in disease prevention is only now being developed. While endogenous ligands for these receptors remain to be identified, the current research focuses on developing synthetic ligands that may contribute to clinical benefits through altered NR4A activities. This is exemplified by the application of para-phenyl diindolylmethane (C-DIM12), which activates NR4A2 to suppress tumorigenesis in urothelial carcinoma cells [111, 112] and in pancreatic cells [113]. Notably, the treatment of normal human epidermal keratinocytes (NHEKs) with 1,1-bis(39-indolyl)-1-(p-chlorophenyl methane) (DIM-D) reduces UV-induced ROS formation and apoptosis via NR4A2 transactivation, and hence cancer formation due to improved DNA repair capacity [114]. Another study has also provided evidence showing that altered NR4A2 receptor activity may contribute to the healing of skin-related diseases, such as psoriasis [115]. Similarly, a role for NR4A1 in wound healing and fibrosis through the upregulation of integrin β4 [116] and the activation of TGF-β signalling pathways has been reported [117], respectively. Taken together, these studies highlight the potential of the NR4A family members as therapeutic targets for pharmacological modulation that could potentially provide additional chemo-preventative strategy for melanoma and non-melanoma skin cancers. While targeting the NR4A receptors may prove to be a useful chemo-prevention strategy, sustained expression of this family down-stream of oncogenic MAPK signalling has been reported [118]. Recent advances that specifically target the MAPK pathway in melanoma have improved patient outcomes, extending 5-year survival rates considerably; however, clinical resistance ultimately develops in the majority of cases [119]. Interestingly, recent genetic screening in BRAF mutant melanoma cells identified the ability of both NR4A1 and NR4A2 genes to confer resistance to BRAF/MEK inhibitors [120]. Together, these data would suggest that elevated NR4A expression in melanoma may be undesirable, and in this context, the inhibition of their function may be an important strategy to prolong the efficacy of these agents with further investigation of their role in melanoma cell function warranted.
The retinoid-related orphan receptor subfamily
The retinoid-related orphan receptors (RORs) are a subfamily of orphan receptors predominantly expressed in the central nervous system, modulating physiological processes, such as immune functions, metabolism, and cerebellar development in a tissue-specific manner [121]. There are three members to this subfamily: RORα, RORβ, and RORγ. Among the RORs, recent studies have shown that RORα and RORγ are widely expressed in all major skin cell populations [122–124]. In mice, the expression of RORα has been linked to a role of epithelial differentiation [123]. Similarly, in human keratinocytes, RORα has been shown to promote differentiation via regulating genes involved in epidermal lipid/barrier functions [125], as well as inducing the expression of the skin barrier protein filaggrin [126]. Based on these observations, it is tempting to speculate that identifying ROR ligands may be useful for treating skin pathologies, including cancer, that are associated with abnormal keratinocyte differentiation.
While the RORs are traditionally recognised as orphan receptors, emerging evidence has identified a number of ligands that interact with the ligand binding domain of RORs, functioning as either agonists or antagonists of ROR transcriptional activity [127–130]. In particular, the activation of vitamin D-signalling pathway leads to the production of two major antagonists or inverse agonists of the RORα and γ receptors in the human skin [131]. These bioactive chemicals have been implicated as effective treatment strategy for autoimmune diseases, such as psoriasis in several independent studies [132, 133].
Pregnane X receptor (PXR) and farnesol X-activated receptors (FXR)
The pregnane X receptor (PXR) is a ligand-dependent transcription factor that is expressed predominantly in the liver. PXR is termed the master regulator of the expression of CYP3A4 enzymes, which are involved in detoxifying environmental pollutants that the human body is exposed to [134]. In skin, pro-carcinogenic effects can be augmented when exposed to these chemicals that carry DNA damaging properties. Interestingly, a recent study has reported that the expression of PXR can be upregulated in epidermal cells with the treatment of chemical compounds [135]. Accordingly, it is likely that the expression and activation of PXR in the skin may be beneficial for combating skin carcinogenesis caused by chemical exposure. PXR is also capable of preventing skin fibrosis via reduced TGFβ signalling [136].
Farnesol X-activated receptor ligands have also been shown to accelerate epidermal development differentiation in utero has been demonstrated suggesting a role for this receptor in the regulation of cutaneous development [87].
Concluding remarks
To date, the nuclear receptor family has proven to be important targets in the treatment of a range of skin pathologies with glucocorticoids, retinoids, and deltanoids commonly prescribed for inflammatory skin conditions, such as psoriasis and dermatitis. Side effects associated with these agents have prompted searches for alternative agents to treat these conditions for the same therapeutic benefit or to co-administer with the mainstay drugs to ameliorate the anticipated side effects. Increasing evidence points to other members of the nuclear receptor family as potential tools with which to achieve these aims, which will undoubtedly continue to expand as the role of these and other nuclear receptors are explored in the context of skin physiology and patho-physiology in the future. Concurrent development of novel modulators that target members of the orphan and ‘adopted’ sub-classes of nuclear receptors will provide an exciting platform of agents for future analysis. While a multitude of cell based and animal studies has provided crucial supportive evidence for the utility of agents targeting adopted and orphan nuclear receptors to treat a range of skin pathologies, at this juncture clinical trials will be crucial to ultimately realise the potential of these agents in the clinic.
References
- 1.Elias PM, et al. Basis for the permeability barrier abnormality in lamellar ichthyosis. Exp Dermatol. 2002;11(3):248–256. doi: 10.1034/j.1600-0625.2001.110308.x. [DOI] [PubMed] [Google Scholar]
- 2.Schmuth M, et al. Structural and functional consequences of loricrin mutations in human loricrin keratoderma (Vohwinkel syndrome with ichthyosis) J Invest Dermatol. 2004;122(4):909–922. doi: 10.1111/j.0022-202X.2004.22431.x. [DOI] [PubMed] [Google Scholar]
- 3.Albanesi C, Pastore S. Pathobiology of chronic inflammatory skin diseases: interplay between keratinocytes and immune cells as a target for anti-inflammatory drugs. Curr Drug Metab. 2010;11(3):210–227. doi: 10.2174/138920010791196328. [DOI] [PubMed] [Google Scholar]
- 4.Lin JY, Fisher DE. Melanocyte biology and skin pigmentation. Nature. 2007;445(7130):843–850. doi: 10.1038/nature05660. [DOI] [PubMed] [Google Scholar]
- 5.Feingold KR, Elias PM. Role of lipids in the formation and maintenance of the cutaneous permeability barrier. Biochim Biophys Acta. 2014;1841(3):280–294. doi: 10.1016/j.bbalip.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Proksch E, Brandner JM, Jensen JM. The skin: an indispensable barrier. Exp Dermatol. 2008;17(12):1063–1072. doi: 10.1111/j.1600-0625.2008.00786.x. [DOI] [PubMed] [Google Scholar]
- 7.van Smeden J, et al. The important role of stratum corneum lipids for the cutaneous barrier function. Biochim Biophys Acta. 2014;1841(3):295–313. doi: 10.1016/j.bbalip.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 8.Rieger S, et al. The role of nuclear hormone receptors in cutaneous wound repair. Cell Biochem Funct. 2015;33(1):1–13. doi: 10.1002/cbf.3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor JS. Biomolecules. The dark side of sunlight and melanoma. Science. 2015;347(6224):824. doi: 10.1126/science.aaa6578. [DOI] [PubMed] [Google Scholar]
- 10.Narayanan DL, Saladi RN, Fox JL. Ultraviolet radiation and skin cancer. Int J Dermatol. 2010;49(9):978–986. doi: 10.1111/j.1365-4632.2010.04474.x. [DOI] [PubMed] [Google Scholar]
- 11.Gilchrest BA, et al. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 1999;340(17):1341–1348. doi: 10.1056/NEJM199904293401707. [DOI] [PubMed] [Google Scholar]
- 12.Soehnge H, Ouhtit A, Ananthaswamy ON. Mechanisms of induction of skin cancer by UV radiation. Front Biosci. 1997;2:d538–d551. doi: 10.2741/A211. [DOI] [PubMed] [Google Scholar]
- 13.Sturm RA. Molecular genetics of human pigmentation diversity. Hum Mol Genet. 2009;18(R1):R9–R17. doi: 10.1093/hmg/ddp003. [DOI] [PubMed] [Google Scholar]
- 14.Cleaver JE. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat Rev Cancer. 2005;5(7):564–573. doi: 10.1038/nrc1652. [DOI] [PubMed] [Google Scholar]
- 15.Nagpal S (2003) An orphan meets family members in skin. J Invest Dermatol 120(2):viii–x [DOI] [PubMed]
- 16.Hengge UR et al (2006) Adverse effects of topical glucocorticosteroids. J Am Acad Dermatol 54(1):1–15 (Quiz 8–16) [DOI] [PubMed]
- 17.Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96(1):23–43. doi: 10.1016/S0163-7258(02)00297-8. [DOI] [PubMed] [Google Scholar]
- 18.Schoepe S, et al. Glucocorticoid therapy-induced skin atrophy. Exp Dermatol. 2006;15(6):406–420. doi: 10.1111/j.0906-6705.2006.00435.x. [DOI] [PubMed] [Google Scholar]
- 19.Sheu HM, et al. Depletion of stratum corneum intercellular lipid lamellae and barrier function abnormalities after long-term topical corticosteroids. Br J Dermatol. 1997;136(6):884–890. doi: 10.1111/j.1365-2133.1997.tb03929.x. [DOI] [PubMed] [Google Scholar]
- 20.Kao JS, et al. Short-term glucocorticoid treatment compromises both permeability barrier homeostasis and stratum corneum integrity: inhibition of epidermal lipid synthesis accounts for functional abnormalities. J Invest Dermatol. 2003;120(3):456–464. doi: 10.1046/j.1523-1747.2003.12053.x. [DOI] [PubMed] [Google Scholar]
- 21.Ashwell JD, Lu FW, Vacchio MS. Glucocorticoids in T cell development and function*. Annu Rev Immunol. 2000;18:309–345. doi: 10.1146/annurev.immunol.18.1.309. [DOI] [PubMed] [Google Scholar]
- 22.Smith AG, Muscat GE. Skeletal muscle and nuclear hormone receptors: implications for cardiovascular and metabolic disease. Int J Biochem Cell Biol. 2005;37(10):2047–2063. doi: 10.1016/j.biocel.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Michalik L, Wahli W. Peroxisome proliferator-activated receptors (PPARs) in skin health, repair and disease. Biochim Biophys Acta. 2007;1771(8):991–998. doi: 10.1016/j.bbalip.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 24.Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab. 2012;23(7):351–363. doi: 10.1016/j.tem.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 25.Braissant O, et al. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology. 1996;137(1):354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- 26.Rivier M, et al. Differential expression of peroxisome proliferator-activated receptor subtypes during the differentiation of human keratinocytes. J Invest Dermatol. 1998;111(6):1116–1121. doi: 10.1046/j.1523-1747.1998.00439.x. [DOI] [PubMed] [Google Scholar]
- 27.Westergaard M, et al. Expression and localization of peroxisome proliferator-activated receptors and nuclear factor kappaB in normal and lesional psoriatic skin. J Invest Dermatol. 2003;121(5):1104–1117. doi: 10.1046/j.1523-1747.2003.12536.x. [DOI] [PubMed] [Google Scholar]
- 28.Fluhr JW, et al. Topical peroxisome proliferator activated receptor activators accelerate postnatal stratum corneum acidification. J Invest Dermatol. 2009;129(2):365–374. doi: 10.1038/jid.2008.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hatano Y, et al. Efficacy of combined peroxisome proliferator-activated receptor-alpha ligand and glucocorticoid therapy in a murine model of atopic dermatitis. J Invest Dermatol. 2011;131(9):1845–1852. doi: 10.1038/jid.2011.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jung K et al (2011) Peroxisome proliferator-activated receptor gamma-mediated suppression of dendritic cell function prevents the onset of atopic dermatitis in NC/Tnd mice. J Allergy Clin Immunol 127(2):420–429 (e1–6) [DOI] [PubMed]
- 31.Mastrofrancesco A, et al. Preclinical studies of a specific PPARgamma modulator in the control of skin inflammation. J Invest Dermatol. 2014;134(4):1001–1011. doi: 10.1038/jid.2013.448. [DOI] [PubMed] [Google Scholar]
- 32.Sheu MY, et al. Topical peroxisome proliferator activated receptor-alpha activators reduce inflammation in irritant and allergic contact dermatitis models. J Invest Dermatol. 2002;118(1):94–101. doi: 10.1046/j.0022-202x.2001.01626.x. [DOI] [PubMed] [Google Scholar]
- 33.Hanley K, et al. Keratinocyte differentiation is stimulated by activators of the nuclear hormone receptor PPARalpha. J Invest Dermatol. 1998;110(4):368–375. doi: 10.1046/j.1523-1747.1998.00139.x. [DOI] [PubMed] [Google Scholar]
- 34.Komuves LG, et al. Ligands and activators of nuclear hormone receptors regulate epidermal differentiation during fetal rat skin development. J Invest Dermatol. 1998;111(3):429–433. doi: 10.1046/j.1523-1747.1998.00296.x. [DOI] [PubMed] [Google Scholar]
- 35.Mao-Qiang M, et al. Peroxisome-proliferator-activated receptor (PPAR)-gamma activation stimulates keratinocyte differentiation. J Invest Dermatol. 2004;123(2):305–312. doi: 10.1111/j.0022-202X.2004.23235.x. [DOI] [PubMed] [Google Scholar]
- 36.Schmuth M, et al. Peroxisome proliferator-activated receptor (PPAR)-beta/delta stimulates differentiation and lipid accumulation in keratinocytes. J Invest Dermatol. 2004;122(4):971–983. doi: 10.1111/j.0022-202X.2004.22412.x. [DOI] [PubMed] [Google Scholar]
- 37.Westergaard M, et al. Modulation of keratinocyte gene expression and differentiation by PPAR-selective ligands and tetradecylthioacetic acid. J Invest Dermatol. 2001;116(5):702–712. doi: 10.1046/j.1523-1747.2001.01329.x. [DOI] [PubMed] [Google Scholar]
- 38.Demerjian M, et al. Topical treatment with thiazolidinediones, activators of peroxisome proliferator-activated receptor-gamma, normalizes epidermal homeostasis in a murine hyperproliferative disease model. Exp Dermatol. 2006;15(3):154–160. doi: 10.1111/j.1600-0625.2006.00402.x. [DOI] [PubMed] [Google Scholar]
- 39.Ellis CN, et al. Troglitazone improves psoriasis and normalizes models of proliferative skin disease: ligands for peroxisome proliferator-activated receptor-gamma inhibit keratinocyte proliferation. Arch Dermatol. 2000;136(5):609–616. doi: 10.1001/archderm.136.5.609. [DOI] [PubMed] [Google Scholar]
- 40.Kim DJ, et al. PPARbeta/delta selectively induces differentiation and inhibits cell proliferation. Cell Death Differ. 2006;13(1):53–60. doi: 10.1038/sj.cdd.4401713. [DOI] [PubMed] [Google Scholar]
- 41.Komuves LG, et al. Keratinocyte differentiation in hyperproliferative epidermis: topical application of PPARalpha activators restores tissue homeostasis. J Invest Dermatol. 2000;115(3):361–367. doi: 10.1046/j.1523-1747.2000.00076.x. [DOI] [PubMed] [Google Scholar]
- 42.Komuves LG, et al. Stimulation of PPARalpha promotes epidermal keratinocyte differentiation in vivo. J Invest Dermatol. 2000;115(3):353–360. doi: 10.1046/j.1523-1747.2000.00073.x. [DOI] [PubMed] [Google Scholar]
- 43.Lee SS, et al. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15(6):3012–3022. doi: 10.1128/MCB.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmuth M, et al. Role of peroxisome proliferator-activated receptor alpha in epidermal development in utero. J Invest Dermatol. 2002;119(6):1298–1303. doi: 10.1046/j.1523-1747.2002.19605.x. [DOI] [PubMed] [Google Scholar]
- 45.Man MQ, et al. Deficiency of PPARbeta/delta in the epidermis results in defective cutaneous permeability barrier homeostasis and increased inflammation. J Invest Dermatol. 2008;128(2):370–377. doi: 10.1038/sj.jid.5701026. [DOI] [PubMed] [Google Scholar]
- 46.Demerjian M, et al. Activators of PPARs and LXR decrease the adverse effects of exogenous glucocorticoids on the epidermis. Exp Dermatol. 2009;18(7):643–649. doi: 10.1111/j.1600-0625.2009.00841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michalik L, et al. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)alpha and PPARbeta mutant mice. J Cell Biol. 2001;154(4):799–814. doi: 10.1083/jcb.200011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Icre G, Wahli W, Michalik L. Functions of the peroxisome proliferator-activated receptor (PPAR) alpha and beta in skin homeostasis, epithelial repair, and morphogenesis. J Investig Dermatol Symp Proc. 2006;11(1):30–35. doi: 10.1038/sj.jidsymp.5650007. [DOI] [PubMed] [Google Scholar]
- 49.Tan NS, et al. Essential role of Smad3 in the inhibition of inflammation-induced PPARbeta/delta expression. EMBO J. 2004;23(21):4211–4221. doi: 10.1038/sj.emboj.7600437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan NS, et al. Critical roles of PPAR beta/delta in keratinocyte response to inflammation. Genes Dev. 2001;15(24):3263–3277. doi: 10.1101/gad.207501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mirza RE, et al. Macrophage PPARgamma and impaired wound healing in type 2 diabetes. J Pathol. 2015;236(4):433–444. doi: 10.1002/path.4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lucas T, et al. Differential roles of macrophages in diverse phases of skin repair. J Immunol. 2010;184(7):3964–3977. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- 53.Mirza R, DiPietro LA, Koh TJ. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am J Pathol. 2009;175(6):2454–2462. doi: 10.2353/ajpath.2009.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bannon P, et al. Diabetes induces stable intrinsic changes to myeloid cells that contribute to chronic inflammation during wound healing in mice. Dis Model Mech. 2013;6(6):1434–1447. doi: 10.1242/dmm.012237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khanna S, et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One. 2010;5(3):e9539. doi: 10.1371/journal.pone.0009539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mirza R, Koh TJ. Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine. 2011;56(2):256–264. doi: 10.1016/j.cyto.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 57.Hong C, Tontonoz P. Coordination of inflammation and metabolism by PPAR and LXR nuclear receptors. Curr Opin Genet Dev. 2008;18(5):461–467. doi: 10.1016/j.gde.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10(5):365–376. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- 59.Ricote M, Glass CK. PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta. 2007;1771(8):926–935. doi: 10.1016/j.bbalip.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tyagi S, et al. The peroxisome proliferator-activated receptor: a family of nuclear receptors role in various diseases. J Adv Pharm Technol Res. 2011;2(4):236–240. doi: 10.4103/2231-4040.90879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391(6662):82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 62.Bongartz T, et al. Treatment of active psoriatic arthritis with the PPARgamma ligand pioglitazone: an open-label pilot study. Rheumatology (Oxford) 2005;44(1):126–129. doi: 10.1093/rheumatology/keh423. [DOI] [PubMed] [Google Scholar]
- 63.Mittal R, et al. Efficacy and safety of combination acitretin and pioglitazone therapy in patients with moderate to severe chronic plaque-type psoriasis: a randomized, double-blind, placebo-controlled clinical trial. Arch Dermatol. 2009;145(4):387–393. doi: 10.1001/archdermatol.2009.5. [DOI] [PubMed] [Google Scholar]
- 64.Robertshaw H, Friedmann PS. Pioglitazone: a promising therapy for psoriasis. Br J Dermatol. 2005;152(1):189–191. doi: 10.1111/j.1365-2133.2005.06369.x. [DOI] [PubMed] [Google Scholar]
- 65.Shafiq N, et al. Pilot trial: pioglitazone versus placebo in patients with plaque psoriasis (the P6) Int J Dermatol. 2005;44(4):328–333. doi: 10.1111/j.1365-4632.2005.02504.x. [DOI] [PubMed] [Google Scholar]
- 66.Behshad R, Cooper KD, Korman NJ. A retrospective case series review of the peroxisome proliferator-activated receptor ligand rosiglitazone in the treatment of atopic dermatitis. Arch Dermatol. 2008;144(1):84–88. doi: 10.1001/archdermatol.2007.22. [DOI] [PubMed] [Google Scholar]
- 67.Boguniewicz M, Leung DY. Atopic dermatitis: a disease of altered skin barrier and immune dysregulation. Immunol Rev. 2011;242(1):233–246. doi: 10.1111/j.1600-065X.2011.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dahten A, et al. Systemic PPARgamma ligation inhibits allergic immune response in the skin. J Invest Dermatol. 2008;128(9):2211–2218. doi: 10.1038/jid.2008.84. [DOI] [PubMed] [Google Scholar]
- 69.Kawakami T, et al. Mast cells in atopic dermatitis. Curr Opin Immunol. 2009;21(6):666–678. doi: 10.1016/j.coi.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tachibana M, et al. Activation of peroxisome proliferator-activated receptor gamma suppresses mast cell maturation involved in allergic diseases. Allergy. 2008;63(9):1136–1147. doi: 10.1111/j.1398-9995.2008.01677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Palmer CN, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38(4):441–446. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 72.Wallmeyer L, et al. Stimulation of PPARalpha normalizes the skin lipid ratio and improves the skin barrier of normal and filaggrin deficient reconstructed skin. J Dermatol Sci. 2015;80(2):102–110. doi: 10.1016/j.jdermsci.2015.09.012. [DOI] [PubMed] [Google Scholar]
- 73.Lee SE, et al. Pseudoceramide stimulates peroxisome proliferator-activated receptor-alpha expression in a murine model of atopic dermatitis: molecular basis underlying the anti-inflammatory effect and the preventive effect against steroid-induced barrier impairment. Arch Dermatol Res. 2015;307(9):781–792. doi: 10.1007/s00403-015-1584-9. [DOI] [PubMed] [Google Scholar]
- 74.Romanowska M, et al. Activation of PPARbeta/delta causes a psoriasis-like skin disease in vivo. PLoS One. 2010;5(3):e9701. doi: 10.1371/journal.pone.0009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hack K, et al. Skin-targeted inhibition of PPAR beta/delta by selective antagonists to treat PPAR beta/delta-mediated psoriasis-like skin disease in vivo. PLoS One. 2012;7(5):e37097. doi: 10.1371/journal.pone.0037097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kippenberger S, et al. Activators of peroxisome proliferator-activated receptors protect human skin from ultraviolet-B-light-induced inflammation. J Invest Dermatol. 2001;117(6):1430–1436. doi: 10.1046/j.0022-202x.2001.01537.x. [DOI] [PubMed] [Google Scholar]
- 77.Thuillier P, et al. Activators of peroxisome proliferator-activated receptor-alpha partially inhibit mouse skin tumor promotion. Mol Carcinog. 2000;29(3):134–142. doi: 10.1002/1098-2744(200011)29:3<134::AID-MC2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 78.Kim DJ, et al. Peroxisome proliferator-activated receptor beta (delta)-dependent regulation of ubiquitin C expression contributes to attenuation of skin carcinogenesis. J Biol Chem. 2004;279(22):23719–23727. doi: 10.1074/jbc.M312063200. [DOI] [PubMed] [Google Scholar]
- 79.Kim DJ, et al. Peroxisome proliferator-activated receptor-beta/delta inhibits epidermal cell proliferation by down-regulation of kinase activity. J Biol Chem. 2005;280(10):9519–9527. doi: 10.1074/jbc.M413808200. [DOI] [PubMed] [Google Scholar]
- 80.Chen D, Auborn K. Fish oil constituent docosahexa-enoic acid selectively inhibits growth of human papillomavirus immortalized keratinocytes. Carcinogenesis. 1999;20(2):249–254. doi: 10.1093/carcin/20.2.249. [DOI] [PubMed] [Google Scholar]
- 81.He G, et al. The effect of PPARgamma ligands on UV- or chemically-induced carcinogenesis in mouse skin. Mol Carcinog. 2005;43(4):198–206. doi: 10.1002/mc.20111. [DOI] [PubMed] [Google Scholar]
- 82.Nicol CJ, et al. PPARgamma influences susceptibility to DMBA-induced mammary, ovarian and skin carcinogenesis. Carcinogenesis. 2004;25(9):1747–1755. doi: 10.1093/carcin/bgh160. [DOI] [PubMed] [Google Scholar]
- 83.Grabacka M, et al. Peroxisome proliferator-activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin Cancer Res. 2006;12(10):3028–3036. doi: 10.1158/1078-0432.CCR-05-2556. [DOI] [PubMed] [Google Scholar]
- 84.Liu Y, et al. Growth inhibition and differentiation induced by peroxisome proliferator activated receptor gamma ligand rosiglitazone in human melanoma cell line A375. Med Oncol. 2006;23(3):393–402. doi: 10.1385/MO:23:3:393. [DOI] [PubMed] [Google Scholar]
- 85.Smith AG, et al. PPARgamma agonists attenuate proliferation and modulate Wnt/beta-catenin signalling in melanoma cells. Int J Biochem Cell Biol. 2009;41(4):844–852. doi: 10.1016/j.biocel.2008.08.037. [DOI] [PubMed] [Google Scholar]
- 86.Grabacka M, et al. Inhibition of melanoma metastases by fenofibrate. Arch Dermatol Res. 2004;296(2):54–58. doi: 10.1007/s00403-004-0479-y. [DOI] [PubMed] [Google Scholar]
- 87.Hanley K, et al. Fetal epidermal differentiation and barrier development In vivo is accelerated by nuclear hormone receptor activators. J Invest Dermatol. 1999;113(5):788–795. doi: 10.1046/j.1523-1747.1999.00743.x. [DOI] [PubMed] [Google Scholar]
- 88.Hanley K, et al. Oxysterols induce differentiation in human keratinocytes and increase Ap-1-dependent involucrin transcription. J Invest Dermatol. 2000;114(3):545–553. doi: 10.1046/j.1523-1747.2000.00895.x. [DOI] [PubMed] [Google Scholar]
- 89.Russell LE, et al. Characterization of liver X receptor expression and function in human skin and the pilosebaceous unit. Exp Dermatol. 2007;16(10):844–852. doi: 10.1111/j.1600-0625.2007.00612.x. [DOI] [PubMed] [Google Scholar]
- 90.Komuves LG, et al. Oxysterol stimulation of epidermal differentiation is mediated by liver X receptor-beta in murine epidermis. J Invest Dermatol. 2002;118(1):25–34. doi: 10.1046/j.0022-202x.2001.01628.x. [DOI] [PubMed] [Google Scholar]
- 91.Man MQ, et al. Basis for improved permeability barrier homeostasis induced by PPAR and LXR activators: liposensors stimulate lipid synthesis, lamellar body secretion, and post-secretory lipid processing. J Invest Dermatol. 2006;126(2):386–392. doi: 10.1038/sj.jid.5700046. [DOI] [PubMed] [Google Scholar]
- 92.Chang KC, et al. Liver X receptor is a therapeutic target for photoaging and chronological skin aging. Mol Endocrinol. 2008;22(11):2407–2419. doi: 10.1210/me.2008-0232. [DOI] [PubMed] [Google Scholar]
- 93.Fowler AJ, et al. Liver X receptor activators display anti-inflammatory activity in irritant and allergic contact dermatitis models: liver-X-receptor-specific inhibition of inflammation and primary cytokine production. J Invest Dermatol. 2003;120(2):246–255. doi: 10.1046/j.1523-1747.2003.12033.x. [DOI] [PubMed] [Google Scholar]
- 94.Cork MJ et al (2006) New perspectives on epidermal barrier dysfunction in atopic dermatitis: gene-environment interactions. J Allergy Clin Immunol 118(1):3–21 (Quiz 22–3) [DOI] [PubMed]
- 95.Ong PY, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347(15):1151–1160. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 96.Proksch E, Jensen JM, Elias PM. Skin lipids and epidermal differentiation in atopic dermatitis. Clin Dermatol. 2003;21(2):134–144. doi: 10.1016/S0738-081X(02)00370-X. [DOI] [PubMed] [Google Scholar]
- 97.Sugarman JL, et al. The objective severity assessment of atopic dermatitis score: an objective measure using permeability barrier function and stratum corneum hydration with computer-assisted estimates for extent of disease. Arch Dermatol. 2003;139(11):1417–1422. doi: 10.1001/archderm.139.11.1417. [DOI] [PubMed] [Google Scholar]
- 98.Hatano Y et al (2010) Murine atopic dermatitis responds to peroxisome proliferator-activated receptors alpha and beta/delta (but not gamma) and liver X receptor activators. J Allergy Clin Immunol 125(1):160–169 (e1–5) [DOI] [PMC free article] [PubMed]
- 99.Zhang W, et al. Liver X receptor activation induces apoptosis of melanoma cell through caspase pathway. Cancer Cell Int. 2014;14(1):16. doi: 10.1186/1475-2867-14-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pencheva N, et al. Broad-spectrum therapeutic suppression of metastatic melanoma through nuclear hormone receptor activation. Cell. 2014;156(5):986–1001. doi: 10.1016/j.cell.2014.01.038. [DOI] [PubMed] [Google Scholar]
- 101.Wang Z, et al. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature. 2003;423(6939):555–560. doi: 10.1038/nature01645. [DOI] [PubMed] [Google Scholar]
- 102.Safe S, et al. Nuclear receptor 4A (NR4A) family—orphans no more. J Steroid Biochem Mol Biol. 2016;157:48–60. doi: 10.1016/j.jsbmb.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mohan HM, et al. Molecular pathways: the role of NR4A orphan nuclear receptors in cancer. Clin Cancer Res. 2012;18(12):3223–3228. doi: 10.1158/1078-0432.CCR-11-2953. [DOI] [PubMed] [Google Scholar]
- 104.Ranhotra HS. The NR4A orphan nuclear receptors: mediators in metabolism and diseases. J Recept Signal Transduct Res. 2015;35(2):184–188. doi: 10.3109/10799893.2014.948555. [DOI] [PubMed] [Google Scholar]
- 105.Newton RA, et al. Activation of the cAMP pathway by variant human MC1R alleles expressed in HEK and in melanoma cells. Peptides. 2005;26(10):1818–1824. doi: 10.1016/j.peptides.2004.11.031. [DOI] [PubMed] [Google Scholar]
- 106.Smith AG, et al. Melanocortin-1 receptor signaling markedly induces the expression of the NR4A nuclear receptor subgroup in melanocytic cells. J Biol Chem. 2008;283(18):12564–12570. doi: 10.1074/jbc.M800480200. [DOI] [PubMed] [Google Scholar]
- 107.de Leseleuc L, Denis F. Nur77 forms novel nuclear structures upon DNA damage that cause transcriptional arrest. Exp Cell Res. 2006;312(9):1507–1513. doi: 10.1016/j.yexcr.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 108.Jagirdar K, et al. The NR4A2 nuclear receptor is recruited to novel nuclear foci in response to UV irradiation and participates in nucleotide excision repair. PLoS One. 2013;8(11):e78075. doi: 10.1371/journal.pone.0078075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Malewicz M, et al. Essential role for DNA-PK-mediated phosphorylation of NR4A nuclear orphan receptors in DNA double-strand break repair. Genes Dev. 2011;25(19):2031–2040. doi: 10.1101/gad.16872411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Malewicz M, Perlmann T. Function of transcription factors at DNA lesions in DNA repair. Exp Cell Res. 2014;329(1):94–100. doi: 10.1016/j.yexcr.2014.08.032. [DOI] [PubMed] [Google Scholar]
- 111.Inamoto T, et al. 1,1-Bis(3′-indolyl)-1-(p-chlorophenyl)methane activates the orphan nuclear receptor Nurr1 and inhibits bladder cancer growth. Mol Cancer Ther. 2008;7(12):3825–3833. doi: 10.1158/1535-7163.MCT-08-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Inamoto T, et al. Cytoplasmic mislocalization of the orphan nuclear receptor Nurr1 is a prognostic factor in bladder cancer. Cancer. 2010;116(2):340–346. doi: 10.1002/cncr.24737. [DOI] [PubMed] [Google Scholar]
- 113.Li X, Lee SO, Safe S. Structure-dependent activation of NR4A2 (Nurr1) by 1,1-bis(3′-indolyl)-1-(aromatic)methane analogs in pancreatic cancer cells. Biochem Pharmacol. 2012;83(10):1445–1455. doi: 10.1016/j.bcp.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Boakye CH, et al. Chemoprevention of skin cancer with 1,1-bis (3′-indolyl)-1-(aromatic) methane analog through induction of the orphan nuclear receptor, NR4A2 (Nurr1) PLoS One. 2013;8(8):e69519. doi: 10.1371/journal.pone.0069519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.O’Kane M, et al. Increased expression of the orphan nuclear receptor NURR1 in psoriasis and modulation following TNF-alpha inhibition. J Invest Dermatol. 2008;128(2):300–310. doi: 10.1038/sj.jid.5701023. [DOI] [PubMed] [Google Scholar]
- 116.Niu G, et al. Orphan nuclear receptor TR3/Nur77 improves wound healing by upregulating the expression of integrin beta4. FASEB J. 2015;29(1):131–140. doi: 10.1096/fj.14-257550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Palumbo-Zerr K, et al. Orphan nuclear receptor NR4A1 regulates transforming growth factor-beta signaling and fibrosis. Nat Med. 2015;21(2):150–158. doi: 10.1038/nm.3777. [DOI] [PubMed] [Google Scholar]
- 118.Smith AG, et al. Regulation of NR4A nuclear receptor expression by oncogenic BRAF in melanoma cells. Pigment Cell Melanoma Res. 2011;24(3):551–563. doi: 10.1111/j.1755-148X.2011.00843.x. [DOI] [PubMed] [Google Scholar]
- 119.Wong DJ, Ribas A. Targeted therapy for melanoma. Cancer Treat Res. 2016;167:251–262. doi: 10.1007/978-3-319-22539-5_10. [DOI] [PubMed] [Google Scholar]
- 120.Johannessen CM, et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013;504(7478):138–142. doi: 10.1038/nature12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal. 2009;7:e003. doi: 10.1621/nrs.07003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Slominski A, et al. On the role of melatonin in skin physiology and pathology. Endocrine. 2005;27(2):137–148. doi: 10.1385/ENDO:27:2:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Steinmayr M, et al. Staggerer phenotype in retinoid-related orphan receptor alpha-deficient mice. Proc Natl Acad Sci USA. 1998;95(7):3960–3965. doi: 10.1073/pnas.95.7.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhu Y, et al. RORA, a large common fragile site gene, is involved in cellular stress response. Oncogene. 2006;25(20):2901–2908. doi: 10.1038/sj.onc.1209314. [DOI] [PubMed] [Google Scholar]
- 125.Dai J, et al. The retinoid-related orphan receptor RORalpha promotes keratinocyte differentiation via FOXN1. PLoS One. 2013;8(7):e70392. doi: 10.1371/journal.pone.0070392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hanyu O, et al. Cholesterol sulfate induces expression of the skin barrier protein filaggrin in normal human epidermal keratinocytes through induction of RORalpha. Biochem Biophys Res Commun. 2012;428(1):99–104. doi: 10.1016/j.bbrc.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 127.Huh JR, Littman DR. Small molecule inhibitors of RORgammat: targeting Th17 cells and other applications. Eur J Immunol. 2012;42(9):2232–2237. doi: 10.1002/eji.201242740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kallen JA, et al. X-ray structure of the hRORalpha LBD at 1.63 A: structural and functional data that cholesterol or a cholesterol derivative is the natural ligand of RORalpha. Structure. 2002;10(12):1697–1707. doi: 10.1016/S0969-2126(02)00912-7. [DOI] [PubMed] [Google Scholar]
- 129.Solt LA, Burris TP. Action of RORs and their ligands in (patho)physiology. Trends Endocrinol Metab. 2012;23(12):619–627. doi: 10.1016/j.tem.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Marciano DP, et al. The therapeutic potential of nuclear receptor modulators for treatment of metabolic disorders: PPARgamma, RORs, and Rev-erbs. Cell Metab. 2014;19(2):193–208. doi: 10.1016/j.cmet.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 131.Slominski AT et al (2014) The role of CYP11A1 in the production of vitamin D metabolites and their role in the regulation of epidermal functions. J Steroid Biochem Mol Biol 144 Pt A:28–39 [DOI] [PMC free article] [PubMed]
- 132.Skepner J, et al. Pharmacologic inhibition of RORgammat regulates Th17 signature gene expression and suppresses cutaneous inflammation in vivo. J Immunol. 2014;192(6):2564–2575. doi: 10.4049/jimmunol.1302190. [DOI] [PubMed] [Google Scholar]
- 133.Keijsers RR, et al. In vivo induction of cutaneous inflammation results in the accumulation of extracellular trap-forming neutrophils expressing RORgammat and IL-17. J Invest Dermatol. 2014;134(5):1276–1284. doi: 10.1038/jid.2013.526. [DOI] [PubMed] [Google Scholar]
- 134.Wang H, LeCluyse EL. Role of orphan nuclear receptors in the regulation of drug-metabolising enzymes. Clin Pharmacokinet. 2003;42(15):1331–1357. doi: 10.2165/00003088-200342150-00003. [DOI] [PubMed] [Google Scholar]
- 135.Elentner A et al (2015) Skin response to a carcinogen involves the xenobiotic receptor pregnane X receptor. Exp Dermatol [DOI] [PMC free article] [PubMed]
- 136.Beyer C, et al. Activation of pregnane X receptor inhibits experimental dermal fibrosis. Ann Rheum Dis. 2013;72(4):621–625. doi: 10.1136/annrheumdis-2012-202476. [DOI] [PubMed] [Google Scholar]