

Abstract

Owing to stereoelectronic effects, lactones often deviate in reactivity from their open-chain ester analogues as demonstrated by the CH acidity (in DMSO) of 3-isochromanone (pKa = 18.8) and 2-coumaranone (pKa = 13.5), which is higher than that of ethyl phenylacetate (pKa = 22.6). We have now characterized the reactivity of the lactone enolates derived from 3-isochromanone and 2-coumaranone by following the kinetics of their Michael reactions with p-quinone methides and arylidenemalonates (reference electrophiles) in DMSO at 20 °C. Evaluation of the experimentally determined second-order rate constants k2 by the Mayr–Patz equation, lg k2 = sN(N + E), furnished the nucleophilicity parameters N (and sN) of the lactone enolates. By localizing their position on the Mayr nucleophilicity scale, the scope of their electrophilic reaction partners becomes predictable, and we demonstrate a novel catalytic methodology for a series of carbon–carbon bond-forming reactions of lactone enolates with chalcones under phase transfer conditions in toluene.
Introduction
The development of new applications of well-known reaction modes is of paramount importance in organic chemistry, which usually stems from the identification of new nucleophiles or electrophiles. When there is the necessity to develop catalytic reactions, a pronucleophile should have suitable Brønsted acidity to be deprotonated under relatively mild conditions to guarantee turnover during the reaction. At the same time, the deprotonated species should be sufficiently nucleophilic to react at a reasonable rate with the electrophilic reaction partner. In this context, monoesters, such as ethyl phenylacetate 1, have rarely been applied as pronucleophiles and required mostly the use of stoichiometric amounts of strong bases to undergo reactions.1 If additional strong electron-withdrawing groups (EWGs) are present on the aromatic ring in the ortho or para position to stabilize the formed ester enolate ion, 2-arylacetate esters become suitable for organocatalytic reactions.2 Another viable strategy includes the addition of isothiourea to activate the 2-phenylacetates as C1 ammonium enolates.3 The observed reactivity of arylacetates in organic syntheses can be ascribed to the relatively low Brønsted acidity of alkyl phenylacetates. For example, a pKa of 22.6 (in DMSO) has been estimated for ethyl phenylacetate 1 (Figure 1).4
Figure 1.

C–H acidities (pKa in DMSO, data from ref (4)) of the ester 1 and the lactones 2a and 3a.
Differently, the structurally related lactones 3-isochromanone (2a) and 2-coumaranone [3a, also known as benzofuran-2(3H)-one] have pKa(DMSO) values of 18.8 and 13.5, respectively, close to the C–H acidity level of methyl p-nitrophenylacetate 1′ (pKa = 15.1 in DMSO2b). The surprisingly high CH acidities of 2a and 3a, which are by 4 and 9 orders of magnitude stronger CH acids than 1, were rationalized as being a consequence of the locked s-(E) conformation of the alkoxy group, which is linked to the carbonyl carbon of the ester (or lactone) moiety. The s-(E) conformation causes ineffective nO → σ*CO interactions that can operate in the better-stabilized s-(Z) conformation of open chain esters.5
A limited number of catalytic reactions of 3-substituted 2-coumaranones 3 have been reported.6 However, to our knowledge, the use of 3-isochromanones (2) as pronucleophiles has so far almost been neglected. While SN2 alkylations at C-4 of 2a as well as Knoevenagel-type condensations with aldehydes have occasionally been studied,7,8 only one example for a conjugate addition to a Michael acceptor has been reported to date. Flintoft and co-workers generated the lithium enolate of lactone 2a, which reacted with nitroethene in THF (−78 °C to r.t., 30 min, 75% yield).9 This void of synthetic application is surprising since applications of benzolactones 2 and 3 can be correlated to natural products modification10,11 and are, thus, synthetically valuable.
As part of our research interest in the development of new methodologies for the synthesis of heterocyclic compounds,12 the aim of this work is the determination of nucleophilicity parameters of the lactone enolates of 2a and 3a in the framework of the Mayr reactivity scales13,14 and the investigation of their synthetic utility in Michael reactions. In particular, chalcones attracted our attention as Michael acceptors because they are an often-met motif in natural products and relevant in medicinal chemistry.15
Results and Discussion
Nucleophilicity of Lactone Enolates
The more than 5 orders of magnitude different pKa(DMSO) values of the lactones 2a and 3a already indicate that significantly different nucleophilic reactivities have to be expected for the corresponding lactone enolate species 4 and 5. Consequently, different sets of reference electrophiles 6 were chosen as the reaction partners for 4 and 5 in the kinetic measurements (Figure 2), which were carried out to quantify the nucleophilic reactivities of the lactone enolates in DMSO solution. The selected quinone methides and arylidenemalonates qualify as reference electrophiles because of their reliably determined electrophilic reactivity E on the Mayr scale16 and their favorable UV–vis absorbance ranges, which enabled us to follow the kinetics of their reactions with the lactone enolates 4 and 5 by photometric methods.
Figure 2.

Reference electrophiles used for the characterization of the nucleophilic lactone enolates 4 and 5. Electrophilicity parameters E are from refs (13a,14,16).
The lactone enolate 4 (counterion: Na+) was quantitatively generated in DMSO solution by deprotonation of 2a with sodium hydride (1.1 equiv).17 The more acidic lactone 3a was quantitatively deprotonated by the milder Brønsted base DBU (2.2 equiv) to generate DMSO solutions of the lactone enolate 5 (counterion: DBU-H+). In a first step, we investigated the products, which were formed in exemplary reactions of the lactone enolates with selected reference electrophiles. As shown in Scheme 1, all selected combinations of lactones and electrophiles reacted in the presence of different bases and solvents to the expected Michael adducts. Diastereomeric mixtures of the Michael adducts 7a–7c and 8, isolated with low diastereoselectivity, were obtained after aqueous workup. As a consequence of the selective and uniform product formations, we assumed the occurrence of analogous Michael additions for all further combinations of lactone enolates with other electrophiles, which were studied in the kinetic experiments.
Scheme 1. Michael Adducts of Reactions of Lactones 2a and 3a with Quinone Methides 6 under Basic Conditions.

The kinetics of adduct formation between 4 or 5 and the reference electrophiles 6 in DMSO (20 °C) were followed spectrophotometrically by utilizing stopped-flow techniques. The nucleophiles were used in at least 10-fold excess to achieve pseudo-first-order conditions, which enabled us to derive the first-order rate constants kobs (s–1) by least-squares fitting of the function At = A0 exp(−kobst) + C to the experimentally observed decay of the time-dependent absorbances of 6. Only for the reaction of 4 with 6a, the kinetic experiments were carried out by using the electrophile 6a in excess (>10 equiv).
For each nucleophile–electrophile combination, kobs was determined at four or five different concentrations of the excess reaction partner, which made it possible to calculate the second-order rate constants k2 (M–1 s–1) from the slope of the linear relationships of kobs with the nucleophile concentration (or electrophile concentration for the reaction of 4 with 6a). Details of all kinetics experiments are given in the Supporting Information, and the determined second-order rate constants k2 are listed in Table 1.
Table 1. Second-Order Rate Constants k2 (M–1 s–1) for the Reactions of Lactone Enolate Ions 4 and 5 with Reference Electrophiles 6 (in DMSO at 20 °C).
| lactone enolates | electrophiles | E | k2 (M–1 s–1) | N (sN)a |
|---|---|---|---|---|
| 4 (enolate of 2a) | 6a | –20.55 | 3.79 × 102 | 25.39 (0.54) |
| 6b | –17.90 | 1.40 × 104 | ||
| 6c | –17.29 | 2.04 × 104 | ||
| 6d | –16.38 | 6.68 × 104 | ||
| 5 (enolate of 3a) | 6c | –17.29 | 5.19 × 101 | 19.60 (0.75) |
| 6e | –16.11 | 3.60 × 102 | ||
| 6f | –15.03 | 3.06 × 103 | ||
| 6g | –14.36 | 1.31 × 104 | ||
| 6h | –13.39 | 3.28 × 104 | ||
| enolate of 1 | 6c | –17.29 | 5.51 × 105b | 27.54 (0.57)b |
| enolate of 1′ | 6c | –17.29 | 7.21 × 101b | 20.00 (0.71)b |
The second-order rate constants k2 were then evaluated by the Mayr–Patz eq 1.
| 1 |
In eq 1, the decadic logarithm of a second-order rate constants k2 for a nucleophile–electrophile reaction is expressed by the three parameters E, N, and sN. Given that the electrophilicities E of the reference electrophiles 6 were reported before, and the second-order rate constants k2 were experimentally determined in this work, the remaining parameters N and sN, which are characteristic for the reactivity of a nucleophile in a specific solvent, can be calculated. Thus, the quantitative descriptors N (and sN) for the nucleophilicities of 4 (N = 25.39, sN = 0.54) and 5 (N = 19.60, sN = 0.75) were derived from the linear correlations of lg k2 with the E parameters of the electrophilic reaction partners (Figure 3).
Figure 3.
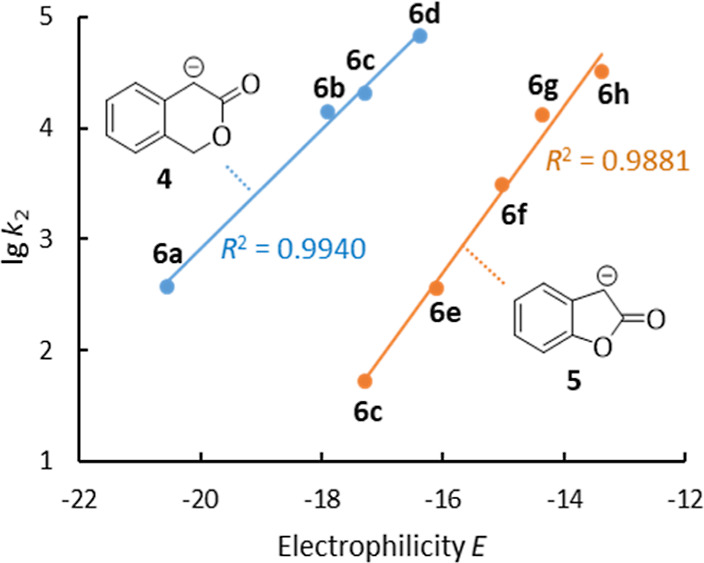
Determination of N and sN for the lactone enolates 4 and 5 from the linear correlations of lg k2 with the electrophilicity parameters E of the reference electrophiles 6.
Interestingly, the determined N parameters for the carbanions derived from 1, 1′, 2a, and 3a correlate linearly with the Brønsted acidities pKa of these CH acids. The scatter in Figure 4a (R2 = 0.9565) is rationalized by the fact that the slope parameters sN, which vary between 0.54 and 0.75 for this set of nucleophiles (see Table 1), were neglected when constructing the graph. Linear correlations of higher precision can be expected if rate constants for reactions of the enolates with a common electrophile are plotted against the pKa values of 1, 1′, 2a, and 3a.
Figure 4.

(a) Correlation of enolate nucleophilicities N with pKa of the corresponding CH acids 1, 1′, 2a, and 3a. (b) The Brønsted plot shows a linear relationship between the enolate reactivities (lg k2) toward 6c and the acidity constants pKa of the corresponding CH acids 1, 1′, 2a, and 3a.
Accordingly, the Brønsted correlation in Figure 4b illustrates that the nucleophilic reactivities (lg k2) of the ester and lactone enolates in reactions with quinone methide 6c correlate excellently with the Brønsted basicities of the CH acids 1, 1′, 2a, and 3a in DMSO (R2 = 0.9740). The slope of 0.476 in Figure 4b is similar to the one reported for an analogous correlation for a series of benzyl anions stabilized by ethoxycarbonyl, nitro, cyano, and sulfonyl groups (slope = 0.438, n = 11),18 which covered 14 orders of magnitude on the pKa scale but showed much higher uncertainties (R2 = 0.74) owing to the structurally more diverse set of carbanions that undergo reactions via variable intrinsic barriers.18
By using the Mayr nucleophilicity parameters N and sN, it is now possible to rationalize published synthetic procedures that involve the investigated lactone enolates. For example, Tominaga and co-workers reported that a mixture of 2-coumaranone 3a, sodium hydroxide, and carbon disulfide in DMSO reacted at 10–15 °C to form an anionic adduct, which was trapped by methylation with iodomethane to form 3-bis(methylthio)methylene-2-coumaranone (Scheme 2a).19 This procedure is in good agreement with a moderately rapid reaction that can be expected from a second-order rate constant of k2 = 27 M–1 s–1 (at 20 °C), which is calculated by using eq 1, the N (and sN) parameter of 5 in DMSO, and the electrophilicity E = −17.70 for CS2.20
Scheme 2. Michael Adducts of Reactions of Lactones 2a and 3a with (a) Carbon Disulfide, (b) Methyl Acrylate (9), (c) But-3-enone (11), and (d) Chalcone (13a).

Yields of isolated product after chromatographic purification.
On the fundament of their nucleophilicity parameters, we attempted to further explore the synthetic scope of 2a and 3a and investigated their reactivity toward prototypical Michael acceptors, such as methyl acrylate (9) or the α,β-unsaturated ketones 11 and 13a. The reactions of 3a with the β-unsubstituted electrophiles 9 (E = −18.84)21 and 11 (E = −16.76),21 both gave the 1:2 adducts 10 and 12, respectively (Scheme 2b,c).
Interestingly, the parent chalcone 13a (E = −19.39)21 and lactone 2a under basic conditions in MeCN formed product 14a in good yield. The 4-monofunctionalized lactone 14a corresponds to the 1:1-Michael adduct and was isolated as a 1:1 mixture of diastereomers (Scheme 2d).
Base-Catalyzed Michael Additions of Lactones to Chalcones
Aiming to develop a catalytic process, we optimized the adduct formation of 2a with the parent chalcone 13a under phase transfer conditions. In entry 1 of Table 2, lactone 2a and chalcone 13a were mixed in toluene in the presence of K2CO3 (1 equiv.). Not unexpectedly, the starting materials were recovered completely after 24 h at ambient temperature (Table 1, entry 1) because of the insufficient solubility of potassium carbonate in toluene. The use of the phase transfer catalyst tetra-n-butyl ammonium bromide (TBAB) led to 14a in 90% yield within 2 h (entry 2), which was both a significantly shorter reaction time and a higher yield of 14a than from the analogous reaction performed in the more polar solvent acetonitrile but without the phase transfer catalyst (Scheme 2d). Comparable yields of 14a were obtained even when decreasing the amounts of both TBAB and K2CO3 to 0.05 and 0.1 equiv, respectively (compare with entries 3 and 4). The catalysts amount was further decreased to 0.01 equiv with a concomitant scale up at 1 mmol, leading to high yield as well but requiring a longer reaction time (entry 5). However, the conditions were adjusted for practical reasons using 0.1 equiv of both TBAB and K2CO3, providing product formation in almost quantitative yield over 5 h (entry 6). These conditions can also be scaled up to 1 mmol with minimal impact on product yield (89% yield). The use of Li2CO3 proved to be less effective, probably because of the lower counteranion exchange rate with TBAB (entry 7). On the other hand, when the significantly less Brønsted acidic ethyl 2-phenylacetate 1 was subjected to the same conditions, even after mixing for 24 h, we recovered the starting materials unreacted. This demonstrates the importance of the pKa of the pronucleophiles’ C–H bonds for the development of effective catalytic reactions (Scheme 3).
Table 2. Optimization of the Michael Adduct Formation between Chalcone (13a) and Lactone 2a.

| entry | NBu4Br (equiv) | base (equiv) | molarity [2a] | time (h) | yield (%)a |
|---|---|---|---|---|---|
| 1 | K2CO3 (1.0) | 0.337 | 24 | -b | |
| 2 | 0.20 | K2CO3 (1.0) | 0.337 | 2 | 90 |
| 3 | 0.05 | K2CO3 (1.0) | 0.337 | 2 | 88 |
| 4 | 0.05 | K2CO3 (0.10) | 0.337 | 2.5 | 86 |
| 5 | 0.01 | K2CO3 (0.01) | 0.675 | 48 | 88 |
| 6 | 0.10 | K2CO3(0.10) | 0.675 | 5 | 97 |
| 7 | 0.10 | Li2CO3 (0.10) | 0.675 | 36 | 80 |
Isolated yield.
Starting materials recovered.
Scheme 3. Open-Chain Ester 1 Did Not React with Chalcone 13a under the Optimal Conditions of Table 2, Entry 6.

The scope of the lactone–chalcone adduct formation was further explored under the optimized conditions of entry 6 of Table 2. To assess the full extent of this reaction, we synthesized several substituted 3-isochromanones with a range of electronic properties incorporated on the aromatic ring [2b–2f, Supporting Information]. Several chalcones 13 reacted with differently substituted 3-isochromanones 2a–2f with both reactants bearing halogens, electron-donating groups and EWGs in different positions on the aromatic rings, showing variable reaction time as detected by thin-layer chromatography (TLC) (used to monitor the disappearance of 2), obtaining 14 in moderate to excellent yields (Table 3). In the case of (E)-3-(2-chlorophenyl)-1-phenylprop-2-en-1-one 13d, a high reaction temperature was required, probably because of the increased steric hindrance (entry 4), while at room temperature, we did not observe any reaction. 7-Nitroisochroman-3-one 2d was not reactive at room temperature, and a higher reaction temperature (110 °C) was necessary to achieve a moderate yield in the Michael addition with the parent chalcone 13a (entry 13). In this case, the nitro group leads to a decrease of the pKa, stabilizing the carbanion by resonance, and its nucleophilicity is lower than that of 4, explaining the observed reactivity. On the chalcone side, even (E)-3-(furan-2-yl)-1-phenylprop-2-en-1-one (13j), the presumably least electrophilic Michael acceptor in Table 3, gave a good yield in the reaction with 2a to give product 14j (entry 10).
Table 3. Scope Analysis for the Base-Catalyzed Michael Addition of Lactones 2 and Chalcones 13.

| entry | lactones | chalcones | Ar | Ar′ | time (h) | yield (%)a | drb |
|---|---|---|---|---|---|---|---|
| 1 | 2a, R = H | 13a | Ph | Ph | 5 | 14a, 97 | 50/50 |
| 2 | 2a, H | 13b | 4-ClC6H4 | Ph | 5 | 14b, 97 | 50/50 |
| 3 | 2a, H | 13c | 2-NO2C6H4 | Ph | 7 | 14c, 68 | 56/44 |
| 4c | 2a, H | 13d | 2-ClC6H4 | Ph | 18 | 14d, 62 | 60/40 |
| 5 | 2a, H | 13e | 4-FC6H4 | Ph | 7 | 14e, 91 | 53/47 |
| 6 | 2a, H | 13f | 4-MeOC6H4 | Ph | 9 | 14f, 82 | 55/45 |
| 7 | 2a, H | 13g | Ph | 3-CF3C6H4 | 6 | 14g, 76 | 53/47 |
| 8 | 2a, H | 13h | Ph | 4-NO2C6H4 | 8 | 14h, 77 | 52/48 |
| 9 | 2a, H | 13i | Ph | 4-MeOC6H4 | 10 | 14i, 73 | 53/47 |
| 10 | 2a, H | 13j | 2-furyl | Ph | 10 | 14j, 65 | 58/42 |
| 11 | 2b, 7-Br | 13a | Ph | Ph | 6 | 14k, 81 | 54/46 |
| 12 | 2c, 6-OMe | 13a | Ph | Ph | 3 | 14l, 96 | 50/50 |
| 13d | 2d, 7-NO2 | 13a | Ph | Ph | 12 | 14m, 58 | 53/47 |
| 14 | 2e, 6-Cl | 13a | Ph | Ph | 9 | 14n, 94 | 52/48 |
| 15 | 2c, 6-OMe | 13f | 4-MeOC6H4 | Ph | 3 | 14o, 93 | 51/49 |
| 16 | 2f, 8-F | 13a | Ph | Ph | 7 | 14p, 90 | 59/41 |
Isolated yield.
Determined by 1H NMR spectroscopic analysis of the crude material.
Reaction temperature at 60 °C.
Reaction temperature at 110 °C.
Low diastereoselectivity was usually observed, but the majority of diastereomeric mixtures were easily separated by chromatography or crystallization. In the case of the chloro-substituted 14b, crystals suitable for single-crystal X-ray analysis were obtained by slow evaporation of a solution of 14b (5 mg) in a dichloromethane/hexane mixture (1 mL, v/v = 1:5) allowing for determination of the relative configuration as (S*,S*) (Figure 5). This was extended to all the series, considering the similarity of the 1H NMR spectra and of the retention factors observed in chromatography.
Figure 5.
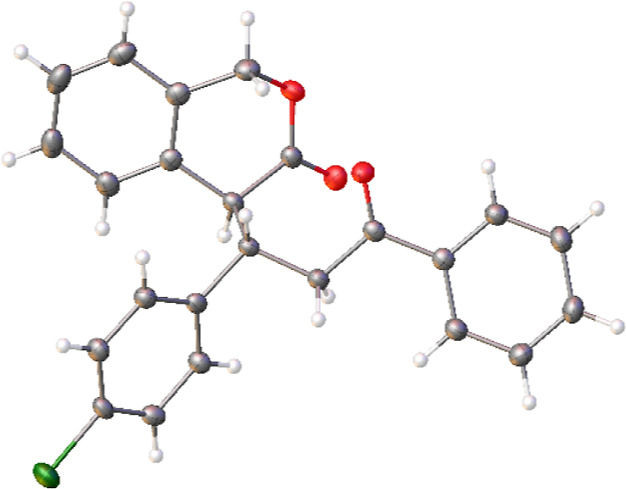
ORTEP diagram of 14b-1.22 Crystals of 14b-1 were obtained by slow evaporation of a solution of 14b-1 (5 mg) in a dichloromethane/hexane mixture (1 mL, 1:5).
Although the enolate of 2-coumaranone 3a is less nucleophilic than that of 3-isochromanone 2a, it reacted successfully with chalcones 13 under the conditions of entry 6 from Table 2. Several substituted 2-coumaranones were reacted with chalcones bearing different substituents on both the aromatic rings as well, leading from good to high yields and from moderate to low diastereoselectivity (Table 4). Of note, 5-chloro-coumaranone 3c required a higher temperature and longer reaction time (Table 4, entry 8). Only 5-nitro-coumaranone 3d did not react at all, even at 80 °C, because of the decrease of its nucleophilicity (entries 9 and 10) by the electron-withdrawing and, therefore, anion-stabilizing properties of the nitro group (Hammett substituent constant σm = 0.7123). In this series, the separation of the diastereomers was more problematic and only in the case of 15d did attempts at crystallization yield crystals suitable for X-ray analysis, depicted in Figure 6, allowing for the determination of the relative configuration as (S*,R*).
Table 4. Analysis of the Scope with 2-Coumaranones 3 and Chalcones 13.

| entry | lactones | chalcones | Ar | Ar′ | time (h) | yield (%)a | d.rb |
|---|---|---|---|---|---|---|---|
| 1 | 3a, R = H | 13a | Ph | Ph | 4 | 15a, 81 | 64/36 |
| 2 | 3a, H | 13b | 4-ClC6H4 | Ph | 5.5 | 15b, 83 | 53/47 |
| 3 | 3a, H | 13f | 4-MeOC6H4 | Ph | 5 | 15c, 87 | 54/46 |
| 4 | 3b, 5-OAc | 13a | Ph | Ph | 2.5 | 15d, 89 | 59/41 |
| 5 | 3a, H | 13i | Ph | 4-MeOC6H4 | 5 | 15e, 83 | 62/38 |
| 6 | 3a, H | 13d | 2-ClC6H4 | Ph | 8.5 | 15f, 81 | 54/46 |
| 7 | 3a, H | 13e | 4-FC6H4 | Ph | 6.5 | 15g, 79 | 64/36 |
| 8c | 3c, 5-Cl | 13a | Ph | Ph | 36 | 15h, 75 | 63/37 |
| 9d | 3d, 5-NO2 | 13a | Ph | Ph | 24 | -e | - |
| 10d | 3d, 5-NO2 | 13f | 4-MeOC6H4 | Ph | 40 | -e | - |
Isolated yield.
Determined by 1H NMR on the crude material.
Reaction temperature at 50 °C.
Reaction temperature at 80 °C.
Starting materials recovered.
Figure 6.
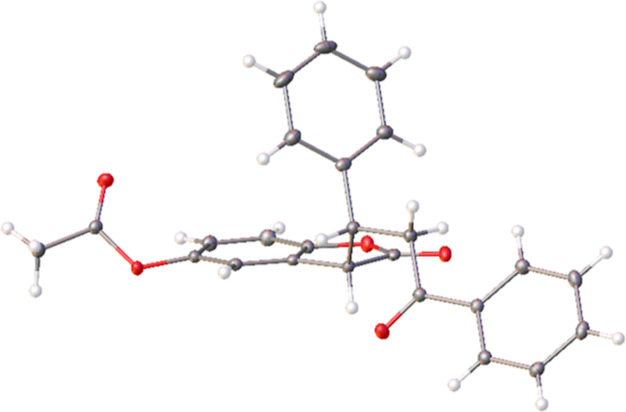
ORTEP diagram of 15d-1.22 Crystals of 15d-1 were obtained by slow evaporation of a solution of 15d (10 mg) in a diethyl ether/isopropanol mixture (1 mL, v/v = 1:4).
Conclusions
In this article, we explored the reactivity of isochroman-3-ones and 2-coumaranones as nucleophiles in Michael reactions. Based on the Mayr–Patz eq 1 and by following the kinetics of the reactions of the respective lactone enolates with a series of reference electrophiles (quinone methides and arylidenemalonates) with known electrophilicity E, the nucleophilicity parameters N (sN) of the corresponding lactone enolates 4 and 5 in DMSO were determined to be 25.39 (0.54) and 19.60 (0.75), respectively. Considering the higher Brønsted acidity of isochroman-3-one and 2-coumaranone in comparison to their noncyclic structurally analogous alkyl phenylacetates, along with their high level of nucleophilic reactivity, an efficient catalytic method for Michael additions was developed. In particular, series of substituted isochroman-3-ones and 2-coumaranones were synthesized and then shown to react with chalcones. In combination with the phase transfer catalyst tetra-n-butylammonium bromide (0.1 equiv), the low-cost Brønsted base K2CO3 (0.1 equiv) could be used in catalytic amounts to activate the lactones in toluene and, thus, widen the scope of this carbon–carbon bond-forming reaction. Based on these findings, further studies about the development of asymmetric versions of the investigated Michael additions and other applications in reactions with carbon-carbon and carbon-heteroatom bond formation are ongoing.
Experimental Section
General Information
Unless otherwise noted, all chemicals, reagents, and solvents for the performed reactions are commercially available. Isochroman-3-one was purchased from fluorochem, and substituted isochroman-3-ones were prepared according to literature procedures (see the Supporting Information for further details). All the reactions were monitored by TLC on precoated silica gel plates (0.25 mm) and visualized by fluorescence quenching at 254 nm. Flash chromatography was carried out using silica gel 60 (70–230 mesh, Merck, Darmstadt, Germany). Preparative TLC (PrepTLC) was carried out on silica gel plates (200 × 200 × 0.5 mm). The products were detected under UV light, extracted with ethyl acetate, and obtained after rotary evaporation. Yields are given for isolated products showing one spot on a TLC plate. The NMR spectra were recorded on Bruker DRX 600, 400, and 300 MHz spectrometers (600 MHz, 1H, 150 MHz, 13C; 400 MHz, 1H, 100.6 MHz; 13C, 300 MHz, 1H, 75.5 MHz, 13C, 250 MHz, 1H, 62.5 MHz, 13C). Internal reference was set to the residual solvent signals (δH 7.26 ppm, δC 77.16 ppm for CDCl3).24 The 13C{1H} NMR spectra were recorded under broad-band proton-decoupling and reported Cq, CH, CH2, or CH3 assignments were based on additional heteronuclear single quantum coherence (HSQC) and heteronuclear multiple bond correlation (HMBC) experiments. 1H NMR and high-resolution mass spectrometry (HRMS) data are reported for all compounds. IR and 13C NMR data are given only for new compounds. The following abbreviations are used to indicate the multiplicity in NMR spectra: s—singlet, d—doublet, t—triplet, q—quartet, dd—doublet of doublets, m—multiplet, brs—broad signal. High-resolution mass spectra were acquired by the Salerno team using a Bruker SolariX XR Fourier transform ion cyclotron resonance mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany) equipped with a 7T refrigerated actively shielded superconducting magnet. For ionization of the samples electrospray ionization (ESI) or matrix-assisted laser desorption/ionization (MALDI) was applied. In Munich, HRMS was performed by using a Thermo Finnigan LTQ FT (ESI) or a Thermo Finnigan MAT 95 instrument (EI). IR spectra were recorded on a IR Bruker Vertex 70v spectrometer.
Kinetics
The kinetics of the reactions of the lactone enolates with the reference electrophiles 6 were followed by UV/vis spectroscopy (Applied Photophysics SX20 stopped-flow spectrophotometer). A constant temperature (20.0 ± 0.2 °C) was maintained through the use of a circulating bath cryostat. All solutions were freshly prepared under an atmosphere of dry argon by using dry DMSO (over molecular sieves, Acros Organics). Solutions of sodium 3-oxoisochroman-4-ide (4) in DMSO were prepared by deprotonation of 3-isochromanone (2a) with sodium hydride. Solutions of 2-oxo-2,3-dihydrobenzofuran-3-ide (5) in DMSO were prepared by deprotonation of 2-coumaranone (3a) with DBU (2.2 equiv.).
The kinetic measurements were initiated by mixing equal volumes of DMSO solutions of the nucleophiles and electrophiles. Selected reactions of 4 with the electrophiles 6 were measured with and without added crown ether (15-crown-5, 1.1 equiv relative to the concentration of 4). In general, nucleophile concentrations were at least ten times higher than electrophile concentrations to achieve pseudo-first-order kinetics. Only for the reaction of 4 with 6a (Table S1), the kinetic experiments were carried out by using the electrophile 6a in excess (>10 equiv). The first-order rate constants kobs (s–1) could be obtained from the decay of the absorbance at or close to the absorption maximum of the reaction partner used in lower concentration by least-squares fitting of the equation At = A0 exp(−kobst) + C to the exponential absorption decay curve. Plots of kobs (s–1) versus the nucleophile concentration (or electrophile concentration for the reaction of 4 with 6a) gave the second-order rate constants k2 (M–1 s–1) as slopes of the linear correlations.
4-((3,5-Di-tert-butyl-4-hydroxyphenyl)(julolidin-9-yl)-λ3-methyl)-4λ3-isochroman-3-one (7a)
Procedure A
3-Isochromanone 2a (31.2 mg, 0.211 mmol), pQM 6b (77.5 mg, 0.199 mmol), and potassium tert-butoxide (30.3 mg, 0.270 mmol) were mixed in DMSO (13 mL) and stirred for 10 min. Then, the reaction mixture was poured on 0.5% aq. acetic acid (50 mL). The phases were separated, and the aqueous phase was extracted with ethyl acetate (3 × 40 mL). The combined organic phases were washed with brine (2 × 40 mL), dried over sodium sulfate, and filtered. Partial crystallization by slow diffusion of pentane into a dichloromethane solution of the diastereomeric mixture of 7a at 4 °C (fridge) delivered a small amount of diastereomerically pure cubic crystals of 7a (13.9 mg, yield: 13%), which were characterized by X-ray crystallography (bv020).22 A solution of the diastereomerically pure crystals in CDCl3 was used for the NMR spectroscopic characterization of 7a-major.
Procedure B
3-Isochromanone 2a (42.5 mg, 0.287 mmol) and sodium hydride (7.1 mg, 95% purity, 0.281 mmol) were dissolved in DMSO (5 mL) under an argon atmosphere at room temperature and stirred for 45 min. Then, a solution of pQM 6b (101 mg, 0.259 mmol) in a DMSO/dichloromethane mixture (4.5 mL/2.5 mL) was added. The reaction mixture was stirred for 1 h and then poured on 0.5% aq. acetic acid (50 mL). The layers were separated, and the aq. phase was extracted with ethyl acetate (3 × 20 mL). The combined organic phases were washed with brine (2 × 20 mL) and dried over MgSO4. After filtration, volatiles were evaporated to obtain the crude product (146 mg). Purification by column chromatography (silica gel, n-pentane/ethyl acetate = 7:1) furnished 7a (92.7 mg, yield: 67%); a mixture of diastereomers, d.r. 1:1.6. NMR spectra of this sample were used to assign 1H and 13C resonances of 7a-minor.
7a-major: 1H NMR (400 MHz, CDCl3, δ) 7.23–7.19 (m, 1H), 7.10–7.07 (m, 1H), 7.04–7.02 (m, 1H), 6.84 (s, 2H), 6.72 (s, 2H), 6.61–6.59 (m, 1H), 5.05 (s, 1H), 4.93 (d, J = 14.3 Hz, 1H), 4.54 (d, J = 14.1 Hz, 1H), 4.37 (d, J = 6.8 Hz, 1H), 4.30 (d, J = 7.0 Hz, 1H), 3.10 (dd, J = 6.3 Hz, 5.0 Hz, 4H), 2.79–2.67 (m, 4H), 2.00–1.93 (m, 4H), 1.28 (s, 18H). 13C{1H} NMR (101 MHz, CDCl3, δ) 172.4 (Cq), 152.8 (Cq), 142.0 (Cq), 135.4 (Cq), 133.6 (Cq), 131.9 (Cq), 130.5 (Cq), 129.0 (CH), 127.7 (CH), 127.6 (Cq), 127.3 (CH), 127.2 (CH), 126.0 (CH), 124.0 (Cq), 121.5 (Cq), 69.9 (CH2), 54.4 (CH, Ar2CH), 53.4 (CH), 50.2 (CH2), 34.3 (Cq), 30.3 (CH3), 27.8 (CH2), 22.3 (CH2). HRMS (ESI): m/z calcd for C36H44NO3+ (M + H+) 538.3316; found: 538.3305.
7a-minor: 1H NMR (400 MHz, CDCl3, δ) 6.57 (s, 2H), 5.08 (s, 1H), 4.87 (d, J = 14.2 Hz, 1H), 4.23 (d, J = 5.6 Hz, 1H), 2.64–2.61 (m, 4H), 1.37 (s, 18H); further resonances could not unequivocally be assigned because of superimposition with resonances of 7a-major. 13C{1H} NMR (101 MHz, CDCl3, δ) 172.0, 152.8, 142.0, 135.6, 134.1, 132.2, 131.4, 128.9, 127.8, 127.7, 127.6, 127.2, 125.7, 123.7, 121.3, 69.9, 56.2, 52.9, 50.2, 34.5, 30.4, 27.8, 22.3.
4-((3,5-Di-tert-butyl-4-hydroxyphenyl)(4-(dimethylamino)phenyl)-λ3-methyl)-4λ3-isochroman-3-one (7b)
The product was obtained from 3-isochromanone 2a (29.6 mg, 0.200 mmol), pQM 6c (65.3 mg, 0.193 mmol), and potassium tert-butoxide (31.4 mg, 0.280 mmol) by analogously following Procedure A (cf. synthesis of 7a). Purification of the crude product by column chromatography (silica gel, pentane/ethyl acetate = 7:1) furnished 7b (17.1 mg, yield: 18%); a mixture of diastereomers, d.r. 1:2.
1H NMR (400 MHz, CDCl3, δ) 7.30–7.28 (m, 4H), 7.25–7.17 (m, 4H), 7.11–7.02 (m, 9H), 6.78–6.76 (m, 5H), 6.72–6.70 (m, 4H), 6.62–6.58 (m, 4H), 5.10 (s, 1 Hminor), 5.07 (s, 2 Hmajor), 4.94 (d, J = 14.2 Hz, 2 Hmajor), 4.89 (d, J = 14.3 Hz, 1 Hminor), 4.57 (d, J = 14.2 Hz, 1 Hmajor), 4.46 (d, J = 6.2 Hz, 1 Hminor), 4.43 (br s, 4 Hmajor), 4.34 (d, J = 6.1 Hz, 1 Hminor), 4.39 (d, J = 14.0 Hz, 1 Hminor), 2.94 (s, 12 Hmajor), 2.91 (s, 6 Hminor), 1.37 (s, 18 Hminor), 1.29 (s, 38 Hmajor); reported integrals refer to 1H (=1.0) of the minor diastereomer. 13C{1H} NMR (101 MHz, CDCl3, δ) 172.2 (Cqmajor), 171.8 (Cqminor), 152.9 (Cqmajor+minor), 149.65 (Cqminor), 149.61 (Cqmajor), 135.7 (Cqminor), 135.5 (Cqmajor), 134.1 (Cqminor), 133.6 (Cqmajor), 132.0 (Cqminor), 131.8 (Cqmajor), 131.2 (Cqminor), 130.5 (Cqmajor), 129.7 (CHminor), 129.4 (CHmajor), 128.91 (CHmajor), 128.88 (CHminor), 128.84 (Cqminor), 128.4 (Cqmajor), 128.0 (CHminor), 127.9 (CHmajor), 127.29 (CHminor), 127.27 (CHmajor), 126.0 (CHmajor), 125.7 (CHminor), 124.1 (CHmajor), 123.9 (CHminor), 112.7 (CHmajor), 112.5 (CHminor), 69.90 (CH2major), 69.88 (CH2minor), 55.9 (CH, lactone-CHminor or Ar2CHminor), 54.4 (CH, lactone-CHmajor or Ar2CHmajor), 53.2 (CH, lactone-CHmajor or Ar2CHmajor), 52.6 (CH, (CH, lactone-CHminor or Ar2CHminor)), 40.75 (CH3minor), 40.74 (CH3major), 34.5 (Cqminor), 34.3 (Cqmajor), 30.4 (CH3minor), 30.3 (CH3major). HRMS (ESI): m/z calcd for C32H40NO3+ (M + H+): 486.3003; found: 486.2995.
4-((4-Hydroxy-3,5-dimethoxyphenyl)(4-methoxyphenyl)-λ3-methyl)-4λ3-isochroman-3-one (7c)
The product was obtained from 3-isochromanone 2a (38.4 mg, 0.259 mmol), sodium hydride (7.1 mg, 95% purity, 0.281 mmol), and pQM 6d (61.0 mg, 0.224 mmol) by analogously following Procedure B (cf. synthesis of 7a). Purification of the crude product by column chromatography (silica gel, n-pentane/ethyl acetate = 1:1) furnished a yellow oil (74.8 mg). The content of residual ethyl acetate in the sample was determined from the integrals in the 1H NMR spectrum (7.9 mg) and the yield of 7c calculated (66.8 mg, yield: 71%); a mixture of diastereomers, d.r. 1:1.3.
1H NMR (599 MHz, CDCl3, δ) 7.29–7.24 (m, 6.6H), 7.16–7.13 (m, 2.4H), 7.11–7.08 (m, 2.4H), 7.04–7.03 (m, 2 Hminor), 6.88–6.85 (m, 2.7 Hmajor), 6.77–6.74 (m, 2 Hminor), 6.73–6.71 (m, 2.3H), 6.52 (s, 2 Hminor), 6.27 (s, 2.6 Hmajor), 5.04 (d, J = 14.2 Hz, 1.3 Hmajor), 5.02 (d, J = 14.1 Hz, 1 Hminor), 4.84 (d, J = 14.3 Hz, 1.3 Hmajor), 4.77 (d, J = 14.3 Hz, 1 Hminor), 4.51–4.49 (m, 2.3 Hmajor+minor), 4.39–4.37 (m, 2.3 Hmajor+minor), 3.81 (s, 6 Hminor), 3.80 (s, 4.0 Hmajor), 3.77 (s, 3 Hminor), 3.69 (s, 7.9 Hmajor); reported integrals refer to 1H (=1.0) of the minor diastereomer. 13C{1H} NMR (151 MHz, CDCl3, δ) 171.5 (Cqmajor), 171.3 (Cqminor), 158.8 (Cqmajor), 158.7 (Cqminor), 147.0 (Cqminor), 146.8 (Cqmajor), 134.0 (Cqminor), 133.8 (Cqmajor), 133.50 (Cqmajor), 133.48 (Cqminor), 132.5 (Cqminor), 132.3 (Cqmajor), 131.65 (Cqminor), 131.61 (Cqmajor), 131.5 (Cqminor), 131.3 (Cqmajor), 129.9 (CHminor), 129.6 (CHmajor), 128.73 (CHminor), 128.71 (CHmajor), 128.3 (CHmajor), 128.2 (CHminor), 127.61 (CHminor), 127.57 (CHmajor), 124.33 (CHmajor), 124.26 (CHminor), 114.0 (CHmajor), 113.8 (CHminor), 105.8 (CHmajor), 105.7 (CHminor), 70.1 (CH2minor), 70.0 (CH2major), 56.5 (CH3, 2 × OMeminor), 56.3 (CH3, 2 × OMemajor), 55.36 (CH3, OMemajor), 55.34 (CH3, OMeminor), 54.7 (CHminor), 54.4 (CHmajor), 52.3 (CHmajor), 52.1 (CHminor); additional resonances at δ 171.6, 60.6, 21.2, and 14.3 are caused by residual ethyl acetate, which could not be removed from the sample. HRMS (EI): m/z calcd for C25H22O6•+ [M – H2]•+: 418.1411; found: 418.1415.
3-((3,5-Di-tert-butyl-4-hydroxyphenyl)(4-methoxyphenyl)methyl)benzofuran-2(3H)-one (8)
Under an atmosphere of dry N2, 2-coumaranone 3a (100 mg, 0.746 mmol) and potassium carbonate (206 mg, 1.49 mmol) were mixed in acetonitrile (2 mL). Then, pQM 6e (242 mg, 0.746 mmol, solution in 8 mL MeCN) was added dropwise. The reaction mixture was stirred for 1.5 h at room temperature. The resulting purple solution was filtered and neutralized with sat. aq NH4Cl solution (ca. 25 mL). The aq phase was extracted with ethyl acetate (3 × 25 mL), and the combined organic phases were dried (MgSO4) and filtered. Evaporation of volatiles and drying under vacuum yielded a foamy, orange solid (360 mg). The content of residual ethyl acetate in the sample was determined from the integrals in the 1H NMR spectrum (25 mg) and the yield of 8 calculated (335 mg, yield: 98%); a mixture of diastereomers, d.r. 1:1.6.
1H NMR (400 MHz, CDCl3, δ) 7.24–7.19 (m, 5.2 Hmajor+minor), 7.02–6.96 (m, 9.7 Hmajor+minor), 6.90–6.87 (m, 3.3 Hmajor), 6.77–6.72 (m, 6.3 Hmajor+minor), 6.68–6.63 (m, 2.0 Hminor), 5.15 (s, 1 Hminor), 5.06 (s, 1.6 Hmajor), 4.81 (d, J = 4.8 Hz, 1 Hminor), 4.78 (d, J = 5.3 Hz, 1.6 Hmajor), 4.52–4.49 (m, 2.6 Hmajor+minor), 3.82 (s, 4.7 Hmajor), 3.76 (s, 3.0 Hminor), 1.36 (s, 18.0 Hminor), 1.26 (s, 29.4 Hmajor); reported integrals refer to 1H (=1.0) of the minor diastereomer. 13C{1H} NMR (101 MHz, CDCl3, δ) 175.7 (Cqminor), 175.6 (Cqmajor), 158.7 (Cqminor), 158.5 (Cqmajor), 154.18 (Cqminor), 154.16 (Cqmajor), 153.0 (Cqmajor), 152.8 (Cqminor), 135.8 (Cqminor), 135.6 (Cqmajor), 133.1 (Cqmajor), 131.6 (Cqminor), 130.7 (CHminor), 130.4 (CHmajor), 129.6 (CHmajor), 129.2 (CHminor or Cqminor), 129.1 (CHminor or Cqminor), 129.0 (Cqmajor), 126.50 (Cqmajor), 126.47 (Cqminor), 126.1 (CHmajor), 125.5 (CHmajor), 125.4 (CHminor), 125.2 (CHminor), 123.81 (CHmajor), 123.75 (CHminor), 113.9 (CHmajor), 113.8 (CHminor), 110.71 (CHminor), 110.69 (CHmajor), 55.4 (CH3major), 55.3 (CH3minor), 51.6 (CH, C–Hminor), 51.4 (CH, C–Hmajor), 49.54 (CH, C–Hminor), 49.52 (CH, C–Hmajor), 34.6 (Cqminor), 34.3 (Cqmajor), 30.4 (CH3minor), 30.3 (CH3major). HRMS (EI): m/z calcd for C30H32O4•+ [M – H2]•+: 456.2295; found: 456.2292.
Dimethyl 3,3′-(2-oxo-2,3-dihydrobenzofuran-3,3-diyl)dipropionate (10)
Procedure C
Under an atmosphere of dry N2, 2-coumaranone 3a (100 mg, 0.746 mmol) was added to a solution of potassium carbonate (206 mg, 1.49 mmol) in acetonitrile (5 mL). Subsequently, methyl acrylate (9) (67 μL, 0.739 mmol) was added dropwise, and the reaction mixture was stirred at room temperature for 30 h (TLC showed complete consumption of 3a). Then, the mixture was washed with sat. aq NH4Cl solution, and the aqueous phase was extracted with ethyl acetate (3 × 30 mL). The combined organic phases were dried over MgSO4 and filtered. Volatiles were removed under vacuum, which left a colorful oily crude product. Purification by column chromatography (silica gel, hexane/CH2Cl2 = 5:1) gave 10 (58.2 mg, yield: 51%) as a gray-green oil.
1H NMR (599 MHz, CDCl3, δ) 7.33–7.31 (m, 1H), 7.20–7.12 (m, 3H), 3.55 (s, 6H), 2.32–2.15 (m, 6H), 1.97 (ddd, J = 16.0, 10.7, 5.0 Hz, 2H). 13C{1H} NMR (151 MHz, CDCl3, δ) 178.7 (Cq), 172.6 (Cq), 153.4 (Cq), 129.6 (CH), 128.1 (Cq), 124.8 (CH), 123.8 (CH), 111.2 (CH), 51.9 (CH3), 50.9 (Cq), 33.0 (CH2), 29.4 (CH2). IR (ATR, neat): 2953, 1798, 1733, 1477, 1461, 1435, 1226, 1196, 1170, 1128, 1089, 1032, 868, 753 cm–1. HRMS (EI): m/z calcd for C16H18O6•+ [M•+]: 306.1098; found: 306.1097.
4,4′-(2-Oxo-2,3-dihydrobenzofuran-3,3-diyl)bis(butan-2-one) (12)
The product was obtained from 2-coumaranone 3a (100 mg, 0.746 mmol) and methyl vinyl ketone (11) (52 mg, 0.742 mmol) by analogously following Procedure C (cf. synthesis of 10). Purification by column chromatography (silica gel, hexane/CH2Cl2 = 5:1) gave 12 (88.7 mg, yield: 87%) as a yellow-brown oil.
1H NMR (599 MHz, CDCl3, δ) 7.34–7.31 (m, 1H), 7.18–7.10 (m, 3H), 2.32–2.28 (m, 2H), 2.26–2.13 (m, 4H), 2.10–2.00 (m, 2H), 1.98 (s, 6H). 13C{1H} NMR (151 MHz, CDCl3, δ) 206.9 (Cq, ketone C=O), 179.1 (Cq, lactone C=O), 153.1 (Cq), 129.4 (CH), 128.8 (Cq), 124.9 (CH), 123.8 (CH), 111.0 (CH), 50.4 (Cq), 38.4 (CH2), 31.6 (CH2), 30.1 (CH3). IR (ATR, neat): 2927, 1798, 1712, 1478, 1461, 1362, 1225, 1164, 1129, 1038, 880, 754, 733 cm–1. HRMS (EI): m/z calcd for C16H18O4•+ [M•+]: 274.1200; found: 274.1199.
K2CO3-Promoted Michael Reaction of 2a with Chalcone 13a (Scheme 2d)
Isochroman-3-one 2 (30 mg, 0.20 mmol), potassium carbonate (28 mg, 0.20 mmol), and chalcone 13a (46 mg, 0.22 mmol) were stirred in MeCN (0.4 mL) for 30 h at room temperature. Purification was carried out directly by chromatography on a silica gel column with eluent hexane/ethyl acetate (95/5 → 70/30) to elute 14a (50 mg, 70% yield) as a mixture of diastereomers.
Catalytic Michael Reactions of 2 or 3 with Chalcones
Procedure D
Isochroman-3-one 2 or 3-coumaranone 3 (0.135 mmol), potassium carbonate (10 mol %), tetrabutyl ammonium bromide (TBAB, 10 mol %), and chalcone (0.148 mmol, 1.1 equiv) were stirred in toluene (0.2 mL) for 5 h at room temperature. Purification was carried out by chromatography on a short pad of silica gel column with eluent hexane/ethyl acetate (95/5 → 70/30) to elute the mixture of diastereomers. Where possible, the diastereomers were separated by PrepTLC using hexane/ethyl acetate (90/10) as the eluent.
4-(3-Oxo-1,3-diphenylpropyl)isochroman-3-one (14a) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 8:2), isolated in a total yield of 97%, and analyzed by HRMS. Subsequently, PrepTLC (silica gel, hexane/ethyl acetate = 85:15) was used to separate the diastereomers of 14b (dr = 50/50). HRMS (MALDI-FT ICR): m/z calcd. for C24H21O3 [M + H]+: 357.1485; found: 357.1475.
The reaction was also scaled up to 1 mmol scale of isochroman-3-one 2a. Product 14a was isolated as a mixture of diastereomers after flash chromatographic purification on silica gel (hexane/ethyl acetate = 8:2) in a total yield of 89% (317 mg).
(R*)-4-((R*)-3-Oxo-1,3-diphenylpropyl)isochroman-3-one (14a-1): white solid (23 mg); mp 143–145 °C. 1H NMR (400 MHz, CDCl3, δ) 8.01 (dd, J = 8.4, 1.3 Hz, 2H), 7.65 (d, J = 6.1 Hz, 2H), 7.56 (t, J = 7.5 Hz, 2H), 7.49 (t, J = 7.6 Hz, 1H), 7.37–7.28 (m, 2H), 7.24 (t, J = 7.3 Hz, 2H), 6.99 (d, J = 6.9 Hz, 2H), 6.95 (d, J = 7.7 Hz, 1H), 4.76 (d, J = 14.4 Hz, 1H), 4.40 (dd, J = 18.3, 9.4 Hz, 1H), 4.22 (d, J = 3.8 Hz, 1H), 4.03–3.94 (m, 1H), 3.60 (d, J = 15.3 Hz, 1H), 3.54 (dd, J = 18.3, 4.4 Hz, 1H). 13C{1H} NMR (151 MHz, CDCl3, δ) 200.0, 172.5, 140.6, 138.5, 135.2, 134.7, 132.4, 131.1, 130.09, 130.06, 129.9, 129.7, 129.5, 129.1, 128.8, 124.7, 71.1, 51.0, 48.4, 42.9. IR (neat): 2926, 1727, 1679, 1595 cm–1.
(R*)-4-((S*)-3-Oxo-1,3-diphenylpropyl)isochroman-3-one (14a-2): white solid (23 mg); mp 137–139 °C. 1H NMR (400 MHz, CDCl3, δ) 8.03 (dd, J = 7.1, 1.3 Hz, 2H), 7.50 (tt, J = 7.4, 1.3 Hz, 1H), 7.39 (t, J = 7.8 Hz, 1H), 7.24–7.09 (m, 4H), 7.08 (d, J = 7.3 Hz, 1H), 7.00 (t, J = 7.6 Hz, 1H), 6.90 (dd, J = 7.4, 2.1 Hz, 2H), 6.42 (d, J = 7.7 Hz, 1H), 5.18 (d, J = 14.1 Hz, 1H), 5.01 (d, J = 14.1 Hz, 1H), 3.91 (ddd, J = 9.1, 7.2, 5.2 Hz, 1H), 3.87 (d, J = 9.1 Hz, 1H), 3.79 (dd, J = 17.8, 7.3 Hz, 1H), 3.42 (dd, J = 17.7, 5.3 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ) 197.8, 172.4, 140.7, 136.8, 133.3, 132.6, 131.7, 128.7, 128.6, 128.5, 128.3, 128.1, 127.7, 127.48, 127.43, 124.5, 69.6, 52.7, 42.6, 42.1. IR (neat): 2994, 1731, 1685, 1597 cm–1.
4-(1-(4-Chlorophenyl)-3-oxo-3-phenylpropyl)isochroman-3-one (14b) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 8:2), isolated in a total yield of 97%, and analyzed by HRMS. Subsequently, PrepTLC (silica gel, hexane/ethyl acetate = 85:15) was used to separate the diastereomers of 14b (dr = 50/50).
HRMS (MALDI-FT ICR): m/z calcd. for C24H20ClO3 [M + H]+: 391.1095; found: 391.1091.
(R*)-4-((R*)-1-(4-Chlorophenyl)-2-oxo-2-phenylethyl)isochroman-3-one (14b-1): white solid (26 mg); mp 174–176 °C. 1H NMR (400 MHz, CDCl3, δ) 8.11 (dd, J = 7.1, 1.5 Hz, 2H), 7.65 (d, J = 6.1 Hz, 2H), 7.56 (t, J = 7.5 Hz, 2H), 7.49 (t, J = 7.6 Hz, 1H), 7.36–7.29 (m, 2H), 7.24 (t, J = 7.3 Hz, 2H), 6.99 (d, J = 6.9 Hz, 2H), 6.95 (d, J = 7.7 Hz, 1H), 4.76 (d, J = 14.4 Hz, 1H), 4.40 (dd, J = 18.3, 9.4 Hz, 1H), 4.22 (d, J = 3.8 Hz, 1H), 4.04–3.94 (m, 1H), 3.60 (d, J = 15.3 Hz, 1H), 3.54 (dd, J = 18.3, 4.4 Hz, 1H). 13C{1H} NMR (63 MHz, CDCl3, δ) 198.1, 170.7, 137.6, 136.7, 133.5, 133.3, 133.2, 130.6, 129.4, 128.6, 128.0, 127.4, 123.4, 69.7, 49.3, 46.2, 41.3. IR (neat): 2925, 1744, 1685, 1590 cm–1.
(R*)-4-((S*)-1-(4-Chlorophenyl)-2-oxo-2-phenylethyl)isochroman-3-one (14b-2): white solid (26 mg); mp 164–166 °C. 1H NMR (400 MHz, CDCl3, δ) 7.95 (dd, J = 7.2, 1.5 Hz, 2H), 7.57 (t, J = 7.3 Hz, 1H), 7.46 (t, J = 7.4 Hz, 2H), 7.25 (t, J = 7.5 Hz, 1H), 7.18 (d, J = 6.1 Hz, 2H), 7.10 (t, J = 7.4 Hz, 1H), 6.91 (d, J = 8.4 Hz, 2H), 6.48 (d, J = 7.6 Hz, 2H), 5.36 (d, J = 14.1 Hz, 1H), 5.13 (d, J = 14.3 Hz, 1H), 3.97–3.79 (m, 3H), 3.47 (dd, J = 17.6, 5.6 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ) 197.4, 171.9, 139.3, 136.7, 133.4, 133.2, 132.4, 131.6, 129.7, 128.7, 128.5, 128.0, 127.9, 127.6, 127.1, 124.7, 69.6, 52.6, 41.9, 36.2. IR (neat): 2924, 1725, 1685, 1582 cm–1.
4-(1-(2-Nitrophenyl)-3-oxo-3-phenylpropyl)isochroman-3-one (14c) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 7:3) and isolated as a white solid (36 mg, yield: 68%), a mixture of diastereomers (dr = 56/44). 1H NMR (400 MHz, CDCl3, δ) 7.99major (dd, J = 7.1, 1.4 Hz, 2H), 7.94major (dd, J = 7.1 Hz, 1.4 Hz, 2H), 7.77–7.70 (m, 3H), 7.68 (d, J = 3.9 Hz, 3H), 7.60 (d, J = 7.4 Hz, 2H), 7.48 (t, J = 9.3 Hz, 5H), 7.45–7.40 (m, 4H), 7.29 (d, J = 5.0 Hz, 2H), 7.15 (d, J = 7.3 Hz, 1H), 7.04 (dt, J = 8.4, 4.4 Hz, 1H), 6.37minor (d, J = 7.6 Hz, 1H), 6.02minor (d, J = 14.2 Hz, 1H), 5.35major (d, J = 14.2 Hz, 1H), 5.08minor (d, J = 14.2 Hz, 1H), 5.01–4.83 (m, 2H), 4.26minor (d, J = 7.6 Hz, 1H), 4.05–3.90 (m, 3H), 3.72major (dd, J = 18.4, 7.3 Hz, 1H), 3.54minor (dd, J = 18.0, 6.7 Hz, 1H), 3.45 (d, J = 2.6 Hz, 1H), 13C{1H} NMR (63 MHz, CDCl3, δ) 196.6, 196.5, 171.3, 170.3, 150.4, 150.2, 136.3, 136.2, 136.0, 134.4, 133.3, 132.7, 132.7, 131.9, 131.8, 131.0, 129.4, 129.2, 128.7, 128.6, 128.5, 128.1, 128.0, 127.9, 127.8, 127.7, 127.2, 124.8, 124.5, 124.4, 69.9, 69.6, 52.6, 50.3, 42.2, 42.0, 37.2, 33.8. HRMS (MALDI-FT ICR): m/z calcd. for C26H24NaO5 [M + Na]+: 439.1516; found: 439.1525.
4-(1-(2-Chlorophenyl)-3-oxo-3-phenylpropyl)isochroman-3-one (14d). The reaction mixture was heated at 60 °C (oil bath). The crude product was purified by flash chromatography (silica gel, hexane/ethyl acetate = 85:15) and isolated as a white solid (32 mg, yield: 62%), a mixture of diastereomers (dr = 60/40). 1H NMR (400 MHz, CDCl3, δ) 8.03minor (d, J = 4.4 Hz, 2H), 8.02major (d, J = 4.2 Hz, 2H), 7.65–7.56 (m, 2H), 7.55–7.48 (m, 4H), 7.39 (d, J = 7.3 Hz, 2H), 7.27 (td, J = 16.1, 8.1 Hz, 6H), 7.19 (d, J = 6.8 Hz, 1H), 7.09–7.01 (m, 2H), 6.36minor (d, J = 7.6 Hz, 1H), 6.03minor (d, J = 14.1 Hz, 1H), 5.34minor (d, J = 14.2 Hz, 1H), 4.95minor (d, J = 14.4 Hz, 1H), 4.75–4.66major (m, 1H), 4.40 (d, J = 14.6 Hz, 1H), 4.25 (d, J = 5.6 Hz, 1H), 4.15 (dd, J = 18.1, 8.2 Hz, 1H), 4.03 (dd, J = 18.3, 6.7 Hz, 1H), 3.96 (d, J = 10.9 Hz, 1H), 3.63major (dd, J = 18.3, 6.0 Hz, 1H), 3.50minor (dd, J = 18.1, 5.5 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ) 197.6, 197.4, 171.9, 171.1, 138.8, 137.6, 136.8, 136.7, 135.1, 134.9, 133.37, 133.32, 132.9, 132.5, 132.0, 130.8, 129.9, 129.7, 129.1, 128.9, 128.7, 128.6, 128.6, 128.3, 128.1, 128.0, 127.8, 127.8, 127.7, 127.5, 127.4, 127.1, 124.5, 124.0, 69.9, 52.6, 50.3, 42.3, 42.0, 40.4, 36.8. HRMS (MALDI-FT ICR): m/z calcd. for C24H20ClO3 [M + H]+: 391.1095; found: 391.1072.
4-(1-(4-Fluorophenyl)-3-oxo-3-phenylpropyl)isochroman-3-one (14e) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 9:1), isolated in a total yield of 91%, and analyzed by HRMS. Subsequently, PrepTLC (silica gel, hexane/ethyl acetate = 92:8) was used to separate the diastereomers (dr = 53/47).
HRMS (MALDI-FT ICR): m/z calcd. for C24H19FO3K [M + K]+: 413.0950; found: 413.0982.
(R*)-4-((R*)-1-(4-fluorophenyl)-3-oxo-3-phenylpropyl)isochroman-3-one (14e-1): white solid (24 mg); mp 137–139 °C. 1H NMR (400 MHz, CDCl3, δ) 8.02 (dd, J = 7.2, 1.4 Hz, 2H), 7.63 (tt, J = 7.4, 1.4 Hz, 1H), 7.52 (t, J = 7.6 Hz, 2H), 7.30 (dd, J = 7.5, 1.2 Hz, 1H), 7.23 (d, J = 6.5 Hz, 1H), 7.15 (t, J = 7.5 Hz, 1H), 7.01–6.93 (m, 4H), 6.54 (d, J = 6.4 Hz, 1H), 5.37 (d, J = 14.2 Hz, 1H), 5.17 (d, J = 14.3 Hz, 1H), 4.07–4.00 (m, 1H), 3.93 (dd, J = 7.1, 6.5 Hz, 1H), 3.87 (d, J = 7.1 Hz, 1H), 3.54 (dd, J = 17.8, 5.9 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ) 197.5, 172.0, 161.8 (d, JC,F = 246.4 Hz), 136.5 (d, JC,F = 13.2 Hz), 133.2, 132.3, 131.4, 129.7 (d, JC,F = 7.9 Hz), 128.5, 128.4, 128.0, 127.7, 127.4, 124.5, 115.5, 115.2, 69.5, 52.6, 42.1, 41.6. 19F{1H} NMR (376 MHz, CDCl3, δ) −114.21. IR (neat): 2924, 1725, 1686, 1597 cm–1.
(R*)-4-((S*)-1-(4-Fluorophenyl)-3-oxo-3-phenylpropyl)isochroman-3-one (14e-2): white solid (22 mg); mp 134–136 °C. 1H NMR (400 MHz, CDCl3, δ) 8.12 (dd, J = 7.1, 1.3 Hz, 2H), 7.64 (t, J = 7.6 Hz, 2H), 7.56 (t, J = 7.6 Hz, 2H), 7.49 (t, J = 7.5 Hz, 1H), 7.33 (d, J = 7.3 Hz, 1H), 7.01–6.92 (m, 5H), 4.84 (d, J = 14.5 Hz, 1H), 4.34 (dd, J = 18.2, 9.1 Hz, 1H), 4.19 (d, J = 3.9 Hz, 1H), 3.98 (dt, J = 8.9, 4.4 Hz, 1H), 3.79 (d, J = 14.5 Hz, 1H), 3.52 (dd, J = 18.2, 4.8 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ) 198.3, 170.9, 162.3 (d, JC,F = 246.7 Hz), 137.0, 135.0, 133.5, 133.4, 130.8, 129.8 (d, JC,F = 7.7 Hz), 128.8, 128.7, 128.1, 127.5, 123.5, 115.6, 115.4, 69.8, 49.5, 46.3, 41.7. 19F{1H} NMR (376 MHz, CDCl3, δ) −114.21. IR (neat): 2925, 1744, 1681, 1511 cm–1.
4-(1-(4-Methoxyphenyl)-3-oxo-3-phenylpropyl)isochroman-3-one (14f) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 8:2), isolated in a total yield of 82%, and analyzed by HRMS. Subsequently, PrepTLC (silica gel, hexane/ethyl acetate = 85:15) was used to separate the diastereomers (dr = 55/45).
HRMS (MALDI-FT ICR): m/z calcd. for C25H21KO4 [M + K]+: 425.1149; found: 425.1155.
(R*)-4-((R*)-1-(4-Methoxyphenyl)-3-oxo-3-phenylpropyl)isochroman-3-one (14f-1): white solid (23 mg); mp 138–141 °C. 1H NMR (400 MHz, CDCl3, δ) 8.13 (dd, J = 7.1, 1.5 Hz, 2H), 7.68–7.61 (m, 2H), 7.55 (t, J = 7.5 Hz, 2H), 7.48 (t, J = 7.5 Hz, 1H), 7.36–7.29 (m, 1H), 6.96 (d, J = 7.6 Hz, 1H), 6.90 (d, J = 8.8 Hz, 2H), 6.77 (d, J = 8.8 Hz, 2H), 4.79 (d, J = 14.4 Hz, 1H), 4.34 (dd, J = 18.2, 9.3 Hz, 1H), 4.18 (d, J = 3.8 Hz, 1H), 3.93 (dt, J = 8.8, 4.2 Hz, 1H), 3.82 (s, 3H), 3.74 (d, J = 14.3 Hz, 1H), 3.50 (dd, J = 18.2, 4.6 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ) 198.7, 171.2, 159.2, 137.2, 134.0, 133.3, 131.2, 131.0, 129.3, 128.7, 128.6, 128.1, 127.3, 123.4, 113.9, 69.9, 55.2, 49.7, 46.3, 41.8. IR (neat): 3065, 1744, 1679, 1513 cm–1.
(R*)-4-((S*)-1-(4-Methoxyphenyl)-3-oxo-3-phenylpropyl)isochroman-3-one (14f-2): white solid (19 mg); mp 121–124 °C. 1H NMR (400 MHz, CDCl3, δ) 8.02 (dd, J = 7.1, 1.5 Hz, 2H), 7.62 (t, J = 7.3 Hz, 1H), 7.51 (t, J = 7.6 Hz, 2H), 7.37–7.26 (m, 1H), 7.22–7.16 (m, 2H), 6.92 (d, J = 8.8 Hz, 2H), 6.79 (d, J = 8.8 Hz, 2H), 6.62 (d, J = 7.9 Hz, 1H), 5.20 (d, J = 14.2 Hz, 1H), 5.11 (d, J = 14.3 Hz, 1H), 4.06–3.95 (m, 1H), 3.87 (d, J = 7.3 Hz, 1H), 3.82 (s, 3H), 3.78 (d, J = 7.3 Hz, 1H), 3.51 (dd, J = 17.7, 5.5 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ) 197.9, 172.4, 158.9, 136.9, 133.2, 132.6, 132.6, 131.7, 129.3, 128.7, 128.6, 128.1, 127.7, 127.4, 124.5, 113.9, 69.7, 55.2, 52.7, 42.3, 42.1. IR (neat): 3061, 1741, 1669, 1523 cm–1.
4-(3-Oxo-1-phenyl-3-(3-(trifluoromethyl)phenyl)propyl)isochroman-3-one (14g) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 85:15), isolated in a total yield of 76%, and analyzed by HRMS. Subsequently, PrepTLC (silica gel, hexane/ethyl acetate = 9:1) was used to separate the diastereomers (dr = 53/47).
HRMS (MALDI-FT ICR): m/z calcd. for C25H19F3KO3 [M + K]+: 463.0918; found: 463.0945.
(R*)-4-((R*)-2-Oxo-1-phenyl-2-(3-(trifluoromethyl)phenyl)ethyl)isochroman-3-one (14g-1): white solid (22 mg); mp 133–135 °C. 1H NMR (400 MHz, CDCl3, δ) 8.35 (dd, J = 18.0, 7.9 Hz, 2H), 7.91 (d, J = 8.1 Hz, 1H), 7.91 (d, J = 8.1 Hz, 1H), 7.64 (d, J = 7.6 Hz, 1H), 7.50 (t, J = 7.6 Hz, 1H), 7.33 (q, J = 7.0 Hz, 2H), 7.25 (t, J = 7.4 Hz, 2H), 6.99 (d, J = 7.3 Hz, 2H), 6.95 (d, J = 7.5 Hz, 1H), 4.77 (d, J = 14.4 Hz, 1H), 4.43 (dd, J = 18.4, 9.5 Hz, 1H), 4.21 (d, J = 3.8 Hz, 1H), 3.99 (dt, J = 9.4, 4.2 Hz, 1H), 3.62 (d, J = 14.4 Hz, 1H), 3.54 (dd, J = 18.4, 4.3 Hz, 1H), 13C{1H} NMR (101 MHz, CDCl3, δ) 197.4, 171.2, 138.9, 137.6, 133.6, 131.4 (q, JC,F = 32.5 Hz), 131.4, 131.1, 129.8, 129.8, 129.4, 128.7, 128.3, 128.1, 127.9, 127.5, 125.0 (q, JC,F = 3.7 Hz), 123.5, 69.8, 49.4, 46.9, 41.7. 19F{1H} NMR (376 MHz, CDCl3, δ) −62.76. IR (neat): 2924, 1725, 1685, 1582 cm–1.
(R*)-4-((S*)-2-Oxo-1-phenyl-2-(3-(trifluoromethyl)phenyl)ethyl)isochroman-3-one (14g-2): white solid (20 mg); mp 114–116 °C. 1H NMR (400 MHz, CDCl3, δ) 8.21 (s, 1H), 8.16 (d, J = 8.0 Hz, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.61 (t, J = 7.7 Hz, 1H), 7.28–7.08 (m, 5H), 7.04 (t, J = 7.4 Hz, 1H), 6.98 (dd, J = 6.5, 3.1 Hz, 2H), 6.39 (d, J = 7.5 Hz, 1H), 5.45 (d, J = 14.2 Hz, 1H), 5.14 (d, J = 14.2 Hz, 1H), 3.97–3.90 (m, 2H), 3.46 (dd, J = 13.5, 6.5 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ) 196.6, 172.4, 140.7, 137.4, 132.7, 131.6, 131.4 (q, JC,F = 32.8 Hz), 131.3, 129.7 (q, JC,F = 3.9 Hz), 129.3, 128.6, 128.6, 128.3, 127.8, 127.6, 127.5, 125.0, 124.9, 124.6, 69.6, 52.8, 42.5. 19F{1H} NMR (376 MHz, CDCl3, δ) −62.64. IR (neat): 2935, 1727, 1689, 1612 cm–1.
(R*)-4-((R*)-3-(4-Nitrophenyl)-3-oxo-1-phenylpropyl)isochroman-3-one (14h) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 7:3) and isolated as a white solid (21 mg, yield: 77%); mp 183–186 °C. 1H NMR (400 MHz, CDCl3, δ) 8.35 (d, J = 8.9 Hz, 2H), 8.23 (d, J = 9.0 Hz, 2H), 7.56 (d, J = 7.7 Hz, 1H), 7.44 (t, J = 7.5 Hz, 1H), 7.26 (t, J = 5.8 Hz, 2H), 7.18 (t, J = 7.2 Hz, 2H), 6.90 (t, J = 6.6 Hz, 3H), 4.71 (d, J = 14.5 Hz, 1H), 4.39 (dd, J = 18.5, 9.5 Hz, 1H), 4.13 (d, J = 3.8 Hz, 1H), 3.91 (dt, J = 8.7, 4.0 Hz, 1H), 3.57–3.43 (m, 2H). 13C{1H} NMR (101 MHz, CDCl3, δ) 197.3, 171.1, 150.6, 141.5, 138.6, 133.4, 131.1, 129.3, 128.8, 128.7, 128.2, 128.1, 128.0, 127.6, 124.0, 123.5, 69.8, 49.3, 46.9, 42.1. IR (neat): 2944, 1729, 1682, 1597 cm–1. HRMS (MALDI-FT ICR): m/z calcd. for C24H19NNaO5 [M + Na]+: 424.1155; found: 424.1177. The other diastereomer was isolated with an unsuitable purity. Therefore, we were not able to perform a full characterization. The overall yield and the dr were determined on the integration of crude NMR.
4-(3-(4-Methoxyphenyl)-3-oxo-1-phenylpropyl)isochroman-3-one (14i) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 8:2), isolated in a total yield of 73%, and analyzed by HRMS. Subsequently, PrepTLC (silica gel, hexane/ethyl acetate = 85:15) was used to separate the diastereomers (dr = 53/47).
HRMS (MALDI-FT ICR): m/z calcd. for C25H22NaO4 [M + Na]+: 409.1410; found: 409.1417.
(R*)-4-((R*)-3-(4-Methoxyphenyl)-3-oxo-1-phenylpropyl)isochroman-3-one (14i-1): white solid (20 mg); mp 146–148 °C. 1H NMR (400 MHz, CDCl3, δ) 8.13 (d, J = 9.0 Hz, 2H), 7.64 (dd, J = 13.2, 7.2 Hz, 2H), 7.55 (t, J = 7.5 Hz, 2H), 7.48 (t, J = 7.5 Hz, 1H), 7.32 (t, J = 7.5 Hz, 1H), 6.96 (d, J = 7.6 Hz, 1H), 6.90 (d, J = 8.8 Hz, 2H), 6.77 (d, J = 8.8 Hz, 2H), 4.79 (d, J = 14.4 Hz, 1H), 4.34 (dd, J = 18.2, 9.3 Hz, 1H), 4.18 (d, J = 3.8 Hz, 1H), 3.93 (dt, J = 8.8, 4.2 Hz, 1H), 3.82 (s, 3H), 3.74 (d, J = 14.3 Hz, 1H), 3.50 (dd, J = 18.2, 4.6 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ) 198.7, 171.3, 159.2, 137.2, 134.0, 133.3, 131.2, 131.1, 129.3, 128.7, 128.7, 128.2, 127.4, 123.4, 113.9, 69.9, 55.3, 49.7, 46.4, 41.8. IR (neat): 2994, 1744, 1679, 1513 cm–1.
(R*)-4-((S*)-3-(4-Methoxyphenyl)-3-oxo-1-phenylpropyl)isochroman-3-one (14i-2): white solid (18 mg); mp 141–143 °C, 1H NMR (400 MHz, CDCl3, δ) 8.02 (d, J = 8.9 Hz, 2H),7.62 (t, J = 7.3 Hz, 1H), 7.51 (t, J = 7.6 Hz, 2H), 7.30 (t, J = 8.3 Hz, 1H), 7.18 (t, J = 7.7 Hz, 2H), 6.92 (d, J = 8.8 Hz, 2H), 6.79 (d, J = 8.8 Hz, 2H), 6.62 (d, J = 7.9 Hz, 1H), 5.20 (d, J = 14.2 Hz, 1H), 5.11 (d, J = 14.2 Hz, 1H), 4.09–3.95 (m, 1H), 3.87 (d, J = 7.3 Hz, 1H), 3.82 (s, 3H), 3.78 (t, J = 6.7 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ) 197.9, 172.5, 158.9, 136.9, 133.3, 132.65, 132.62, 131.8, 129.3, 128.7, 128.6, 128.1, 127.8, 127.4, 124.5, 113.9, 69.7, 55.3, 52.7, 42.3, 42.1. IR (neat): 2991, 1740, 1681, 1599 cm–1.
((Furan-2-yl)-3-oxo-3-phenylpropyl)isochroman-3-one (14j) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 80:20) and isolated as a white solid (30 mg, yield: 65%), a mixture of diastereomers (dr = 58/42). 1H NMR (400 MHz, CDCl3, δ) 8.10major (dd, J = 7.1, 1.4 Hz, 2H), 8.04minor (dd, J = 7.1, 1.4 Hz, 2H), 7.64 (d, J = 7.3 Hz, 2H), 7.58–7.51 (m, 5H), 7.45 (t, J = 7.4 Hz, 2H), 7.40–7.33 (m, 3H), 7.31–7.16 (m, 3H), 7.08major (d, J = 7.7 Hz, 1H), 6.70minor (d, J = 7.7 Hz, 1H), 6.30major (dd, J = 3.2, 1.9 Hz, 1H), 6.26minor (dd, J = 3.2, 1.9 Hz, 1H), 5.89minor (d, J = 3.2 Hz, 1H), 5.84major (d, J = 3.2 Hz, 1H), 5.54minor (d, J = 14.3 Hz, 1H), 5.38major (s, 1H), 5.24minor (d, J = 14.3 Hz, 1H), 5.02major (d, J = 14.5 Hz, 1H), 4.31major (d, J = 13.4 Hz, 1H), 4.19–4.07 (m, 3H), 3.86–3.77major (m, 1H), 3.63–3.51 (m, 3H), 13C{1H} NMR (75 MHz, CDCl3, δ) 198.6, 197.8, 171.7, 170.5, 140.6, 138.9, 137.0, 136.7, 133.8, 133.5, 133.4, 133.1, 133.0, 131.9, 130.9, 130.2, 129.9, 128.8, 128.8, 128.7, 128.6, 128.3, 128.2, 128.1, 128.0, 127.8, 127.7, 127.7, 126.5, 121.4, 121.2, 68.9, 68.8, 52.3, 49.0, 47.1, 42.6, 42.1, 41.4. HRMS (MALDI-FT ICR): m/z calcd. for C22H18NaO4 [M + Na]+: 369.1097; found: 369.1093.
7-Bromo-4-(3-oxo-1,3-diphenylpropyl)isochroman-3-one (14k) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 80:20) and isolated as a white solid (47 mg, yield: 81%), a mixture of diastereomers (dr = 54/46). 1H NMR (400 MHz, CDCl3, δ) 8.14major (dd, J = 7.1, 1.4 Hz, 2H), 8.03minor (d, J = 7.1, 1.4, 2H), 7.71–7.48 (m, 8H), 7.41–7.21 (m, 9H), 7.12–6.97 (m, 5H), 6.36minor (d, J = 7.9 Hz, 1H), 5.35minor (d, J = 14.3 Hz, 1H), 5.10 minor (d, J = 14.3 Hz, 1H), 4.69major (d, J = 14.5 Hz, 1H), 4.43major (dd, J = 18.4, 9.8 Hz, 1H), 4.19major (d, J = 3.5 Hz, 1H), 4.02–3.88 (m, 3H), 3.55–3.45 (m, 4H). 13C{1H} NMR (75 MHz, CDCl3, δ) 198.6, 197.8, 171.7, 170.5, 140.6, 138.9, 137.0, 136.7, 133.8, 133.5, 133.4, 133.1, 133.0, 131.9, 130.9, 130.2, 129.9, 128.8, 128.8, 128.7, 128.6, 128.3, 128.2, 128.1, 128.0, 127.8, 127.74, 127.7, 126.5, 121.4, 121.2, 68.9, 68.8, 52.3, 49.0, 47.1, 42.6, 42.1, 41.4. HRMS (MALDI-FT ICR): m/z calcd. for C24H19BrNaO3 [M + Na]+: 457.0410; found: 457.0419.
6-Methoxy-4-(3-oxo-1,3-diphenylpropyl)isochroman-3-one (14l) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 75:25), isolated in a total yield of 96%, and analyzed by HRMS. Subsequently, PrepTLC (silica gel, hexane/ethyl acetate = 80:20) was used to separate the diastereomers (dr = 50/50).
HRMS (MALDI-FT ICR): m/z calcd. For C25H21KO4 [M + K]+: 425.1149; found: 425.1189.
(R*)-6-Methoxy-4-((S*)-3-oxo-1,3-diphenylpropyl)isochroman-3-one (14l-1): white solid (25 mg); mp 157–159 °C. 1H NMR (400 MHz, CDCl3, δ) 8.13 (dd, J = 7.0, 1.5 Hz, 2H), 7.65 (t, J = 7.4 Hz, 2H), 7.33–7.29 (m, 1H), 7.24 (t, J = 7.3 Hz, 2H), 7.14 (s, 1H), 7.04 (d, J = 6.9 Hz, 2H), 6.86 (s, 2H), 4.38 (dd, J = 18.3, 9.4 Hz, 1H), 4.16 (d, J = 3.9 Hz, 1H), 4.01 (dt, J = 9.1, 4.2 Hz, 1H), 3.95 (s, 3H), 3.59 (d, J = 13.9 Hz, 1H), 3.53 (dd, J = 18.3, 4.4 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ) 198.6, 171.2, 159.9, 139.3, 137.1, 135.3, 133.3, 128.7, 128.6, 128.4, 128.2, 127.8, 124.7, 123.4, 114.1, 112.3, 69.5, 55.5, 50.0, 46.8, 41.5. IR (neat): 3061, 1734, 1685, 1598 cm–1.
(R*)-6-Methoxy-4-((S*)-3-oxo-1,3-diphenylpropyl)isochroman-3-one (14l-2): white solid (25 mg); mp 146–148 °C. 1H NMR (400 MHz, CDCl3, δ) 8.03 (dd, J = 7.1, 1.5 Hz, 2H), 7.51 (t, J = 7.6 Hz, 2H), 7.30–7.26 (m, 3H), 7.11 (d, J = 8.4 Hz, 1H), 7.07 (d, J = 3.6 Hz, 2H), 6.81 (dd, J = 8.4, 2.6 Hz, 1H), 5.95 (d, J = 2.6 Hz, 1H), 5.39–5.29 (m, 1H), 5.11 (d, J = 13.8 Hz, 1H), 3.99 (d, J = 7.9 Hz, 1H), 3.93 (d, J = 7.9 Hz, 1H), 3.88 (d, J = 12.4 Hz, 1H), 3.58 (d, J = 4.7 Hz, 1H), 3.53 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3, δ) 197.9, 172.4, 159.0, 141.0, 136.9, 134.2, 133.3, 128.7, 128.6, 128.5, 128.1, 127.4, 125.7, 123.7, 114.0, 113.1, 69.4, 55.2, 53.1, 42.5, 42.2. IR (neat): 3065, 1724, 1681, 1612 cm–1.
7-Nitro-4-(3-oxo-1,3-diphenylpropyl)isochroman-3-one (14m). The reaction mixture was heated at 110 °C (oil bath). The crude product was purified by flash chromatography (silica gel, hexane/ethyl acetate = 7:3) to afford a white solid (31 mg, yield: 58%), a mixture of diastereomers (dr = 53/47). 1H NMR (400 MHz, CDCl3, δ) 8.15major (d, J = 6.5 Hz, 2H), 8.04minor (d, J = 7.7 Hz, 2H), 7.94major (t, J = 10.2 Hz, 2H), 7.87minor (s, 1H), 7.67major (dt, J = 14.7, 7.2 Hz, 2H), 7.59–7.51 (m, 3H), 7.26–7.38 (m, 7H), 7.05 (d, J = 3.6 Hz, 2H), 6.98 (d, J = 7.2 Hz, 2H), 6.58minor (d, J = 8.4 Hz, 1H), 5.66major (d, J = 14.3 Hz, 1H), 5.32major (d, J = 14.6 Hz, 1H), 4.84 (d, J = 14.9 Hz, 1H), 4.51 (dd, J = 18.9, 10.3 Hz, 1H), 4.37 (d, J = 3.4 Hz, 1H), 4.11–3.95 (m, 4H), 3.53–3.46 (m, 3H). 13C{1H} NMR (101 MHz, CDCl3, δ) 199.9, 199.1, 172.2, 171.0, 148.6, 142.9, 139.9, 138.2, 135.0, 135.0, 134.0, 131.1, 131.0, 130.4, 130.4, 130.2, 130.1, 129.7, 129.6, 129.5, 129.4, 125.2, 124.1, 121.4, 120.3, 70.2, 70.0, 54.4, 50.6, 48.8, 44.0, 43.5, 42.7. HRMS (MALDI-FT ICR): m/z calcd for C24H19NNaO5 [M + Na]+: 424.1155; found: 424.1180.
6-Chloro-4-(3-oxo-1,3-diphenylpropyl)isochroman-3-one (14n) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 85:15), isolated in a total yield of 94%, and analyzed by HRMS. Subsequently, PrepTLC (silica gel, hexane/ethyl acetate = 90:10) was used to separate the diastereomers (dr = 52/48).
HRMS (MALDI-FT ICR): m/z calcd. For C24H19ClNaO3 [M + Na]+: 413.0914; found: 413.0919.
(R*)-6-Chloro-4-((R*)-3-oxo-1,3-diphenylpropyl)isochroman-3-one (14n-1): white solid (25 mg); mp 146–148 °C. 1H NMR (400 MHz, CDCl3, δ) 8.14 (dd, J = 7.0, 1.5 Hz, 2H), 7.69 (d, J = 2.1 Hz, 1H), 7.65 (dt, J = 7.4, 1.4 Hz, 1H), 7.56 (t, J = 7.5 Hz, 2H), 7.33–7.28 (m, 2H), 7.25 (t, J = 7.3 Hz, 2H), 7.02 (s, 1H), 7.00 (d, J = 1.6 Hz, 1H), 6.88 (d, J = 8.1 Hz, 1H), 4.71 (d, J = 14.5 Hz, 1H), 4.42 (dd, J = 18.4, 9.8 Hz, 1H), 4.19 (d, J = 3.7 Hz, 1H), 3.96 (dt, J = 9.8, 3.8 Hz, 1H), 3.54–3.50 (m, 1H), 3.48 (d, J = 4.0 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3, δ) 198.6, 171.2, 159.9, 139.3, 137.1, 135.3, 133.4, 128.7, 128.6, 128.4, 128.2, 127.8, 124.7, 123.4, 114.1, 112.3, 69.5, 55.5, 50.0, 46.8, 41.5. IR (neat): 3029, 1766, 1680, 1600 cm–1.
(R*)-6-Chloro-4-((S*)-3-oxo-1,3-diphenylpropyl)isochroman-3-one (14n-2): white solid (23 mg); mp 129–132 °C. 1H NMR (400 MHz, CDCl3, δ) 8.03 (dd, J = 7.1, 1.5 Hz, 2H), 7.63 (t, J = 7.4 Hz, 1H), 7.52 (t, J = 7.6 Hz, 2H), 7.33–7.29 (m, 3H), 7.26 (dd, J = 8.1, 2.0 Hz, 1H), 7.15 (d, J = 8.1 Hz, 1H), 7.04 (d, J = 2.2 Hz, 1H), 7.02 (d, J = 3.7 Hz, 1H), 6.44 (d, J = 2.1 Hz, 1H), 5.35 (d, J = 14.3 Hz, 1H), 5.13 (d, J = 14.3 Hz, 1H), 4.02–3.97 (m, 1H), 3.94–3.89 (m, 2H), 3.53 (dd, J = 12.8, 4.4 Hz, 1H). 13C{1H} NMR (75 MHz, CDCl3, δ) 197.7, 171.5, 140.3, 136.6, 134.6, 133.5, 133.3, 130.0, 128.6, 128.1, 128.0, 127.6, 127.4, 125.7, 68.8, 52.5, 42.4, 41.8. IR (neat): 3029, 1734, 1685, 1598 cm–1.
6-Methoxy-4-(1-(4-methoxyphenyl)-3-oxo-3-phenylpropyl)isochroman-3-one (14o) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 8:2) and isolated as a white solid (52 mg, yield: 93%), a mixture of diastereomers (dr = 51/49). 1H NMR (400 MHz, CDCl3, δ) 8.11 (dd, J = 7.1 Hz, 1.5 Hz, 2H), 8.02 (dd, J = 7.1 Hz, 1.5 Hz, 2H), 7.68–7.57 (m, 2H), 7.52 (dt, J = 14.5, 7.5 Hz, 4H), 7.12 (s, 1H), 7.09 (d, J = 8.4 Hz, 1H), 6.96–6.92 (m, 5H), 6.87 (m, 2H), 6.85–6.69 (m, 5H), 5.18 (d, J = 13.8 Hz, 1H), 5.06 (d, J = 13.9 Hz, 1H), 4.73 (d, J = 13.9 Hz, 1H), 4.32 (dd, J = 18.3, 9.3 Hz, 1H), 4.12 (d, J = 3.8 Hz, 1H), 4.05–3.95 (m, 1H), 3.94 (s, 3H), 3.90 (dd, J = 8.2, 2.7 Hz, 1H), 3.85 (d, J = 7.9 Hz, 1H), 3.80 (s, 6H), 3.69 (d, J = 14.0 Hz, 1H), 3.58 (s, 3H), 3.53 (dd, J = 7.0, 5.1 Hz, 1H), 3.48 (dd, J = 7.7, 5.2 Hz, 1H). 13C{1H} NMR (75 MHz, CDCl3, δ) 198.7, 198.0, 172.5, 171.3, 159.9, 159.2, 159.0, 158.9, 137.2, 136.9, 135.4, 134.1, 133.3, 133.3, 132.7, 131.2, 129.4, 129.3, 128.7, 128.7, 128.2, 128.1, 125.6, 124.8, 123.8, 123.3, 114.0, 113.9, 113.3, 112.3, 69.7, 69.4, 55.5, 55.3, 55.3, 55.2, 53.0, 50.1, 46.0, 42.3, 42.0, 41.8. HRMS (MALDI-FT ICR): m/z calcd. for C26H24NaO5 [M + Na]+: 439.1516; found: 439.1525.
8-Fluoro-4-(-3-oxo-1,3-diphenylpropyl)isochroman-3-one (14p) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 80:20), isolated in a total yield of 90%, and analyzed by HRMS. Subsequently, PrepTLC (silica gel, hexane/ethyl acetate = 85:15) was used to separate the diastereomers (dr = 59/41).
HRMS (MALDI-FT ICR): m/z calcd. For C24H20FO3 [M + H]+: 375.1391; found: 375.1386.
(R*)-8-Fluoro-4-((R*)-3-oxo-1,3-diphenylpropyl)isochroman-3-one (14p-1): white solid (30 mg); mp 119–121 °C. 1H NMR (400 MHz, CDCl3, δ) 8.07 (dd, J = 7.1, 1.5 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.54–7.41 (m, 2H), 7.43–7.38 (m, 2H), 7.25 (d, J = 7.3 Hz, 1H), 7.19 (t, J = 7.9 Hz, 2H), 6.99–6.92 (m, 1H), 6.91 (d, J = 7.0 Hz, 2H), 4.95 (d, J = 15.0 Hz, 1H), 4.35 (dd, J = 18.4, 9.7 Hz, 1H), 4.18 (d, J = 3.6 Hz, 1H), 3.89 (dt, J = 9.7, 3.8 Hz, 1H), 3.44 (dd, J = 18.4, 4.1 Hz, 1H), 3.36 (d, J = 15.0 Hz, 1H). 13C NMR (63 MHz, CDCl3, δ) 198.4, 170.3, 157.2 (d, JC,F = 247.2 Hz), 138.7, 136.8, 136.5, 136.4, 133.3, 130.0 (d, JC,F = 8.1 Hz), 128.6, 128.1, 128.0, 127.9, 123.5 (d, JC,F = 3.3 Hz), 118.7 (d, JC,F = 16.6 Hz), 113.7 (d, JC,F = 20.2 Hz), 64.2 (d, JC,F = 3.7 Hz), 48.8, 47.1, 41.2. 19F{1H} NMR (376 MHz, CDCl3, δ) −120.74. IR (neat): 2947, 1755, 1656, 929 cm–1.
(R*)-8-Fluoro-4-((S*)-3-oxo-1,3-diphenylpropyl)isochroman-3-one (14p-2): white solid (21 mg); mp 113–115 °C. 1H NMR (300 MHz, CDCl3, δ) 7.97 (dd, J = 7.0, 1.5 Hz, 2H), 7.57 (tt, J = 7.4, 1.5 Hz, 1H), 7.46 (t, J = 7.4 Hz, 2H), 7.24–7.19 (m, 3H), 7.09–7.01 (m, 1H), 6.99–6.89 (m, 3H), 6.29 (d, J = 7.6 Hz, 1H), 5.40 (d, J = 13.8 Hz, 1H), 5.03 (d, J = 14.8 Hz, 1H), 4.00–3.95 (m, 2H), 3.85 (ddd, J = 17.6, 5.0, 2.7 Hz, 1H), 3.45 (ddd, J = 17.5, 3.4, 1.3 Hz, 1H). 13C NMR (63 MHz, CDCl3, δ) 197.6, 171.6, 157.8 (d, JC,F = 247.4 Hz), 140.3, 136.6, 135.2, 133.2, 129.1 (d, JC,F = 8.2 Hz), 128.55, 128.50, 128.1, 127.9, 127.5, 124.1, 119.1 (d, JC,F = 16.3 Hz), 114.0 (d, JC,F = 20.4 Hz), 63.4 (d, JC,F = 3.7 Hz), 52.0, 42.8, 41.8. 19F{1H} NMR (376 MHz, CDCl3, δ) −119.96. IR (neat): 2942, 1749, 1627, 925 cm–1.
3-(3-Oxo-1,3-diphenylpropyl)benzofuran-2(3H)-one (15a) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 75:25) and isolated as a white solid (37 mg, yield: 81%), a mixture of diastereomers (dr = 64/36).
1H NMR (400 MHz, CDCl3, δ) 8.11major (dd, J = 7.0, 1.5 Hz, 2H), 8.02minor (dd, J = 7.0, 1.5 Hz, 2H) 7.64 (q, J = 8.1 Hz 2H), 7.52–7.56 (m, 4H), 7.44major (d, J = 7.2 Hz, 1H), 7.31 (d, J = 7.7 Hz, 4H), 7.17–7.25 (m, 9H), 7.06 (d, J = 7.9 Hz, 1H), 6.92major (d, J = 7.9 Hz, 1H), 6.76minor (d, J = 7.9 Hz, 1H), 4.24–4.28 (m, 4H), 4.20major (d, J = 7.3 Hz, 1H), 4.04–4.07 (m, 1H), 4.00minor (d, J = 6.6 Hz, 1H), 3.71minor (d, J = 6.7 Hz, 1H), 3.57–3.68 (m, 2H). 13C{1H} NMR (75 MHz, CDCl3, δ) 198.3, 197.5, 176.1, 175.5, 153.7, 153.4, 139.9, 138.7, 136.7, 136.6, 133.4, 133.2, 129.0, 128.7, 128.6, 128.5, 128.3, 128.05, 128.00, 127.7, 127.5, 127.3, 126.0, 125.7, 125.1, 124.5, 123.9, 123.7, 110.6, 110.3, 47.9, 47.5, 42.8, 41.9, 41.4, 39.5. HRMS (MALDI-FT ICR) m/z: calcd for C23H18NaO3 [M + Na]+: 365.1148, found: 365.1140.
3-(1-(4-Chlorophenyl)-2-oxo-2-phenylethyl)benzofuran-2(3H)-one (15b) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 75:25) and isolated as a white solid (42 mg, yield: 83%), a mixture of diastereomers (dr = 53/47). 1H NMR (400 MHz, CDCl3, δ) 8.10major (d, J = 7.1 Hz, 2H), 8.00minor (d, J = 7.1 Hz, 1H), 7.66major (q, J = 7.6 Hz, 2H), 7.58–7.51 (m, 4H), 7.45 (d, J = 7.5 Hz, 1H), 7.39–7.30minor (m, 2H), 7.27major (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.6 Hz, 1H), 7.19–7.12major (m, 4H), 7.12–7.07minor (m, 3H), 6.96 (d, J = 8.1 Hz, 1H), 6.89 (d, J = 7.6 Hz, 1H), 4.31–4.15 (m, 4H), 4.08minor (q, J = 7.4 Hz, 1H), 3.93major (dd, J = 17.3, 6.3 Hz, 1H), 3.72–3.56 (m, 2H). 13C{1H} NMR (75 MHz, CDCl3, δ) 197.9, 197.1, 175.8, 175.2, 153.7, 153.3, 138.2, 137.1, 136.5, 136.4, 133.5, 133.4, 133.1, 129.7, 129.3, 129.2, 128.9, 128.6, 128.5, 128.0, 127.9, 125.7, 125.2, 124.9, 124.3, 124.0, 123.8, 110.8, 110.5, 47.9, 47.5, 42.2, 41.3, 41.1, 39.5. HRMS (MALDI-FT ICR) m/z: calcd for C23H17ClNaO3 [M + Na]+: 399.0758, found: 399.0762.
3-(1-(4-Methoxyphenyl)-2-oxo-2-phenylethyl)benzofuran-2(3H)-one (15c) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 70:30) and isolated as a white solid (43 mg, yield: 87%), a mixture of diastereomers (dr = 54/46). 1H NMR (400 MHz, CDCl3, δ) 8.11major (dd, J = 7.0, 1.4 Hz, 2H), 8.01minor (dd, J = 7.0, 1.4 Hz, 2H), 7.64 major (q, J = 8.4 Hz, 2H), 7.54 (q, J = 8.4 Hz, 4H), 7.45 (d, J = 7.3 Hz, 1H), 7.32 (t, J = 7.3 Hz, 1H), 7.26 (t, J = 7.4 Hz, 1H), 7.20 (td, J = 7.6, 1.2 Hz, 1H), 7.09 (dd, J = 15.9, 8.8 Hz, 6H), 6.94 minor (d, J = 9.2 Hz, 1H), 6.87–6.80 (m, 3H), 6.73 (d, J = 8.8 Hz, 2H), 4.30–4.20 (m, 3H), 4.17 (d, J = 7.4 Hz, 1H), 4.06–3.91 (m, 2H), 3.82minor (s, 3H), 3.76 major (s, 3H), 3.65major (dd, J = 16.9, 7.3 Hz, 1H), 3.58minor (d, J = 12.6 Hz, 1H). 13C{1H} NMR (75 MHz, CDCl3, δ) 198.5, 197.8, 176.2, 175.7, 158.9, 158.7, 153.9, 153.6, 136.9, 136.8, 133.4, 133.3, 131.9, 130.8, 129.5, 129.1, 128.8, 128.7, 128.2, 128.1, 126.4, 125.9, 125.3, 124.6, 124.0, 123.8, 113.9, 113.8, 110.8, 110.5, 55.2, 55.1, 48.3, 47.9, 42.3, 41.8, 41.4, 39.9. HRMS (MALDI-FT ICR) m/z: calcd for C24H20NaO4 [M + Na]+: 395.1234, found: 395.1253.
2-Oxo-3-(2-oxo-1,2-diphenylethyl)-2,3-dihydrobenzofuran-5-yl acetate (15d) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 75:25) and isolated as a white solid (48 mg, yield: 89%), a mixture of diastereomers (dr = 59/41). 1H NMR (400 MHz, CDCl3, δ) 8.10major (dd, J = 7.1, 1.5 Hz, 2H), 8.02 (dd, J = 7.1, 1.5 Hz, 2H), 7.64major (q, J = 7.5 Hz, 2H), 7.59–7.50 (m, 3H), 7.42minor (d, J = 7.0 Hz, 1H), 7.38–7.28 (m, 2H), 7.26–7.20 (m, 5H), 7.18 (d, J = 7.9 Hz, 2H), 7.04–6.99 (m, 1H), 6.96 (dd, J = 8.6, 2.4 Hz, 1H), 6.90 (d, J = 8.6, 1H), 6.55 (m, 1H), 4.36–4.23major (m, 3H), 4.21minor (d, J = 8.8 Hz, 1H), 4.07minor (q, J = 7.1 Hz, 1H), 3.97minor (dd, J = 17.1, 6.9 Hz, 1H), 3.68minor (dd, J = 17.2, 7.0 Hz, 1H), 3.59major (dd, J = 17.1, 3.8 Hz, 1H), 2.38major (s, 3H), 2.31minor (s, 3H). 13C{1H} NMR (75 MHz, CDCl3, δ) 198.3, 197.6, 175.9, 175.6, 169.6, 169.5, 151.3, 150.9, 146.9, 146.6, 139.6, 138.5, 136.8, 136.7, 133.5, 133.4, 130.0, 129.4, 128.77, 128.74, 128.6, 128.5, 128.2, 128.2, 127.8, 127.6, 127.2, 126.7, 122.3, 121.9, 119.2, 118.6, 111.2, 111.0, 48.5, 48.2, 43.0, 42.1, 41.4, 39.5, 21.1, 21.0. HRMS (MALDI-FT ICR) m/z: calcd for C25H20KO5 [M + K]+: 439.0942, found: 439.0967.
One diastereomer was separated by crystallization from a mixture of isopropanol/ether = 1:5 (1 mL):
(R*)-2-Oxo-3-((S*)-3-oxo-1,3-diphenylpropyl)-2,3-dihydrobenzofuran-5-yl acetate: white solid (28 mg); mp 116.5–117.7 °C. 1H NMR (400 MHz, CDCl3, δ) 8.10 (dd, J = 7.1, 1.5 Hz, 2H), 7.66 (q, J = 7.4, 1H), 7.55 (t, J = 7.6 Hz, 2H), 7.26–7.16 (m, 6H), 6.96 (dd, J = 8.6, 2.4 Hz, 1H), 4.34–4.21 (m, 3H), 3.59 (dd, J = 17.2, 4.0 Hz, 1H), 2.38 (s, 3H). 13C{1H} NMR (75 MHz, CDCl3, δ) 197.3, 171.2, 150.6, 141.5, 138.6, 133.4, 131.0, 129.3, 128.8, 128.7, 128.2, 128.1, 128.0, 127.6, 124.0, 123.5, 69.8, 49.3, 46.9, 42.1. IR (KBr): 2923, 1786, 1769, 1688, 1643, 1487 cm–1. HRMS (MALDI-FT ICR) m/z: calcd for C25H20KO5 [M + K]+: 439.0942, found: 439.0967.
3-(3-(4-Methoxyphenyl)-3-oxo-1-phenylpropyl)benzofuran-2(3H)-one (15e) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 70:30) and isolated as a white solid (41 mg, yield: 83% yield), a mixture of diastereomers (dr = 62/38). 1H NMR (400 MHz, CDCl3, δ) 8.09major (d, J = 9.0 Hz, 2H), 8.00minor (d, J = 8.9 Hz, 2H), 7.44major (d, J = 7.3 Hz, 1H), 7.35–7.25 (m, 3H), 7.24–7.14 (m, 8H), 7.10–7.04major (m, 1H), 7.00 (t, J = 9.5 Hz, 3H), 6.91major (d, J = 8.7 Hz, 1H), 6.84minor (d, J = 7.4 Hz, 1H), 4.32–4.23 (m, 3H), 4.21major (d, J = 8.6 Hz, 1 H), 4.07minor (q, J = 7.1 Hz, 1H), 3.93major (s, 3H), 3.92minor (s, 3H), 3.63minor (dd, J = 17.0, 7.3 Hz, 1H), 3.54major (dd, J = 16.6, 4.1 Hz, 1H), 13C{1H} NMR (75 MHz, CDCl3, δ) 196.9, 196.1, 176.2, 175.7, 163.8, 163.7, 153.9, 153.6, 140.1, 139.0, 130.5, 130.4, 130.0, 129.9, 129.1, 128.8, 128.6, 128.5, 128.4, 128.1, 127.6, 127.4, 126.3, 125.9, 125.3, 124.7, 124.0, 123.8, 113.9, 110.7, 110.4, 55.6, 48.1, 47.8, 43.2, 42.3, 41.1, 39.2. HRMS (MALDI-FT ICR) m/z: calcd for C24H20KO4 [M + K]+: 411.0993, found: 411.0973.
3-(1-(2-Chlorophenyl)-3-oxo-3-phenylpropyl)benzofuran-2(3H)-one (15f) was purified by flash chromatography (silica gel, hexane/ethyl acetate = 75:25) and isolated as a white solid (41 mg, yield: 81% yield), a mixture of diastereomers (dr = 54/46). 1H NMR (400 MHz, CDCl3, δ) 8.02major (dd, J = 7.1, 1.4 Hz, 2H), 7.96minor (d, J = 7.1, 1.4 Hz, 2H), 7.62 (q, J = 8.0 Hz, 2H), 7.54–7.46 (m, 4H), 7.40 (dd, J = 5.8, 3.7 Hz, 1H), 7.37–7.31 (m, 3H), 7.29 (dd, J = 8.5, 5.0 Hz, 3H), 7.24–7.06 (m, 5H), 7.03 (d, J = 8.2 Hz, 1H), 6.84minor (d, J = 7.0 Hz, 1H), 5.01major (td, J = 7.4, 4.8 Hz, 1H), 4.59minor (q, J = 7.4 Hz, 1H), 4.45major (d, J = 4.8 Hz, 1H), 3.97major (dd, J = 18.0, 7.7 Hz, 1H), 3.81minor (dd, J = 17.6, 8.8 Hz, 1H), 3.69minor (dd, J = 17.7, 5.0 Hz, 1H), 3.48major (dd, J = 18.0, 6.9 Hz, 1H). 13C{1H} NMR (75 MHz, CDCl3, δ) 197.5, 197.0, 176.2, 175.2, 153.6, 153.4, 137.8, 137.3, 136.4, 134.3, 134.1, 133.3, 133.2, 130.1, 130.0, 129.2, 128.9, 128.6, 128.5, 128.4, 128.0, 127.9, 127.6, 127.03, 127.00, 125.9, 125.2, 125.0, 124.7, 124.1, 123.7, 110.6, 110.3, 46.4, 46.2, 39.1, 39.0, 37.1. HRMS (MALDI-FT ICR) m/z: calcd for C23H17ClKO3 [M + K]+: 415.0497, found: 415.0499.
3-(1-(4-Fluorophenyl)-3-oxo-3-phenylpropyl)benzofuran-2(3H)-one (15g). The reaction mixture was heated at 50 °C (oil bath). The crude product was purified by flash chromatography (silica gel, hexane/ethyl acetate = 75:25) and isolated as a white solid (38 mg, yield: 79%), a mixture of diastereomers (dr = 64/36). 1H NMR (400 MHz, CDCl3, δ) 8.11major (dd, J = 7.2, 1.3 Hz, 2H), 8.01minor (dd, J = 7.2, 1.3 Hz, 2H), 7.65major (q, J = 7.8 Hz, 2H), 7.58–7.51 (m, 3H), 7.46major (d, J = 6.5 Hz, 1H), 7.34minor (t, J = 7.9 Hz, 1H), 7.26 (d, J = 8.6 Hz, 1H), 7.22 (dd, J = 7.5, 1.2 Hz, 1H), 7.17minor (dd, J = 8.9, 5.4 Hz, 1H), 7.12major (dd, J = 9.0, 5.4 Hz, 3H), 7.07minor (d, J = 8.0 Hz, 1H), 6.98 (t, J = 8.7 Hz, 1H), 6.94 (d, J = 7.9 Hz, 1H), 6.88 (t, J = 8.7 Hz, 3H), 4.33–4.21 (m, 4H), 4.18 (d, J = 7.2 Hz, 1H), 4.09minor (q, J = 7.3 Hz, 1H), 3.94major (dd, J = 17.3, 6.4 Hz, 1H), 3.73–3.55 (m, 2H). 13C{1H} NMR (101 MHz, CDCl3, δ) 198.2, 197.4, 176.0, 175.5, 163.2, 160.7, 153.9, 153.5, 136.8, 136.7, 135.6, 134.5, 133.6, 133.5, 130.1 (d, JC,F = 7.7 Hz), 129.7 (d, JC,F = 7.7 Hz), 129.3, 129.0, 128.8 (2C), 128.2, 128.1, 126.1, 125.5, 125.1, 124.5, 124.2, 123.9, 115.5 (d, JC,F = 21.2 Hz), 115.3 (d, JC,F = 21.2 Hz), 110.9, 110.6, 48.2, 47.9, 42.3, 41.5, 41.5, 39.8. 19F{1H} NMR (376 MHz, CDCl3, δ) −114.53, −114.84. HRMS (MALDI-FT ICR) m/z: calcd for C23H17FNaO3 [M + Na]+: 383.1053, found: 383.1055.
5-Chloro-3-(3-oxo-1,3-diphenylpropyl)benzofuran-2(3H)-one (15h). The reaction mixture was heated at 50 °C (oil bath). The crude product was purified by flash chromatography (silica gel, hexane/ethyl acetate = 80:20) and isolated as a white solid (38 mg, yield: 75%), a mixture of diastereomers (dr = 63/37). 1H NMR (300 MHz, CDCl3, δ) 8.04major (d, J = 8.1 Hz, 2H), 7.95minor (d, J = 8.2 Hz, 2H), 7.73minor (d, J = 8.1 Hz, 1H), 7.58major (d, J = 6.7 Hz, 2H), 7.49major (t, J = 7.8 Hz, 3H), 7.38major (d, J = 6.6 Hz, 3H), 7.25 (d, J = 7.6 Hz, 4H), 7.19–7.07 (m, 8H), 7.03 (d, J = 7.3 Hz, 2H), 6.87minor (d, J = 8.0 Hz, 2H), 6.79–6.67major (m, 1H), 4.29–4.11 (m, 4H), 3.98major (d, J = 3.9 Hz, 1H), 3.92minor (d, J = 6.5 Hz, 1H), 3.67–3.49 (m, 2H). 13C{1H} NMR (75 MHz, CDCl3, δ) 198.3, 197.5, 176.0, 175.5, 153.7, 153.4, 139.9, 138.7, 136.7, 136.6, 133.3, 133.2, 129.2, 129.0, 128.6, 128.5, 128.3, 127.5, 127.3, 126.0, 125.7, 125.1, 124.5, 123.9, 123.7, 110.6, 110.3, 47.9, 47.6, 42.9, 41.9, 41.5, 39.5. HRMS (MALDI-FT ICR) m/z: calcd for C23H17ClNaO3 [M + Na]+: 399.0758, found: 399.0771.
Acknowledgments
A.M. thanks the University of Salerno and MUR for financial support (FARB). The authors also thank Dr. Patrizia Iannece (Salerno) for performing some of the HRMS, Nathalie Hampel (LMU) for experimental support, and Anna Stahuber (LMU) for initial kinetic experiments. This research was funded by the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 101024710 “SImCat” (MSCA-IF-2020 to M.J.H.).
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.4c00277.
The authors declare no competing financial interest.
Supplementary Material
References
- Heathcock C. H.; Buse C. T.; Kleschick W. A.; Pirrung M. C.; Sohn J. E.; Lampe J. Acyclic stereoselection. 7. Stereoselective synthesis of 2-alkyl-3-hydroxy carbonyl compounds by aldol condensation. J. Org. Chem. 1980, 45, 1066–1081. 10.1021/jo01294a030. [DOI] [Google Scholar]
- a Seo S. W.; Kim S.-G. Enantioselective organocatalytic Michael addition of 2-arylacetates and 2-arylacetonitriles having an electron-withdrawing group to α,β-unsaturated aldehydes. Tetrahedron Lett. 2012, 53, 2809–2812. 10.1016/j.tetlet.2012.03.102. [DOI] [Google Scholar]; b Petrov É. S.; Tsvetkov E. N.; Mesyats S. P.; Shatenshtein A. N.; Kabachnik M. I. Equilibrium CH acidity of organophosphorus compounds. Russ. Chem. Bull. 1976, 25, 762–766. 10.1007/BF00922375. [DOI] [Google Scholar]
- a McLaughlin C.; Smith A. D. Generation and Reactivity of C(1)-Ammonium Enolates by Using Isothiourea Catalysis. Chem. - Eur. J. 2020, 27, 1533–1555. 10.1002/chem.202002059. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Stockhammer L.; Weinzierl D.; Bögl T.; Waser M. Enantioselective α-Chlorination Reactions of in Situ Generated C1 Ammonium Enolates under Base-Free Conditions. Org. Lett. 2021, 23, 6143–6147. 10.1021/acs.orglett.1c02256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang X.-M.; Bordwell F. G. Acidities and Homolytic Bond Dissociation Enthalpies (BDEs) of the Acidic H-A Bonds in Acyclic and Cyclic Alkoxycarbonyl Compounds (Esters and Carbamates). J. Org. Chem. 1994, 59, 6456–6458. 10.1021/jo00100a058. [DOI] [Google Scholar]; b Bordwell F. G.; Zhang S.; Zhang X.-M.; Liu W.-Z. Homolytic Bond Dissociation Enthalpies of the Acidic H–A Bonds Caused by Proximate Substituents in Sets of Methyl Ketones, Carboxylic Esters, and Carboxamides Related to Changes in Ground State Energies. J. Am. Chem. Soc. 1995, 117, 7092–7096. 10.1021/ja00132a008. [DOI] [Google Scholar]
- For stereoelectronic effects in lactones and esters:; a Corral-Bautista F.; Mayr H. Quantification of the Nucleophilic Reactivities of Cyclic β-Keto Ester Anions. Eur. J. Org Chem. 2015, 2015, 7594–7601. 10.1002/ejoc.201501107. [DOI] [Google Scholar]; b Alabugin I. V.; Kuhn L.; Medvedev M. G.; Krivoshchapov N. V.; Vil’ V. A.; Yaremenko I. A.; Mehaffy P.; Yarie M.; Terent’ev A. O.; Zolfigol M. A. Stereoelectronic power of oxygen in control of chemical reactivity: the anomeric effect is not alone. Chem. Soc. Rev. 2021, 50, 10253–10345. 10.1039/D1CS00386K. [DOI] [PubMed] [Google Scholar]
- a Zhu C.-L.; Fu X.-Y.; Wei A.-J.; Cahard D.; Ma J.-A. P-Spiro phosphonium salts catalyzed asymmetric fluorination of 3-substituted benzofuran-2(3H)-ones. J. Fluorine Chem. 2013, 150, 60–66. 10.1016/j.jfluchem.2013.03.007. [DOI] [Google Scholar]; b Cassani C.; Tian X.; Escudero-Adán E. C.; Melchiorre P. Multiple approaches to enantiopure spirocyclic benzofuranones using organocatalytic cascade reactions. Chem. Commun. 2011, 47, 233–235. 10.1039/C0CC01957G. [DOI] [PubMed] [Google Scholar]; c Li X.; Xi Z.; Luo S.; Cheng J.-P. Highly Enantioselective Michael Addition Reactions of 3-Substituted Benzofuran-2(3H)-ones to Chalcones Catalyzed by a Chiral Alkyl-Substituted Thiourea. Adv. Synth. Catal. 2010, 352, 1097–1101. 10.1002/adsc.201000106. [DOI] [Google Scholar]
- a Jefford C. W.; Bernardinelli G.; Wang Y.; Spellmeyer D. C.; Buda A.; Houk K. N. Torquoselectivity in the Electrocyclic Conversion of Benzocyclobutenes to o-Xylylenes. J. Am. Chem. Soc. 1992, 114, 1157–1165. 10.1021/ja00030a005. [DOI] [Google Scholar]; b Jones D. W.; Nongrum F. M. Intramolecular Diels-Alder additions to 2-benzopyran-3-ones; endo-selective additions and some reactions of the adducts. J. Chem. Soc., Perkin Trans. 1 1996, 705–713. 10.1039/P19960000705. [DOI] [Google Scholar]; c Nagao Y.; Nakazawa N.; Tagami K.; Iimori H.; Sano S.; Ishikawa T. Copper(I) Iodide-promoted Hydroxylation onto the Lithium or Potassium Enolate of Lactones and Lactams. Heterocycles 2001, 55, 2157–2170. 10.3987/COM-01-9330. [DOI] [Google Scholar]; d Zhao T.-Y.; Li K.; Yang L.-L.; Zhu S.-F.; Zhou Q.-L. Nickel-Catalyzed Desymmetrizing Cyclization of 1,6-Dienes to Construct Quaternary Stereocenters. Org. Lett. 2021, 23, 3814–3817. 10.1021/acs.orglett.1c00796. [DOI] [PubMed] [Google Scholar]
- a Barbier M. Direct Access to 4-Substituted 3-Isochromanones by the pH Controlled Addition of Aldehydes. Heterocycles 1987, 26, 421–425. 10.3987/R-1987-02-0421. [DOI] [Google Scholar]; b Barbier M. Syntheses of Substituted 2-Phenyl α β-Unsaturated Aliphatic Acids. Synth. Commun. 1989, 19, 1661–1667. 10.1080/00397918908051064. [DOI] [Google Scholar]; c Lóránd T.; Forgó P.; Földesi A.; Ôsz E.; Prókai L. Improved Solvent-Free Synthesis and Structure Elucidation of (E)- and (Z)-4-(Arylmethylene)-3-isochromanones. Eur. J. Org Chem. 2002, 2002, 2996–3003. . [DOI] [Google Scholar]; d Huszár M.; Varga A.; Horváth A.; Loránd T.; Agocs A.; Idei M.; Mandl J.; Vántus T.; Kéri G. Comparative Characterization of Experimental and Calculated Lipophilicity and Anti-Tumour Activity of Isochromanone Derivatives. Curr. Med. Chem. 2010, 17, 321–333. 10.2174/092986710790192703. [DOI] [PubMed] [Google Scholar]; e Bhowmik A.; Das S.; Sarkar W.; Saidalavi K. M.; Mishra A.; Roy A.; Deb I. Diastereoselective Spirocyclization via Intramolecular C(sp3)–H Bond Functionalization Triggered by Sequential [1,5]-Hydride Shift/Cyclization Process: Approach to Spiro-tetrahydroquinolines. Adv. Synth. Catal. 2021, 363, 826–832. 10.1002/adsc.202001011. [DOI] [Google Scholar]
- Flintoft R. J.; Buzby J. C.; Tucker J. A. Alkylation of Ketone and Ester Lithium Enolates with Nitroethylene. Tetrahedron Lett. 1999, 40, 4485–4488. 10.1016/S0040-4039(99)00814-X. [DOI] [Google Scholar]
- a Habtemariam S. Anti-Inflammatory Therapeutic Mechanisms of Natural Products: Insight from Rosemary Diterpenes, Carnosic Acid and Carnosol. Biomedicines 2023, 11, 545–571. 10.3390/biomedicines11020545. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rukachaisirikul V.; Khamthong N.; Sukpondma Y.; Phongpaichit S.; Hutadilok-Towatana N.; Graidist P.; Sakayaroj J.; Kirtikara K. Cyclohexene, Diketopiperazine, Lactone and Phenol Derivatives from the Sea Fan-derived Fungi Nigrospora sp. PSU-F11 and PSU-F12. Arch. Pharm. Res. 2010, 33, 375–380. 10.1007/s12272-010-0305-3. [DOI] [PubMed] [Google Scholar]
- a Watanabe K.; Taniguchi E. Systematic Syntheses and Structure-Activity Relationships of Substituted and Nonsubstituted Isochroman-3-ones as Plant Growth Regulators. J. Pestic. Sci. 1994, 19, 209–219. 10.1584/jpestics.19.3_209. [DOI] [Google Scholar]; b Zhao Z.; Kang K.; Yue J.; Ji X.; Qiao H.; Fan P.; Zheng X. Research progress in biological activities of isochroman derivatives. Eur. J. Med. Chem. 2021, 210, 113073. 10.1016/j.ejmech.2020.113073. [DOI] [PubMed] [Google Scholar]
- Recent examples from the Salerno group about the investigation of new electrophiles:; a Serusi L.; Di Mola A.; Massa A. A facile access to 1-substituted and unsubstituted 3-isoquinolinones via Mannich or Sn2 initiated cascade reactions under catalyst-free conditions. RSC Adv. 2023, 13, 6557–6563. 10.1039/D3RA00378G. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Macchia A.; Summa F. F.; Monaco G.; Eitzinger A.; Ofial A. R.; Di Mola A.; Massa A. Access to β-Alkylated γ-Functionalized Ketones via Conjugate Additions to Arylideneisoxazol-5-ones and Mo(CO)6-Mediated Reductive Cascade Reactions. ACS Omega 2022, 7, 8808–8818. 10.1021/acsomega.1c07081. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Serusi L.; Palombi L.; Pierri G.; Di Mola A.; Massa A. Asymmetric cascade Aza-Henry/lactamization reaction in the highly enantioselective organocatalytic synthesis of 3-(nitromethyl)isoindolin-1-ones from α-amido sulfones. J. Org. Chem. 2022, 87, 8420–8428. 10.1021/acs.joc.2c00518. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Macchia A.; Summa F. F.; Di Mola A.; Tedesco C.; Pierri G.; Ofial A. R.; Monaco G.; Massa A. Base-Promoted Cascade Reactions for the Synthesis of 3,3-Dialkylated Isoindolin-1-ones and 3-Methyleneisoindolin-1-ones. J. Org. Chem. 2021, 86, 15128–15138. 10.1021/acs.joc.1c01794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lucius R.; Loos R.; Mayr H. Kinetic Studies of Carbocation-Carbanion Combinations: Key to a General Concept of Polar Organic Reactivity. Angew. Chem., Int. Ed. 2002, 41, 91–95. . [DOI] [PubMed] [Google Scholar]; b Mayr H. Reactivity Scales for Quantifying Polar Organic Reactivity: The Benzhydrylium Methodology. Tetrahedron 2015, 71, 5095–5111. 10.1016/j.tet.2015.05.055. [DOI] [Google Scholar]; c Mayr H.; Ofial A. R. A Quantitative Approach to Polar Organic Reactivity. SAR QSAR Environ. Res. 2015, 26, 619–646. 10.1080/1062936X.2015.1078409. [DOI] [PubMed] [Google Scholar]
- For a freely accessible database of reactivity parameters N, sN, and E, see: https://www.cup.lmu.de/oc/mayr/reaktionsdatenbank2/ (accessed Feb 27, 2024). [Google Scholar]
- Zhuang C.; Zhang W.; Sheng C.; Zhang W.; Xing C.; Miao Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. 10.1021/acs.chemrev.7b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Richter D.; Hampel N.; Singer T.; Ofial A. R.; Mayr H. Synthesis and Characterization of Novel Quinone Methides: Reference Electrophiles for the Construction of Nucleophilicity Scales. Eur. J. Org Chem. 2009, 2009, 3203–3211. 10.1002/ejoc.200900299. [DOI] [Google Scholar]; b Kaumanns O.; Lucius R.; Mayr H. Determination of the Electrophilicity Parameters of Diethyl Benzylidenemalonates in Dimethyl Sulfoxide: Reference Electrophiles for Characterizing Strong Nucleophiles. Chem. - Eur. J. 2008, 14, 9675–9682. 10.1002/chem.200801277. [DOI] [PubMed] [Google Scholar]
- A solution of 4 in d6-DMSO was sufficiently stable to characterize the enolate 4 by 1D and 2D NMR spectroscopy (Supporting Information, Figure S1).
- Corral-Bautista F.; Mayr H. Quantification of the Nucleophilic Reactivities of Ethyl Arylacetate Anions. Eur. J. Org Chem. 2013, 2013, 4255–4261. 10.1002/ejoc.201300265. [DOI] [Google Scholar]
- Tominaga Y.; Takada S.; Kohra S.; Shigemitsu Y. Synthesis and [3 + 2] Cycloaddition Reaction of 3-[(Trimethylsilylmethylamino)(methylthio)-methylene]heterocyclic Compounds. J. Heterocycl. Chem. 2001, 38, 1143–1151. 10.1002/jhet.5570380519. [DOI] [Google Scholar]
- Li Z.; Mayer R. J.; Ofial A. R.; Mayr H. From Carbodiimides to Carbon Dioxide: Quantification of the Electrophilic Reactivities of Heteroallenes. J. Am. Chem. Soc. 2020, 142, 8383–8402. 10.1021/jacs.0c01960. [DOI] [PubMed] [Google Scholar]
- Allgäuer D. S.; Jangra H.; Asahara H.; Li Z.; Chen Q.; Zipse H.; Ofial A. R.; Mayr H. Quantification and Theoretical Analysis of the Electrophilicities of Michael Acceptors. J. Am. Chem. Soc. 2017, 139, 13318–13329. 10.1021/jacs.7b05106. [DOI] [PubMed] [Google Scholar]
- Deposition number 2320508 (for 7a-major, bv020), 2314553 (for 14b-1) and 2314554 (for 15d-1) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- Hansch C.; Leo A.; Taft R. W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- Fulmer G. R.; Miller A. J. M.; Sherden N. H.; Gottlieb H. E.; Nudelman A.; Stoltz B. M.; Bercaw J. E.; Goldberg K. I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. 10.1021/om100106e. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.