

Summary
Mechanisms by which Porphyromonas gingivalis (P. gingivalis) infection enhances oral tumor growth or resistance to cell death remain elusive. Here, we determined that P. gingivalis infection mediates therapeutic resistance via inhibiting lethal mitophagy in cancer cells and tumors. Mechanistically, P. gingivalis targets the LC3B-ceramide complex by associating with LC3B via bacterial major fimbriae (FimA) protein, preventing ceramide-dependent mitophagy in response to various therapeutic agents. Moreover, ceramide-mediated mitophagy is induced by Annexin A2 (ANXA2)-ceramide association involving the E142 residue of ANXA2. Inhibition of ANXA2-ceramide-LC3B complex formation by wild-type P. gingivalis prevented ceramide-dependent mitophagy. Moreover, a FimA-deletion mutant P. gingivalis variant had no inhibitory effects on ceramide-dependent mitophagy. Further, 16S rRNA sequencing of oral tumors indicated that P. gingivalis infection altered the microbiome of the tumor macroenvironment in response to ceramide analog treatment in mice. Thus, these data provide a mechanism describing the pro-survival roles of P. gingivalis in oral tumors.
Subject areas: Bacteriology, Cancer, Cell biology, Microbiology, Molecular biology
Graphical abstract
Highlights
-
•
P. gingivalis infection mediates therapeutic resistance in oral tumors
-
•
Ceramide-mediated mitophagy is induced by the ANXA2-ceramide-LC3B complex
-
•
P. gingivalis targets the ANXA2-ceramide-LC3B complex via bacterial FimA protein
-
•
P. gingivalis inhibits ceramide-dependent mitophagy, leading to therapy resistance
Bacteriology; Cancer; Cell biology; Microbiology; Molecular biology
Introduction
Oral squamous cell carcinomas (referred to here as OSCC) most commonly affect the base of the tongue, soft palate, tonsils, and posterior pharyngeal wall.1,2 These tumors are distinct from oral cavity squamous cell carcinomas, which affect the labial and buccal mucosa, floor of the mouth, anterior tongue, and the gingiva.3,4 Porphyromonas gingivalis (P. gingivalis) is a gram-negative, asaccharolytic anaerobe and a major pathogen of adult chronic periodontitis, an inflammatory condition causing gingiva recession and alveolar bone resorption resulting in tooth loss.5,6,7 P. gingivalis is notable for its ability to invade human primary gingival epithelial and endothelial cells to evade immune clearance and successfully replicate within host cells.8,9,10,11,12 This is partly accomplished by binding of β-1-integrin on the host cell surface with P. gingivalis major fimbriae, which induces cytoskeletal rearrangements that internalize the bacterium.13,14 P. gingivalis is then able to modulate host signaling through a variety of virulence factors (gingipains, fimbriae, lipopolysaccharide, outer membrane vesicles, and nucleoside diphosphate kinase) affecting cell growth, migration, and pro-inflammatory cytokine production that promotes tumorigenesis and/or resistance to therapy.15,16,17 P. gingivalis was found to increase the expression of pro-tumorigenic molecules such as cyclin D1, matrix metalloproteinases (MMP-9), and heparinase, increasing cell migration and proliferation. P. gingivalis was also found to upregulate the expression of B7 homolog 1 (B7-H1) and B7-dendritic cell programmed cell death ligand 2 (DC, PD-L2) in OSCC cell lines and in human primary epithelial cells.18 B7-H1 and B7-DC receptors interact with programmed cell death protein 1 (PD1) receptors on tumor infiltrating lymphocytes, inducing apoptosis, and inhibiting the immune system’s ability to control OSCC.19,20,21 Importantly, P. gingivalis was detected at a higher concentration in OSCC tissue compared to adjacent normal gingiva.22 Severity of OSCC mortality was also correlated with serum antibody levels to P. gingivalis and periodontal disease.23,24 These results indicate that P. gingivalis may be an additional risk factor influencing the severity and development of OSCC in patients.
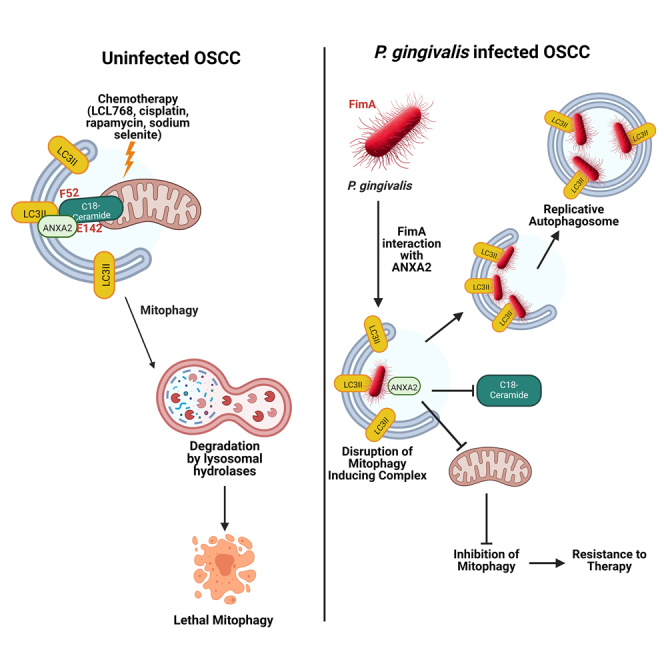
Once inside the host gingival epithelial cell, P. gingivalis induces the formation of ER-derived autophagosomal vesicles that incorporate and protect the bacterium from cytosolic ubiquitin-labeling and lysosomal degradation.25 These double-membrane autophagosomal vesicles (also called replicative autophagosomes) provide a nutrient-rich environment for continued and protected replication inside the host cell.25 Formation and elongation of autophagosomes are regulated by various stress-mediated lipid metabolic pathways, including bioactive sphingolipids.26,27,28 Sphingolipids are membrane lipids that play an important role in disease pathogenesis and the stress-induced signal transduction of multiple cellular pathways.29,30 Endogenous ceramide generation, especially ceramide synthase 1 (CerS1)-generated C18-ceramide,31 acts as an inducer for lethal mitophagy.26,27,28 It has been shown that CerS1-mediated C18-ceramide generation is repressed in OSCC by a mechanism involving epigenetic and micro-RNA crosstalk.32 However, CerS1/C18-ceramide can be activated in response to many stress stimuli, like radiotherapy or chemotherapeutic agents, in OSCC cells and mouse models or in patients.33,34 One of the downstream biological responses that CerS1/C18-ceramide signaling exerts is induction of lethal mitophagy in OSCC, which is enhanced by HPV-E7 signaling.35 However, whether P. gingivalis inhibits ceramide-dependent lethal mitophagy signaling, resulting in resistance to lipid-mediated OSCC cell death and/or tumor suppression, has not been studied previously. Thus, in this study, we set out experiments to understand the roles and mechanisms whereby P. gingivalis infection mediates therapeutic resistance via targeting ceramide-mediated mitophagy in OSCC cells and tumors.
Results
P. gingivalis inhibits lethal mitophagy by LC3 association, disrupting the ceramide-LC3 complex
P. gingivalis infection is associated with poor therapeutic response and clinical outcomes in patients with OSCC.24 We first aimed to validate these data in an HPV-negative human OSCC cell line, UM-SCC-1A, in response to ceramide analog drug, LCL768, a pyridinium ceramide derivative, which is known to induce lethal mitophagy.26 We optimized infection in this cell line by infecting with wildtype P. gingivalis (ATCC 33277) at various multiplicity of infection (MOIs) and time points (50, 100, and 500 MOIs for 1, 3, 6, and 24 h) and analyzing via antibody-labeled immunofluorescence (IF). An infection of 100 MOI was most optimal for a 6 h incubation (Figure S1A, left and right panels). P. gingivalis infection also resulted in resistance to lethal mitophagy induced by LCL768 compared to uninfected controls, which is supported by previous reports revealing infection association with chemotherapeutic resistance (Figure S1B).36,37 To determine the mechanisms by which P. gingivalis infection mediates resistance to lethal mitophagy, we examined markers of ceramide-mediated lethal mitophagy in infected and uninfected UM-SCC-1A cells. We started by measuring LC3-Tom20, LAMP1-Tom20, or ceramide-LC3 co-localizations, known markers of ceramide-mediated mitophagy,38 by IF. P. gingivalis infection inhibited the colocalization of LC3-Tom20 (∼62%), Tom20-LAMP1 (∼60%), and ceramide-LC3 (∼50%) in response to LCL768 compared to uninfected controls (Figures 1A–1C). Similarly, inhibition of LCL768-mediated mitophagy by P. gingivalis infection was consistent with the attenuation of LC3-LAMP1 and ceramide-Tom20 co-localization (∼40% and ∼50%, respectively) (Figures S1C and S1D). Moreover, P. gingivalis infection inhibited mitophagy ∼50–60%, measured by Mtphagy dye and MtKeima protein autofluorescence, in response to LCL768, cisplatin, rapamycin or sodium selenite, therapeutic agents that are known to induce lethal mitophagy, in human OSCC cells (Figures 1H–1J). These data support the hypothesis that P. gingivalis infection results in therapeutic resistance to lethal mitophagy in response to ceramide analog LCL768 and other conventional therapeutic agents in human OSCC cells.
Figure 1.

P gingivalis inhibits mitophagy through disruption of ceramide, LC3, and Annexin A2 (ANXA2) binding
(A) Confocal images of UMSCC1A cells infected/uninfected with P. gingivalis (100 MOI, 6 h) and treated with vehicle or LCL768 (30 μM, 2 h) were stained for LC3 (green) and Tom20 (red).
(B) Confocal images of UMSCC1A cells infected/uninfected with P. gingivalis and treated with vehicle or LCL768, labeled for LAMP1 (green) and Tom20 (red).
(C and D) Similarly, confocal images of ceramide (green) and LC3 (red), and LC3 (green) and P. gingivalis (red) are shown. Yellow shows colocalization. Images represent three independent experiments. Scale bars are 100 μm (throughout the manuscript unless specifically noted). Quantification of colocalization was estimated via Pearson’s correlation coefficient, completed with ImageJ Fiji software, and normalized to uninfected, untreated control. Values indicate mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(E) Western blotting was used to detect the protein abundance of stably knocked down LC3 and transiently transfected tagged mutant or wildtype LC3 in UMSCC1A cells. Actin was used as the loading control.
(F) UMSCC1A cells stably expressing shRNA against LC3 and transiently transfected with FLAG-wildtype LC3, V5-F52A LC3, or V5-G120A LC3 pcDNA3.1+ plasmids were labeled with ceramide antibody and tag antibody (FLAG or V5) for confocal microscopy. Quantification of colocalization was estimated using Pearson’s correlation coefficient and normalized to uninfected, untreated control using ImageJ Fiji software. Images represent three independent experiments. Data are means ± SD (n = 3, ns, not significant, ∗∗∗∗p < 0.0001).
(G) UMSCC1A cells stably expressing shRNA against LC3 were transiently transfected with FLAG-wildtype LC3, V5-F52A LC3, or V5-G120A LC3 plasmids before infection with P. gingivalis (6 h, 100 MOI) and treatment with 30 μM LCL768 or vehicle for 3 h. Cells were dual labeled with P. gingivalis antibody and the appropriate tag antibody (FLAG or V5) for confocal microscopy. Quantification of colocalization, shown via Pearson’s correlation coefficient, are means ± SD (n = 3, ns, not significant, ∗∗∗∗p < 0.0001).
(H) Live UMSCC1A cells were stained using Mtphagy dye (red) in which fluorescence intensity increases with mitophagy induction, following treatment with 5 μM cisplatin, 8 μM rapamycin, or 8 μM sodium selenite for 3 h. Images represent three independent experiments. The bottom panels show the quantification of Mtphagy fluorescence intensity using ImageJ Fiji software. Data are means ± SD (n = 3, ∗∗∗∗p < 0.0001).
(I) UMSCC1A cells were transiently transfected with pHAGE-MtKeima plasmid, in which auto-fluorescent Keima protein conjugated to mitochondrial-targeting sequence (COX8) fluoresces green at neutral pH (>pH 6) and red at acidic pH (<pH 5). Live cells were then treated (2 h) with vehicle control, LCL768, or alternative mitophagy-inducing drugs following infection/no infection with P. gingivalis (6 h). Images represent three independent experiments.
(J and K) Quantification of colocalization in cells infected with wildtype or FimA deletion mutant P. gingivalis (ΔFimA) are shown via Pearson’s correlation coefficient as means ± SD (∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001).
Previous studies have shown that lethal mitophagy is induced through the direct binding of CerS1-generated C18-ceramide (on the outer mitochondrial membrane) and LC3 (on the autophagosomal membrane). This direct interaction then facilitates the recruitment of autophagosomes to damaged mitochondria to induce mitophagy (1). It has been reported previously that P. gingivalis is associated with LC3 to hijack autophagosomes in primary gingival epithelial cells25 for intracellular survival, which is hypothesized to prevent ceramide-LC3 binding, resulting in the inhibition of lethal mitophagy. We first validated that P. gingivalis is associated with LC3 in UM-SCC-1A cells using immunofluorescence in the absence or presence of LCL768 (Figure 1D). There was no significant colocalization between P. gingivalis and Tom20, ceramide, or LAMP1 in these cells (Figures S1E–S1H). Also, to validate that the induction of LCL768-mediated mitophagy is dependent on CerS1 expression, we knocked down CerS1 expression using shRNAs and measured its effects on mitophagy via co-localization of Tom20-LAMP1 through IF in UM-SCC-1A cells. The data demonstrated that shRNA-mediated CerS1 knockdown, confirmed by Western blotting, blunted LCL768-induced mitophagy compared to scrambled (SCR)-shRNA transfected controls treated with LCL768 (Figures S2A–S2C). Moreover, inhibition of mitophagy by P. gingivalis in response to LCL768 treatment was validated by co-immunoprecipitation studies (co-IP) in UM-SCC-1A cells (Figures S2D and S2E). Co-localization of P. gingivalis with LC3 was also detected in human HPV+ UM-SCC-47 OSCC cells, consistent with the prevention of LC3-Tom20 and Tom20-LAMP1 co-localization (Figure S2F). These data support that LCL768 induces ceramide-dependent mitophagy and P. gingivalis inhibits this process via LC3 association in human OSCC cells independently of their HPV status.
We then examined if P. gingivalis infection inhibits ceramide-mediated mitophagy by interfering with ceramide-LC3 association and if P. gingivalis association with LC3 requires ceramide association. Therefore, we transiently expressed FLAG-WT-LC3, mutant LC3 with F52A conversion (V5-LC3-F52A) that prevents ceramide binding, or G120A conversion (V5-LC3-G120A) that inhibits LC3 activation, used as a negative control, in UM-SCC-1A cells in which endogenous LC3 expression was down-regulated by shRNAs (Figure 1E). The data demonstrated that P. gingivalis almost completely inhibited the association between ceramide and FLAG-WT-LC3, whereas it had no effects on ceramide-V5-F52A-LC3 or ceramide-V5-G120A-LC3 co-localizations (Figure 1F). Moreover, P. gingivalis co-localized with FLAG-WT-LC3, but not with V5-F52A-LC3 or V5-G120A-LC3, which indicates that the bacterium may interact with LC3 associated with ceramide (Figure 1G). Overall, these data demonstrate that P. gingivalis associates with LC3 and inhibits ceramide-LC3 binding, resulting in the inhibition of ceramide-dependent mitophagy in response to LCL768. These data also suggest that P. gingivalis-LC3 association requires LC3 lipidation via G120 and P. gingivalis-LC3 interaction has some overlap within the F52 residue of LC3, like ceramide-LC3 binding.
P. gingivalis-LC3 association is dependent on bacterial major fimbriae (FimA)
To determine the bacterial virulence factors that are involved in P. gingivalis-LC3 association and inhibition of mitophagy, we focused on Fimbriae A (FimA), a unique major fimbriae present on the surface of P. gingivalis that is partly responsible for mediating bacterial entry into host cells via β1 integrin.13 We utilized WT-P. gingivalis and fimA-deleted isogenic mutant P. gingivalis (ΔFimA) strains. Results showed that while WT-P. gingivalis inhibited LCL768-mediated co-localization of LAMP1-Tom20 and LC3-Tom20, ΔFimA mutant-P. gingivalis did not prevent their co-localization in UM-SCC-1A cells (Figures S2H and S2I). These data were supported when the MtKeima fluorescent shift was observed in ΔFimA mutant-P. gingivalis infected cells compared to WT-P. gingivalis infected OSCC, indicating restored mitophagy (Figure 1K). In addition, while treatment of UMSCC1A cells with LCL768 significantly reduced recovery and proliferation of uninfected cells, wildtype P. gingivalis displayed no effect on colony growth with LCL768 in a clonogenic assay. Infection of cells with ΔFimA P. gingivalis reversed this trend, with colony formation significantly reduced in response to LCL768 treatment (Figures S1I and S1J). As a control, we used infections with another well-characterized isogenic mutant strain of P. gingivalis with nucleoside diphosphate kinase (NDK) deletion (ΔNDK) mutant or its NDK-complemented strain (ΔNDK+NDK).5,17,39 The data showed no differences in P. gingivalis-LC3 association between WT-, ΔNDK-mutant- or ΔNDK+NDK-complemented-P. gingivalis in the absence/presence of LCL768 in these cells (Figure S2G). These data suggest that bacterial FimA plays a key role in the association of P. gingivalis with LC3 in OSCC cells to prevent ceramide-mediated mitophagy.
P. gingivalis FimA disrupts the Annexin A2 (ANXA2)-ceramide-LC3 complex to prevent mitophagy
To identify LC3-binding proteins in UM-SCC-1A cells infected/uninfected with P. gingivalis in the presence/absence of LCL768, we immunoprecipitated endogenous LC3B and separated LC3B-bound protein bands via SDS-PAGE for proteomics analysis (Figure S3A). Among the multiple LC3B-associated proteins identified (Figure S3A), we focused our attention on Annexin A2 (ANXA2) due to its known roles in calcium-dependent phospholipid-binding, participation in endocytosis/exocytosis, and activity linking F-actin cytoskeleton to the plasma membrane.40,41,42 We validated LC3B-ANXA2 association by co-IP and IF using anti-LC3B and anti-ANXA2 antibodies (Figures 2A and 2B). Interestingly, these results showed that P. gingivalis infection blunted LC3B-associated ANXA2 levels ∼75–80% with or without LCL768 in UM-SCC-1A cells (Figures 2A and 2B) without any changes in overall intracellular protein levels of ANXA2 (Figure 2A, input panel). We did not detect any major differences in the association between P. gingivalis-ANXA2, FimA-ANXA2, Atg3-ANXA2, or S100A10-ANXA2 in response to P. gingivalis infection with/without LCL768 compared to controls in these cells (Figures S3B–S3E). However, colocalization between ANXA2 and FimA was observed in P. gingivalis infected cells (Figure S3C). The S100A10-ANXA2 association was used as a positive control in these experiments as S100A1043 is a known protein partner of ANXA2 (31,32). However, ceramide-ANXA2 and ANXA2-Atg7 co-localizations were prevented by P. gingivalis (∼70% or 85%, respectively) compared to uninfected UM-SCC-1A cells in the presence of LCL768 (Figures 2C and 2D). To further explore how FimA impacted these associations, we directly transfected recombinant FimA protein into uninfected UM-SCC-1A cells compared to sham control protein. It was observed that P. gingivalis FimA significantly repressed both ceramide-ANXA2 and Tom20-LC3 associations via immunofluorescence (Figures 2E and 2F), confirming the data obtained with FimA mutant P. gingivalis infections in UM-SCC-1A cells, which had no impact on ceramide-mediated mitophagy. To this end, we also measured the internalization of WT vs. FimA mutant P. gingivalis into UM-SCC-1A cells. Data showed that FimA-deficient mutant P. gingivalis was internalized into UM-SCC-1A cells at comparable levels to wild-type P. gingivalis (Figure 2H). To further validate the direct association between FimA and ANXA2, we performed far Western blotting using purified recombinant FimA and ANXA proteins. These results showed that recombinant human ANXA2 (rANXA2) and P. gingivalis FimA (rFimA) proteins directly bind each other (top left) compared to no binding seen between recombinant human ANXA2 and P. gingivalis nucleoside diphosphate kinase (rNDK) protein, used as a negative control (bottom left, Figure 2G). These data suggest that P. gingivalis inhibits the LC3B-ceramide-ANXA2 complex through FimA to ablate mitophagy via FimA-ANXA2 interaction.
Figure 2.

ANXA2 association with mitophagy markers
(A) UMSCC1A cells were lysed for immunoprecipitation with LC3 (right) or species-matched IgG control antibody (left). Co-immunoprecipitation of ANXA2 is shown via western blot, and input controls were normalized to GAPDH. Images represent at least three independent experiments. Quantification of co-immunoprecipitated ANXA2 western blot bands from was normalized to uninfected, untreated control. Data are means ± SD (n = 3, ∗p < 0.05, ∗∗∗p < 0.001).
(B) Quantification of confocal images of UMSCC1A cells dual-labeled with LC3 and ANXA2 antibody, infected with P. gingivalis, and treated with LCL768. Pearson’s correlation coefficient was normalized to uninfected, untreated control. Values indicate mean ± SD of n = 3 independent experiments. ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
(C) Confocal images of ceramide (green) and ANXA2 (red) in UMSCC1A cells infected with P. gingivalis and treated with LCL768 are shown.
(D) Confocal images of Atg7 (green) and ANXA2 (red) in UMSCC1A cells infected with P. gingivalis and treated with LCL768.
(E and F) Uninfected UM-SCC-1A cells were transfected with either sham or P. gingivalis FimA virulence factor proteins and treated with LCL768 or vehicle control. Confocal images of ceramide (green) and ANXA2 (red), and LC3 (green) and Tom20 (red), in uninfected cells are shown. Yellow shows colocalization. Images represent three independent experiments. Quantification of colocalization was estimated via Pearson’s correlation coefficient, completed with ImageJ Fiji software, and normalized to uninfected, untreated control. Values indicate mean ± SD of n = 3 independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(G) Far western blotting analysis was conducted by running human recombinant ANXA2 (rANXA2) on a gel and testing direct protein-protein binding with recombinant P. gingivalis virulence factors (rFimA, rNDK). The top left blot shows direct binding between ANXA2 and FimA recombinant proteins, identified via FimA antibody. No direct binding between human recombinant ANXA2 and P. gingivalis recombinant NDK was observed in the bottom left blot. The top right blot confirms recombinant ANXA2 presence in the blot. The bottom right blot indicates that there is no cross-reactivity between recombinant ANXA2 and FimA antibody.
(H) Immunofluorescence confirmation of wildtype and FimA-deficient (ΔFimA) P. gingivalis internalization into UM-SCC-1A cells with anti-P. gingivalis antibody is shown. Quantification of mean fluorescent intensity was completed with ImageJ Fiji software and normalized to uninfected, untreated control. Values indicate mean ± SD of n = 3 independent experiments. Ns, not significant, ∗∗∗∗p < 0.0001.
We then hypothesized that if resistance to ceramide-mediated mitophagy by P. gingivalis is linked to the disruption of the LC3B-ceramide-ANXA2 complex, then down-regulation of ANXA2 should similarly prevent mitophagy in uninfected cells. This hypothesis was tested in UM-SCC-1A cells which were stably transfected with SCR- or ANXA2-shRNAs, which resulted in ∼90% knockdown in ANXA2 expression compared to SCR-shRNA transfected controls (Figure 3A, upper panel). We then measured the effects of ANXA2 knockdown on ceramide-dependent mitophagy in response to LCL768. Knockdown of ANXA2 attenuated Tom20-LC3 association, measured by co-IP and IF (∼%85) (Figures 3B and 3C), and ceramide-Tom20 co-localization measured by IF (60%) (Figure 3D) in the presence of LCL768 compared to SCR-shRNA transfected controls. These data were also consistent with the inhibition of ceramide-LC3 and LAMP1-Tom20 co-localization (∼50–60%) in response to ANXA2 knockdown in the presence of LCL768 compared to SCR-shRNA transfected and vehicle-treated controls (Figures 3E and 3F). ANXA2 downregulation almost completely inhibited the interaction between ceramide-LC3 or ceramide-Tom20 in response to LCL768, measured using the proximity ligation assay (PLA) with anti-ceramide, anti-LC3 or anti-Tom20 antibodies (Figures 3H–3J). Taken together these data indicate that ANXA2 knockdown mimics P. gingivalis infection mediated phenotype to prevent ceramide-dependent lethal mitophagy.
Figure 3.

ANXA2 knockdown inhibits mitophagy
(A and B) Immunoprecipitation with Tom20 or species-matched IgG antibody in uninfected UMSCC1A cells stably expressing shRNA against ANXA2 or scrambled control. The left panel shows input (pre-immunoprecipitation) ANXA2 and LC3 via western blotting, normalized to Actin control bands. The right panel shows co-immunoprecipitated LC3 in cells treated with LCL768 or vehicle control. Images represent at least three independent experiments. Quantification of co-immunoprecipitated LC3 western blot bands was normalized to scrambled shRNA expressing, untreated control. Data are means ± SD (n = 3, ∗p < 0.05, ∗∗∗∗p < 0.0001).
(C and D) Quantification of confocal images of uninfected UMSCC1A cells stably expressing shRNA against ANXA2 or scrambled control and treated with LCL768 or vehicle control. Colocalization between LC3 and Tom20 (C), or ceramide and Tom20 (D), was estimated via Pearson’s correlation coefficient and normalized to uninfected, untreated control. Values indicate mean ± SD of n = 3 independent experiments. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(E and F) Confocal images of uninfected UMSCC1A cells stably expressing shRNA against ANXA2 or scrambled control, treated with LCL768 or vehicle control and dual labeled with ceramide (green) and LC3 (red), or LAMP1 (green) and Tom20 (red) antibodies.
(G) Quantification of confocal images in (E) and (F), shown via Pearson’s correlation coefficient. Values indicate mean ± SD of n = 3 independent experiments. ∗p < 0.05, ∗∗p < 0.01.
(H and I) Proximity ligation assay was performed on uninfected UMSCC1A cells stably expressing shRNA against ANXA2 or scrambled control for ceramide and LC3, or ceramide and Tom20. Confocal images show DAPI nuclei staining (blue) and PLA signal (red) indicating interaction of targets within close proximity (<40 nm).
(J) Quantification of the average number of PLA signal foci per cell in (H) and (I) was completed with ImageJ Fiji. Values indicate mean ± SD of n = 3 independent experiments. Ns, not significant, ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(K) Live, uninfected UMSCC1A cells stably expressing shRNA against ANXA2, LC3, or scrambled control were stained with Mtphagy (red) and LysoGreen dyes following treatment with LCL768 or vehicle control. Images represent three independent experiments. Yellow indicates colocalization.
(L) Quantification of Mtphagy fluorescence intensity in (K) was done using ImageJ Fiji software. Data are means ± SD (n = 3, ∗∗∗∗p < 0.0001).
(M) Live, uninfected UMSCC1A cells stably expressing shRNA against ANXA2, or scrambled control, were treated with increasing concentrations of LCL768 for 2 h and stained with trypan blue cell exclusion dye. Cell viability (percent live, unstained cells) is shown as means ± SD (n = 3, ∗∗p < 0.01, ∗∗∗∗p < 0.0001).
To measure mitophagy directly, we also performed live cell imaging using Mtphagy dye in SCR-, ANXA2-or LC3B-shRNA transfected UM-SCC-1A cells in the absence/presence of LCL768. The data showed that while LCL768 induced mitophagy in SCR-controls, knockdown of ANXA2 and LC3B (negative control)),26,28 did not induce mitophagy in UM-SCC-1A cells (Figures 3K–3L). Trypan blue cell viability assay also showed that knockdown of ANXA2 resulted in resistance to lethal mitophagy induced by increasing concentrations of LCL768 (p < 0.05) compared to SCR-shRNA-transfected and vehicle-treated cells, increasing the IC50 concentration from 24 to 30 μM (Figure 3M). Overall, these data demonstrate that downregulation of ANXA2 inhibits ceramide-dependent mitophagy by preventing ceramide-LC3 association in response to LCL768.
ANXA2-ceramide-LC3 complex plays key roles in ceramide-dependent mitophagy, targeted by P. gingivalis FimA
Our IF studies suggest that P. gingivalis FimA disrupts the ANXA2-ceramide-LC3B complex to inhibit ceramide-dependent mitophagy by associating with ANXA2. This was also consistent with in silico molecular modeling, which indicated a strong interaction between monomeric FimA (i.e., fimbrillin) and ANXA2, but not between monomeric FimA and LC3 (Figures S4A and S4B). Moreover, our molecular modeling results (Figures S5A and S5B) showed that there is a strong association between ANXA2 and ceramide, and previously reported ceramide and LC3B, providing a multi-protein-lipid complex in which C18-ceramide appears to play a bridge role between ANXA2 and LC3B (Figures S5A, S5B, and 4A). Specifically, molecular docking and simulation analyses suggested that ceramide-ANXA2 interaction involves the E142 residue of ANXA2 (Figures S5A, S5B, 4B, and 4C). To validate these models, we generated a tagged ANXA2 variant with a point mutation for E142A conversion (V5-ANXA2-E142A) and measured its effects compared to V5-ANXA2-WT on ceramide association and ceramide-induced mitophagy in UM-SCC-1A cells that were stably transfected with ANXA2-shRNA. After validation of ANXA2 knockdown and tag expression by Western blotting (Figure 4D), IF experiments showed that E142A point mutation successfully prevented ceramide-V5-ANXA2 co-localization compared to V5-ANXA2-WT in response to LCL768 (Figure 4E). In addition, E142A mutation attenuated ceramide-dependent mitophagy via IF co-localization of ceramide-LC3 and LC3-Tom20, which were inhibited ∼50–62%, in response to LCL768 compared to vehicle-treated V5-ANXA2-WT expressing controls (Figures 4F and 4G). Our results were supported by colocalization between FimA and V5-ANXA2-WT (∼48%) in P. gingivalis infected UM-SCC-1A compared to vehicle-treated controls (Figure 4H). However, there was no co-localization detected between FimA and V5-ANXA2-E142A in response to LCL768 in these cells (Figure 4I). Uninfected cells (no P. gingivalis) were used as negative controls for FimA-V5-ANXA2 co-localization studies (Figures 4H and 4I). These experiments indicate that ceramide-dependent mitophagy is regulated by the ANXA2-ceramide-LC3 complex, and ANXA2-ceramide interaction involves the E142 residue of ANXA2. Also, these data demonstrate that P. gingivalis FimA targets the ANXA2-ceramide-LC3 complex by preventing ceramide-ANXA2 association to inhibit ceramide-dependent mitophagy.
Figure 4.

Mutation of the projected ceramide-binding site on ANXA2 (E142A) inhibited ceramide-mediated mitophagy induction and FimA association
(A) Trimolecular modeling of ANAX2, ceramide, and LC3 in MOE.
(B) Docking of ceramide onto ANXA2. Bimolecular modeling of ANXA2 and ceramide was refined from 100 poses in MOE and CLUSPRO, revealing a preference for ceramide to bind ANXA2 on one face of the enzyme, interacting specifically with Glu142 (E142).
(C) Skeletal model of ceramide interacting with residues of interest, particularly E142, on ANXA2.
(D) Western blotting was performed to detect the protein abundance of stably knocked down ANXA2, and transiently transfected V5-tagged mutant or wildtype ANXA2. Actin was used as the loading control.
(E–G) Uninfected UMSCC1A cells stably expressing shRNA against endogenous ANXA2 were transiently transfected with V5-wiltype ANXA2 or V5-E142A ANXA2 pcDNA3.1+ plasmid and treated with LCL768 (30 μM, 2 h) or vehicle control. Cells were dual labeled with ceramide (green) and V5 (red) antibody for confocal imagery. Similarly, ceramide (green) and LC3 (red), and LC3 (green) and Tom20 (red) confocal images are shown. Images represent three independent experiments. Yellow indicates colocalization. Quantification of colocalization in confocal images was estimated via Pearson’s correlation coefficient and normalized to exogenous wildtype ANXA2 expressing, untreated control cells. Values indicate mean ± SD of n = 3 independent experiments. ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(H and I) UMSCC1A cells stably expressing shRNA against endogenous ANXA2 were transiently transfected with either V5-wildtype ANXA2 or V5-E142A ANXA2 pcDNA3.1+ plasmid, infected with WT P. gingivalis for 6 h (100 MOI), and treated with LCL768 (30 μM, 2 h) or vehicle control. Confocal microscopy was conducted on cells dual-labelled with FimA and V5-tag antibody. Quantification of colocalization via Pearson’s correlation coefficient was normalized to exogenous wildtype ANXA2 expressing, untreated, uninfected control cells. Values indicate mean ± SD of n = 3 independent experiments. Ns not significant, ∗∗p < 0.01.
Sh-RNA-mediated knockdown of LC3B or P. gingivalis infection prevents LCL768-induced mitophagy in OSCC tumors in vivo
P. gingivalis targets the ANXA2-ceramide-LC3B complex to prevent ceramide-mediated mitophagy, so we aimed to determine the effects of LCL768 on the inhibition of OSCC tumor growth in vivo. First, we measured the effects of LCL768 on the growth/proliferation of UM-SCC-1A-derived xenografts in NUDE mice after injection of SCR-, ANXA2-, or LC3B-shRNA-transfected cells into the flanks. After the OSCC xenograft-derived tumors became palpable (23 days post-injection), mice were treated with LCL768 (10 mg/kg) or vehicle control intraperitoneally for 37 days (every other day/week, n = 8 mice). Our results show that while LCL768 inhibited tumor growth and increased survival in SCR-shRNA-transfected xenograft tumors compared to vehicle-treated controls (Figures 5A and 5B), knockdown of LC3B almost completely attenuated the growth inhibitory effects of LCL768 (Figure 5A). ANXA2-shRNA-transfection also ablated tumor growth in both vehicle and LCL768 treated NUDE mice (Figure 5A). Mice with ANXA2-or LC3-shRNA xenograft tumors exhibited protection of mitophagy, measured by TOM20-LC3 and ceramide-LC3 co-localization in response to LCL768 compared to SCR-shRNA tumors (Figure 5C, upper panels). Down regulation of ANXA2 and LC3 also prevented Tom20-LAMP1 and ceramide-ANXA2 co-localization in response to LCL768 compared to SCR-shRNA-transfected controls (Figure 5C, lower panels), consistent with ceramide-dependent mitophagy inhibition. Although survival of mice did not change, there was no reduction in Ki67+ staining (indicating proliferation) in response to LCL768 treatment when ANXA2 or LC3 were knocked down (Figures 5B, 5D, and 5E). On the other hand, SCR-shRNA-transfected tumor burdened mice showed significantly better survival outcomes and reduced Ki67+ staining when treated with LCL768 compared to SCR-transfected and vehicle-treated controls (Figures 5B, 5D, and 5E). It should be noted that there was a sharp drop in tumor size in some control animals bearing SCR-shRNA-transfected oral tumors that were treated with vehicle (SCR-VEH) between days 38 and 42. (Figure 5A). This was mainly associated with tumor necrosis that occurs in the hypoxic center of solid tumors due to large tumor sizes. These data suggest that LCL768 inhibits OSCC-xenograft-derived tumor growth via LC3B-mediated mitophagy in vivo. It also indicates that ANXA2 and LC3B are necessary for the induction of ceramide-dependent lethal mitophagy in vivo, supporting in vitro results.
Figure 5.

ANXA2 and LC3 knockdown in OSCC xenograft tumors resulted in attenuation of mitophagy
(A) ANXA2, LC3, or scrambled control (SCR) shRNA were stably expressed in UMSCC1A cells. Cells were then injected into the midline flanks of nude mice and allowed to incubate for approximately 23 days before intraperitoneal treatment with LCL768/vehicle control (10 mg/kg, n = 8). Flank tumors were measured using calipers, and measurements indicate mean ± SD of n = 8 independent experiments. Student’s t test was conducted to examine significance in growth response curves (SCR VEH vs. LCL ∗∗p < 0.01, ANXA2KD VEH vs. LCL Ns not significant, LC3KD vs. LCL Ns not significant).
(B) Survival Kaplan-Meier curves of nude mice displaying xenograft tumors induced by injection with UM-SCC-1A OSCC cells stably expressing shRNA for ANXA2, LC3, or scrambled (SCR) control and treated/untreated with LCL768 (n = 8 mice). ∗p < 0.05.
(C) Immunofluorescence was completed on sections of nude mouse xenograft flank tumors stably expressing either ANXA2, LC3, or scrambled (SCR) control shRNA and treated with LCL768 or vehicle control intraperitoneally (10 mg/kg). Quantification of confocal images of LC3 and Tom20, ceramide and LC3, Tom20 and LAMP1, and ceramide and ANXA2 association are shown. Pearson’s correlation coefficient values indicate mean ± SD of n = 3 independent experiments, normalized to uninfected, untreated control. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(D) Mouse xenograft flank tumors injected with cells stably expressing ANXA2, LC3, or SCR shRNA and treated/untreated with LCL768 were stained with Ki67 antibody, indicating proliferating tumor cells.
(E) Quantification of Ki67+ nuclei staining in tumor sections was performed in inform Automated Image Analysis software and represents a mean ± SD of n = 3 independent experiments.
Since P. gingivalis is a resident member of the oral microbiota, to measure the effects of infection on ceramide-dependent mitophagy in a clinically relevant environment, we injected murine MOC2 OSCC cells intramuscularly into the floor of the mouth (mylohyoid) of C57BL/6 mice to generate orthotopic tumors (Figure 6A). The resident oral microbiota was removed via antibiotic washout before reconstituting with 109 colony forming unit (CFU) wildtype P. gingivalis. After the treatment and growth studies were completed, tumors were excised and subjected to a variety of ex vivo assays. First, P. gingivalis infection in orthotopic OSCC tumors was confirmed by fluorescent in situ hybridization (FISH) assay using P. gingivalis-specific 16S rRNA oligonucleotide probe (Figures 6B and 6C). LCL768 treatment did not seem to affect P. gingivalis infection in these OSCC orthotopic tumors compared to uninfected OSCC tumors, used as a negative control (Figures 6B and 6C). Furthermore, while LCL768 induced Tom20-LC3, ceramide-LC3, and Tom20-LAMP1 co-localization in uninfected control OSCC tumors, P. gingivalis infection almost completely blunted association of these mitophagy markers in response to LCL768 compared to vehicle-treated tumors (Figures 6D–6F). Our data showed that while LCL768 increased ceramide-ANXA2 co-localization about 6-fold in uninfected OSCC tumors, P. gingivalis infection prevented this association compared to vehicle-treated controls (Figure 6G). Measurements of orthotopic tumors of the floor of the mouth using calipers could not be performed for accurate measurements due to growth of the tumors deeper into the oral cavity and neck, so we elected to measure Ki67 expression using IHC to show proliferation. These data exhibit that P. gingivalis-infected OSCC tumors showed higher Ki67 expression in response to LCL768 treatment compared to uninfected tumors, indicating resistance to LCL768-mediated inhibition of proliferation (Figures 6H and 6I). As a control, we did not detect any major changes in the tumor infiltrating immune cells in P. gingivalis-infected versus uninfected OSCC tumors with/without LCL768 treatment, as MOC2 are aggressive OSCC cells derived from CXCR3-deficient C57BL/6 mice (Figure 6J). These data demonstrate that P. gingivalis infection inhibits ceramide-dependent mitophagy and enhances resistance to LCL768 concomitant with targeting the ANXA2-ceramide-LC3B complex in orthotopic OSCC tumors in C57BL/6 mice.
Figure 6.

Orthotopic mouse oral squamous cell carcinoma tumors exhibit inhibited mitophagy when orally infected with P gingivalis
(A) Orthotopic oral squamous cell carcinoma tumors were generated in female C57BL/6 mice (6–8 weeks old) by injecting MOC2 cells intramuscularly into the mylohyoid. Tumors developed over at least 2.5 weeks before uninfected (NP) mice were treated with 10 mg/kg LCL768 intraperitoneally every other day for 2.5 weeks. Otherwise, the resident mouse oral microbiota was treated with antibiotic for 3 days in drinking water prior to starting concurrent P. gingivalis infection (109 CFU, oral gavage) and LCL768/vehicle intraperitoneal treatment every other day. P. gingivalis infection via oral gavage was performed four times total.
(B) Fluorescent in situ hybridization (FISH) was conducted on mouse OSCC tumor sections infected/uninfected with P. gingivalis and treated with LCL768 or vehicle control to confirm the presence of the bacterium in tumor samples. Insets (right panels) show higher magnifications marked with white squares in the middle panels.
(C) Quantification of P. gingivalis signal foci mean fluorescence intensity in (B) was completed with ImageJ Fiji and normalized to uninfected, untreated control. Values indicate mean ± SD of n = 3 independent experiments. Ns, not significant, ∗∗∗∗p < 0.0001.
(D–G) Immunofluorescence was completed on sections of female C57BL/6 mouse orthotopic oral squamous carcinoma tumor infected or uninfected with P. gingivalis (109 CFU) via oral feeding and treated with LCL768 or vehicle control intraperitoneally (10 mg/kg). Quantification of confocal images of LC3 and Tom20 (D), ceramide and LC3 (E), Tom20 and LAMP1 (F), ANXA2 and ceramide (G) association are shown. Pearson’s correlation coefficient values indicate mean ± SD of n = 3 independent experiments, normalized to uninfected, untreated control. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(H) Mouse OSCC tumors were stained with Ki67 antibody, indicating proliferating tumor cells.
(I) Quantification of Ki67+ nuclei staining in mouse orthotopic tumor sections in (H).
(J) Basal percentage of tumor infiltrating lymphocytes in mouse orthotopic tumors.
Finally, to examine whether P. gingivalis infection affects the microbiome composition within the OSCC tumor macroenvironment, we performed 16S rRNA sequencing in orthotopic P. gingivalis-infected or uninfected OSCC tumors in the presence/absence of LCL768. These mice were initially subjected to antibiotic washout to repress the resident, healthy oral microbiota to study the effects of prevalent P. gingivalis infection in OSCC tumors. The data showed that infection with P. gingivalis and resistance to ceramide-induced mitophagy in response to LCL768 was also associated with an increase in the presence of bacteria, including Gordonia, Stenotrophomonas, and Herbaspirillum (Figure S6A). The effects of these bacteria on tumor growth, resistance to chemotherapeutics, and immune response remain to be elucidated. However, it may be noted that 16S rRNA analysis of human OSCC tumors deposited in the bacteria in the Cancer (BIC) database revealed that Porphyromonas abundance was significantly higher at Stage I than in normal control tissue (Figure S6B), other tumor stages, and alternative bacterial species abundance, which indicate possible P. gingivalis-induced changes in oral microbiome constitution with OSCC progression. These data suggest that P. gingivalis infection alters the constitution of the oral microbiota within OSCC tumors correlating with resistance to ceramide-dependent mitophagy and enhanced proliferation in vivo.
Discussion
In this work, we defined the roles and mechanisms by which an opportunistic oral pathogen, P. gingivalis, mediates therapeutic resistance via inhibiting lethal mitophagy in OSCC cells and orthotopic tumors. Our data demonstrated that P. gingivalis targets the LC3B-ceramide complex in OSCC by associating with ANXA2 via bacterial FimA protein, preventing ceramide-dependent lethal mitophagy in response to ceramide analog drug, LCL768, and cisplatin, rapamycin, and sodium selenite. These studies also revealed that the ceramide-LC3B association is regulated by ANXA2-ceramide interaction, which involves the E142 residue of ANXA2, and is required for the formation of the ANXA2-ceramide-LC3B complex that induces lethal mitophagy in OSCC cells and tumors in vitro and in vivo. Inhibition of the ANXA2-ceramide-LC3B complex formation by P. gingivalis FimA prevented ceramide-dependent mitophagy in response to LCL768, resulting in resistance to therapy in OSCC cells, xenograft-derived OSCC tumors in NUDE mice, and allograft-derived orthotopic oral tumors in C57BL/6 mice. Thus, these data provide a mechanism describing the pro-survival roles of P. gingivalis in oral tumors by inhibiting ceramide-dependent lethal mitophagy.
Increased abundance of P. gingivalis has been detected in OSCC tumor tissues in human patients, which are associated with poor clinical outcomes.22,24 However, the roles and mechanisms of how P. gingivalis infection exhibits pro-survival effects in OSCC cells or tumors, particularly in response to chemotherapeutic treatment, remains an ongoing area of study. Our data are consistent with these clinical observations about the pro-survival/pro-proliferative roles of P. gingivalis in OSCCs and provide an understanding of how P. gingivalis targets the ANXA2-ceramide-LC3B complex through bacterial FimA-ANXA2 association, resulting in therapeutic resistance via inhibiting ceramide-dependent mitophagy in response to LCL768 (Figures S5A and S5B). The effects of P. gingivalis infection on evading cellular detection and autophagic lysosomal degradation in human primary gingival epithelial and other cell types have been shown previously, modulating inflammation in culture and in mouse models.25,44,45 However, the roles of P. gingivalis in mitophagy regulation have not been described previously. Our data showed that the association between P. gingivalis and LC3B appeared to play a key role in targeting and inhibiting ceramide-LC3B binding, which involves the F52 residue of LC3B,28 known to recruit autophagosomes to mitochondria for degradation in cancer cells. This work provides previously unknown details of the ceramide-LC3B complex, which includes ANXA2,46 as decreased ANXA2-ceramide association by P. gingivalis or shRNA-mediated knockdown prevented ceramide-dependent mitophagy. Our data also showed that bacterial FimA protein47,48 is key to targeting and inhibiting the formation of ANXA2-ceramide-LC3B by FimA-ANXA2 interaction in response to P. gingivalis infection in OSCC cells. FimA is a unique type V fimbria distinct to P. gingivalis, which is used in biofilm formation, adhesion to host molecules, host cell invasion, and enhancing proliferation in human primary epithelial cells.49,50,51 Our findings add to the importance of FimA, and loss of FimA ablated the association of P. gingivalis with LC3B or ANXA2 in response to LCL768, thereby restoring ceramide-dependent mitophagy in OSCC cells. Although the roles of P. gingivalis FimA in the regulation of resistance to lethal mitophagy in OSCC cells or tumors have not been previously reported, our data suggest that targeting FimA or pili formation in P. gingivalis might improve therapeutic responses in OSCC patients with concomitant infection. This therapeutic approach, however, needs to be explored in future studies.
Our data also showed that ANXA2 is a key component of the ceramide-LC3B complex that recruits autophagosomes to mitochondria to induce hyper/lethal mitophagy in OSCC. Interestingly, high ANXA2 expression is reported in various human tumors, particularly OSCC.52,53,54,55,56 ANXA2 is a multi-functional protein that is involved in lipid binding, as well as endo- or exocytosis regulation. It seems that knockdown of ANXA2 mimics P. gingivalis infection to inhibit ceramide-dependent lethal mitophagy by attenuating the formation of the LC3B-ceramide complex. However, it remains unknown whether ANXA2 is involved in ceramide trafficking from the inner to outer leaflet of the mitochondrial membrane, or from the endoplasmic reticulum (ER) to mitochondria. It has been previously reported that ANXA2 is able to bind and extract lipids from membranes,42,57,58,59,60 which suggest that ANXA2 might also be able to induce ceramide trafficking from ER to mitochondria, then triggering initial mitochondrial ceramide signaling. Mitochondrial accumulation of ceramide is known to induce the generation of reactive oxygen species that in turn activates Drp1 and CerS1-p17/PERMIT trafficking to induce hyper-mitophagy,26 which then triggers lethal mitophagy.26,28,61,62,63,64,65,66,67 These ideas, however, need to be tested in more detailed studies.
It has been shown previously that P. gingivalis produces unusual sphingolipids, like phosphoglycerol dihydroceramides and phosphoethanolamine dihydroceramides, which alter alveolar bone homeostasis at periodontal disease sites.68 In this study, although we did not measure P. gingivalis-generated phosphoglycerol dihydroceramides or phosphoethanolamine dihydroceramides after infection of OSCC cells, P. gingivalis infection prevented ceramide-dependent mitophagy in both xenograft-derived human OSCC tumors and allograft-derived murine orthotopic OSCC tumors in response to LCL768 in vivo. Further, our 16S rRNA sequencing data showed an increased presence of Gordonia,69 Stenotrophomonas,70,71,72,73 and Herbaspirillum74,75 in P. gingivalis-infected tumors that showed resistance to LCL768-induced mitophagy in in vivo orthotopic OSCC tumors. Whether increased levels of these bacteria in the tumor macroenvironment play any independent or synergistic roles in the modulation of lethal mitophagy in response to LCL768 in OSCC tumors is unknown.
Limitations of the study
There are some limitations in our studies. For example, whether inhibition of FimA or pili formation using specific antibodies or small molecules (such as pilicides)76 improves therapeutic response in P. gingivalis-infected OSCC tumors needs to be determined. Nevertheless, this study supports that LCL768 induces ceramide-dependent lethal mitophagy and inhibits OSCC tumor growth/proliferation in vivo. In this study, we only utilized the 33277 strain (a type strain), which has been one of the most characterized strains for the host pathogenesis of P. gingivalis.48,56 Therefore, future studies that use low-passage clinical strains might be warranted to reach deeper conclusions. Nevertheless, our study provides a mechanism by which P. gingivalis infection results in resistance to ceramide-dependent mitophagy by targeting the ANXA2-ceramide-LC3B complex via bacterial FimA-ANXA2 association. These data also suggest mechanism-based therapeutic strategies to inhibit FimA function that could potentially improve the therapeutic responses of P. gingivalis-infected oral cancers regardless of their HPV status.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Tom20 | Santa Cruz Biotechnology | Cat#sc-17764; RRID: AB_628381 |
| Rabbit anti-Tom20 | Cell Signaling Technology | Cat#42406S; RRID: AB_2687663 |
| Rabbit anti-LC3 | Cell Signaling Technology | Cat#2775S; RRID: AB_915950 |
| Rabbit anti-LC3 | Novus Biologicals | Cat# NB100-2220 |
| Mouse anti-LAMP1 | Santa Cruz Biotechnology | Cat#SC20011; RRID: AB_626853 |
| Rabbit anti-PDI | Cell Signaling Technology | Cat#3501S; RRID: AB_2156433 |
| Rabbit anti-GM130 | Cell Signaling Technology | Cat#12480S; RRID: AB_2797933 |
| Mouse anti-GAPDH | Millipore Sigma | Cat# MAB374; RRID: AB_2107445 |
| Rabbit anti-CerS1 | Novus Biologicals | Cat# NBP1-59733 |
| Mouse anti-Ceramide | Enzo Life Sciences | Cat# ALX-804-196-T050; RRID: AB_2051116 |
| Mouse anti-S100A10 | Invitrogen | Cat# MA5-24769 |
| Rabbit anti-P-Y-1000 MultiMab | Cell Signaling Technology | Cat#8954S; RRID: AB_2687925 |
| Mouse anti-Tim23 | ThermoFisher | Cat#67535-1-IG |
| Rabbit anti-ANXA2 | ProteinTech | Cat#11256-1-AP; RRID: AB_2057311 |
| Mouse anti-ANXA2 | ProteinTech | Cat#60051-1-IG; RRID: AB_2057309 |
| Mouse anti-V5-tag | Invitrogen | Cat#37-7500 |
| Rabbit anti-V5-tag | Cell Signaling Technology | Cat#13202; RRID: AB_2687461 |
| Rabbit anti-DYKDDDDK (FLAG)-tag | Cell Signaling Technology | Cat#14793S; RRID: AB_2572291 |
| Mouse anti-DYKDDDDK (FLAG)-tag | Cell Signaling Technology | Cat#8146S; RRID: AB_10950495 |
| Rabbit anti-Actin | Sigma-Aldrich | Cat# A2066; RRID: AB_476693 |
| Mouse Normal IgG | Millipore Sigma | Cat#12–371; RRID: AB_145840 |
| Rabbit Normal IgG | Millipore Sigma | Cat#12–370; RRID: AB_145841 |
| Rabbit/Mouse Anti-Porphyromonas gingivalis | Dr. Özlem Yilmaz lab | Lee et al.6,25 |
| Rabbit anti-FimA | Dr. Özlem Yilmaz lab | Yilmaz et al.13 |
| Rabbit anti-Nucleoside diphosphate kinase (NDK) | Dr. Özlem Yilmaz lab | Atanasova et al.17 |
| Rabbit HRP-conjugated secondary antibody | Cell Signaling Technology | Cat#7074; RRID: AB 2099233 |
| Bacterial and virus strains | ||
| Porphyromonas gingivalis 33277 strain | ATCC | Cat#33277 |
| Porphyromonas gingivalis ΔfimA mutant of 33277 strain | Dr. Özlem Yilmaz lab | Yilmaz et al.13 |
| Porphyromonas gingivalis Δndk mutant and Δndk- complemented 33277 strains | Dr. Özlem Yilmaz lab | Yilmaz et al.13 |
| Chemicals, peptides, and recombinant proteins | ||
| LCL768 | Laboratory of Zdzislaw Szulc | N/A |
| Rapamycin | Sigma-Aldrich | Cat#R0395 |
| Sodium Selenite | Thermo Fisher Scientific | Cat#18-613-846 |
| Ethanol absolute (200 proof) | Koptec | Cat#64-17-5 |
| DMEM | Cytiva | Cat# SH30243.01 |
| PBS pH 7.4 (1x) | Gibco | Cat# 10010-023 |
| Fetal Bovine Serum | Avantor | Cat # 97068-085 |
| MEM Non-essential Amino Acids (100X) | Gibco | Cat#11140050 |
| Penicillin-Streptomycin (100X) | Corning | Cat # 30-002-CI |
| PlasmocinTM Prophylactic | InvivoGen | Cat# ant-mpp |
| 0.05% trypsin-0.02% EDTA | Corning | Cat# 25-052-CI |
| Trypan Blue solution | MilliporeSigma | Cat# T8154 |
| MTT Cell Proliferation Assay | ATCC | 30-1010K |
| 4% paraformaldehyde (PFA) | Boster | Cat# AR1068 |
| G418 | InvivoGen | Cat#ant-gn-1 |
| Puromycin | InvivoGen | Cat#ant-pr-1 |
| Polyethylenimine (PEI, 1 mg/mL) | Laboratory of Besim Ogretmen | N/A |
| Effectene transfection reagent | Qiagen | Cat#301425 |
| Polybrene | Sigma-Aldrich | Cat#TR-1003-G |
| 4X Laemmli Dye | Laboratory of Besim Ogretmen | N/A |
| Pierce RIPA buffer | Thermo Fisher Scientific | Cat# 89900 |
| Pierce IP Lysis Buffer | Thermo Fisher Scientific | Cat# 87787 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# P8340 |
| HALT Phosphatase Inhibitor | ThermoFisher Scientific | Cat# 78428 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | Thermo Fisher Scientific | Cat# 34580 |
| Protein Assay Dye Reagent | Bio-Rad | Cat#5000006 |
| Precision Plus Protein Dual Color Standard | Bio-Rad | Cat#1610374 |
| Magnetic SureBeads protein A or G | Bio-Rad | Cat#1614833 |
| Polyvinylidene Difluoride (PVDF) Membrane | Cytivia | Cat#10600021 |
| 1X Tris-Glycine Transfer Buffer | Novex | Cat#LC3675 |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat#A7906-500G |
| Saponin | Thermo Fisher Scientific | Cat#A18820.14 |
| Pierce Silver Stain Kit | Thermo Fisher Scientific | Cat#24612 |
| Bio-Safe Coomassie Brilliant Blue | Bio-Rad | Cat #1610786 |
| Duolink in situ hybridization | Sigma-Aldrich | Cat#DUO92101 |
| Antigen Unmasking Solution, Citrate-Based (pH 6.0) | Vector Laboratories | Cat# H-3300 |
| Vectashield Antifade Mounting Media with DAPI | Vector Laboratories | Cat#H-1200-10 |
| Duolink In Situ PLA Probe | Millipore Sigma | Cat# DUO92002 |
| MtPhagy Dye | Dojindo | Cat#MT02 |
| LysoTracker Green Dye | Thermo Fisher Scientific | Cat#L7526 |
| QuikChange XL Site-Directed Mutagenesis kit | Agilent Technologies | Cat#200516 |
| Sulfamethoxazole | Thermo Fisher | Cat#S036125G |
| Trimethoprim | Thermo Fisher | Cat#AC455120050 Cat#C5678-500G |
| Carboxymethylcellulose (CMC) | MilliporeSigma | N/A |
| Human recombinant ANXA2 protein | R&D Systems | Cat#9409-AN-050 |
| Porphyromonas gingivalis recombinant FimA protein | Laboratory of Dr. Özlem Yilmaz | Yilmaz et al.13 |
| Porphyromonas gingivalis recombinant NDK protein | Laboratory of Dr. Özlem Yilmaz | Atanasova et al.17 |
| Pierce Protein Transfection Reagent | Thermo Fisher Scientific | Cat#89850 |
| DPBS | Thermo Fisher Scientific | Cat#28344 |
| Critical commercial assays | ||
| Mitochondrial Isolation Kit for Cultured Cells | Thermo Fisher Scientific | Cat#89874 |
| DNeasy Blood and Tissue Kit | Qiagen | Cat#69506 |
| Deposited data | ||
| Original, uncropped western blot image files | ||
| Proteomics data | ProteomeXchange | PXD051568 |
| Experimental models: Cell lines | ||
| Human: UMSCC1A | Millipore Sigma | Cat#SCC070 |
| Human: UMSCC22A | Millipore Sigma | Cat#SCC076 |
| Human: 293T | ATCC | Cat# CRL-3216 |
| Mouse: MOC2 | Kerafast | Cat# EWL002-FP |
| Human: UMSCC47 | Millipore Sigma | Cat#SCC071 |
| Human: UMSCC104 | Millipore Sigma | Cat#SCC072 |
| Human: SCC152 | ATCC | Cat# CRL-3240 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Strain#000664; RRID:IMSR_JAX:000664 |
| Mouse: Nu/J | The Jackson Laboratory | Strain#002019; RRID:IMSR_JAX:002019 |
| Oligonucleotides | ||
| Human E142A ANXA2 Forward Primer GAAGACACCTGCTGCGTATGACGC |
Integrated DNA Technologies, IDT | N/A |
| Human E142A ANXA2 Reverse Primer GCTCAGAAGCGTCATACGCAGCAGG |
Integrated DNA Technologies, IDT | N/A |
| Human K49A LC3 Forward Primer GCTGCCCGTCCTGGACGCGACCAAGTTTTTGTTC |
Laboratory of Dr. Besim Ogretmen | N/A |
| Human K49A LC3 Reverse Primer CCGGGACCAAAAACTTGGTCGCGTCCAGGACGGG |
Laboratory of Dr. Besim Ogretmen | N/A |
| Human F52A LC3 Primers | Laboratory of Dr. Besim Ogretmen | Dany et al.27 |
| Human G120A LC3 Primers | Laboratory of Dr. Besim Ogretmen | Dany et al.27 |
|
P. gingivalis 16S rRNA FISH Probe 5′-CAATACTCGTATCGCCCGTTATTC-3′ |
Laboratory of Dr. Özlem Yilmaz | Lee et al.6 |
| Recombinant DNA | ||
| ANXA2 Human shRNA | Millipore Sigma | TRCN0000056145 (Clone ID), 302 (Gene ID), NM_001002857 (RefSeq), pLKO.1 vector |
| LC3 Human shRNA | Laboratory of Dr. Besim Ogretmen | Dany et al.27 |
| CerS1 Human shRNA | Laboratory of Dr. Besim Ogretmen | Olenik et al.26 |
| psPAX2 Lentiviral Packaging Plasmid | Laboratory of Dr. Besim Ogretmen | Addgene plasmid #12260, http://n2t.net/addgene:12260, RRID:12260 |
| pMD2.G VSV-G Envelope Expressing Plasmid | Laboratory of Dr. Besim Ogretmen | Addgene plasmid #12259, http://n2t.net/addgene:12259, RRID:12259 |
| pHAGE-mt-mKeima | Laboratory of Dr. Richard Youle | Addgene plasmid #131626, http://n2t.net/addgene:131626, RRID:131626 |
| Software and algorithms | ||
| ImageJ Version 1.54days | NIH Image | https://imagej.net/ij/index.html |
| Fiji | NIH Image | http://imagej.net/Fiji |
| Prism 8.0.1 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| BioRender | BioRender | https://www.biorender.com/ |
| Duolink Image Tool Software | Olink Bioscience | N/A |
| FV10i | Olympus Corp. | FV10-ASW http://www.grapecity.com |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Besim Ogretmen (ogretmen@musc.edu). The reagents solely relating to P. gingivalis should be directed to the co-corresponding author, Özlem Yilmaz (yilmaz@musc.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
-
•
Original, uncropped western blot image files have been deposited at Mendeley Data, and they will be publicly available at: $https://data.mendeley.com/datasets/zbppjnyyrr/2.
-
•
Original 16S rDNA files have been deposited in the NCBI Sequence Read Archive (SRA) and are publicly available as of the date of publication at Submission ID SUB14165627, BioProject ID PRJNA1067697, BioSample accessions SAMN39543253, SAMN39543254, SAMN39543255, SAMN39543256.
-
•
Original proteomics data are available via ProteomeXchange with the identifier PXD051568.
-
•
This paper does not report the original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Animals
Six-to eight-week-old female C57BL/6 mice or Nu/J mice were purchased from the Jackson Laboratory and used in this study. The animal procedures described below were approved by the Institutional Animal Care and Use Committee (IACUC) at the Medical University of South Carolina and conducted following accepted veterinary and regulatory standards.
Cell lines and culture conditions
UMSCC1A (HNSCC cell line isolated from a tumor located on the floor of the mouth of a male patient), 293T (female human fetal kidney cell line), and MOC2 (female C57BL/6 mouse oral cavity squamous cell carcinoma cell line) cell lines were cultured in DMEM (Cytiva, high glucose) with 10% FBS (Avantor Seradigm Premium Grade, USA) and 1% penicillin/streptomycin (100X, Corning). Human papillomavirus-positive cells, UMSCC47, were cultured in DMEM (Cytiva, high glucose) with 10% FBS, 1% penicillin/streptomycin, and 1% non-essential amino acids (100X, Gibco). Cell lines were grown at 37°C and 5% CO2. All cell lines were authenticated and tested for mycoplasma contamination periodically in Ogretmen and Yilmaz laboratories.
P. gingivalis bacterial growth conditions
Wildtype P. gingivalis (ATCC 33277) or an isogenic fimA deletion mutant (ΔfimA) strain was grown to mid-log phase in Trypticase soy broth (TSB) supplemented with yeast extract (1 μg/mL), menadione (1 μg/mL) and hemin (5 μg/mL), at 37°C under anaerobic conditions and the number of bacteria was determined by using a Klett–Summerson photometer as described previously. Erythromycin (10 μg/mL) was added to the media for culture of the mutant strains (ΔfimA or Δndk), and the media of the Δndk-complemented strain was supplemented with erythromycin (10 μg/mL), and tetracycline (3 μg/mL).5,13,17,39,51
Method details
P. gingivalis in vitro infection
Target cells were grown in antibiotic-free DMEM media with 10% FBS at 37°C and 5% CO2. Cells were then infected with wildtype or ΔfimA P. gingivalis strains at 100 MOI for 6 h (unless otherwise specified). Subsequently, cells were treated with 30 μM C18-ceramide analog LCL768 or 0.2% ethanol vehicle control for 2 h.
Cell lysis
Cells were scraped or trypsinized (0.5% trypsin, 0.53 mM EDTA, Corning), collected, and spun down at 1,300 rpm for 5 min at room temperature. The supernatant was removed, and cell pellets were washed with 1X PBS, then centrifuged again at the same speed and temperature. The supernatant was again discarded, and cells were resuspended in IP Lysis buffer (Pierce #87788) or RIPA buffer containing 1:1000 phosphatase inhibitor (ThermoFisher Scientific Halt Phosphatase Inhibitor #78428) and 1:1000 protease inhibitor (Sigma-Aldrich Protease Inhibitor Cocktail #P8340). Cells were mechanically lysed via 28-gauge insulin syringe five times and incubated on ice for 20 min. Lysed cells were then centrifuged at 13,000xg for 15 min at 4°C. The cell lysis supernatant was saved, and pellet was discarded. A bicinchoninic acid assay (BCA) was used to determine the protein content of the cell lysate for downstream applications. Cell lysate was stored at −80°C.
Bicinchoninic acid assay (BCA)
Protein standards were prepared using bovine serum albumin (BSA) and protein assay dye reagent concentration (Bio-Rad #5000006). Cell lysate protein content was determined by diluting 5 μL cell lysate in 795 μL deionized MilliQ water and 200 μL Bio-Rad dye reagent (#5000006). Standards and samples were aliquoted in a 96-well plate, and absorbance was measured at 630 nm on the Biotech Synergy HT Microplate Reader.
Trypan blue cell viability assay
Cells were grown in 6-well plates at 37°C and 5% CO2 and treated with drug or vehicle control before being lifted with a cell scraper. Cells were collected in 15 mL tubes, and wells were washed with 1X PBS. Cells were centrifuged at 1,300 rpm for 5 min before removing as much media as possible and resuspending in 100–200 μL 1X PBS. Cells were then counted using trypan blue (1:1 dilution) on the TC20 Automated Cell Counter (Bio-Rad) or using a hemacytometer.
Immunoprecipitation
Protein A or Protein G SureBeads (for rabbit or mouse antibodies, respectively, Bio-Rad #161–4013, #161–4023) were washed with 1X PBST at room temperature, three times using a Bio-Rad magnetic rack (#161–4916). 2–10 μg antibody was added to the beads in 500 μL 1X PBST and agitated for 1 h at 4°C. A species-matched IgG control bead duplicate was prepared for each sample. Beads were washed three times at room temperature with 1X PBST to remove the primary antibody. 150–250 μg cell lysate was added to both target antibody beads and 1gG control beads, and samples were agitated overnight at 4°C. Beads were transferred to a new 1.5 mL Eppendorf and washed three times with 1X PBST. Finally, immunoprecipitated proteins were eluted from the beads using 1X Laemmli buffer and boiling at 100°C for 3 min. Cell lysate not used for immunoprecipitation was utilized as the input control for target proteins.77
Western blotting
Cell lysate (15–30 μg protein) was diluted in 4X Laemmli dye and boiled for 3 min at 100°C. Samples were run on a 4–20% polyacrylamide gradient gel with an adjacent ladder (Bio-Rad Precision Plus Protein Dual Color Standard # 1610374) in 1X SDS Running Buffer. Samples were run at 170 V for about 30 min, and the gel was transferred onto a polyvinylidene difluoride (PVDF) membrane (Cytiva #10600021) at 25 V (2.0 A) for 20 min in 1X Tris-Glycine Transfer Buffer (Novex #LC3675). Blots were then blocked with 5% BSA in 1X PBST for 1 h at room temperature. Primary antibody (1:1000) was added to the blot in 5% BSA, 1X PBST agitating overnight at 4°C. Western blots were then washed 3 times with 1X PBST at 37°C, and a secondary antibody was added (1:10,000) in 5% BSA, 1X PBST at room temperature. Finally, blots were washed again 3 times with 1X PBST and exposed to ECL solution (ThermoFisher SuperSignal West Pico Plus Chemiluminescent Substrate #34580) for imaging on the Bio-Rad ChemiDoc Imager.
Far western overlay analysis
Five Micrograms (5 μg) of recombinant human ANXA2 (R&D Systems, #9409-AN-050) was resolved by SDS-PAGE on 4–20% gradient polyacrylamide gels (Bio-Rad, #4568094) as previously described in Far Western methodologies.78,79 The SDS-PAGE resolved protein samples were then transferred to a 0.22 μm PVDF membrane and were blocked overnight in 5% non-fat milk. The blots were washed and incubated with 10 μg of recombinant FimA13 for 1 h. PVDF membranes were probed for FimA binding to ANXA2 using a rabbit polyclonal antibody specific to FimA (1:500)13,80 followed by an anti-rabbit HRP-conjugated secondary antibody (Cell Signaling Technology, #7074; 1:2000). Protein bands were viewed via enhanced chemiluminescence (ECL). Control western blots were performed to examine ANXA2 presence and serve as loading controls using rabbit anti-ANXA2 antibody (#11256-1-AP, ProteinTech; 1:1000). To reconfirm the specificity of the interactions between FimA and ANXA2 and serve as a negative control, ANXA2 blots were also incubated with 10 μg of recombinant P. gingivalis Nucleoside diphosphate kinase (NDK), which were then probed with a rabbit polyclonal Anti-NDK antibody (1:500).10,17 Anti-rabbit HRP-conjugated secondary antibody (Cell Signaling Technology, 7076; 1:2000) was then utilized to confirm that there were no interactions between NDK and ANXA2. Antibody cross-reactivity was accounted for via probing blots loaded with recombinant ANXA2 with rabbit polyclonal Anti-FimA antibody (1:500). Far Western blots were performed at least in duplicate.
Immunofluorescence
Cells were grown in 4 well slides and treated at 37°C and 5% CO2. Cells transiently expressing or knocking down protein were grown first in 60 mm plates and then transferred to 4 well slides (to prevent nonspecific DAPI staining of condensed plasmid with polyethyleneimine in media). Once treatment was concluded, cells were washed once with 1X PBS at room temperature and fixed with 4% paraformaldehyde for 15 min. Wells were washed once with 1X PBS and then blocked for 1 h with 5% BSA and 0.1% saponin in 1X PBS at room temperature. Primary antibodies (1:100) were added in 5% BSA, 0.1% saponin solution agitating overnight at 4°C. Wells were washed once again with 1X PBS, and secondary Alexa Fluor antibodies were added in 5% BSA and 0.1% saponin (1X PBS) at room temperature for 1 h. Finally, the wells were washed with 1X PBS once, and the well separators were removed. 8–10 μL mounting media containing DAPI (Vectashield Antifade Mounting Media with DAPI #H-1200-10) was added to each well, and slides were stored overnight in the dark (room temperature) for imaging on the Olympus FV10i (Cell & Molecular Imaging Shared Resource, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313).
Mtphagy live-cell staining
Cells were grown at 37°C and 5% CO2 in 35 mm MatTek glass-bottom petri dishes before treating with a vehicle or drug for 2 h. Mtphagy dye (mitochondria) was added to cell media according to the manufacturer’s instructions (Dojindo #MT02 Mtphagy Dye) prior to imaging on the Olympus Fv10i. Lysotracker green dye (lysosome, ThermoFisher Scientific #L7526) was optionally added to show colocalization between mitochondria and lysosomes in live cells.
ImageJ quantification
ImageJ was utilized to quantify western blots and immunofluorescence colocalization. Normalization was done based on loading control for western blots (Actin or GAPDH) or immunoprecipitated protein (ex. Tom20). Normalization for immunofluorescence colocalization (Pearson’s colocalization coefficient) was done based on untreated, uninfected controls.
Proximity ligation assay (PLA)
Cells were grown in 4-well slides at 37°C and 5% CO2 before being treated with a vehicle or drug and fixed with 4% paraformaldehyde (PFA). The proximity ligation assay (Millipore Sigma Duolink in situ PLA Probe # DUO92002) was performed as per the manufacturer’s instructions.77
Antibodies
The antibodies used in this study are as follows: rabbit Tom20 (42406S, Cell Signaling Technology), mouse Tom20 (sc-17764, Santa Cruz Biotechnology), rabbit LC3 (2775S, Cell Signaling Technology), mouse LAMP1 (SC20011, Santa Cruz Biotechnology), rabbit PDI (3501S, Cell Signaling Technology), rabbit GM130 (12480S, Cell Signaling Technology), mouse GAPDH (MAB374, Millipore Sigma), rabbit CerS1 (NBP1-59733, Novus Biologicals), ceramide (ALX-804-196-T050, Enzo Life Sciences), mouse S100A10 (MA5-24769, Invitrogen), rabbit P-Y-1000 MultiMab (8954S, Cell Signaling Technology), mouse Tim23 (67535-1-IG, ThermoFisher), rabbit ANXA2 (11256-1-AP, ProteinTech), mouse ANXA2 (60051-1-IG, ProteinTech), mouse V5-tag (37–7500, Invitrogen), rabbit V5-tag (13202, Cell Signaling Technology), rabbit FLAG tag (14793S, Cell Signaling Technology), mouse FLAG tag (8146S, Cell Signaling Technology), rabbit Actin (A2066, Sigma-Aldrich). The following antibodies were used for immunoprecipitation (5–10 μg): mouse Normal IgG (12–371, Millipore Sigma), rabbit Normal IgG (12–370, Millipore Sigma), rabbit LC3 (NB100-2220, Novus Biologicals), mouse Tom20 (sc-17764, Santa Cruz Biotechnology). Polyclonal antibodies targeted at P. gingivalis (ATCC 33277), NDK, and FimA, along with recombinant FimA and NDK proteins, were provided, as previously described by Dr. Özlem Yilmaz’s lab.13,17,25 Secondary Alexa Fluor antibodies used in immunofluorescence were provided by Jackson ImmunoResearch.
Plasmids and shRNA
Wildtype FLAG-LC3, V5-F52A LC3,27 V5-G120A LC3,27 and FLAG-K49A LC3 pcDNA3.1+ were generated in the Ogretmen lab. Wildtype V5-ANXA2 pcDNA3.1+ was prepared by GenScript (#U5568EI030), and site-directed mutagenesis (Agilent Technologies, #200516) was conducted to produce V5-E142A ANXA2 pcDNA3.1+ plasmid (Forward: 5′- GAAGACACCTGCTGCGTATGACGC-3′, Reverse: 5′- GCTCAGAAGCGTCATACGCAGCAGG-3′). pcDNA3.1+ empty (Addgene plasmid #V790-20), pLenti empty (Addgene plasmid # 39481), and pLKO.1 empty (Addgene plasmid #10878) were used as controls where appropriate. psPAX2 (Addgene plasmid #12260) and pMD2.G (Addgene plasmid #12259) were gifts from Dr. Bob Weinberg. MtKeima fluorescence was analyzed in cells via transient transfection with pHAGE-MtKeima plasmid (Addgene plasmid #131626). ANXA2 shRNA (TRCN0000056145) was ordered from the MUSC shRNA Technology Shared Resource. LC3 shRNA27 and CerS1 shRNA26 were provided by the Ogretmen lab. Confirmational sequencing of control and mutant plasmids was completed by Genewiz.
Mass spectrometry-based proteomics
Mass spectrometry analysis of samples was completed by the Mass Spectrometry Facility at the MUSC Proteomics Center. Lysate or immunoprecipitated was run on a 4–12% polyacrylamide gel and then stained with Coomassie blue dye according to the manufacturer’s instructions (Bio-Safe Coomassie Brilliant Blue #1610786). For better visualization of faint bands, silver staining was conducted following the manufacturer’s instructions (ThermoFisher Pierce Silver Stain Kit # 24612). Gel bands of interest were selected, excised, and placed in a microcentrifuge tube with ddH2O for overnight shipment to the core.26 The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE81 partner repository with the dataset identifier PXD051568.
Site-directed mutagenesis
Primers were designed based on the coding sequence of the canonical gene of interest. The coding sequence was converted into the amino acid codon sequence using Expasy for ease of mutation of the amino acid(s) of interest. About 15–20 amino acids upstream and downstream of the mutated codon were selected, and the New England Biolabs Tm Calculator was used to adjust the primer length, projected annealing temperature, and GC content. The Harvard Reverse Complement Tool was used to produce the reverse primer sequence. Primers were then ordered from Integrated DNA Technologies, including 5′-phosphorylation for plasmid ligation. The template plasmid (containing the wildtype gene of interest for mutation) and the primers were then used with the QuikChange XL Site-Directed Mutagenesis kit (Agilent Technologies, #200516) as per the manufacturer’s instructions. The successful mutation was confirmed via sequencing of the plasmid (Genewiz, Azenta Life Sciences).26
In vitro transient transfection
Cells at 50–60% confluency were washed with 1X PBS and incubated in serum-free (ThermoFisher Gibco Opti-MEM Reduced Serum Media # 31985070) media for at least 15 min at 37°C and 5% CO2. Concurrently, plasmid DNA was incubated for 15 min at room temperature with polyethyleneimine (PEI, 1 mg/mL stock) at a 1:4 ratio (0.5 μg DNA: 2 μg PEI) in serum-free media (Optimem). The DNA solution was then added directly to target cells and incubated for 6 h at 37°C and 5% CO2. After 6 h, cells were washed with 1X PBS, and normal growth media was added. Transiently transfected cells were collected or treated for downstream applications the following day.
In vitro lentiviral stable transfection
293T cells were grown to 50–70% confluency before adding target DNA, pCMV-psPAX2, and pMD2 plasmids with Effectene transfection reagent (Qiagen #301425) as per manufacturer’s instructions. Transfected 293T cells were incubated at 37°C and 5% CO2 for 48 h. The viral media supernatant was collected and centrifuged at 1,300 rpm for 5 min at room temperature. The media was then filtered through a 10 mL 10cc syringe with a 0.45 μm filter. Target cells (UMSCC1A) were grown to 50–80% confluency, and the filtered viral media was added directly to the cells with 8 μg/mL polybrene (Sigma-Aldrich #TR-1003-G) to enhance transduction efficiency. After 48 h at 37°C and 5% CO2, the viral media was replaced with normal growth media containing the selection drug, with selection media being replaced every other day. Knockdown or overexpression was validated via western blotting, and cryostocks were prepared as soon as cells approached 90% confluency.
In vitro protein transfection
Pierce protein transfection reagent (Thermo Scientific #89850) was used to transfect UM-SCC-1A cells with recombinant FimA (rFimA) protein described before following the manufacturer’s protocol. In brief, Pierce reagent was resuspended in 250 μL of methanol, and 2.5 μL was aliquoted into a microcentrifuge tube. Following evaporating the methanol under a laminar flow for 4 h, 1 μg of rFimA in a 20 μL DPBS (Thermo Scientific #28344) was added to hydrate the dried Pierce reagent and incubated at room temperature for 5 min. DPBS without rFimA was used as a control. Next, serum-free DMEM medium was added to bring the final delivery volume up to 250 μL. UM-SCC-1A cells (1.5 × 105 cells/well) pre-seeded in 4-well chamber slides were washed once with DPBS and once with serum-free DMEM medium, followed by transferring the delivery mix onto the cells. After incubating the cells for 4 h at 37°C, 250 μL of DMEM medium with 20% FBS was added and incubated for an additional 20 h before treatment with LCL768 or vehicle control.
Clonogenic assay
In vitro clonogenic assay was conducted to detect the proliferation of UMSCC1A cells able to form colonies with at least 50 cells (minimum 6 cell divisions) following pretreatment with/without P. gingivalis infection (100 MOI, 6 h) and subsequent treatment with LCL768 (15 μM, 3 h) or vehicle control. Cells were incubated at 37°C and 5% CO2 prior to infection and treatment, as previously described. Cells were then collected via scraping, and single cells were isolated through trypsinization (0.05% trypsin, 0.53 mM EDTA) and repeated pipetting. Single-cell suspensions were then added to 6 well plates (3 biological replicates) and allowed to grow at 37°C and 5% CO2 for about 2 weeks. Cell colonies were then counted and normalized to colonies formed from uninfected, untreated cells.
Establishment of orthotopic oral squamous cell carcinoma tumors in C57BL/6 female mice
Female C57BL/6 mice (6–8 weeks old) were purchased from the Jackson Laboratory. The orthotopic base of mouth/tongue tumors were induced by injecting 3,000 MOC2 cells resuspended in 50 μL 1X PBS intramuscularly into the mylohyoid, allowing at least 2.5 weeks for the tumors to develop. Mice were treated with 10 mg/kg LCL768 in 100 μL 1X PBS (or 100 μL 1X PBS vehicle only, maximum tolerated dose, n = 10) intraperitoneally every other day for the duration of the 2.5-week treatment period. For P. gingivalis infected and uninfected animals, the mouse resident oral microbiota was removed with 700 μg/mL sulfamethoxazole and 400 μg/mL trimethoprim for 3 days in drinking water prior to LCL768/vehicle intraperitoneal treatment. Oral feeding of 109 colony forming unit (CFU) wildtype P. gingivalis (ATCC 33277) strain in 100 μL of 2% carboxymethylcellulose (CMC) in PBS was done four times at 2-day intervals, concurrently with LCL768/vehicle treatment. To prevent washout or ingestion of inoculated bacteria, food and water are removed for 2 h prior and following infection. After 2.5 weeks of treatment or if body weight and conditions indicated earlier endpoint, mice were sacrificed, and blood and tumors were collected for downstream applications.77
16S sequencing of bacterial species in mouse orthotopic OSCC tumor tissue
Resected mouse orthotopic OSCC tumors were homogenized mechanically in lysis buffer (Pierce #87787), flash frozen, and tissue DNA was isolated (Qiagen #69506) as per manufacturer’s instructions. Isolated tissue DNA was resuspended in nuclease-free water and shipped overnight to LCScience. Bacterial species were identified via sequencing of the prokaryotic 16S rDNA transcript using conserved V3 and V4 hypervariable region primers. Statistical analysis of results was completed by LCScience.82
Establishment of human oral squamous cell carcinoma flank tumors in female nude mice
Female Nu/J mice (6–8 weeks old) were purchased from the Jackson Laboratory. UMSCC1A cells stably expressing SCR, ANXA2, or LC3 shRNA were utilized. Five hundred thousand cells resuspended in 100 μL 1X PBS were injected subcutaneously into the midline flanks of nude mice (n = 16 mice per group: SCR, ANXA2KD, LC3KD), and tumors were incubated until they were at least 3 mm in diameter. Mice were treated intraperitoneally with 10 mg/kg LCL768 in 100 μL 1X PBS (or 1X PBS vehicle only) every other day for 2.5 weeks (n = 8 vehicle/LCL768). After 2.5 weeks of treatment or if body weight and conditions indicated earlier endpoint, mice were sacrificed, and blood and tumors were collected for downstream applications.77
Immunohistochemistry
Immunohistochemistry was performed on resected tumor samples fixed in formalin by embedding with paraffin and sectioning the paraffin block (5 μm slices). Embedding, sectioning, and Ki67 proliferation staining were done by the MUSC Histology and Immunohistochemistry Shared Resource. Formalin-fixed paraffin-embedded (FFPE) tumor slices were deparaffinized with xylene and rehydrated with decreasing concentrations of ethanol in water (Abcam IHC deparaffinization protocol). According to the manufacturer’s instructions, antigens were demasked using a citric-acid-based unmasking solution (Vector Laboratories # H-3300-250). Sectioned slides were blocked using 0.1% saponin and 5% BSA prepared in 1X PBS for 1 h at room temperature. Slides were washed with 1X PBS before adding 1:100 primary antibody in BSA solution (0.1% saponin, 5% BSA, 1X PBS), shaking overnight at 4°C in a humidity chamber. Slides were again washed with 1X PBS before adding 1:500 secondary Alexa Fluor antibody (Jackson ImmunoResearch) for 1 h at room temperature in the dark (in a humidity chamber). Slides were mounted, and nuclei were stained (Vectashield Antifade Mounting Media with DAPI #H-1200-10) before imaging on the Olympus FV10i (Cell & Molecular Imaging Shared Resource, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313).
Fluorescence in situ hybridization
Tumor tissue sections of orthotopic OSCC with/without infection with P. gingivalis wild-type strain were used to detect P. gingivalis by fluorescence in situ hybridization (FISH) assay following the previous method.6 In brief, tissue sections were mounted on microscope glass slides and deparaffinized, followed by blocking with 1% BSA and 0.1% Triton X-100 in PBS at RT for 30 min. The slides were then washed with distilled water and hybridized with 5nM of Pg-488 probe (P. gingivalis’ 16S rRNA-specific probe 5ʹ-CAATACTCGTATCGCCCGTTATTC-3ʹ labeled with Alexa Fluor 488 at the 5ʹ-end)6 for 4 h in the dark at 46°C. DAPI (1:500) was used to stain nuclear material for 5 min at 37°C. Finally, the slides were dehydrated, mounted with nonfluorescent immersion oil, and covered with coverslips for imaging. Slides were observed under the Olympus FV10i confocal microscope (Cell & Molecular Imaging Shared Resource, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313).
Synthesis of C18-ceramide analog drug LCL768
Selenium-C18-ceramide analog drug (LCL768) was produced by the synthetic unit of the Lipidomics Shared Resource (MUSC).
In silico molecular modeling
Molecular Operating Environment (MOE) and ClusPro 2.0 protein-protein docking programs were utilized by Dr. Yuri Peterson to model and refine protein interactions in silico. Both programs were used to compare projected protein-protein interactions and analyze the consistency of modeling between programs.28 Protein-protein and protein-lipid interaction predictions were done in a sequential fashion. We began with our previous model of C18-ceramide interaction with LC3 (Sentelle et al.28). This simulation used MOE docking to interact C18-ceramide with LC3 (PDB:1V49). This dimer model was turned into a monomer pdf file, and then the dimer interaction with ANXA2 (PDB:4HRE) was probed using ClusPro (i.e., LC3-C18-ceramide was protein docked to ANXA2) (PMCID: PMC5540229). A similar biomolecular docking simulation looked at ANXA2 interaction with FimA (PDB:6KMF). The top-scoring consensus pose (i.e., the site with the greatest number of poses using the balanced scoring method) was used for subsequent analysis. These models were then superposed using LC3 as the overlay to create the assembled LC3 (C18-ceramide)-ANXA2-FimA complex.
Quantification and statistical analysis
All data are presented as means ± SD (Standard Deviation) of at least three independent studies (n ≥ 3). Group comparisons were performed using Graph Pad Prism using either two-tailed unpaired t-tests (and nonparametric tests) or a one-way ANOVA (and nonparametric or mixed). In animal studies, the Kruskal-Wallis’s test was used to compare continuous outcomes among three groups due to relatively smaller sample sizes. Because a significant result was discovered, all possible pairwise comparisons were performed using the Wilcoxon rank-sum test. p < 0.05 (∗) was considered significant. Mouse studies were conducted with 6–8-week-old animals randomly assigned to each treatment group (n = 10–12). For clinical analyses, the significance was calculated by the Log rank test. For correlation analysis, Pearson correlation coefficients were calculated using GraphPad Prism Software, 8.0.1.26,77
Acknowledgments
We thank the members of the Ogretmen Laboratory for their technical support and helpful discussions. Funding: This work was supported by research funding from the National Institutes of Health (AG069769, CA214461, CA214461-S1, DE016572, DE016572-S1, and P01 CA203628 to B.O.), the National Institute of Dental and Craniofacial Research (NIDCR) grant R01DE030313 to Ö.Y., and the NIDCR T32 training grant (T32DE017551) to M.S. The core facilities utilized are supported by NIH (C06 RR015455), Hollings Cancer Center Support Grant (P30 CA138313), or Center of Biomedical Research Excellence (Cobre) in Lipidomics and Pathobiology (P30 GM103339). In addition, the Zeiss 880 microscope was funded by a Shared Instrumentation Grant (S10 OD018113).
Ethics Statement: This study was performed in accordance with the guidelines in the Guide for the Care and the Use of Laboratory Animals of the National Institutes of Health. All the animals were handled according to the approved Institutional Animal Care and Use Committee (IACUC) protocol (2018-00412) of the Medical University of South Carolina.
Author contributions
Conception and design: M.S., Ö.Y., and B.O.; Methodology development: M.S., N.C., B.W., N.O., M.F.K., S.P., H.G.L., M.E., Ö.Y., and B.O. Data acquisition: M.S., N.C., B.W., N.O., M.F.K., S.P., H.G.L., M.E., H.A., Z.M.S., Ö.Y., and B.O.; Data analysis and interpretation: M.S., N.C., M.E., Ö.Y., and B.O.; Manuscript writing, review, and revision: M.S., Ö.Y., and B.O.; Administrative, technical, or material support: M.S., N.C., M.E., Ö.Y., and B.O.; Study supervision: M.E., Ö.Y., and B.O.
Declaration of interests
B.O is the CEO and co-founder of Lipo-Immuno Tech, LLC, which has a licensing agreement for the commercialization and marketing of a ceramide analog drug LCL768. The rest of the authors declare no competing interests.
Published: April 30, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109860.
Contributor Information
Özlem Yilmaz, Email: yilmaz@musc.edu.
Besim Ogretmen, Email: ogremten@musc.edu.
Supplemental information
References
- 1.Knochelmann H.M., Horton J.D., Liu S., Armeson K., Kaczmar J.M., Wyatt M.M., Richardson M.S., Lomeli S.H., Xiong Y., Graboyes E.M., et al. Neoadjuvant presurgical PD-1 inhibition in oral cavity squamous cell carcinoma. Cell Rep. Med. 2021;2 doi: 10.1016/j.xcrm.2021.100426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Graillon N., Iocca O., Carey R.M., Benjamin K., Cannady S.B., Hartner L., Newman J.G., Rajasekaran K., Brant J.A., Shanti R.M. What has the National Cancer Database taught us about oral cavity squamous cell carcinoma? Int. J. Oral Maxillofac. Surg. 2022;51:10–17. doi: 10.1016/j.ijom.2021.03.015. [DOI] [PubMed] [Google Scholar]
- 3.Graboyes E.M., Gross J., Kallogjeri D., Piccirillo J.F., Al-Gilani M., Stadler M.E., Nussenbaum B. Association of Compliance With Process-Related Quality Metrics and Improved Survival in Oral Cavity Squamous Cell Carcinoma. JAMA Otolaryngol. Head Neck Surg. 2016;142:430–437. doi: 10.1001/jamaoto.2015.3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilardi M., Saddawi-Konefka R., Wu V.H., Lopez-Ramirez M.A., Wang Z., Soto F., Ramms D.J., Proietto M., Mikulski Z., Miki H., et al. Microneedle-mediated Intratumoral Delivery of Anti-CTLA-4 Promotes cDC1-dependent Eradication of Oral Squamous Cell Carcinoma with Limited irAEs. Mol. Cancer Ther. 2022;21:616–624. doi: 10.1158/1535-7163.MCT-21-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi C.H., Spooner R., DeGuzman J., Koutouzis T., Ojcius D.M., Yilmaz Ö. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cell Microbiol. 2013;15:961–976. doi: 10.1111/cmi.12089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee J.S., Spooner R., Chowdhury N., Pandey V., Wellslager B., Atanasova K.R., Evans Z., Yilmaz Ö. In Situ Intraepithelial Localizations of Opportunistic Pathogens, Porphyromonas gingivalis and Filifactor alocis, in Human Gingiva. Curr. Res. Microb. Sci. 2020;1:7–17. doi: 10.1016/j.crmicr.2020.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spooner R., Weigel K.M., Harrison P.L., Lee K., Cangelosi G.A., Yilmaz Ö. In Situ Anabolic Activity of Periodontal Pathogens Porphyromonas gingivalis and Filifactor alocis in Chronic Periodontitis. Sci. Rep. 2016;6 doi: 10.1038/srep33638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts J.S., Atanasova K.R., Lee J., Diamond G., Deguzman J., Hee Choi C., Yilmaz Ö. Opportunistic Pathogen Porphyromonas gingivalis Modulates Danger Signal ATP-Mediated Antibacterial NOX2 Pathways in Primary Epithelial Cells. Front. Cell. Infect. Microbiol. 2017;7:291. doi: 10.3389/fcimb.2017.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spooner R., DeGuzman J., Lee K.L., Yilmaz O. Danger signal adenosine via adenosine 2a receptor stimulates growth of Porphyromonas gingivalis in primary gingival epithelial cells. Mol. Oral Microbiol. 2014;29:67–78. doi: 10.1111/omi.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee J., Roberts J.S., Atanasova K.R., Chowdhury N., Yilmaz Ö. A novel kinase function of a nucleoside-diphosphate-kinase homologue in Porphyromonas gingivalis is critical in subversion of host cell apoptosis by targeting heat-shock protein 27. Cell Microbiol. 2018;20 doi: 10.1111/cmi.12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J., Roberts J.S., Atanasova K.R., Chowdhury N., Han K., Yilmaz Ö. Human Primary Epithelial Cells Acquire an Epithelial-Mesenchymal-Transition Phenotype during Long-Term Infection by the Oral Opportunistic Pathogen, Porphyromonas gingivalis. Front. Cell. Infect. Microbiol. 2017;7:493. doi: 10.3389/fcimb.2017.00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yilmaz O., Jungas T., Verbeke P., Ojcius D.M. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect. Immun. 2004;72:3743–3751. doi: 10.1128/IAI.72.7.3743-3751.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yilmaz O., Watanabe K., Lamont R.J. Involvement of integrins in fimbriae-mediated binding and invasion by Porphyromonas gingivalis. Cell Microbiol. 2002;4:305–314. doi: 10.1046/j.1462-5822.2002.00192.x. [DOI] [PubMed] [Google Scholar]
- 14.Yilmaz Ö., Young P.A., Lamont R.J., Kenny G.E. Gingival epithelial cell signalling and cytoskeletal responses to Porphyromonas gingivalis invasion. Microbiology (Read.) 2003;149:2417–2426. doi: 10.1099/mic.0.26483-0. [DOI] [PubMed] [Google Scholar]
- 15.Yamada C., Ho A., Nusbaum A., Xu R., Davey M.E., Nichols F., Mao C., Movila A. Inhibitory effect of Porphyromonas gingivalis-derived phosphoethanolamine dihydroceramide on acid ceramidase expression in oral squamous cells. J. Cell Mol. Med. 2023;27:1290–1295. doi: 10.1111/jcmm.17722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ha N.H., Park D.G., Woo B.H., Kim D.J., Choi J.I., Park B.S., Kim Y.D., Lee J.H., Park H.R. Porphyromonas gingivalis increases the invasiveness of oral cancer cells by upregulating IL-8 and MMPs. Cytokine. 2016;86:64–72. doi: 10.1016/j.cyto.2016.07.013. [DOI] [PubMed] [Google Scholar]
- 17.Atanasova K., Lee J., Roberts J., Lee K., Ojcius D.M., Yilmaz Ö. Nucleoside-Diphosphate-Kinase of P. gingivalis is Secreted from Epithelial Cells In the Absence of a Leader Sequence Through a Pannexin-1 Interactome. Sci. Rep. 2016;6 doi: 10.1038/srep37643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atanasova K.R., Yilmaz O. Looking in the Porphyromonas gingivalis cabinet of curiosities: the microbium, the host and cancer association. Mol. Oral Microbiol. 2014;29:55–66. doi: 10.1111/omi.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu J., Hu Y., Tang Z., Zhang C., Jin L., Gu M., Yang Y. Porphyromonas gingivalis lipopolysaccharide enhances the proliferation of human periodontal ligament cells via upregulation of cyclin D1, cyclin A and cyclin B1. Exp. Ther. Med. 2022;23:2. doi: 10.3892/etm.2021.10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen T.Y., Kuo P.J., Lin C.Y., Hung T.F., Chiu H.C., Chiang C.Y., Shih K.C., Fu E. Porphyromonas gingivalis lipopolysaccharide and gingival fibroblast augment MMP-9 expression of monocytic U937 cells through cyclophilin A. J. Periodontol. 2022;93:449–457. doi: 10.1002/JPER.19-0740. [DOI] [PubMed] [Google Scholar]
- 21.Gaddis D.E., Maynard C.L., Weaver C.T., Michalek S.M., Katz J. Role of TLR2-dependent IL-10 production in the inhibition of the initial IFN-gamma T cell response to Porphyromonas gingivalis. J. Leukoc. Biol. 2013;93:21–31. doi: 10.1189/jlb.0512220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katz J., Onate M.D., Pauley K.M., Bhattacharyya I., Cha S. Presence of Porphyromonas gingivalis in gingival squamous cell carcinoma. Int. J. Oral Sci. 2011;3:209–215. doi: 10.4248/IJOS11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park D.G., Woo B.H., Lee B.J., Yoon S., Cho Y., Kim Y.D., Park H.R., Song J.M. Serum Levels of Interleukin-6 and Titers of Antibodies Against Porphyromonas gingivalis Could Be Potential Biomarkers for the Diagnosis of Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20112749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahn J., Segers S., Hayes R.B. Periodontal disease, Porphyromonas gingivalis serum antibody levels and orodigestive cancer mortality. Carcinogenesis. 2012;33:1055–1058. doi: 10.1093/carcin/bgs112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee K., Roberts J.S., Choi C.H., Atanasova K.R., Yilmaz Ö. Porphyromonas gingivalis traffics into endoplasmic reticulum-rich-autophagosomes for successful survival in human gingival epithelial cells. Virulence. 2018;9:845–859. doi: 10.1080/21505594.2018.1454171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oleinik N., Kim J., Roth B.M., Selvam S.P., Gooz M., Johnson R.H., Lemasters J.J., Ogretmen B. Mitochondrial protein import is regulated by p17/PERMIT to mediate lipid metabolism and cellular stress. Sci. Adv. 2019;5 doi: 10.1126/sciadv.aax1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dany M., Gencer S., Nganga R., Thomas R.J., Oleinik N., Baron K.D., Szulc Z.M., Ruvolo P., Kornblau S., Andreeff M., Ogretmen B. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood. 2016;128:1944–1958. doi: 10.1182/blood-2016-04-708750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sentelle R.D., Senkal C.E., Jiang W., Ponnusamy S., Gencer S., Selvam S.P., Ramshesh V.K., Peterson Y.K., Lemasters J.J., Szulc Z.M., et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012;8:831–838. doi: 10.1038/nchembio.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer. 2018;18:33–50. doi: 10.1038/nrc.2017.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hannun Y.A., Obeid L.M. Author Correction: Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018;19:673. doi: 10.1038/s41580-018-0046-6. [DOI] [PubMed] [Google Scholar]
- 31.Pewzner-Jung Y., Ben-Dor S., Futerman A.H. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: Insights into the regulation of ceramide synthesis. J. Biol. Chem. 2006;281:25001–25005. doi: 10.1074/jbc.R600010200. [DOI] [PubMed] [Google Scholar]
- 32.Meyers-Needham M., Ponnusamy S., Gencer S., Jiang W., Thomas R.J., Senkal C.E., Ogretmen B. Concerted functions of HDAC1 and microRNA-574-5p repress alternatively spliced ceramide synthase 1 expression in human cancer cells. EMBO Mol. Med. 2012;4:78–92. doi: 10.1002/emmm.201100189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koybasi S., Senkal C.E., Sundararaj K., Spassieva S., Bielawski J., Osta W., Day T.A., Jiang J.C., Jazwinski S.M., Hannun Y.A., et al. Defects in cell growth regulation by C18:0-ceramide and longevity assurance gene 1 in human head and neck squamous cell carcinomas. J. Biol. Chem. 2004;279:44311–44319. doi: 10.1074/jbc.M406920200. [DOI] [PubMed] [Google Scholar]
- 34.Karahatay S., Thomas K., Koybasi S., Senkal C.E., Elojeimy S., Liu X., Bielawski J., Day T.A., Gillespie M.B., Sinha D., et al. Clinical relevance of ceramide metabolism in the pathogenesis of human head and neck squamous cell carcinoma (HNSCC): attenuation of C(18)-ceramide in HNSCC tumors correlates with lymphovascular invasion and nodal metastasis. Cancer Lett. 2007;256:101–111. doi: 10.1016/j.canlet.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas R.J., Oleinik N., Panneer Selvam S., Vaena S.G., Dany M., Nganga R.N., Depalma R., Baron K.D., Kim J., Szulc Z.M., Ogretmen B. HPV/E7 induces chemotherapy-mediated tumor suppression by ceramide-dependent mitophagy. EMBO Mol. Med. 2017;9:1030–1051. doi: 10.15252/emmm.201607088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song J.M., Woo B.H., Lee J.H., Yoon S., Cho Y., Kim Y.D., Park H.R. Oral Administration of Porphyromonas gingivalis, a Major Pathogen of Chronic Periodontitis, Promotes Resistance to Paclitaxel in Mouse Xenografts of Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20102494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woo B.H., Kim D.J., Choi J.I., Kim S.J., Park B.S., Song J.M., Lee J.H., Park H.R. Oral cancer cells sustainedly infected with Porphyromonas gingivalis exhibit resistance to Taxol and have higher metastatic potential. Oncotarget. 2017;8:46981–46992. doi: 10.18632/oncotarget.16550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheridan M., Ogretmen B. The Role of Ceramide Metabolism and Signaling in the Regulation of Mitophagy and Cancer Therapy. Cancers. 2021;13 doi: 10.3390/cancers13102475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yilmaz O., Yao L., Maeda K., Rose T.M., Lewis E.L., Duman M., Lamont R.J., Ojcius D.M. ATP scavenging by the intracellular pathogen Porphyromonas gingivalis inhibits P2X7-mediated host-cell apoptosis. Cell Microbiol. 2008;10:863–875. doi: 10.1111/j.1462-5822.2007.01089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morel E., Gruenberg J. Annexin A2 binding to endosomes and functions in endosomal transport are regulated by tyrosine 23 phosphorylation. J. Biol. Chem. 2009;284:1604–1611. doi: 10.1074/jbc.M806499200. [DOI] [PubMed] [Google Scholar]
- 41.Morel E., Parton R.G., Gruenberg J. Annexin A2-dependent polymerization of actin mediates endosome biogenesis. Dev. Cell. 2009;16:445–457. doi: 10.1016/j.devcel.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 42.Sveeggen T.M., Abbey C.A., Smith R.L., Salinas M.L., Chapkin R.S., Bayless K.J. Annexin A2 modulates phospholipid membrane composition upstream of Arp2 to control angiogenic sprout initiation. FASEB J. 2023;37 doi: 10.1096/fj.202201088R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He K.L., Deora A.B., Xiong H., Ling Q., Weksler B.B., Niesvizky R., Hajjar K.A. Endothelial cell annexin A2 regulates polyubiquitination and degradation of its binding partner S100A10/p11. J. Biol. Chem. 2008;283:19192–19200. doi: 10.1074/jbc.M800100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arjunan P., Swaminathan R., Yuan J., Al-Shabrawey M., Espinosa-Heidmann D.G., Nussbaum J., Martin P.M., Cutler C.W. Invasion of Human Retinal Pigment Epithelial Cells by Porphyromonas gingivalis leading to Vacuolar/Cytosolic localization and Autophagy dysfunction In-Vitro. Sci. Rep. 2020;10:7468. doi: 10.1038/s41598-020-64449-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.El-Awady A.R., Miles B., Scisci E., Kurago Z.B., Palani C.D., Arce R.M., Waller J.L., Genco C.A., Slocum C., Manning M., et al. Porphyromonas gingivalis evasion of autophagy and intracellular killing by human myeloid dendritic cells involves DC-SIGN-TLR2 crosstalk. PLoS Pathog. 2015;10 doi: 10.1371/journal.ppat.1004647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mohammad H.S., Kurokohchi K., Yoneyama H., Tokuda M., Morishita A., Jian G., Shi L., Murota M., Tani J., Kato K., et al. Annexin A2 expression and phosphorylation are up-regulated in hepatocellular carcinoma. Int. J. Oncol. 2008;33:1157–1163. [PubMed] [Google Scholar]
- 47.Gao S., Liu K., Jiao Y., Chen P., Gu B., Liu Y., Liang G., Shi L., Zhou F., Lamont R.J., et al. Selective activation of TGFbeta signaling by P. gingivalis-mediated upregulation of GARP aggravates esophageal squamous cell carcinoma. Am. J. Cancer Res. 2023;13:2013–2029. [PMC free article] [PubMed] [Google Scholar]
- 48.Wielento A., Bereta G.P., Szczęśniak K., Jacuła A., Terekhova M., Artyomov M.N., Hasegawa Y., Grabiec A.M., Potempa J. Accessory fimbrial subunits and PPAD are necessary for TLR2 activation by Porphyromonas gingivalis. Mol. Oral Microbiol. 2023;38:334–346. doi: 10.1111/omi.12427. [DOI] [PubMed] [Google Scholar]
- 49.Nagano K., Abiko Y., Yoshida Y., Yoshimura F. Genetic and antigenic analyses of Porphyromonas gingivalis FimA fimbriae. Mol. Oral Microbiol. 2013;28:392–403. doi: 10.1111/omi.12032. [DOI] [PubMed] [Google Scholar]
- 50.Shibata S., Shoji M., Okada K., Matsunami H., Matthews M.M., Imada K., Nakayama K., Wolf M. Structure of polymerized type V pilin reveals assembly mechanism involving protease-mediated strand exchange. Nat. Microbiol. 2020;5:830–837. doi: 10.1038/s41564-020-0705-1. [DOI] [PubMed] [Google Scholar]
- 51.Kuboniwa M., Hasegawa Y., Mao S., Shizukuishi S., Amano A., Lamont R.J., Yilmaz O. P. gingivalis accelerates gingival epithelial cell progression through the cell cycle. Microbes Infect. 2008;10:122–128. doi: 10.1016/j.micinf.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herrera-Lopez E.E., Guerrero-Escalera D., Aguirre-Maldonado I., Lopez-Hernandez A., Montero H., Gutierrez-Nava M.A., Del Pozo-Yauner L., Arellanes-Robledo J., Camacho J., Perez-Carreon J.I. Annexins A2 and A5 are potential early biomarkers of hepatocarcinogenesis. Sci. Rep. 2023;13:6948. doi: 10.1038/s41598-023-34117-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang Y., Jia M., Yang X., Han H., Hou G., Bi L., Yang Y., Zhang R., Zhao X., Peng C., Ouyang X. Annexin A2: The diversity of pathological effects in tumorigenesis and immune response. Int. J. Cancer. 2022;151:497–509. doi: 10.1002/ijc.34048. [DOI] [PubMed] [Google Scholar]
- 54.Abdelraouf E.M., Hussein R.R.S., Shaaban A.H., El-Sherief H.A.M., Embaby A.S., Abd El-Aleem S.A. Annexin A2 (AnxA2) association with the clinicopathological data in different breast cancer subtypes: A possible role for AnxA2 in tumor heterogeneity and cancer progression. Life Sci. 2022;308 doi: 10.1016/j.lfs.2022.120967. [DOI] [PubMed] [Google Scholar]
- 55.Cardoso C.M., de Jesus S.F., de Souza M.G., Queiroz L.D.R.P., Santos E.M., Dos Santos E.P., Oliveira L.P., Cordeiro Santos C.K., Santos S.H.S., de Paula A.M.B., et al. High levels of ANXA2 are characteristic of malignant salivary gland tumors. J. Oral Pathol. Med. 2019;48:929–934. doi: 10.1111/jop.12932. [DOI] [PubMed] [Google Scholar]
- 56.Ma Y., Wang H. Clinical significance of Annexin A2 expression in oral squamous cell carcinoma and its influence on cell proliferation, migration and invasion. Sci. Rep. 2021;11:5033. doi: 10.1038/s41598-021-84675-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Valapala M., Vishwanatha J.K. Lipid raft endocytosis and exosomal transport facilitate extracellular trafficking of annexin A2. J. Biol. Chem. 2011;286:30911–30925. doi: 10.1074/jbc.M111.271155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Illien F., Piao H.R., Coué M., di Marco C., Ayala-Sanmartin J. Lipid organization regulates annexin A2 Ca(2+)-sensitivity for membrane bridging and its modulator effects on membrane fluidity. Biochim. Biophys. Acta. 2012;1818:2892–2900. doi: 10.1016/j.bbamem.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 59.Drucker P., Pejic M., Galla H.J., Gerke V. Lipid segregation and membrane budding induced by the peripheral membrane binding protein annexin A2. J. Biol. Chem. 2013;288:24764–24776. doi: 10.1074/jbc.M113.474023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hankins J.L., Ward K.E., Linton S.S., Barth B.M., Stahelin R.V., Fox T.E., Kester M. Ceramide 1-phosphate mediates endothelial cell invasion via the annexin a2-p11 heterotetrameric protein complex. J. Biol. Chem. 2013;288:19726–19738. doi: 10.1074/jbc.M113.481622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang X., Song M., Li X., Su C., Yang Y., Wang K., Liu C., Zheng Z., Jia Y., Ren S., et al. CERS6-derived ceramides aggravate kidney fibrosis by inhibiting PINK1-mediated mitophagy in diabetic kidney disease. Am. J. Physiol. Cell Physiol. 2023;325:C538–C549. doi: 10.1152/ajpcell.00144.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vos M., Klein C. Ceramide-induced mitophagy impairs ss-oxidation-linked energy production in PINK1 deficiency. Autophagy. 2022;18:703–704. doi: 10.1080/15548627.2022.2027193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vos M., Dulovic-Mahlow M., Mandik F., Frese L., Kanana Y., Haissatou Diaw S., Depperschmidt J., Böhm C., Rohr J., Lohnau T., et al. Ceramide accumulation induces mitophagy and impairs beta-oxidation in PINK1 deficiency. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2025347118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vaena S., Chakraborty P., Lee H.G., Janneh A.H., Kassir M.F., Beeson G., Hedley Z., Yalcinkaya A., Sofi M.H., Li H., et al. Aging-dependent mitochondrial dysfunction mediated by ceramide signaling inhibits antitumor T cell response. Cell Rep. 2021;35 doi: 10.1016/j.celrep.2021.109076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Poole L.P., Macleod K.F. Mitophagy in tumorigenesis and metastasis. Cell. Mol. Life Sci. 2021;78:3817–3851. doi: 10.1007/s00018-021-03774-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morad S.A.F., MacDougall M.R., Abdelmageed N., Kao L.P., Feith D.J., Tan S.F., Kester M., Loughran T.P., Jr., Wang H.G., Cabot M.C. Pivotal role of mitophagy in response of acute myelogenous leukemia to a ceramide-tamoxifen-containing drug regimen. Exp. Cell Res. 2019;381:256–264. doi: 10.1016/j.yexcr.2019.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dany M., Ogretmen B. Ceramide induced mitophagy and tumor suppression. Biochim. Biophys. Acta. 2015;1853:2834–2845. doi: 10.1016/j.bbamcr.2014.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y.H., Jiang J., Zhu Q., AlAnezi A.Z., Clark R.B., Jiang X., Rowe D.W., Nichols F.C. Porphyromonas gingivalis lipids inhibit osteoblastic differentiation and function. Infect. Immun. 2010;78:3726–3735. doi: 10.1128/IAI.00225-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kofteridis D.P., Valachis A., Scoulica E., Christidou A., Maraki S., Samonis G. Hickman catheter-related bacteremia caused by Gordonia sputi in a patient with breast cancer. J. Infect. Dev. Ctries. 2012;6:188–191. doi: 10.3855/jidc.1770. [DOI] [PubMed] [Google Scholar]
- 70.Goncalves M.F.M., Pina-Vaz T., Fernandes A.R., Miranda I.M., Silva C.M., Rodrigues A.G., Lisboa C. Microbiota of Urine, Glans and Prostate Biopsies in Patients with Prostate Cancer Reveals a Dysbiosis in the Genitourinary System. Cancers. 2023;15 doi: 10.3390/cancers15051423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krcmery V., Jr., Pichna P., Oravcova E., Lacka J., Kukuckova E., Studena M., Grausova S., Stopkova K., Krupova I. Stenotrophomonas maltophilia bacteraemia in cancer patients: report on 31 cases. J. Hosp. Infect. 1996;34:75–77. doi: 10.1016/s0195-6701(96)90129-2. [DOI] [PubMed] [Google Scholar]
- 72.Sharma A.K., DeBusk W.T., Stepanov I., Gomez A., Khariwala S.S. Oral Microbiome Profiling in Smokers with and without Head and Neck Cancer Reveals Variations Between Health and Disease. Cancer Prev. Res. 2020;13:463–474. doi: 10.1158/1940-6207.CAPR-19-0459. [DOI] [PubMed] [Google Scholar]
- 73.Sarkar P., Malik S., Laha S., Das S., Bunk S., Ray J.G., Chatterjee R., Saha A. Dysbiosis of Oral Microbiota During Oral Squamous Cell Carcinoma Development. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.614448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Elkholy A., Avuthu N., Abdalla M., Behring M., Bajpai P., Kim H.G., Header D., Abo Elwafa R.A., Saed H., Embaby A., et al. Microbiome diversity in African American, European American, and Egyptian colorectal cancer patients. Heliyon. 2023;9 doi: 10.1016/j.heliyon.2023.e18035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chemaly R.F., Dantes R., Shah D.P., Shah P.K., Pascoe N., Ariza-Heredia E., Perego C., Nguyen D.B., Nguyen K., Modarai F., et al. Cluster and sporadic cases of herbaspirillum species infections in patients with cancer. Clin. Infect. Dis. 2015;60:48–54. doi: 10.1093/cid/ciu712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu S., Fan X., Zheng S., Lin L., Liu J., Pan Y., Li C. The sialidase inhibitor, DANA, reduces Porphyromonas gingivalis pathogenicity and exerts anti-inflammatory effects: An in vitro and in vivo experiment. J. Periodontol. 2021;92:286–297. doi: 10.1002/JPER.19-0688. [DOI] [PubMed] [Google Scholar]
- 77.Janneh A.H., Kassir M.F., Atilgan F.C., Lee H.G., Sheridan M., Oleinik N., Szulc Z., Voelkel-Johnson C., Nguyen H., Li H., et al. Crosstalk between pro-survival sphingolipid metabolism and complement signaling induces inflammasome-mediated tumor metastasis. Cell Rep. 2022;41 doi: 10.1016/j.celrep.2022.111742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Burgess R.R., Arthur T.M., Pietz B.C. Mapping protein-protein interaction domains using ordered fragment ladder far-western analysis of hexahistidine-tagged fusion proteins. Methods Enzymol. 2000;328:141–157. doi: 10.1016/s0076-6879(00)28396-1. [DOI] [PubMed] [Google Scholar]
- 79.Edmondson D.G., Roth S.Y. Identification of protein interactions by far Western analysis. Curr Protoc Mol Biol Chapter. Curr. Protoc. Mol. Biol. 2001;20 doi: 10.1002/0471142727.mb2006s55. Unit 20.6. [DOI] [PubMed] [Google Scholar]
- 80.Zhang W., Ju J., Rigney T., Tribble G. Integrin alpha5beta1-fimbriae binding and actin rearrangement are essential for Porphyromonas gingivalis invasion of osteoblasts and subsequent activation of the JNK pathway. BMC Microbiol. 2013;13:5. doi: 10.1186/1471-2180-13-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Perez-Riverol Y., Bai J., Bandla C., García-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M., et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Engevik M.A., Danhof H.A., Auchtung J., Endres B.T., Ruan W., Bassères E., Engevik A.C., Wu Q., Nicholson M., Luna R.A., et al. Fusobacteriumnucleatum Adheres to Clostridioides difficile via the RadD Adhesin to Enhance Biofilm Formation in Intestinal Mucus. Gastroenterology. 2021;160:1301–1314.e8. doi: 10.1053/j.gastro.2020.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Original, uncropped western blot image files have been deposited at Mendeley Data, and they will be publicly available at: $https://data.mendeley.com/datasets/zbppjnyyrr/2.
-
•
Original 16S rDNA files have been deposited in the NCBI Sequence Read Archive (SRA) and are publicly available as of the date of publication at Submission ID SUB14165627, BioProject ID PRJNA1067697, BioSample accessions SAMN39543253, SAMN39543254, SAMN39543255, SAMN39543256.
-
•
Original proteomics data are available via ProteomeXchange with the identifier PXD051568.
-
•
This paper does not report the original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.