

Abstract
Cancer cells release extracellular vesicles known as extracellular vesicles (EVs), containing tumor-derived DNA, RNA and proteins within their cargo, into the circulation. Circulating tumor-derived extracellular vesicles (TEV) can be used in the context of serial “liquid biopsies” for early detection of cancer, for monitoring disease burden in patients, and for assessing recurrence in the post-resection setting. Nonetheless, isolating sufficient TEV by ultracentrifugation-based approaches, in order to enable molecular assessment of EVs cargo, can be an arduous, time-consuming process and is inconsistent in the context of yield and purity among institutions. Herein, we describe a microfluidic platform, which we have named MITEV (Microfluidic Isolation of Tumor-derived Extracellular Vesicles) for the rapid isolation of TEV from the plasma of pancreatic cancer patients. The device, which has 100,000 pillars placed in a zigzag pattern and is coated with antibodies against generic EV surface proteins (anti-CD63, -CD9, and -CD81 antibodies) or the TEV specific anti-Epithelial Cell Adhesion Molecule (EpCAM) antibody, is capable of high-throughput EVs isolation and yields sufficient DNA (total of ~2–14 ng from 2-ml plasma) for downstream genomic analysis. Using two independent quantitative platforms, droplet digital polymerase chain reaction (ddPCR) and molecular barcoding using nanoString nCounter® technology, we can reliably identify KRAS mutations within isolated TEV of treatment-naïve metastatic pancreatic cancer patients. Our study suggests that the MITEV device can be used for point-of-care applications, such as in the context of monitoring residual or recurrent tumor presence in pancreatic cancer patients undergoing therapy.
Key terms: Extracellular Vesicles, Microfluidics, Pancreatic Cancer, KRAS, ddPCR
Introduction:
As the third leading cause of cancer death in the United states, the incidence of pancreatic ductal adenocarcinoma (PDAC) is rising annually1. Owing to the retroperitoneal location of the pancreas and non-specific symptoms, the vast majority of PDAC patients have locally advanced or distant metastatic disease that renders the tumor inoperable2,3. In contrast, patients with localized tumors that can be resected have increasingly longer median survival, thanks to improvements in surgical techniques, and neoadjuvant and adjuvant therapies. Thus, earlier detection of PDAC remains one of the greatest unmet needs in oncology3,4.
Although imaging studies such as computed tomography or magnetic resonance imaging are the standard for PDAC diagnosis4, these studies are expensive, and their interpretation requires special expertise3,5. Therefore, imaging studies to assess patients for PDAC should be supported by a molecular diagnostic assay that can be conducted in parallel to clinical setting, which would add a different dimension to the diagnosis. However, the only biomarker approved by the U.S. Food and Drug Administration for PDAC diagnosis, the glycoprotein CA19–9, has markedly sub-optimal sensitivity for early-stage disease6. Early diagnosis of PDAC requires not only biomarkers of higher sensitivity and specificity, but also a technique that can be readily and widely adapted in the clinical setting.
Liquid biopsies provide a promising approach for the early, non-invasive diagnosis of PDAC7,8. In addition, liquid biopsies also provide an opportunity for longitudinal monitoring of patients on treatment, and assessment of tumor recurrence in the minimal residual disease setting post-surgery9,10. Liquid biopsies are comprised of circulating tumor DNA (ctDNA), circulating tumor cells (CTCs) and CTC clusters, and extracellular vesicles and have received great attention over recent years for cancer diagnosis11,12. Due to their cargo protection and preservation of high molecular weight DNA, EVs are of special interest for clinical applications. EVs, with a size 30–500nm, are shed by endosomal pathway or ectocytosis from plasma membranes13–15. Circulating EVs comprise both tumor-derived EVs (TEV) and non-tumor-derived EVs (NTEV), and the latter can skew genomic assessment in liquid biopsies. Isolating TEV from plasma would facilitate genomic assessment for the early diagnosis and longitudinal monitoring of disease response in PDAC. Thus, the challenge here is how to isolate only TEX from the plasma16.
The many reported methods for isolating EVs can be categorized into one of three categories: 1) density-based separation such as ultracentrifugation or centrifugation using density solutions, 2) size-based separation including chromatography and filtration, and 3) affinity-based separation using antibody coating on magnetic beads or microfluidic devices17. The gold standard for EVs isolation, ultracentrifugation (UC, typically performed at 100,000x g), is laborious and time-consuming, with markedly low throughput, and results in a non-specific pellet of EVs contaminated with proteins and NTEV, which interfere with vesicle counts17. Size-based technologies such as filtration and size exclusion chromatography (e.g., qEV isolation columns [Izon Science]) can provide higher-purity samples18–20. However, because filtration is prone to clogging, and chromatography can yield diluted samples, these approaches are more suitable as adjuncts to other EVs isolation methodologies than as stand-alone methodologies. In addition, newer size-based methods that use external fields such as dielectrophoresis21 or acoustic waves22 have been found to yield smaller samples of moderate purity. Most importantly, none of these techniques can be readily adapted as a point of care test (POCT) device in the outpatient setting for rapid isolation of TEV from limited liquid biopsy samples.
In terms of sample purity, affinity-based methods using antibodies against tetraspanins (e.g., anti-CD9, -CD63, and -CD81) appear to be more promising23–25. Microfluidic devices, in particular, can use immobilized anti-tetraspanin antibodies and cancer-specific antibodies to capture EVs16,25,26. However, these devices yield samples in which the TEV counts are below the detection threshold of most sequencing methods (e.g. droplet digital polymerase chain reaction (ddPCR) and hence are insufficient for the detection of cancer mutations.
Herein, we introduce a new microfluidic device - Microfluidic Isolation of Tumor-derived EVs or MITEV - that can yield enough DNA for downstream genomic analysis to confirm tumor mutational burden. This device has 100,000 pillars positioned in a zigzag pattern that causes chaos in the flow stream, thus leading to more interaction between the pillars and circulating TEV in plasma. The pillars are coated with antibodies specific to EVs surface proteins (CD63, CD9, and CD81) or a cancer-specific antigen (EpCAM)27 and the captured EVs are later released using an acidic solution. Using a single MITEV device, we were able to collect enough DNA (total of ~2–14ng from 2-ml of plasma) to run quantitative mutation detection assays on two independent platforms, and detect KRAS mutations in TEV from PDAC patients.
Materials and Methods
MITEV design and manufacture
The MITEV device has 3 sections: two flow-distribution sections (one at the inlet and one at the outlet) and TEV-capture section containing 100,000 pillars (Figure 1A). The device is 75.75 mm by 50.5 mm, the size of a glass slide (Figure 1B). Internally, each flow-distribution section is 11.73 ± 0.64 mm by 22.76 ± 0.53 mm, and the pillared section is 45.83 ± 0.57 mm by 22.76 ± 0.53 mm; the internal height is 10 ± 0.86 μm. The pillars are oblong (81 ± 1.35 μm long and 32 ± 0.96 μm wide) and separated on all sides by 45 ± 1.24 μm of space. The pillars are distributed in a zigzag pattern in 45° angles to perturb the flow of plasma, thereby increasing their interaction with floating EVs (small SEM frame in Figure 1A). 45° angles were chosen to provide an equal chance of EVs capture on either side of the pillars. We used 100,000 pillars to maximize the perturbation of the plasma flow compared to a bare channel (with no pillars).
Figure 1.

Design and manufacturing of the microfluidic device. A) AutoCAD design of the device, which has three sections, two flow distribution sections for inlet and outlet and one pillars section. In the small frame, a SEM image of a section of pillars in the device is shown. The bar shows 100μm. B) a bright-field image of the device, beside a penny for size comparison. C) a simulation of fluid flow inside the device. D) a schematic of the coating layers and captured EVs. The coating procedure is adopted from Ref. 15. E) pH of variant solutions of Glycin (pH = 2.1) to Tris (pH = 8.5).
We simulated flow in a section of MITEV device using COMSOL Multiphysics (V5.2, Burlington, MA) software (Figure 1C). A 2-dimensional simulation for water and Polydimethylsiloxane (PDMS) was performed assuming a laminar time-independent flow. The boundary conditions of the simulation were the same as those used during the experiments presented in this study (inlet flow velocity: 2.1 mm/s, no-slip on the walls, outlet pressure = 0 Pa). The simulation confirms agitation of flow around the pillars, which can provide more interaction between EVs and coated antibodies, and therefore a higher capture efficiency.
To manufacture the MITEV device, we used the standard soft lithography methodology28. Molds were fabricated by spin-coating SU8–5 negative photoresist (MicroChem, Westborough, MA) on a 3-inch diameter silicon wafer with a 7.63 cm diameter. The thickness of the spin-coated layer corresponded to the height of the channels. Polydimethylsiloxane (PDMS), a transparent biocompatible polymer, was poured into the molds, degassed in desiccators, and baked for 2 h at 80°C. The PDMS was then cut and peeled from the molds, and inlet and outlet holes were punched using Harris Uni-Core sampling tools (1-mm outer diameter, Ted Pella, INC, Redding, CA). The MITEV devices were then bonded to glass cover slides by exposing the surfaces to air plasma (Plasma Cleaner, Harrick Plasma, Ithaca, NY).
MITEV device coating
For coating the MITEV device, using an approach adapted from Reategui et al16, 3-mercaptopropyl trimethoxysilane (4% in ethanol; Sigma Aldrich, St. Louis, MO), a silane with a sulfhydryl group, was injected into the device and incubated for 1h (Figure 1D). Next, N-γ-maleimidobutyryl-oxysuccinimide ester (GMBS) (0.01μg/ml in ethanol; Sigma Aldrich), a crosslinker between sulfhydryl and amine groups, was injected and incubated for 30 mins. Next, 10μg/ml neutravidin (Thermo Fisher Scientific, Waltham, MA) was injected and incubated for 1h (neutravidin, as a protein, has amine groups that can bind to GMBS, and it can, in turn, act as a substrate for biotin groups).
Two biochemical functional layers were then injected into the MITEV device and incubated for 1h each. The first injection was of biotin polystyrene nanoparticles (0.01% w/v in molecular biology grade water), which attach to neutravidin owing to the presence of biotin on the nanolayer. The second injection was avidin polystyrene nanoparticles (0.01% w/v in molecular biology grade water), which attach to biotins on the first nanolayer, were injected. The thickness of the nanoparticle layers was measured by SEM images as less than 1μm. These dual nanolayers help increase the internal surface area in the devices. The devices were refrigerated at 4°C until antibody coating and EVs isolation. Finally, on the day of experiment, biotinylated antibodies with a 2-kDa polyethylene glycol spacer (biotinylating was performed according to previously established protocols15) were injected into the device and incubated for 1h. All antibodies were mouse polyclonal specific to human antigens and purchased from Abcam (Cambridge, MA).
MITEV experimental setup for TEV capture and release
To achieve the highest yield of TEV capture and release, we started with proof of concept experiments to optimize the MITEV device. We used Pa04C and Panc1 cell lines (established PDAC cell lines cultured in serum deprived media) for optimization of MITEV, followed by plasma samples from treatment naïve metastatic pancreatic cancer patients (N= 3 samples) for further validation. Three patients were recruited from MD Anderson Cancer Center through informed written consent following institutional review board (IRB, protocols #PA14–0867 and PA15–0014). After the blood samples were collected in 3 ACD tubes, the plasma was spun down using double centrifugation, first at 1000 rpm for 5 min followed by 5000 rpm for 10 mins. The samples were stored at −80C until they could be used for EVs isolation in MITEV. Each sample was pre-filtered through 0.45μm pore syringe filters. To identify the flow rate that would yield the most EVs, we used syringe pumps (kdScientific, Holliston, MA) to inject samples into the MITEV device at flow rates of 10, 30, or 50 μl/min at room temperature and in a culture hood. After samples were run, the devices were washed with phosphate-buffered saline (PBS) filtered through 0.2μm pores to remove any remaining non-specific components. Then, DNase I (0.1 mg/ml) was added to the device and incubated for 10 min to remove any non-EVs DNA (e.g. cell-free or surface-bound DNA), without impacting TEV cargo DNA.
To release captured EVs from the MITEV, we injected an acidic solution of glycine in hydrochloric acid (180μl, 0.2 M, pH 2.1, filtered through 0.2 μm pores) into the devices at the optimal flow rate of 30μl/min to detach the captured TEV. The acidic glycine solution facilitated disengagement of the antibody antigen bindings and released the captured TEV from the bound surfaces on the MITEV device. We used Tris solution (pH 8.5) to raise the pH back to the neutral point (pH=7.4). To find the neutral pH, the pH values of various ratios of glycine to Tris (from 5:1 to 30:1) were measured (Figure 1E). From these measurements, a glycin:Tris ratio of 9:1, which is around the same range reported before27, was determined as equivalent to pH 7.4. Thus, during the EVs-release process, we injected 180μl of glycine solution into the device at 50μl/min and collected the EVs-containing glycine solution in tubes containing 20μl of Tris solution. The detached TEV were then collected from the outlet in small PCR tubes containing 18μl Tris (pH 8.5) to neutralize the acidic solution and raise the pH to 7.4.
Scanning electron microscopy
To visually confirm EVs capture within the MITEV device, we first used scanning electron microscopy (SEM). After EVs capture, sectioned portions (cut in pieces of 5 mm by 5 mm) of the device were fixed in a solution containing 3% glutaraldehyde plus 2% paraformaldehyde in 0.1M cacodylate buffer, pH 7.3. The fixed sections were then washed with 0.1M cacodylate buffer, pH 7.3, post-fixed with 1% cacodylate-buffered osmium tetroxide, washed with 0.1M cacodylate buffer, and then washed with distilled water. The samples were dehydrated with a graded series of increasing concentrations of ethanol and then transferred to graded series of increasing concentrations of hexamethyldisilazane and air-dried overnight. Samples were mounted on double-stick carbon tabs (Ted Pella, Inc., Redding, CA), which had been previously mounted on glass microscope slides. The samples were placed under vacuum, coated with a 25 nm layer of platinum alloy using a Balzer MED 010 evaporator (Technotrade International, Manchester, NH), and immediately flash carbon-coated. The samples were stored in a desiccator until examination. Samples were imaged with a JSM-5910 scanning electron microscope (JEOL USA, Inc., Peabody, MA) at an accelerating voltage of 5 kV.
Confocal microscopy
Confocal microscopy was used for visual confirmation of TEV capture. For this analysis, media from Pa04C cells expressing CD63-conjugated green fluorescent protein (GFP, established in our laboratory) was processed through the MITEV device. The cells were cultured in serum-free media in a 75mm flask for 2 days. We injected 2ml of EVs-containing media (prefiltered by 0.2μm filters) into the device at a rate of 30μl/min. The devices were then subjected to confocal microscopy with a LSM 880 Confocal Laser Scanning Microscope (Zeiss, Oberkochen, Germany), and photographs were captured using an oil immersion objective (63x magnification).
Quantitative assessment of captured TEV
To count the number of captured TEV on the MITEV device, a fraction of the eluate was evaluated using the ZetaView particle tracking analyzer (Particle Metrix, Meerbusch, Germany). This system works by imaging frames of a sample at different spots (11 spots were chosen). After diluting each sample 5–10x, a final solution of ~0.5–1 ml was injected into the ZetaView, after its calibration with molecular grade water. The software (ZetaView, version 8.02.30) generated a size distribution graph, with the peak showing the mean size of the captured TEV.
Ultracentrifugation
Ultracentrifugation was used as a control method to compare the MITEV efficiency of EVs isolation. We divided 2 ml of plasma into two 1.5 ml ultra-microtubes (Thermo Fisher Scientific) then added to each tube 500 μl of cold PBS, 1x without calcium and magnesium (Corning, Inc., Corning, NY). The samples were centrifuged at 34,000 rpm for 12 h in a Thermo Sorvall WX 80 ultracentrifuge (Thermo Fisher Scientific). The pellets were combined and resuspended in DNase I (0.1 mg/ml), incubated on ice for 10 min and then diluted in 1 ml of cold PBS. A second centrifugation cycle at 34,000 rpm for 2 h was completed, and the pellet was again resuspended in 400 μL of cold PBS.
EVs DNA extraction and measurement
Extracellular vesicle DNA (evDNA) was isolated using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Isopropanol precipitation was performed on the extracted evDNA sample and eluted into a final volume of 12μl. DNA concentration was determined by a Qubit fluorometer (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions.
Droplet digital PCR analysis
Droplet digital PCR (ddPCR) was applied to 8μl of evDNA. The KRAS G12/G13 multiplex assay (BioRad, Hercules, CA) was used for the highly sensitive and quantitative detection of the most common KRAS “hotspot” mutations (G12V, G12D, G12C, G12S, and G13D) in PDAC29,30. Samples were amplified for forty cycles. Panc1 and water were used as positive and negative controls respectively. The droplets were generated in an automated Biorad QX200/QX100 Droplet Generator and were detected in a QX200 Droplet Reader. Results were analyzed by the QuantaSoft software v.1.6 (BioRad).
Single nucleotide variant profiling by molecular barcoding (nCounter assay)
Single nucleotide variant (SNV) profiling was performed on the nCounter FLEX Instrument using nanoString Technologies Vantage 3D DNA SNV kit (nanoString Technologies, Seattle, WA). Briefly, extracted DNA was quantified on the Qubit system (Life Technologies, Carlsbad, CA) using the DNA High Sensitivity kit. 5 ng of DNA were amplified using nanoString proprietary primers and then hybridized to gene-specific, fluorescent-labeled probes, which were then purified on the nCounter Prep Station. The purified, fluorescent-labeled products were then scanned on the nCounter Digital Analyzer. The data was analyzed using nSolver 4.0 software (nanoString Technologies).
For statistical analysis, the nonparametric Kolmogorov-Smirnov test (ks test, a nonparametric test) was performed in MATLAB software (R2015b) and p-values <0.05 were considered significant.
Results and Discussion
Quality control experiments for TEV isolation and release
Direct visualization of the MITEV using two independent microscopy platforms confirmed the presence of bound TEV on the device. Firstly, confocal microscopy of CD63-GFP labeled PA04C EVs (from 3 devices) revealed that before glycine treatment, TEV were captured on the peripheries of the pillars, whereas after glycine treatment, almost no EVs remained (Figure 2A). This differential fluorescence demonstrates the efficient capture of TEV and then the expected dissociation of antibody-EVs binding and release of bound TEV using the acidic glycine solution. It also shows that the device can efficiently capture, and subsequently release TEV into the eluate. The same method of dissociation has been used in other studies that observed similar outcomes26. Further, using Panc-1 EVs-rich media and PDAC plasma (N = 3) and antibodies directed against EVs surface generic proteins (anti-CD63, CD9 and CD81 antibodies) for device coating, scanning electron microscopy confirmed the presence of individual TEV bound to the MITEV device (Figure 2B).
Figure 2.

Imaging of EVs captured and released A) i) fluorescent confocal imaging of the surface and surrounding area of the posts with captured EVs that express CD63-GFP, from Pa04C cell line. ii) Fluorescent images of the device after adding glycine, which released the EVs. B, i-ii) SEM images of the EVs captured on the surface of the posts. The left frame shows a snapshot from Panc1 sample and the right frame shows a snapshot from PDAC plasma. First scale bar shows 500 nm and second 1μm.
Optimization experiments
Next, in order to assess the impact of antibody coating of the MITEV device on the efficiency of TEV capture, we assessed the device with versus without antibody coating (Figure 3A). The eluate from both experiments were measured and compared using the ZetaView particle tracking analyzer. The distribution of the particle sizes for a representative sample of Panc1 media is shown in Figure 3A, in which the peak shows the mean EVs size. We found that the mean EVs sizes for Panc1 media samples (115.9±17.6, N = 3) and PDAC plasma samples (97.1±22.6 nm, N = 3) did not differ significantly (Figure 3B), which supports that the MITEV device can reliably capture and release TEV from “real world” clinical samples.
Figure 3.
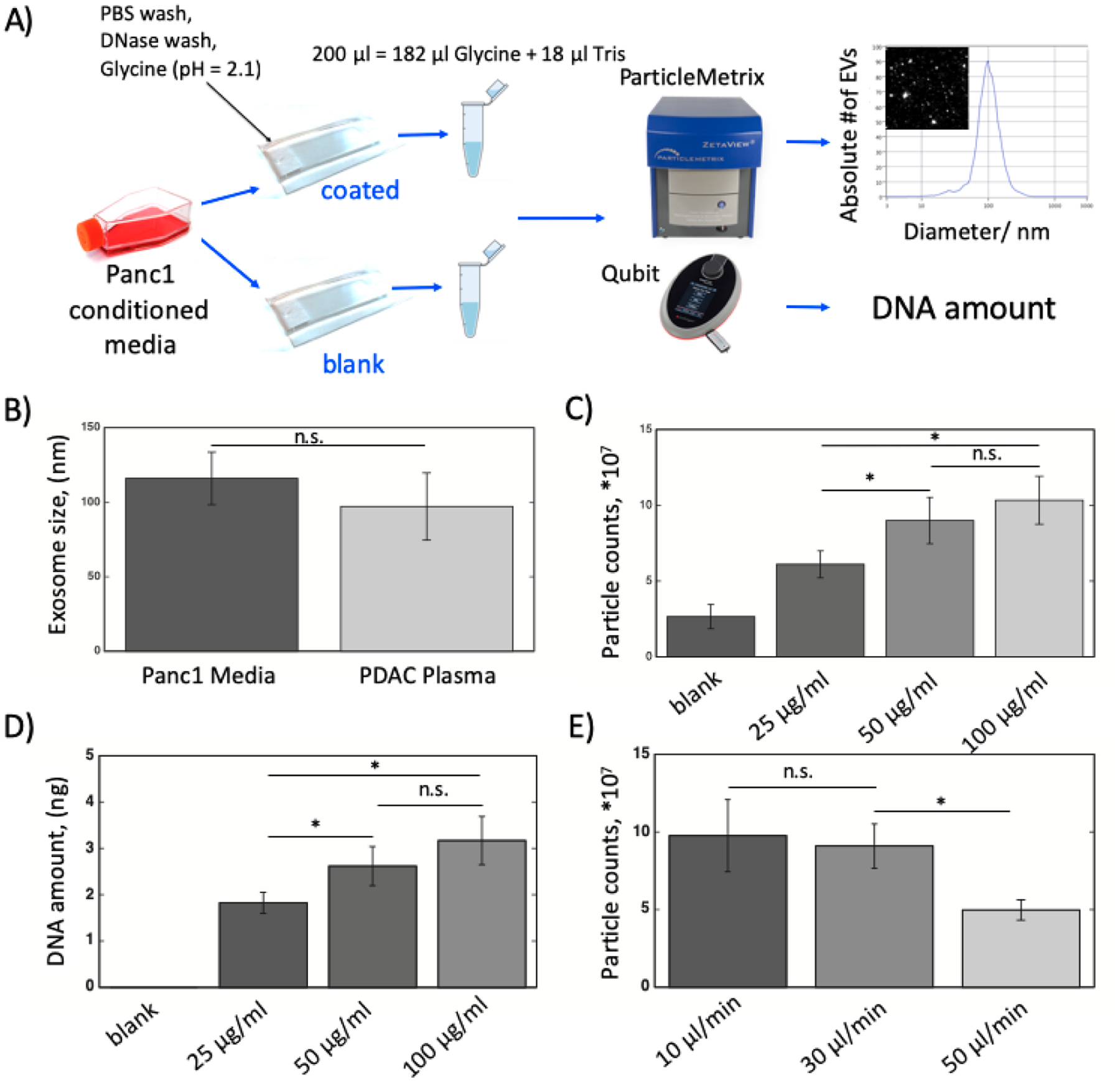
The optimization experiments for best condition of MITEV device. A) The schematic of how the quality control and optimization experiments were run. In the far right, a size distribution graph generated by ZetaView is shown. The peak shows the mean EVs size. B) The average EVs size for Panc1 media vs. PDAC plasma. C) The number of particles for different concentrations of antibody (anti-CD63) coating. D) The evDNA amount for different concentrations of antibody (anti-CD63) coating. E) The number of particles for different flow rates of Panc1 EV-rich media. Number of devices=3 for all experiments. * shows p-value<0.05. n.s. shows non-significance.
To identify the antibody concentration that generated the highest yield of EVs, we used anti-CD63 antibody as a baseline antibody in concentrations of 25, 50, and 100 μg/ml (Figure 3C). The MITEV device without antibody coating yielded a small number of particles (2.65±0.80*107). We speculated that these particles are not TEV but merely the particles present in the elution media, as the number of particles in the eluate collected from the device outlet are around the same range as the number of particles for elution media (1.87±0.62 *107, p-value>0.05). Using ZetaView, we found that doubling the antibody concentration from 25 to 50 μg/ml significantly increased the number of EVs. In contrast, doubling the antibody concentration from 50 to 100 μg/ml marginally increased the EVs number, but not significantly (Figure 3C). The same pattern was observed for DNA concentrations extracted from captured EVs, i.e., DNA amounts were the highest, and their differences statistically non-significant, for the antibody concentrations of 50 and 100 μg/ml (2.62±0.42 and 3.17±0.52 ng/ml from 1 ml media, respectively) (Figure 3D). Based on this result, an antibody concentration of 50 μg/ml was used for subsequent experiments. Even though this concentration might seem higher than what has been reported by similar devices before (~3–10 μg/ml16,26), when the smaller inner volume of the device is accounted (~10 μl, ~10 times smaller), the total amount of antibody required by the device is around the same range or lower.
To identify the optimal flow rate of plasma, rates of 10, 30, and 50 μl/min were tested (Figure 3E). The 10 and 30 μl/min flow rates yielded the most particles, whereas 50 μl/min yielded the fewest particles. Considering the total time of operation, we chose 30 μl/min as the flow rate for the experiments.
Efficiency of the MITEV device
To ensure that we obtained the highest yield of EVs isolation, we added a second MITEV device in a series to the first device so that the output of the first MITEV device was directly injected into the second one. To compare the efficiency of these devices, we assessed the number of particles (Figure 4A) and amount of evDNA (Figure 4B) in each device. For this experiment, two different sources of samples were used: conditioned media from Panc1 (in triplicate) and PDAC plasma (N=3). For both sets of specimens, the first device yielded significantly more particles and more DNA than the second device did. In other words, the percentage of the DNA yield from the second device compared to the first was less than 20% (17.7% for Panc1 media and 8.9% for plasma). Even though the number of particles from second device seems to be significantly high, we speculate most of them are contamination due to change of device and background particles in PBS. Based on these results and considering that each device takes approximately one hour to be run, we decided to use only one device in our downstream experiments to reduce the total operation time.
Figure 4.

Comparison of MITEV with themselves and with Ultracentrifugation. A) The number of particles (EVs) for two devices in series with Panc1 EVs-rich media vs. PDAC plasma. M stands for MITEV device. B) The amount of evDNA for two devices in series with Panc1 EVs-rich media vs. PDAC plasma. C) The number of particles for ultracentrifugation method vs. MITEV. D) The amount of DNA for ultracentrifugation methods vs. our method. Number of devices = 3 for all samples. * shows p-value<0.05. n.s. shows non-significance.
We then compared our method coated with generic antibodies with ultracentrifugation by using PDAC plasma samples (N = 3) and assessing the number of particles and the amount of DNA each method yielded. The conventional ultracentrifugation method yielded significantly more particles than the MITEV (p<0.05; Figure 4C); however, both methods yielded approximately the same amount of DNA (~10 ng; Figure 4D). This might have been due to the presence of particles other than EVs (such as floating proteins) inside the ultracentrifugation pellet16. However, as the yield of evDNA was comparable, the MITEV device may provide a purer output than the ultracentrifugation pellet. Also, ultracentrifugation was run for 14h, whereas the MITEV device only required approximately 1.5 h total to capture and release the EVs. Thus, compared with ultracentrifugation, the MITEV device yielded an equivalent amount of DNA in significantly lesser time, justifying its potential use as a POCT device. It is also worth noting that other microfluidic devices that are reported to outperform ultracentrifugation16,26, have an ultracentrifugation for 2 hours, which is way lower than a typical time required for an ultracentrifugation protocol for EV isolation and yields fewer total EVs.
Quantitative detection of KRAS mutations in MITEV captured evDNA
Our ultimate goal in isolating EVs is to reliably detect cancer mutations in serial liquid biopsies obtained from PDAC patients, since it may lead to actionable clinical decision-making in patient care. Our previous work has shown our capabilities for detecting mutant KRAS in evDNA and circulating free DNA in liquid biopsies from PDAC patients27,31,32. To ensure sufficient genomic material for downstream testing of cancer mutations in evDNA by the MITEV device, we used 2 ml of plasma as input volume for the MITEV. We used ddPCR to detect KRAS mutations (the most common gene mutated in PDAC, occurring in ~90% of cases3,4), with a multiplex probe mix that includes G12V, G12D, G12C, G12S, and G13D mutant alleles3,30. By coating two sets of devices with antibodies against either generic EVs surface proteins (CD63, CD9, and CD81) or a cancer-specific antigen (EpCAM)27, we were able to collect 10.3±2.2 and 6.7±1.7 ng DNA, respectively, from 2-ml plasma specimens from patients with metastatic PDAC (N=3) (Figure 5A). The difference between the two DNA amounts confirms that the absolute number of TEV in plasma is a fraction of cumulative EV. Hisley et al26, similarly observed lower counts of EVs when using anti-EpCAM (isolating TEV) as a “bait” versus anti-CD9 (isolating NTEV) for high grade serum ovarian cancer (HGSOC).
Figure 5.
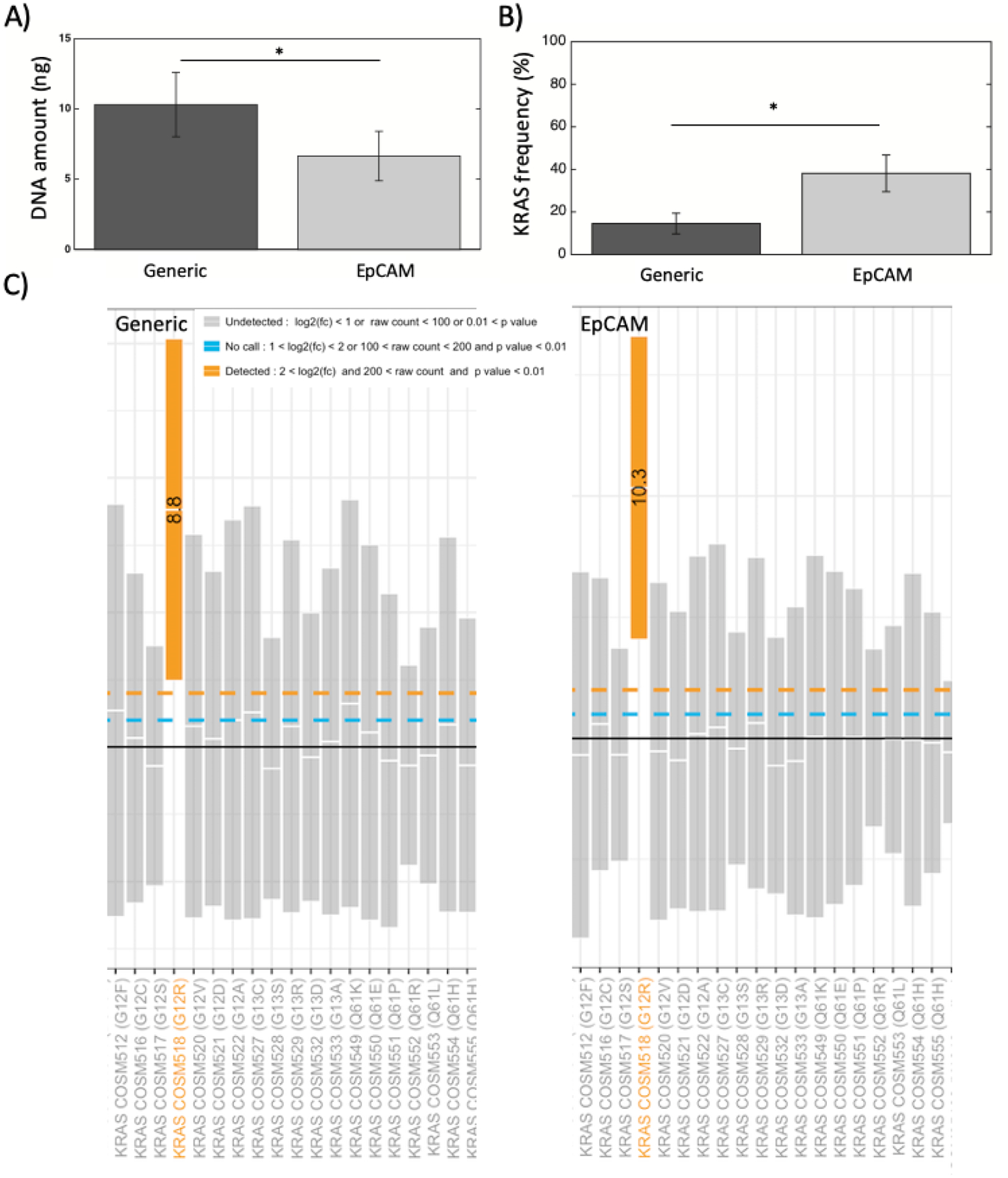
Genomic analysis of evDNA in clinical samples. A) The amount of evDNA measured by ddPCR for EVs isolated with generic antibodies vs. EpCAM from PDAC plasma. B) The KRAS allele frequency measured by ddPCR for EVs isolated with generic antibodies vs. EpCAM from PDAC plasma. * shows p-value<0.05. Number of devices = 3 for all samples. C) The nanostring SNV mutation panel for evDNA from one PDAC patient, isolated in a MITEV device coated with generic antibodies versus a MITEV device coated with EpCAM antibody.
ddPCR analysis of our DNA samples detected KRAS mutations in both conditioned media from Panc1 cell-line and PDAC plasma (Supplementary Fig. 1). Compared with the antibodies directed against generic EVs surface proteins, the anti-EpCAM antibody isolated lower amount of total DNA but had a significantly higher KRAS mutant allele frequency in PDAC Plasma samples (14.53±4.88 vs 38.13±8.63, n=3, p<0.05) (Figure 5B). Furthermore, as an additional quantitative readout on KRAS mutation, we used the NanoString nCounter platform on evDNA from one sample of metastatic PDAC, to interrogate a panel of mutations in different codons of KRAS. This panel was able to detect KRAS mutation on codon G12R in this sample, confirming the mutation detection from ddPCR (Figure 5C).
Taken together, these results suggest a single MITEV device was able to provide sufficient evDNA to perform mutation assessment and reliably identify KRAS mutation in PDAC patients. Most similar microfluidic devices yield TEV counts that are below the detection threshold of common sequencing methods. For example, in order to obtain sufficient yield of EVs isolation, Reategui et al.16 used 4 of their isolation devices in a series. In addition, because MITEV has a small internal volume (~10 μl), it requires only a small amount of antibody for coating, reducing its total cost. Nevertheless, one of the limitations of our study is low numbers of patient samples (N=3). Our aim was to use these few samples to just validate MITEV ability in TEV isolation; however, for a comprehensive clinical study, the next step will be to test this device in a larger cohort and for monitoring patients under therapy. Another limitation was the use of only one cancer specific antibody, anti-EpCAM that can be addressed by expanding the library of tumor specific antibodies for PDAC EVs.
Conclusion
In this study we developed a microfluidic device (MITEV) that can efficiently capture and release EVs from the plasma of PDAC patients, in a relatively short amount of time (1.5 hours cumulative for isolation and release) for mutational assessment of captured TEV. Each MITEV device is equipped with 100,000 pillars in zigzag arrays and coated with several layers prior to antibody coating, including two layers of nanoparticles that increased the surface area of the device. A single MITEV yielded sufficient evDNA (in total ~2–14ng from 2-ml plasma) to run mutation detection using quantitative platforms, like droplet digital PCR or nanoString nCounter® platform. Using these two independent quantitative platforms, we could also reliably identify KRAS mutations within isolated TEV of treatment naïve PDAC patients. Our study suggests that the MITEV device can be used for point-of-care applications, such as in the context of early detection in high risk patients on surveillance, or in monitoring residual or recurrent disease volume in pancreatic cancer patients on therapy.
Supplementary Material
Acknowledgements
Scanning electron microscopy was performed at the High-Resolution Electron Microscopy Facility, while nanoString analysis was performed at the Sequencing and Microarray facility (SMF) in MD Anderson Cancer Center (Houston, TX), both of which are supported by the NCI Cancer Center Support Grant (P30CA016672). N.K. was supported by the CPRIT Research Training Program (RP170067).
Conflict of interest
A.M. discloses receiving royalties from Hangzhou Guangkeande (Cosmos) Biotechnology Company LTD for being a co-inventor on a license related to pancreatic cancer early detection, and this financial relationship is managed by the MD Anderson Conflict of Interest Committee. There are no other conflicts of interests to declare.
Abbreviations, symbols, and terminology:
- TEV
Tumor associated Extracellular Vesicles
- NTEV
Non-Tumor associated Extracellular Vesicles
- MITEV
Microfluidic Isolation of Tumor-derived EVs
- PDAC
pancreatic ductal adenocarcinoma
- EV
Extracellular Vesicles
- EpCAM
Epithelial Cell Adhesion Molecule
- ddPCR
droplet digital polymerase chain reaction
References
- 1.Ionescu-Tirgoviste C et al. A 3D map of the islet routes throughout the healthy human pancreas. Sci Rep 5, 14634, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gillen S, Schuster T, Meyer Zum Buschenfelde C, Friess H & Kleeff J Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med 7, e1000267, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamisawa T, Wood LD, Itoi T & Takaori K Pancreatic cancer. Lancet 388, 73–85, 2016. [DOI] [PubMed] [Google Scholar]
- 4.Kleeff J et al. Pancreatic cancer. Nat Rev Dis Primers 2, 16022, 2016. [DOI] [PubMed] [Google Scholar]
- 5.Tang S et al. Usefulness of 18F-FDG PET, combined FDG-PET/CT and EUS in diagnosing primary pancreatic carcinoma: a meta-analysis. Eur J Radiol 78, 142–150, 2011. [DOI] [PubMed] [Google Scholar]
- 6.Chan A et al. Validation of biomarkers that complement CA19.9 in detecting early pancreatic cancer. Clin Cancer Res 20, 5787–5795, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen JD et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 359, 926–930, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamyabi N, Bernard V & Maitra A Liquid biopsies in pancreatic cancer. Expert Rev Anticancer Ther 19, 869–878, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sausen M et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun 6, 7686, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tjensvoll K et al. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol Oncol 10, 635–643, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crowley E, Di Nicolantonio F, Loupakis F & Bardelli A Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol 10, 472–484, 2013. [DOI] [PubMed] [Google Scholar]
- 12.Kamyabi N et al. A microfluidic device for label-free isolation of tumor cell clusters from unprocessed blood samples. Biomicrofluidics 13, 044111, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu R et al. Extracellular vesicles in cancer - implications for future improvements in cancer care. Nat Rev Clin Oncol, 2018. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 527, 100–104, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeppesen DK et al. Reassessment of Exosome Composition. Cell 177, 428–445 e418, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reategui E et al. Engineered nanointerfaces for microfluidic isolation and molecular profiling of tumor-specific extracellular vesicles. Nat Commun 9, 175, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pariset E, Agache V & Millet A Extracellular Vesicles: Isolation Methods. Advanced Biosystems 1, 1700040, 2017. [DOI] [PubMed] [Google Scholar]
- 18.Woo HK et al. Exodisc for Rapid, Size-Selective, and Efficient Isolation and Analysis of Nanoscale Extracellular Vesicles from Biological Samples. ACS Nano 11, 1360–1370, 2017. [DOI] [PubMed] [Google Scholar]
- 19.Liu F et al. The Exosome Total Isolation Chip. ACS Nano 11, 10712–10723, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grant R et al. A filtration-based protocol to isolate human plasma membrane-derived vesicles and exosomes from blood plasma. J Immunol Methods 371, 143–151, 2011. [DOI] [PubMed] [Google Scholar]
- 21.Lewis JM et al. Integrated Analysis of Exosomal Protein Biomarkers on Alternating Current Electrokinetic Chips Enables Rapid Detection of Pancreatic Cancer in Patient Blood. ACS Nano 12, 3311–3320, 2018. [DOI] [PubMed] [Google Scholar]
- 22.Wu M et al. Isolation of exosomes from whole blood by integrating acoustics and microfluidics. Proc Natl Acad Sci U S A 114, 10584–10589, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen C et al. Microfluidic isolation and transcriptome analysis of serum microvesicles. Lab Chip 10, 505–511, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanwar SS, Dunlay CJ, Simeone DM & Nagrath S Microfluidic device (ExoChip) for on-chip isolation, quantification and characterization of circulating exosomes. Lab Chip 14, 1891–1900, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ko J et al. Combining Machine Learning and Nanofluidic Technology To Diagnose Pancreatic Cancer Using Exosomes. ACS Nano 11, 11182–11193, 2017. [DOI] [PubMed] [Google Scholar]
- 26.Hisey CL, Dorayappan KDP, Cohn DE, Selvendiran K & Hansford DJ Microfluidic affinity separation chip for selective capture and release of label-free ovarian cancer exosomes. Lab Chip 18, 3144–3153, 2018. [DOI] [PubMed] [Google Scholar]
- 27.Castillo J et al. Surfaceome profiling enables isolation of cancer-specific exosomal cargo in liquid biopsies from pancreatic cancer patients. Ann Oncol 29, 223–229, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia Y & Whitesides GM SOFT LITHOGRAPHY. Annual Review of Materials Science 28, 153–184, 1998. [Google Scholar]
- 29.Biankin AV et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bailey P et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52, 2016. [DOI] [PubMed] [Google Scholar]
- 31.Allenson K et al. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann Oncol 28, 741–747, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernard V et al. Circulating Nucleic Acids Are Associated With Outcomes of Patients With Pancreatic Cancer. Gastroenterology 156, 108–118 e104, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.