

Abstract
GDF15 (growth differentiation factor 15) is a stress cytokine with several proposed roles, including support of stress erythropoiesis. Higher circulating GDF15 levels are prognostic of mortality during acute respiratory distress syndrome, but the cellular sources and downstream effects of GDF15 during pathogen-mediated lung injury are unclear. We quantified GDF15 in lower respiratory tract biospecimens and plasma from patients with acute respiratory failure. Publicly available data from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection were reanalyzed. We used mouse models of hemorrhagic acute lung injury mediated by Pseudomonas aeruginosa exoproducts in wild-type mice and mice genetically deficient for Gdf15 or its putative receptor, Gfral. In critically ill humans, plasma levels of GDF15 correlated with lower respiratory tract levels and were higher in nonsurvivors. SARS-CoV-2 infection induced GDF15 expression in human lung epithelium, and lower respiratory tract GDF15 levels were higher in coronavirus disease (COVID-19) nonsurvivors. In mice, intratracheal P. aeruginosa type II secretion system exoproducts were sufficient to induce airspace and plasma release of GDF15, which was attenuated with epithelial-specific deletion of Gdf15. Mice with global Gdf15 deficiency had decreased airspace hemorrhage, an attenuated cytokine profile, and an altered lung transcriptional profile during injury induced by P. aeruginosa type II secretion system exoproducts, which was not recapitulated in mice deficient for Gfral. Airspace GDF15 reconstitution did not significantly modulate key lung cytokine levels but increased circulating erythrocyte counts. Lung epithelium releases GDF15 during pathogen injury, which is associated with plasma levels in humans and mice and can increase erythrocyte counts in mice, suggesting a novel lung–blood communication pathway.
Keywords: GDF15, GFRAL, stress erythropoiesis, acute respiratory failure, cytokines
Clinical Relevance
We demonstrate that lung epithelial cells in humans and mice release GDF15 (growth differentiation factor 15), a stress cytokine with proposed roles in stress erythropoiesis, into airspaces and blood in response to pathogen injury. Airspace reconstitution of GDF15 during hemorrhagic lung injury in knockout mice increased plasma levels of GDF15 and circulating erythrocyte counts. We speculate that GDF15 release by the lung may act as an extrapulmonary stress response hormone, suggesting a previously unrecognized pathway of lung–blood communication.
Growth differentiation factor 15 (GDF15), initially described as macrophage inhibitory cytokine-1, is a member of the TGF (transforming growth factor) superfamily (1). GDF15, which is also known NAG-1 or MIC-1, among others (2), is expressed by a variety of cell types, and expression can increase during states of cellular stress such as aging, injury, and inflammation. Currently, the only widely accepted receptor for GDF15 is GFRAL (glial cell–derived neurotrophic factor receptor family receptor α-like), which is primarily restricted to the hindbrain in mice and humans and requires RET (Ret proto-oncogene) as a coreceptor (3, 4). Reported roles for GDF15 are wide-ranging and include metabolic regulation (5), metabolic resilience during inflammation (6), modulation of cellular recruitment and cytokines (7), and support for stress erythropoiesis (8–10). GDF15 has been implicated as a prognostic biomarker in a wide range of human diseases such as cardiovascular disease (11), diabetes mellitus (12), and malignancy (13). Given its pleiotropic roles as a presumed stress cytokine, there is increasing recognition of potential roles for GDF15 during lung disease and critical illness (2, 14).
In the lung, GDF15 expression has been described in lung epithelium and endothelium and has been linked to numerous lung diseases, including pulmonary vascular disease, chronic obstructive pulmonary disease, and pulmonary fibrosis, as reviewed previously (2). Despite this growing interest for GDF15 in the lung, few studies have investigated GDF15 during pneumonia and pathogen-mediated lung injury, which is the primary risk factor for acute respiratory distress syndrome (ARDS) (15). In a prior observational study, circulating GDF15 levels were increased in patients with ARDS compared with healthy controls, and higher GDF15 levels were prognostic of increased mortality during ARDS (16). Yet, there is limited understanding of the source, role, and significance of GDF15 during pneumonia and pathogen-mediated acute lung injury. Therefore, we conducted a translational investigation of GDF15 using biospecimens from an observational cohort of patients receiving mechanical ventilation for acute respiratory failure, publicly available proteomic and single-cell RNA sequencing data from patients with coronavirus disease (COVID-19), as well as mouse models of acute lung injury caused by Pseudomonas aeruginosa exoproducts during genetic deficiency of GDF15 to dissect its potential significance during pathogen-mediated acute lung injury.
Methods
Patient Population
We performed retrospective analyses in a subset of critically ill adult patients (n = 103 with plasma, n = 110 with endotracheal aspirate [ETA], n = 44 with both; Figure E1 in the data supplement) prospectively enrolled in the University of Pittsburgh Acute Lung Injury Registry and Biospecimen Repository (ALIR) between 2012 and 2018 with acute respiratory failure not caused by COVID-19 (17–19). We performed additional analyses in a separate subset (n = 46 patients with a total of 76 patient time points; see Figure E1) limited to patients mechanically ventilated as a result of COVID-19 prospectively enrolled in ALIR between April 2020 and November 2021 (20). Both patient subsets, before COVID-19 and with COVID-19, were derived from consecutively enrolled subjects based on sample availability (see data supplement for additional details on ALIR). The ALIR study is approved by the University of Pittsburgh Institutional Review Board (STUDY19050099), and written informed consent was provided by all participants or their legally authorized representatives. All research was conducted according to the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Patient Biomarkers
Plasma and ETA were collected and processed using a standardized protocol as previously described (20, 21). Before assays, ETA was initially diluted with Sputasol for patients without COVID-19 or Zymo DNA/RNA shield for those with COVID-19 per institutional biosafety protocols, then further diluted with PBS solution (20). GDF15 levels in human biospecimens were quantified by sandwich ELISA (DGD150; R&D Systems). Other protein levels were previously quantified by multiplex assay (21). ETA biomarker levels were normalized to total protein levels quantified in ETA by a bicinchoninic acid (BCA) assay (20). From a separate aliquot of ETA, we previously extracted bacterial DNA and performed microbiota profiling with 16S RNA gene sequencing as previously described (see data supplement) (17, 22). We classified patients into biomarker-based subphenotypes as previously described (see data supplement) (23).
Animals
All animal studies were approved by the institutional animal care and use committee of the University of Pittsburgh. Wild-type C57BL/6J mice were obtained from Jackson Laboratories. GDF15 global knockout mice were obtained from the Knockout Mouse Project (www.mmrrc.org) and back-crossed to C57BL/6J mice for at least three generations. The original strain was a knockout first allele (Gdf15−/− hereafter) that was subsequently bred to an FLP recombinase–expressing line [B6.129S4-Gt(ROSA)26Sortm2(FLP*)Sor/J; Jax strain 012930] to generate Gdf15fl/fl mice. Mice with lung epithelial-specific genetic deficiency of Gdf15 (pan-epithelial knockout) were generated by breeding Gdf15fl/fl with SFTPC-Cre transgenic mice obtained thanks to a gift from Brigid Hogan (24, 25). Gdf15fl/fl mice were used as experimental controls for Gdf15fl/fl;SFTPC-Cre mice. All mice were maintained on a C57BL/6J background. Mice globally deficient for the GDF15 central nervous system receptor Gfral were created by removing the fifth exon of Gfral using CRISPR/Cas9 genome editing (see data supplement for a detailed description) (26). In vivo experiments were performed in age-matched (8–16 wk) and sex-matched mice by personnel blinded to the experimental groups.
Animal Experimental Procedures
Mice were intratracheally inoculated with the supernatant of P. aeruginosa strains (PA SN) or vehicle under direct visualization after isoflurane anesthesia as previously described (27). PA14 and PA14 ΔxcpQ strains and their cell-free SN were prepared as previously described (27, 28). At the time of necropsy approximately 20 hours after inoculation, mice were euthanized with ketamine/xylazine. Plasma was collected via a 1-ml syringe after laceration of the inferior vena cava in the thoracic cavity, placed into K2-EDTA microtubes, and centrifuged at 1,500 × g for 15 minutes at 4°C before preservation at −80°C. BAL fluid was collected after cannulation of the trachea and administration of a total volume of 0.75 ml of PBS solution that was lavaged through the lungs three times as previously described (14). Raw BAL fluid was used for cell counts, preparation of Cytospins for differential cell counts, and measurement of absorbance at 540 nm (OD540) (28). BAL was centrifuged at 10,000 × g for 10 minutes at 4°C, and the cell-free SN was removed and stored at −80°C before assays.
In a dedicated experiment to collect lung RNA and protein, the left lung was clamped, BAL was obtained from the right lung, and the left lung was immediately removed and snap-frozen in liquid nitrogen before storage at −80°C. In select experiments to determine the contribution of airspace GDF15, recombinant murine GDF15 (rmGDF15; no. 8944; R&D Systems) or vehicle was simultaneously administered at the time of intratracheal inoculation with PA SN. BAL protein was determined by BCA assay and IgM levels by sandwich ELISA (Bethyl). In select experiments, mouse cell counts were performed in EDTA plasma using Hemavet 950 (Drew Scientific) (28).
RNA Sequencing of the Lung
Snap-frozen lung obtained at necropsy was homogenized in Trizol (Life Sciences), and RNA was isolated per the manufacturer instructions. RNA cleanup was performed using a Qiagen RNeasy Mini Kit per the manufacturer instructions. Quality control and mRNA library preparation of isolated lung RNA were performed by Novogene, followed by paired-end RNA sequencing using an Illumina NovaSeq platform. FASTQ files provided by Novogene were quality-controlled with FastQC, trimmed, and aligned using STAR (29), and RNA counts were determined with the quantMode Gene Counts option. Raw data and sequencing files are available at the Gene Expression Omnibus database with accession code GSE253234. Differential expression analysis was performed using DEseq2 in R. Gene set overrepresentation and expression analysis were performed in R.
Reanalysis of Publicly Available Data
Protein levels in cell lysates from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)–infected or sham-infected human lung alveolar epithelial cells in polarized coculture with human endothelial cells were obtained from published data (30). Single-cell RNA sequencing data from BAL fluid from patients with SARS-CoV-2 infection or healthy controls were downloaded from the Gene Expression Omnibus database (accession code GSE145926) (31) and analyzed with Seurat in R.
Isolation of Lung Protein
A portion of snap-frozen lung was homogenized in extraction buffer composed of sterile culture-grade PBS solution with protease inhibitor cocktail (Roche cOmplete ULTRA, 1 tab per 10 ml) and 1 mM EDTA (32, 33). Homogenized lung was then centrifuged at 13,000 × g for 10 minutes at 4°C, and the SN was removed and stored at −80°C before assays. Protein content of the homogenate was quantified by a BCA assay, which was used to normalize lung homogenate protein values.
Quantification of RNA and Protein Levels
Quantitative PCR was performed on a CFX96 real-time instrument (BioRad) using SYBR Green (BioRad) and primers for mouse Gdf15 and Hprt as previously described (14). In select experiments, BAL and plasma protein levels were quantified by a multiplex assay (M60009RDPD; BioRad) according to the manufacturer instructions. Otherwise, mouse protein levels were quantified using sandwich ELISA kits (R&D Systems): GDF15 (DY6385), granulocyte colony–stimulating factor (G-CSF; DY414), TNF-α (DY410), and CXCL10 (DY466). Neutrophil elastase activity was quantified as previously described and is detailed in the data supplement (27, 34).
Statistics
In human studies, statistical comparisons were performed in R using Wilcoxon rank-sum testing for two groups or a Kruskal-Wallis test with Benjamini-Hochberg correction for three or more groups. In mouse studies, a nonparametric Mann-Whitney test in GraphPad Prism (version 9.5) was used to compare groups. Adjustment of multiplex ELISA results for multiple comparisons was performed using Benjamini-Hochberg correction in R.
Results
Increased Plasma GDF15 Levels Are Associated with Worse Clinical Outcomes, Lower Respiratory Tract Dysbiosis, and Lower Respiratory Tract GDF15 Levels
We quantified plasma GDF15 levels at the time of enrollment in a convenience sample of 103 critically ill patients with acute respiratory failure prospectively enrolled in ALIR. The median age was 56.3 years, 49 (47.5%) were female, and 16 (15.5%) died by 30 days (Table E1 in the data supplement). Demographic characteristics and burden of comorbid disease did not significantly differ between patients with or at risk for ARDS (n = 51) and patients who were mechanically ventilated for nonpulmonary etiologies (e.g., encephalopathy, “airway controls”; n = 52). Baseline plasma GDF15 levels were higher in patients with or at risk for ARDS (P < 0.001; Figure 1A) compared with airway controls. Additionally, patients with a harmful “hyperinflammatory” biomarker-based phenotype exhibited increased plasma GDF15 levels (P < 0.001; Figure 1B) (23). Nonsurvivors also exhibited higher plasma GDF15 levels (median, 7,026 pg/ml; IQR, 4,655–17,586; Figure 1C) compared with survivors (median, 3,502 pg/ml; IQR, 1,551–6,231 pg/ml), which is in line with a prior report limited to patients with ARDS (16). Plasma GDF15 levels were correlated with several host response cytokines (Figure E2A) (21). Probabilistic graphical modeling revealed associations with clinical diagnosis of pneumonia as well as biomarkers of lung injury (see Figure E2B). Patients with lower respiratory tract bacterial profiles classified in a harmful “low-diversity” cluster, indicative of lower respiratory tract infection or dysbiosis (17, 22), exhibited higher plasma levels of GDF15 (Figure 1D) compared with patients with nonpathogenic lower respiratory tract microbial clusters, which suggests that plasma GDF15 may reflect pathogen-driven biological processes in the lung.
Figure 1.
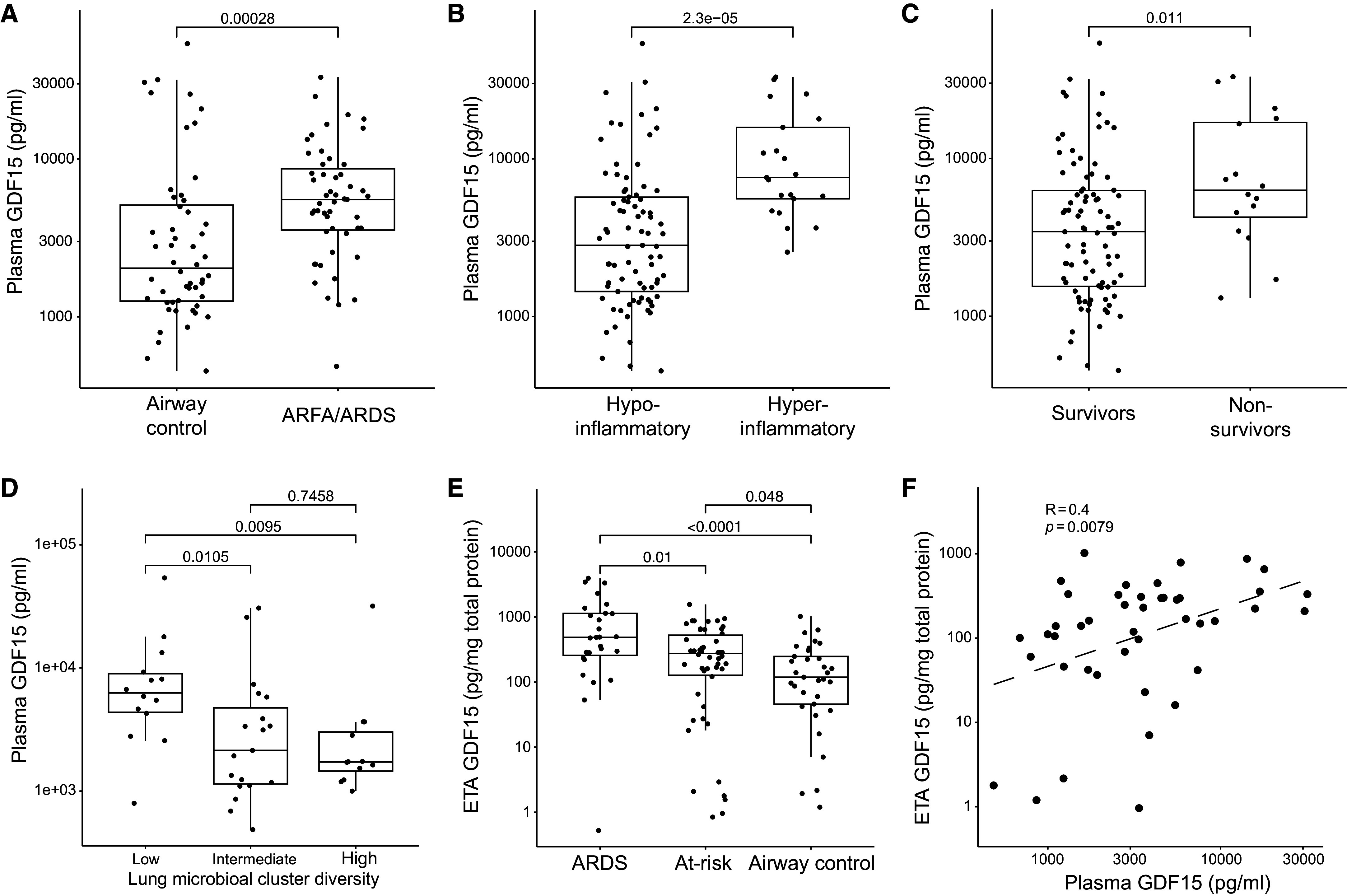
Higher plasma GDF15 (growth differentiation factor 15) levels are associated with worse clinical outcomes and lower respiratory tract dysbiosis and positively correlate with lower respiratory tract GDF15 levels. (A–C) Plasma GDF15 levels from critically ill patients (n = 103) with acute respiratory failure by clinical group: patients with risk factors for (ARFA), or a diagnosis of, acute respiratory distress syndrome (ARDS) and airway controls ventilated for non-pulmonary indications (A). (B) Host response phenotypes as classified by a four-variable model consisting of angiopoietin-2, soluble TNF receptor-1, procalcitonin, and bicarbonate and (C) survival status at 30 days. (D) Plasma GDF15 levels are displayed for a subset of patients (n = 45) separated by Dirichlet multinomial model clusters of bacterial composition. Clusters had significant differences in bacterial abundance and α-diversity characterized as low (n = 14), intermediate (n = 19), and high (n = 12) α-diversity. The low-diversity cluster, characterized by a high abundance of typical pathogenic bacteria indicative of lower respiratory tract infection or dysbiosis, showed significantly higher plasma GDF15 levels. (E) Endotracheal aspirate (ETA) GDF15 levels are presented stratified by clinical classification of ARDS, at-risk for ARDS, or airway control mechanically ventilated for non-pulmonary illness (n = 110). Range displayed with box plot displaying median and IQR. P values represent differences by Kruskal-Wallis test with Dunn’s post hoc test using Benjamini-Hochberg correction for multiple comparisons. (F) Scatter plot of plasma and ETA GDF15 levels normalized to total protein level (n = 44, Spearman ρ displayed). Each point represents a single patient, and the P value represents significance by Wilcoxon rank sum test unless otherwise stated.
Given that plasma levels of GDF15 were associated with markers of lung injury and pneumonia, we also quantified lower respiratory tract GDF15 in patients with banked ETA specimens collected at enrollment (n = 110 total, n = 44 with plasma and ETA; see Figure E1). Patient clinical characteristics were similar to those in the plasma cohort and are detailed further in Table E2. We normalized ETA GDF15 levels to total protein as previously described (median normalized level, 248 pg/mg total protein; IQR, 99–494 pg/mg) (20). We found significantly higher ETA GDF15 levels in patients with ARDS (n = 27; Figure 1E) compared with patients who were at risk for ARDS (n = 50) and airway controls (n = 33). In patients with GDF15 quantified in plasma and ETA (n = 44), we found a moderate but highly significant correlation between plasma and lower respiratory tract GDF15 levels (Spearman ρ = 0.40, P < 0.01; Figure 1F).
Mice Release Airspace and Plasma GDF15 during Lung Injury Induced by P. aeruginosa Exoproducts
Given the striking associations between plasma and lower respiratory tract GDF15 levels during pneumonia and lung injury in humans, we used a model of pathogen-mediated acute lung injury induced by P. aeruginosa exoproducts (i.e., PA SN) in mice (27, 28) to probe the determinants of GDF15 release into the airspaces and plasma. Wild-type C57Bl/6J mice exhibit severe lung injury marked by hemorrhage after intratracheal PA SN induction, which was attenuated in mice that received the cell-free SN of P. aeruginosa with genetic deletion of the xcpQ chaperone protein (ΔxcpQ SN) that mediates extrabacterial transport of type II secretion system toxins (Figure 2A) (27, 28, 35, 36). It is important to note that ΔxcpQ SN contains LPS but is deficient for toxins such as LasB and exotoxin A. PA SN also elicited marked increases in BAL protein levels (Figure 2B), plasma GDF15 levels (Figure 2C), and BAL GDF15 levels (Figure 2D) that were significantly attenuated with ΔxcpQ SN. This finding suggests that direct pathogen-mediated lung injury is sufficient to induce airspace and plasma release of GDF15 in mice. To probe potential sources of GDF15, we treated murine lung epithelial cells with PA SN and observed a marked increase in mRNA levels by quantitative PCR (Figure 2E), as well as extracellular release (Figure 2F) of GDF15 by ELISA at 4 hours and 24 hours, respectively. We also found that mice with pan-epithelial genetic deficiency of Gdf15 exhibit decreased BAL levels of GDF15 (Figure 2G) compared with control mice during PA SN injury, which confirmed that lung epithelial cells are a source of airspace GDF15. Plasma GDF15 levels were also significantly decreased in the epithelial-specific knockout (Figure 2H), although the magnitude of decrease was attenuated compared with the airspaces, suggesting that lung endothelium or other cells may also contribute to plasma levels during acute lung injury (37).
Figure 2.

Mice release GDF15 during lung injury induced by Pseudomonas aeruginosa type II secretion system exoproducts. Wild-type (WT) C57Bl/6J mice were intratracheally inoculated with vehicle (n = 6) or the supernatant (SN) of P. aeruginosa grown in culture. Parent P. aeruginosa (P. aeruginosa cell-free SN [PA SN], n = 8) and a genetically modified strain (ΔxcpQ SN, n = 6) deficient for the type II secretion system chaperone protein xcpQ were used. Necropsy was performed 20 hours after inoculation. (A) Gross appearance of BAL fluid, (B) BAL protein concentrations, (C) plasma GDF15 levels (vehicle group, n = 5), and (D) BAL GDF15 levels. (E) Murine lung epithelial cells (MLE12) were exposed in vitro to vehicle or PA SN. Cell lysates were collected 4 hours after exposure, and Gdf15 transcripts (E) and protein (F) were measured. (G) Gdf15fl/fl mice (“control”) or epithelial-specific knockout (KO) Gdf15fl/fl;SFTPC-Cre (“epithelial KO”) mice were exposed to PA SN (n = 9 control and n = 14 epithelial KO), and BAL (G) and plasma (H) GDF15 was measured. In H, plasma GDF15 concentration was normalized to control to compare across experiments. Each tube or point represents an individual mouse, and the group median is displayed. Groups were compared using a Mann-Whitney test.
Human Lung Epithelium Releases GDF15 in Response to SARS-CoV-2 Infection
We next tested if GDF15 release from lung epithelial cells also occurs in humans during pathogen-mediated lung injury by using publicly available data from SARS-CoV-2 infection. In human lung alveolar epithelial cells in polarized coculture with human microvascular endothelial cells, GDF15 is the second most upregulated protein in alveolar epithelial cell lysates after infection with SARS-CoV-2 as measured by label-free quantification using mass spectrometry (infected:sham ratio, 5.39; Figure 3A) (30). Interestingly, GDF15 is also significantly upregulated in cocultured endothelial cells, albeit to a lesser extent (infected:sham ratio, 1.55). Similarly, GDF15 RNA levels were significantly increased in epithelial cells identified using single-cell RNA sequencing in BAL fluid from patients with severe COVID-19 compared with healthy controls (P < 6.9 × 10−12; Figure 3B) (31, 38).
Figure 3.
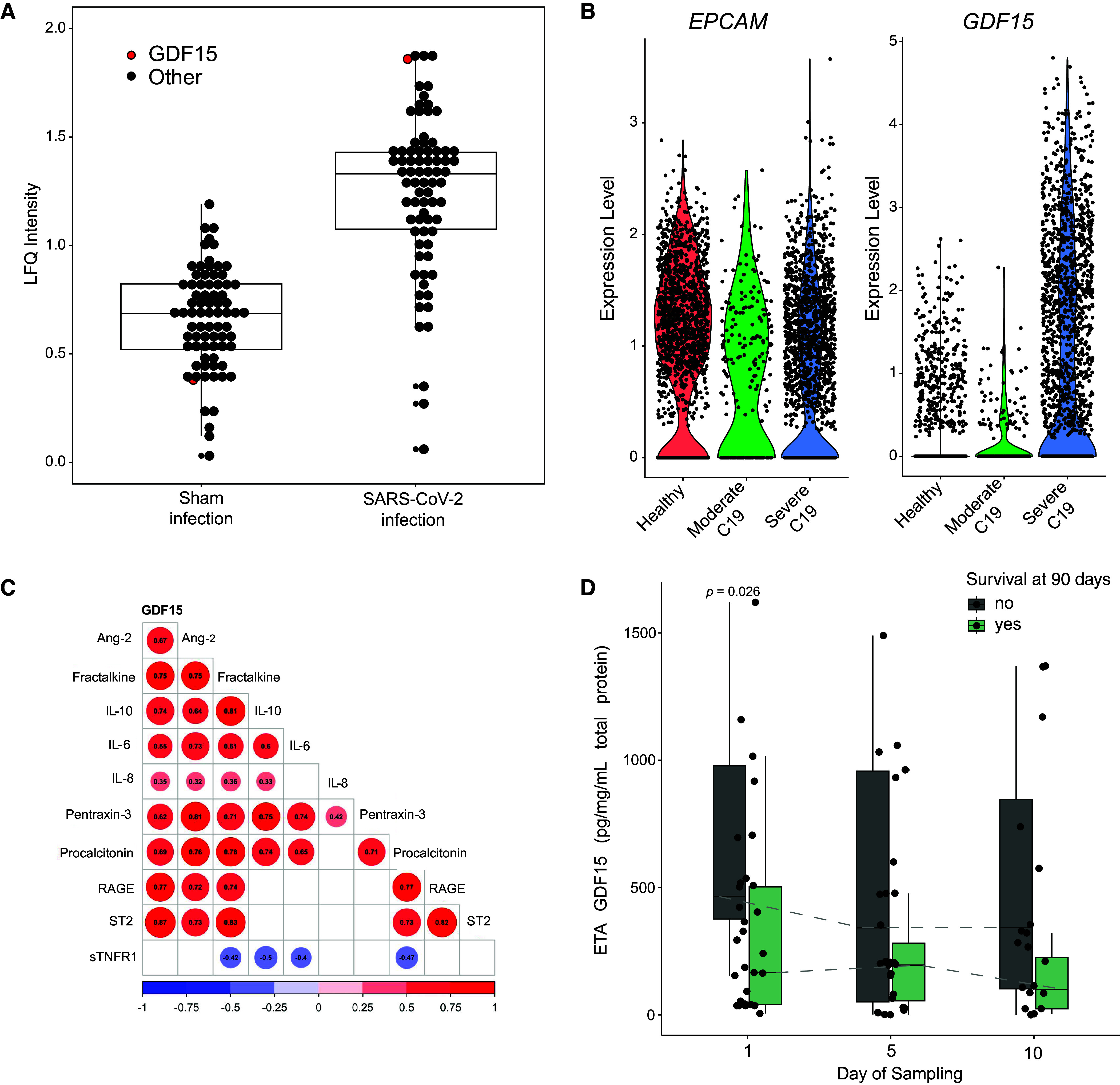
Human lung epithelium releases GDF15 in response to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. (A) Human lung alveolar epithelial cells infected with SARS-CoV-2 (n = 3) reveal increased levels of GDF15 by label-free quantification (LFQ) compared with control sham infection (n = 3) (30). Each point represents the median protein expression of upregulated proteins. GDF15 is red, whereas all other proteins are black. Box plot displays the median and IQR. (B) Publicly available single-cell RNA sequencing data from BAL fluid collected from patients infected with severe coronavirus disease (COVID-19) pneumonia (n = 6) or moderate COVID-19 pneumonia (n = 3) and healthy controls (n = 3; from Gene Expression Omnibus database accession GSE145926). Cells identified as lung epithelium by cluster analysis display significantly increased GDF15 RNA levels (P < 6.9 × 10−120 for severe COVID compared with healthy controls). (C) Correlogram of associations of ETA GDF15 levels (46 patients with 76 total time points on sampling Days 1, 5, and 10) with host inflammatory response biomarkers during COVID-19 pneumonia requiring mechanical ventilation. All ETA protein levels are normalized to total protein. The number in the circle displays the Spearman correlation. The direction and magnitude of correlation are displayed by color, nonsignificant correlations are blank, and P values are adjusted for multiple comparisons by Benjamini-Hochberg correction with significance set to P < 0.05. (D) Box-and-whisker plots showing ETA GDF15 levels stratified by sampling day (n = 29 on Day 1, n = 27 on Day 5, and n = 20 on Day 10; total n = 46) and comparing nonsurvivors (dark gray) and survivors (light green). Boxes represent the IQRs, horizontal lines represent the medians, and vertical lines extend from minimum to maximum values. Dashed lines connect the median values for each group at each sampling day to show longitudinal trends in GDF15 levels. Ang-2 = angiopoietin-2; ARFA = at risk for ARDS; C19 = coronavirus disease; LFQ = label-free quantification; Procal = procalcitonin; RAGE = soluble receptor for advanced glycation end-products; ST2 = suppression of tumorigenicity-2 (also known as IL-1 receptor ligand-1 or IL-33 receptor); sTNFR1 = soluble TNF receptor-1.
We validated these findings in a subset of ALIR patients (n = 46; 76 patient time points) with severe COVID-19 pneumonia (see Figure E1 and Table E3). We observed increased levels of GDF15 in ETA (baseline median normalized level, 294 pg/mg of total protein; IQR, 54–518 pg/mg) during severe COVID-19 pneumonia managed with mechanical ventilation. GDF15 levels were strongly correlated with ST2, RAGE, fractalkine, and IL-10; moderately correlated with procalcitonin, angipoietin-2, pentraxin-3, and IL-6; and weakly correlated with IL-8 (adjusted P < 0.05 for all associations; Figure 3C) levels in ETA after normalization to total protein. ETA GDF15 levels were higher in nonsurvivors compared with survivors (P = 0.026 at Day 1; Figure 3D). Together, these data strongly support that GDF15 is secreted by mouse and human lung epithelial cells in response to infection.
Global Deficiency of Gdf15 Is Marked by an Altered Airspace Cytokine Profile during Lung Injury Induced by P. aeruginosa Exoproducts
To further assess the role of airspace GDF15 during pathogen-mediated injury, we returned to our mouse model of acute lung injury induced by PA SN, using mice globally deficient for Gdf15. At necropsy 20 hours after intratracheal instillation of PA SN, we noted decreased BAL hemorrhage in Gdf15−/− mice by gross examination (Figure 4A) and OD540 (Figure 4B), which is a surrogate measure for hemoglobin (28). However, we did not find any difference in alveolar–capillary barrier disruption as measured by BAL IgM (Figure 4C). Given the striking association between GDF15 levels and several other cytokines during COVID-19 pneumonia, we wondered whether Gdf15−/− mice would exhibit differences in airspace and/or plasma cytokine levels. Using a discovery approach with a multiplex assay, we found significantly decreased levels of several BAL proteins in Gdf15−/− mice, including TNF-α, G-CSF (Figures 4D and 4E), and macrophage inflammatory protein-1α and -1β (Figure E3) after correction for multiple comparisons. Despite the strong difference in BAL TNF-α, there was no difference in plasma levels (Figure 4F). In fact, the only plasma cytokine that was different was G-CSF (Figure 4G and Figure E4), which remained significantly decreased in Gdf15−/− mice after correction for multiple comparisons. Although there was decreased G-CSF in the airspaces and blood of Gdf15−/− mice, we did not identify any difference in airspace neutrophil recruitment as measured by BAL polymorphonuclear cells (Figure 4H). Because G-CSF also can influence neutrophil functions such as elastase release (39), we performed a functional assay of neutrophil elastase, identifying significantly decreased neutrophil elastase activity in BAL from Gdf15−/− mice compared with wild-type mice (Figure 4I).
Figure 4.

Global deficiency of Gdf15 is marked by an altered airspace cytokine profile during lung injury induced by P. aeruginosa exoproducts. In two experiments, WT (n = 19) and Gdf15−/− mice (n = 16) were intratracheally inoculated with PA SN, and BAL fluid and plasma were collected 20 hours after infection. (A) Gross images of BAL hemorrhage from each experiment. (B) BAL absorbance (optical density [OD]) at 540 nm (OD540) normalized to the WT median value to compare across experiments, (C) BAL IgM normalized to the WT median value to compare across experiments, (D) TNF-α, (E) BAL TNF-α, (F) plasma granulocyte colony–stimulating factor, (G) BAL granulocyte colony–stimulating factor, (H) BAL polymorphonuclear leukocytes counts normalized to the WT median value to compare across experiments, and (I) BAL neutrophil elastase activity in WT (n = 24) and Gdf15−/− mice (n = 21) normalized to the WT median to compare across experiments. Each tube or point represents a single mouse. Groups were compared using a Mann-Whitney test. Nonsignificant comparisons are not displayed. G-CSF = granulocyte colony–stimulating factor. PMN = polymorphonuclear leukocyte.
Global Genetic Deficiency of Gfral Does Not Recapitulate the Lung Phenotype Seen in Gdf15−/− Mice during Lung Injury Induced by P. aeruginosa Exoproducts
GFRAL is a putative coreceptor for GDF15 in the hindbrain in mice and humans (3, 4). Therefore, we investigated whether genetic deficiency of Gfral would recapitulate the phenotype of decreased airway hemorrhage and decreased levels of TNF-α, G-CSF, and neutrophil elastase activity demonstrated in Gdf15−/− mice during acute lung injury elicited by PA SN. In contrast to Gdf15−/− mice, Gfral−/− mice had subtly increased levels of BAL hemorrhage on gross examination (Figure 5A) and quantification by OD540 (Figure 5B). Similar to Gdf15−/− mice, Gfral−/− mice exhibited no difference in cell counts, airspace recruitment of polymorphonuclear leukocytes, or lung injury as measured by BAL total protein (Figures 5C–5E). In anticipation of a potential compensatory response due to Gfral deficiency, we measured GDF15 levels in BAL and plasma. We found no difference in BAL levels of GDF15 (Figure 5F), but there was a trend toward increased GDF15 in the plasma (Figure 5G) of Gfral−/− mice that did not reach statistical significance. In contrast to Gdf15−/− mice, Gfral−/− mice exhibited no difference in levels of BAL or plasma G-CSF (Figures 5H and 5I), BAL neutrophil elastase activity (Figure 5J), or BAL TNF-α (Figure 5K). Because genetic deficiency of Gfral did not recapitulate genetic deficiency of Gdf15 during intratracheal PA SN challenge, we examined whether GDF15 may act locally to modulate the lung during pathogen-mediated injury.
Figure 5.

Global genetic deficiency of Gfral does not recapitulate the lung phenotype seen in Gdf15−/− mice during lung injury induced by P. aeruginosa exoproducts. WT (n = 7) and mice globally deficient for GFRAL (Gfral−/−; n = 6) were intratracheally inoculated with PA SN. (A) Gross images of BAL hemorrhage. (B) BAL OD540 showing increased hemorrhage in Gfral−/− mice. (C) BAL cellularity. (D) Polymorphonuclear cell counts in BAL assessed by manual differential count. (E) BAL total protein by bicinchoninic acid assay. (F–I) The indicated proteins were measured by ELISA in BAL or plasma. (J) BAL neutrophil elastase activity (OD400 at 24 h) and (K) BAL TNF-α. Each tube or point represents a single mouse. Groups were compared using a Mann-Whitney test. P values are displayed for all comparisons.
Global Genetic Deficiency of Gdf15 Alters the Lung Transcriptional Profile during Lung Injury Induced by P. aeruginosa Exoproducts
To probe potential local roles of Gdf15 in the lung during pathogen-mediated injury, we used a discovery approach with bulk RNA sequencing of snap-frozen lung obtained from wild-type mice and mice globally deficient for Gdf15 20 hours after intratracheal PA SN challenge. We found substantial differences in the transcriptional response in Gdf15−/− mice compared with wild-type mice. A total of 3,005 transcripts were differentially regulated (adjusted P < 0.05), or 15.8% of 19,064 total transcripts with read counts greater than 5. Volcano plot visualization suggested an overall downregulation in the transcriptional response in Gdf15−/− mice (Figure 6A). Heat map visualization of the top differentially transcribed genes revealed fewer RNA counts in Gdf15−/− mice for key genes such as Cxcl10 (Figure 6B). To help understand the potential consequences of this change in the transcriptional program, we first performed overrepresentation analysis for Gene Ontology for molecular functions in statistically significant (adjusted P < 0.01; q < 0.05) gene sets in upregulated (Figure 6C) and downregulated (Figure 6D) transcripts. Gene sets with overrepresentation in upregulated genes were notable for metabolic functions, whereas downregulated transcripts were overrepresented in a number of gene sets with functions in DNA binding and transcriptional regulation. We further performed gene set enrichment analysis using hallmark gene sets and identified statistically significantly (adjusted P < 0.05) increased enrichment for several gene sets in metabolic pathways (Figure 6E). In contrast, we noted decreased enrichment in gene sets for TNF-α signaling via NF-κB, KRAS signaling, IFN responses, and myogenesis. Together, these suggest a marked change in the host transcriptional response in the lung in the genetic absence of GDF15. Cxcl10 was among the most differentially regulated transcripts (−3.0 log2 fold change, adjusted P = 1.32 × 10−7 in Gdf15−/− mice), so we validated RNA sequencing at the protein level by quantifying CXCL10 levels in lung homogenate after normalization to total protein levels (Figure 6F) as well as in BAL fluid (Figure 6G). We found a more blunted response at the protein level, with an approximately twofold decrease in CXCL10 levels in lung homogenate and BAL, which was smaller than the approximately eightfold decrease at the RNA level. We then sought to understand whether airspace protein reconstitution of GDF15 could induce local changes in the lung.
Figure 6.

Global genetic deficiency of Gdf15 alters the lung transcriptional profile during lung injury elicited by intratracheal instillation of P. aeruginosa exoproducts. WT mice (n = 5) and mice globally deficient for GDF15 (Gdf15−/−; n = 5) were intratracheally inoculated with PA SN, and RNA was collected 20 hours after infection. (A) Volcano plot of normalized RNA counts with annotations showing 3,005 differentially expressed genes with adjusted P value <0.05. (B) Heat map of top differentially expressed genes after regularized log transformation of RNA counts and removal of pseudogenes clustered by row with genotype by column. (C and D) Dot plots of overrepresentation analysis for Gene Ontology molecular functions (P < 0.01, q < 0.05) in the 20 most upregulated (C) and downregulated (D) gene sets using all genes with statistically significant differential RNA counts (adjusted P < 0.05). (E) Gene set enrichment analysis of Hallmark pathways (adjusted P < 0.05) of all transcripts from control and Gdf15−/− mice. (F and G) CXCL10 was quantified by ELISA in lung homogenate (F) and BAL fluid (G). Each dot represents a single mouse. Groups were compared using a Mann-Whitney test.
Intratracheal Reconstitution Increases Plasma GDF15 Levels and Circulating Erythrocyte Counts during Lung Injury Induced by P. aeruginosa Exoproducts
To test the role of released GDF15 protein in the lung, we intratracheally administered equivalent volumes of vehicle (n = 9) or approximately 650 ng of recombinant murine GDF15 (rmGDF15; n = 12) to Gdf15−/− mice at the time of intratracheal challenge with PA SN. We used wild-type mice challenged with PA SN and vehicle as reference controls. Even 20 hours after intratracheal administration, BAL levels of GDF15 remained high in Gdf15−/− mice that received rmGDF15 (range, 4.9–31.5 ng/ml; Figure 7A), whereas the levels were undetectable in Gdf15−/− mice that received vehicle. Despite the high levels of GDF15 protein in the airspaces, we found only modest and nonsignificant increases in levels of lung homogenate CXCL10 (median increase, 18.3%; Figure 7B), BAL CXCL10 (median increase, 46.7%; Figure 7C), and BAL TNF-α (median increase, 4.3%; Figure 7D) in Gdf15−/− mice that received rmGDF15 compared with vehicle. Given the lack of local effects of protein reconstitution of GDF15 in the airspaces of knockout mice, we tested whether airspace GDF15 may have extrapulmonary roles.
Figure 7.

Intratracheal recombinant murine GDF15 at the time of instillation of P. aeruginosa exoproducts increases plasma levels of GDF15 and circulating erythrocyte counts in mice globally deficient for Gdf15. In two experiments, WT and Gdf15−/− mice were intratracheally administered recombinant murine GDF15 (rmGDF15) or an equivalent volume of vehicle (n = 11 WT with vehicle, n = 9 Gdf15−/− mice with vehicle, and n = 12 Gdf15−/− mice with rmGDF15) at the time of intratracheal inoculation with PA SN. Note that two mice were removed from the Gdf15−/− vehicle group and one mouse was removed from the Gdf15−/− rmGDF15 group because of an absence of lung injury observed at necropsy. BAL fluid, plasma, and lung tissue were collected 20 hours after infection. (A) BAL GDF15. (B) CXCL10 levels in homogenate of lavaged left lung (adjusted to total protein in homogenate) after normalization to the median of the Gdf15−/− vehicle group. (C) BAL CXCL10 levels after normalization to the median of the Gdf15−/− group. (D) BAL TNF-α levels after normalization. (E) Plasma GDF15. (F–H) Hemavet counts of plasma neutrophils (F), monocytes (G), and erythrocytes (H) after normalization. Each point represents a single mouse. Gdf15−/− vehicle and Gdf15−/− rmGDF15 groups were compared using a Mann-Whitney test. WT is displayed as reference. All P values for statistical comparisons are displayed.
We demonstrated that lower respiratory tract GDF15 levels correlate with plasma GDF15 levels in humans (Figure 1F), that PA SN–induced lung injury is sufficient to increase plasma GDF15 levels in mice (Figure 2C), and that lung epithelial deficiency of Gdf15 attenuates the increase in plasma GDF15 levels (Figure 2H). Indeed, intratracheal reconstitution of rmGDF15 was sufficient to dramatically increase the plasma levels of GDF15 (range, 0.5–2.1 ng/ml; Figure 7E), although the levels were approximately an order of magnitude lower than in the airspaces. GDF15 protein is required for stress erythropoiesis in human cells in vitro (8) and in mouse models of acute anemia (9). Therefore, we assessed whether intratracheal administration of GDF15 could modulate circulating blood counts. Intratracheal administration of GDF15 did not change circulating neutrophils (Figure 7F) or monocytes (Figure 7G), but significantly increased circulating erythrocyte counts (Figure 7H). Together, these data support that lung epithelial GDF15 can influence the systemic response to pathogen injury by modulating circulating erythrocyte counts.
Discussion
We performed a translational investigation of the role of GDF15 in pathogen-mediated lung injury in humans and mice. We found higher levels of GDF15 in the plasma of patients at risk for or diagnosed with ARDS as well as in nonsurvivors, consistent with previous reports that have found increased GDF15 in the context of injury or disease (2). We also found higher levels of GDF15 in the lower respiratory tracts of patients with ARDS compared with those intubated for nonpulmonary etiologies and demonstrated that levels of GDF15 in the lower respiratory tract correlate with plasma levels, which is the first such report to our knowledge. Furthermore, using publicly available data, we demonstrated that GDF15 is transcribed and translated by human lung epithelial cells during SARS-CoV-2 infection and that lower respiratory tract levels were higher in nonsurvivors in severe COVID-19. Together, our findings strongly support that the lung epithelium is a systemic source of GDF15, that GDF15 is produced in distinct infectious contexts, and that its production may be useful as a biomarker of severe injury.
To begin to dissect the functional role of secreted GDF15 during lung injury, we investigated its function in a mouse model of pathogen-mediated injury. Intratracheal instillation of P. aeruginosa exoproducts was sufficient to induce airspace and plasma GDF15 release, which were both significantly attenuated with lung epithelial–specific deficiency of Gdf15, further supporting the lung epithelium as a primary source of GDF15. When mice globally deficient for Gdf15 were treated with P. aeruginosa exoproducts, we found significantly decreased airspace hemorrhage without associated changes in barrier function. We also found a modulated airspace cytokine profile, including decreased TNF-α, G-CSF, and neutrophil elastase activity. Moreover, Gdf15 deficiency led to widespread transcriptional differences in lung tissues, including marked decreases in cytokine signaling pathways and Gene Ontology molecular function gene sets for nucleic acid binding (Figure 6D). The decreased hemorrhage phenotype and suppressed cytokine response of Gdf15−/− mice was not recapitulated in mice deficient for the central nervous system receptor for GDF15, Gfral, suggesting that these functions were mediated by a Gfral-independent signaling mechanism. Reconstitution of recombinant GDF15 into Gdf15−/− mice during P. aeruginosa challenge did not significantly alter the cytokine response in the lung. However, airspace GDF15 reconstitution increased plasma GDF15 levels and significantly increased erythrocyte counts, which is consistent with existing literature describing a role for GDF15 in stress erythropoiesis (8, 9, 40).
Steady-state erythropoiesis is highly regulated to balance production and removal of erythrocytes (40). In contrast, stress erythropoiesis can be induced during inflammation to rapidly increase erythrocyte counts (40). Stress erythropoiesis is thought to be independent of erythropoietin and to occur outside the bone marrow in the liver and spleen in mice (40, 41). Importantly, GDF15 protein is required to support the formation of stress erythropoiesis colonies in human bone marrow cells in vitro (8) and in murine spleen cells (9). We describe a novel finding of increased erythrocyte counts in Gdf15−/− mice after intratracheal administration of recombinant murine GDF15 that significantly increased plasma levels of GDF15 and led to erythrocyte counts similar to those in wild-type mice. We speculate that decreased airspace hemorrhage in Gdf15−/− mice during PA SN injury may result from decreased erythrocyte counts. Taken together, we propose that airspace release of GDF15 during pathogen-mediated injury increases plasma GDF15 levels to support stress erythropoiesis in mice, yielding a previously unrecognized lung–blood signaling pathway.
Despite our intriguing findings that suggest that lung GDF15 may have roles in lung–blood signaling, the receptor(s) by which GDF15 may operate in the lung and at extrapulmonary sites other than the brain remain(s) undefined. We generated animals lacking the putative GDF15 receptor, GFRAL, but found that they do not recapitulate the pulmonary phenotypes seen in mice lacking Gdf15. Several additional receptors have been proposed, including TGF-β receptors (42, 43), but definitive data supporting the interaction between GDF15 and alternative receptors are lacking. Given the lack of a well-defined receptor outside of the brain, the currently available data on GDF15 in the lung are insufficient to understand its suggested pleiotropic roles. We have used mouse models of genetic deficiency to explore the roles of GDF15, whereas others have used antibody neutralization (6, 44). Despite the difference in approach, there are similarities between our findings of decreased cytokine signaling in the airspaces during acute lung injury and the attenuation of lung fibrosis and fibroblast activation demonstrated in bleomycin-injured mouse lungs with neutralizing antibody–mediated GDF15 deficiency (44). To attempt to synthesize the possible roles for GDF15 in the lung during injury, we and others have noted lung epithelial expression of GDF15 (14, 44, 45), increased lung and lower respiratory tract levels during injury (44, 45), increased plasma levels (14), and modulation of local and remote functions such as cytokine signaling, modulation of fibroblast and macrophage activation (44, 46, 47), and stress erythropoiesis (8, 9). Further work is needed to understand the contribution of other cellular sources of GDF15 during pathogen injury such as lung endothelium and the mechanism(s) by which GDF15 may execute its roles.
We further propose that future research should consider whether GDF15 may have a bifurcation of functional roles between the cleaved, mature, secreted dimeric extracellular hormone acting upon GFRAL and potentially other sites such as stress erythroid progenitor cells versus the intracellular full-length propeptide, with roles that are currently poorly defined. For example, we noted decreased representation of transcriptional pathways in molecular function gene sets in Gdf15−/− mice during PA SN injury (Figure 6D). Others have described similarly meaningful differences in transcriptional programs with genetic deficiency of GDF15 (46, 48), and others have suggested that pro-GDF15 possesses a nonclassical nuclear localization sequence and can interact with Smad binding to DNA (49). Therefore, it remains poorly defined whether GDF15 may execute intracellular roles in the lung and what underlying mechanism(s) may account for any such roles, which are topics worthy of further investigation.
We note a specific limitation of our human studies, as we are unable to account for potential effects of GDF15 genotype on measured GDF15 levels in plasma and ETA obtained with a commercially available human ELISA kit (50). We anticipate an overall limited effect because of the relatively low frequency of the affected allele, and there should be no potential effect of genotype on the key finding that circulating and lower respiratory tract GDF15 levels are significantly correlated in individual patients. Additionally, we have not defined whether GDF15 may contribute to roles in lung host defense or whether other pathogens may elicit GDF15 release, and we have not determined whether GDF15 may have roles in the airspaces before injury.
In conclusion, we demonstrate in this translational investigation that lung epithelium transcribes and releases GDF15 in humans and mice in response to pathogen-mediated injury. Lower respiratory tract GDF15 levels are significantly correlated with circulating GDF15 levels during lung injury in humans and mice. In a mouse model of pathogen-mediated lung injury with genetic deficiency of GDF15, airspace reconstitution with recombinant GDF15 is sufficient to increase airspace and circulating GDF15 levels as well as circulating erythrocyte counts, which is consistent with prior reports of GDF15 roles in stress erythropoiesis. We speculate that GDF15 release by the lung may act as a hormone to modulate the extrapulmonary stress response, including stress erythropoiesis, thereby providing a previously unrecognized pathway of lung–blood communication that is of uncertain significance.
Acknowledgments
Acknowledgment
The authors thank the patients and patient families that have enrolled in the University of Pittsburgh Acute Lung Injury Registry; the physicians, nurses, respiratory therapists, and other staff at the University of Pittsburgh Medical Center Presbyterian, Shadyside, and East Hospital ICUs for assistance with coordination and collection of biospecimens; Haopu Yang, M.D., of the Tsinghua University School of Medicine (Beijing, China) for assistance with data analysis; the staff of the Translational Research Core Lab at the Division of Pulmonary, Allergy, Critical Care, and Sleep Medicine of the University of Pittsburgh for processing and banking of clinical samples and biomarker analysis associated with this study.
Footnotes
Supported by IK2 BX004886 from the U.S. Department of Veterans Affairs Biomedical Laboratory R&D Service (W.G.B.); National Heart, Lung, and Blood Institute of the National Institutes of Health awards R01 HL135062 (J.K.A.), K23 GM122069 (F.A.S.), R38HL150207 (N.J.T.), R03 HL162655 (G.D.K.), R01 HL159805 and R01 AA028436 (P.V.B.), T32 HL007563 (K.R.), R35 HL161177 (A.C.S.), R01 HL146519 (O.E.), K08 HL163324 (T.L.S.), P01HL114453 (J.S.L., P.R., and B.J.M.), and R01 HL136143, R01 HL142084, and K24 HL143285 (J.S.L.).
Author Contributions: F.A.S., J.K.A., and W.G.B. conceived, designed, performed, analyzed, and interpreted the data and wrote the manuscript. H.B., M.J., M.T., R.v.d.G., N.J.T., M.K., Z.X., N.A.-Y., K.S., and K.R. performed the experiments and analyzed the data. K.S. and P.V.B. conducted probabilistic graphical modeling, interpreted the data, and revised the work for important intellectual content. J.E., M.D.N., M.E.S., O.E., P.R., C.D.C., J.B., B.J.M., A.C.S., M.J.J., and T.L.S. interpreted the data and revised the work for important intellectual content. Y.Z., K.C., G.D.K., and J.S.L. designed and interpreted the data and revised the work for important intellectual content.
A data supplement for this article is available via the Supplements tab at the top of the online article.
Originally Published in Press as DOI: 10.1165/rcmb.2023-0429OC on February 1, 2024
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc Natl Acad Sci USA . 1997;94:11514–11519. doi: 10.1073/pnas.94.21.11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Verhamme FM, Freeman CM, Brusselle GG, Bracke KR, Curtis JL. GDF-15 in pulmonary and critical care medicine. Am J Respir Cell Mol Biol . 2019;60:621–628. doi: 10.1165/rcmb.2018-0379TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mullican SE, Lin-Schmidt X, Chin C-N, Chavez JA, Furman JL, Armstrong AA, et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat Med . 2017;23:1150–1157. doi: 10.1038/nm.4392. [DOI] [PubMed] [Google Scholar]
- 4. Emmerson PJ, Wang F, Du Y, Liu Q, Pickard RT, Gonciarz MD, et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat Med . 2017;23:1215–1219. doi: 10.1038/nm.4393. [DOI] [PubMed] [Google Scholar]
- 5. Xie B, Murali A, Vandevender AM, Chen J, Silva AG, Bello FM, et al. Hepatocyte-derived GDF15 suppresses feeding and improves insulin sensitivity in obese mice. iScience . 2022;25:105569. doi: 10.1016/j.isci.2022.105569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luan HH, Wang A, Hilliard BK, Carvalho F, Rosen CE, Ahasic AM, et al. GDF15 is an inflammation-induced central mediator of tissue Tolerance. Cell . 2019;178:1231–1244.e11. doi: 10.1016/j.cell.2019.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Santos I, Colaço HG, Neves-Costa A, Seixas E, Velho TR, Pedroso D, et al. CXCL5-mediated recruitment of neutrophils into the peritoneal cavity of Gdf15-deficient mice protects against abdominal sepsis. Proc Natl Acad Sci USA . 2020;117:12281–12287. doi: 10.1073/pnas.1918508117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xiang J, Wu DC, Chen Y, Paulson RF. In vitro culture of stress erythroid progenitors identifies distinct progenitor populations and analogous human progenitors. Blood . 2015;125:1803–1812. doi: 10.1182/blood-2014-07-591453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hao S, Xiang J, Wu D-C, Fraser JW, Ruan B, Cai J, et al. Gdf15 regulates murine stress erythroid progenitor proliferation and the development of the stress erythropoiesis niche. Blood Adv . 2019;3:2205–2217. doi: 10.1182/bloodadvances.2019000375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramirez J-M, Schaad O, Durual S, Cossali D, Docquier M, Beris P, et al. Growth differentiation factor 15 production is necessary for normal erythroid differentiation and is increased in refractory anaemia with ring-sideroblasts. Br J Haematol . 2009;144:251–262. doi: 10.1111/j.1365-2141.2008.07441.x. [DOI] [PubMed] [Google Scholar]
- 11. Wollert KC, Kempf T, Wallentin L. Growth differentiation factor 15 as a biomarker in cardiovascular disease. Clin Chem . 2017;63:140–151. doi: 10.1373/clinchem.2016.255174. [DOI] [PubMed] [Google Scholar]
- 12. Bao X, Borné Y, Muhammad IF, Nilsson J, Lind L, Melander O, et al. Growth differentiation factor 15 is positively associated with incidence of diabetes mellitus: the Malmö Diet and Cancer-Cardiovascular Cohort. Diabetologia . 2019;62:78–86. doi: 10.1007/s00125-018-4751-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tanno T, Lim Y, Wang Q, Chesi M, Bergsagel PL, Matthews G, et al. Growth differentiating factor 15 enhances the tumor-initiating and self-renewal potential of multiple myeloma cells. Blood . 2014;123:725–733. doi: 10.1182/blood-2013-08-524025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Y, Jiang M, Nouraie M, Roth MG, Tabib T, Winters S, et al. GDF15 is an epithelial-derived biomarker of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol . 2019;317:L510–L521. doi: 10.1152/ajplung.00062.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. LUNG SAFE Investigators; ESICM Trials Group Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA . 2016;315:788–800. doi: 10.1001/jama.2016.0291. [DOI] [PubMed] [Google Scholar]
- 16. Clark BJ, Bull TM, Benson AB, Stream AR, Macht M, Gaydos J, et al. ARDS Network Investigators Growth differentiation factor-15 and prognosis in acute respiratory distress syndrome: a retrospective cohort study. Crit Care . 2013;17:R92. doi: 10.1186/cc12737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kitsios GD, Yang H, Yang L, Qin S, Fitch A, Wang XH, et al. Respiratory tract dysbiosis is associated with worse outcomes in mechanically ventilated patients. Am J Respir Crit Care Med . 2020;202:1666–1677. doi: 10.1164/rccm.201912-2441OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bain W, Li H, van der Geest R, Moore SR, Olonisakin TF, Ahn B, et al. Increased alternative complement pathway function and improved survival during critical illness. Am J Respir Crit Care Med . 2020;202:230–240. doi: 10.1164/rccm.201910-2083OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Evankovich J, Lear T, Baldwin C, Chen Y, White V, Villandre J, et al. Toll-like receptor 8 stability is regulated by ring finger 216 in response to circulating microRNAs. Am J Respir Cell Mol Biol . 2020;62:157–167. doi: 10.1165/rcmb.2018-0373OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kitsios GD, Nouraie SM, Qin S, Zhang Y, Ray P, Ray A, et al. Distinct profiles of host responses between plasma and lower respiratory tract during acute respiratory failure. ERJ Open Res . 2023;9:00743-2022. doi: 10.1183/23120541.00743-2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kitsios GD, Yang L, Manatakis DV, Nouraie M, Evankovich J, Bain W, et al. Host-response subphenotypes offer prognostic enrichment in patients with or at risk for acute respiratory distress syndrome. Crit Care Med . 2019;47:1724–1734. doi: 10.1097/CCM.0000000000004018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitsios GD, Sayed K, Fitch A, Yang H, Britton N, Shah F, et al. Prognostic insights from longitudinal multicompartment study of host-microbiota interactions in critically ill patients. Res Sq . 2023. https://www.researchsquare.com/article/rs-3338762/v1
- 23. Drohan CM, Nouraie SM, Bain W, Shah FA, Evankovich J, Zhang Y, et al. Biomarker-based classification of patients with acute respiratory failure into inflammatory subphenotypes: a single-center exploratory study. Crit Care Explor . 2021;3:e0518. doi: 10.1097/CCE.0000000000000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Okubo T, Knoepfler PS, Eisenman RN, Hogan BLM. Nmyc plays an essential role during lung development as a dosage-sensitive regulator of progenitor cell proliferation and differentiation. Development . 2005;132:1363–1374. doi: 10.1242/dev.01678. [DOI] [PubMed] [Google Scholar]
- 25. Jiang M, Roth MG, Chun-On P, Sullivan DI, Alder JK. Phenotypic diversity caused by differential expression of SFTPC-Cre-transgenic alleles. Am J Respir Cell Mol Biol . 2020;62:692–698. doi: 10.1165/rcmb.2019-0416MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pelletier S, Gingras S, Green DR. Mouse genome engineering via CRISPR-Cas9 for study of immune function. Immunity . 2015;42:18–27. doi: 10.1016/j.immuni.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qu Y, Olonisakin T, Bain W, Zupetic J, Brown R, Hulver M, et al. Thrombospondin-1 protects against pathogen-induced lung injury by limiting extracellular matrix proteolysis. JCI Insight . 2018;3:e96914. doi: 10.1172/jci.insight.96914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bain W, Olonisakin T, Yu M, Qu Y, Hulver M, Xiong Z, et al. Platelets inhibit apoptotic lung epithelial cell death and protect mice against infection-induced lung injury. Blood Adv . 2019;3:432–445. doi: 10.1182/bloodadvances.2018026286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics . 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang P, Luo R, Zhang M, Wang Y, Song T, Tao T, et al. A cross-talk between epithelium and endothelium mediates human alveolar-capillary injury during SARS-CoV-2 infection. Cell Death Dis . 2020;11:1042. doi: 10.1038/s41419-020-03252-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med . 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 32. Palmer LD, Minor KE, Mettlach JA, Rivera ES, Boyd KL, Caprioli RM, et al. Modulating isoprenoid biosynthesis increases lipooligosaccharides and restores Acinetobacter baumannii resistance to host and antibiotic stress. Cell Rep . 2020;32:108129. doi: 10.1016/j.celrep.2020.108129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Trammell RA, Liberati TA, Toth LA. Host genetic background and the innate inflammatory response of lung to influenza virus. Microbes Infect . 2012;14:50–58. doi: 10.1016/j.micinf.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 34. Peñaloza HF, Olonisakin TF, Bain WG, Qu Y, van der Geest R, Zupetic J, et al. Thrombospondin-1 restricts interleukin-36γ-mediated neutrophilic inflammation during Pseudomonas aeruginosa pulmonary infection. MBio . 2021;12:e03336-20. doi: 10.1128/mBio.03336-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Filloux A. Protein secretion systems in Pseudomonas aeruginosa: an essay on diversity, evolution, and function. Front Microbiol . 2011;2:155. doi: 10.3389/fmicb.2011.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jyot J, Balloy V, Jouvion G, Verma A, Touqui L, Huerre M, et al. Type II secretion system of Pseudomonas aeruginosa: in vivo evidence of a significant role in death due to lung infection. J Infect Dis . 2011;203:1369–1377. doi: 10.1093/infdis/jir045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nickel N, Jonigk D, Kempf T, Bockmeyer CL, Maegel L, Rische J, et al. GDF-15 is abundantly expressed in plexiform lesions in patients with pulmonary arterial hypertension and affects proliferation and apoptosis of pulmonary endothelial cells. Respir Res . 2011;12:62. doi: 10.1186/1465-9921-12-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lipskaia L, Maisonnasse P, Fouillade C, Sencio V, Pascal Q, Flaman J-M, et al. Evidence that SARS-CoV-2 induces lung cell senescence: potential impact on COVID-19 lung disease. Am J Respir Cell Mol Biol . 2022;66:107–111. doi: 10.1165/rcmb.2021-0205LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Castellani S, D’Oria S, Diana A, Polizzi AM, Di Gioia S, Mariggiò MA, et al. G-CSF and GM-CSF modify neutrophil functions at concentrations found in cystic fibrosis. Sci Rep . 2019;9:12937. doi: 10.1038/s41598-019-49419-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Paulson RF, Hariharan S, Little JA. Stress erythropoiesis: definitions and models for its study. Exp Hematol . 2020;89:43–54.e2. doi: 10.1016/j.exphem.2020.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lenox LE, Shi L, Hegde S, Paulson RF. Extramedullary erythropoiesis in the adult liver requires BMP-4/Smad5-dependent signaling. Exp Hematol . 2009;37:549–558. doi: 10.1016/j.exphem.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johnen H, Lin S, Kuffner T, Brown DA, Tsai VW-W, Bauskin AR, et al. Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat Med . 2007;13:1333–1340. doi: 10.1038/nm1677. [DOI] [PubMed] [Google Scholar]
- 43. Artz A, Butz S, Vestweber D. GDF-15 inhibits integrin activation and mouse neutrophil recruitment through the ALK-5/TGF-βRII heterodimer. Blood . 2016;128:529–541. doi: 10.1182/blood-2016-01-696617. [DOI] [PubMed] [Google Scholar]
- 44. Radwanska A, Cottage CT, Piras A, Overed-Sayer C, Sihlbom C, Budida R, et al. Increased expression and accumulation of GDF15 in IPF extracellular matrix contribute to fibrosis. JCI Insight . 2022;7:e153058. doi: 10.1172/jci.insight.153058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Verhamme FM, Seys LJM, De Smet EG, Provoost S, Janssens W, Elewaut D, et al. Elevated GDF-15 contributes to pulmonary inflammation upon cigarette smoke exposure. Mucosal Immunol . 2017;10:1400–1411. doi: 10.1038/mi.2017.3. [DOI] [PubMed] [Google Scholar]
- 46. Al-Mudares F, Cantu Gutierrez M, Cantu A, Jiang W, Wang L, Dong X, et al. Loss of growth differentiation factor 15 exacerbates lung injury in neonatal mice. Am J Physiol Lung Cell Mol Physiol . 2023;325:L314–L326. doi: 10.1152/ajplung.00086.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Weng J-H, Koch PD, Luan HH, Tu H-C, Shimada K, Ngan I, et al. Colchicine acts selectively in the liver to induce hepatokines that inhibit myeloid cell activation. Nat Metab . 2021;3:513–522. doi: 10.1038/s42255-021-00366-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Deng M, Su D, Xiao N, Zhang Z, Wang Y, Zong F, et al. Gdf15 deletion exacerbates acute lung injuries induced by intratracheal inoculation of aerosolized ricin in mice. Toxicology . 2022;469:153135. doi: 10.1016/j.tox.2022.153135. [DOI] [PubMed] [Google Scholar]
- 49. Min KW, Liggett JL, Silva G, Wu WW, Wang R, Shen RF, et al. NAG-1/GDF15 accumulates in the nucleus and modulates transcriptional regulation of the Smad pathway. Oncogene . 2016;35:377–388. doi: 10.1038/onc.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Karusheva Y, Ratcliff M, Mörseburg A, Barker P, Melvin A, Sattar N, et al. The common H202D variant in GDF-15 does not affect its bioactivity but can significantly interfere with measurement of its circulating levels. J Appl Lab Med . 2022;7:1388–1400. doi: 10.1093/jalm/jfac055. [DOI] [PubMed] [Google Scholar]