

Abstract
Background
Immune checkpoint blockade (ICB) has revolutionized the treatment of various cancer types. Despite significant preclinical advancements in understanding mechanisms, identifying the molecular basis and predictive biomarkers for clinical ICB responses remains challenging. Recent evidence, both preclinical and clinical, underscores the pivotal role of the extracellular matrix (ECM) in modulating immune cell infiltration and behaviors. This study aimed to create an innovative classifier that leverages ECM characteristics to enhance the effectiveness of ICB therapy.
Methods
We analyzed transcriptomic collagen activity and immune signatures in 649 patients with cancer undergoing ICB therapy. This analysis led to the identification of three distinct immuno‐collagenic subtypes predictive of ICB responses. We validated these subtypes using the transcriptome data from 9,363 cancer patients from The Cancer Genome Atlas (TCGA) dataset and 1,084 in‐house samples. Additionally, novel therapeutic targets were identified based on these established immuno‐collagenic subtypes.
Results
Our categorization divided tumors into three subtypes: “soft & hot” (low collagen activity and high immune infiltration), “armored & cold” (high collagen activity and low immune infiltration), and “quiescent” (low collagen activity and immune infiltration). Notably, “soft & hot” tumors exhibited the most robust response to ICB therapy across various cancer types. Mechanistically, inhibiting collagen augmented the response to ICB in preclinical models. Furthermore, these subtypes demonstrated associations with immune activity and prognostic predictive potential across multiple cancer types. Additionally, an unbiased approach identified B7 homolog 3 (B7‐H3), an available drug target, as strongly expressed in “armored & cold” tumors, relating with poor prognosis.
Conclusion
This study introduces histopathology‐based universal immuno‐collagenic subtypes capable of predicting ICB responses across diverse cancer types. These findings offer insights that could contribute to tailoring personalized immunotherapeutic strategies for patients with cancer.
Keywords: collagen deposition, immune infiltration, immunotherapy, pan‐cancer, tumor microenvironment
List of abbreviations
- ICB
immune checkpoint blockade
- PD‐1
programmed cell death 1
- PD‐L1
programmed cell death 1 ligand 1
- CTLA4
cytotoxic T‐lymphocyte associated antigen 4
- TME
tumor microenvironment
- TMB
tumor mutational burden
- ECM
extracellular matrix
- CAF
cancer‐associated fibroblast
- TIME
tumor immune microenvironment
- TCGA
the Cancer Genome Atlas
- OS
overall survival
- GSEA
gene sets enrichment analysis
- GSVA
Gene Set Variation Analysis
- NSCLC
non‐small cell lung cancer
- TMA
tissue microarray
- TNBC
triple negative breast cancer
- FPKM
Fragments Per Kilobase of exon model per Million mapped reads
- HE
hematoxylin and eosin
- TIIC
tumor‐infiltrating immune cell
- CPS
Combined Positive Score
- V
volume
- PBS
phosphate belanced solution
- GR1
lymphocyte antigen 6 complex, locus G/C
- scRNA‐seq
single‐cell RNA‐sequencing
- 4NQO
4‐Nitroquinoline N‐oxide
- PCA
principal component analysis
- PC
principal component
- DCN
decorin
- ANOVA
one‐way analysis of variance
- ROC
receiver‐operating characteristic
- AUC
area under the ROC curve
- TGF‐β
transforming growth factor‐β
- DDR1
discoidin domain receptor tyrosine kinase 1
- MHC
major histocompatibility complex
- NTNBC
non‐TNBC
- PD‐L1 IC score
PD‐L1 expression score in immune cell
- PD‐L1 TC score
PD‐L1 expression score in tumor cell
- FAP
fibroblast activation protein
- MDSC
myeloid‐derived suppressor cell
- MSI‐H
microsatellite instability‐high
- dMMR
mismatch repair deficient
- EBV
Epstein‐Barr virus
- FGFR
fibroblast growth factor receptor
- PDGFR
platelet‐derived growth factor receptor
- B7‐H3
B7 homolog 3
- KDR
kinase insert domain receptor
- ERBB
erb‐b2 receptor tyrosine kinase
- PVRL4
poliovirus receptor‐related 4
- FOLR
folate receptor
- ACC
adrenocortical carcinoma
- BLCA
bladder urothelial carcinoma
- BRCA
breast invasive carcinoma
- CESC
cervical & endocervical cancer
- CHOL
cholangiocarcinoma
- COAD
colon adenocarcinoma
- ESCA
esophageal carcinoma
- GBM
glioblastoma multiforme
- HNSC
head and neck squamous cell carcinoma
- KICH
kidney chromophobe
- KIRC
kidney renal clear cell carcinoma
- KIRP
kidney renal papillary cell carcinoma
- LGG
brain lower grade glioma
- LIHC
liver hepatocellular carcinoma
- LUAD
lung adenocarcinoma
- LUSC
lung squamous cell carcinoma
- MESO
mesothelioma
- OV
ovarian serous cystadenocarcinoma
- PAAD
pancreatic adenocarcinoma
- PCPG
pheochromocytoma and paraganglioma
- PRAD
prostate adenocarcinoma
- READ
rectum adenocarcinoma
- SARC
sarcoma
- SKCM
skin cutaneous melanoma
- STAD
stomach adenocarcinoma
- TGCT
testicular germ cell tumor
- THCA
thyroid carcinoma
- THYM
thymoma
- UCEC
uterine corpus endometrial carcinoma
- UCS
uterine carcinosarcoma;
- UVM
uveal melanoma.
1. BACKGROUND
Cancer continues to pose a significant global health challenge, accounting for nearly 20 million new cases and 10 million cancer‐related deaths annually [1]. Genetic mutations in tumor cells lead to the production of tumor antigens, such as melanoma‐associated antigen, which can trigger recognition by the immune system due to the instability in these cells [2]. The immune system's role in both surveillance against tumors and inhibiting their growth is well recognized and has been translated into clinical therapies, notably immune checkpoint blockade (ICB). This approach targets immune inhibitory molecules such as programmed cell death 1 (PD‐1), programmed cell death‐ligand 1 (PD‐L1), or cytotoxic T‐lymphocyte‐associated antigen 4 (CTLA4). Despite significant progresses in various cancer treatments, the clinical application of ICB faces challenges such as limited response rates, unclear underlying mechanisms, and immune‐related adverse events [3, 4]. Extensive efforts have been undertaken to identify factors aiding patient selection for ICB. PD‐L1 expression has been a focus in numerous clinical trials [5, 6, 7], but conflicting studies showed that even patients with negative PD‐L1 expression could benefit from ICB [8, 9]. An accurate, reliable, and user‐friendly predictive biomarker remains elusive in this field.
Recent knowledge emphasizes the significance of immune cell types, density, function, and distribution within the tumor microenvironment (TME) for patient classification. Histopathology‐based immune status surpasses the classical tumor‐node‐metastasis staging in accurately defining cancer's immune status and its responsiveness to ICB [10, 11]. For instance, the cytotoxic T cell‐based immunoscore [12, 13] has been widely utilized to categorize tumors as “hot” or “cold”, representing T cell inflamed and non‐inflamed tumors. While these classifications are widely adopted in clinical settings, further enhancements are crucial to better stratify patients and guide treatment selection. Besides the cytotoxic T cell landscape, numerous biomarkers such as B cell infiltration [14], interferon‐driven signaling [15], tumor mutational burden (TMB) [16], specific chemokines and adhesion molecules [17], angiogenesis [18], and the combination of biomarkers [19], have been proposed for their ICB response predictive potential. Nevertheless, the quest for reliable clinical biomarkers remains challenging, constrained by tumor types and patient sample sizes.
Solid tumors possess an extracellular matrix (ECM) markedly different from healthy tissues, characterized by its abundance, high density, and stiffness. The tumoral ECM, predominantly composed of collagens, is produced by both tumor cells and cancer‐associated fibroblasts (CAFs) [20]. These collagens form a barrier around tumor cell clusters, shielding them from nutrients, therapeutic agents, or cytotoxic immune cells [21, 22]. Growing evidence suggests that these collagens impede T cell infiltration and dictate the distribution of immune cells in various cancer types [23, 24]. Despite the close association between the ECM and immune cells in the TME, its contribution to patient stratification and ICB response prediction remains poorly understood.
This study aimed to evaluate the interaction of collagen deposition and the tumor immune microenvironment (TIME) and its clinical implications. Data of 649 patients with cancer receiving ICB from public cohorts, 9,363 patients from The Cancer Genome Atlas (TCGA) dataset, and 1,084 in‐house patients were collected and analyzed. A novel subtyping strategy was developed and validated according to collagen deposition and immune activity in large‐scale cohorts. By combining analyses of large‐scale public and in‐house cohorts, we hope our exploration provides some insights into the collagen‐immune interaction in the TME and clinically reliable pan‐cancer biomarkers for predicting the ICB response.
2. MATERIALS AND METHODS
2.1. Overall study design
In this study, we aimed to evaluate the interaction of collagen deposition and the TIME and its clinical value. We collected data from 649 cancer patients receiving ICB, 9,363 patients from the TCGA dataset, and 1,084 in‐house patients. We developed and validated a novel subtyping strategy according to collagen deposition and immune activity in the above large‐scale cohorts. In addition, based on the mutually exclusive pattern between collagen deposition and immune cell infiltration, we also proposed that anti‐collagen deposition was a significant strategy to sensitize immunotherapy (Figure 1).
FIGURE 1.
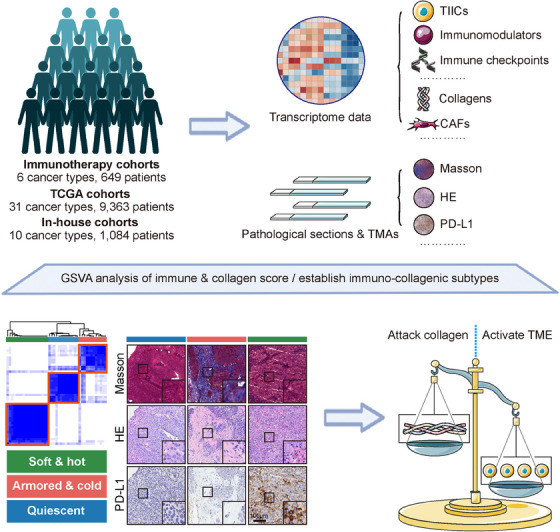
Schematic overview in this study. Clinical cohorts: Immunotherapy cohorts representing 6 cancer types and 649 patients, The Cancer Genome Atlas cohorts representing 31 cancer types and 9,363 patients, and in‐house cohorts representing 10 cancer types and 1,084 patients. Methods: Assessing the transcriptional profile and the histopathologic status in the above clinical cohorts to develop and validate novel immuno‐collagenic subtypes. Clinical outcomes: Patients were categorized into three subtypes with distinct features: soft & hot (low collagen activity and high immune infiltration), armored & cold (high collagen activity and low immune infiltration), and quiescent (low collagen activity and immune infiltration). Clinical effects: Predicting the immunotherapeutic responses and proposing targets highly expressed in armored & cold tumors. Abbreviations: CAF, cancer‐associated fibroblast; HE, hematoxylin and eosin; PD‐L1, programmed cell death 1 ligand 1; TCGA, The Cancer Genome Atlas; TIIC, tumor‐infiltrating immune cells; TMA, tumor microarray.
2.2. Transcriptome data from public portals
Transcriptome datasets and clinical annotations of 9,363 cancer patients for 31 solid tumor types in the TCGA dataset were acquired through the University of California Santa Cruz Xena platform (https://xenabrowser.net/datapages/). Samples with available overall survival (OS) data were specifically chosen for further analysis. In certain cancer types, somatic mutation data were sourced from the TCGA database (http://cancergenome.nih.gov/) and used to compute the TMB using the R package “maftools” [25]. For the stomach adenocarcinoma (STAD) dataset, we referenced the latest four published molecular classifications of STAD samples [26].
For mouse tumor analysis, we obtained the GSE168846 dataset [27] containing transcriptomic data from tumors of diverse syngeneic mouse models (such as melanoma CM3, colorectal cancer CT26, melanoma B16F10, urothelial carcinoma MB49, colorectal cancer MC38, bladder cancer MBT2, lung cancer LL2, renal cell carcinoma RENCA, breast cancer EMT6, and breast cancer 4T1) subjected to ICB treatment from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/).
2.3. Transcriptome data from cancer patients receiving ICB
A panel of public immunotherapy datasets, including the GSE173839 dataset [28], the GSE194040 dataset [29], the PRJEB25780 dataset [30], the PRJEB23709 dataset [31], the GSE126044 dataset [32], the GSE135222 dataset [33], comprising transcriptome data from cancer patients receiving ICB were downloaded from the GEO or the Tumor Immune Dysfunction and Exclusion databases (http://tide.dfci.harvard.edu/). The MEDI4736 dataset was obtained from Dr. Lajos Pusztai in Yale University School of Medicine (New Haven, CT, USA) [34]. The transcriptome data and clinical information of the IMvigor210 cohort [35] were obtained from the corresponding website (http://research‐pub.gene.com/IMvigor210CoreBiologies/). By combining these datasets, we created cohorts with 6 tumor types. For each tumor type, the “removeBatchEffect” function in the “limma” package [36] was applied to remove batch effects of datasets with various data formats. The case number and characteristics of these cohorts are summarized in Supplementary Table S1.
2.4. Enrichment analysis of ICB response‐related genes
To further describe the biological progresses of ICB response‐related genes, “limma” package [36] was utilized to identify ICB response‐related genes firstly. Subsequently, the enrichment analysis was performed by the “gene sets enrichment analysis (GSEA)” function in the “clusterProfiler” package [37] in terms of the “REACTOME” analysis [38].
2.5. Definition of immune signature and collagen signature
Gene markers of various immune signature and collagen signature were collected from various studies (Supplementary Table S2). The enrichment scores of these signatures were estimated using the GSEA algorithm by the “gene set variation analysis (GSVA)” R package [39]. Immune score and collagen score were assessed based on the average values of immune signature and collagen signature.
2.6. Transcriptomic patient stratification
To examine immuno‐collagenic heterogeneity, we utilized consensus clustering (employing the “ConsensusClusterPlus” package [40] in R software; 1,000 iterations, 80% resampling) to categorize subtypes based on the abundance of immune and collagen signatures. Across each transcriptome dataset, patients were stratified into three subtypes, each characterized by distinct features: “soft & hot” (low collagen activity and high immune infiltration), “armored & cold” (high collagen activity and low immune infiltration), and “quiescent” (low collagen activity and immune infiltration).
2.7. Clinical samples
Two cohorts comprising a total of 62 patients with non‐small cell lung cancer (NSCLC) who received ICB between January 2019 and December 2021 were retrospectively collected. These cohorts were obtained from The Affiliated Wuxi People's Hospital of Nanjing Medical University (Wuxi, Jiangsu, China) and Xuzhou Central Hospital (Xuzhou, Jiangsu, China), following ethical approval from The Clinical Research Ethics Committees at Nanjing Medical University (No. 2022‐383) and Xuzhou Central Hospital (No. XZXY‐LK‐20230610‐086). Tumor tissue samples were obtained via biopsy before immunotherapy. The therapeutic response was evaluated using RECIST 1.1 criteria [41], which include complete response, partial response, stable disease, and progressive disease.
Additionally, paraffin‐embedded cancer tissue microarrays (TMA) consisting of 1,012 cases for 10 cancer types used in some experiments were obtained from the National Engineering Center for Biochip (Outdo Biotech, Shanghai, China) with approval from the Outdo Biotech Clinical Research Ethics Committee. Due to the minority of samples losses during multiple staining procedures, the 1,012 samples are the total number of cases after excluding the losing samples. Detailed clinicopathological and survival data for these in‐house cohorts are summarized in Supplementary Table S3.
2.8. RNA‐sequencing (RNA‐seq) for paraffin‐embedded tumor tissues
A total of 10 paraffin‐embedded triple‐negative breast cancer (TNBC) samples were collected from Wuxi Maternal and Child Health Hospital (Wuxi, Jiangsu, China) under the ethical approval of The Clinical Research Ethics Committees at Wuxi Maternal and Child Health Hospital (No. 2021‐01‐0927‐28) and submitted to Shanghai Biotechnology Corporation (Shanghai, China) for RNA‐seq using the Illumina NovaSeq 6000 sequenator (model: PE150). RNA‐seq data was standardized to Fragments Per Kilobase of exon model per Million mapped reads (FPKM) format [42]. The cohort was used to check correlations between transcriptome‐based scores and histology‐based scores.
2.9. Histochemistry and immunohistochemistry analyses
Human and mouse paraffin‐embedded tissues were sectioned into 4‐µm‐thick sections. These tissue slides underwent staining with Masson staining, hematoxylin and eosin (HE) staining, and anti‐PD‐L1 staining. The bladder urothelial carcinoma (BLCA) TMA (HBlaU079Su01) was also stained with anti‐B7 homolog 3 (B7‐H3). Standard operating procedures were employed for immunohistochemistry and HE staining. Primary antibodies, including a ready‐to‐use anti‐PD‐L1 antibody (1:1, GT2280, GeneTech, Shanghai, China) and an anti‐B7‐H3 antibody (1:20,000, ab219648, Abcam, Cambridge, UK), were utilized. Samples were visualized using EnVision™ FLEX+ (K8009, Dako, Copenhagen, Denmark). Masson staining was conducted using the Trichrome Stain Kit (FH115100, FreeThinking, Nanjing, Jiangsu, China) following the manufacturer's instructions. Sections were captured using a slide scanner (Aperio GT 450 DX, Leica, Wetzlar, Germany).
Assessment of tumor‐infiltrating immune cells (TIICs) was conducted by two senior pathologists using the criteria established by TCGA Network [43]. Pathological parameters for each case included TIIC distribution (0, no TIICs; 1, <25% tissue involvement; 2, 25%‐50% tissue involvement; 3, >50% tissue involvement) and TIIC density (0 = absent, 1 = mild, 2 = moderate, 3 = severe). The TIIC score, the sum of distribution and density scores (0‐6), was calculated per case. Masson staining evaluation involved determining positively stained area percentages using the HALO software (v3.4.2986, Indica Labs, Albuquerque, NM, USA). Two experienced pathologists scored PD‐L1 and B7‐H3 staining. PD‐L1 staining was quantitatively assessed based on the combined positive score (CPS), while B7‐H3 expression used a modified CPS criterion: B7‐H3 CPS = (membrane‐positive tumor cells, immune cells, fibroblasts, and endothelial cells) / (total tumor cells) × 100 [44]. The TIIC score's cutoff value referred to previous research [43]. The collagen area's cut‐off value was determined by averaging across all samples, approximating 10%. Tumor samples were categorized based on TIIC score and collagen area: soft & hot tumors (TIIC score ≥ 3 and collagen area < 10%), armored & cold tumors (TIIC score ≤ 2 and collagen area ≥ 10%), and quiescent tumors (TIIC score ≤ 2 and collagen area < 10%).
2.10. Animals, cell culture, tumor‐bearing mouse model, and drug treatment
Female BALB/c mice (5‐6 weeks old) from the Shanghai Laboratory Animal Center (Shanghai, China) were housed in specific pathogen‐free facilities at 20‐24°C under 12‐hour light/dark cycles. All mouse studies were approved by the Laboratory Animal Ethics Committee of Nanjing Medical University (IACUC‐2305039). The murine breast cancer cell line 4T1 (KG338, KeyGEN Biotech, Nanjing, Jiangsu, China) was cultured in Dulbecco's minimum essential medium (KGM12800N‐500, KeyGEN Biotech) supplemented with 10% fetal bovine serum (10099141C, Gibco, Waltham, MA, USA) at 37°C with 5% CO2. All cell lines were free from mycoplasma and authenticated recently by short tandem repeat profiling. The mouse breast cancer model was established by subcutaneously injecting approximately 5 × 106 cells into each BALB/c mouse. Tumor size was monitored every 2‐3 days using calipers, and tumor volume (V) was calculated using the formula V = (length × width2) / 2.
Upon tumors reaching an average size of approximately 100 mm3, tumor‐bearing mice were randomly assigned to groups. The control group received oral administration of phosphate belanced solution (PBS). The talabostat group received daily oral administration of talabostat (HY‐13233A, MedChemExpress, Shanghai, China) at 20 µg/mouse. The anti‐PD‐1 group was injected intraperitoneally with the neutralizing antibody InVivoMAb anti‐mouse PD‐1 (BE0273, BioXCell, Lebanon, NH, USA) at 200 µg/mouse three times a week. The combination group received daily oral talabostat at 20 µg/mouse and intraperitoneal injections of the anti‐PD‐1 antibody at 200 µg/mouse three times a week. The tumors were removed from the unconscious animals at day 21 after the initiation of the treatment, which was subsequently documented and weighed.
2.11. Flow cytometry
Flow cytometry was performed following previously outlined procedures [45]. In summary, freshly dissected mouse tumor tissues underwent digestion and labeling using anti‐CD3 (1 µg/test, 100203, Biolegend, San Diego, CA, USA), anti‐CD8 (0.25 µg/test, 100711, Biolegend), anti‐lymphocyte antigen 6 complex, locus G/C (Ly6g/c, GR1; 0.25 µg/test, 108427, Biolegend), and anti‐CD11b antibodies (0.25 µg/test, 101207, Biolegend). A CytoFLEX flow cytometer (Beckman Coulter, Brea, CA, USA) was employed for analysis, and FlowJo (v7.6.5, https://www.flowjo.com/) was used for data interpretation.
2.12. Cellular reprogramming after anti‐B7‐H3 therapy
To investigate cellular reprogramming post anti‐B7‐H3 treatment, single‐cell RNA‐sequencing (scRNA‐seq) datasets from 4‐nitroquinoline N‐oxide (4NQO)‐induced mice following anti‐B7‐H3 therapy were obtained from the GSE164817 dataset [46]. Cells were excluded based on mitochondrial gene expression exceeding 10% or if they displayed fewer than 300 or more than 5,000 detected genes. Subsequently, 15,931 cells were analyzed (control: 9,561 cells; anti‐B7‐H3: 6,370 cells). The “RunHarmony” function [47] was utilized to mitigate batch effects between the two samples. Principal component analysis was conducted on the top 4,000 variable genes, using the first 40 principal components to reduce dimensionality. Cells were annotated into five major cell types based on established markers such as epithelial cell adhesion molecule for tumor cells and decorin for fibroblasts. Recognizing the significance of cell‐cell communications mediated by ligand‐receptor complexes in diverse biological processes, the “CellPhoneDB” tool was used to describe interactions among cell types in both control and anti‐B7‐H3 samples. Ligand‐receptor pairs with a P value < 0.05 were retained to assess relationships among different cell clusters. Furthermore, to evaluate the status of CD8+ T cells, the “AddModuleScore” function was employed to calculate the cytotoxic and exhausted scores of CD8+ T cells from both control and anti‐B7‐H3 samples.
2.13. Statistical analysis
Statistical analyses and figure presentations were conducted using R language 4.0.2 and GraphPad Prism 6.0 (https://www.graphpad‐prism.cn/). All R packages utilized in this study are summarized in Supplementary Table S4. Group differences were assessed using the Student t‐test or Mann‐Whitney test for two groups, while one‐way analysis of variance or the Kruskal‐Wallis test with multiple comparisons was utilized for multiple groups. Categorical variables were assessed using the chi‐square test or Fisher exact probability test. Pearson's or Spearman's correlation tests were employed to evaluate correlations between variables. Receiver‐operating characteristic (ROC) curve analysis was utilized to determine the specificity and sensitivity of candidate indicators, generating the area under the ROC curve for diagnostic biomarkers. Prognostic values of categorical variables were assessed via log‐rank test and Cox regression analysis. A P value of < 0.05 was considered statistically significant for all analyses.
3. RESULTS
3.1. Immuno‐collagenic subtypes were associated with ICB response in NSCLC
NSCLC is one of the most progressive cancer types, and NSCLC patients exhibit a notable enhancement in survival and quality of life through the application of ICB [48, 49, 50]. In our pursuit to comprehend the molecular mechanism identifying patients who derive the greatest benefits from ICB, we consolidated a combined NSCLC cohort treated with anti‐PD‐1/PD‐L1 immunotherapy [32, 33]. A comprehensive transcriptome analysis encompassing 43 patients with NSCLC was conducted, focusing on biological pathway analysis employing the peer‐reviewed REACTOME database [38]. This examination scrutinized pathways involving classical intermediary metabolism, signaling, and apoptosis. Notably, a connection was established between ICB responses and anti‐tumor immunity pathways. Intriguingly, inadequate responses to ICB were linked with pathways related to collagen deposition (Figure 2A). These prominent biological processes associated with ICB responses captivated our interest and thus we underwent further investigation.
FIGURE 2.
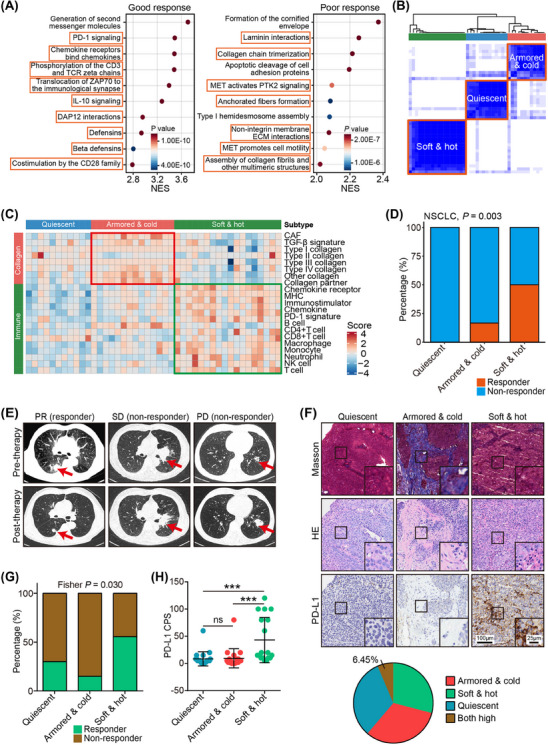
Establish the immuno‐collagenic subtypes for immune checkpoint blockade response prediction in non‐small cell lung cancer. (A) Gene set enrichment analysis of genes associated with good immunotherapeutic response (left) and poor immunotherapeutic response (right) in the merged dataset of the GSE126044 dataset and the GSE135222 dataset. Orange boxes indicate immune‐related pathways and collagen‐related pathways. (B) Heatmap exhibiting consensus clustering solution for collagen‐ and immune‐related genes in NSCLC samples. (C) Heatmap showing scores of collagen‐ and immune‐related gene modules in the three immuno‐collagenic subtypes. (D) Difference in the immunotherapeutic response rate in the three immuno‐collagenic subtypes in the merged NSCLC cohort. Significance was calculated using the Fisher exact probability test. (E) Representative images uncovering the collagen area, TIIC infiltration, and PD‐L1 expression in tumors with various subtypes and corresponding immunotherapeutic responses. (F) Proportion of four subtypes divided by collagen area and TIIC infiltration. The proportion of co‐high collagen and TIIC was quietly limited as shown on the pie chart. (G) Difference in the immunotherapeutic response rate in the three immuno‐collagenic subtypes in the in‐house NSCLC immunotherapy cohort. Significance was calculated using the Fisher exact probability test. (H) Difference in the PD‐L1 expression in three immuno‐collagenic subtypes in the in‐house NSCLC immunotherapy cohort. Data are presented as mean ± standard deviation. Significance was calculated using the Kruskal‐Wallis test with Dunn's multiple‐comparison test. ns: non‐significance, ***: P < 0.001. Abbreviations: CAF, cancer‐associated fibroblast; CPS, Combined Positive Score; GSEA, gene sets enrichment analysis; HE, hematoxylin and eosin; ICB, immune checkpoint blockade; MHC, major histocompatibility complex; NSCLC, non‐small‐cell lung cancer; PD, progressive disease; PD‐1, programmed cell death 1; PD‐L1, programmed cell death 1 ligand 1; PR, partial response; TGF‐β, transforming growth factor‐β; SD, stable disease; TIIC, tumor‐infiltrating immune cells.
To delineate the role of collagen deposition and the TIME in NSCLC, we compiled 8 functional modules representing collagen signatures. These encompassed all collagen genes, CAF signatures [20], stimulatory signals for CAFs such as transforming growth factor‐β signaling [51], and signals stimulating collagen, such as discoidin domain receptor tyrosine kinase 1 (DDR1) [23] (Supplementary Table S3). Additionally, 13 functional modules representing immune signatures, inclusive of common markers for tumor‐infiltrating immune cells [52], immunomodulators such as PD‐1 signaling, major histocompatibility complex molecules, and chemokine signaling [53], were compiled (Supplementary Table S3). The correlation matrix illustrated an inverse relationship between these anti‐tumor immune signature modules and collagen modules (Supplementary Figure S1). We next evaluated the combined utility of these two factors in predicting ICB response in patients with NSCLC. A correlation matrix was established based on the distribution of gene modules across samples, revealing three distinct subtypes characterized by immuno‐collagenic scores (Figure 2B‐C). These subtypes were closely linked to ICB response (Figure 2D). These findings suggest a novel stratification of ICB‐treated patients with NSCLC based on transcriptome analysis.
The high cost associated with transcriptome analysis has limited the practical application of immuno‐collagenic subtypes. Consequently, we explored an affordable, simple, and convenient alternative to transcriptomic stratification. To achieve this, we gathered 10 TNBC samples and subjected them to RNA‐seq analysis. Additionally, paraffin‐embedded sections underwent HE and Masson staining. We then compared the correlations between the collagen expression score obtained from RNA‐seq data and the collagen levels assessed by Masson staining, as well as between the TIIC score derived from RNA‐seq data and the TIIC score determined through HE staining. Positive correlations were observed between the transcriptome‐based scores and histology‐based scores (Supplementary Figure S2). This user‐friendly approach holds promise for significantly broadening the scope of the newly defined immuno‐collagenic subtypes.
In an effort to assess whether this simplified method is adequate for predicting ICB response, we conducted Masson and HE staining on NSCLC tissues from 62 patients with different ICB responses (Figure 2E). We used anti‐PD‐L1 staining as a validation for ICB biomarkers (Figure 2F). The results showed that samples with both high PD‐L1 and collagen were accounted for only 6.45%, which were very limited (Figure 2F). Notably, soft & hot tumors, armored & cold tumors, and quiescent tumors demonstrated associations with ICB responses (Figure 2G). Particularly, the soft & hot subtype exhibited high PD‐L1 expression (Figure 2H). These subtypes displayed enhanced sensitivity and specificity compared to conventional PD‐L1 expression (Supplementary Figure S3), indicating the potential of these subtypes as clinical biomarkers.
3.2. Conserved immuno‐collagenic subtypes predicted ICB response
To investigate the applicability of these subtypes in addition to NSCLC, we aggregated several publicly available cohorts, creating 5 cohorts encompassing patients with melanoma, BLCA, non‐TNBC (NTNBC), TNBC, and STAD, who exhibited various ICB responses, with some cohorts providing survival data. Intriguingly, ICB responses were associated with anti‐tumor immunity in melanoma, NTNBC, TNBC, and STAD, while poor ICB responses were linked to collagen deposition in BLCA, TNBC, and STAD (Supplementary Tables S5‐S9). Across all 5 cancer types, anti‐tumor immunity modules consistently displayed an inverse correlation with collagen modules (Supplementary Figure S4). By integrating information on anti‐tumor immunity and collagen deposition, the immuno‐collagenic subtypes were identified through consensus analysis (Figure 3A). These subtypes were associated with ICB responses and clinical outcomes in melanoma and BLCA (Figure 3B‐F). Moreover, they showed associatons with established biomarkers, such as immuno‐subtypes (Figure 3G), PD‐L1 expression scores in immune cells (PD‐L1 IC score), and PD‐L1 expression scores in tumor cells (PD‐L1 TC score; Supplementary Figure S5). Similarly, these subtypes categorized ICB responses in NTNBC, TNBC, and STAD (Figure 3H‐J). It is worth noting that neither anti‐tumor immunity nor collagen deposition predicted ICB response across all the mentioned cancer types, whereas the immuno‐collagenic subtypes consistently demonstrated a strong association with ICB response in all 6 cancer types (Supplementary Table S10), suggesting their potential as a universal, reliable biomarker for solid tumors.
FIGURE 3.
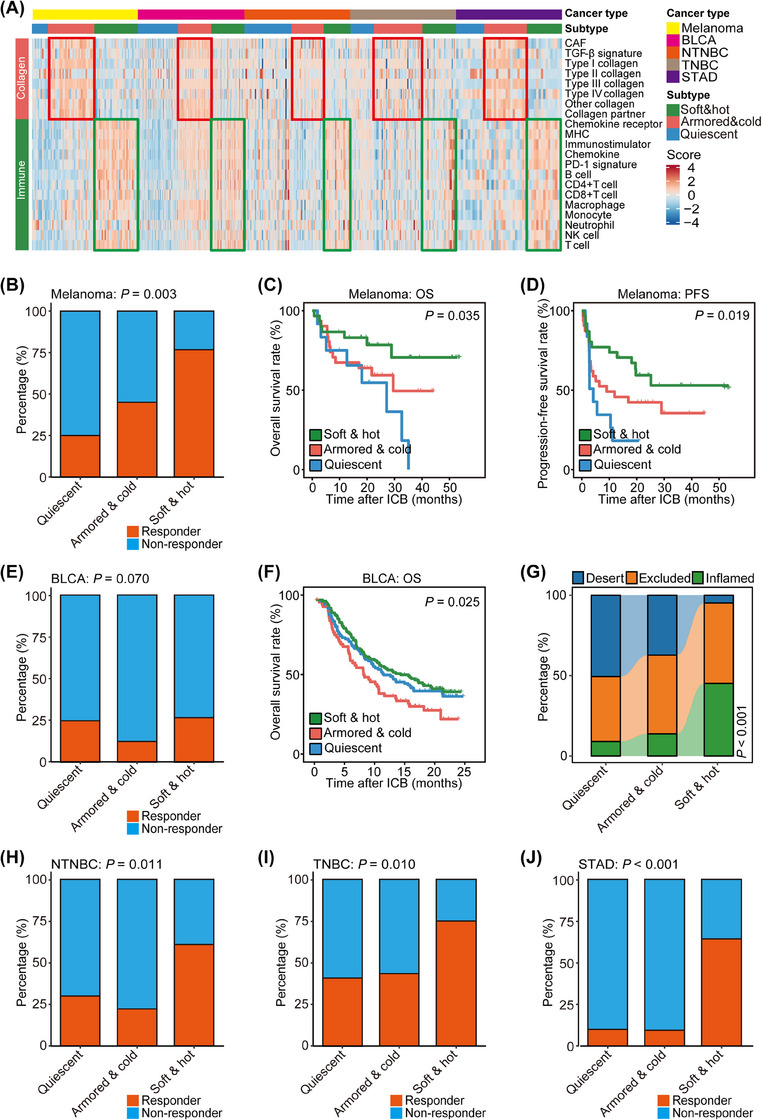
Immuno‐collagenic subtypes predict immune checkpoint blockade response in five cancer types. (A) Heatmap showing scores of collagen‐ and immune‐related gene modules in three immuno‐collagenic subtypes in five cancer types. (B) Difference in the immunotherapeutic response rate in three immuno‐collagenic subtypes in the melanoma cohort (the PRJEB23709 cohort). Significance was calculated using the Fisher exact probability test. (C, D) Difference in overall survival (C) and progression‐free survival (D) in three immuno‐collagenic subtypes in the melanoma cohort. Significance was calculated using the log‐rank test. (E) Difference in the immunotherapeutic response rate in three immuno‐collagenic subtypes in the BLCA cohort (the IMvigor210 cohort). Significance was calculated using the chi‐square test. (F) Difference in overall survival in three immuno‐collagenic subtypes in the BLCA cohort. Significance was calculated using the log‐rank test. (G) Difference in immune subtype distribution in three immuno‐collagenic subtypes in the BLCA cohort. (H) Difference in the immunotherapeutic response rate in three immuno‐collagenic subtypes in the non‐triple negative breast cancer cohort (the merged cohort of the GSE173839 dataset and the GSE194040 dataset). Significance was calculated using the chi‐square test. (I) Difference in the immunotherapeutic response rate in three immuno‐collagenic subtypes in the triple negative breast cancer cohort (the merged cohort of the GSE173839 dataset and the GSE194040 dataset and the MEDI4736 dataset). Significance was calculated using the chi‐square test. (J) Difference in the immunotherapeutic response rate in three immuno‐collagenic subtypes in the STAD cohort (the PRJEB25780 cohort). Significance was calculated using the Fisher exact probability test. Abbreviations: BLCA, bladder urothelial carcinoma; CAF, cancer‐associated fibroblast; ICB, immune checkpoint blockade; MHC, major histocompatibility complex; NTNBC, non‐triple negative breast cancer; OS, overall survival; PD‐1, programmed cell death 1; PFS, progression‐free survival; STAD, stomach adenocarcinoma; TGF‐β, transforming growth factor‐β; TNBC, triple negative breast cancer.
3.3. Collagen inhibition improved ICB response in preclinical models
We hypothesized that inhibiting collagen deposition might enhance the effectiveness of anti‐PD‐1 immunotherapy, particularly in armored & cold tumors known to resist immunotherapy. Our investigation focused on collagen and immune modules within various murine tumors. Surprisingly, analysis of tumor tissues derived from 4T1 cells indicated the highest collagen score and a low immune score (Figure 4A‐B), suggesting that the 4T1 tumor exhibited characteristics of being armored & cold. To inhibit collagen within the TME in vivo, we administered talabostat, a fibroblast activation protein (FAP) inhibitor known for significantly reducing collagen production in CAFs [54], to BALB/c mice bearing 4T1 tumors (Figure 4C). Monitoring the time course of tumor volume and weight revealed that talabostat effectively inhibited tumor growth (Figure 4D‐E). To assess the effect of collagen inhibition on the response to ICB, we administered anti‐mouse PD‐1 antibody to these models. As expected, ICB resulted in a 50% inhibition of tumor growth (Figure 4D‐E). Surprisingly, the combination of talabostat with ICB further suppressed tumor growth, achieving an inhibition rate of approximately 75% (Figure 4D‐E). This suggests that inhibiting collagen improves the efficacy of ICB. Within the TME, talabostat strongly inhibited collagen, whereas anti‐PD‐1 treatment did not (Figure 4F, Supplementary Figure S6). Notably, flow cytometry revealed increased infiltration of CD8+ T cells and reduced myeloid‐derived suppressor cells (MDSCs) following talabostat or anti‐PD‐1 treatment (Figure 4G). The combination of talabostat and anti‐PD‐1 further enhanced T cell infiltration while suppressing MDSC infiltration (Figure 4G). These findings indicate that targeting collagen triggers an inflammatory response in the TIME, thus enhancing the efficacy of ICB in preclinical models.
FIGURE 4.
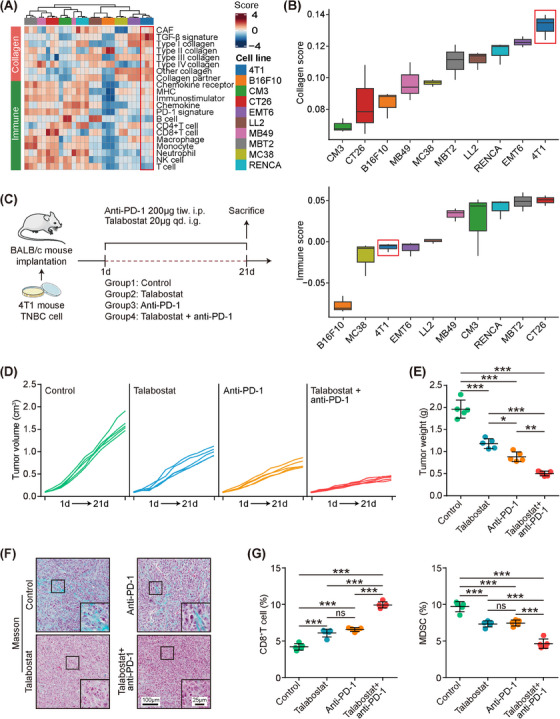
Inhibition of collagen deposition activates tumor immune microenvironment and boosts immune checkpoint blockade efficacy in preclinical models. (A, B) Heatmap and boxplot showing scores of collagen‐ and immune‐related gene modules in three immuno‐collagenic subtypes in mouse tumors (the GSE168846 dataset) receiving no antitumor therapy. (C) Schematic protocol of the combination of PD‐1 monoclonal antibody (mAb) and talabostat in BALB/c mice bearing 4T1 cells. (D) Effect of PD‐1 mAb and talabostat on tumor volume in BALB/c mice bearing 4T1 cells. (E) Effect of PD‐1 mAb and talabostat on tumor weight in BALB/c mice bearing 4T1 cells and quantitative analysis. Data are presented as mean ± standard deviation. Significance was calculated using the ANOVA with Tukey's multiple‐comparison test. ***: P < 0.001. (F) Representative images uncovering collagen areas in tumors from BALB/c mice. (G) CD8+ T cells and MDSCs in tumors from BALB/c mice examined by flow cytometry. Data are presented as mean ± standard deviation. Significance was calculated with one‐way ANOVA with Tukey's multiple‐comparison test. ns: non‐significance, ***: P < 0.001. Abbreviations: ANOVA, analysis of variance; CAF, cancer‐associated fibroblast; ICB, immune checkpoint blockade; MDSC, myeloid‐derived suppressor cell; MHC, major histocompatibility complex; PD‐1, programmed cell death 1; TGF‐β, transforming growth factor‐β; TIME, tumor immune microenvironment; TNBC, triple negative breast cancer.
3.4. Immuno‐collagenic subtypes were conserved across a wide range of cancers
To investigate whether immuno‐collagenic subtypes are prevalent across various cancers, transcriptomic data from 31 solid tumor types were obtained from the TCGA dataset. These data were scored based on 21 functional gene modules as mentioned earlier. GSVA, a model centered on biological pathways, was employed to decipher the immuno‐collagenic landscape. As anticipated, all 31 types of solid tumors could be categorized according to the immuno‐collagenic classification (Figure 5A, Supplementary Figure S7). Taking skin cutaneous melanoma as an example, a detailed analysis revealed distinct features of the three subtypes. The armored & cold subtype, associated with ICB resistance in other cancers, exhibited a lower TMB level (Figure 5B‐C). Notably, there was no discernible difference in TMB between soft & hot subtypes and quiescent types (Figure 5B‐C), suggesting that TMB alone is inadequate for predicting ICB response. Lower TMB has been linked to ICB resistance in melanoma [55], indicating that using TMB as a sole predictor of response is confounded by melanoma subtypes [56]. Similar to melanoma, immuno‐collagenic subtypes were strongly associated with immunotherapy indicators across cancer types but were not associated with TMB levels (Supplementary Figure S8). Specific biomarkers have been proposed for ICB response in certain tumor types, such as microsatellite instability‐high (MSI‐H; or mismatch repair deficient [dMMR]) for colon adenocarcinoma (COAD) [57], the triple‐negative subtype for BRCA [58], and Epstein‐Barr virus (EBV) infection in STAD [59]. Our analysis revealed that MSI‐H was enriched in the soft & hot subtype in COAD, the triple‐negative subtype was enriched in the soft & hot subtype in BRCA, and the EBV rate was highest in the soft & hot subtype in STAD (Figure 5D‐F), supporting the notion that immuno‐collagenic subtypes hold predictive value. Subsequent investigation showed that the immuno‐collagenic subtypes predicted survival benefit in 13 cancer types, with the soft & hot subtype indicating a favorable prognosis (Figure 5G, Supplementary Figure S9). Additionally, we examined the prognostic value of either collagen or immune score across various cancers. The results demonstrated that the predictive value of immuno‐collagenic subtypes for prognosis surpassed that of either collagen or immune score (Supplementary Figure S10). However, immuno‐collagenic subtypes were not associated with prognosis in several challenging‐to‐treat tumor types, including pancreatic adenocarcinoma (PAAD), glioblastoma multiforme (GBM), and liver hepatocellular carcinoma (LIHC) (Supplementary Figure S9).
FIGURE 5.
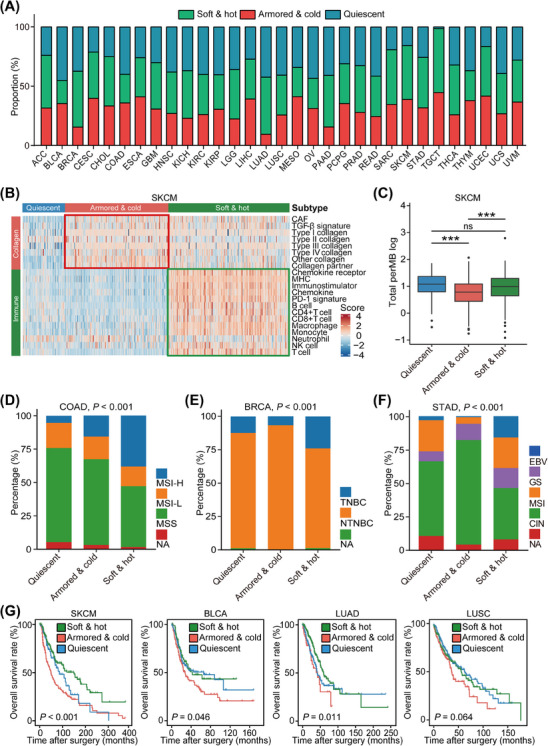
Immuno‐collagenic subtypes across a broad array of cancer transcriptomic data. (A) Stacked bar plots depicting the relative proportion of each immuno‐collagenic subtype in all solid cancers queried. The proportion of all tumors of a particular cancer type belonging to individual immuno‐collagenic subtypes was calculated. (B) Heatmap showing scores of collagen‐ and immune‐related gene modules in three immuno‐collagenic subtypes in the TCGA‐SKCM cohort. (C) Difference in TMB levels in three immuno‐collagenic subtypes in the TCGA‐SKCM cohort. Horizontal lines in the boxplots represent the median value, the lower and upper hinges correspond to the first and third quartiles, respectively. Significance was calculated using the ANOVA with Tukey's multiple‐comparison test. ns: non‐significance, ***: P < 0.001. (D) Difference in microsatellite instability status in three immuno‐collagenic subtypes in the TCGA‐COAD cohort. Significance was calculated using the chi‐square test. (E) Difference in PAM50 subtypes in three immuno‐collagenic subtypes in the TCGA‐BRCA cohort. Significance was calculated using the chi‐square test. (F) Difference in molecular subtypes in three immuno‐collagenic subtypes in the TCGA‐STAD cohort. Significance was calculated using the chi‐square test. (G) Difference in overall survival in three immuno‐collagenic subtypes in the TCGA‐SKCM, TCGA‐BLCA, TCGA‐LUAD, and TCGA‐LUSC cohorts. Significance was calculated using the log‐rank test. Abbreviations: ACC, adrenocortical carcinoma; ANOVA, analysis of variance; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CAF, cancer‐associated fibroblast; CIN, chromosomally unstable; CESC, cervical & endocervical cancer; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; EBV, Epstein‐Barr virus; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; GS, genomically stable; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; TGF‐β, transforming growth factor‐β; MESO, mesothelioma; MHC, major histocompatibility complex; MSI, microsatellite instability; MSI‐H, microsatellite instability‐high; MSI‐L, microsatellite instability‐low; MSS, microsatellite stability; NTNBC, non‐triple negative breast cancer; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PAM50, Prediction Analysis of Microarray 50; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TCGA, The Cancer Genome Atlas; TGCT, testicular germ cell tumor; THCA, thyroid carcinoma; THYM, thymoma; TNBC, triple negative breast cancer; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma.
We collected a total of 1,012 patient samples from 10 cancer types to investigate the suitable cancer types for immuno‐collagenic subtypes. Our histopathological analysis revealed significant variation in collagen infiltration among these cancer types. Notably, PAAD exhibited the highest infiltration, whereas glioma showed the lowest (Figure 6A‐B). These distinct characteristics in collagen expression created challenges in stratification, potentially explaining why the immuno‐collagenic subtypes did not associate with prognosis in these tumor types. To refine our subtyping, we utilized Masson, HE, and anti‐PD‐L1 staining. Generally, samples displaying high levels of both TIICs (TIIC‐high) and collagen (collagen‐high) were limited (Figure 6C, Supplementary Figure S11). In specific cancers such as NSCLC, BLCA, ovarian serous cystadenocarcinoma (OV), cervical & endocervical cancer (CESC), and COAD, the immuno‐collagenic subtypes accurately predicted prognosis and exhibited associations with PD‐L1 expression or dMMR status (Figure 6D‐G, Supplementary Figure S11). Conversely, in BRCA and STAD, while these subtypes did not predict prognosis, they still associated with PD‐L1 expression, the triple‐negative subtype, or dMMR (Supplementary Figure S12). Notably, collagen score alone predicted prognosis in only two cancer types, while TIIC score alone predicted prognosis in 5 cancer types (Supplementary Figure S13), emphasizing the significant prognostic value of the immuno‐collagenic subtypes. These subtypes were validated in a comprehensive pan‐cancer analysis and were associated with immuno‐activity and prognosis in 13 cancer types (Supplementary Table S11). Collectively, our TCGA analysis and clinical samples provide a comprehensive framework for subtype stratification and prognostic prediction across various common cancer types.
FIGURE 6.
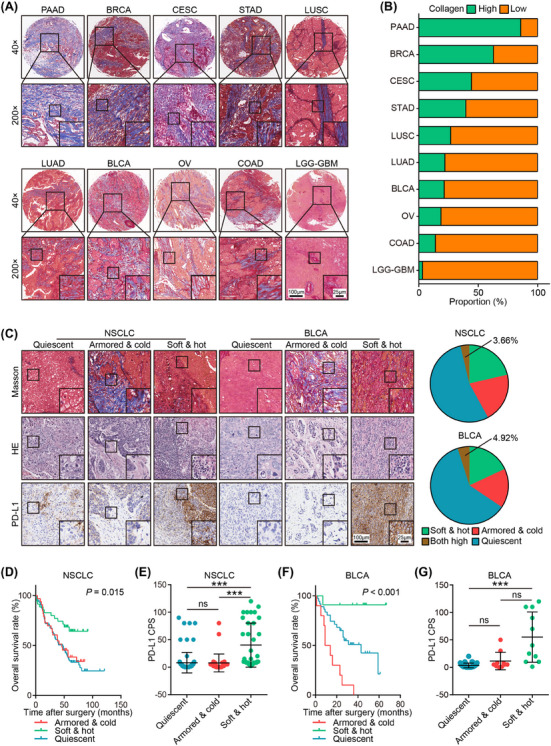
Pan‐cancer analysis of collagen distribution landscape and immuno‐collagenic subtypes. (A) Representative images uncovering various collagen distribution in 10 common cancer types. (B) Proportion of low and high collagen distribution in 10 common cancer types. (C) Representative images uncovering collagen area, TIIC infiltration, and PD‐L1expression in tumors of various subtypes in NSCLC (the merged cohort of the HLugA180Su06 and HLugS180Su01 cohorts) and BLCA (the HBlaU079Su01 cohort), and the proportion of four subtypes divided by collagen area and TIIC infiltration. The proportion of co‐high collagen and TIIC was quietly limited. (D) Difference in overall survival in three immuno‐collagenic subtypes in the in‐house NSCLC cohort. Significance was calculated using the log‐rank test. (E) Difference in PD‐L1 expression in three immuno‐collagenic subtypes in the in‐house NSCLC cohort. Data are presented as mean ± standard deviation. Significance was calculated using the Kruskal‐Wallis test with Dunn's multiple‐comparison test. ns: non‐significance, ***: P < 0.001. (F) Difference in overall survival in three immuno‐collagenic subtypes in the in‐house BLCA cohort. Significance was calculated using the log‐rank test. (G) Difference in PD‐L1 expression in three immuno‐collagenic subtypes in the in‐house BLCA cohort. Data are presented as mean ± standard deviation. Significance was calculated using the Kruskal‐Wallis test with Dunn's multiple‐comparison test. ns: non‐significance, *: P < 0.05, ***: P < 0.001. Abbreviations: BLCA, bladder urothelial carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical & endocervical cancer; COAD, colon adenocarcinoma; CPS, Combined Positive Score; GBM, glioblastoma multiforme; HE, hematoxylin and eosin; LGG, brain lower grade glioma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; NSCLC, non‐small‐cell lung cancer; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PD‐L1, programmed cell death 1 ligand 1; STAD, stomach adenocarcinoma; TIIC, tumor‐infiltrating immune cells.
3.5. Identification of B7‐H3 as a potential target for ICB response
To determine potential therapeutic targets for armored & cold tumor types, we conducted a pan‐cancer analysis to assess the expression of various drug targets. To enhance clinical relevance, we focused on targets from cancer drugs either in use or entering phase II clinical trials. Intriguingly, targets such as fibroblast growth factor receptor (FGFR), platelet‐derived growth factor receptor (PDGFR), and immune inhibition‐associated targets such as CD276 (B7‐H3) and angiogenesis‐related targets exhibited high expression in armored & cold subtype, contrasting with lower expression in quiescent or soft & hot subtype (Figure 7A). These findings support our subtype categorization. Among these targets, B7‐H3 demonstrated the most significant association with subtypes in our pan‐cancer analysis (Figure 7A, Supplementary Figure S14), exhibiting consistent associations with collagen signatures across various cancer types (Figure 7B). Notably, B7‐H3 was consistently overexpressed in tumor tissues across different cancer types (Figure 7C), and its high expression was associated with poor prognosis in the pan‐cancer analysis (Figure 7D). Validation using our in‐house BLCA cohort confirmed the upregulation of B7‐H3 in tumor tissues, especially in armored & cold tumors (Figure 7E‐F). Moreover, high B7‐H3 expression was associated with poor prognosis in this cohort (Figure 7G).
FIGURE 7.
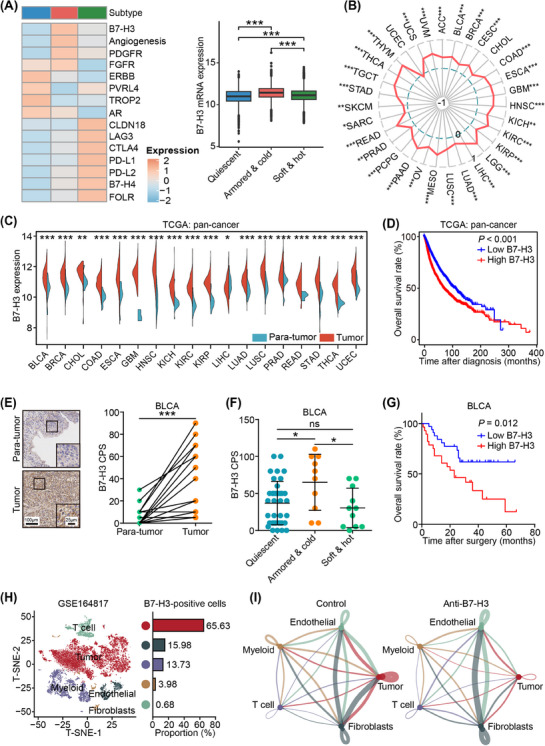
B7‐H3 as a therapeutic target in armored & cold tumors. (A) Heatmap showing expression levels of 15 drug targets based on their mean expression level in three immuno‐collagenic subtypes in pan‐cancer analysis (left), and boxplot showing high expression of B7‐H3 in armored & cold tumors in pan‐cancer analysis. Horizontal lines in the boxplots represent the median value, and the lower and upper hinges correspond to the first and third quartiles, respectively. Significance was calculated using the ANOVA with Tukey's multiple‐comparison test. ns: non‐significance, *: P < 0.05, **: P < 0.01. (B) Radar plot showing the positive correlation between B7‐H3 expression and collagen score in most cancer types. Significance was calculated using Pearson test. (C) Expression of B7‐H3 in tumor and nontumor tissues in The Cancer Genome Atlas (TCGA) dataset. Cancer types with tumor or nontumor samples over 5 are showed. Significance was calculated using the Student t test. (D) Prognostic value of B7‐H3 in pan‐cancer samples from the TCGA dataset. Significance was calculated using the log‐rank test. (E) Representative images uncovering B7‐H3 expression in tumor and non‐tumor samples in the in‐house BLCA cohort and quantitative analysis. Significance was calculated using the paired Student t test. ***: P < 0.001. (F) Difference in B7‐H3 expression in three immuno‐collagenic subtypes in the in‐house BLCA cohort. Data are presented as mean ± standard deviation. Significance was calculated using the one‐way ANOVA with Tukey's multiple‐comparison test. ns: non‐significance, *: P < 0.05. (G) Difference in overall survival in patients with low and high B7‐H3 expression in the in‐house BLCA cohort. Significance was calculated using the log‐rank test. (H) t‐SNE visualization of cell types annotated by classical gene markers, and the proportion of B7‐H3‐positive cells in various cell types. (I) Cell‐cell communications analysis in the control and anti‐B7‐H3 therapy groups. Abbreviations: B7‐H3, B7 homolog 3; TCGA, The Cancer Genome Atlas; ANOVA, analysis of variance; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; STAD, stomach adenocarcinoma; THCA, thyroid carcinoma; UCEC, uterine corpus endometrial carcinoma; CPS, Combined Positive Score.
Further investigation into the alterations within the tumor cells and TME following anti‐B7‐H3 treatment involved analyzing primary scRNA‐seq data from murine head and neck squamous cell carcinoma tissues treated with either vehicles or anti‐B7‐H3 antibodies for 4 weeks [46]. In the tumor tissues, B7‐H3‐positive cells primarily constituted tumor cells (Figure 7H). Strikingly, anti‐B7‐H3 therapy not only reduced B7‐H3 expression in tumor cells but also suppressed their proliferation activity (Supplementary Figure S15A‐B). Additionally, targeting B7‐H3 increased the populations of naive and cytotoxic T cells while decreasing exhausted T cells (Supplementary Figure S15C‐E), indicating an activation of the immuno‐cold TME. Of equal significance was our analysis of cell‐cell communication, revealing that anti‐B7‐H3 therapy notably impeded communication between tumor cells and fibroblasts (Figure 7I, Supplementary Figure S16). Remarkably, these communications in the control group primarily involved collagen receptors (Supplementary Table S12), suggesting that anti‐B7‐H3 therapy hindered collagen‐mediated tumor progression. Overall, these findings underscore the association of B7‐H3 with immuno‐collagenic subtypes and its potential as a promising target to enhance ICB response.
4. DISCUSSION
Herein, we propose a framework that integrates the collagen features with immune signatures to predict responses to ICB treatment. Tumors obtained from a large cohort of patients receiving ICB were categorized into three distinct subtypes: soft & hot (low collagen activity and high immune infiltration), armored & cold (high collagen activity and low immune infiltration), and quiescent (low collagen activity and immune infiltration). Among these subtypes, armored & cold tumors exhibited the highest response to ICB treatment across multiple cancer types. In preclinical models, collagen inhibition improved response to ICB treatment. Additionally, these subtypes were validated in a comprehensive pan‐cancer analysis and were associated with immuno‐activity and prognosis in 13 cancer types. Moreover, B7‐H3, a clinically available drug target, was strongly expressed in armored & cold tumors and was associated with poor prognosis, which could be a potential candidate target for the therapy of armored & cold tumors.
Collagen, a vital component of the ECM, is anomalously deposited in the TME, encapsulating tumor cell clusters. This action has several consequences: (1) it obstructs physical contact between immune cells and tumor cells [60]; (2) it mechanically restrains the cytotoxicity of immune cells [61]; and (3) it releases bioactive fragments that regulate immune cell activity [62]. Notably, specific collagen markers have been linked to immunotherapy response and prognosis [63]. Given the close interaction between collagen and immune cells, incorporating collagen signatures into the classical ICB response predictor emerges as an enticing strategy for enhancing ICB response prediction. In this study, tumors were classified into three subtypes: soft & hot, armored & cold, and quiescent, which effectively predicted ICB response. To validate the hypothesis that collagen impedes the inflammatory TIME, we demonstrated that inhibiting collagen using an FAP inhibitor sensitized ICB in preclinical models, proposing a potential combination therapy strategy for ICB‐resistant tumors. While talabostat revealed alterations in collagen‐induced TIME, understanding the detailed molecular mechanism underlying this combination therapy necessitates further investigation. Additionally, an unbiased pan‐cancer drug target analysis confirmed the collagen‐immune axis. Targets such as fibroblast activation‐associated FGFR, PDGFR, and immune inhibition‐associated B7‐H3, angiogenesis associated with immuno‐collagenic subtypes. Despite validating only the most highly associated target, B7‐H3, we assert that targeting the collagen‐immune axis through clinically available targets holds clinical relevance and presents a potential combination therapy strategy for ICB‐resistant tumors.
It is crucial to note that quiescent tumors, resistant to ICB therapy, exhibited poor prognosis in certain cancer types, with high expression of erb‐b2 receptor tyrosine kinase (ERBB) signaling and poliovirus receptor‐related 4 (PVRL4). ERBB signaling is recognized for promoting immune escape [64], while PVRL4 associates with cancer progression and poor prognosis [65]. However, whether ERBB signaling and PVRL4 sustain malignancy and immune evasion in quiescent cancers demands further exploration. In soft & hot tumors, aside from classical immune checkpoints, we observed high expression of folate receptor, which represents an appealing therapeutic target in tumors [66]. To succinctly portray the impact of immuno‐collagenic subtypes on clinical treatment, we summarized the molecular and clinical features in Figure 8.
FIGURE 8.
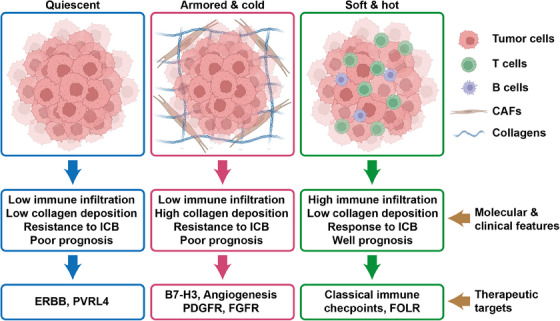
Schematic description of the features associated with the three pan‐cancer immuno‐collagenic subtypes. A framework was developed to stratify tumors according to collagen deposition and immune activity. Soft & hot tumors are accompanied by high immune infiltration and low collagen deposition, which showed good prognosis and responded to immune checkpoint blockade (ICB) therapy. These tumors highly express classical immune checkpoints and folate receptor. Armored & cold tumors are accompanied by low immune infiltration and high collagen deposition, which showed poor prognosis and resistance to ICB therapy. These tumors highly express B7‐H3, angiogenesis‐related markers, platelet‐derived growth factor receptor, and fibroblast growth factor receptor. Quiescent tumors are accompanied by both low immune infiltration and low collagen deposition, which showed poor prognosis and resistance to ICB therapy. These tumors highly express erb‐b2 receptor tyrosine kinase and PVRL4. Abbreviations: B7‐H3, B7 homolog 3; ERBB, erb‐b2 receptor tyrosine kinase; FGFR, fibroblast growth factor receptor; FOLR, folate receptor; ICB, immune checkpoint blockade; PDGFR, platelet‐derived growth factor receptor; PVRL4, poliovirus receptor‐related 4.
Our study yielded a striking discovery: the existence of proposed subtypes across all solid tumor types, associating with the prognosis in 13 tumor types. These universal subtypes likely stem from the widespread expression of collagen in solid tumors. Unlike specific cancer‐centric biomarkers such as TMB or MSI associated with ICB response, these immuno‐collagenic subtypes assist in identifying ICB‐responsive patients in various common cancer types, encompassing the majority of patients with cancer. This research demonstrates the interrelationship between collagen and immune cell infiltrations. Notably, while 13 tumor types have shown these associations, large‐scale clinical validation is pending to determine the predictive value of these subtypes.
Current biomarkers typically rely on transcriptomic and genomic data to predict ICB response. However, performing deep genome sequencing and transcriptome sequencing for every clinical sample is impractical due to the high economic burden on patients. In pursuit of clinical relevance, we revisited transcriptome‐based immuno‐collagenic subtypes and found that a simple histochemistry method can replicate these subtypes with satisfactory similarity. This simplified approach transforms the bioinformatics‐driven patient stratification into an affordable pathological task, manageable by an experienced pathologist. Together, this provides a universal, accurate, and user‐friendly predictive biomarker for ICB response across cancer types, benefiting a broad range of patients with cancer undergoing ICB treatment.
Although this study sheds light on the crucial association between collagen and anti‐tumor immune responses in the TME, several limitations warrant acknowledgment. Our primary analysis extracted TME features from heterogeneous bulk RNA‐seq data, necessitating further validation using high‐resolution techniques such as scRNA‐seq and spatial transcriptomics to mitigate confounding variables from the heterogeneous TME. Furthermore, the mechanisms responsible for the negative correlation between collagen and immune cell infiltration remain largely unclear. To effectively translate these findings, standard clinical trials across different cancer types are imperative to scrutinize the targets highlighted in this research.
5. CONCLUSIONS
In summary, leveraging the biological association between collagen deposition and immune activity in the TME, we developed a framework with exceptional predictive value for stratifying patients who respond favorably to ICB therapy. Additionally, it proposes potential therapeutic strategies for those resistant to ICB therapy. Furthermore, immuno‐collagenic subtypes surpass previous predictive biomarkers and exhibit consistency across tumor types. Moreover, the ease of measuring Masson staining and HE‐based detection of TIIC score sets the stage for the practical translation of these findings.
DECLARATIONS
AUTHOR CONTRIBUTIONS
Yongmei Yin, Yan Zhang, Yichao Zhu, and Yunlong Yang conceived the study and participated in the study design, performance, coordination, and project supervision. Jie Mei, Yun Cai, Rui Xu, and Yuxin Shi collected the public data and conducted the bioinformatics analysis. Yongmei Yin, Yan Zhang, Jie Mei, Qing Li, Junying Xu, Yuxin Shi, Di Li, and Shuai Liang collected the tumor samples and the clinical data. Jie Mei, Rui Xu, Qing Li, Jiahui Chu, Zhiwen Luo, Yaying Sun, Yuxin Shi, Ying Jiang, Jiayu Liu, Zhiwen Qian, Jiaofeng Zhou, Mengyun Wan performed in vivo experiments and the tissues staining. Jie Mei, Yun Cai, Rui Xu, and Qing Li wrote the draft. Yichao Zhu and Yunlong Yang revised the manuscript. Yongmei Yin, Yan Zhang, Yichao Zhu, and Junying Xu received financial supports. All authors approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
There are no competing interests.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Ethical approval for the use of TMAs was granted by the Clinical Research Ethics Committee in Outdo Biotech (No. SHYJS‐CP‐1910002, SHYJS‐CP‐2001007, SHYJS‐CP‐1910016, SHYJS‐CP‐1801003, SHYJS‐CP‐1804003, SHYJS‐CP‐1707004, SHYJS‐CP‐1807017, SHYJS‐CP‐1810010, SHYJS‐CP‐1804019, and SHYJS‐CP‐1710003). Ethical approval for the collection of the immunotherapy cohort from The Affiliated Wuxi People's Hospital of Nanjing Medical University was granted by the Clinical Research Ethics Committee in Nanjing Medical University (No. 2022‐383). Ethical approval for the collection of the immunotherapy cohort from Xuzhou Central Hospital was granted by the Clinical Research Ethics Committee in Xuzhou Central Hospital (No. XZXY‐LK‐20230610‐086). All animal experiments were approved by the Laboratory Animal Ethics Committee at Nanjing Medical University (IACUC‐2305039).
Supporting information
Supporting information
ACKNOWLEDGEMENTS
We thank Bullet Edits Limited for the linguistic editing and proofreading of the manuscript. This work was supported by the National Key Research and Development Program of China (ZDZX2017ZL‐01), the National Natural Science Foundation of China (82073194, 81972484), the High‐level Innovation Team of Nanjing Medical University (JX102GSP201727), the Precision Medicine Project of Wuxi Municipal Health Commission (J202106), and the Project of Wuxi Medical Center of Nanjing Medical University (WMCC202319).
Mei J, Cai Y, Xu R, Li Q, Chu J, Luo Z, et al. Conserved immuno‐collagenic subtypes predict response to immune checkpoint blockade. Cancer Commun. 2024;44:554–575. 10.1002/cac2.12538
Jie Mei, Yun Cai, Rui Xu, and Qing Li contributed equally to this work.
Contributor Information
Yunlong Yang, Email: yunlongyang@fudan.edu.cn.
Yichao Zhu, Email: zhuyichao@njmu.edu.cn.
Yan Zhang, Email: fuyou2007@126.com.
Yongmei Yin, Email: ymyin@njmu.edu.cn.
DATA AVAILABILITY STATEMENT
All data are available from the authors upon reasonable request.
REFERENCES
- 1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–249. [DOI] [PubMed] [Google Scholar]
- 2. Keenan TE, Burke KP, Van Allen EM. Genomic correlates of response to immune checkpoint blockade. Nature medicine. 2019;25(3):389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018;8(9):1069–1086. [DOI] [PubMed] [Google Scholar]
- 4. Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol. 2017;14(8):463–482. [DOI] [PubMed] [Google Scholar]
- 5. Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, et al. PD‐L1 as a biomarker of response to immune‐checkpoint inhibitors. Nat Rev Clin Oncol. 2021;18(6):345–362. [DOI] [PubMed] [Google Scholar]
- 6. Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ, et al. Pembrolizumab versus chemotherapy for previously untreated, PD‐L1‐expressing, locally advanced or metastatic non‐small‐cell lung cancer (KEYNOTE‐042): a randomised, open‐label, controlled, phase 3 trial. Lancet. 2019;393(10183):1819–1830. [DOI] [PubMed] [Google Scholar]
- 7. Brahmer JR, Rodriguez‐Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Health‐related quality‐of‐life results for pembrolizumab versus chemotherapy in advanced, PD‐L1‐positive NSCLC (KEYNOTE‐024): a multicentre, international, randomised, open‐label phase 3 trial. Lancet Oncol. 2017;18(12):1600–1609. [DOI] [PubMed] [Google Scholar]
- 8. Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non‐small‐cell lung cancer (OAK): a phase 3, open‐label, multicentre randomised controlled trial. Lancet. 2017;389(10066):255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous‐Cell Non‐Small‐Cell Lung Cancer. N Engl J Med. 2015;373(2):123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, Meatchi T, et al. Histopathologic‐based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol. 2011;29(6):610–618. [DOI] [PubMed] [Google Scholar]
- 11. Pages F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391(10135):2128–2139. [DOI] [PubMed] [Google Scholar]
- 12. Galon J, Costes A, Sanchez‐Cabo F, Kirilovsky A, Mlecnik B, Lagorce‐Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. [DOI] [PubMed] [Google Scholar]
- 13. Galon J, Angell HK, Bedognetti D, Marincola FM. The continuum of cancer immunosurveillance: prognostic, predictive, and mechanistic signatures. Immunity. 2013;39(1):11–26. [DOI] [PubMed] [Google Scholar]
- 14. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–795. [DOI] [PubMed] [Google Scholar]
- 15. Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman‐Saint Victor C, et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell. 2016;167(6):1540–1554 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science. 2015;348(6230):124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, et al. Biomolecular network reconstruction identifies T‐cell homing factors associated with survival in colorectal cancer. Gastroenterology. 2010;138(4):1429–1440. [DOI] [PubMed] [Google Scholar]
- 18. Subramanian M, Kabir AU, Barisas D, Krchma K, Choi K. Conserved angio‐immune subtypes of the tumor microenvironment predict response to immune checkpoint blockade therapy. Cell Rep Med. 2023;4(1):100896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan‐tumor genomic biomarkers for PD‐1 checkpoint blockade‐based immunotherapy. Science. 2018;362(6411):eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Caligiuri G, Tuveson DA. Activated fibroblasts in cancer: Perspectives and challenges. Cancer Cell. 2023;41(3):434–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cox TR. The matrix in cancer. Nat Rev Cancer. 2021;21(4):217–238. [DOI] [PubMed] [Google Scholar]
- 22. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer‐associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20(1):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun X, Wu B, Chiang HC, Deng H, Zhang X, Xiong W, et al. Tumour DDR1 promotes collagen fibre alignment to instigate immune exclusion. Nature. 2021;599(7886):673–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kolesnikoff N, Chen CH, Samuel MS. Interrelationships between the extracellular matrix and the immune microenvironment that govern epithelial tumour progression. Clin Sci (Lond). 2022;136(5):361–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28(11):1747–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cancer Genome Atlas Research N . Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Georgiev P, Muise ES, Linn DE, Hinton MC, Wang Y, Cai M, et al. Reverse Translating Molecular Determinants of Anti‐Programmed Death 1 Immunotherapy Response in Mouse Syngeneic Tumor Models. Mol Cancer Ther. 2022;21(3):427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pusztai L, Yau C, Wolf DM, Han HS, Du L, Wallace AM, et al. Durvalumab with olaparib and paclitaxel for high‐risk HER2‐negative stage II/III breast cancer: Results from the adaptively randomized I‐SPY2 trial. Cancer Cell. 2021;39(7):989–998 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wolf DM, Yau C, Wulfkuhle J, Brown‐Swigart L, Gallagher RI, Lee PRE, et al. Redefining breast cancer subtypes to guide treatment prioritization and maximize response: Predictive biomarkers across 10 cancer therapies. Cancer Cell. 2022;40(6):609–623 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim ST, Cristescu R, Bass AJ, Kim KM, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD‐1 inhibition in metastatic gastric cancer. Nature Med. 2018;24(9):1449–1458. [DOI] [PubMed] [Google Scholar]
- 31. Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, et al. Distinct Immune Cell Populations Define Response to Anti‐PD‐1 Monotherapy and Anti‐PD‐1/Anti‐CTLA‐4 Combined Therapy. Cancer Cell. 2019;35(2):238–255 e6. [DOI] [PubMed] [Google Scholar]
- 32. Cho JW, Hong MH, Ha SJ, Kim YJ, Cho BC, Lee I, et al. Genome‐wide identification of differentially methylated promoters and enhancers associated with response to anti‐PD‐1 therapy in non‐small cell lung cancer. Exp Mol Med. 2020;52(9):1550–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jung H, Kim HS, Kim JY, Sun JM, Ahn JS, Ahn MJ, et al. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat Commun. 2019;10(1):4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blenman KRM, Marczyk M, Karn T, Qing T, Li X, Gunasekharan V, et al. Predictive Markers of Response to Neoadjuvant Durvalumab with Nab‐Paclitaxel and Dose‐Dense Doxorubicin/Cyclophosphamide in Basal‐Like Triple‐Negative Breast Cancer. Clin Cancer Res. 2022;28(12):2587–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD‐L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gillespie M, Jassal B, Stephan R, Milacic M, Rothfels K, Senff‐Ribeiro A, et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022;50(D1):D687–D692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC bioinformatics. 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26(12):1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schwartz LH, Litiere S, de Vries E, Ford R, Gwyther S, Mandrekar S, et al. RECIST 1.1‐Update and clarification: From the RECIST committee. Eur J Cancer. 2016;62:132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA‐Seq. Nat Methods. 2008;5(7):621–628. [DOI] [PubMed] [Google Scholar]
- 43. Cancer Genome Atlas N . Genomic Classification of Cutaneous Melanoma. Cell. 2015;161(7):1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mei J, Cai Y, Zhu H, Jiang Y, Fu Z, Xu J, et al. High B7‐H3 expression with low PD‐L1 expression identifies armored‐cold tumors in triple‐negative breast cancer. NPJ Breast Cancer. 2024;10(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mao W, Cai Y, Chen D, Jiang G, Xu Y, Chen R, et al. Statin shapes inflamed tumor microenvironment and enhances immune checkpoint blockade in non‐small cell lung cancer. JCI Insight. 2022;7(18):e161940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang C, Li Y, Jia L, Kim Jk, Li J, Deng P, et al. CD276 expression enables squamous cell carcinoma stem cells to evade immune surveillance. Cell Stem Cell. 2021;28(9):1597–1613.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single‐cell data with Harmony. Nat Methods. 2019;16(12):1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Couzin‐Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342(6165):1432–1433. [DOI] [PubMed] [Google Scholar]
- 49. Howlader N, Forjaz G, Mooradian MJ, Meza R, Kong CY, Cronin KA, et al. The Effect of Advances in Lung‐Cancer Treatment on Population Mortality. N Engl J Med. 2020;383(7):640–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Calles A, Aguado G, Sandoval C, Alvarez R. The role of immunotherapy in small cell lung cancer. Clin Transl Oncol. 2019;21(8):961–976. [DOI] [PubMed] [Google Scholar]
- 51. Principe DR, Timbers KE, Atia LG, Koch RM, Rana A. TGFbeta Signaling in the Pancreatic Tumor Microenvironment. Cancers. 2021;13(20):5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Racle J, de Jonge K, Baumgaertner P, Speiser DE, Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife. 2017;6:e26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mei J, Cai Y, Xu R, Zhu Y, Zhao X, Zhang Y, et al. Protocol to identify novel immunotherapy biomarkers based on transcriptomic data in human cancers. STAR Protocols. 2023;4(2):102258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cunningham CC. Talabostat. Expert Opin Investig Drugs. 2007;16(9):1459–1465. [DOI] [PubMed] [Google Scholar]
- 55. Eroglu Z, Zaretsky JM, Hu‐Lieskovan S, Kim DW, Algazi A, Johnson DB, et al. High response rate to PD‐1 blockade in desmoplastic melanomas. Nature. 2018;553(7688):347–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby‐Arnon L, et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med. 2019;25(12):1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Andre T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in Microsatellite‐Instability‐High Advanced Colorectal Cancer. N Engl J Med. 2020;383(23):2207–2218. [DOI] [PubMed] [Google Scholar]
- 58. Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im SA, Yusof MM, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple‐negative breast cancer (KEYNOTE‐355): a randomised, placebo‐controlled, double‐blind, phase 3 clinical trial. Lancet. 2020;396(10265):1817–1828. [DOI] [PubMed] [Google Scholar]
- 59. Kubota Y, Kawazoe A, Sasaki A, Mishima S, Sawada K, Nakamura Y, et al. The Impact of Molecular Subtype on Efficacy of Chemotherapy and Checkpoint Inhibition in Advanced Gastric Cancer. Clin Cancer Res. 2020;26(14):3784–3790. [DOI] [PubMed] [Google Scholar]
- 60. Salmon H, Franciszkiewicz K, Damotte D, Dieu‐Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122(3):899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. O'Connor RS, Hao X, Shen K, Bashour K, Akimova T, Hancock WW, et al. Substrate rigidity regulates human T cell activation and proliferation. J Immunol. 2012;189(3):1330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hurkmans DP, Jensen C, Koolen SLW, Aerts J, Karsdal MA, Mathijssen RHJ, et al. Blood‐based extracellular matrix biomarkers are correlated with clinical outcome after PD‐1 inhibition in patients with metastatic melanoma. J ImmunoTher Cancer. 2020;8(2):e001193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kumagai S, Koyama S, Nishikawa H. Antitumour immunity regulated by aberrant ERBB family signalling. Nat Rev Cancer. 2021;21(3):181–197. [DOI] [PubMed] [Google Scholar]
- 65. MR M, Cabaud O, Josselin E, Finetti P, Castellano R, Farina A, et al. Nectin‐4: a new prognostic biomarker for efficient therapeutic targeting of primary and metastatic triple‐negative breast cancer. Ann Oncol. 2017;28(4):769–776. [DOI] [PubMed] [Google Scholar]
- 66. Matulonis UA, Lorusso D, Oaknin A, Pignata S, Dean A, Denys H, et al. Efficacy and Safety of Mirvetuximab Soravtansine in Patients With Platinum‐Resistant Ovarian Cancer With High Folate Receptor Alpha Expression: Results From the SORAYA Study. J Clin Oncol. 2023;41(13):2436–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
All data are available from the authors upon reasonable request.