

Abstract
The design of new hydrogel-based biomaterials with tunable physical and biological properties is essential for the advancement of applications related to tissue engineering and regenerative medicine. For instance, interpenetrating polymer network (IPN) and semi-IPN hydrogels have been widely explored to engineer functional tissues due to their characteristic microstructural and mechanical properties. Here, we engineered IPN and semi-IPN hydrogels comprised of a tough pectin grafted polycaprolactone (pectin-g-PCL) component to provide mechanical stability, and a highly cytocompatible gelatin methacryloyl (GelMA) component to support cellular growth and proliferation. IPN hydrogels were formed by calcium ion (Ca2+)-crosslinking of pectin-g-PCL chains, followed by photocrosslinking of the GelMA precursor. Conversely, semi-IPN networks were formed by photocrosslinking of the pectin-g-PCL and GelMA mixture, in the absence of Ca2+ crosslinking. IPN and semi-IPN hydrogels synthesized with varying ratios of pectin-g-PCL to GelMA, with and without Ca2+-crosslinking, exhibited a broad range of mechanical properties. For semi-IPN hydrogels, the aggregation of microcrystalline cores led to formation of hydrogels with compressive moduli ranging from 3.1 to 10.4 kPa. For IPN hydrogels, the mechanistic optimization of pectin-g-PCL, GelMA, and Ca2+ concentrations resulted in hydrogels with comparatively higher compressive modulus, in the range of 39 kPa–5029 kPa. Our results also showed that IPN hydrogels were cytocompatible in vitro and could support the growth of three-dimensionally (3D) encapsulated MC3T3-E1 preosteoblasts in vitro. The simplicity, technical feasibility, low cost, tunable mechanical properties, and cytocompatibility of the engineered semi-IPN and IPN hydrogels highlight their potential for different tissue engineering and biomedical applications.
Introduction
The extracellular matrix (ECM) is a collection of acellular components such as water, proteins and polysaccharides that provides physicochemical cues essential for tissue homeostasis, morphogenesis, and differentiation.1 The compositional and architectural features of the ECM vary greatly, and are characteristic of each type of tissue and organ in the body.2 For this reason, naturally-derived hydrogels are extensively used for tissue engineering, as their physical properties and composition can be tuned to mimic the native features of the ECM.3,4 However, the precise control over the structural and functional properties in the design of hydrogel-based biomaterials remains technically challenging. This is particularly significant in the field of tissue engineering, since different properties of hydrogels including their swellability, porosity, degradation rate, and mechanical properties have been shown to play a key role in tissue homeostasis.5,6 In this regard, various groups have reported the development of hydrogels synthesized using different naturally derived, as well as synthetic-based polymers.7,8 Among naturally derived polymers, gelatin methacryloyl (GelMA) hydrogels have been extensively employed for the engineering of cardiac,9 epidermal,10 neuromuscular,11 and vascular12 tissue constructs, as well as wound healing13 and other biomedical applications. This is mainly due to the remarkable biocompatibility, processability, tunability, and low immunogenicity of GelMA, as well as its ability to promote bioactive responses in vivo, such as cell migration and angiogenesis.14 Moreover, the emergence of different microfabrication techniques, such as 3D bioprinting and soft lithography has allowed the development of GelMA-based scaffolds with controllable geometries and microarchitectures.15
The ability to easily modulate the degree of functionalization of GelMA and the composition of the hydrogel precursors have enabled the engineering of hydrogels with a broad spectrum of elastic moduli (i.e., 3.3–110 kPa).16 Despite this wide range of mechanical properties, tissue-engineered scaffolds for musculoskeletal and osteochondral applications often require significantly higher mechanical strength to adequately mimic those of native tissues.17 Therefore, previous works have explored the incorporation of additional materials with increased physical compliance to improve the mechanical performance of GelMA-based hydrogels. For instance, the incorporation of silk fibroin into GelMA hydrogels was shown to yield composite hydrogels with compressive moduli up to 80 kPa,18 whereas the addition of dextran glycidyl methacrylate was shown to increase the modulus of GelMA-based hydrogels from 10 kPa to 24 kPa.19 The incorporation of reduced graphene oxide (rGO)20 and carbon nanotubes21 has also been reported to increase the Young’s modulus of GelMA-based hydrogels from 2.0 to 22.6 kPa, and from 12.5 to 50.0 kPa, respectively. Furthermore, the addition of gold nanoparticles could not only improve the mechanical strength of GelMA-based hydrogels, but also promoted the proliferation and osteoblastic differentiation of human adipose-derived stem cells (ADSCs) in vitro.22 Other studies have reported the development of GelMA-hydroxyapatite hybrid hydrogels to enhance implant osseointegration during bone repair, and GelMA-hyaluronic acid methacrylate for cartilage tissue engineering.23 Therefore, the precise modulation of the mechanical properties and the incorporation of biological cues or bioactive materials into GelMA-based hydrogels could yield composites with increased bioactivity and enhance their potential for different tissue engineering applications.
Pectin is a polyuronate heteropolysaccharide naturally found in the primary cell walls of terrestrial plants, which consists of partially esterified D-galacturonic acid residues in α-(1 → 4) chains.24 Due to its simple, fast, and cytocompatible gelling mechanism, pectin has been widely used for drug delivery25–27 and bone tissue engineering applications.28–32 Pectin composites have been shown to exhibit high cytocompatibility in vitro, and to support the proliferation of various cell types such as NIH3T333 and L929 mouse fibroblasts,34 as well as human dermal fibroblasts (HDF).35 Pectin-based biomaterials could also support the proliferation of metabolically active MC3T3-E1 preosteoblasts and induce their osteogenic differentiation in vitro.36 This intrinsic osteogenic activity has been attributed mainly to the presence of bioactive rhamnogalacturonan moieties, located in the branched regions of the pectin chains.30,31 Another material that is commonly used to enhance the functionality of hydrogel-based scaffolds is polycaprolactone (PCL). PCL is a semi-crystalline aliphatic polymer that has been extensively used for the development of tissue engineering scaffolds, due to its combined properties of biocompatibility, biodegradability, and high mechanical strength.37–39 However, in contrast to GelMA and pectin, the ability of PCL to support cell adhesion and proliferation is greatly limited, mainly due to the lack of bioactive functional groups and its intrinsic hydrophobicity.40,41 Because of this, different modification strategies have been developed to circumvent the limitations associated with the surface characteristics of PCL-based scaffolds, which includes coating or blending PCL with other biocompatible materials.42,43 Thus, we hypothesized that the incorporation of pectin and PCL into GelMA hydrogels could be used to engineer biocompatible scaffolds with improved mechanical strength and intrinsic bioactivity for tissue engineering.
Another approach to develop hydrogels with enhanced mechanical stability is the formation of interpenetrating polymer networks (IPNs), in which one component is cross-linked in the presence of another.44 Hence, this strategy could be used to incorporate blends of polymers with different advantageous characteristics into a single composite biomaterial with enhanced functionality. Here, we engineered IPN and semi-IPN hydrogels based on a highly cytocompatible GelMA network to support cellular growth, combined with an inherently bioactive pectin-g-PCL network with high mechanical strength. The chemical composition of pectin-g-PCL was evaluated using 1H-nuclear magnetic resonance (1H-NMR) and Fourier-transform infrared spectroscopy (FTIR), as well as differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). The mechanical properties of GelMA/pectin-g-PCL hydrogels were characterized using tensile and compression tests. We also evaluated the porosity of the hydrogels through scanning electron microscopy (SEM), as well as their in vitro degradation rate. Lastly, we investigated the capability of GelMA/pectin-g-PCL hydrogels to support the proliferation of three-dimensionally (3D)-encapsulated MC3T3-E1 preosteoblasts in vitro. The engineered hydrogels can be used as an injectable material that could be readily delivered and rapidly photocrosslinked in situ. In addition, pectin-g-PCL GelMA hydrogels could also be used to develop prefabricated scaffolds with enhanced mechanical stiffness and intrinsic bioactivity for tissue engineering.
Experimental
Materials
Low methoxyl pectin from citrus peel (galacturonic acid ≥74.0% on dried basis) from Sigma-Aldrich. The degree of esterification of pectin was ≅60% with an average molecular weight of 30 000–100 000 g mol−1. Methanesulfonic acid (MeSO3H, >99.5%), ε-caprolactone monomer (>99%), fish gelatin, and methacrylic anhydride were also purchased from Sigma Aldrich. Dulbecco’s phosphate-buffered saline (DPBS) and the 2-hydroxy-1-(4-(hydroxyethoxy) phenyl)-2-methyl-1-propanone (Irgacure 2959) photoinitiator were purchased from HyClone™ and CIBA Chemicals, respectively. Mono- and di-basic phosphate buffer were used to adjust the pH. Deionized and distilled water (DI) were used for all experiments. Lastly, all other reagents were of analytical grade and used as without any additional modifications.
Chemical and analytical characterization
1H-Nuclear magnetic resonance.
A 400 MHz 1H-NMR Bruker Biospin Spectrometer was used to analyze chemical shifts in pectin-g-PCL using CD3COCD3 and D2O solvents.
Fourier transform infrared spectroscopy.
A Shimadzu IRAffinity–1 FTIR spectrophotometer was utilized to detect the functional groups of pectin and pectin-g-PCL. Spectra were recorded in the range of 4000–400 cm−1 using KBr pellets.45
Differential scanning calorimetry.
Samples were heated from 30 °C to 275 °C under 70 ml min−1 inert N2 gas at a heating rate of 10 °C min−1 using a Netzch 204 F1 Phoenix DSC equipped with an intra-cooler system. Indium was used for the calibration of temperature enthalpy scale values. A second-heating run was performed to confirm the melting point endotherm of PCL, which was set as the top peak of the melting endotherm. The degree of crystallinity was then determined using eqn (1):
| (1) |
where ΔHm and ΔH100% stand for the enthalpy of fusion of PCL samples (in J g−1), and 100% crystallinity (139.5 J g−1), respectively.38 The enthalpies of fusion were calculated from the area under the endotherm curve using the software of the Netzch instrument.
Thermogravimetric analysis.
The samples were scanned from 25 °C to 900 °C under flow of inert N2 gas at 10 °C min−1 to evaluate the thermal stability and decomposition temperature of pectin and pectin-g-PCL samples using a Netzsch Proteus thermogravimetric analyzer.46 The degradation temperature was estimated as the midpoint of the 1st derivative peak of the TGA thermograms for both pectin and pectin-g-PCL (ESI Fig. 2†).
Scanning electron microscopy.
To visualize the microstructure of IPN and semi-IPN networks, hydrogels were mounted on aluminum stabs, then coated with gold ion through the sputtering method, and scanned using a Hitachi S-4800 SEM.
Synthesis of pectin-g-PCL via ring opening polymerization
Synthesis of PCL-grafted pectin (pectin-g-PCL) was carried out via bulk ring-opening polymerization (ROP).39 Briefly, 1.0 g pectin and 10 mL of methanesulfonic acid were charged in a flame-dried 50 mL round-bottom flask, which was equipped with a stir bar (Teflon-coated) under inert N2 atmosphere. Step-wise addition of 5 equivalents (5.0 g) of ε-caprolactone monomer was performed. The reaction mixture was stirred for 4 h at 40 °C under inert N2 atmosphere. After the reaction was complete, the mixture was transferred to a 250 mL beaker with 100 mL of 0.2 M KH2PO4, 16 mL of 10 M NaOH, and ice. Monobasic phosphate buffer was used to neutralize excessive unreacted methanesulfonic acid and to prevent uncontrolled changes in pH during the quenching process of the reaction. The ice was used to preserve pectin from chain degradation due to excessive heat release. The precipitated pectin-g-PCL compound was filtered using Whatman filter paper and washed four times with 100 mL DI water to remove the excessive unreacted materials. The final mixture was then dried at room temperature and stored until used.
Synthesis of GelMA
GelMA was synthesized according to previously published method.47 Briefly, 10 g of cold fish gelatin was dissolved in 100 mL DPBS (Gibco) under continuous stirring at 60 °C. Then, methacrylic anhydride (Sigma Aldrich) was added to the solution dropwise (8.0 mL). The reaction was stirred vigorously at 50 °C for 3 h and diluted three times with warm DPBS to stop the reaction. The solution was then dialyzed against DI water using a dialysis tubing (cutoff: 12–14 kDa) at 40 °C for one week to remove salts and unreacted methacrylic anhydride. The GelMA solution was then frozen for one day at −80 °C, and then freeze-dried at 8.0–9.0 Pa and −52 °C for four days. The obtained porous GelMA foam was stored until used.
Synthesis of semi-IPN and IPN GelMA/pectin-g-PCL hydrogels
IPN hydrogels were prepared using varying concentrations of GelMA (10, 15, 20, and 25% (w/v)), pectin-g-PCL (10, 15, 20, and 25% (w/v)), and Ca2+ (0.5, 1, 2, and 5% (w/v)). Briefly, 1.0 mg of Irgacure 2959 photoinitiator was dissolved in 100 μL DPBS, followed by addition of varying concentrations of calcium chloride. 100 μL of pectin-g-PCL solution was added to the mixture to yield a final concentration of 1.15%. The solution was stirred to achieve physical crosslinking of pectin chains with Ca2+ for 10 min, until a homogenous viscous solution was obtained. Different concentrations of GelMA precursor were dissolved in the crosslinked pectin solution and vortexed vigorously for 5 min to allow mixing of GelMA chains within the physically crosslinked pectin network. The viscous solution was poured in a polydimethylsiloxane (PDMS) mold and photocrosslinked using 7.7 mW cm−2 UV irradiation (360–480 nm) for 240 s to form IPN GelMA/pectin-g-PCL hydrogels. For semi-IPN hydrogels, the same procedure was carried out in the absence of Ca2+ crosslinker.
Physical characterization of GelMA/pectin-g-PCL hydrogels
Tensile tests.
Tensile stress–strain curves were obtained using an Instron 5542 mechanical tester. For tensile tests, 12.5 mm × 5.5 mm × 1.0 mm rectangular hydrogels were prepared as described previously.48 Hydrogels were stretched at a constant rate of 1.1 mm min−1 at room temperature. The tensile moduli were calculated from the slope of the linear region of the stress–strain curve up to 10% strain. Each point represented the mean ± SD (n = 3).
Compression tests.
Compressive stress–strain curves were obtained using an Instron 5542 mechanical tester. For compression tests, 7 mm × 2.5 mm cylindrical hydrogels were prepared as described previously.48 The specimens were compressed at a rate of 1.1 mm min−1 at room temperature. The compressive moduli were calculated by taking the slope of the linear region of the stress–strain curve, up to 10% strain. Each point represented the mean ± SD (n = 3).
Evaluation of in vitro degradation
In vitro degradation was evaluated using both DPBS and DPBS supplemented with 10% fetal bovine serum (FBS, Lonza) as described previously.49 Briefly, cylindrical hydrogels (8 mm × 2 mm) were prepared, lyophilized, and weighed. Degradation was determined by placing the hydrogels in solution at 37 °C for 2 weeks. The samples were retrieved at fixed time intervals (0, 1, 4, 7 and 14 days), freeze-dried, and weighed. The percentage degradation (D%) of the hydrogels was calculated using eqn (2):
| (2) |
where W0 is the initial weight and Wt is the weight after time t.
3D encapsulation of MC3T3-E1 preosteoblasts in GelMA/pectin-g-PCL hydrogels
MC3T3-E1 preosteoblasts (ATCC) were cultured in an incubator at 37 °C in Minimum Essential Medium Alpha (MEM-α) Medium (Life Technologies), supplemented with 10% FBS, and 1% penicillin–streptomycin (Thermo Fisher Scientific). For 3D cell encapsulation, MC3T3 cells were mixed with the hydrogel precursor at a density of 3 × 106 cells per ml. A 7 μl drop of the mixture was then pipetted on a Petri dish and sandwiched using a 3-(trimethoxysilyl) propyl methacrylate (TMSPMA)-coated glass slide, on top of two 150 μm spacers. Hydrogels were then photocrosslinked upon exposure to UV light for 20 s. The resulting hydrogels were washed several times with warm culture medium to eliminate residual uncrosslinked reagents and incubated at 37 °C for 7 days.
Determination of in vitro cell viability and spreading
Cell viability was determined using a commercial calcein AM/ethidium homodimer live/dead kit (Invitrogen) after 1, 3 and 5 days. Briefly, culture medium was removed from the wells, and samples were washed three times with DPBS. Next, samples were placed in a solution containing 2 μM calcein AM and 4 μM ethidium homodimer and incubated for 15 min in the dark at 37 °C. Cell viability was calculated as the percentage ratio of live cells to the total number of cells.50
Cell spreading inside the hydrogels was evaluated via fluorescent staining on days 1, 3 and 5 post-encapsulation. Actin filaments were stained with Alexa-Fluor 488 phalloidin (Invitrogen), and 4′,6-diamidino-2-phenylindole (DAPI, Sigma) was used to stain the cell nuclei. The samples were first fixed using a solution of 4% paraformaldehyde (Sigma) in DPBS for 20 min. Then, a solution of 1% bovine serum albumin (BSA) in DPBS was used to block the samples for 20 min. Afterwards, a 1/40 solution of Alexa Fluor 488 phalloidin in 0.1% BSA was prepared and added to the samples, followed by a 45 min incubation at 37 °C. For DAPI per cell nuclei staining, a 0.1% (v/v) DAPI solution in DPBS was added to the samples and incubated for 5 min at 37 °C. Samples were then washed and imaged using a Zeiss Axio Observer Z1 inverted microscope.
Results and discussion
In this study, we developed IPN and semi-IPN hydrogels for tissue engineering by combining a biocompatible soft GelMA polymer with a tough polymeric network of pectin-g-PCL (Fig. 1). In addition to the enhanced structural integrity of these hybrid hydrogels, the intrinsic bioactivity of pectin combined with the cell supportive nature of GelMA could be remarkably advantageous in the development of scaffolds for musculoskeletal and osteochondral tissue engineering.
Fig. 1.
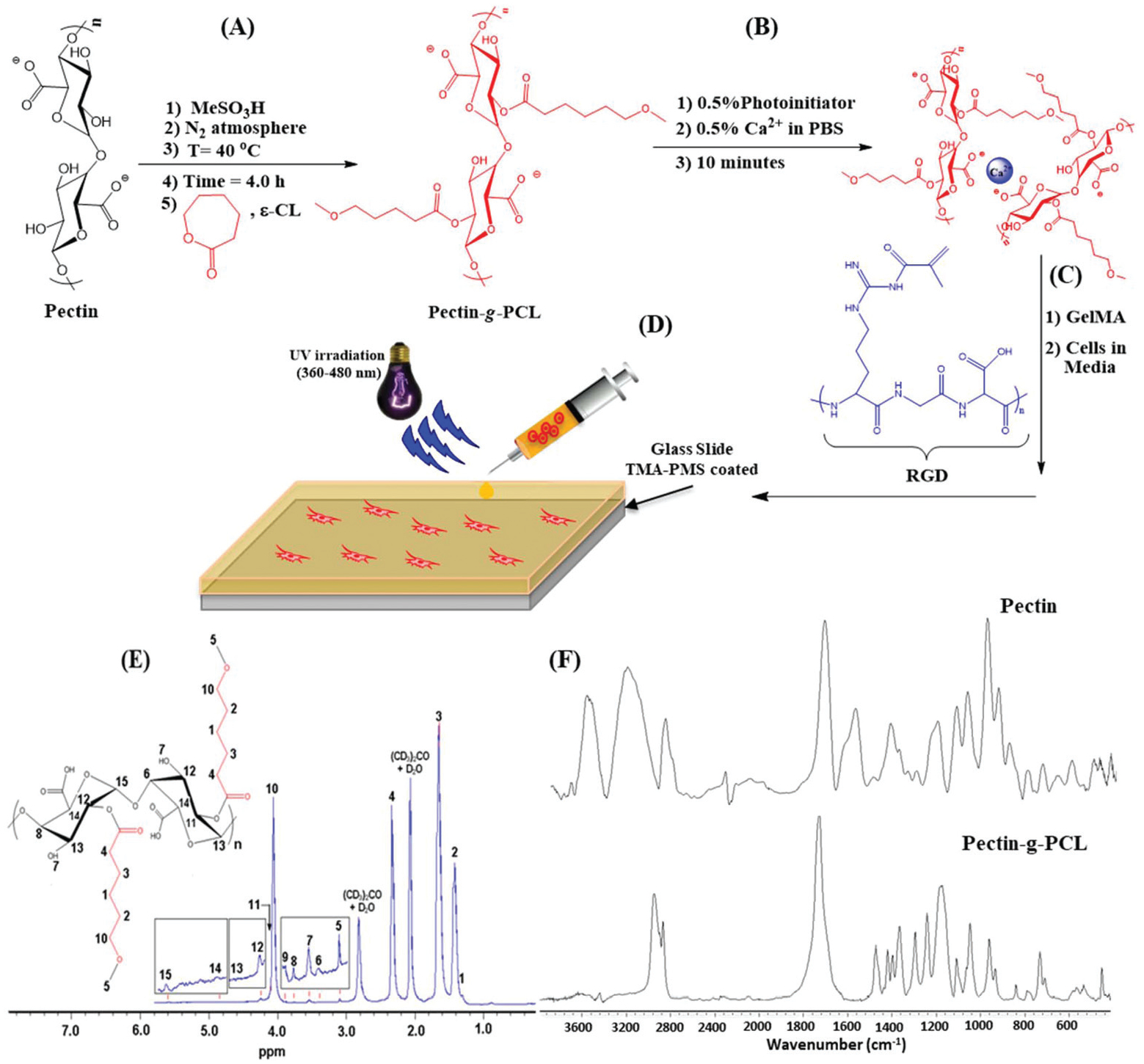
Schematic and structural analysis of semi-IPN and IPN GelMA/pectin-g-PCL hydrogels. Schematic representation of (A) the synthesis of pectin-g-PCL via ROP, (B) crosslinking of pectin-g-PCL chains via Ca2+, and (C) synthesis of IPN and semi-IPN GelMA/pectin-g-PCL hydrogels; (D) schematic of 3D cell encapsulation using GelMA/pectin-g-PCL hydrogels. (E) 1H-NMR spectrum of pectin-g-PCL, demonstrating chemical grafting of PCL onto the surface of pectin; (F) FTIR spectra of pectin and pectin-g-PCL hydrogels showing characteristic functional groups.
Synthesis and structural analysis of pectin-g-PCL
GelMA was synthesized from cold fish gelatin due to its lower melting point, which allowed hydrogel fabrication at room temperature.51,52 On the other hand, ε-caprolactone (CL) was grafted onto the surface of pectin via ROP using methanesulfonic acid as a catalyst at 40 °C (Fig. 1A). The resulting pectin-g-PCL was crosslinked via Ca2+ (Fig. 1B), and then mixed with the GelMA precursor (Fig. 1C) followed by photocrosslinking with UV light (Fig. 1D) to form IPN hydrogels.
We first aimed to characterize the chemical structure of the engineered hydrogels via spectrophotometric techniques. 1H-NMR and FTIR are widely used for chemical identification, as well as functional group and conformational analyses of chemical reactions. The 1H-NMR spectrum of pectin-g-PCL confirmed successful ROP and grafting of ε-caprolactone to the surface of the pectin polysaccharide (Fig. 1E). PCL and pectin constituents showed sharp distinct moieties in the spectrum. For the PCL moiety, CH2 peaks of the opened ring caprolactone monomer corresponded to δ = 1.31 ppm (peak 1), 1.42 ppm (peak 2), 1.65 ppm (peak 3), 2.33 ppm (peak 4), and 3.10 ppm (peak 5). CH2 peak bound to oxygen atom in PCL backbone corresponded to δ = 4.07 ppm (peak 10). On the other hand, the pectin moiety showed characteristic CH peaks; 3.39 ppm (peak 6), 3.75 ppm (peak 8), 3.88 ppm (peak 9), 4.20 ppm (peak 11), 4.25 ppm (peak 12), 4.63 ppm (peak 13), 4.89 ppm (peak 14), and 5.61 ppm (peak 15). Lastly, the hydroxyl group (OH) on the backbone of pectin appeared as a peak at δ = 3.55 ppm (peak 7). The observed peaks were compared to pectin and PCL controls, which were also in agreement with values reported previously in the literature.53,54 It is important to note that the peaks corresponding to pectin and PCL showed small shifts, mainly due to chemical interactions between them in the pectin-g-PCL sample.
FTIR is used to determine the stretching and bending vibrations of different functional groups, which could be used to confirm the chemical structure of the analyzed compound. Fig. 1F shows the FTIR spectra of pectin-g-PCL, as well as the pectin control. The detailed wavenumber, normalized absorbance and vibrational assignments for each characteristic functional group of pectin-g-PCL and pectin control are shown in ESI Table 1.† For the pectin control, in the region of 4000–2000 cm−1, characteristic peaks can be observed associated with OH stretching of the secondary alcohol, as well as symmetric and asymmetric CH stretching. In the region of 1900–1000 cm−1 characteristic peaks of C=O, symmetric and asymmetric COO−, and COC stretching appeared as typical vibrational peaks of pectin.55,56 For the pectin-g-PCL sample, the presence of the characteristic peaks of pectin confirmed the non-degradation of this polymer during the quenching process. Moreover, the peak corresponding to C=O stretching of PCL at 1729 cm−1 (ref. 57) was overlapped with C=O of pectin at 1744 cm−1, which formed an unresolved and broadened peak with high intensity. Other characteristic peaks of PCL like asymmetric and symmetric COC stretching at 1240 and 1170 cm−1 could also be observed.57 The intense peak at 1293 cm−1 corresponding to C–O and C–C in the crystalline phase of PCL indicated the presence of crystalline regions in PCL.58 Furthermore, the high crystallinity of PCL was further supported by the results of the DSC analysis (ESI Fig. 1†). Lastly, the large decay of the OH peak at 3269 cm−1 with respect to COC stretching at 1147 cm−1 was indicative of the successful grafting process through the hydroxyl groups on the surface of pectin.
DSC and TGA can be used to determine the melting point, enthalpy of fusion, degree of crystallinity, thermal stability, and degradation temperature of hydrogels. Here, we used DSC to estimate the enthalpy of fusion and the degree of crystallinity of pectin-g-PCL hydrogels. The DSC thermograms of hydrogels synthesized using four different pectin-to-PCL ratios are shown in ESI Fig. 1.† Our results suggested that the enthalpy of fusion decreased with increasing concentrations of PCL, which strongly influenced the degree of crystallinity (Table 1). This behavior could be explained in part owing to the interactions that occur due to the chemical structures of pectin and PCL. For instance, pectin contains three functional groups in every repeating unit of its linear structure (i.e., hydroxyl, carboxylate ion, and ester, Fig. 1A). In turn, the presence of these groups would strongly influence the orientation of the PCL chains on the surface of pectin, mainly due to the establishment of electrostatic interactions between them. Hence, the optimization of these interactions (i.e., at an optimal ratio of pectin to PCL) would promote a better alignment of the polymeric chains, which would then favor the formation of microcrystalline cores. In addition, the high degradation temperatures of pectin and PCL measured by TGA thermograms (ESI Fig. 2†) indicated high thermal stability at elevated temperatures. The determination of the degree of crystallization (i.e., Xc(%)) from the DSC thermogram in Table 1 indicated that, as the concentration of caprolactone increased, the degree of crystallization decreased significantly. This observation suggested that, as the number of repeating units of PCL decreased (i.e., at lower concentrations of caprolactone) a higher degree of crystallization was obtained. This could be explained in part due to the optimization of electrostatic and H-bonding interactions occurring between pectin and PCL, when the 1 : 1 ratio was used. This ratio could have favored the alignment and organization of pectin and PCL chains on top each other, leading to a higher degree of crystallization (i.e., Xc(%) = 234). In contrast, for hydrogels synthesized using the 1 : 10 ratio, the resulting longer chains of PCL could have favored the establishment of more random and irregular hydrophobic interactions. This in turn could have impaired their organization and lead to a lower degree of crystallization (i.e., Xc(%) = 84).
Table 1.
Characterization of the thermal properties of pectin-g-PCL synthesized using different ratios of pectin to PCL
| Polymer | ΔHma (J g−1) | Tma (°C) | Xcb (%) | Degradation temperaturec | |
|---|---|---|---|---|---|
| Pectin (°C) | PCL (°C) | ||||
| Pectin | — | — | — | 228 | — |
| Pectin-g-PCL (1:1) | 326.7 | 66.6 | 234 | 386 | 404 |
| Pectin-g-PCL (1:3) | 168.0 | 59.0 | 120 | 391 | 394 |
| Pectin-g-PCL (1: 5) | 141.0 | 63.9 | 101 | 398 | 407 |
| Pectin-g-PCL (1: 10) | 117.3 | 63.1 | 84 | 400 | 407 |
Determined from DSC thermogram performed under inert gas atmosphere at a 10 °C min−1 heating rate. The melting point was considered as the midpoint of the peak.
Degree of crystallization was calculated from DSC using eqn (1).
Determined from DTG thermogram performed under inert N2 gas atmosphere at a 10 °C min−1 heating rate.
Engineering semi-IPN and IPN GelMA/pectin-g-PCL hydrogels
The development of biocompatible hydrogels with high mechanical stiffness and tunable physicochemical properties remains technically challenging. One strategy towards the inclusion of all these features into a single biomaterial is the use of multiple component IPN systems, based on the combination of synthetic and naturally derived polymers. The synthetic component provides increased mechanical strength and elasticity, while also allowing the modulation of the porosity and swellability of the composite. Meanwhile, the incorporation of a cytocompatible and bioactive component provides biological cues that modulate cell behavior and allows the bio-degradation of the system when implanted in vivo. Here, we synthesized IPN and semi-IPN hydrogels that consist of a flexible and cell-supportive GelMA network and a tough pectin-g-PCL network. The engineered semi-IPN and IPN hydrogels demonstrated hydrogels reported previously in the literature.16,59,60
Synthesis and mechanical characterization of semi-IPN GelMA/pectin-g-PCL hydrogels
Semi-IPN systems are comprised of polymeric networks interlaced with each other at the molecular level, where the linear or branched polymeric chains can be separated without breaking bonds.41 The success in the formation of semi-IPN hydrogels depends on many factors such as compatibility, thermodynamic and kinetic interactions between components, and crosslinking density.40 We first synthesized hydrogels using different formulations containing 0.4–2.0% pectin-g-PCL and 10% GelMA through photopolymerization via UV light exposure. The photoinitiation process crosslinked the GelMA chains in presence of linear pectin-g-PCL chains. As shown in Fig. 2, the incorporation of pectin-g-PCL into the GelMA network significantly influenced the mechanical properties of the resulting hydrogels. For instance, the tensile modulus of GelMA/pectin-g-PCL hydrogels synthesized using 10% GelMA, decreased from 35.9 ± 3.0 kPa to 14.6 ± 1.5 kPa as the pectin-g-PCL concentration increased from 0.4 to 2% (Fig. 2A). In contrast, the elongation at break was increased from 16.3% to 55.9%, as the concentration of pectin-g-PCL was increased from 0.4% to 2.0% (Fig. 2B). Similar to the elastic modulus, the compressive modulus also decreased from 10.4 ± 1.1 kPa to 3.1 ± 0.4 kPa by increasing the concentration of pectin-g-PCL from 0.4 to 2.0% (Fig. 2C). While GelMA is a hydrophilic polymer, the pectin-g-PCL copolymer consists of a hydrophilic pectin domain and hydrophobic microcrystalline cores of PCL (Table 1). This data suggested that the introduction of increasing concentrations of pectin-g-PCL led to hydrophobic aggregation of PCL within the GelMA chains. In turn, such aggregates could have delayed or prevented the complete crosslinking of GelMA, which could then lead to a weakened microstructure and the lower elastic and compressive moduli observed (Fig. 2). However, despite the lower mechanical performance, semi-IPN hydrogels have been previously shown to better maintain rapid kinetic response rates to pH or temperature, mainly due to the lack of a restricting interpenetrating elastic network.61 This characteristic, together with the comparatively more efficient drug loading properties of IPN hydrogels, could result highly advantageous in the development of drug delivery systems for pharmacological applications.
Fig. 2.
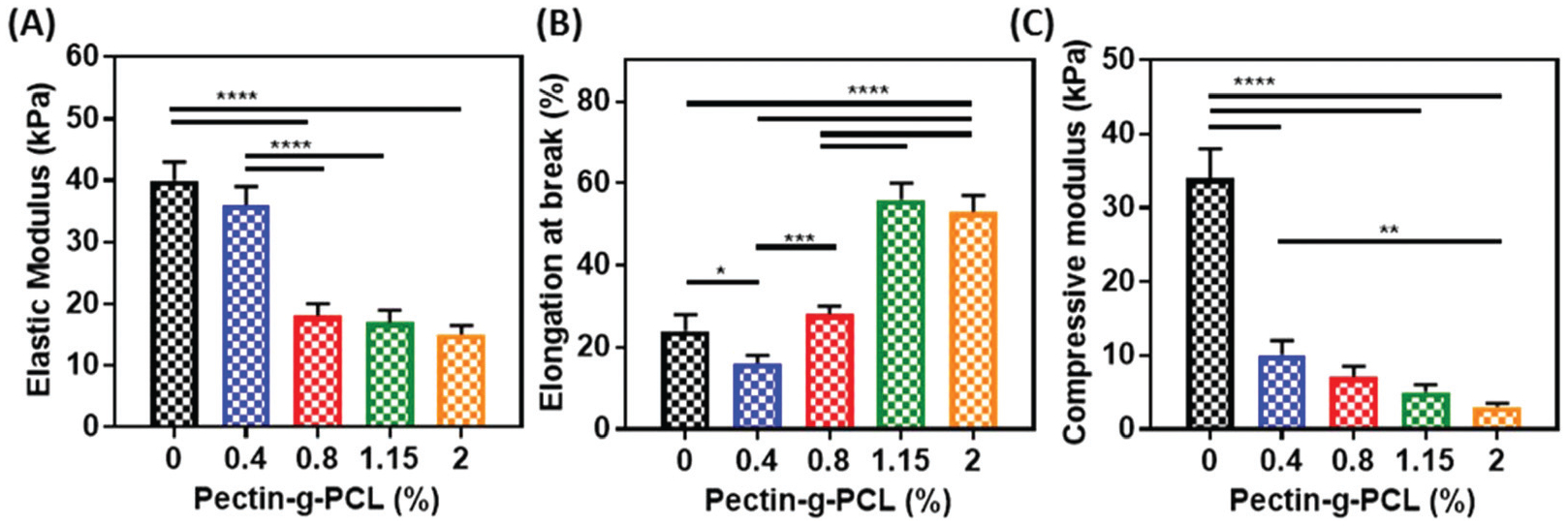
Mechanical properties of semi-IPN GelMA/pectin-g-PCL hydrogels. (A) elastic modulus, (B) elongation at break, and (C) compressive modulus of GelMA/pectin-g-PCL hydrogels synthesized by using different concentrations of pectin-g-PCL (i.e., 0, 0.4, 0.8, 1.15, and 2%), at 10% GelMA and 4 min UV exposure time.
Synthesis and characterization of IPN GelMA/pectin-g-PCL hydrogels
Mechanical characterization revealed that semi-IPN hydrogels exhibited poor structural integrity, as indicated by the low values obtained for the elastic (15–36 kPa) (Fig. 2A) and compressive moduli (3–10 kPa) (Fig. 2C). One approach to improve the mechanical properties of these hydrogels is the formation of IPNs of GelMA/pectin-g-PCL. IPN systems are comprised of two or more networks interlaced with each other at the molecular level, but not covalently bound to each other.62,63 The main technical advantages of IPNs are associated with the formation of relatively denser hydrogel matrices with higher mechanical properties, and a more precise control of the physical characteristics of these hydrogels.61 Here, we synthesized IPN hydrogels via physical crosslinking of pectin-g-PCL chains through Ca2+ crosslinking, followed by chemical crosslinking of GelMA via photoinitiation. Physical crosslinking occurred through the alignment of pectin chains via Ca2+ crosslinking, based on the “egg-box”-like model reported previously in the literature.64–66 GelMA was then added to the solution and photopolymerized via UV radiation to form the second network. This mechanistic procedure allowed consistent sequential interlacements that yielded IPN hydrogels, which were consistently more structurally stable than semi-IPN hydrogels. Therefore, we aimed to evaluate the effect of varying concentrations of pectin-g-PCL, GelMA, and Ca2+ on the mechanical properties of IPN hydrogels.
Optimization of pectin-g-PCL, GelMA, and Ca2+ concentrations.
To evaluate the effect of varying concentrations of pectin-g-PCL, we fixed the concentrations of GelMA and Ca2+ at 15% and 0.5%, respectively (Fig. 3A–C). Our results showed that increasing the concentration of pectin-g-PCL from 0.4 to 1.15% led to a consistent increase in the compressive (Fig. 3A) and tensile moduli (Fig. 3B), as well as in the elongation at break (Fig. 3C). This behavior could be due to the consistent interlacement of the “egg-box” structure of pectin-g-PCL within GelMA chains.64–66 However, when the pectin-g-PCL concentration was increased to 2.0%, these values were sharply reduced to 40 ± 5 kPa and 30 ± 3 kPa, respectively. Moreover, these values were similar to those obtained for pure GelMA hydrogels (Fig. 2). This observation suggested that the aggregation of PCL microcrystalline cores prevented the interlacement with GelMA and led to a decrease in the mechanical properties of IPN hydrogels.
Fig. 3.
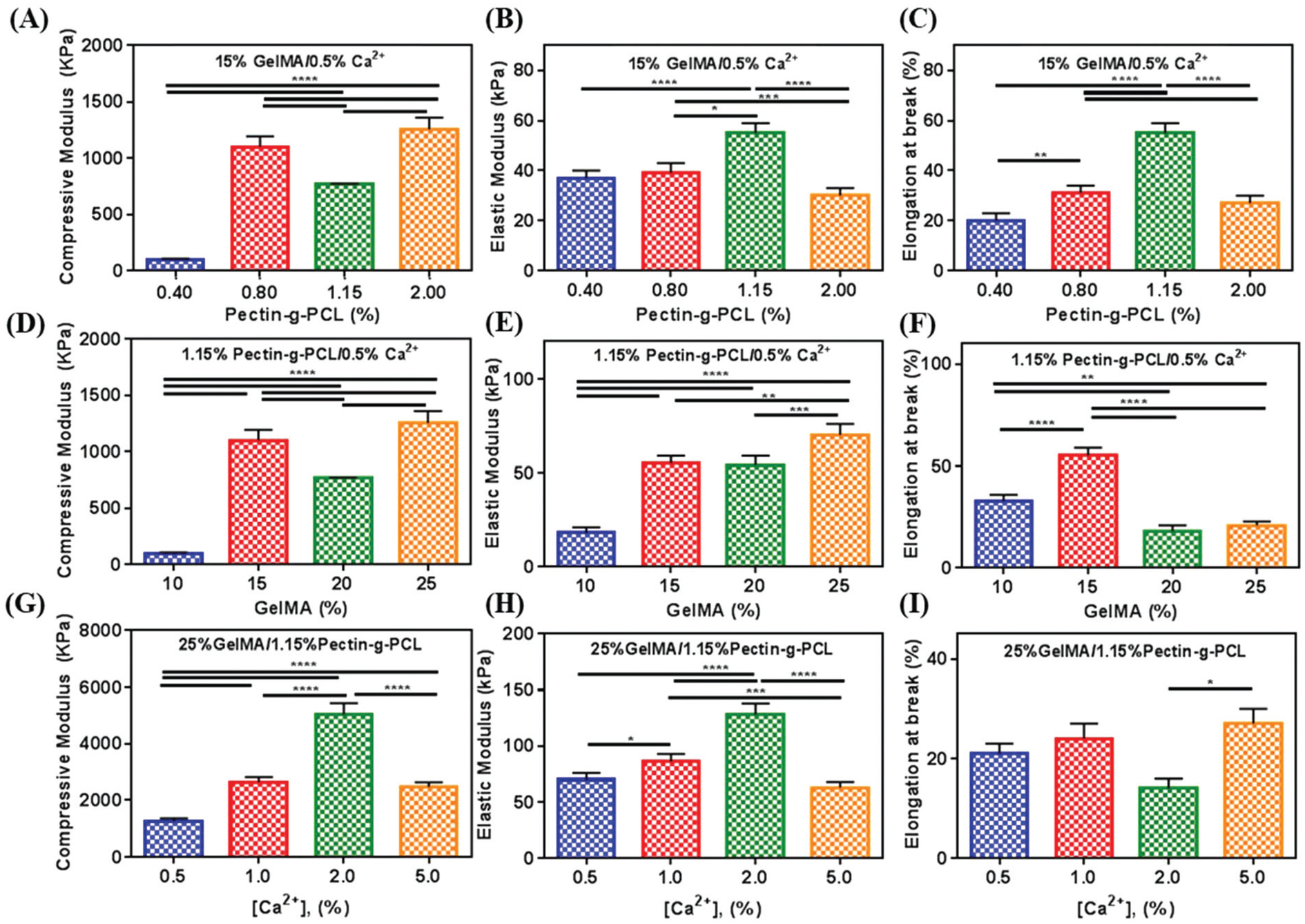
Mechanical characterization of IPN GelMA/pectin-g-PCL hydrogels. (A) Compressive modulus, (B) elastic modulus, and (C) elongation at break of IPN GelMA/pectin-g-PCL hydrogels synthesized using 15% GelMA, 0.5% Ca2+, and varying concentrations of pectin-g-PCL (i.e., 0.4, 0.8, 1.15, and 2%). (D) compressive modulus, (E) elastic modulus, and (F) elongation at break of IPN GelMA/pectin-g-PCL hydrogels synthesized using 1.5% pectin-g-PCL, 0.5% Ca2+, and varying concentrations of GelMA (i.e., 10, 15, 20, and 25%). (G) compressive modulus, (H) elastic modulus, and (I) elongation at break of IPN GelMA/pectin-g-PCL hydrogels synthesized using 25% GelMA, 1.5% pectin-g-PCL, and varying concentrations of Ca2+ crosslinker (i.e., 0.5, 1.0, 2, and 5%). Data is represented as mean ± SD (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n ≥ 3).
To evaluate the effect of varying concentrations of GelMA, we fixed the concentrations of pectin-g-PCL and Ca2+ to 1.15% and 0.5%, respectively (Fig. 3D–F). For these experiments, we used four different concentrations of GelMA (i.e., 10, 15, 20 and 25%). Moreover, we used the 1.15% pectin-g-PCL concentration, as this formulation exhibited the highest elongation at break. Our results showed that the highest compressive (Fig. 3D) and elastic (Fig. 3E) moduli were observed for the formulation containing 25% GelMA (i.e. 1256 ± 105 kPa and 70 ± 6 kPa, respectively). However, similar to what we observed by increasing the concentration of pectin-g-PCL, this also led to a decrease in the elongation at break at higher concentrations of GelMA (Fig. 3F).
Lastly, to evaluate the effect of the Ca2+ concentration, we used the concentrations of pectin-g-PCL and GelMA that showed the highest elastic moduli (i.e. 1.15% and 25%, respectively) (Fig. 3G–I). For this, four different Ca2+ concentrations were used (i.e. 0.5, 1.0, 2.0 and 5.0%). Our results showed that the compressive (Fig. 3G) and tensile (Fig. 3H) moduli increased consistently (5029 ± 408 kPa and 128 ± 10 kPa, respectively) as the concentration of Ca2+ increased up to 2.0%. Although these values decreased when the concentration of Ca2+ was increased at 5%, the elongation at break increased to its maximum value at 26 ± 3 kPa (Fig. 3I). These observations could be explained in part due to the fact that the Ca2+ concentration can directly affect the crosslinking of pectin-g-PCL chains, by binding and dimerizing two carboxylate ions of d-galacturonic acid residues that are present in the pectin chains. Moreover, the alignment of the pectin chains and the crosslink density of the hydrogel also depend on the Ca2+ concentration. Therefore, increasing the concentration of Ca2+ could have led to better alignment of free chains of pectin, and a more ordered microstructure with higher compressive and tensile moduli. However, at higher Ca2+ concentrations (i.e., 5%), pectin chains could have aligned with each other, allowing for partial precipitation of the crosslinked pectin microstructures, and the formation of a weaker IPN.
Taken together, these results demonstrated that the inclusion of pectin-g-PCL led to enhanced mechanical properties, when compared to pristine GelMA hydrogels. These results were in accordance to previous studies showing that the introduction of IPNs into single component systems yields hydrogels with increased mechanical performance. For instance, a previous study showed that the incorporation of a network of polyacrylonitril led to a 50-fold increase in the compression strength and elasticity of super porous hydrogels synthesized from various vinyl monomers.67 Other studies have demonstrated that hydrogels based on double networks of mechanically weak materials such as poly(2-acrylamido-2-methylpropanesulfonic acid) and polyacrylamide show mechanical strengths many times higher than hydrogels based on single component networks.68 The enhanced mechanical performance of IPN hydrogels has been attributed in part to the effective relaxation of locally applied stress, as well as the dissipation of energy through a combination of networks with different microstructures and cross-link densities.69 Therefore, the combination of a cross-linked rigid network of pectin-g-PCL with a ductile GelMA network is an efficient strategy to achieve synergistic improvements in the mechanical performance of single component hydrogels.
Microstructure and in vitro degradation of semi-IPN and IPN GelMA/pectin-g-PCL hydrogels.
We characterized the microstructure of semi-IPN and IPN GelMA/pectin-g-PCL hydrogels through SEM analysis (Fig. 4). The results revealed that the surface of semi-IPN hydrogels presented scattered and sporadic aggregates of pectin-g-PCL within the GelMA network (Fig. 4A). In contrast, the surface of the IPN hydrogels showed a more porous and aligned flaked microstructure, due to the interlacement of GelMA and pectin-g-PCL (Fig. 4B). Moreover, the rate of in vitro degradation of the IPN hydrogels was shown to be comparatively slower than semi-IPN hydrogels (Fig. 4C). The micro- and nanoporous architecture of GelMA/pectin-g-PCL IPN hydrogels could allow for the adequate diffusion of nutrients and metabolites across the scaffold and to enable autologous tissue ingrowth when implanted in vivo. Moreover, IPN hydrogels have been widely used for the delivery of bioactive molecules in controlled and targeted drug delivery applications.70 This is mainly due to the ability to modulate the porosity of single component hydrogels through the introduction of a second polymer network, which in turn yields hydrogels with faster response rates required for drug release systems.71 Furthermore, IPNs have been used extensively in the development of smart drug delivery systems since they allow the delivery of drugs with a zero-order profile (i.e., sustained release) with minimum fluctuations.72 Therefore, the tunable microarchitecture and degradation rate of GelMA/pectin-g-PCL IPN hydrogels could be highly advantageous for the engineering of these types of drug delivery platforms.
Fig. 4.
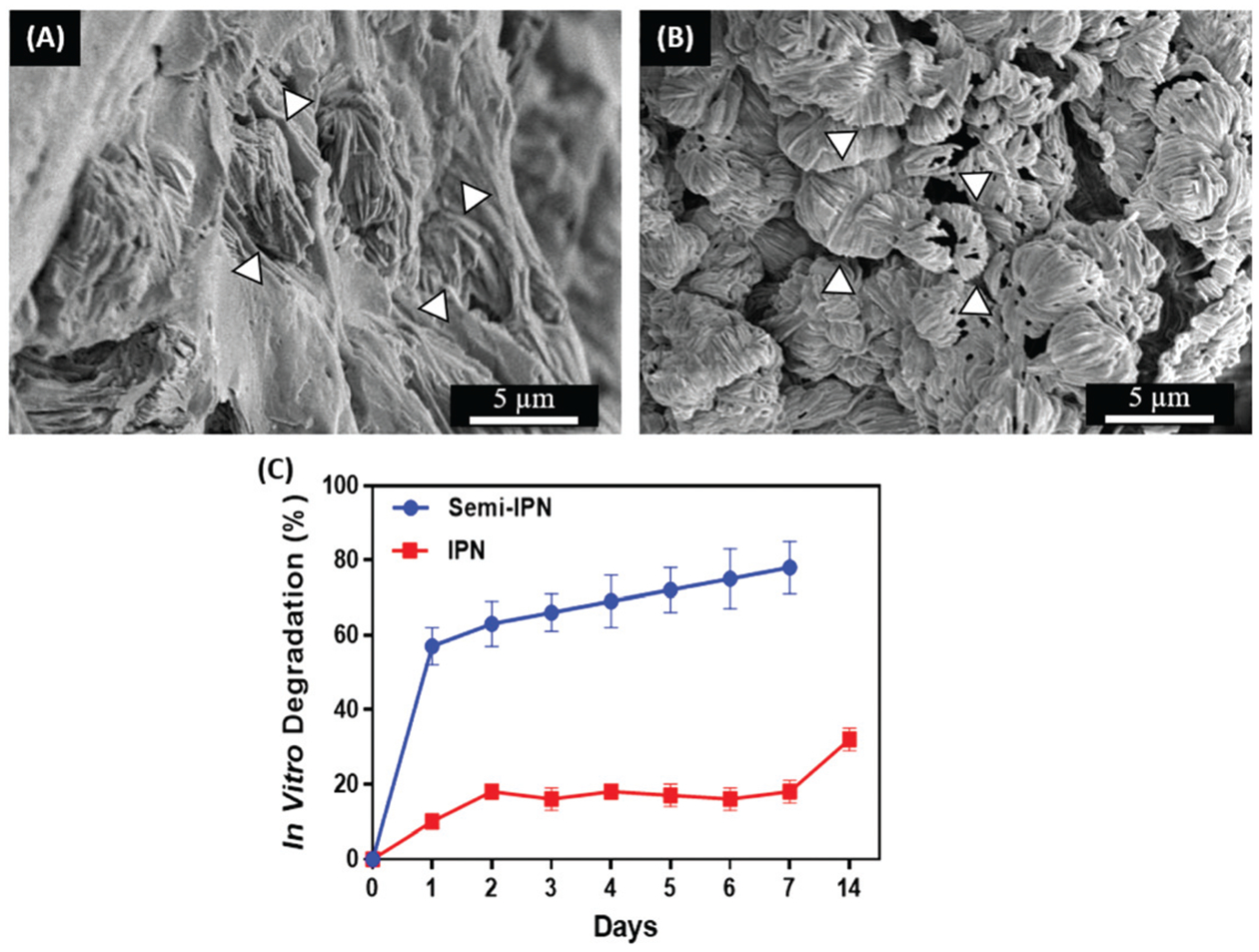
Microstructure and in vitro degradation of semi-IPN and IPN GelMA/pectin-g-PCL hydrogels. Representative SEM images of (A) semi-IPN hydrogels synthesized using 10% GelMA, and 1.15% pectin-g-PCL, and (B) IPN hydrogels synthesized using 10% GelMA, 1.15% pectin-g-PCL, and 0.5% Ca2+. White arrows indicate pectin-g-PCL aggregates within the GelMA network. (C) In vitro degradation rate of semi-IPN and IPN GelMA/pectin-g-PCL hydrogels.
Evaluation of cell viability, proliferation and spreading
Hydrogels used for 3D cell encapsulation should allow the attachment and growth of cells within the polymeric structure, without eliciting any cytotoxic response. Therefore, we evaluated the ability of GelMA/pectin-g-PCL IPN to support the growth and spread of 3D encapsulated MC3T3-E1 preosteoblasts using a commercial live/dead assay (Fig. 5A–D) and fluorescent F-actin/DAPI staining (Fig. 5E–H). Our results demonstrated that the viability of 3D encapsulated cells remained higher than 80% for both GelMA/pectin-g-PCL and GelMA control hydrogels up to 5 days of culture (Fig. 5I). However, the semi-IPN hydrogels rapidly lost structural integrity and were degraded after 3 days of culture, which made them unsuitable for further in vitro experimentation (ESI Fig. 3†). In addition, quantitative analysis of fluorescent F-actin/DAPI images revealed that cell spreading in IPN hydrogels was significantly lower than pure GelMA controls (Fig. 5J). This behavior could be explained in part due to differences in composition and the increased mechanical stiffness of GelMA/pectin-g-PCL IPN scaffolds. Recent evidence has shown that the stiffness of a substratum is a powerful mediator of cell fate and migration, which are important for the infiltration of host tissue cells into the scaffold.73 Previous works have demonstrated that MC3T3-E1 cells display cuboidal morphologies and undergo growth arrest under differentiating conditions.74 However, other studies have shown that different cell types such as 3T3 fibroblasts exhibit comparatively better cell spreading on stiffer matrixes.75 Therefore, the lower spreading observed for GelMA/pectin-g-PCL IPN hydrogels could also be due to the lesser number of cell-binding motifs available, which in turn results from the lower GelMA concentration in composite scaffolds.
Fig. 5.
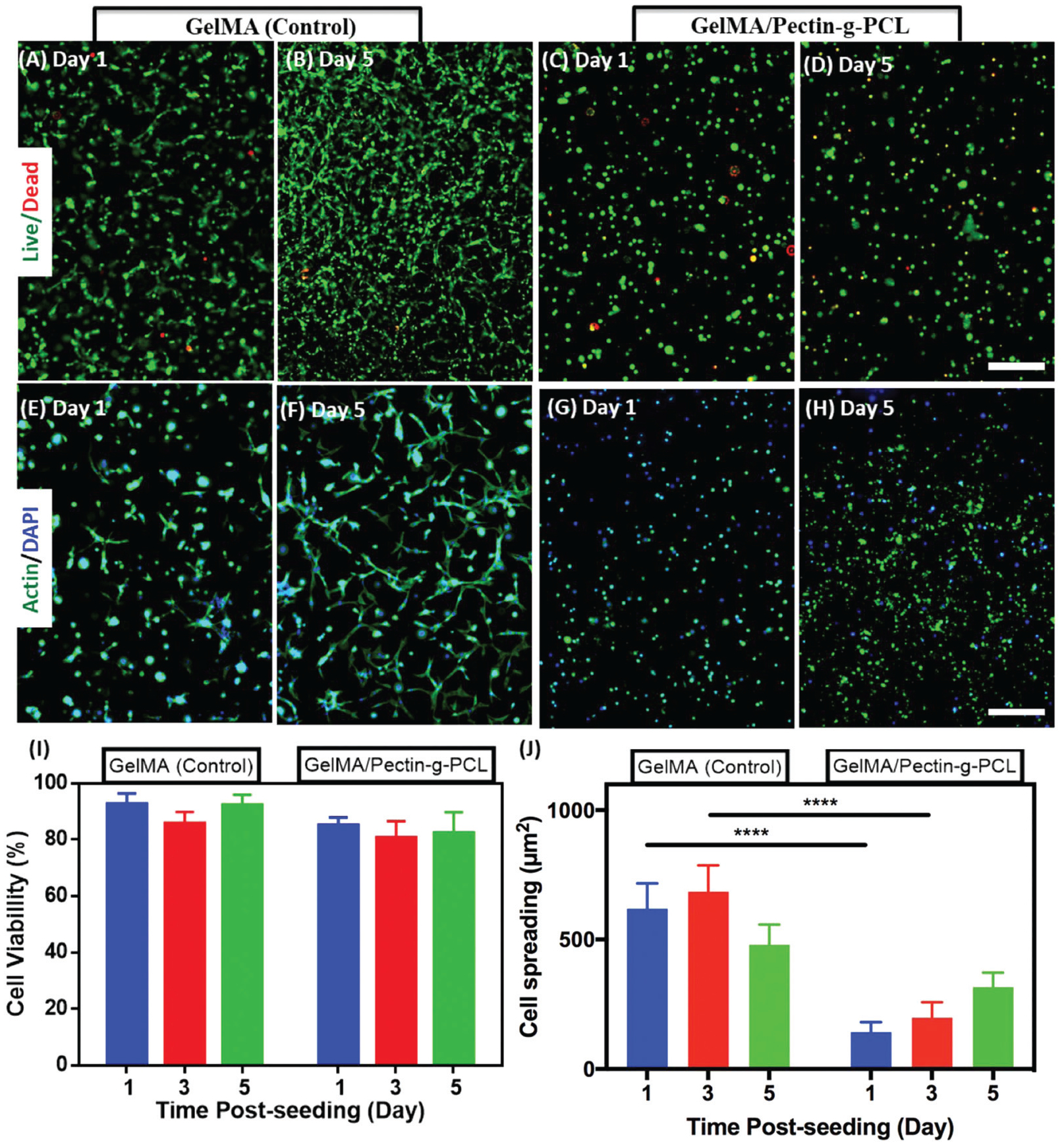
In vitro 3D cell encapsulation of MC3T3-E1 cells in GelMA and GelMA/pectin-g-PCL hydrogels. Representative live/dead images of MC3T3-E1 cells encapsulated in (A, B) GelMA hydrogels and (C, D) IPN GelMA/pectin-g-PCL hydrogels, on days 1 and 5. (E–G) Representative fluorescent F-actin/DAPI images of MC3T3-E1 cells encapsulated in (E, F) GelMA hydrogels, and (G, H) IPN GelMA/pectin-g-PCL hydrogels, at 1 and 5 days post-encapsulation (scale bar = 200 μm). Quantification of (I) cell viability and (J) cell spreading (per each cell) of MC3T3-E1 encapsulated in GelMA and IPN GelMA/pectin-g-PCL hydrogels at 1, 3, and 5 days post-encapsulation. IPN hydrogels were synthesized using 10% GelMA, 1.15% pectin-g-PCL, and 0.5% Ca2+. Control hydrogels were synthesized using 10% GelMA. Error bars indicate standard error of the means, asterisks mark significance levels of p < 0.0001 (****).
GelMA/pectin-g-PCL IPN hydrogels could be used to develop highly biocompatible and bioactive hydrogel-based scaffolds, mainly due to the presence of bioactive pectin moieties and the remarkable cell supportive nature of GelMA. For instance, previous groups have reported the engineering of gelatin/collagen-pectin composite hydrogels for bone tissue engineering applications.36,76 These studies showed that the combination of the intrinsic bioactivity of pectin with the remarkable cytocompatibility of GelMA resulted in hydrogels with enhanced osteogenic and osteoinductive properties for bone regeneration. Here, we demonstrated that the incorporation of pectin-g-PCL significantly enhanced the mechanical stiffness of GelMA/pectin-g-PCL IPN hydrogels. This is important, since scaffolds with high mechanical stiffness have been associated with enhanced induction of osteogenic differentiation in human mesenchymal stem cells in vitro.77,78 Therefore, although 3D encapsulated MC3T3-E1 cells showed comparatively lower spreading in IPN hydrogels, the high stiffness of the scaffold could potentially lead to the induction of osteogenic pathways. However, the ability of GelMA/pectin-g-PCL hydrogels to induce osteogenic differentiation in vitro is outside the scope of this paper and will be further investigated in future works.
Conclusion
In this study, we described the engineering of semi-IPN and IPN GelMA/pectin-g-PCL hydrogels with high mechanical strength for tissue engineering. Pectin-g-PCL was synthesized through ROP chemistry and were characterized via 1H-NMR, FTIR, TGA, and DSC. IPN hydrogels were synthesized by physical crosslinking of pectin-g-PCL via Ca2+, followed by chemical photocrosslinking of GelMA. On the other hand, the semi-IPN hydrogels were synthesized by inclusion of pectin-g-PCL in the GelMA precursor solution, and subsequent photocrosslinking in the absence of Ca2+ crosslinker. The engineered semi-IPN and IPN GelMA/pectin-g-PCL hydrogels exhibited a wide spectrum of physical properties, which can be readily tuned for different tissue engineering and biomedical applications. For semi-IPN hydrogels, low elastic moduli were obtained due to the aggregation of microcrystalline PCL cores. For IPN hydrogels, the optimization of pectin-g-PCL, GelMA, and Ca2+ concentrations resulted in the synthesis of robust hydrogels with a wide range of tunable compressive and tensile modului. IPN hydrogels were shown to support the growth of 3D-encapsulated MC3T3-E1 preosteoblasts in vitro. Moreover, previous studies have shown that both pectin-based biomaterials36 and scaffolds with high mechanical stiffness77,78 could induce osteogenic differentiation of progenitor cells in vitro. Hence, GelMA/pectin-g-PCL IPN hydrogels hold great potential for the development of scaffolds with intrinsic osteogenic activity for bone tissue engineering. The simplicity, feasibility, low cost, and tunable mechanical properties of the engineered hydrogels hold remarkable potential for various tissue engineering and biomedical applications.
Supplementary Material
Acknowledgements
M. M. Fares acknowledges Jordan University of Science & Technology for sabbatical leave. N. A. acknowledges the funding from National Institutes of Health (NIH) (R01EB023052; R01HL140618) and the American Heart Association (AHA, 16SDG31280010).
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm00474a
Conflicts of interest
There are no conflicts to declare.
References
- 1.Frantz C, Stewart KM and Weaver VM, J. Cell Sci, 2010, 123, 4195–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harburger DS and Calderwood DA, J. Cell Sci, 2009, 122, 159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pina S, Oliveira JM and Reis RL, Adv. Mater, 2015, 27, 1143–1169. [DOI] [PubMed] [Google Scholar]
- 4.Gasperini L, Mano JF and Reis RL, J. R. Soc., Interface, 2014, 11, 20140817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naahidi S, Jafari M, Logan M, Wang YJ, Yuan YF, Bae H, Dixon B and Chen P, Biotechnol. Adv, 2017, 35, 530–544. [DOI] [PubMed] [Google Scholar]
- 6.Vedadghavami A, Minooei F, Mohammadi MH, Khetani S, Rezaei Kolahchi A, Mashayekhan S and Sanati-Nezhad A, Acta Biomater, 2017, 62, 42–63. [DOI] [PubMed] [Google Scholar]
- 7.Zhang YS and Khademhosseini A, Science, 2017, 356, eaaf3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bryant SJ and Vernerey FJ, Adv. Healthcare Mater, 2017, 7, 1700605. [Google Scholar]
- 9.Noshadi I, Walker BW, Portillo-Lara R, Sani ES, Gomes N, Aziziyan MR and Annabi N, Sci. Rep, 2017, 7, 4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao X, Lang Q, Yildirimer L, Lin ZY, Cui W, Annabi N, Ng KW, Dokmeci MR, Ghaemmaghami AM and Khademhosseini A, Adv. Healthcare Mater, 2016, 5, 108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ostrovidov S, Ahadian S, Ramon-Azcon J, Hosseini V, Fujie T, Parthiban SP, Shiku H, Matsue T, Kaji H, Ramalingam M, Bae H and Khademhosseini A, J. Tissue Eng. Regener. Med, 2017, 11, 582–595. [DOI] [PubMed] [Google Scholar]
- 12.Lin RZ, Chen YC, Moreno-Luna R, Khademhosseini A and Melero-Martin JM, Biomaterials, 2013, 34, 6785–6796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao X, Sun X, Yildirimer L, Lang Q, Lin ZY, Zheng R, Zhang Y, Cui W, Annabi N and Khademhosseini A, Acta Biomater, 2017, 49, 66–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yue K, Trujillo-de Santiago G, Alvarez MM, Tamayol A, Annabi N and Khademhosseini A, Biomaterials, 2015, 73, 254–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S and Khademhosseini A, Biomaterials, 2010, 31, 5536–5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hassanzadeh P, Kazemzadeh-Narbat M, Rosenzweig R, Zhang X, Khademhosseini A, Annabi N and Rolandi M, J. Mater. Chem. B, 2016, 4, 2539–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heo DN, Castro NJ, Lee SJ, Noh H, Zhu W and Zhang LG, Nanoscale, 2017, 9, 5055–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao W, He J, Nichol JW, Wang L, Hutson CB, Wang B, Du Y, Fan H and Khademhosseini A, Acta Biomater, 2011, 7, 2384–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Zhou L, Liao J, Tan Y, Ouyang K, Ning C, Ni G and Tan G, J. Mater. Sci. Mater. Med, 2014, 25, 2173–2183. [DOI] [PubMed] [Google Scholar]
- 20.Shin SR, Zihlmann C, Akbari M, Assawes P, Cheung L, Zhang KZ, Manoharan V, Zhang YS, Yuksekkaya M, Wan KT, Nikkhah M, Dokmeci MR, Tang XW and Khademhosseini A, Small, 2016, 12, 3677–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahadian S, Ramon-Azcon J, Estili M, Liang XB, Ostrovidov S, Shiku H, Ramalingam M, Nakajima K, Sakka Y, Bae H, Matsue T and Khademhosseini A, Sci. Rep, 2014, 4, 4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heo DN, Ko WK, Bae MS, Lee JB, Lee DW, Byun W, Lee CH, Kim EC, Jung BY and Kwon IK, J. Mater. Chem. B, 2014, 2, 1584–1593. [DOI] [PubMed] [Google Scholar]
- 23.Camci-Unal G, Cuttica D, Annabi N, Demarchi D and Khademhosseini A, Biomacromolecules, 2013, 14, 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nunes C, Silva L, Fernandes AP, Guine RPF, Domingues MRM and Coimbra MA, Carbohydr. Polym, 2012, 87, 620–626. [DOI] [PubMed] [Google Scholar]
- 25.Mashingaidze F, Choonara YE, Kumar P, du Toit LC, Maharaj V, Buchmann E and Pillay V, Int. J. Pharm, 2016, 503, 16–28. [DOI] [PubMed] [Google Scholar]
- 26.Fares MM, Assaf SM and Abul-Haija YM, J. Appl. Polym. Sci, 2010, 117, 1945–1954. [Google Scholar]
- 27.Assaf SM, Abul-Haija YM and Fares MM, J. Macromol. Sci., Part A: Pure Appl.Chem, 2011, 48, 493–502. [Google Scholar]
- 28.Kumar PT, Ramya C, Jayakumar R, Nair S and Lakshmanan VK, Colloids Surf., B, 2013, 106, 109–116. [DOI] [PubMed] [Google Scholar]
- 29.Ninan N, Muthiah M, Park IK, Elain A, Thomas S and Grohens Y, Carbohydr. Polym, 2013, 98, 877–885. [DOI] [PubMed] [Google Scholar]
- 30.Kokkonen HE, Ilvesaro JM, Morra M, Schols HA and Tuukkanen J, Biomacromolecules, 2007, 8, 509–515. [DOI] [PubMed] [Google Scholar]
- 31.Kokkonen H, Verhoef R, Kauppinen K, Muhonen V, Jorgensen B, Damager I, Schols HA, Morra M, Ulvskov P and Tuukkanen J, J. Biomed. Mater. Res., Part A, 2012, 100, 111–119. [DOI] [PubMed] [Google Scholar]
- 32.Munarin F, Guerreiro SG, Grellier MA, Tanzi MC, Barbosa MA, Petrini P and Granja PL, Biomacromolecules, 2011, 12, 568–577. [DOI] [PubMed] [Google Scholar]
- 33.Tummalapalli M, Berthet M, Verrier B, Deopura BL, Alam MS and Gupta B, Int. J. Biol. Macromol, 2016, 82, 104–113. [DOI] [PubMed] [Google Scholar]
- 34.Munarin F, Petrini P, Barcellona G, Roversi T, Piazza L, Visai L and Tanzi MC, Mater. Sci. Eng., C, 2014, 45, 154–161. [DOI] [PubMed] [Google Scholar]
- 35.Deters AM, Meyer U and Stintzing FC, J. Ethnopharmacol, 2012, 142, 438–444. [DOI] [PubMed] [Google Scholar]
- 36.Amirian J, Linh NT, Min YK and Lee BT, Int. J. Biol. Macromol, 2015, 76, 10–24. [DOI] [PubMed] [Google Scholar]
- 37.Keothongkham K, Charoenphandhu N, Thongbunchoo J, Suntornsaratoon P, Krishnamra N, Tang IM and Pon-On W, Mater. Sci. Eng., C, 2017, 74, 47–54. [DOI] [PubMed] [Google Scholar]
- 38.He Y, Wang W, Tang X and Liu X, Dent. Mater. J, 2017, 36, 325–332. [DOI] [PubMed] [Google Scholar]
- 39.Thi Hiep N, Chan Khon H, Dai Hai N, Byong-Taek L, Van Toi V and Thanh Hung L, J. Biomater. Sci., Polym. Ed, 2017, 28, 864–878. [DOI] [PubMed] [Google Scholar]
- 40.Yildirim ED, Ayan H, Vasilets VN, Fridman A, Guceri S and Sun W, Plasma Processes Polym, 2008, 5, 58–66. [Google Scholar]
- 41.Chen G, Zhou P, Mei N, Chen X, Shao Z, Pan L and Wu C, J. Mater. Sci.: Mater. Med, 2004, 15, 671–677. [DOI] [PubMed] [Google Scholar]
- 42.Marra KG, Szem JW, Kumta PN, DiMilla PA and Weiss LE, J. Biomed. Mater. Res, 1999, 47, 324–335. [DOI] [PubMed] [Google Scholar]
- 43.Kang YG, Wei J, Shin JW, Wu YR, Su J, Park YS and Shin JW, Int. J. Nanomed, 2018, 13, 1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sperling LH and Hu R, in Polymer Blends Handbook, ed. Utracki LA, Springer; Netherlands, Dordrecht, 2003, pp. 417–447, DOI: 10.1007/0-306-48244-4_6. [DOI] [Google Scholar]
- 45.Arshadi M, Abdolmaleki M, Eskandarloo H, Azizi M and Abbaspourrad A, ACS Sustainable Chem. Eng, 2018, 6, 11662–11676. [Google Scholar]
- 46.Azizi M, Kierulf A, Lee MC and Abbaspourrad A, Food Chem, 2018, 246, 448–456. [DOI] [PubMed] [Google Scholar]
- 47.Noshadi I, Hong S, Sullivan KE, Shirzaei Sani E, Portillo-Lara R, Tamayol A, Shin SR, Gao AE, Stoppel WL, Black Iii LD, Khademhosseini A and Annabi N, Biomater. Sci, 2017, 5, 2093–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Annabi N, Rana D, Shirzaei Sani E, Portillo-Lara R, Gifford JL, Fares MM, Mithieux SM and Weiss AS, Biomaterials, 2017, 139, 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soucy JR, Shirzaei Sani E, Portillo Lara R, Diaz D, Dias F, Weiss AS, Koppes AN, Koppes R and Annabi N, Tissue Eng, 2018, DOI: 10.1089/ten.TEA.2017.0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pajoumshariati SR, Azizi M, Wesner D, Miller PG, Shuler ML and Abbaspourrad A, ACS Appl. Mater. Interfaces, 2018, 10, 9235–9246. [DOI] [PubMed] [Google Scholar]
- 51.Yoon HJ, Shin SR, Cha JM, Lee SH, Kim JH, Do JT, Song H and Bae H, PLoS One, 2016, 11, e0163902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karim AA and Bhat R, Food Hydrocolloids, 2009, 23, 563–576. [Google Scholar]
- 53.Rosenbohm C, Lundt I, Christensen TI and Young NG, Carbohydr. Res, 2003, 338, 637–649. [DOI] [PubMed] [Google Scholar]
- 54.Sinnwell S, Inglis AJ, Davis TP, Stenzel MH and Barner-Kowollik C, Chem. Commun, 2008, 2052–2054, DOI: 10.1039/B718180A. [DOI] [PubMed] [Google Scholar]
- 55.Kowalonek J, Int. J. Biol. Macromol, 2017, 103, 515–524. [DOI] [PubMed] [Google Scholar]
- 56.Monsoor MA, Kalapathy U and Proctor A, J. Agric. Food Chem, 2001, 49, 2756–2760. [DOI] [PubMed] [Google Scholar]
- 57.Elzein T, Nasser-Eddine M, Delaite C, Bistac S and Dumas P, J. Colloid Interface Sci, 2004, 273, 381–387. [DOI] [PubMed] [Google Scholar]
- 58.Mandal A and Chakrabarty D, Carbohydr. Polym, 2015, 134, 240–250. [DOI] [PubMed] [Google Scholar]
- 59.Assmann A, Vegh A, Ghasemi-Rad M, Bagherifard S, Cheng G, Sani ES, Ruiz-Esparza GU, Noshadi I, Lassaletta AD, Gangadharan S, Tamayol A, Khademhosseini A and Annabi N, Biomaterials, 2017, 140, 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shirzaei Sani E, Portillo-Lara R, Spencer A, Yu W, Geilich BM, Noshadi I, Webster TJ and Annabi N, ACS Biomater. Sci. Eng, 2018, 4, 2528–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoare TR and Kohane DS, Polymer, 2008, 49, 1993–2007. [Google Scholar]
- 62.Aleman J, Chadwick AV, He J, Hess M, Horie K, Jones RG, Kratochvil P, Meisel I, Mita I, Moad G, Penczek S and Stepto RFT, Pure Appl. Chem, 2007, 79, 1801–1827. [Google Scholar]
- 63.Jenkins AD, Kratochvil P, Stepto RFT and Suter UW, Polimery, 1998, 43, 559–564. [Google Scholar]
- 64.Sun JY, Zhao X, Illeperuma WR, Chaudhuri O, Oh KH, Mooney DJ, Vlassak JJ and Suo Z, Nature, 2012, 489, 133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liang B, Zhao H, Zhang Q, Fan Y, Yue Y, Yin P and Guo L, ACS Appl. Mater. Interfaces, 2016, 8, 28816–28823. [DOI] [PubMed] [Google Scholar]
- 66.Yang CH, Wang MX, Haider H, Yang JH, Sun JY, Chen YM, Zhou JX and Suo ZG, ACS Appl. Mater. Interfaces, 2013, 5, 10418–10422. [DOI] [PubMed] [Google Scholar]
- 67.Qiu Y and Park K, AAPS PharmSciTech, 2003, 4, E51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tanaka Y, Gong JP and Osada Y, Prog. Polym. Sci, 2005, 30, 1–9. [Google Scholar]
- 69.Gong JP, Katsuyama Y, Kurokawa T and Osada Y, Adv. Mater, 2003, 15, 1155–1158. [Google Scholar]
- 70.Lohani A, Singh G, Bhattacharya SS and Verma A, J. Drug Delivery, 2014, 2014, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dragan ES, Chem. Eng. J, 2014, 243, 572–590. [Google Scholar]
- 72.Aminabhavi TM, Nadagouda MN, More UA, Joshi SD, Kulkarni VH, Noolvi MN and Kulkarni PV, Expert Opin. Drug Delivery, 2015, 12, 669–688. [DOI] [PubMed] [Google Scholar]
- 73.Ghasemi-Mobarakeh L, Prabhakaran MP, Tian L, Shamirzaei-Jeshvaghani E, Dehghani L and Ramakrishna S, World J. Stem Cells, 2015, 7, 728–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Quarles LD, Yohay DA, Lever LW, Caton R and Wenstrup RJ, J. Bone Miner. Res, 1992, 7, 683–692. [DOI] [PubMed] [Google Scholar]
- 75.Breuls RG, Jiya TU and Smit TH, Open Orthop. J, 2008, 2, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wenpo F, Gaofeng L, Shuying F, Yuanming Q and Keyong T, Int. J. Biol. Macromol, 2015, 74, 218–223. [DOI] [PubMed] [Google Scholar]
- 77.Olivares-Navarrete R, Lee EM, Smith K, Hyzy SL, Doroudi M, Williams JK, Gall K, Boyan BD and Schwartz Z, PLoS One, 2017, 12, e0170312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yao QQ, Cosme JGL, Xu T, Miszuk JM, Picciani PHS, Fong H and Sun HL, Biomaterials, 2017, 115, 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.