

Abstract
Intracellular surveillance for systemic microbial components during homeostasis and infections governs host physiology and immunity. However, a long-standing question is how circulating microbial ligands become accessible to intracellular receptors. Here, we show a role for host-derived extracellular vesicles (EVs) in this process; human and murine plasma- and cell culture-derived EVs have an intrinsic capacity to bind bacterial lipopolysaccharide (LPS). Remarkably, circulating host EVs capture blood-borne LPS in vivo, and the LPS-laden EVs confer cytosolic access for LPS, triggering noncanonical inflammasome activation of GSDMD and pyroptosis. Mechanistically, the interaction between the lipid bilayer of EVs and the lipid A of LPS underlies EV capture of LPS, and the intracellular transfer of LPS by EVs is mediated by CD14. Overall, this study demonstrates that EVs capture and escort systemic LPS to the cytosol licensing inflammasome responses, uncovering EVs as a previously unrecognized link between systemic microbial ligands and intracellular surveillance.
Introduction
The commensal and pathogenic microbial products commonly reach the circulation and, subsequently, the intracellular space1–5. Intracellular surveillance by pattern recognition receptors (PRR) for systemic microbial components elicits specialized responses that play crucial homeostatic and inflammatory roles in shaping immunity, tissue repair, metabolism, and brain functions1–4. Whereas these intracellular PRRs and their signaling have been relatively well-defined, how circulating microbial products access the host cell cytosol remains least understood.
A prototypical microbial product that translocates to the cytosol is bacterial lipopolysaccharide (LPS). The cytosolic localization of LPS is consequential as it evokes responses distinct from the predominantly transcriptional responses evoked by extracellular LPS through TLR46–8. Cytosolic LPS is sensed by the noncanonical inflammasome (comprising caspase-4 in humans and caspase-11 in rodents), triggering, in turn, the pore-forming activity of gasdermin D (GSDMD) that culminates in lytic cell death (pyroptosis) and IL-1β and IL-18 maturation9–13. During infections, bacterial invasion of the cytosol and outer membrane vesicles (OMVs) secreted by bacteria enable cytosolic localization of LPS8,14. However, it is amply evident that in addition to bacteria- or OMV-bound forms, free LPS is also present in circulation in many instances15–17. Bacterial lysis by complement, antimicrobial peptides, and antibiotics liberates LPS from bacteria into the circulation17–19. Gut microbiome-derived systemic LPS is frequently noted in patients with HIV, inflammatory bowel disease, leaky gut and liver diseases15,17,20.
Such bacteria-free blood-borne LPS attains cytosolic localization and activates caspase-11-GSDMD-mediated pyroptosis9,10. Moreover, the cytosolic translocation of free LPS occurs exclusively in vivo but not in vitro9,10,14. Whereas free or purified LPS cannot passively diffuse across the plasma/endosomal membranes and become cytosolic in cultured cells in vitro, it does reach the cytosol robustly in mice following systemic administration9,14,21. Although intracellular localization of bacteria-free LPS and its pathophysiological significance have been known, how circulating LPS gains cytosolic access, triggering caspase-11 activation exclusively in vivo, remains a long-standing question. Intriguingly, this study found that host-derived extracellular vesicles capture systemic LPS and transfer it to the cytosol licensing noncanonical inflammasome responses, uncovering a fundamental paradigm in cytosolic immune surveillance.
Results
Circulating host-derived EVs capture LPS during endotoxemia
From the in vivo-specific nature of cytosolic translocation of free or purified LPS, it can be reasoned that certain in vivo-rich host factors enable the cytosolic access of LPS. Host-derived extracellular vesicles (EVs) are membrane-encapsulated structures abundantly released by living cells and are emerging as a crucial mode of intercellular transfer of biomolecules22–25. EVs are highly abundant in the blood (>1010–11 particles/ml), and circulating EVs come into intimate contact with blood-borne substances of self and foreign origins. EVs can intracellularly deliver cargoes that cannot translocate into cells on their own. These characteristics and the presence of LPS-binding proteins such as LBP and HMGB1 on EVs26–30 prompted us to examine whether host EVs capture and facilitate the cytosolic access of LPS.
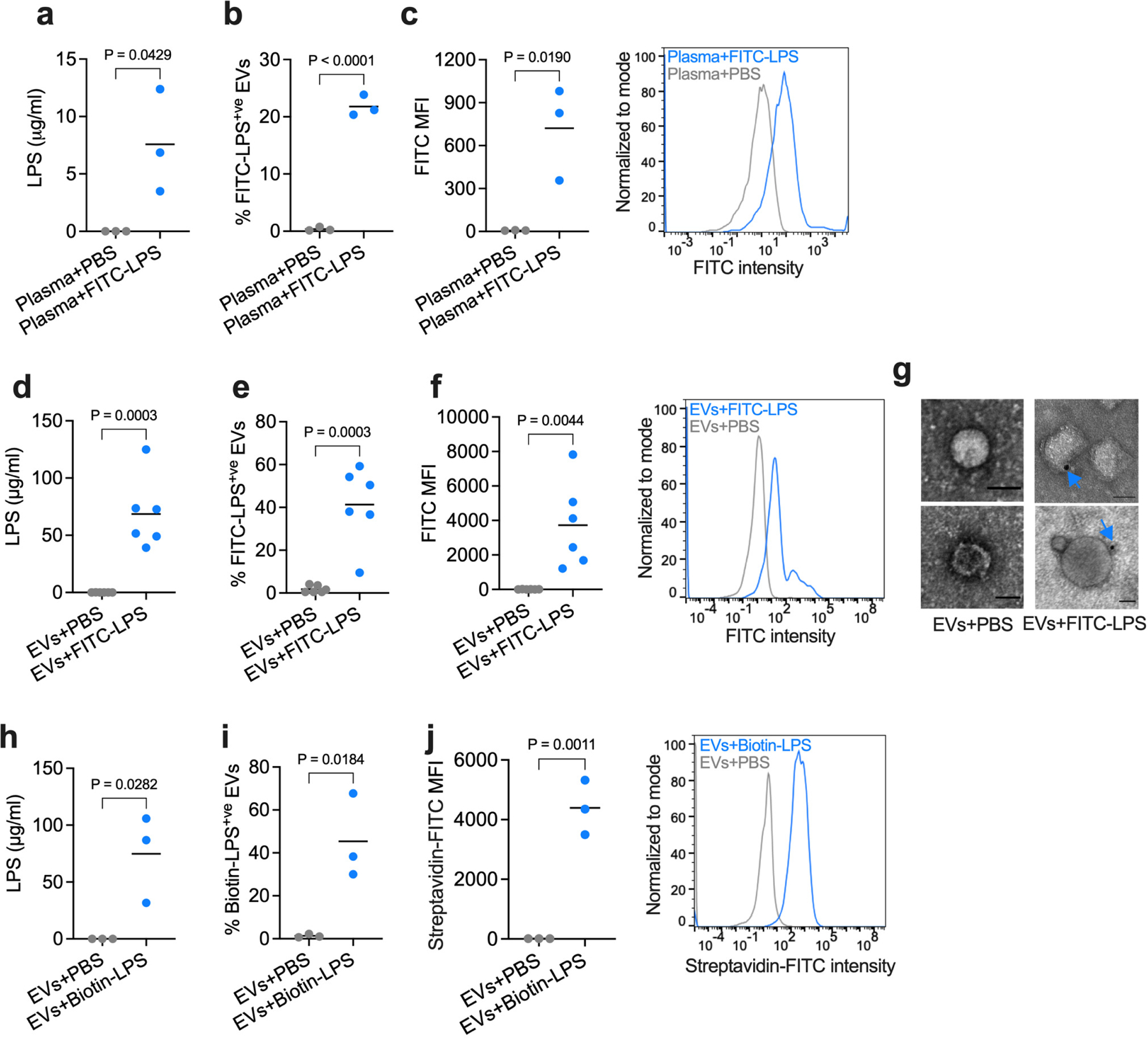
To test if host-derived EVs bind LPS, mice were intraperitoneally (i.p.) administered PBS or LPS, and EVs were isolated from the plasma by ultracentrifugation. EV preparations were verified by nanotracking analysis for size distribution, immunoblotting for EV markers (CD9 and CD63), and transmission electron microscopy (TEM) for morphology (Fig. 1a–c). Circulating EV levels were comparable between PBS- and LPS-injected mice (Extended Data Fig. 1a). EVs were then subjected to the Limulus Amebocyte Lysate (LAL) and HEK-Blue TLR4 reporter cell assays, standard assays for LPS detection. Remarkably, a substantial amount of LPS was present in EVs obtained from LPS- but not PBS-injected mice (Fig. 1d,e). Next, we took a complementary approach to assess LPS association with EVs. EVs isolated from mice that received unlabeled LPS or FITC-labeled LPS were stained with antibodies to EV-specific markers—CD9, CD63, and CD81 present on EVs but not on circulating lipoproteins such as HDL23,24,31—and analyzed by ImageStream imaging flow cytometry. This analysis showed a significant FITC-LPS signal from CD9/CD63/CD81+ve EVs originating from mice injected with FITC-labeled LPS but not the control unlabeled LPS (Fig. 1f,g). To further confirm and visualize LPS in EVs, EVs isolated from the plasma of mice injected with biotin-LPS or PBS were subjected to staining with streptavidin gold particles and TEM. We observed that EVs isolated from biotin-LPS-injected mice were decorated with LPS staining (Fig. 1h). The accessibility of biotin-LPS for streptavidin gold particles and the localization of the LPS staining on the rim of the EVs are suggestive of LPS binding to the EV surface. Importantly, EVs isolated from LPS-injected Casp11−/− mice also had LPS, indicating that EV binding of LPS is not a consequence of LPS-induced noncanonical inflammasome activation and pyroptosis (Fig. 1i–l).
Fig. 1: Circulating host-derived EVs capture LPS.
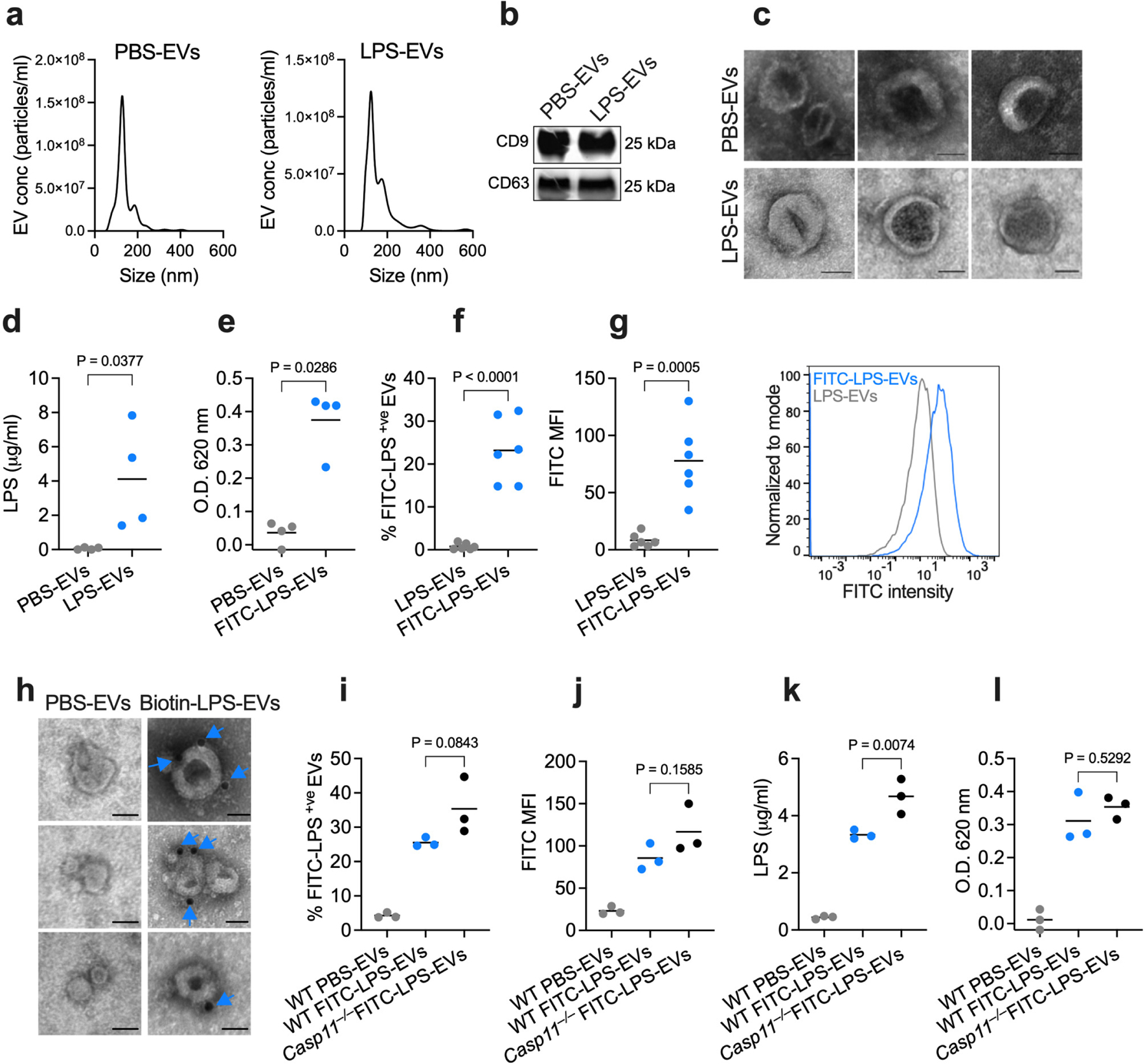
a, Nanoparticle tracking analysis (NTA) of the size distribution of EVs isolated from the plasma of wild-type (WT) mice injected with PBS or LPS (25 mg/kg) 1.5 h post-injection via ultracentrifugation. b, Immunoblotting analysis of EVs isolated from the plasma of PBS- or LPS-injected WT mice for CD9 and CD63. c, Negative staining transmission electron microscopy (TEM) of EVs isolated from the plasma of PBS- or LPS-injected WT mice as in (a). d,e, LPS content of the EVs isolated from WT mice injected with PBS, LPS or FITC-LPS (25 mg/kg) 1.5 h post-injection via ultracentrifugation as assessed by the LAL (n=4) (d) and HEK-Blue TLR4 reporter cell (e) assays (n=4). f,g, Percentage of FITC-LPS+ve EVs (f) and FITC histogram and mean fluorescence intensity (MFI) of EVs (g) isolated from mice injected with unlabeled LPS or FITC-LPS 1.5 h post-injection via ultracentrifugation as assessed by ImageStream flow cytometry (n=6). h, TEM of EVs isolated from the plasma of mice injected with PBS- or biotin-LPS 1.5 h post-injection and stained with streptavidin-gold particles. Arrows indicate LPS. i,j, Percentage of FITC-LPS+ve EVs (i) and FITC MFI of EVs (j) isolated from WT and Casp11−/− mice injected with PBS or FITC-LPS 1.5 h post-injection via ultracentrifugation as assessed by ImageStream flow cytometry (n=3). k,l, LPS content of the EVs isolated from WT and Casp11−/− mice injected with PBS or FITC-LPS 1.5 h post-injection via ultracentrifugation as assessed by the LAL (k) and HEK-Blue TLR4 reporter cell (l) assays (n=3). Combined data from three (i–l) four (d,e) or six (f,g) independent experiments or data from one experiment of representative of two (b,c and h) or three (a) are shown. Each circle represents a mouse, and the horizontal lines represent the mean (d–g,i–l). P values were determined by unpaired two-tailed t-test (d–g), one-way ANOVA with Dunnett’s post-test (i–l). Scale bar, 50 nm (c,h).
To independently validate EV-LPS binding, we employed additional EV isolation methods that rely on different principles. First, we used density gradient fractionation23 to isolate EVs from the plasma (Extended Data Fig. 1b–d). Subsequent fluorescent staining for EV markers and imaging flow cytometric analysis revealed LPS association with EVs, also confirmed by the LAL assay (Extended Data Fig. 1e–g). Next, we isolated EVs from the plasma of PBS- and LPS-injected mice by size exclusion chromatography (SEC) using the IZON qEV columns31–33 (Fig. 2a–c). SEC-isolated EVs from FITC-LPS-injected mice also contained substantial LPS, as shown by the LAL, reporter cell and imaging flow cytometric assays (Fig. 2d–g). Additionally, LPS was visualized on SEC-isolated EVs from FITC-LPS-injected mice by TEM following staining with a gold-conjugated anti-FITC antibody (Fig. 2h). SEC-isolated EVs from LPS-injected WT and Casp11−/− mice displayed similar LPS-binding (Fig. 2i–k). Finally, we used a direct immunoaffinity capture (DIC) method that uses EV-specific magnetic beads to isolate EVs without involving ultracentrifugation23; here, magnetic bead-conjugated CD9, CD63, and CD81 antibodies were added directly to the plasma from PBS- or FITC-LPS-injected mice and EVs were purified using MACS® columns. Consistent with the EVs isolated by previous methods, the DIC-isolated EVs from LPS-, but not PBS-injected mice contained LPS (Extended Data Fig. 1h–l). Overall, four independent EV isolation methods combined with multiple LPS detection assays rigorously demonstrate the association of LPS with circulating host-derived EVs in vivo.
Fig. 2: Host-derived EVs bind LPS in vivo.
a, Nanoparticle tracking analysis (NTA) of the size distribution of EVs isolated from the plasma of wild-type (WT) mice injected with PBS or LPS (25 mg/kg) 1.5 h post-injection via the IZON qEV column-based size exclusion chromatography (SEC). b, Immunoblotting analysis of EVs isolated as described above for CD9 and CD63. c, Negative staining TEM of EVs isolated from the plasma of PBS- or LPS-injected WT mice as described above. d,e, LPS content of the EVs isolated from PBS- or FITC-LPS-injected mice by the SEC method 1.5 h post-injection as assessed by the LAL (d; n=10) and HEK-Blue TLR4 reporter cell (e; n=7) assays. f,g, Percentage of FITC-LPS+ve EVs (f) and FITC histogram and mean fluorescence intensity (MFI) of EVs (g) isolated from mice injected with PBS or FITC-LPS as assessed by ImageStream analysis (n=6). h, TEM of EVs isolated as described above and stained with gold-conjugated anti-FITC antibody. Arrows indicate LPS. i, LPS content of the EVs isolated from WT and Casp11−/− mice injected with PBS or FITC-LPS (25 mg/kg) 1.5 h post-injection via SEC as assessed by the LAL assay (n=4). j,k, Percentage of FITC-LPS+ve EVs (j) and FITC MFI of EVs (k) isolated from WT and Casp11−/− mice injected with PBS or FITC-LPS 1.5 h post-injection via SEC as assessed by ImageStream flow cytometry (n=3). Combined data from three (j,k) four (i), six (f,g) or seven (d,e) independent experiments or data from one experiment representative of two (a–c,h) are shown. Each circle represents a mouse, and the horizontal lines represent the mean (d–g,i–k). P values were determined by unpaired two-tailed t-test (d–g) or one-way ANOVA with Dunnett’s post-test (i,j,k). Scale bar, 50 nm (c,h).
EVs are heterogeneous populations of different subcellular origins and sizes. Multivesicular body-derived exosomes and plasma membrane-derived ectosomes/microvesicles are key subtypes. In this context, considering that CD9, CD63, and CD81 are typically found on exosomes and some smaller ectosomes23,31,34,35, the detection of LPS on CD9+, CD63+, or CD81+ EVs in the flow cytometric analysis and the capture of LPS-bearing EVs by anti-CD9, -CD63, and -CD81 magnetic beads in the DIC method suggest that exosomes and smaller ectosomes bind LPS. We also isolated the larger ectosomes, commonly called microvesicles, from PBS or FITC-LPS-injected mice by low-speed ultracentrifugation and assessed LPS binding. These larger vesicles expressing annexin A1, a putative marker for microvesicles23,35, also bound FITC-LPS (Extended Data Fig. 2a,b). Thus, different subpopulations of EVs were found to bind LPS in vivo.
LPS binding is common to EVs of in vivo and in vitro origins
We further tested LPS binding by EVs under various ex vivo and in vitro conditions. First, the plasma collected from naïve WT mice was incubated with PBS or FITC-LPS ex vivo, and EVs were then isolated by SEC. The subsequent imaging flow cytometry and LAL analysis showed that EV binding of LPS can occur ex vivo (Extended Data Fig. 3a–c). To understand if any factor(s) in the blood, such as soluble plasma proteins, are necessary for the binding of LPS to EVs, EVs were first isolated from the plasma of naïve mice and then incubated with PBS, FITC-LPS, or biotin-LPS in vitro, followed by SEC-mediated repurification. These purified EVs also bound LPS without requiring any blood components (Extended Data Fig. 3d–j). Furthermore, LPS binding was also observed with EVs isolated from human plasma (Extended Data Fig. 4a– h). Next, to assess the LPS binding capacity of EVs of cell culture origin and to exclude any involvement of blood-borne lipoprotein complexes such as HDL and LDL, EVs were isolated from murine endothelial cells (bEnd.3) and human epithelial cells (HeLa) cultured in serum-free conditions and tested for LPS binding. Like plasma EVs, EVs isolated from cultured endothelial and epithelial cell lines bind LPS (Extended Data Fig. 4i–p). Together, these data indicate that the LPS binding capacity is intrinsic and common to EVs of plasma and cell culture origins.
EVs transfer LPS to myeloid and endothelial cell cytosol
That EVs have a remarkable capacity for intracellular delivery of their cargos prompted us to test whether LPS association with EVs (Fig. 1–2, and Extended Data Fig. 1–4) facilitates its access to the cytosol. We tested this idea first in macrophages; it is well-known that free LPS does not translocate to the cytosol of bone marrow-derived macrophages (BMDMs) in vitro9–11,14,21, allowing us to ask if the cytosol of BMDMs, inaccessible for free LPS, becomes accessible for EV-bound LPS. Therefore, we treated Casp11−/− BMDMs (the choice of which was to avoid pyroptosis of cells following cytosolic entry of LPS) with LPS-bound EVs and extracted the cytosol by digitonin fractionation14. A significant quantity of LPS was detected in the cytosol extracted from LPS-EVs-treated but not PBS-EVs- or free LPS-treated BMDMs (Fig. 3a,b). Additionally, whereas the intracellular localization of free FITC-LPS was poor, EV-associated FITC-LPS localizes robustly in the intracellular space in Casp11−/− iBMDMs (Fig. 3c,d and Extended Data Fig. 5). Furthermore, Casp11−/− iBMDMs treated with FITC-LPS, FITC-LPS-EVs, or PBS-EVs were stained with a gold-labeled anti-FITC antibody and examined by TEM. Intracellular LPS staining in organelle- and membrane-free areas suggestive of cytosolic localization was noticeable in cells treated with FITC-LPS-EVs but not FITC-LPS or PBS-EVs (Fig. 3e). Moreover, LPS-EVs, but not free LPS, induced endolysosomal perturbation evidenced by galectin-3 accumulation, which may explain the cytosolic access of EV-bound LPS (Extended Data Fig. 6).
Fig. 3: Cytosolic delivery of LPS by host-derived EVs.

a,b, LPS levels in the cytosol extracted from Casp11−/− BMDMs stimulated for 15 h with purified LPS, PBS-EVs, or LPS-EVs as assessed by the LAL (a) and HEK-Blue TLR4 reporter cell (b) assays (n=3). c,d, Confocal microscopy of Casp11−/− iBMDMs stimulated for 5–6 h with PBS-EVs or FITC-LPS-EVs and stained with anti-CD45 and anti-FITC antibodies to visualize plasma membrane and LPS, respectively (c) and the quantification of FITC intensity (d). e, TEM of Casp11−/− iBMDMs stimulated for 15 h with PBS-EVs, FITC-LPS, or FITC-LPS-EVs and stained with gold-conjugated anti-FITC antibody. Arrows indicate cytosolic localization of LPS. f, LPS levels in the cytosol of splenic myeloid cells from Casp11−/− mice injected with PBS-EVs or FITC-LPS-EVs 5 h post-injection as assessed by the LAL assay (n=3). g,h, ImageStream flow cytometric analysis of intracellular localization of LPS in peritoneal lavage cells of PBS-EV- or FITC-LPS-EV-injected Casp11−/− mice 5 h post-injection stained with anti-CD45 and anti-FITC antibodies (g) to visualize the plasma membrane and LPS, respectively and the quantification of intracellular FITC intensity (h). i, TEM of peritoneal lavage cells isolated from PBS-EV- or FITC-LPS-EV-injected Casp11−/− mice and stained with gold-conjugated anti-FITC antibody. Arrows indicate cytosolic localization of LPS. j,k, ImageStream flow cytometric analysis of intracellular localization of LPS in splenic endothelial cells isolated from PBS-EV- or FITC-LPS-EV-injected Casp11−/− mice 5 h post-injection and stained with anti-CD31 and anti-FITC antibodies (j) to visualize the plasma membrane and LPS, respectively and the quantification of intracellular FITC intensity (k). l, LPS levels in the cytosol of splenic endothelial cells isolated from PBS-EV- or FITC-LPS-EV-injected Casp11−/− mice 5 h post-injection as assessed by the LAL assay (n=4). Combined data from two (c,d,g,h,i) or three (a,b,e,f,j,k,l) independent experiments are shown. Each circle represents a mouse (f,l), cell (d,h,k), or individual experiments (a,b) and the horizontal lines represent the mean. P values were determined by one-way ANOVA with Dunnett’s post-test (a,b) or unpaired two-tailed t-test (d,f,h,k,l). Scale bar, 5 µm (c) or 200 nm (e,i). BF, brightfield.
Next, we examined whether EVs mediate the cytosolic translocation of LPS in vivo. Casp11−/− mice were injected with FITC-LPS-EVs or PBS-EVs, and the cytosol was extracted from splenic myeloid cells. The LAL assay showed the presence of LPS in the cytosol of splenic myeloid cells sorted from FITC-LPS-EVs-injected mice (Fig. 3f). Furthermore, intracellular localization of LPS was visualized in peritoneal lavage cells of mice that received FITC-LPS-EVs (Fig. 3g,h). More importantly, TEM analysis of peritoneal lavage cells stained with gold-conjugated anti-FITC antibody showed the presence of LPS in the cytosol of cells from FITC-LPS-EVs-injected mice (Fig. 3i). EVs also delivered LPS into endothelial cells as revealed by imaging and biochemical analyses of endothelial cells isolated from the spleens of mice receiving FITC-LPS-EVs (Fig. 3j–l). Collectively, these data show that LPS-laden EVs deliver LPS to the cytosol of myeloid and endothelial cells, key cell types sensing cytosolic LPS during sepsis36,37
Cytosolic LPS delivery by EVs activates caspase-11 and GSDMD
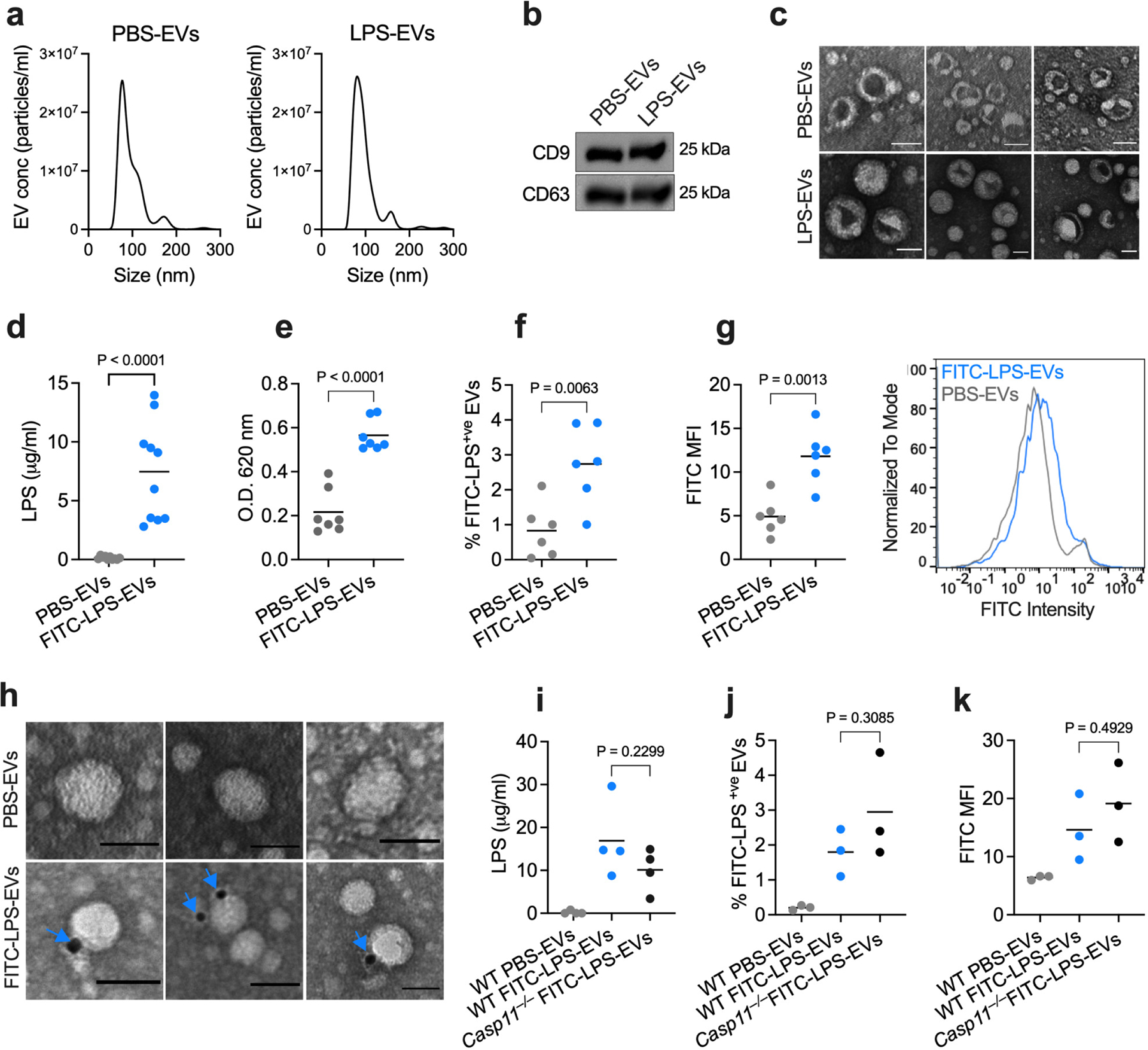
The binding and cytosolic delivery of LPS by EVs suggests that EV-associated LPS should become accessible to caspase-11. Towards this, unlike free LPS, EV-associated LPS triggered GSDMD cleavage and pyroptosis in BMDMs in an EV-dose-dependent manner (Fig. 4a,b) and in human THP1 monocytes (Extended Data Fig. 7a,b). Notably, LPS-EVs induced GSDMD activation, pyroptosis, and caspase-1 and IL-1β activation only in WT but not in Casp11−/− macrophages (Fig. 4c,d and Extended Data Fig. 7c,d). Furthermore, LPS-EVs induced GSDMD activation and pyroptosis in Nlrp3−/− and Aim2−/− macrophages and RAW macrophages inherently lacking ASC for canonical inflammasome signaling (Extended Data Fig. 7e–j). Together, these data indicate that LPS-EVs activate the noncanonical inflammasome in macrophages in vitro.
Fig. 4: EV-mediated cytosolic LPS delivery activates the noncanonical inflammasome.
a,b, GSDMD (FL, full length; NT, N-terminal domain), caspase-11 and β-actin in the lysates (a) and cell death (LDH release; b) of IFN-γ-primed (10 ng/ml) WT BMDMs stimulated with medium, varying doses of PBS-EVs or LPS-EVs (6.25–50 μg/ml), LPS or LPS transfection for 16 h. c,d, GSDMD, caspase-11, and GAPDH in the lysates (c) and cell death (LDH release; d) of IFN-γ-primed (10 ng/ml) WT and Casp11−/− BMDMs stimulated as indicated for 16 h. e, GSDMD, caspase-11, and β-actin in the lysates of spleen and liver of poly(I:C)-primed (150 μg) WT mice injected i.p with SEC-isolated PBS-EVs or LPS-EVs for 15 h (n=3). f–i, IL-18 (f), IL-1β (g) , IL-1α (h) and galectin-1 (i) in the plasma of poly(I:C)-primed WT mice injected with SEC-isolated PBS-EVs or LPS-EVs for 15 h (n=9). j,k, IL-18 and IL-1β in the plasma of poly(I:C)-primed WT mice injected with UC-isolated PBS-EVs or LPS-EVs for 6 h (n=7). l,m, IL-18 and IL-1β in the plasma of poly(I:C)-primed WT mice administered with UC-isolated EVs from LPS-injected WT and Casp1/11−/− mice (n=4). n, GSDMD, caspase-11, and β-actin in the lysates of liver and spleen of poly(I:C)-primed WT and Casp11−/− mice injected with SEC-isolated LPS-EVs for 15 h (n=3). o,p, IL-18 and IL-1β in the plasma of WT and Casp11−/− mice treated as described in (n) (n=6). q, Survival of poly(I:C)-primed WT and Casp11−/− mice injected with SEC-isolated LPS-EVs (n=9). Combined data from two (j–m,o,p) or three (b,d,f,g,h,i,q) independent experiments or representative data of two (a,c,n) or three (e) experiments are shown. Each circle represents a mouse and the horizontal lines represent the mean (f-m,o,p). Each lane represents a mouse (e,n). Data are presented as mean±s.e.m (b,d). P values were determined by unpaired two-tailed t-test (f-m,o,p), one-way ANOVA with Dunnett’s post-test (b), two-way ANOVA with Sidak’s post-test (d) or Mantel-Cox test (q).
We next assessed if EVs carrying LPS enable cytosolic LPS sensing in vivo. PBS-EVs or LPS-EVs isolated by SEC were injected into WT mice, and inflammasome responses were analyzed. Strikingly, LPS-EVs but not PBS-EVs induced GSDMD cleavage in the spleen and liver, IL-18 and IL-1β secretion into the plasma, and the release of DAMPs (IL-1α and galectin-138,39) (Fig. 4e–i). Furthermore, LPS-EVs but not PBS-EVs isolated by ultracentrifugation also induced IL-18 and IL-1β secretion into the plasma (Fig. 4j,k). Notably, EVs isolated from LPS-injected WT and Casp1−/−Casp11−/− mice elicited comparable IL-18 and IL-1β responses in WT recipient mice (Fig. 4l,m), excluding the possibility of pyroptotic debris and IL-1 cytokines from LPS-EV donor mice contributing to inflammasome responses in LPS-EV recipient mice. Importantly, LPS-EV-induced GSDMD activation in the liver and spleen and IL-18 and IL-1β secretion were significantly reduced in Casp11−/− mice (Fig. 4n–p). Consistently, Casp11−/− mice were also protected from LPS-EV-induced lethality (Fig. 4q). Contrastingly, LPS-EVs were able to induce lethality in Tlr4−/− mice even though LPS-EVs activate TLR4 in vitro and in vivo (Extended Data Fig. 7k–m). All these findings clearly indicate that LPS-EVs mediate cytosolic translocation of LPS leading to caspase-11 activation.
Given that there are no mutant mice lacking EVs, we used GW4869 (a commonly used inhibitor of EV production40,41) to reduce circulating EV levels and assess its effect on intracellular LPS sensing. Circulating EV levels were expectedly reduced in GW4869-treated mice (Fig. 5a). Correspondingly, intracellular localization of LPS in splenic myeloid cells and peritoneal lavage cells—as shown by the LAL and reporter assays, ImageStream analysis or immunogold TEM (Fig. 5b–f)—was reduced in GW4869-treated mice. Moreover, the systemic activation of GSDMD, IL-18, and IL-1β was also impaired in GW4869-treated mice (Fig. 5g–i and Extended Data Fig. 7n). Additionally, EV depletion with GW4869 ameliorated LPS-induced lethality in Tlr4−/− mice (Extended Data Fig. 7o) but expectedly didn’t affect the survival of Casp11−/− mice (Extended Data Fig. 7p). Another EV inhibitor, nexinhib2041, also reduced noncanonical inflammasome responses to LPS (Extended Data Fig. 7q–s). Finally, exogenous administration of LPS-EVs rescued noncanonical inflammasome responses in GW4869-treated mice (Fig. 5j–l). These data collectively suggest a likely involvement of EVs in cytosolic sensing of LPS in vivo.
Fig. 5: The effect of EV depletion on intracellular LPS sensing.

a, Mice were injected with DMSO or the EV inhibitor (2.5 μg/g GW4869) on days 1 and 2, and plasma numbers of EVs were measured by NTA on day 3 (n=6). b,c, LPS levels in the cytosol of splenic myeloid cells from Casp11−/− mice pretreated with DMSO or GW4869 as described in (a) and injected i.p. with FITC-LPS on day 3 for 5 h as assessed by the LAL (b) and HEK-Blue TLR4 reporter cell (c) assays (n=7). d,e, ImageStream analysis of intracellular LPS in splenic myeloid cells isolated from Casp11−/− mice pretreated with DMSO or GW4869 as in (a) and injected with FITC-LPS on day 3 and stained with anti-CD45 and anti-FITC antibodies (d) and the quantification of intracellular FITC intensity (e). f, TEM of peritoneal lavage cells isolated from Casp11−/− mice pretreated with DMSO or GW4869 on days 1 and 2 and injected i.p. with FITC-LPS on day 3 and stained with gold-conjugated anti-FITC antibody. Arrows indicate cytosolic localization of LPS. g–i, GSDMD and caspase-11 in the liver and spleen (g; n=3) and IL-18 and IL-1β in the plasma (h,i; n=8) of WT mice pretreated with DMSO or GW4869 as in (a) and injected i.p. with LPS on day 3 for 8 h. (j–l) GSDMD, caspase-11 and β-actin in the liver and spleen (j; n=3) and IL-18 and IL-1β in the plasma (k,l; n=8) of WT mice pretreated with DMSO or GW4869 as in (a) and injected i.p. on day 3 with LPS or LPS followed by LPS-EVs 2 h later. Combined data from two or three independent experiments (a–c,h,i,k,l) or a representative experiment of two (d,e,f) or three (g,j) are shown. Each circle represents a mouse (a,b,c,h,i,k,l) or cell (e) and the horizontal lines represent the mean. Each lane represents a mouse (g,j). P values were determined by unpaired two-tailed t-test (a,b,c,e,h,i) or one-way ANOVA with Dunnett’s post-test (k,l). Scale bar: 200 nm (f). BF, brightfield.
EV lipid bilayer-lipid A binding underlies EV capture of LPS
It is possible that EVs can adsorb LPS by virtue of their surface-exposed proteins with LPS binding properties, namely CD14, HMGB1, and LPS-binding protein (LBP)26–30. However, EVs isolated from WT and Tlr4−/− Cd14−/− mice and incubated with FITC-LPS in vitro (Fig. 6a–c) and EVs isolated from FITC-LPS-injected WT and Tlr4−/− Cd14−/− mice (Fig. 6d,e) displayed comparable LPS binding, thus ruling out a role for TLR4 and CD14 in EV binding of LPS in vitro and in vivo. Also, blocking HMGB1 or LBP with anti-HMGB1 or anti-LBP neutralizing antibodies, respectively, did not prevent EVs from binding LPS (Fig. 6f–i and Extended Data Fig. 8a–d). Interestingly, even the shaving off EV surface proteins with trypsin didn’t reduce LPS association with EVs in vitro (Fig. 6j–k and Extended Data Fig. 9a–d) and in vivo (Extended Data Fig. 9e,f).
Fig. 6: The EV lipid bilayer-lipid A interaction underlies EV capture of LPS.

a–c, LPS binding by EVs isolated from WT and Tlr4−/−Cd14−/− mice and incubated in vitro with FITC-LPS for 45 min followed by SEC re-purification as assessed by the LAL (n=3) (a) and ImageStream (n=3) (b,c) analysis. d,e, Percentage of FITC-LPS+ve EVs (d) and FITC MFI (e) of EVs isolated from FITC-LPS-injected WT and Tlr4−/−Cd14−/− mice (n=3). f–i, LPS binding by EVs preincubated with isotype, anti-HMGB1 (f,g), or anti-LBP (h,i) antibodies (Ab) and incubated with FITC-LPS for 45 min followed by SEC re-purification as assessed by the LAL (n=4) (f,h) and ImageStream (n=3) (g,i) analysis. j,k, LPS binding by unshaved and surface protein-shaved EVs incubated with FITC-LPS followed by SEC re-purification as assessed by the LAL (n=3) (j) and ImageStream (n=3) (k) analysis. l–n, LPS binding by EV mimic liposomes incubated with PBS or biotin-LPS for 45 min as assessed by the LAL (l; n=4) and ImageStream analysis (Percentage of biotin-LPS+ve EV mimic liposomes (m; n=4) and streptavidin-FITC MFI of EV mimic liposomes (n; n=3)). o,p, Percentage of FITC-LPS+ve EV mimic liposomes (o; n=6) and FITC MFI of EV mimics (p; n=5) isolated from mice 1.5 h post-injection with DiD-labeled EV mimics followed by FITC-LPS. q, Cell death in indicated BMDMs stimulated as indicated for 16 h. r–t, The LAL (r) and ImageStream analysis [streptavidin-FITC+ve EVs (s) and streptavidin-FITC-MFI (t)] of EVs incubated with biotin-LPS or biotin-lipid A for 45 min followed by SEC re-purification (n=4). u–x, Lipid A binding by unshaved and surface protein-shaved EVs (u,v) or EV mimic liposomes (w,x) incubated with PBS or lipid A for 45 min followed by SEC re-purification as assessed by the LAL (u,w) and TLR4 reporter (v,x) assays (n=3). Combined data from two (o,p), three (a–e,g,i,j,k,n,q,u–x) or four (f,h,l,m,r,s,t) independent experiments are shown. Horizontal lines represent the mean (a–p,r–x). Data are presented as mean±s.e.m (q). P values were determined by unpaired two-tailed t-test (a-p, r–x) or two-way ANOVA with Sidak’s post-test (q).
This result—LPS binding by protein-shaved EVs—suggested that the EV capture of LPS is mediated not by surface proteins but rather likely by their lipid bilayer. To test this, we formulated EV-mimicking liposomes with DOPC/SM/Chol/DOPS/DOPE as described previously42 and tested if these EV-mimics devoid of proteins can bind LPS. Remarkably, EV-mimic liposomes were found to bind LPS upon incubation with biotin-LPS in vitro (Fig. 6l–n). Furthermore, FITC-LPS administered into mice injected with fluorescently labeled EV-mimic liposomes was captured by EV-mimic liposomes (Fig. 6o,p). These data lend evidence to the idea that the lipid membrane of EV is sufficient for LPS binding. Consistent with these observations, LPS-bound EV-mimic liposomes induced caspase-11-dependent pyroptosis in macrophages (Fig. 6q). LPS comprises lipid A and core/outer carbohydrate chains43. We noticed comparable binding of lipid A and full-length LPS to EVs (Fig. 6r–t), suggesting that lipid A moiety of LPS is sufficient for EV binding. Furthermore, lipid A of LPS was able to bind to both surface protein-shaved EVs (Fig. 6u,v) and EV mimic liposomes (Fig. 6w,x). These findings collectively suggest that the association of lipid A with the lipid bilayer of EV underlies EV capture of LPS.
CD14 mediates the intracellular transfer of LPS by EVs
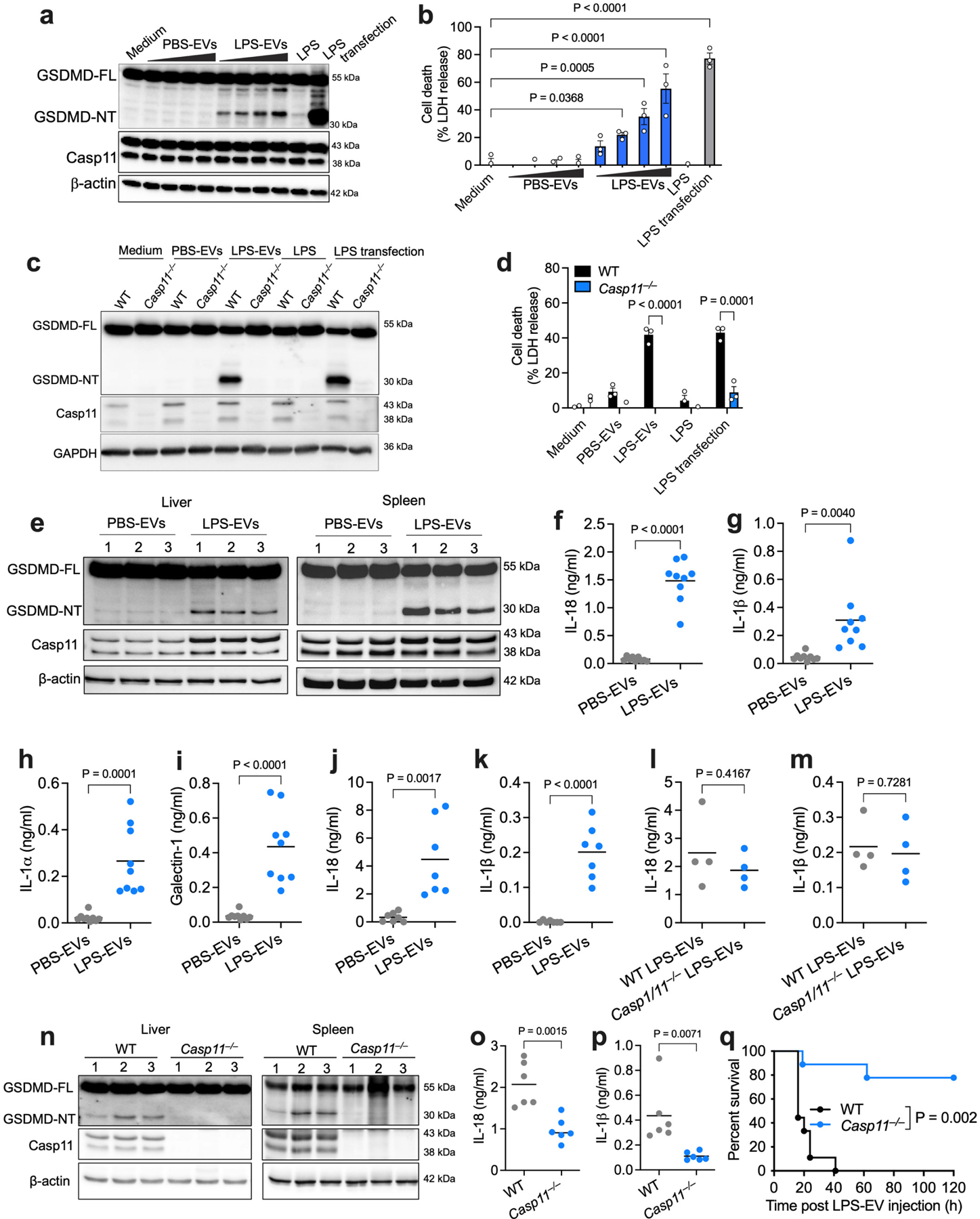
How do LPS-laden EVs deliver LPS intracellularly? Receptor-mediated endocytosis is a common mode of internalization of EVs, facilitating the intracellular transfer of their cargoes24,34,44. CD14 is a TAXI (transporters associated with the execution of inflammation) protein that binds LPS and drives the endocytosis of the TLR4-MD2 complex45–48. Importantly, our latest work shows a key role for CD14 in the cytosolic localization of free LPS in vivo and caspase-11 activation21. Considering these findings, we hypothesized that the intracellular transfer of LPS by EVs is mediated by CD14-dependent receptor-mediated endocytosis. Supporting this, we observed the colocalization of FITC-LPS-EVs with CD14 and the cellular uptake of FITC-LPS-EVs (Extended Data Fig. 10). Further, WT and Cd14−/− mice were injected with FITC-LPS-EVs, and cytosolic localization of LPS in splenic myeloid cells was examined by biochemical, imaging and immunogold TEM assays. All these assays showed reduced cytosolic localization of LPS in Cd14−/− myeloid cells (Fig. 7a–e). Consistent with the reduced cytosolic delivery of LPS by EVs in the absence of CD14, caspase-11 activation by LPS-EVs was impaired in Cd14−/− mice—primed with IFN-γ to negate any noncanonical inflammasome priming issues21—as evidenced by reduced GSDMD activation and IL-18, IL-1β, IL-1α, and galectin-1 release (Fig. 7f–j). Overall, these data clearly indicate that CD14 mediates EV-facilitated cytosolic transfer of LPS (Fig. 7k).
Fig. 7: EV-mediated cytosolic delivery of LPS is CD14-dependent.
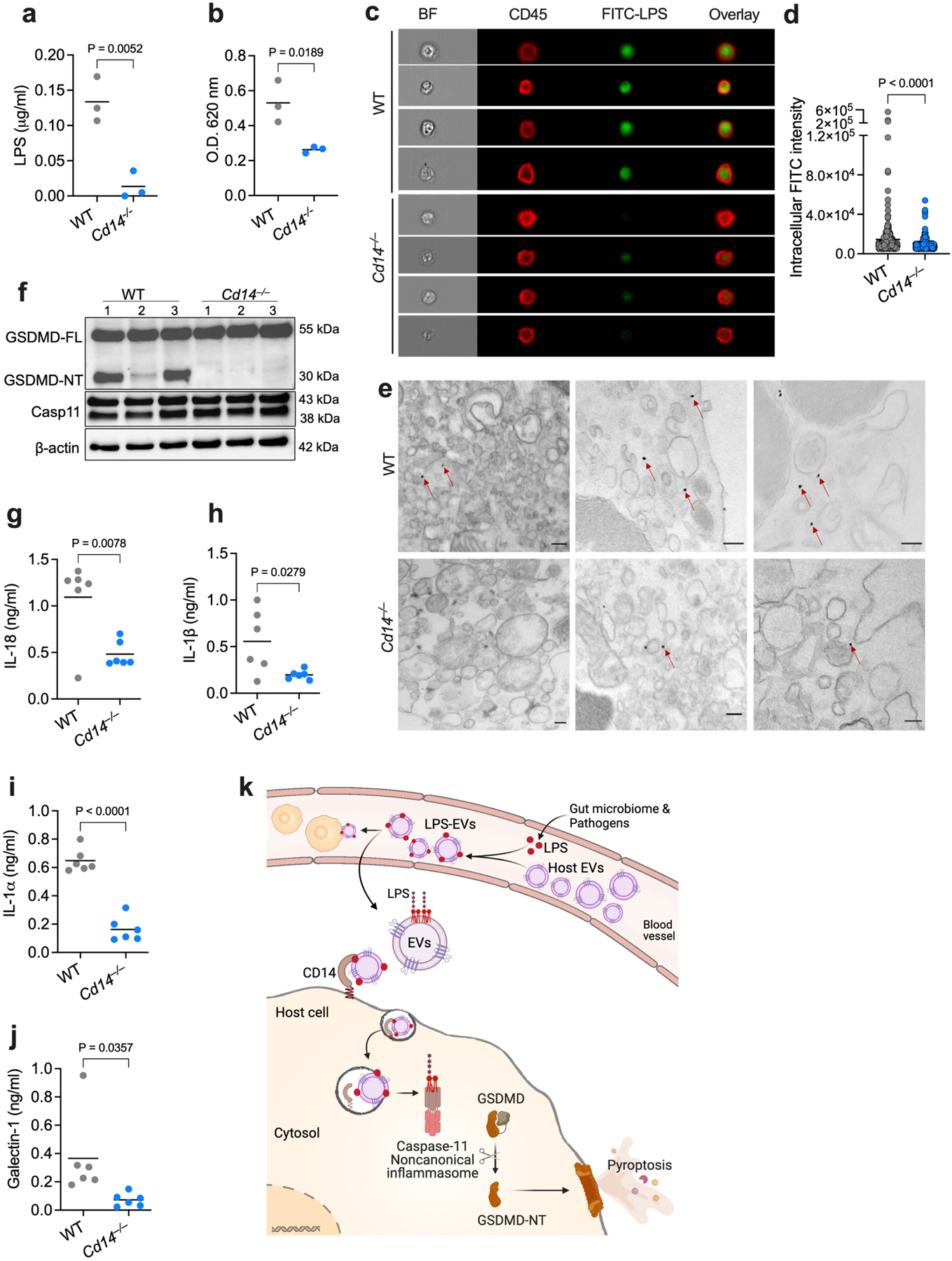
a,b, LPS levels in the cytosol of splenic myeloid cells from WT and Cd14−/− mice injected with FITC-LPS-EVs for 5 h as assessed by the LAL (a) and HEK-TLR4 reporter cell (b) assays (n=3). c,d, ImageStream flow cytometric analysis of intracellular localization of LPS in splenic myeloid cells of FITC-LPS-EV-injected WT and Cd14−/− mice and stained with anti-CD45 and anti-FITC antibodies (c) and the quantification of intracellular FITC intensity (d). e, TEM of splenic myeloid cells isolated from FITC-LPS-EV-injected WT and Cd14−/− mice and stained with gold-conjugated anti-FITC antibody. Arrows indicate cytosolic localization of LPS. f–j, GSDMD, caspase-11 and β-actin in the spleen (f; n=3) and IL-18, IL-1β, IL-1α and galectin-1 in the plasma (g–j; n=6) of IFN-γ (1 μg)-primed WT and Cd14−/− mice injected with LPS-EVs for 6 h. k, Working model for the host EV capture and cytosolic release of LPS and noncanonical inflammasome activation (created with Adobe Illustrator and BioRender.com). Combined data from two (g–j) or three (a,b) independent experiments or representative data of two (c–f) independent experiments are shown. Each circle represents a mouse (a,b,g–j) or cell (d) and the horizontal lines represent the mean. Each lane represents a mouse (f). P values were determined by unpaired two-tailed t-test. Scale bar, 200 nm (e). BF, brightfield.
Discussion
Through extensive analysis integrating independent methods of EV isolation and LPS detection, we show that EVs capture systemic LPS and transfer it into the cytosol via a CD14-dependent route for caspase-11 sensing, expanding EVs’ immune-relevant functions28,32,49–51. Even though free LPS attaches to the plasma membrane, it fails to cross the plasma and endosomal membranes and reach the cytosol9,10,14. Therefore, it is conceivable that once LPS gets adsorbed onto EVs, it could be delivered to the cytosol like a typical EV cargo.
We recently reported a role for CD14 in LPS internalization leading to caspase-11 activation21. Considering our findings from this work, namely the cytosolic transfer of LPS by EVs and the CD14-dependency of this process, the form of LPS that gets transferred to the cytosol via CD14 is EV-bound LPS rather than free LPS. As CD14’s role is limited to endocytosis of the cargo, further trafficking of the cargo from endosomes to the cytosol is, in part, driven by the biophysical properties of the cargo. In this regard, following CD14-mediated uptake, cargoes such as EVs, but not free LPS, would undergo fusion with the endosomal membranes on account of the fusogenic capacity of their lipid bilayer. Such mixing of LPS-laden EVs with endosomal membranes and the consequent endosomal membrane disruption is likely responsible for the eventual exposure of EV membrane-associated LPS to the cytosol. Thus, EV-bound LPS internalized via CD14 is far more likely to traffic to the cytosol than the free LPS.
LPS has been known to associate with circulating lipoprotein complexes such as HDL and LDL. However, LPS association with lipoprotein complexes and EVs differ in two key aspects: binding characteristics and functional outcomes. Whereas LPS binding by lipoprotein complexes was mediated by LBP52,53, LPS binding of EVs in this study is not dependent on LBP. More importantly, it is well-established that the consequence of lipoprotein binding and sequestering of LPS is LPS clearance in the liver, thus limiting the bioavailability of LPS to elicit inflammatory responses52,54–56. In contrast, the EV-binding of LPS mediates its cytosolic entry and activates inflammatory responses.
HMGB1 also binds LPS and mediates its cytosolic access57. However, unlike EVs that are abundant in extracellular fluids in steady-state, HMGB1 is predominantly nuclear and becomes extracellular only after cell rupture58–60. Therefore, considering the chronology that cytosolic entry of LPS occurs first and is a prerequisite for HMGB1 release in the context of the noncanonical inflammasome58, a likely scenario is that EVs already present in the circulation bind and deliver LPS intracellularly, leading to pyroptosis, and HMGB1 thus released contributes to the intracellular LPS localization subsequently.
Persistent low-grade inflammation driven by circulating free LPS is a risk factor for inflammatory, autoimmune, and metabolic disorders16,61–64. Therefore, it is of interest to determine if host EV-facilitated cytosolic access of LPS and the ensuing inflammasome responses contribute to those chronic diseases. Future studies are also warranted to elucidate if host-derived EVs capture additional microbial products and promote their access to cytosolic surveillance mechanisms.
Methods
Mice
C57BL/6J, Cd14−/−, Tlr4−/− , Nlrp3−/− and Casp1−/−Casp11−/− mice from the Jackson Laboratory (Bar Harbor, ME), Casp11−/− and Aim2−/− mice from Genentech (kind gifts of Dr. Vishva Dixit), and Tlr4−/−Cd14−/− described previously21 were bred and maintained in specific pathogen-free conditions in the animal facility of UConn Health. Eight-24-week-old male and female mice were used. All mice used in this study were housed at an ambient temperature of ~22 °C, a humidity of 40–60%, and a light/dark cycle of 12 h. All experiments were carried out in accordance with the guidelines set forth by the UConn Health Institutional Animal Care and Use Committee.
EV isolation from the plasma
Mice were intraperitoneally (i.p.) injected either with PBS, LPS (25 mg/kg; catalog no. L2630; Sigma), FITC-LPS (25 mg/kg; catalog no. F3665; Sigma) or biotin-LPS (25 mg/kg; catalog no. tlrl-lpsbiot; invivogen) and the blood was harvested in lithium-heparin tubes after 90 min. Plasma was isolated by spinning the blood at 1500 x g for 10 min, followed by 10,000 x g for 10 min at room temperature. Human plasma was purchased from Innovative Research and precleared by spinning at 10,000 x g for 10 min at 4 oC. Plasma was either used afresh or saved at −80 oC for future use. EVs were isolated from the plasma by four methods: ultracentrifugation (UC), OptiPrep density gradient (DG), size exclusion chromatography (SEC), and direct immunocapture (DIC).
In UC method, an equal volume of plasma from each mouse was added to ultracentrifuge tubes, and the volume was made up to 3.5–4 ml with ice-cold filtered PBS. Samples were spun for 2 h at 100,000 x g at 4 oC in a TLA-100.3 rotor (Optima MAX-XP ultracentrifuge, Beckman Coulter), and the EV pellet was resuspended in 50 μl of PBS with gentle vortexing. To assess size distribution and concentration, EVs were subjected to nanoparticle tracking analysis (NTA) on Nanosight NS300 (Malvern Panalytical). In DG method, EV isolated through UC, as described above, was kept at the bottom of the tube and layered from bottom to top with 700 μl of 36%, 30%, 24%, 18%, 12%, and 6% OptiPrep solution. Next, samples were spun in an MLS-50 rotor (Optima MAX-XP ultracentrifuge, Beckman Coulter) for 18 h at 120,000 x g at 4 oC. The EV-rich fractions from the top 6%, 12%, and 18% were washed with PBS at 100,000 x g for 2 h at 4 oC in a TLA-100.3 rotor (Optima MAX-XP ultracentrifuge, Beckman Coulter).
In SEC method, EVs were isolated from the plasma using IZON SEC columns per the manufacturer’s instructions. Briefly, 500 μl or 2 ml of plasma was added to qEVoriginal or qEV2 70nm columns, respectively. After discarding the first 6 (from qEVoriginal) and 8 (from qEV2) void fractions, the rest of the fractions were collected using an IZON automated fraction collector. The EVs were enriched in the fractions 7–9 (qEVoriginal) and 9 and 10 (qEV2) and were pooled before use. In DIC method, plasma EVs were isolated using the Pan Exosome isolation kit (catalog number: 130-117-039; Miltenyi Biotec); briefly, 500 μl plasma was diluted to 2 ml with PBS, and Miltenyi MicroBeads recognizing the exosome proteins CD9, CD63, and CD81 were mixed with the diluted plasma and incubated for 1 h at room temperature. The microbead-labeled EVs were then isolated using a µ column and µMACS separator following the manufacturer’s protocol. Microvesicles were isolated by ultracentrifugation of the plasma from PBS- or FITC-LPS-injected mice at 20,000 x g at 4 oC for 1 h (Optima MAX-XP ultracentrifuge, Beckman Coulter). MVs in the pellet were resuspended in PBS for further analysis.
EV isolation from cell culture supernatants
bEnd.3 cells and HeLa cells were seeded in 15 cm dishes to 90% confluency in serum-free DMEM for 24 h. The supernatant was collected and precleared by spinning at 8000 x g for 30 min at 4 oC. EVs were isolated by ultracentrifugation as described above. EVs isolated from 40 ml supernatants were resuspended in 1 ml PBS.
EV and LPS binding in vitro and in vivo
Naïve plasma or EVs isolated from the plasma or bEnd.3 and HeLa cell culture supernatants were mixed with either 100 μl of PBS or 100 μl of FITC-LPS (500 μg) or 100 μl Biotin LPS (100 μg). This mixture was incubated at 37 oC for 45 min and then applied to qEVoriginal 70nm columns as described above to repurify EVs, which were subjected to downstream assays to detect EVs and LPS such as imaging flow cytometry, the LAL assay, and the HEK-Blue TLR4 reporter assay. In some experiments, EVs were incubated with biotinylated-lipid A (10 μg) at 37 oC for 45 min followed by SEC fractionation through qEVoriginal 70nm columns described above. To shave off EV surface proteins, EVs harvested from WT mice by SEC were incubated with trypsin (50 μg/ml) at 37 oC for 2 h and with PMSF at 4o C for 1 h to inactivate trypsin and repurified by SEC. Shaving was confirmed by immunoblotting for CD9 and CD63 and TEM, and the shaved and unshaved EVs were then subjected to in vitro LPS binding as described above. Additionally, Casp11−/− mice were injected with FITC-LPS and EVs isolated from the plasma of these mice were subjected or not to surface protein shaving with trypsin as described above. LPS associated with unshaved and shaved EVs was assessed by the LAL assay, the HEK-Blue TLR4 reporter assay, or imaging flow cytometry as indicated. To assess the role of LBP and HMGB1, EVs isolated by SEC were first incubated for 2 h at room temperature with the neutralizing antibody (10 μg/ml) against LBP (clone mAb M330-19; catalog no. HM1026; HyCult Biotech) or HMGB1 (Clone 3E8; catalog no. 651402; BioLegend) or the isotype control antibody rat IgG2α (Clone RTK2758; catalog no. 400543; BioLegend) or mouse IgG2b (Clone MCP11; catalog no. 400347; BioLegend) and then subjected to FITC-LPS binding as described above. After LPS binding, EVs were repurified by SEC and subjected to LPS detection assays.
EV mimic liposome binding of LPS in vitro and in vivo
EV mimic liposomes (6×1012 liposomes/ml) formulated with DOPC/SM/Chol/DOPS/DOPE (21/17.5/30/14/17.5 mol/mol) as described previously42 were mixed with either PBS or 20 μg of Biotin-LPS and incubated at 37 oC for 45 min followed by SEC fractionation through qEVoriginal 70nm as described above. EV mimic liposomes were subjected to imaging flow cytometry, the LAL assay, and the HEK-Blue TLR4 reporter assay for LPS detection. Furthermore, Casp11−/− mice were first injected i.p. with DiD-labelled EV mimic liposomes and then with PBS or FITC-LPS (500 μg) 15 min later. After 1.25 h, EV mimic liposomes were isolated using the ultracentrifugation method as described above and imaging flow cytometry was used to assess FITC-LPS signal associated with DiD-labelled EV mimic liposomes.
Assessment of EV binding of LPS
EVs or MVs isolated from PBS, LPS, or FITC-LPS injected mice or EVs or EV mimic liposomes incubated with LPS, FITC-LPS, biotin-LPS, or biotin-lipid A were tested for LPS binding using the LAL assay kit (Associates of Cape Cod) according to the manufacturer’s instructions, ImageStream analysis, and the HEK-Blue TLR4 reporter cell assay. In the latter, HEK293 cells expressing hTLR4 (Invivogen) were cultured with HEK-Blue Detection and HEK-Blue Selection (Invivogen) in a 96-well plate (40,000 cells/well), and EVs were added to the cell culture medium and SEAP activity was assessed by measuring absorbance at 620 nm 10–12 h later. In ImageStream analysis, EVs were mixed with 1:200 dilution of a cocktail of PE-conjugated CD9 (Clone MZ3; catalog no. 124805; BioLegend), CD63 (Clone NVG-2; catalog no. 143904; BioLegend), and CD81 (Clone Eat-2; catalog no. 104905; BioLegend) antibodies and incubated at 4 oC for 30 min. Microvesicles were mixed with 1:200 dilution of anti-annexin A1 (clone no. BL28553; catalog no. 855301; BioLegend) and incubated at 4 oC for 30 min, followed by incubation with CF647-anti-mouse secondary antibody (catalog no. 20281; Biotium). In experiments where EVs were incubated with biotin-LPS, labeling with FITC-labeled streptavidin (catalog no. 11-4317-87; Invitrogen) was also done to detect LPS. ImageStream and flow cytometric (for EVs isolated by DIC) analyses for FITC (for LPS), PE (for EVs) or CF647 (for MVs) were done with ImageStream®X Mark II Imaging Flow Cytometer (Cytek Biosciences) or BD LSRII, respectively. FACS data was analyzed in FlowJo.
Cell stimulations and inflammasome assays
WT, Casp11−/−, Aim2−/−, and Nlrp3−/− BMDMs, RAW macrophages and THP1 monocytes were primed with IFN-γ (1 ng/ml; R&D Systems) for 3 h and treated with PBS-EVs or LPS-EVs (25 μg/ml) prepared by incubating SEC-isolated EVs with PBS or LPS, respectively, as described above. Cells were also stimulated with 1 μg LPS or transfected with lipofectamine 2000-complexed LPS. IFN-γ-primed WT and Casp11−/− BMDMs were also stimulated with PBS- or LPS-bound EV mimic liposomes prepared as described above. After 16 h, cell death was analyzed by measuring LDH release in the supernatant with the LDH cytotoxicity kit (catalog no. MK401; Takara) and secreted cytokine levels in the supernatant were analyzed by ELISA according to the manufacturer’s protocol using the BMG Labtech CLARIOstar microplate reader. To assess GSDMD activation, cells were lysed in RIPA buffer with a protease inhibitor cocktail (catalog no. 1861279; Invitrogen), and the lysates were subjected to GSDMD immunoblotting as described later. Unprimed WT and Tlr4−/− BMDMs were stimulated with LPS, PBS-EVs and LPS-EVs for 16 h and TNF and IL-6 secretion was assessed by ELISA.
In vivo stimulations with EVs or LPS
Mice were primed with 150 μg of HMW poly(I:C) (catalog no. tlrl-pic-5; InvivoGen) for 6 h, followed by i.p. injection with PBS-EVs or LPS-EVs as indicated. Each recipient mouse received EVs from approximately 1 donor mouse. Plasma, liver, and spleen were harvested at indicated times post-EV treatment. The liver and spleen were homogenized in PBS with a protease inhibitor cocktail (catalog no. 1861279; Invitrogen). Liver and spleen homogenates were incubated with an equal volume of RIPA lysis buffer for 1 h at 4 oC and centrifuged at 20,000 x g for 10 min at 4 oC. The lysates were subjected to immunoblotting for GSDMD and control proteins. IL-1β, IL-18, IL-6, and TNF cytokine levels in plasma were analyzed by ELISA as described previously14. Unprimed WT and Tlr4−/− mice were injected i.p. with LPS-EVs for 6 h and plasma levels of TNF and IL-6 were assessed by ELISA. In some experiments, Casp11−/− mice were injected with DMSO or EV inhibitor (2.5 μg/g GW4869 [catalog no. 13127; Cayman chemical]) dissolved in Trappsol (catalog no. THPB-P; CDT Inc) on days 1 and 2, and then injected i.p. with FITC-LPS on day 3. After 5 h, splenocytes and peritoneal lavage cells were isolated for cytosol extraction, ImageStream flow cytometric analysis or immunogold EM analysis as described below. Furthermore, WT mice were injected with DMSO or EV inhibitor 2.5 μg/g GW4869 or 25 μg/g Nexinhib20 (catalog no. 29899; Cayman chemical) dissolved in Trappsol on days 1 and 2, and then injected i.p. LPS (25 mg/kg) on day 3 and plasma, liver, and spleen were harvested 6 h post-LPS injection to assess IL-1β and IL-18 secretion and GSDMD activation. In certain experiments, GW4869-treated LPS-injected mice were administered with LPS-EVs and GSDMD activation and IL-1β and IL-18 secretion were analyzed as described above. WT and Casp11−/− mice were injected i.p. with DMSO or 2.5 μg/g GW4869 on days 1, 2 and 3 and injected i.p. with LPS (25 mg/kg) after 6 h and observed for survival. Tlr4−/− mice were injected with DMSO or 2.5 μg/g GW4869 on days 1, 2 and 3, primed with 150 μg of poly(I:C) on day 3 and injected i.p. with LPS (25 mg/kg) after 6 h and observed for survival.
Transmission Electron Microscopy
Casp11−/− BMDMs were primed with IFN-γ (10 ng/ml; R&D Systems) and treated with SEC-isolated PBS-EVs, FITC-LPS EVs, or FITC-LPS alone for 6 h. Also, mice were injected with SEC-isolated PBS-EVs or FITC-LPS-EVs, and peritoneal lavage cells and splenic Cd11b+ cells were collected, fixed, and stained as previously described21. Briefly, after fixing and permeabilizing, the cells were stained with a nano-gold conjugated antibody against FITC (catalog no. 25581; Electron microscopy science), and the gold particle size was enhanced using the GoldEnhance EM kit (catalog no. 2113-8ML; Nanoprobes) to visualize cytosolic LPS. Next, the samples were stained with osmium tetraoxide and uranyl acetate, followed by embedding in resin. Finally, ultrathin sections of the resin block were imaged on Hitachi H-7650 Transmission Electron Microscope at 80kV.
Cytosolic fractionation
Casp11−/− iBMDMs were primed with IFN-γ (10 ng/ml) for 3 h and treated with SEC-isolated PBS-EVs or FITC-LPS-EVs as described above, and the cytosolic fraction was isolated using digitonin as described previously14,21. For in vivo cytosolic fractionation, Casp11−/− mice were injected with SEC-isolated PBS-EVs or FITC-LPS-EVs. The spleen was collected 2.5 or 5 h post-EV injection and digested using Collagenase with DNase I in BSS at 37°C for 25 min. RBC was lysed using ACK Lysing Buffer (catalog no. A10492-01; Gibco). Myeloid cells were enriched by negative selection using B220 magnetic beads (clone RA3-6B2; catalog no. 551513; BD IMag) followed by positive selection with CD11b magnetic beads (clone M1/70; catalog no. 558013; BD IMag). To isolate endothelial cells, CD45+ve leukocytes were depleted from the single cell suspensions of splenocytes prepared as above using biotinylated-CD45.2 monoclonal antibody- (clone 104; catalog no. 13-0454-85, Invitrogen) and dynabeads biotin binder (catalog no. 11047; Invitrogen). Then these cells were stained with APC-conjugated anti-CD31 antibody (clone MEC13.3; catalog no. 102509; BioLegend) and PE-conjugated anti-CD45 antibody (clone 104; catalog no. 109807; BioLegend) and sorted for CD45-ve CD31+ve endothelial cells. Cytosolic fraction was collected using digitonin as described previously14,21. The cytosolic LPS levels were measured by the LAL and HEK-Blue TLR4 reporter cell assays as described above.
Confocal microscopy and imaging flow cytometry
Casp11−/− iBMDMs were primed with IFN-γ (10 ng/ml; R&D Systems) for 3 h and stimulated with SEC-isolated PBS-EVs, FITC-LPS-EVs, or FITC-LPS (as control) for 5–6 h. The cells were washed with PBS, fixed and permeabilized with 4% PFA and 0.1% Triton X-100, respectively, and blocked with 10% goat serum. The cells were then stained with APC-conjugated anti-CD45 (clone 104; catalog no. 17-0454-82; Invitrogen) and CF488A-conjugated anti-FITC (clone 1F8.1E4; catalog no. 20210; Biotium) antibodies, to visualize the plasma membrane and intracellular LPS, respectively21. Images were taken on a Zeiss LSM880 microscope, and intracellular FITC fluorescence was quantified using ImageJ. In some experiments, EVs isolated from WT mice via the UC method were labeled with CellBrite™Steady 550 (Catalog no. 30107; Biotium) or CellBrite™Steady 488 (Catalog no. 30106; Biotium) for 30 min at 37 oC in the dark according to the manufacturer’s instructions. After washing, CellBriteTMSteady 550-labeled EVs were incubated with FITC-LPS (500 μg) as described above and subjected to purification by SEC and concentration by UC. IFN-γ-primed Casp11−/− iBMDMs were treated with FITC-LPS-EVs or CellBrite 550-labeled FITC-LPS-EVs or CellBrite 488-labeled EVs. After 4.5 h, cells were washed, fixed, permeabilized, and blocked with 10% goat serum as described above. Subsequently, cells were stained with fluorophore-conjugated antibodies against mouse CD14 (clone Sa 2–8; catalog no. 17-0141-81; Invitrogen), EAA1 (clone 45B10; catalog no. 3288S; Cell Signaling Technology) and FITC (clone 1F8.1E4; catalog no. 20210; Biotium) and subjected to confocal imaging as described above. WT BMDMs were either stimulated with LPS or LPS-EVs for 6h, followed by fixation, permeabilization and blocking as described above. Cells were stained with lamp1 (clone eBio1D4B; catalog no. 14-1071-85; eBioscience), galectin-3 (clone D4I2R; catalog no. 12733; Cell Signaling) and Rab5A (clone E6N8S; catalog no. 46449S; Cell Signaling) antibodies and the respective fluorophore-conjugated secondary antibodies. Splenic myeloid and endothelial cells isolated as described above and peritoneal lavage cells from FITC-LPS-injected mice were stained with CD45 (for myeloid and peritoneal cells) or CD31 (endothelial cells) antibody and anti-FITC antibody to visualize the plasma membrane and LPS, respectively, and subjected to ImageStream flow cytometric analysis to assess intracellular localization of LPS.
Immunoblotting
Proteins were quantified with a BCA assay kit by Pierce BCA Protein Assay Kit (23227). Total protein (50 μg) was mixed with NuPAGE LDS sample buffer (Invitrogen) and run on polyacrylamide gels and then transferred onto nitrocellulose membranes using the Trans-Blot Turbo Transfer System (Bio-Rad Laboratories). Membranes were blocked in 2.5% milk and probed with the appropriate primary and secondary antibodies. Blots were visualized for proteins using the Bio-Rad Clarity-ECL HRP substrate on a Syngene or BioRAD gel documentation system. Immunoblot analysis was done with antibodies to mouse GSDMD (clone EPR 19828; catalog no. Ab209845; Abcam), caspase-11 (clone 17D9; catalog no. 14340S; Cell Signaling Technology), caspase-1 p20 (clone casper1; catalog no. AG-20B-0042-C100; Adipogen), IL-1β (catalog no. AF-401-NA; R&D Systems), CD9 (clone C-4; catalog no. SC-13118; Santa Cruz Biotechnology), CD63 (clone MX-49.129.5; catalog no. SC-5275; Santa Cruz Biotechnology), GAPDH (clone D16H11; catalog no. 5174; Cell Signaling,), and β-actin (clone 8H10D10; catalog no. 3700; Cell Signaling).
Statistics and Reproducibility
All in vitro and in vivo experiments were performed three times, unless specified otherwise in the figure legends. Data from in vitro experiments are presented as mean±s.e.m. In graphs showing in vivo data, each circle represents a mouse, and the horizontal lines represent the mean. Immunoblots presented are from one experiment representative of indicated number of experiments. The n values are provided in the legends. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications21,37. No randomization was performed. Sex- and age-matched mice were assigned to different experimental groups. Data collection and analysis were not performed blind to the conditions of the experiments. No data were excluded from the analyses. Data distribution was assumed to be normal but this was not formally tested. Statistical analysis was performed on GraphPad Prism using unpaired two-tailed t-test, one-way ANOVA, or two-way ANOVA as indicated in the figure legends. Survival studies were analyzed using the Mantel-Cox test. The exact P values were included in the graphs.
Extended Data
Extended Data Fig. 1: Host-derived EVs bind LPS in vivo.

a, Plasma EV levels in wild-type (WT) mice injected with PBS or LPS (10 mg/kg) for indicated times as assessed by nanoparticle tracking analysis (NTA). b, Size distribution of EVs isolated from the plasma of WT mice injected with PBS or LPS (25 mg/kg) 1.5 h post-injection via OptiPrep density gradient method as assessed by NTA. c, Immunoblotting analysis of EVs isolated from the plasma of PBS- or LPS-injected WT mice as in (b). d, Negative staining TEM of EVs isolated from the plasma of PBS or LPS-injected WT mice as in (b). e, LPS content of the EVs isolated from WT mice injected with PBS or LPS as assessed by the LAL assay (n=4). f,g, Percentage of FITC-LPS+ve EVs (f) and FITC histogram and mean fluorescence intensity (MFI) of EVs (g) isolated from mice injected with PBS or FITC-LPS (25 mg/kg) 1.5 h post-injection as assessed by ImageStream flow cytometry (n=3). h, Immunoblotting analysis of EVs isolated by the DIC method using CD9, CD63, and CD81 antibodies-coated magnetic beads from the plasma of PBS- or FITC-LPS (25 mg/kg)-injected WT mice. i,j, LPS content of the DIC-isolated EVs from PBS- or FITC-LPS-injected mice 1.5 h post-injection as assessed by the LAL (n=3) (i) and HEK-Blue TLR4 reporter cell (n=3) (j) assays. k,l, Percentage of FITC-LPS+ve EVs (k) and FITC histogram and mean fluorescence intensity (MFI) of EVs (l) isolated by the DIC method from mice injected with PBS or FITC-LPS as assessed by flow cytometry (n=3). Combined data from two (a) or three (e,f,g,i,j,k,l) or representative data of two (b,c,d,h) independent experiments are shown. Data are shown as mean±s.e.m (a). Each circle represents a mouse, and the horizontal lines represent mean (a, e–g and i–l). P values were determined by one-way ANOVA with Dunnett’s post-test (a) or unpaired two-tailed t-test (e–g and i– l). Scale bar, 50 nm (d).
Extended Data Fig. 2: Microvesicles bind LPS in vivo.

(a–b) Percentage of FITC-LPS+ve microvesicles (MVs; annexin A1+ve) and LPS content of MVs isolated from Casp11−/− mice injected with PBS or FITC-LPS via ultracentrifugation as assessed by ImageStream flow cytometry (a) and the LAL assay (b). Each circle represents pooled sample from two mice. Combined data from two independent experiments are shown.
Extended Data Fig. 3: Plasma EVs bind LPS independent of blood components.
a, LPS content of the EVs isolated by the SEC method from the plasma incubated with PBS or FITC-LPS (500 μg) at 37 oC for 45 min as assessed by the LAL assay (n=3). b,c, Percentage of FITC-LPS+ve EVs (b) and FITC histogram and mean fluorescence intensity (MFI) of EVs (c) isolated by the SEC method from the plasma incubated with PBS or FITC-LPS in vitro as assessed by ImageStream flow cytometry (n=3). d–f and h–j, EVs isolated from the plasma by the SEC method were incubated with PBS, FITC-LPS, or biotin-LPS as indicated at 37 oC for 45 min in vitro, and the LPS binding was assessed by the LAL assay (n=3 or 6 as indicated) (d,h) and ImageStream flow cytometry (n=3 or 6 as indicated) (e,f,i,j). g, TEM of EVs isolated and treated as described above and stained with anti-FITC-gold particles. Arrows indicate LPS. Combined data from three (a–c and h–j) or six (d–f) experiments or one representative of two experiments (g) are shown. Each circle represents a mouse, and the horizontal lines represent the mean in a–f and h–j. P values were determined by unpaired two-tailed t-test. Scale bar, 50 nm (g).
Extended Data Fig. 4: Human plasma EVs and endothelial and epithelial cell EVs bind LPS.

a–h, EVs isolated from the human plasma by the SEC method were incubated with PBS, FITC-LPS or biotin-LPS as indicated at 37 oC for 45 min, and the LPS binding was assessed by the LAL (a,e) and HEK-Blue TLR4 reporter cell (b,f) assays and ImageStream flow cytometry (c,d,g,h) (n=3). i–p, EVs isolated from bEnd.3 endothelial (i–l) and HeLa (m–p) cells cultured in serum-free conditions by the UC method were incubated with PBS or FITC-LPS (500 μg) as indicated at 37 oC for 45 min followed by re-purification via SEC, and the LPS binding was assessed by the LAL (i,m) and HEK-Blue TLR4 reporter cell (j,n) assays and ImageStream flow cytometry (k,l,o,p) (n=3). Combined data from three experiments (a–p) are shown. Each circle represents an independent experiment, and the horizontal lines represent the mean. P values were determined by unpaired two-tailed t-test (a–p).
Extended Data Fig. 5: Intracellular localization of LPS is EV-dependent.

(a–d) Confocal microscopy of Casp11−/− iBMDMs stimulated for 4 h with unlabeled EVs (a), FITC-LPS (b), FITC-LPS-EVs (c), or CellBrite Steady Membrane 488-labeled-EVs (d) and stained with anti-CD45, anti-EEA1 and anti-FITC antibodies to visualize plasma membrane, early endosomes and LPS, respectively. Arrows indicate intracellular localization of EV-bound FITC-LPS (c) or CellBrite 488-labeled-EVs (d). Images representative of two experiments are shown. Scale bar: 5 μm.
Extended Data Fig. 6: LPS-laden host EVs induce endolysosomal membrane disruption.
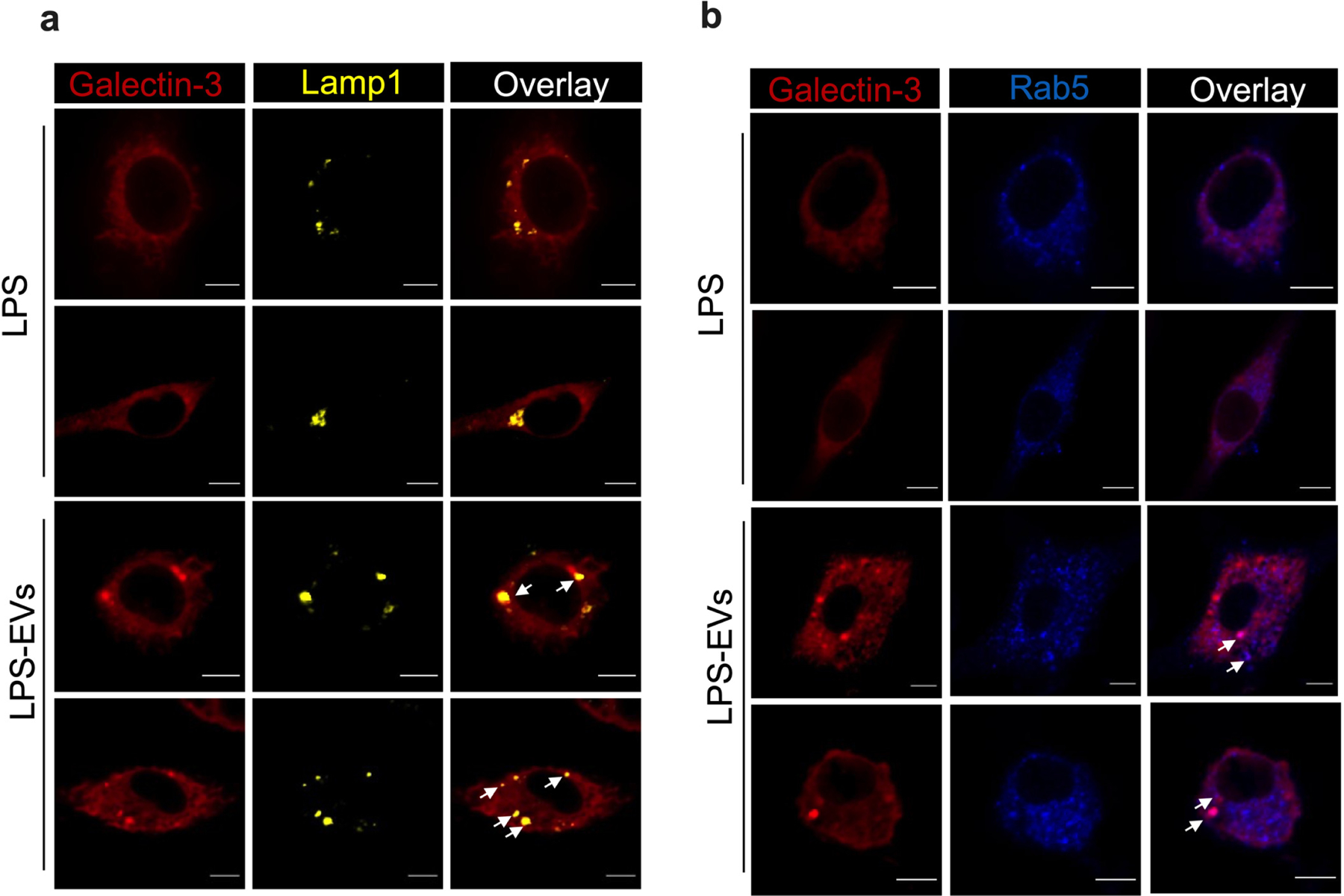
Confocal images of WT BMDMs stimulated with LPS or LPS-EVs for 6 h and stained for galectin-3 (red), lamp1 (yellow; a), and Rab5 (blue; b). Scale bar: 5 μM. White arrows indicate the colocalization of galectin-3 and lamp1 or Rab5. Images representative of two experiments are shown.
Extended Data Fig. 7: EV-bound LPS activates the noncanonical inflammasome.
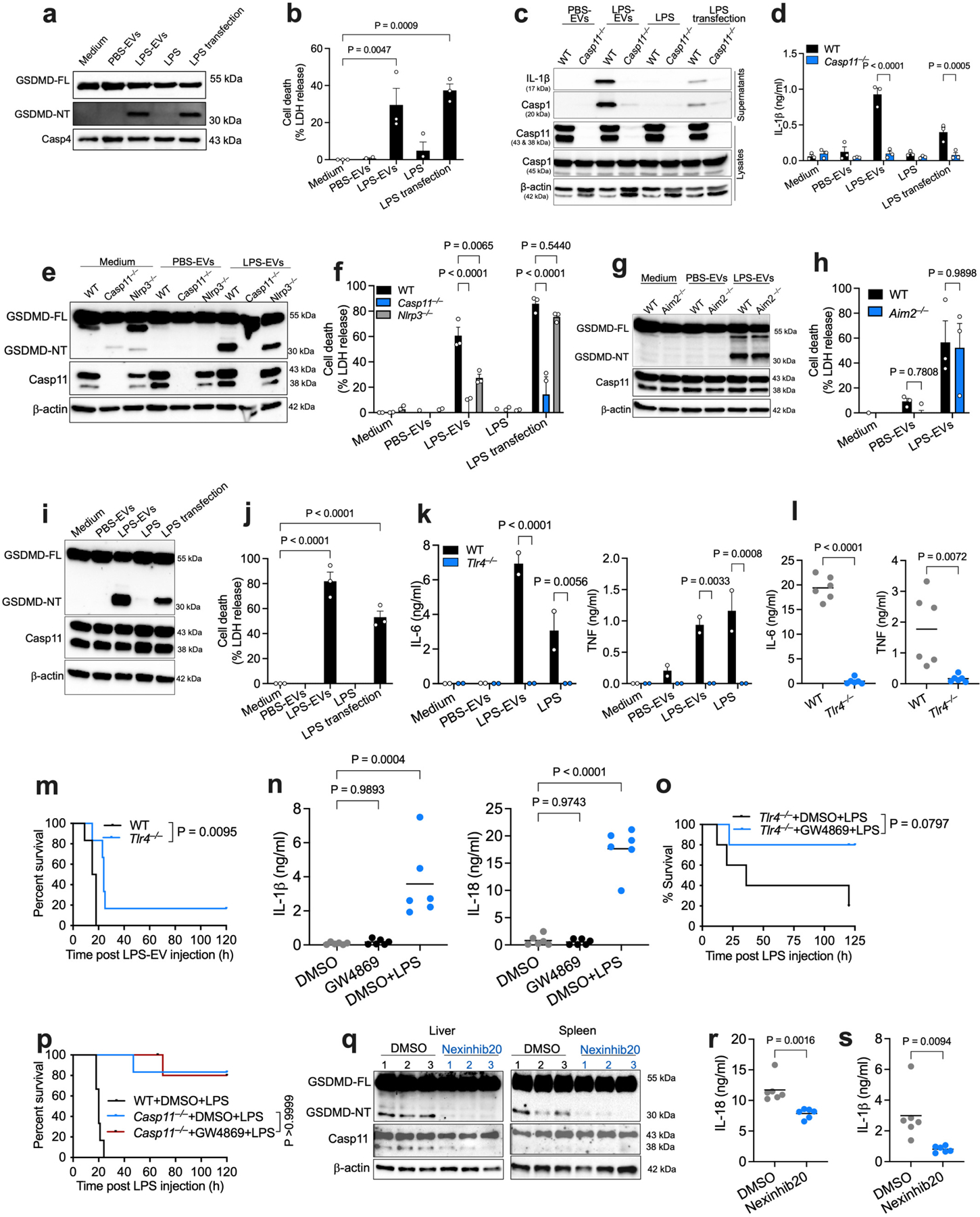
a,b, GSDMD and caspase-4 in lysates (a) and cell death (b) of IFN-γ-primed (10 ng/ml) THP1 monocytes stimulated as indicated for 16 h. c,d, Cleaved IL-1β (p17) and caspase-1 (p20) in the supernatants and indicated proteins in lysates of (c) and IL-1β secretion (d) by IFN-γ-primed WT and Casp11−/− BMDMs stimulated as indicated for 16 h. e–h, GSDMD, caspase-11 and β-actin in lysates (e,g) and cell death (f,h) of IFN-γ-primed indicated BMDMs stimulated as indicated for 16 h. i,j, Indicated proteins in lysates (i) and cell death (j) of IFN-γ-primed RAW macrophages stimulated as indicated for 16 h. k, IL-6 and TNF secretion by indicated BMDMs stimulated with PBS-EVs, LPS-EVs (2.5 μg LPS/ml) or LPS (2.5 μg/ml) for 16 h. l, Plasma IL-6 and TNF in indicated mice administered with LPS-EVs for 6 h (n=6). m, Survival of poly(I:C)-primed WT and Tlr4−/− mice injected with LPS-EVs (n=6). n, Baseline IL-1β and IL-18 in the plasma of WT mice injected with DMSO (vehicle) or GW4869 (2.5 μg/g) twice 24 h apart. DMSO-treated LPS-injected mice are shown as controls (n=6). o, Survival of Tlr4−/− mice injected with DMSO or GW4869 (2.5 μg/g) on days 1–3, primed with poly(I:C) on day 3 and injected with LPS (25 μg/g) 6 h later (n=5). p, Survival of indicated mice injected with DMSO or GW4869 (2.5 μg/g) on days 1–3 and injected with LPS (25 μg/g) on day 3 (n=6). q–s, GSDMD, caspase-11 and β-actin in the liver and spleen (q) and IL-18 and IL-1β in the plasma (r,s; n=6) of WT mice pretreated with DMSO (2.5 μg/g) or Nexinhib20 (25 μg/g) on days 1 and 2 and injected with LPS (25 μg/g) on day 3 for 8 h. Each circle represents a mouse, and the horizontal lines represent mean (l,n,r,s). Each lane represents a mouse (q). Data are presented as mean±s.e.m (b,d,f,h,j,k). Combined data from two (k–p,r,s) or three (b,d,f,h,j) experiments or one experiment representative of two (a,c,e,g,i,q) are shown. P values were determined by one-way ANOVA with Dunnett’s post-test (b,j,n), two-way ANOVA with Sidak’s post-test (d,f,h,k), unpaired two-tailed t-test (l,r,s) or Mantel-Cox test (m,o,p).
Extended Data Fig. 8: EV binding of LPS is not mediated by common LPS binding proteins.

a–d, LPS binding by EVs preincubated with isotype control, anti-HMGB1 or anti-LBP antibodies (Ab) and incubated with FITC-LPS (500 μg) at 37 oC for 45 min as assessed by ImageStream flow cytometry (a,c) and the HEK-Blue TLR4 reporter cell assay (b,d). Combined data from three experiments are shown, and the horizontal lines represent mean. P values were determined by unpaired two-tailed t-test.
Extended Data Fig. 9: EV binding of LPS is not mediated by EV surface proteins.

a,b, Immunoblotting (a) and negative staining TEM (b) of unshaved EVs and EVs subjected to surface protein shaving with trypsin (shaved EVs). c,d, Unshaved and shaved EVs incubated with FITC-LPS at 37 oC for 45 min were subjected to NTA (c), and the HEK-Blue TLR4 reporter cell assay (d). e,f, Percentage of FITC-LPS+ve EVs (e) and FITC MFI (f) of EVs isolated from FITC-LPS-injected mice and subjected or not to surface-protein shaving (n=3). Combined data from three (c,d) or four (e,f) experiments or one experiment representative of two (a,b) are shown, and horizontal lines represent mean (c–f). P values were determined by unpaired two-tailed t-test.
Extended Data Fig. 10: LPS-bound EVs colocalize with CD14.

a,b, Casp11−/− iBMDMs were stimulated with FITC-LPS-EVs unlabeled (a) or labeled with CellBrite Steady 550 dye (b). After 4 h, cells were stained with anti-CD14, anti-EEA1, and anti-FITC antibodies and subjected to confocal imaging. In (b), light blue arrows indicate colocalization of FITC-LPS-EVs (CellBrite-labeled) with CD14 (top two rows) and their internalization (bottom two rows). Images are representative of two experiments. Scale bar, 5 µm (a,b).
Acknowledgements:
We thank V. Dixit and K. Fitzgerald for Casp11−/− mice and N. Frank for the HEK-Blue TLR4 reporter cell line. This work was supported by the National Institutes of Health grant nos. R01AI119015 and R01AI148491 (V.A.R.) and R01AI132850 (S.K.V.). V.F.A., I.R., and M.B. were supported by the Deutsche Forschungsgemeinschaft (German Research Foundation) under Germany’s Excellence Strategy (EXC 2051: Balance of the Microverse, project number 390713860). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Competing interests statement: Y.Z.’s company FormuMax is interested in the sales of exosome-mimicking liposomes. The remaining authors declare no competing interests.
Data availability:
Source data and uncropped immunoblot images are included in the paper as supplementary information. All other data supporting the findings of the paper are available from the corresponding author upon reasonable request.
References
- 1.Gabanyi I et al. Bacterial sensing via neuronal Nod2 regulates appetite and body temperature. Science 376, eabj3986 (2022). [DOI] [PubMed] [Google Scholar]
- 2.Clarke TB et al. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med 16, 228–231 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farrokhi V et al. Bacterial lipodipeptide, Lipid 654, is a microbiome-associated biomarker for multiple sclerosis. Clin Transl Immunol 2, e8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang Z et al. Antibody neutralization of microbiota-derived circulating peptidoglycan dampens inflammation and ameliorates autoimmunity. Nat Microbiol 4, 766–773 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Moltke J von, Ayres, J. S., Kofoed, E. M., Chavarría-Smith, J. & Vance, R. E. Recognition of Bacteria by Inflammasomes. Annu Rev Immunol 31, 73–106 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Kawai T & Akira S The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11, 373–384 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Poltorak A et al. Defective LPS Signaling in C3H/HeJ and C57BL/10ScCr Mice: Mutations in Tlr4 Gene. Science 282, 2085–2088 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Rathinam VAK, Zhao Y & Shao F Innate immunity to intracellular LPS. Nat Immunol 20, 527–533 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kayagaki N et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 341, 1246–1249 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Hagar JA, Powell DA, Aachoui Y, Ernst RK & Miao EA Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 341, 1250–1253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi J et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Shi J et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Kayagaki N et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Vanaja SK et al. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 165, 1106–1119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo Z et al. Variation in blood microbial lipopolysaccharide (LPS) contributes to immune reconstitution in response to suppressive antiretroviral therapy in HIV. Ebiomedicine 80, 104037 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohammad S & Thiemermann C Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front Immunol 11, 594150 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kell DB & Pretorius E On the translocation of bacteria and their lipopolysaccharides between blood and peripheral locations in chronic, inflammatory diseases: the central roles of LPS and LPS-induced cell death. Integr Biol 7, 1339–1377 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Prins JM, Deventer S. J. van, Kuijper EJ& Speelman P Clinical relevance of antibiotic-induced endotoxin release. Antimicrob Agents Ch 38, 1211 1218 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuura M Structural Modifications of Bacterial Lipopolysaccharide that Facilitate Gram-Negative Bacteria Evasion of Host Innate Immunity. Front Immunol 4, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukui H Endotoxin and Other Microbial Translocation Markers in the Blood: A Clue to Understand Leaky Gut Syndrome. Cell Mol Medicine Open Access 2, (2016). [Google Scholar]
- 21.Vasudevan SO, Russo AJ, Kumari P, Vanaja SK & Rathinam VA A TLR4-independent critical role for CD14 in intracellular LPS sensing. Cell Reports 39, 110755 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colombo M, Raposo G & Théry C Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu Rev Cell Dev Bi 30, 1–35 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Jeppesen DK et al. Reassessment of Exosome Composition. Cell 177, 428–445.e18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalluri R & LeBleu VS The biology, function, and biomedical applications of exosomes. Science 367, eaau6977 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Brien K, Breyne K, Ughetto S, Laurent LC & Breakefield XO RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biology 21, 585–606 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim D-K et al. EVpedia: a community web portal for extracellular vesicles research. Bioinformatics 31, 933 939 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang N et al. Circulating exosomes contain protein biomarkers of metastatic non‐small‐cell lung cancer. Cancer Sci 109, 1701–1709 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li W et al. LPS Induces Active HMGB1 Release From Hepatocytes Into Exosomes Through the Coordinated Activities of TLR4 and Caspase-11/GSDMD Signaling. Front Immunol 11, 229 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cosme J, Guo H, Hadipour-Lakmehsari S, Emili A & Gramolini AO Hypoxia-Induced Changes in the Fibroblast Secretome, Exosome, and Whole-Cell Proteome Using Cultured, Cardiac-Derived Cells Isolated from Neonatal Mice. J Proteome Res 16, 2836–2847 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Jin M, Drwal G, Bourgeois T, Saltz J & Wu HM Distinct proteome features of plasma microparticles. Proteomics 5, 1940–1952 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Kugeratski FG et al. Quantitative proteomics identifies the core proteome of exosomes with syntenin-1 as the highest abundant protein and a putative universal biomarker. Nat Cell Biol 23, 631–641 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Budden CF et al. Inflammasome‐induced extracellular vesicles harbour distinct RNA signatures and alter bystander macrophage responses. J Extracell Vesicles 10, e12127 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lobb RJ et al. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J Extracell Vesicles 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathieu M, Martin-Jaular L, Lavieu G & Théry C Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat Cell Biol 21, 9–17 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Jeppesen DK, Zhang Q, Franklin JL & Coffey RJ Extracellular vesicles and nanoparticles: emerging complexities. Trends Cell Biol (2023) doi: 10.1016/j.tcb.2023.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng KT et al. Caspase-11–mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest 127, 4124–4135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumari P, Russo AJ, Wright SS, Muthupalani S & Rathinam VA Hierarchical cell-type-specific functions of caspase-11 in LPS shock and antibacterial host defense. Cell Rep. 35, 109012 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Russo AJ et al. Intracellular immune sensing promotes inflammation via gasdermin D–driven release of a lectin alarmin. Nat Immunol 22, 154–165 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gong T, Liu L, Jiang W & Zhou R DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol 20, 95–112 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Catalano M & O’Driscoll L Inhibiting extracellular vesicles formation and release: a review of EV inhibitors. J Extracell Vesicles 9, 1703244 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang H, Lu J, Liu J, Zhang G & Lu A Advances in the discovery of exosome inhibitors in cancer. J Enzym Inhib Med Ch 35, 1322–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu M et al. Comparison of exosome-mimicking liposomes with conventional liposomes for intracellular delivery of siRNA. Int J Pharmaceut 550, 100–113 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Raetz CRH, Reynolds CM, Trent MS & Bishop RE Lipid A Modification Systems in Gram-Negative Bacteria. Annu Rev Biochem 76, 295–329 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valadi H et al. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9, 654 659 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Zanoni I et al. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 147, 868 880 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ & Mathison JC CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249, 1431 1433 (1990). [DOI] [PubMed] [Google Scholar]
- 47.Kitchens RL & Munford RS CD14-dependent internalization of bacterial lipopolysaccharide (LPS) is strongly influenced by LPS aggregation but not by cellular responses to LPS. J Immunol 160, 1920–8 (1998). [PubMed] [Google Scholar]
- 48.Tan Y, Zanoni I, Cullen TW, Goodman AL & Kagan JC Mechanisms of Toll-like Receptor 4 Endocytosis Reveal a Common Immune-Evasion Strategy Used by Pathogenic and Commensal Bacteria. Immunity 43, 909–922 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keller MD et al. Decoy exosomes provide protection against bacterial toxins. Nature 579, 260–264 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robbins PD & Morelli AE Regulation of immune responses by extracellular vesicles. Nat Rev Immunol 14, 195 208 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Théry C, Ostrowski M & Segura E Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 9, 581–593 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Vreugdenhil ACE et al. Lipopolysaccharide (LPS)-Binding Protein Mediates LPS Detoxification by Chylomicrons. J Immunol 170, 1399–1405 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Vreugdenhil ACE, Snoek AMP, Veer C van ‘t, Greve, J.-W. M. & Buurman, W. A. LPS-binding protein circulates in association with apoB-containing lipoproteins and enhances endotoxin-LDL/VLDL interaction. J Clin Invest 107, 225–234 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berbée JFP, Havekes LM & Rensen PCN Apolipoproteins modulate the inflammatory response to lipopolysaccharide. J Endotoxin Res 11, 97–103 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Yao Z et al. Blood-Borne Lipopolysaccharide Is Rapidly Eliminated by Liver Sinusoidal Endothelial Cells via High-Density Lipoprotein. J Immunol 197, 2390–2399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Topchiy E et al. Lipopolysaccharide Is Cleared from the Circulation by Hepatocytes via the Low Density Lipoprotein Receptor. Plos One 11, e0155030 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deng M et al. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 49, 740 753.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kayagaki N et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Lamkanfi M et al. Inflammasome-Dependent Release of the Alarmin HMGB1 in Endotoxemia. J Immunol 185, 4385–4392 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kayagaki N et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591, 131–136 (2021). [DOI] [PubMed] [Google Scholar]
- 61.Minihane AM et al. Low-grade inflammation, diet composition and health: current research evidence and its translation. Brit J Nutr 114, 999–1012 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Glaros TG et al. Causes and consequences of low grade endotoxemia and inflammatory diseases. Frontiers in bioscience (Scholar edition) 5, 754 765 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Geng S et al. The persistence of low-grade inflammatory monocytes contributes to aggravated atherosclerosis. Nat Commun 7, 13436 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pasternak BA et al. Lipopolysaccharide Exposure is Linked to Activation of the Acute Phase Response and Growth Failure in Pediatric Crohn’s Disease and Murine Colitis. Inflamm Bowel Dis 16, 856 869 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Source data and uncropped immunoblot images are included in the paper as supplementary information. All other data supporting the findings of the paper are available from the corresponding author upon reasonable request.