

Abstract
The microporous annealed particle (MAP) scaffold platform is a subclass of granular hydrogels composed of an injectable slurry of microgels that can form a structurally stable scaffold with cell-scale porosity in situ following a secondary light-based chemical crosslinking step (i.e., annealing). MAP scaffold has shown success in a variety of regenerative medicine applications, including dermal wound healing, vocal fold augmentation, and stem cell delivery. This paper describes the methods for synthesis and characterization of poly(ethylene glycol) (PEG) microgels as the building blocks to form an MAP scaffold. These methods include the synthesis of a custom annealing macromer (MethMAL), determination of microgel precursor gelation kinetics, microfluidic device fabrication, microfluidic generation of microgels, microgel purification, and basic scaffold characterization, including microgel sizing and scaffold annealing. Specifically, the high-throughput microfluidic methods described herein can produce large volumes of microgels that can be used to generate MAP scaffolds for any desired application, especially in the field of regenerative medicine.
SUMMARY:
This protocol describes a set of methods for synthesizing the microgel building blocks for microporous annealed particle scaffold, which can be used for a variety of regenerative medicine applications.
INTRODUCTION:
The MAP scaffold platform is an injectable biomaterial composed entirely of hydrogel microparticles (microgels) that, when crosslinked together, provide cell-scale microporosity, which allows for degradation-independent cell migration and bulk tissue integration1. Due to its ability to quickly integrate with host tissue and inherently low immunogenicity, the MAP scaffold platform has demonstrated preclinical applicability for a wide variety of regenerative medicine therapies2–10, including accelerating dermal wound healing1,3,11, revascularizing brain stroke cavity7, delivering mesenchymal stem cells2, and providing tissue bulking to treat glottic insufficiency6. MAP has also been shown to convey anti-inflammatory effects to host tissue through the recruitment of M2 macrophages3 and can even be tuned to promote a Th2 “tissue repair” immune response8. These favorable properties of the MAP scaffold platform allow it to be expanded to a diverse range of clinical applications.
Previously published methods for generating microgels for MAP scaffold formation have included flow-focusing droplet-microfluidics1,4,7,9, electrospraying5,12, and overhead spinning with batch emulsion6, 10. The droplet microfluidic method can produce particles with high monodispersity but uses very slow flow rates that produce low yields of particles (µL/h). Alternatively, the electrospraying and batch emulsion methods can produce a high volume of particles, but with high particle polydispersity. This protocol uses a high-throughput microfluidic method to produce microgels with a monodisperse population, based on work by de Rutte et al13. This method utilizes soft lithography techniques to make a polydimethylsiloxane (PDMS) microfluidic device from a photomask, which is then bonded to a glass slide. The device design relies on step-emulsification to generate a high volume of microgel particles (mL/h). The monodispersity that can be achieved with this method provides superior control of porosity compared to other techniques, since monodisperse microgels can form scaffolds with more uniform pore sizes2.
The methods for synthesizing and characterizing the individual microgels that can act as building blocks for MAP scaffolds are outlined within this manuscript, specifically in terms of the creation of microgels consisting of a PEG backbone with a maleimide (MAL) group, which readily participates in efficient Michael-type addition with thiol-functionalized crosslinkers for microgel gelation. To decouple microgel gelation from MAP scaffold annealing, this manuscript also describes how to synthesize a published14 custom annealing macromer, MethMAL, which is a heterofunctional methacrylamide/maleimide 4-arm PEG macromer. The methacrylamide functional groups readily participate in free-radical photopolymerization (for microgel annealing), while remaining relatively inert to the conditions that promote Michael-type addition for the MAL functional groups.
Additionally, this manuscript outlines the protocols for creating PDMS microfluidic devices, determining microgel gelation kinetics, and characterizing microgel size. The final part of the manuscript details MAP scaffold annealing, which is when the microgels are transitioned in situ into a solid scaffold through a secondary, photo-initiated crosslinking step that covalently bonds the surfaces of the microgels together. It is important to note that there are other annealing methods that can be implemented in MAP scaffold systems that do not rely on light-based chemistries, such as enzyme-mediated annealing, as previously described1. Overall, these methods can be used directly or used with different hydrogel formulation chemistries (e.g., hyaluronic acid-based) to generate MAP scaffolds for any application.
PROTOCOL:
1. MethMAL annealing macromer synthesis
NOTE: This protocol is specifically for modifying 1 g of PEG-maleimide but can be scaled up to make larger batches.
1.1. Add 1 g of 4-arm 20 kDa PEG-maleimide to 10 mL of 1x phosphate-buffered saline (PBS, pH 7.4) in a small glass beaker with a stir bar. Stir the solution at 300 rpm until the PEG is fully dissolved (~30 min).
1.2. Add 14.65 mg of 2-aminoethanethiol (0.67:1 thiol [SH] to maleimide [MAL] molar ratio) to the reaction with stirring at 300 rpm.
NOTE: The thiol groups on 2-aminoethanethiol will be added to approximately three arms of the PEG-MAL via Michael-type addition, leaving amine end groups.
1.2.1. Observe the following example calculation for the amount of 2-aminoethanethiol to add:
NOTE: To avoid measuring a very small amount, dissolve 100 mg of 2-aminoethanethiol in 1 mL of 1x PBS (pH 7.4) and add 146.5 µL of that solution to the reaction.
1.3. Wait for 1 h and 10 min after step 1.2., then mix 160.1 mg of 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM, 12:1 molar ratio to PEG) and 32.77 µL of methacrylic acid (8:1 molar ratio to PEG) in 5 mL of 1x PBS (pH 7.4) in a new glass beaker and react for 50 min with stirring at 300 rpm.
NOTE: At this step, the methacrylic acid reacts with DMTMM to form a highly reactive ester that can then be coupled to the available amine groups on the PEG.
1.3.1. Observe the following example calculation for calculating the amount of DMTMM to add:
1.3.2. Observe the following example calculation for calculating the amount of methacrylic acid to add:
1.4. Add the methacrylic acid/DMTMM solution to the beaker with the 20 kDa PEG-MAL/2-aminoethanethiol solution.
1.5. Add 53.56 µL of triethylamine (1:1 molar ratio to methacrylic acid) to the beaker and allow to react overnight with stirring at 300 rpm. Cover the beaker with foil to prevent dust and other contaminants from entering the beaker.
1.5.1. Observe the following example calculation for the amount of triethylamine to add:
1.6. Transfer the reaction to snakeskin dialysis tubing (molecular weight cut-off: 3,500 Da).
1.7. Place the dialysis tubing in a large beaker with 1 M NaCl in deionized (DI) water (volume should cover tubing entirely) for 3 days with stirring at 300 rpm. Change the 1 M NaCl solution 2x daily for a total of six washes.
1.8. Dialyze for 6 h in DI water. Change the DI water each hour for a total of six washes.
1.9. Transfer the reaction to a 50 mL conical tube and freeze at −80 °C.
1.10. Lyophilize the tube for at least 72 h at a start temperature of −70 °C and a temperature ramp of 0.01 °C/min until 0 °C.
1.11. Prepare the sample for 1H-NMR by dissolving 25 mg of MethMAL in 700 µL of chloroform-d and transfer to an NMR tube.
1.12. Acquire the 1H-NMR spectra.
NOTE: The spectra in this protocol were acquired using a 500 MHz spectrometer with the following parameters: sweep width = 6,467.3 Hz, time delay = 13.1 s, acquisition time = 5.1 s, pulse time = 5.1 µs, and number of scans = 8.
1.13. For analysis, integrate the maleimide peak as a reference (~6.76 ppm) and then integrate the two methacrylamide peaks (~5.35 ppm and 5.6 ppm). Divide the ratio of the methacrylamide peak areas by the total area of all three peaks to obtain the percentage of arms modified with methacrylamide.
NOTE: An acceptable modification is 67%–75% methacrylamide functional group substitution. An example 1H-NMR spectrum for MethMAL is shown in Figure 1.
Figure 1: Chemical structure and 1H-NMR spectrum of MethMAL.

(A) Chemical structure: the MethMAL annealing macromer is composed of 20 kDa 4-arm poly(ethylene glycol) modified with three methacrylamide arms. (B) This structure generates peaks at 5.36 ppm (3) and 5.76 ppm (2) not present in PEG-MAL spectra, and one maleimide arm, which generates a peak at 6.71 ppm (1). The solvent, chloroform, generated a peak at 7.26 ppm, and residual water in this sample generated a peak at 2.2 ppm (labeled on spectra). In the MethMAL spectra, the maleimide peak had an integrated area of 0.27, and the sum of the methacrylamide peaks areas was 0.73 (0.37 + 0.36). The methacrylamide percentage modification was 73% (0.73/(0.27 + 0.73)). This figure is from Pfaff et al.14. Copyright (2021) American Chemical Society.
1.13.1. Use equation (1) as the degree of substitution equation:
| (1) |
2. Microgel precursor gelation kinetics
NOTE: Gelation time can be modified by adjusting the pH of the buffer used to dissolve the gel precursor components. For PEG-maleimide hydrogels, a more acidic pH typically corresponds to slower gelation time since the thiolate concentration is decreased at lower pH15.
2.1. Predetermine the desired concentrations of polymer, crosslinker, and other gel precursor components (i.e., peptides, glycosaminoglycans). Dissolve the PEG-MAL, RGD, and MethMAL (PEG-backbone) in 10x PBS (pH 1.5) and the MMP-2 crosslinker in 1x PBS (pH 7.4).
NOTE: In this protocol, the gel precursor solution is a published formulation3, which consists of 45.88 mg/mL 4-arm PEG-MAL (10 kDa), 0.82 mg/mL RGD, 8.06 mg/mL MethMAL, and 4.62 mg/mL MMP-2 crosslinker (enzymatically degradable crosslinker).
2.2. Prepare a viscometer, or an equivalent instrument, to monitor storage and loss moduli using a gap size of 0.4 mm, a shear strain of 1.0, a frequency of 1.0 Hz, and a duration of 10,800 s.
2.2.1. Attach a 35 mm plate rotor (P35/Ti) geometry. Use a humidified chamber or a damp sponge around the geometry to maintain a humidified environment.
2.2.2. Mix the PEG-backbone and MMP-crosslinker in a 1:1 volumetric ratio.
2.2.3. Pipette 400 µL of the gel precursor solution onto the center of the viscometer stage.
2.2.4. Slowly lower the geometry onto the stage until it reaches the predetermined gap size of 0.4 mm and immediately begin monitoring storage (G’) and loss (G’’) moduli for up to 6 h at room temperature.
NOTE: Gelation is considered to begin when the storage modulus becomes greater than the loss modulus, and gelation is considered complete when the storage modulus plateaus. A representative gelation kinetics curve is shown in Figure 2.
Figure 2: Representative curve of gelation kinetics of an MAP gel precursor solution (pH 4.5) determined by a viscometer.

Gelation begins at the rapid increase in storage (G’) modulus, and gelation completes when the G’ curve plateaus. G’’ indicates the loss modulus. This figure is from Pruett et al.3. Copyright (2021) Wiley-VCH GmBH.
3. Microfluidic device fabrication
NOTE: This protocol describes device fabrication of a microfluidic step-emulsification device design adapted from de Rutte et al.13, which can be seen in Figure 3A. However, this protocol can be used with any device design that is etched into a SU-8 wafer. It is recommended to outsource the SU-8 silicon wafer master fabrication, unless the appropriate cleanroom facilities are available for fabrication.
Figure 3: Microfluidic PDMS device.
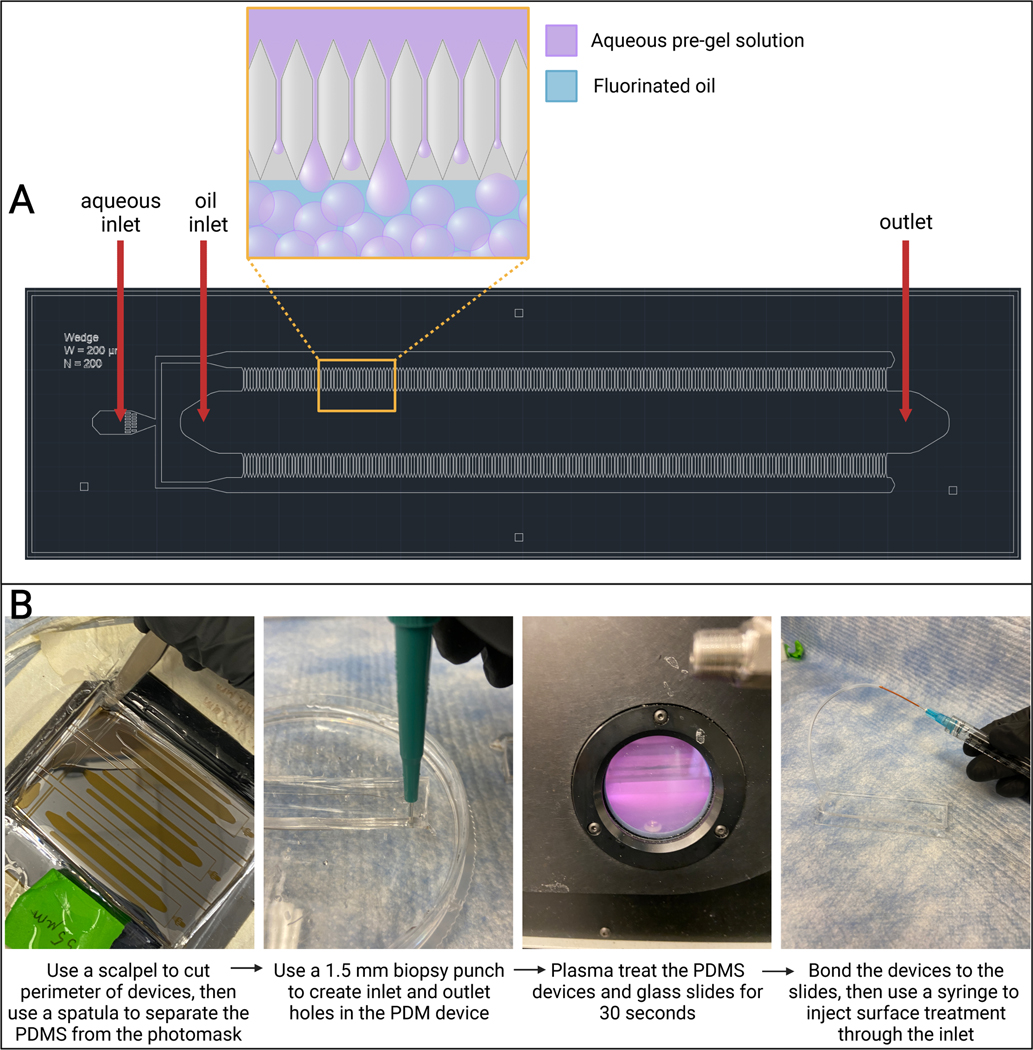
(A) Computer-assisted design (AutoCAD) drawing of microfluidic device design. Microgel droplet formation occurs in the channels on either side of the oil channel, as seen in the magnified outcropping. (B) Overview of PDMS device fabrication. Abbreviation: PDMS = polydimethylsiloxane.
3.1. Create the first polydimethylsiloxane (PDMS) microfluidic device from an SU-8 wafer photomask.
3.1.1. Prepare ~200 g of PDMS by mixing the base and curing agent in a 10:1 w/w ratio (see the Table of Materials).
Table of Materials
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
|---|---|---|---|
| 2-aminoethanethiol hydrochloride | Acros Organics | AC153770250 | MW: 113.61Da |
| 35 mm plate rotor | HAAKE | P35/Ti | Geometry for HAAKE viscometer |
| 4-arm PEG-Maleimide (10kDa) | NOF AMERICA Corporation | SUNBRIGHT PTE-100MA | For microgel precursor solution |
| 4-arm PEG-Maleimide (20kDa) | NOF AMERICA Corporation | SUNBRIGHT PTE-200MA | Molecular weight specific to each batch. |
| BD Syringe with Luer-Lok Tips | Becton Dickinson | Disposable plastic syringes | |
| Biopsy punch | Mitex | MLTX33–31A-P/25 | 1.5 mm diameter |
| Chloroform-d | Acros Organics | AC209561000 | For MethMal Synthesis |
| Collimated LED Light Source | ThorLabs | M365LP1-C1 | 365nm |
| Culture dish (15 cm) | Corning | CLS430599 | 150 mm x 25 mm |
| morpholinium chloride) | Oakwood Chemical | 151882 | MW: 276.72Da |
| Fluorosurfactant | Ran Technologies | 008-Flurosurfactant-5wtH-200G | 5 weight percent of 008-Flurosurfactant in HFE7500 |
| FreeZone Triad Freeze Dry System | Labconco | 7400000 Series | Lyophilizer |
| Glass slides | Fisher Scientific | 12–550-A3 | Plain glass slides, uncoated |
| HAAKE Rheowin viscometer | HAAKE | ||
| KDS Legato 210 Dual Prong Syringe Pump | Kd Scientific | ||
| LED Driver | ThorLabs | DC2200 | |
| Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) | Sigma-Aldrich | 900889 | Photoinitiator |
| Methacrylic Acid | Sigma Aldrich | 155721 | MW: 86.09Da |
| Microfluidic device SU8-Si master wafer | FlowJem | N/A | Custom-made, with silanization |
| MMP-2 degradable crosslinker | WatsonBio Sciences | Sequence: Ac-GCGPQGIAGQDGCG-NH2 | |
| Needles (25 G) | BD | 305122 | Gauge: 0.5 mm |
| Novec 7500 | 3M | 7100025016 | Fluorinated oil |
| Oxygen | Praxair | UN1072 | Compressed |
| Peek tubing | Trajan Scientific | 03–350-523 | 1/32" Outer Diameter; 0.02" Inner Diameter; 10' Length |
| PFOCTS (trichloro(1H,1H,2H,2H-perfluorooctyl)silane) | Sigma-Aldrich | 448931 | For surface treatment |
| Phosphate Buffered Saline | Fisher BioReagants | BP3994 | Diluted to 1X in ultrapure water, pH=7.4 |
| Plasma cleaner | Harrick Plasma | PDC-001-HP | |
| Razor blade | Fisher Scientific | 12–640 | |
| RGD cell adhesive peptide | WatsonBio Sciences | Sequence: Ac-RGDSPGGC-NH2 | |
| Rheowin software | HAAKE | Software compatible with HAAKE viscometer | |
| Scalpel blade | Bard-Parker | 371210 | Size: #10 |
| Scalpel handle | Bard-Parker | 371030 | Size: #3 |
| Sodium Chloride | Fisher BioReagents | BP358–1 | MW: 58.44Da |
| Sylgard 184 silicone elastomer kit | DOW Chemical | 2065622 | Base and curing agent |
| Triethylamine | Fisher Scientific | O4884–100 | MW: 101.19Da |
| Tygon tubing | Saint Gobain Performance Plastics | AAD04103 | ID: 0.51 mm OD: 1.52 mm |
| Varian Inova 500 Spectrometer | Varian | NMR Located in the UVA Biomolecular Magnetic Resonance Facility |
3.1.2. Place the SU-8 wafer in a culture dish (tape the edges of the wafer to the dish), with the microfluidic design facing up.
3.1.3. Pour PDMS into the dish to create a 1–1.5 cm thick layer over the wafer. Place the dish under vacuum to remove any bubbles.
3.1.4. Let the PDMS cure until solidified (24 h at room temperature or ~2 h at 60 °C).
3.1.5. Once the PDMS has cured, carefully use a razor blade or a scalpel to cut a rectangle through the PDMS, such that there is a 0.5–1 cm margin around the design on the wafer.
NOTE: Do not use too much pressure and be very gentle to avoid damaging (i.e., cracking, scratching, etc.) the wafer.
3.1.6. Wedge a spatula into one of the cuts and slide it along the cuts to separate the PDMS from the wafer.
NOTE: As the PDMS separates from the wafer, an air bubble will begin to form under the PDMS (refer to Figure 3B).
3.1.7. Use the spatula to gently lift the rectangle of PDMS out of the dish.
NOTE: The resultant gap in the PDMS above the wafer will be the mold for the microfluidic devices.
3.2. Create subsequent PDMS microfluidic devices from the SU-8 wafer photomask.
3.2.1. Prepare ~15–20 g of PDMS per mold by mixing the base and curing agent in a 10:1 ratio.
3.2.2. Pour PDMS into the rectangular gap in the mold to create a 5 mm layer of PDMS above the wafer. Put the mold under vacuum to remove any bubbles.
3.2.3. Let the PDMS cure until fully solidified (24 h at room temperature or 2 h at 60 °C).
3.2.4. Once the PDMS has cured, use a razor blade or a scalpel to cut the PDMS around the edges of the rectangle.
NOTE: Do not use too much pressure and be very gentle to avoid damaging (i.e., cracking) the wafer.
3.2.5. Wedge a spatula into one of the cuts and slide it along the cuts to separate the PDMS from the wafer.
NOTE: As the PDMS separates from the wafer, an air bubble will begin to form under the PDMS.
3.2.6. Use a 1.5 mm biopsy punch to create holes through the PDMS rectangle at the two inlets and outlet (refer to Figure 3B).
3.2.7. If there are multiple devices on a single wafer, use a spatula or razor blade to lightly score the PDMS between each device, then gently fold the PDMS along the scored lines so that the PDMS separates very cleanly on its own.
3.2.8. Store the PDMS devices in a dust-free container.
3.3. Bond the PDMS microfluidic devices to glass slides.
3.3.1. Prepare one glass slide per PDMS microfluidic device. Use tape, filtered air, or isopropyl alcohol (IPA) washes to remove any dust from the slide. Make sure the slides are completely dry before moving on to the next step.
3.3.2. Place a glass slide and a PDMS device, design-side up, next to each other on a small plastic tray (a 96-well plate lid works well for this) and place it in a plasma cleaner. Close the door and air-flow valve and turn on the vacuum pump. Let it run for at least 30 s, then turn it off.
3.3.3. Connect the oxygen tank gas tube to the air-flow valve. Let the plasma chamber fill with oxygen for 30 s, then turn off the oxygen, and close the air-flow valve.
3.3.4. Turn the vacuum pump on and set the radiofrequency (RF) level to high. Wait until the chamber turns a violet-pink color (refer to Figure 3B). Allow 30 s to pass.
3.3.5. When the timer goes off, turn off the plasma and vacuum. Then, slowly open the air-flow valve to release the vacuum. Remove the tray from the plasma cleaner.
3.3.6. Gently flip the PDMS device onto the glass slide to bond them. As bonding happens, observe the slight difference in the transparency of the PDMS.
NOTE: For the best results, store the bonded devices at 60 °C until immediately before use.
3.4. Surface-treating the PDMS microfluidic devices
3.4.1. Prepare the surface treatment by diluting PFOCTS (trichloro(1H,1H,2H,2H-perfluorooctyl)silane) in Novec oil (1:50). Use 1 mL for 3–4 devices. Transfer the volume to a 1 mL syringe and attach a 25 G needle.
NOTE: The needles used in this protocol are beveled, so take care when handling sharps. Blunt needles can also be used if desired.
3.4.2. Cut a 10–12 cm piece of Tygon tubing per device that will be treated.
3.4.3. Cut a piece of PEEK tubing, ~1 inch in length. Insert a couple of millimeters of the PEEK tubing into the end of Tygon tubing, as shown in Figure 4A, to help prevent the needle from puncturing the Tygon tubing inlets.
Figure 4: Microfluidic setup.
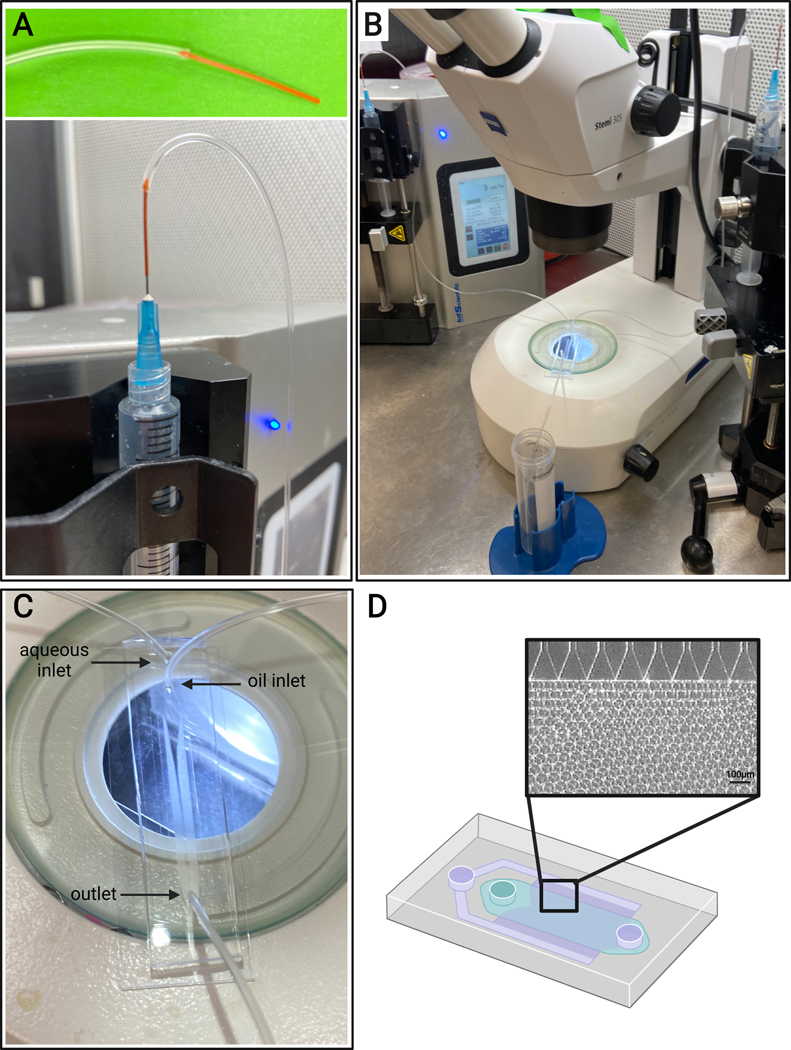
(A) Depiction of the method for connecting PEEK tubing (top) and Tygon tubing to a 25 G needle on a syringe (bottom). (B) Microfluidic setup with syringe pumps, tubing, device, and microscope. (C) Image of microfluidic device setup, with two inlets (aqueous and oil) and one outlet. (D) Schematic of the microfluidic device and representative brightfield image of expected microgel formation from the channels in a step-emulsification device.
3.4.4. Take the device(s) out of the heated chamber and insert the non-PEEK end of the Tygon tubing into the aqueous inlet hole.
3.4.5. Insert the needle of the surface treatment syringe into the PEEK tubing and cover the oil chamber outlet hole (refer to Figure 3B).
3.4.6. Inject the treatment into the device slowly and ensure that it fills the device, without bubbles. Wait for the aqueous chambers to fill first, followed by the smaller channels, and then the oil chamber. Remove the Tygon tubing from the device. Once the device has been filled, let it rest for 10 min at room temperature.
3.4.7. Fill a 5 mL syringe with oil only (no silane) and attach a 25 G needle.
3.4.8. Aspirate the surface treatment out of the device through the inlets and outlet. Insert Tygon tubing into the aqueous inlet, insert the syringe with oil into the PEEK tubing, and flush each device with oil. Aspirate the oil out of the device.
3.4.9. Repeat the oil flush 2x more. Remove the Tygon tubing.
NOTE: The device is ready to be used.
4. Microfluidic generation of microgels
4.1. Predetermine the desired concentrations of polymer, crosslinker, and other gel precursor components (i.e., peptides, glycosaminoglycans). Dissolve the PEG-MAL, RGD, and MethMAL (PEG-backbone) in 10x PBS (pH 1.5) and the MMP-2 crosslinker along with 5 µM of biotin-maleimide in 1x PBS (pH 7.4).
NOTE: In this protocol, the gel precursor solution is a published formulation3 consisting of 45.88 mg/mL 4-arm PEG-MAL (10 kDa), 0.82 mg/mL RGD, 8.06 mg/mL MethMAL, and 4.62 mg/mL MMP-2 crosslinker (enzymatically degradable crosslinker). The methods outlined below will yield ~3 mL of microgels.
4.2. Prepare 6 mL of surfactant solution by diluting the stock 5% FluoroSurfactant to at least 1% in Novec oil. Add this solution to a 10 mL syringe.
NOTE: Fluorinated surfactant is insoluble with water, which allows it to be easily removed during the gel transition to the aqueous phase during the PBS washes in the purification step.
4.3. Cut three pieces of Tygon tubing that are an appropriate length for the syringe pump height.
4.4. Cut two pieces of PEEK tubing, ~1 inch in length. Insert a couple of millimeters of the PEEK tubing into the end of two pieces of Tygon tubing, as shown in Figure 4A, to help prevent the needle from puncturing the Tygon tubing inlets.
4.5. Insert the non-PEEK end of the Tygon tubing into the inlets of the microfluidic devices. Insert the remaining piece of Tygon tubing (with no PEEK tubing on the end) into the microfluidic device outlet, as shown in Figure 4C.
4.6. Add at least 3 mL of oil to a 5 mL plastic syringe and attach it to a 25 G needle. Carefully insert the needle into the PEEK tubing on one of the Tygon inlets. Gently flush the tubing and device with oil. Collect the oil from the outlet in a conical tube. Repeat the oil flush on the other Tygon inlet.
4.7. Set the syringe pumps to the desired flow rates.
NOTE: This protocol uses 3 mL/h for the aqueous flow rate and 6 mL/h for the oil flow rate. It may be required to use two separate syringe pumps.
4.8. Connect the syringe containing the surfactant to the oil inlet via a 25 G needle (refer to Figure 4A) and gently dispense enough oil to prime the tubing and the oil channel of the microfluidic device.
4.9. Once the device and oil inlets have been set up, add 0.5 mL of oil to a new 5 mL syringe, which will contain the gel precursor. Use this small amount of oil to help flush the precursor solution through the microfluidic device.
4.10. In a conical tube, combine 1.5 mL of the PEG-backbone solution and 1.5 mL of the crosslinker solution. Vortex for 30 s and quickly transfer the combined gel precursor solution to the 5 mL syringe.
4.11. Connect the syringe with the gel precursor solution to the aqueous inlet via a 25 G needle. Gently dispense enough solution to prime the tubing and the aqueous channel.
4.12. Clamp the syringes onto the respective syringe pumps and press run (refer to Figure 4B). Ensure that there is liquid flowing through both the aqueous and oil channels.
NOTE: It is recommended to use a microscope to visualize microgel formation within the PDMS device.
4.13. Look for particles of uniform size from the channels (refer to Figure 4D). Collect the microgels from the outlet in a conical tube.
5. Purification and sterilization of microgels
5.1. Once gelation is complete (determined as time to storage modulus plateau in gelation kinetics characterization), use a pipette to carefully remove the oil phase from the bottom of the tube (refer to Figure 5). Deposit this into an appropriate waste container for fluorinated waste.
Figure 5: Overview of the microgel purification procedure.

Abbreviations: PBS = phosphate-buffered saline; IPA = isopropyl alcohol.
NOTE: The gelation time can be accelerated by adding an organic soluble base to the collection tube (e.g., triethylamine), but it is important to note that the addition of a strong base may hydrolyze any unreacted maleimides. If it is desired to functionalize microgels through excess maleimides post gelation, skip the addition of triethylamine.
5.2. Add more oil into the microgel collection tube in a 1:1 ratio. Mix by gently inverting the collection tube. Do not vortex.
5.3. Let the collection tube settle for ~5 min to allow the phases to separate. Look for the oil phase at the bottom and the aqueous phase (the microgels) on the top (refer to Figure 5).
5.4. Repeat the oil washes at least 2x more.
5.5. Add more oil in a 1:1 ratio with the gel and add 1x PBS in a 4:1 PBS to gel ratio. Invert to mix several times. To separate the layers, centrifuge the tube at ~2,000 x g for ~30 s. Look for the oil phase at the bottom of the tube, gel in the middle, and PBS on top (refer to Figure 5).
5.6. Remove the oil phase with a pipette and discard in a waste container.
NOTE: Do not remove the PBS. Use a larger conical tube to continue washes if the original collection tube was 15 mL.
5.7. Repeat oil and PBS washes 2x more. Look for the gel to transition from opaque to clear by the final wash, as shown in Figure 5, indicating that the surfactant was removed and the gel is in the PBS phase.
5.8. Remove all oil. Do not remove PBS from the conical tube.
5.9. In a chemical fume hood, use a glass pipette to add hexanes to the tube at an equal volume to the PBS. Vortex the conical tube for 30 s or until mixed thoroughly. Centrifuge at 4,696 x g for 5 min.
5.10. After separation, look for hexanes in the top layer, PBS in the middle, and gel at the bottom (refer to Figure 5). Remove the hexane layer and discard it in a container for organic waste. Aspirate the PBS.
5.11. Repeat hexane and PBS washes at least 2x more or until the gel appears nearly translucent (refer to Figure 5). Wash the gel with PBS 1x more so any remaining hexanes are removed. Centrifuge at 4,696 x g for 5 min. Aspirate the PBS layer. Take care not to disturb the gel pellet.
5.11.1. To cap, or quench, any unreacted maleimides in the microgels, prepare a 100 mM solution of N-acetyl-L-cysteine in 1x PBS and add this solution to the gel. Place on a tube rotator at 37 °C overnight, followed by many washes with PBS to remove unreacted N-acetyl-L-cysteine.
5.11.2. For long-term storage (up to 1 year), resuspend the microgels in 70% IPA and store at 4 °C to help prevent the growth of bacteria on the microgels.
5.11.3. To sterilize the microgels, add 70% IPA to the gel in a 4:1 v/v ratio in a biosafety hood. Vortex the tube for 30 s, then centrifuge at 4,696 × g for 5 min. Aspirate the IPA supernatant from the gel pellet in a biosafety hood. Perform 2x more IPA washes followed by 3x washes with sterile 1x PBS.
NOTE: All IPA should be removed before using the gel with cells or animals.
6. Microgel size characterization
NOTE: It is recommended to allow microgel particles to equilibrate in 1x PBS overnight at 37 °C to swell to their final diameter before sizing.
6.1. Image gel particles.
6.1.1. Spin down the MAP gel at 4,696 × g for 5 min and aspirate the supernatant.
6.1.2. Using a positive displacement pipette, remove 5 µL of microgels from the gel pellet and dilute in 1 mL of PBS in a microcentrifuge tube (1:200 dilution). Adjust this dilution as needed.
NOTE: During the formulation of the gel precursor solution, 5 µM of biotin-maleimide can be added and used as an alternative to labeling microgels with a fluorophore. In this case, a streptavidin-fluorophore can be added at a 1:300 dilution (from 1 mg/mL stock). Allow incubation with streptavidin for at least 15 min prior to imaging.
6.1.3. Using a positive displacement pipette, transfer 100 µL of the diluted microgels to the wells of a clear 96-well plate.
6.1.4. Use a widefield or confocal microscope to image the microgels with a 10x objective.
6.1.5. See Figure 6 for representative confocal images of microgels.
Figure 6: Representative images of microgels.

(A) Fluorescent confocal image of microgel population A, (B) image of thresholded microgels, and (C) particle outlines after ImageJ analysis. (D) Fluorescent confocal image of microgel population B and (E) transmitted light image of microgels (microgels are nearly translucent). (F) Depiction of representative results from the ImageJ analysis outlined in this protocol. Both microgel populations have relatively monodisperse PDIs. Both populations of microgels were synthesized with a 3 mL/h aqueous flow rate and a 6 mL/h oil flow rate. However, the difference in microgel size is due to differences in microfluidic device step size. For example, microgel population A was synthesized with a microfluidic device with a channel step size of 11 µm, and microgel population B was synthesized in a device with a step size of 40 µm. Scale bars = 100 µm. Abbreviation: PDI = polydispersity index.
6.2. Sizing particles with ImageJ
6.2.1. Open the image files from the microscope in ImageJ.
6.2.2. Select Analyze | Set Scale and set the image scale according to the microscope objective.
6.2.3. Select Image | Type | 8-bit.
6.2.4. Select Image | Adjust | Threshold and then select the “Otsu” auto-threshold option from the dropdown box.
6.2.5. Click on Analyze | Set Measurements and select Feret’s Diameter and limit to threshold.
6.2.6. Click on Analyze | Analyze Particles and enter the area size range (in pixel^2) expected for the microgels (to exclude small debris from being analyzed). Change circularity to 0.75–1.00 and select Show | Outlines. Check Display Results and Exclude on Edges.
NOTE: The circularity shape filter excludes microgels on the image border that may yield an inaccurate diameter measurement.
6.2.7. Run the Analyze Particles module.
6.2.8. Wait for the output, which is the Feret’s diameter of each particle, and export these results to a spreadsheet.
6.2.9. In this protocol, calculate the polydispersity index (PDI) to determine the heterogeneity in microgel size. Analyze at least 100 microgels to define a population: a PDI range of 1.00–1.05 defines a monodisperse population, and a PDI greater than 1.05 defines a polydisperse population. Use equation (2), equation (3), and equation (4) to calculate the PDI, as described below.
| (2) |
| (3) |
| (4) |
7. Microporous annealed particle (MAP) scaffold annealing
7.1. Create a stock solution of 2 mM lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) in 1x PBS (pH 7.4).
7.2. Dilute the LAP stock solution to 0.2 mM in a volume of 1x PBS equivalent to the gel volume. If using the LAP photoinitiator for cell studies or animal studies, be sure to sterilize the solution before use.
7.3. Spin down the MAP gel at 4,696 × g for 5 min and aspirate the supernatant.
7.4. Using a positive displacement pipette, transfer the desired volume of gel into a microcentrifuge tube.
7.5. Add 0.2 mM LAP to the gel in a 1:1 volumetric ratio (final LAP concentration is 0.1 mM).
7.6. Vortex the mixture and incubate for at least 15 min in the dark.
7.7. Centrifuge the mixture at 18,000 × g for 5 min to pellet the gel.
7.8. Carefully remove the supernatant from the gel pellet.
7.9. Transfer the MAP gel to the target location with a positive displacement pipette.
7.10. Apply focused light (365 nm, 8.66mW/cm2) to the sample for 113 s to anneal the scaffold.
NOTE: The annealing time of 113 s has been optimized as previously published for LAP concentration of 0.1 mM14, but additional optimization for different photoinitiator concentrations may be needed.
REPRESENTATIVE RESULTS:
The aim of this protocol is to outline all the steps necessary for synthesizing microgel building blocks to be used in a MAP scaffold. The MethMAL annealing macromer is highly selective and efficient and is compatible with multiple polymer backbones14. It is important that at least 67%–75% of the 20 kDa PEG-maleimide is modified with methacrylamide functional groups to ensure a high annealing efficiency. Percent modification can be most easily determined by analyzing 1H-NMR spectra peaks, as shown in Figure 1. The gelation kinetics, determined by a viscometer, is an important metric to consider for each gel formulation. This protocol uses a gel precursor solution consisting of a PEG backbone with a MAL group, which efficiently reacts with thiol-functionalized crosslinkers for microgel gelation. However, many hydrogel chemistries can be used to fabricate microgels via the high-throughput microfluidic method described herein. The time to onset of gelation will provide insight into the duration of microfluidic microgel generation. It is recommended to choose a gel precursor pH that can initiate gelation between 30 min (Figure 2) and 2 h.
If the gelation time is too fast, the gel precursor solution will begin to polymerize within the microfluidic device and clog the channels. Additionally, it is important to note that changing thiolated ligand concentrations (e.g., RGD) may have impacts on network formation during gelation and may need to be accounted for by adjusting the formulation. The microfluidic device fabrication steps can be tedious, but representative results of a successfully bonded device are shown in Figure 3. This protocol uses a high-throughput, parallelized, step-emulsification microfluidic device that was adapted from a design by de Rutte et al.13, and the silicon wafer fabrication was outsourced to a microfluidic technology company. However, the steps outlined in this protocol can be used with any device design etched on a SU-8 silicon wafer photomask. It is important to note that the step size of the channels on the photomask must be optimized during device fabrication, since it will impact the size of the microgel particles.
The flow rates for the microfluidic generation of microgels should be optimized for each gel formulation based on factors such as gelation time, desired particle size, and microfluidic device design. If using the high-throughput device, the flow rates for the aqueous phase can go as high as 5 mL/h. Figure 4B shows the setup for high-throughput devices used in this protocol. If the device is running correctly, the microgel formation should look similar to that shown in Figure 4D. Before purification, the microgels will be opaque. After completing the various oil, PBS, and hexane washes, the gel should look clear like the representative image in Figure 5. If incorporating a fluorophore into the microgels, the purified product may have a slight colored tint but should still be close to translucent. After purification and swelling, the microgels should be very uniform in size and have a PDI between 1.00 and 1.05, as shown in Figure 6. Various photoinitiators may be used for photoannealing MAP scaffolds. If using an alternative to LAP, described herein, one must determine the annealing kinetics as previously described14. Additionally, various light sources may be used for photoannealing, as long as the light source corresponds with the photoinitiator. One must be sure to calibrate and focus the light source. The annealing time and light intensity may need to be optimized based on gel formulation and photoinitiator concentration. The annealing method outlined in this protocol can be used for in vitro and in vivo studies. After annealing, the microgels will form a porous scaffold that can be visualized with two-photon microscopy (Figure 7B-C).
Figure 7: MAP scaffold annealing.

(A) Schematic of MAP scaffold annealing. When exposed to a photoinitiator and light, the methacrylamide functional groups on the MethMAL macromer undergo a click photopolymerization reaction, which bonds the surfaces of the microgels together. (B) Depiction of a 3D rendering (Imaris) of a two-photon microscope image of MAP microgels (green) annealed together in a 3D puck shape, with dextran (red) in the pores. (C) Depiction of a 3D rendering (Imaris) of a two-photon microscope image showing the porosity of a MAP scaffold that has been perfused with fluorescent 70 kDa dextran (red). Scale bars = (B) 100 µm, (C) 70 µm. Abbreviations: MAP = microporous annealed particle; MethMAL = custom annealing macromer.
DISCUSSION:
This protocol describes methods for synthesizing and characterizing microgels, which serve as the building blocks for microporous annealed particle (MAP) scaffolds. This protocol uses a high-throughput microfluidic approach to generate large volumes of uniform microgels, which cannot be achieved with other methods such as flow-focusing microfluidics1,4,7,9 (high monodisperisty, low yield), batch emulsion6,10, and electrospraying5,12 (low monodispersity, high yield). With the methods described herein, monodisperse microgels can be made for use in MAP scaffolds that can be used for a variety of regenerative medicine applications (e.g., cell delivery, wound healing).
A critical step of this protocol is the creation of the PDMS microfluidic devices. If the devices are not made correctly, this could have negative downstream effects on microgel formation and monodispersity. It is important to prevent the introduction of artifacts (i.e., bubbles, dust) into the PDMS before it cures, since this could clog the channels and significantly impact microgel formation. To mitigate this as much as possible, one should use tape to remove any dust, store the devices in a dust-free container, and work in a dust-free hood, if possible. It is also recommended to store the devices at 60 °C for the best results with the surface treatment.
When pouring the PDMS devices, it is important to maintain a uniform thickness that is about equal to or less than the length of the biopsy punch. If the device is too thick, the biopsy punch will be unable to penetrate all the way through. It is also crucial to not tear the PDMS device inlets/outlet while punching with the biopsy punch and/or inserting tubing. A tear in the PDMS device will cause leaking from the inlets/outlet, which can cause loss of the gel precursor solution. If there is leaking in a PDMS device, the best solution is to replace it with a new device as quickly as possible.
When plasma-treating the device, the use of pure oxygen and plasma-treating for 30 s has produced the best results for adhering PDMS to the glass slide. If the device does not bond correctly (i.e., the PDMS can still be lifted from the glass slide after plasma treatment), one should double-check that the plasma treater is operating correctly and that the device and slides have been cleaned thoroughly. It is also important to use the correct silane surface treatment, and, for best results, the PDMS devices should be surface-treated directly before use. Other methods of surface treatment, such as chemical vapor deposition, could also be used.
Another crucial step is using the PDMS microfluidic devices correctly for microgel formation. It is recommended to use a flow rate ratio of at least 2:1 (this protocol uses a 6 mL/h oil flow rate and a 3 mL/h aqueous flow rate), but this can be tuned to achieve the desired microgel size. The pH of the microgel precursor solution is also an important metric to optimize to prevent clogging of the device. Phosphate-buffered saline (PBS) accelerates thiolate formation in Michael-type addition chemistry, and the PBS concentrations used in this protocol yield the best results for microgel gelation in the microfluidic devices. Once the syringe pumps are started, there may be some bubbles in the microfluidic channels, but this should equilibrate after a few minutes. It is recommended to monitor microgel formation with a microscope. If the flow does not look similar as in this video and/or there are a few channels producing large particles, this is likely due to issues with the surface treatment step. The best solution is to replace the device with one that has been freshly surface-treated.
If the microgels appear to be coalescing, this may be due to an insufficient concentration of FluoroSurfactant. The recommended solution is to increase the wt% of the surfactant in the oil phase. However, one limitation to using high concentrations of surfactant is that it can be more difficult to remove during the purification step. It is recommended to use microfluidic devices once only, but the devices can be reused if flushed with Novec oil immediately after use to remove any aqueous solution that could gel in the device and clog the channels. While one microfluidic device can produce a high-throughput volume of microgels (mL/h), this production rate can be scaled by using multiple microfluidic devices in parallel.
The annealing step of MAP scaffold assembly relies on the use of a light-activated photoinitiator of radical polymerization, and the photoinitiator can be selected based on the desired application. For example, LAP photoinitiator has fast annealing times (<30 s) when using long-wave UV light, which has minimal impact on cell viability in vitro14. However, this wavelength is highly absorbed by tissue16 and may not have as high an annealing efficacy in vivo as in vitro.
Eosin Y is another photoinitiator activated by visible wavelengths (505 nm) and has deeper penetration into tissue, which enhances the capability of the MAP scaffold to be annealed beneath tissue. However, the long light exposure times needed for Eosin Y annealing may prolong cell exposure to free radicals and impact cell viability in vitro14. Using these methods for high-throughput generation of highly uniform microgel building blocks will accelerate MAP scaffold-focused research and advance knowledge in the field of injectable porous materials for regenerative medicine.
ACKNOWLEDGMENTS:
The authors would like to acknowledge Joe de Rutte and the Di Carlo Lab at the University of California, Los Angeles, for their assistance with the original microfluidic device design that the reported device was developed from, as well as their early guidance in PDMS device fabrication and troubleshooting. Figure schematics were created with Biorender.com.
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/64119.
DISCLOSURES:
The authors have no conflicts of interest to disclose.
REFERENCES:
- 1.Griffin DR, Weaver WM, Scumpia PO, di Carlo D, Segura T. Accelerated wound healing by injectable microporous gel scaffolds assembled from annealed building blocks. Nature Materials. 14 (7), 737–744 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koh J et al. Enhanced in vivo delivery of stem cells using microporous annealed particle scaffolds. Small. 15 (39), e1903147 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pruett LJ, Jenkins CH, Singh NS, Catallo KJ, Griffin DR. Heparin microislands in microporous annealed particle scaffolds for accelerated diabetic wound healing. Advanced Functional Materials. 31 (35), 2104337 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Darling NJ, Sideris E, Hamada N, Carmichael ST, Segura T. Injectable and spatially patterned microporous annealed particle (MAP) hydrogels for tissue repair applications. Advanced Science. 5 (11), 1801046 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isaac A et al. Microporous bio-orthogonally annealed particle hydrogels for tissue engineering and regenerative medicine. ACS Biomaterials Science and Engineering. 5 (12), 6395–6404 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pruett L et al. Development of a microporous annealed particle hydrogel for long-term vocal fold augmentation. Laryngoscope. 130 (10), 2432–2441 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Nih LR, Sideris E, Carmichael ST, Segura T. Injection of microporous annealing particle (MAP) hydrogels in the stroke cavity reduces gliosis and inflammation and promotes NPC migration to the lesion. Advanced Materials. 29 (32), doi: 10.1002/adma.201606471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffin DR et al. Activating an adaptive immune response from a hydrogel scaffold imparts regenerative wound healing. Nature Materials. 20 (4), 560–569 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sideris E et al. Particle hydrogels based on hyaluronic acid building blocks. ACS Biomaterials Science and Engineering. 2 (11), 2034–2041 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Schaeffer C et al. Injectable microannealed porous scaffold for articular cartilage regeneration. Annals of Plastic Surgery. 84 (6S Suppl 5), S446–S450 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Pruett L, Ellis R, McDermott M, Roosa C, Griffin D. Spatially heterogeneous epidermal growth factor release from microporous annealed particle (MAP) hydrogel for improved wound closure. Journal of Materials Chemistry B. 9 (35), 7132–7139 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jivan F, Alge DL. Bio-orthogonal, site-selective conjugation of recombinant proteins to microporous annealed particle hydrogels for tissue engineering. Advanced Therapeutics. 3 (1), 1900148 (2020). [Google Scholar]
- 13.de Rutte JM, Koh J, di Carlo D. Scalable high-throughput production of modular microgels for in situ assembly of microporous tissue scaffolds. Advanced Functional Materials. 29 (25), 1900071 (2019). [Google Scholar]
- 14.Pfaff BN et al. Selective and improved photoannealing of microporous annealed particle (MAP) scaffolds. ACS Biomaterials Science and Engineering. 7 (2), 422–427 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Darling NJ, Hung YS, Sharma S, Segura T. Controlling the kinetics of thiol-maleimide Michael-type addition gelation kinetics for the generation of homogenous poly(ethylene glycol) hydrogels. Biomaterials. 101, 199–206 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Sandell JL, Zhu TC. A review of in-vivo optical properties of human tissues and its impact on PDT. Journal of Biophotonics. 4 (11–12), 773–787 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]