

Abstract
Introduction
Patients with psoriatic arthritis (PsA) require treatment providing durable long-term efficacy in different disease domains as well as safety. We present 100-week efficacy and safety results of risankizumab in patients with active PsA and previous inadequate response/intolerance to ≥ 1 conventional synthetic disease-modifying antirheumatic drug (csDMARD-IR).
Methods
KEEPsAKE 1 (NCT03675308) is a global phase 3 study, including a 24-week, double-blind, placebo-controlled and ongoing open-label extension periods. Patients were randomized 1:1 to receive risankizumab 150 mg or placebo at baseline and weeks 4 and 16. After week 24, all patients received open-label risankizumab every 12 weeks thereafter. Patients were evaluated through 100 weeks. Endpoints included achieving ≥ 20% reduction in American College of Rheumatology criteria for symptoms of rheumatoid arthritis (ACR20), minimal disease activity (MDA; defined as ≥ 5/7 criteria of low disease activity and extent), and other measures.
Results
Overall, 828/964 (85.9%) patients completed week 100. For patients receiving continuous risankizumab, 57.3%, 70.6%, and 64.3% achieved ACR20 at weeks 24, 52, and 100, respectively. For the placebo/risankizumab cohort, 33.5% achieved ACR20 at week 24 but increased after switching to active treatment at weeks 52 (63.7%) and 100 (62.1%). In ACR20 responders at week 52, 81.2% of both treatment cohorts maintained response at week 100. MDA was achieved by 25.0%, 38.3%, and 38.2% of the continuous risankizumab cohort at weeks 24, 52, and 100. In the placebo/risankizumab cohort, 10.2% achieved MDA at week 24, increasing at weeks 52 (28.0%) and 100 (35.2%). MDA response was maintained at week 100 in week 52 responders in the continuous risankizumab (75.5%) and placebo/risankizumab cohorts (78.2%). Similar trends were observed for other efficacy measures. Risankizumab was generally well tolerated through 100 weeks.
Conclusions
For patients with active PsA who are csDMARD-IR, risankizumab demonstrated durable long-term efficacy and was generally well tolerated, with a consistent long-term safety profile.
Trial Registration
ClinicalTrials.gov identifier, NCT03675308.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40744-024-00654-5.
Keywords: csDMARD-IR, KEEPsAKE 1, IL-23, Long-term treatment, Psoriatic arthritis, Risankizumab
Plain Language Summary
Psoriatic arthritis (PsA) often affects individuals with the skin condition psoriasis. A biologic disease-modifying antirheumatic drug can help control inflammation and regulate the immune system to ease symptoms and slow progression of PsA. The ongoing KEEPsAKE 1 study is evaluating the efficacy and safety of risankizumab in patients with active PsA who previously have not had success with ≥ 1 conventional disease-modifying antirheumatic drug. Patients were initially treated with risankizumab 150 mg (continuous risankizumab group) or inactive drug (inactive drug/risankizumab group). After 24 weeks, all received risankizumab for the rest of the study. At week 100, 64% (continuous risankizumab group) and 62% (inactive drug/risankizumab group) of patients had ≥ 20% improvement in PsA symptoms (measured using American College of Rheumatology [ACR20] criteria). Both groups showed similar percentages at week 52 and improvement from week 24. In patients who achieved ACR20 at week 52, 81% maintained their ACR20 response at week 100. Minimal disease activity was defined as a combination of joint and skin symptoms, affected body surface area, pain, and physical functioning. At week 100, 38% of the continuous risankizumab group and 35% of the inactive drug/risankizumab group achieved minimal disease activity. Percentages were similar at week 52 and higher than week 24 in both groups. In patients who achieved minimal disease activity at week 52, 81% maintained response at week 100. All other measures of treatment responses showed similar patterns from the start of risankizumab through week 100. Risankizumab was considered generally safe by the treating physicians.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40744-024-00654-5.
Key Summary Points
| Why carry out this study? |
| Because most patients with psoriatic arthritis (PsA) do not maintain minimal disease activity on current therapies, there is an unmet need for safe and effective long-term treatment. |
| This study reports the 100-week efficacy and safety results of the KEEPsAKE 1 study for patients with active PsA treated with risankizumab 150 mg every 12 weeks. |
| Patients enrolled in this study had previous inadequate response/intolerance to ≥ 1 conventional synthetic disease-modifying antirheumatic drug. |
| What was learned from the study? |
| At week 100, risankizumab demonstrated durable improvement in the signs and symptoms of PsA and was generally well tolerated with a stable safety profile. |
Introduction
The systemic, inflammatory disease psoriatic arthritis (PsA) has multiple clinical manifestations, including arthritis, dactylitis, and enthesitis [1]. An estimated 0.05–0.25% of the general population and 6–41% of patients with psoriasis develop PsA [1]; however, PsA remains underdiagnosed with as many as 30% of patients with psoriasis being diagnosed with PsA after assessment by rheumatologists at dermatology centers [2]. Patients with PsA may experience permanent joint damage and reduced health-related quality of life, and the disease burden has been associated with higher total healthcare costs [3–5].
Many different classes of treatments are available for PsA, including nonsteroidal anti-inflammatory drugs, glucocorticoid injections, systemic therapies with conventional synthetic disease-modifying antirheumatic drugs (csDMARDs), targeted synthetic DMARDs, and biologic therapies [6]. However, despite the variety of treatments available, just 33% of women and 50% of men with PsA achieve sustained minimal disease activity (MDA), a validated and well-accepted treatment goal for PsA [7, 8], and in some patients, the disease remains resistant or intolerant to therapies [6], demonstrating a continued unmet need for long-term efficacious treatments.
The biologic therapy risankizumab is a humanized immunoglobulin G1 monoclonal antibody that acts as an interleukin-23 antagonist, with specific binding to the p19 subunit of interleukin-23 [9, 10]. The efficacy and safety of risankizumab continue to be evaluated in patients with PsA in two ongoing global, phase 3, multicenter studies that began in 2019, KEEPsAKE 1 and KEEPsAKE 2, which are being conducted in parallel. In the KEEPsAKE 1 (NCT03675308) study, enrolled patients had active PsA with an inadequate response or intolerance to ≥ 1 csDMARD therapy (csDMARD-IR), and the KEEPsAKE 2 (NCT03671148) study enrolled patients had active PsA that was csDMARD-IR and/or had an inadequate response or intolerance to one or two biologic therapies for PsA [11, 12]. Previous reports from the KEEPsAKE 1 and 2 studies have demonstrated improved PsA signs and symptoms compared with placebo at week 24 [11–13] and efficacy at week 52 [14, 15]. In this report, we present the 100-week long-term efficacy, safety, and tolerability of risankizumab using data from the KEEPsAKE 1 study.
Methods
The study design and patient selection criteria for the KEEPsAKE 1 study have been previously reported [11, 14]. Briefly, the KEEPsAKE study 1 is an ongoing, global study comprising two treatment periods. Period 1 had a 24-week, double-blind, placebo-controlled, parallel-group treatment design, and period 2 permitted patients to continue in an open-label extension period starting from week 24. At baseline of period 1, patients were randomized 1:1 to receive either placebo or risankizumab 150 mg with doses administered subcutaneously at weeks 0, 4, and 16. To maintain blinding before entering open-label period 2, at week 24, patients were administered one dose of the opposite therapy (i.e., those patients randomized to placebo were administered a blinded dose of risankizumab, and those patients who were initially in the risankizumab cohort received blinded placebo). Beginning at week 28 and every 12 weeks thereafter, all patients received open-label risankizumab 150 mg. For the remainder of this report, the treatment cohorts will be called “continuous risankizumab” for patients initially randomized to risankizumab in period 1 and “placebo/risankizumab” for patients initially randomized to placebo in period 1 and switched to risankizumab starting at week 24.
Beginning at week 32, patients with < 20% improvement in tender or swollen joint count over two consecutive visits compared with baseline were discontinued from the study drug: week 36 was the first possible visit for a patient to discontinue because of lack of efficacy.
The study protocol, informed consent forms, and recruitment materials were approved by the independent ethics committee or institutional review board at each study site before patient enrollment began (Supplementary Material Table 1). The studies were conducted in accordance with the International Conference for Harmonisation guidelines, applicable regulations, and the Declaration of Helsinki. All patients provided written informed consent before entering the study.
Assessments
Efficacy
Efficacy was evaluated in all patients who received ≥ 1 dose of risankizumab. Measures of change in PsA signs and symptoms included the proportion of patients achieving ≥ 20%, ≥ 50%, and ≥ 70% improvement from baseline in American College of Rheumatology criteria (ACR20/ACR50/ACR70); change from baseline in PsA-modified Total Sharp Score (PsA-mTSS; total score range 0–528) [16]; and the proportion of patients with no radiographic progression, defined as change from baseline in PsA-mTSS ≤ 0 and ≤ 0.5. In addition, resolution of enthesitis (Leeds Enthesitis Index score = 0 among patients with Leeds Enthesitis Index > 0 at baseline) and dactylitis (Leeds Dactylitis Index score = 0 among patients with Leeds Dactylitis Index > 0 at baseline) was reported following a prespecified analysis of pooled data from the KEEPsAKE 1 and KEEPsAKE 2 studies.
As a measure of skin clearance, patients who had a psoriasis-affected body surface area (BSA) ≥ 3% at baseline were assessed for achieving a 90% improvement from baseline in Psoriasis Area and Severity Index (PASI 90; score range ≤ 3 represents mild disease, > 3 to 15 represents moderate disease, and > 15 represents severe disease). To assess nail clearance in patients with nail psoriasis at baseline, changes from baseline scores were evaluated using the modified Nail Psoriasis Severity Index (mNAPSI; score range 0–140, with lower scores reflecting less nail psoriasis) [17] and Physician’s Global Assessment of Fingernail Psoriasis (PGA-F, with lower scores reflecting less fingernail psoriasis) [18].
The proportion of patients who achieved a status of MDA was evaluated; MDA was defined as fulfilling ≥ 5 of the following seven criteria: a tender joint count ≤ 1, a swollen joint count ≤ 1, PASI ≤ 1 or affected BSA ≤ 3%, patient’s assessment of pain as measured by Visual Analog Scale (VAS) ≤ 15, patient’s global assessment of disease activity VAS ≤ 20, Health Assessment Questionnaire–Disability Index (HAQ-DI) score ≤ 0.5, and/or Leeds Enthesitis Index score ≤ 1.
Evaluated patient-reported outcomes were change from baseline in HAQ-DI as a measure of physical function (lower scores reflect less disability), proportion of patients achieving clinically meaningful improvement in HAQ-DI (≥ 0.35 decrease from baseline) [19], pain measured by a 100-mm horizontal VAS (score range 0–100; lower scores reflect less pain), 36-item Short Form Physical Component Summary (SF-36 PCS; higher scores reflect less physical impairment), and Functional Assessment of Chronic Illness Therapy–Fatigue (FACIT-Fatigue; higher scores reflect less fatigue). All efficacy outcomes were assessed through week 100. Maintenance of response at week 100 for patients who achieved ACR20, ACR50, ACR70, PASI 90, MDA, and clinically meaningful improvements in patient’s assessment of pain (defined as a decrease from baseline assessment of pain of ≥ 10 mm in pain VAS) was also reported.
Safety
All patients who received ≥ 1 dose of the study drug were included in safety assessments. Safety was evaluated throughout the study and included assessments of treatment-emergent adverse events (TEAEs; coded using the Medical Dictionary for Regulatory Activities version 24.1), vital sign measurements, and physical examinations. Results were reported at week 24 and through the cutoff date of March 21, 2022, which includes data through week 100. TEAEs were classified by having an onset date on or after the first dose of the study drug and up to 140 days after the last dose (if the patient discontinued the study drug prematurely).
Statistical Analysis
For binary efficacy endpoints, data through week 24 were reported using nonresponder imputation where patients with missing data due to COVID-19 were handled by multiple imputations, and patients with missing data due to any other reason were treated as nonresponders. Additionally, patients who initiated concomitant medications for PsA that could meaningfully impact efficacy assessments or who received rescue therapy before week 24 were treated as nonresponders. For binary efficacy endpoints after week 24, patients with missing data due to COVID-19 or geopolitical conflict in Ukraine and Russia were handled by multiple imputations; all other missing data were treated as nonresponders, including missing data and patients who received rescue therapy, except for PGA-F which was reported as observed. For all time points, continuous nonradiographic efficacy endpoints were analyzed using a mixed-effect model for repeated measures; all as observed data were included in the model. Data for mTSS are reported as observed.
TEAEs were summarized using exposure-adjusted event rates (events [E]/100 patient-years [PY]).
Results
Patient Disposition and Characteristics
Overall, 964 patients were randomized, with 939 (97.4%) eligible patients choosing to enter period 2 to receive open-label risankizumab. Of all randomized patients, 828 (85.9%) were enrolled at the week 100 data cutoff date (continuous risankizumab, n = 412; placebo/risankizumab, n = 416). As previously reported, baseline characteristics were similar between treatment groups [11]. The most common reasons for study discontinuation in study period 2 were withdrawal of consent (n = 36, 3.7%) and < 20% improvement in tender/swollen joint count for two consecutive visits compared with baseline, as mandated by study protocol (n = 23, 2.4%).
Efficacy
Joint-Related Endpoints Through Week 100
As previously reported, significantly more patients receiving risankizumab achieved the primary endpoint of ACR20 at week 24 (57.3%) compared with placebo (33.5%; p < 0.001; Fig. 1) [11].
Fig. 1.

Achievement over time of ACR improvement endpoints and MDA. ACR20/50/70 ≥ 20%/≥ 50%/≥ 70% improvement in American College of Rheumatology criteria for symptoms of rheumatoid arthritis, CI confidence interval, MDA minimal disease activity, NRI-C, nonresponder imputation incorporating multiple imputations to handle missing data due to COVID-19, NRI-MI nonresponder imputation incorporating multiple imputations to handle missing data due to COVID-19 and geopolitical conflict, and all other missing data treated as nonresponders, including missing data and patients who received rescue therapy, PBO placebo, RZB risankizumab
At week 100, 64.3% and 62.1% of patients achieved ACR20 in the continuous risankizumab cohort and placebo/risankizumab cohort, respectively, which were consistent with results observed at week 52 in both groups (70.6% and 63.7%) (Fig. 1).
Durable efficacy was also achieved for ACR50 and ACR70 through week 100. The proportion of patients achieving ACR50 at week 100 was 42.4% in the continuous risankizumab cohort and 44.2% in the placebo/risankizumab cohort; the corresponding proportion of patients achieving ACR50 at week 52 was 43.6% and 38.3% and at week 24 it was 33.4% and 11.3%, respectively (Fig. 1) [11]. The proportion of patients achieving ACR70 was durable at week 100 (26.7% and 27.0%) from week 52 (25.9% and 20.5%); these proportions were greater than week 24 (15.3% and 4.7%) in both the continuous risankizumab and placebo/risankizumab cohorts (Fig. 1) [11].
Similar findings in ACR20, ACR50, and ACR70 were reported in both treatment cohorts when analyzed using the as observed data approach (Supplementary Material Fig. 1). In patients with ACR20 response at week 52, 81.2% of both cohorts maintained their response at week 100.
Maintenance of response at week 100 was also observed in 72.5% and 79.7% of week 52 ACR50 responders and in 69.1% and 71.2% of week 52 ACR70 responders in the continuous risankizumab and placebo/risankizumab cohorts, respectively (Supplementary Material Table 2).
Maintenance of response at week 100 for ACR20, ACR50, and ACR70 in week 52 responders was also seen in both cohorts when evaluated using the as observed approach (Supplementary Material Table 2).
Mean change from baseline in PsA-mTSS in the KEEPsAKE 1 study indicated low radiographic progression from baseline to week 100 (0.34 for the continuous risankizumab cohort and 0.45 for the placebo/risankizumab cohort), which remained stable from week 52 (0.21 and 0.27) and week 24 (0.23 and 0.32) in both treatment cohorts, respectively (Table 1). Most patients at week 100 (90.6% for continuous risankizumab and 86.9% for placebo/risankizumab) achieved change from baseline in PsA-mTSS ≤ 0, with no radiographic progression defined as PsA-mTSS ≤ 0 and PsA-mTSS ≤ 0.5, similar to week 52 (91.7% and 89.9%) and week 24 (92.4% and 87.7%) (Table 1). Similar numerical results were reported in the proportion of patients achieving change from baseline in PsA-mTSS ≤ 0.5 at week 100, with a comparable pattern of results across weeks 52 and 24 (Table 1). Similar findings in change from baseline in PsA-mTSS were reported when analyzed using as observed data approach (Supplementary Material Table 3).
Table 1.
Efficacy assessments
| Parameter | Week 24 (period 1)a | Week 52 (period 1 and 2)b | Week 100 (period 1 and 2) | |||
|---|---|---|---|---|---|---|
| RZB (n = 483) | PBO (n = 481) | Continuous RZB (n = 483) | PBO/RZB (n = 481) | Continuous RZB (n = 483) | PBO/RZB (n = 481) | |
| Change from baseline PsA-mTSS,c mean [n/N] (95% CI) |
0.23 [451/458] (0.02, 0.44) |
0.32 [451/457] (0.11, 0.53) |
0.21 [375/377] (0.04, 0.38) |
0.27 [385/390] (0.10, 0.44) |
0.34 [372/377] (0.17, 0.52) |
0.45 [383/390] (0.28, 0.62) |
| PsA-mTSS ≤ 0,c,d % [n/N] (95% CI) |
92.4 [423/458] (89.9, 94.8)* |
87.7 [401/457] (84.7, 90.8) |
91.7 [344/375] (88.9, 94.5) |
89.9 [346/385] (86.9, 92.9) |
90.6 [337/372] (87.6, 93.6) |
86.9 [333/383] (83.6, 90.3) |
| PsA-mTSS ≤ 0.5,c,d % [n/N] (95% CI) |
93.7 [429/458] (91.4, 95.9) |
90.4 [413/457] (87.7, 93.1) |
93.6 [351/375] (91.1, 96.1) |
92.5 [356/385] (89.8, 95.1) |
91.9 [342/372] (89.2, 94.7) |
89.3 [342/383] (86.2, 92.4) |
| Change from baseline in PGA-F,e mean (95% CI) |
− 0.8 (− 1.0, − 0.7)*** |
− 0.4 (− 0.5, − 0.3) |
− 1.2 (− 1.3, − 1.1) |
− 1.1 (− 1.2, − 1.0) |
− 1.4 (− 1.5, − 1.3) |
− 1.3 (− 1.4, − 1.2) |
| Clinically meaningful reduction from baseline in HAQ-DI (≥ 0.35),f % [n/N] (95% CI) |
50.3 [208/414] (45.5, 55.1)*** |
27.9 [117/419] 23.5, 32.2) |
57.9 [240/414] (53.1, 62.6) |
46.4 [194/419] (41.6, 51.2) |
54.9 [227/414] (50.0, 59.7) |
48.4 [203/419] (43.5, 53.3) |
| Change from baseline in Patient’s Assessment of Pain VAS,g mean (95% CI) |
− 21.0 (− 23.2, − 18.8)*** |
− 10.2 (− 12.5, − 8.0) |
− 26.5 (− 28.6, − 24.5) |
− 22.6 (− 24.7, − 20.6) |
− 26.9 (− 29.0, − 24.9) |
− 25.0 (− 27.1, − 22.9) |
| Change from baseline in SF-36 PCS, mean [n/N] (95% CI) |
6.5 [474/482] (5.8, 7.2)*** |
3.2 [470/477] (2.5, 3.9) |
8.4 [443/476] (7.8, 9.1) |
7.3 [437/473] (6.7, 8.0) |
8.4 [403/476] (7.7, 9.1) |
7.5 [398/473] (6.8, 8.2) |
| Change from baseline in FACIT-Fatigue, mean (95% CI) |
6.5 (5.6, 7.3)*** |
3.9 (3.1, 4.7) |
8.0 (7.2, 8.8) |
6.5 (5.7, 7.3) |
7.8 (6.9, 8.6) |
6.9 (6.0, 7.7) |
The continuous RZB cohort was randomized to RZB 150 mg (period 1) and remained on RZB (period 2). The PBO/RZB cohort was randomized to placebo (period 1) and switched to RZB 150 mg (period 2). All changes are least squares mean change from baseline. Results are based on the full analysis set and use NRI-C at week 24, where missing data due to COVID-19 were imputed by multiple imputation, and NRI-MI at weeks 52 and 100, where missing data due to COVID-19 and geopolitical conflict were handled by multiple imputation and all other missing data were treated as nonresponders, including missing data and patients who received rescue therapy; MMRM was used for continuous endpoints, excluding mTSS
CI confidence interval, FACIT-Fatigue Functional Assessment of Chronic Illness Therapy–Fatigue, HAQ-DI Health Assessment Questionnaire–Disability Index, MMRM mixed-effect model for repeated measures, NRI-C nonresponder imputation incorporating multiple imputations to handle missing data due to COVID-19, NRI-MI nonresponder imputation incorporating multiple imputations to handle missing data due to COVID-19 and geopolitical conflict, and all other missing data treated as nonresponders, including missing data and patients who received rescue therapy, PBO placebo, PGA-F Physician’s Global Assessment of Fingernail Psoriasis, PsA-mTSS psoriatic arthritis-modified Total Sharp Score, RZB risankizumab, SF-36 PCS 36-item Short Form Physical Component Summary, VAS Visual Analog Scale
aData previously reported [11]
bBinary endpoint data previously reported using nonresponder imputation [14]
cResults for PsA-mTSS were recorded from the third reading session (weeks 52 and 100)
dResults for PsA-mTSS ≤ 0 and ≤ 0.5 were reported using as observed data with no imputation for missing data
eAmong patients with nail psoriasis at baseline (RZB, n = 309; PBO/RZB, n = 338)
fIn patients with baseline HAQ-DI ≥ 0.35. Number of responders (n) is calculated on the basis of the total number of patients and estimated response rate, rounding to the nearest integer
gAmong patients with a baseline Patient’s Assessment of Pain (VAS) score (RZB, n = 482; PBO/RZB, n = 479)
*Nominal p < 0.05
***Nominal p < 0.001
In a prespecified pooled analysis of the KEEPsAKE 1 and KEEPsAKE 2 studies, most patients (57.7% for continuous risankizumab and 58.9% for placebo/risankizumab) with enthesitis at baseline achieved enthesitis resolution at week 100 (Fig. 2). Resolution of enthesitis was durable from earlier time points (week 52: 55.0% and 57.4%, respectively; week 24: 48.4% and 34.8%) (Fig. 2). Similarly, the majority of patients in the KEEPsAKE 1 and KEEPsAKE 2 studies with dactylitis at baseline experienced resolution at week 100 (continuous risankizumab, 76.2%; placebo/risankizumab, 75.3%), which was durable from week 52 (76.6% and 72.8%) and week 24 (68.1% and 51.0%) (Fig. 2).
Fig. 2.

Achievement over time of resolution of enthesitis, resolution of dactylitis, improvement in PASI 90, and reduction in mNAPSI. PASI 90 reported only for patients with psoriasis-affected BSA ≥ 3 at baseline. mNAPSI reported only for patients with nail psoriasis at baseline. Resolution of enthesitis and dactylitis reported as pooled results from the KEEPsAKE 1 and 2 studies for patients with enthesitis (LEI > 0) or dactylitis (LDI > 0) at baseline. BSA body surface area, CI confidence interval, MMRM mixed-effect model for repeated measures, LDI Leeds Dactylitis Index, LEI Leeds Enthesitis Index, mNAPSI modified Nail Psoriasis Severity Index, NRI-C nonresponder imputation incorporating multiple imputations to handle missing data due to COVID-19, NRI-MI nonresponder imputation incorporating multiple imputations to handle missing data due to COVID-19 and geopolitical conflict, and all other missing data treated as nonresponders, including missing data and patients who received rescue therapy, PASI 90 ≥ 90% improvement from baseline in Psoriasis Area and Severity Index, PBO placebo, RZB risankizumab
Change from baseline in resolution of enthesitis and resolution of dactylitis was similar when analyzed using the as observed data (Supplementary Material Fig. 2).
Skin and Nail Clearance Endpoints Through Week 100
In patients who had a psoriasis-affected BSA ≥ 3% at baseline, the proportion who achieved PASI 90 was improved in both the continuous risankizumab and placebo/risankizumab cohorts at week 100 (71.3% and 67.8%, respectively), and the proportion of patients was sustained from week 52 (67.9% and 60.7%) and increased from week 24 (52.3% and 9.9%) (Fig. 2) [11]. Findings were similar for change from baseline in PASI 90 for both cohorts when data were evaluated using an as observed data approach (Supplementary Material Fig. 2).
Reduction from baseline in mNAPSI scores by 14.3 points and 13.5 points was achieved at week 100 in the continuous risankizumab and placebo/risankizumab cohorts, respectively, which was durable from week 52 and increased compared with the week 24 results (Fig. 2) [11]. Change from baseline in mNAPSI results was similar for both cohorts when data were analyzed using an as observed data approach (Supplementary Material Fig. 2).
Reductions from baseline in PGA-F scores were observed with continuous risankizumab and with placebo/risankizumab cohorts at week 100 (− 1.4 and − 1.3, respectively), which were similar to the reductions observed at week 52 (− 1.2 and − 1.1) and increased compared with reductions at week 24 (− 0.8 and − 0.4) (Table 1). In addition, the proportion of patients who achieved “clear” or “minimal” nail psoriasis at week 100 (PGA-F score of 0 or 1) and improvement ≥ 2 grades was 69.9% in the continuous risankizumab cohort and 67.9% in the placebo/risankizumab cohort, which was more than the proportion of patients achieving “clear” or “minimal” nail psoriasis at week 52 (58.0% and 49.2%) and week 24 (37.8% and 15.9%).
MDA Achievement Through Week 100
The proportion of patients achieving MDA at week 100 in the continuous risankizumab cohort was 38.2%, which was stable from week 52 (38.3%) and more than week 24 (25.0%) (Fig. 1) [11]. The proportion of patients in the placebo/risankizumab cohort achieving MDA was increased at week 100 (35.2%) compared with week 52 (28.0%) and week 24 (10.2%), supporting the durability of efficacy with risankizumab [11] (Fig. 1). The overall findings in achieving MDA were similar when data were evaluated as observed (Supplementary Material Fig. 1). Maintenance of MDA response at week 100 in week 52 MDA responders was achieved by 75.5% and 78.2% of the continuous risankizumab and placebo/risankizumab cohorts, respectively; maintenance of MDA response was similar when data were evaluated as observed (Supplementary Material Table 2).
Patient-Reported Outcomes and Health-Related Quality of Life Through Week 100
Patients reported a reduction in HAQ-DI scores from baseline at week 100 in the continuous risankizumab cohort and the placebo/risankizumab cohort (− 0.41 and − 0.36, respectively), which was sustained from week 52 (− 0.41 and − 0.32) and greater than week 24 (− 0.31 and − 0.11) [11] (Fig. 3). At week 100, clinically meaningful reductions from baseline in HAQ-DI (≥ 0.35 decrease from baseline) were achieved by 54.9% of the continuous risankizumab cohort and 48.4% of the placebo/risankizumab population, which were similar to the proportions of patients at week 52 (57.9% and 46.4%) and increased from week 24 (50.3% and 27.9%) [11] (Table 1).
Fig. 3.
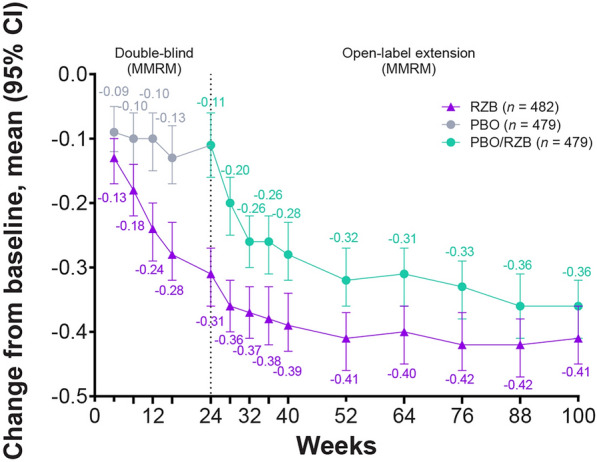
Change from baseline over time in HAQ-DI. Reported using MMRM. CI confidence interval, HAQ-DI Health Assessment Questionnaire–Disability Index, MMRM mixed-effect model for repeated measures, PBO placebo, RZB risankizumab
Results for patient’s assessment of pain also showed evidence of durability, with 26.9- and 25.0-point reductions from baseline observed at week 100 for patients in the continuous risankizumab cohort and the placebo/risankizumab cohort, respectively, which were similar to week 52 (− 26.5 and − 22.6) and increased from week 24 (− 21.0 and − 10.2) [11] (Table 1). Maintenance of achievement of clinically meaningful improvements in patient’s assessment of pain was observed at week 100 in 80.5% and 81.0% of week 52 responders in the continuous risankizumab and placebo/risankizumab cohorts (Supplementary Material Table 2). Patients also reported improved health-related quality of life based on the SF-36 PCS; the continuous risankizumab cohort experienced an 8.4-point reduction from baseline, and the placebo/risankizumab cohort had a 7.5-point reduction from baseline at week 100, which was similar to the results at week 52 (8.4 and 7.3, respectively) and greater than the results at week 24 (6.5 and 3.2) (Table 1).
At week 100, FACIT-Fatigue scores improved from baseline by 7.8 for patients receiving continuous risankizumab and 6.9 for those receiving placebo/risankizumab, which were durable from week 52 (8.0 and 6.5, respectively) and greater than week 24 (6.5 and 3.9) (Table 1). When data were evaluated using an as observed approach, the results for mean changes from baseline in clinically meaningful reduction in HAQ-DI (Supplementary Material Fig. 3), patient’s assessment of pain VAS, SF-36 PCS, and FACIT-Fatigue were similar for both treatment cohorts (Supplementary Material Table 3).
Safety
Long-term safety data included 946 patients who received risankizumab (either during the double-blind or the open-label period regardless of their original randomization cohort), representing 1708.4 PY of exposure (Table 2). The long-term safety cutoff date included ≥ 100 weeks of exposure, with an exposure-adjusted event rate for any TEAEs in patients receiving risankizumab of 130.1 E/100 PY; these event rates decreased compared with week 24 (177.6 E/100 PY) (Table 2).
Table 2.
Safety
| Events (E/100 PY) | Week 24 | Long-terma | ||
|---|---|---|---|---|
| RZB 150 mg (n = 483; PY = 224.1) | Any RZB 150 mg (n = 946; PY = 1708.4) | |||
| Any TEAE | 398 | (177.6) | 2223 | (130.1) |
| Serious TEAE | 15 | (6.7) | 130 | (7.6) |
| TEAE leading to discontinuation of study drug | 6 | (2.7) | 36 | (2.1) |
| COVID-19-related TEAE | 1 | (0.4) | 137 | (8.0) |
| Adjudicated MACE | 0 | 2 | (0.1) | |
| Any serious infection | 6 | (2.7) | 39 | (2.3) |
| Opportunistic infections | ||||
| Infections excluding TB and herpes zoster | 0 | 1 | (< 0.1) | |
| Active TB | 0 | 0 | ||
| Herpes zoster | 2 | (0.9) | 4 | (0.2) |
| Any hypersensitivity | 12 | (5.4) | 43 | (2.5) |
| Malignant tumors | ||||
| NMSC | 0 | 2 | (0.1) | |
| Malignancies excluding NMSC | 0 | 7 | (0.4) | |
| All deathsb | 1 | (0.4) | 7 | (0.4) |
E events, MACE major adverse cardiovascular event, NMSC nonmelanoma skin cancer, PY patient-years, RZB risankizumab, TB tuberculosis, TEAE treatment-emergent adverse event
aSafety reported through data cutoff date (March 21, 2022), which includes data though week 100 and all patients who received any RZB 150 mg (those patients who started on RZB 150 mg at randomization and those patients who switched from placebo to RZB 150 mg after week 24)
bIn the long-term safety analysis set, six total treatment-emergent deaths were reported. Death in two patients was related to COVID-19. One patient died as a result of complications related to acute leukemia. One patient with anemia died from diverticulosis and multiorgan failure due to complications of a hemicolectomy. One patient previously reported at the week 24 cutoff date, aged 81 years with dementia and hospitalized for pneumonia, developed urosepsis and complications resulting in death. One patient previously reported at the week 52 cutoff date was hospitalized for anxiety and depression 1 week after being discharged and was readmitted with septicemia. The patient was found deceased at home 2 weeks after discharge, and the death was subsequently adjudicated as sudden cardiac death. TEAEs were defined as an adverse event with an onset date on or after the first dose of RZB and up to 140 days after the last dose of RZB if the patient prematurely discontinued study drug
The most common TEAEs through week 100 (≥ 4.0 E/100 PY) were COVID-19 infections (8.0 E/100 PY; Table 2) and nasopharyngitis (4.3 E/100 PY). COVID-19-related TEAEs had increased from 0.4 E/100 PY at week 24, likely due to the global COVID-19 pandemic that occurred during the study. TEAEs leading to discontinuation of the study drug were 2.1 E/100 PY through week 100 (Table 2). The most common TEAE leading to discontinuation of the study drug through week 100 was psoriatic arthropathy with seven reported events (0.4 E/100 PY).
Serious TEAEs through week 100 occurred at a rate of 7.6 E/100 PY (Table 2). A total of 39 serious infections (2.3 E/100 PY) were reported, with COVID-19 or COVID-19 pneumonia accounting for 18 of these 39 events. Two major adverse cardiovascular events (0.1 E/100 PY) were reported between weeks 52 and 100 (Table 2), including one sudden cardiac death and one nonfatal myocardial infarction; both events were assessed as having no reasonable possibility of being associated with the study drug by the investigator. Through week 100, there were no cases of active tuberculosis, and no new opportunistic infections had occurred since the one event (< 0.1 E/100 PY) of opportunistic infection (oropharyngeal candidiasis) reported at the week 52 cutoff date [14] (Table 2). There were 43 events of hypersensitivity (2.5 E/100 PY; Table 2), with none of these events being classified as serious. A total of nine malignant tumors were reported (0.5 E/100 PYs), including two patients with nonmelanoma skin cancer (NMSC) and seven patients with malignant tumors excluding NMSC (Table 2). Between the week 52 data cutoff date and the week 100 data cutoff date, there were no new cases of NMSC, and three patients were reported with new malignant tumors excluding NMSC. As previously reported in an analysis of the week 52 data cutoff date, two deaths occurred, one patient with urosepsis on study day 96 [11] and one sudden death on study day 502 [14], both deemed unrelated to the study drug. The sudden death reported at the week 52 cutoff date occurred in a patient who died at home and had “natural causes” stated as the primary cause of death on the death certificate; this was later adjudicated as a sudden cardiac death and defined as a major adverse cardiovascular event in the week 100 data analysis. After that period and in the current analysis, four additional patients died, including two patients with COVID-19; one patient with complications related to acute leukemia; and one patient with anemia, diverticulosis, and multiorgan failure due to complications of a hemicolectomy. The deaths were deemed unrelated to the study drug.
Discussion
Through 100 weeks of treatment in the ongoing KEEPsAKE 1 study, risankizumab demonstrated durable efficacy for the treatment of PsA in patients with previous inadequate response to csDMARDs across multiple disease domains. The proportion of patients with improvements in the clinical signs and symptoms of PsA and patient-reported outcomes (including improved joint outcomes [ACR20/50/70] and improvements in skin [PASI 90] and nail symptoms [mNAPSI and PGA-F]) was stable from week 52, both in patients originally randomized to risankizumab and among those patients who switched from placebo to risankizumab at week 24. The rates of resolution in soft-tissue manifestations of enthesitis and dactylitis were also durable over the 100-week treatment period and approximately 90% of patients showed no signs of radiographic progression with continuous risankizumab treatment for 2 years.
At week 100, 38.2% and 35.2% of patients receiving continuous risankizumab and placebo/risankizumab, respectively, achieved MDA, which were similar to the results at week 52. MDA is a measure that incorporates multiple disease domains including joint, skin, and health-related quality of life measures, enabling a more generalized view of the patient’s disease state. MDA is recommended by the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis/Outcome Measures in Rheumatology Group for patient assessment [20].
In addition, there was a clear maintenance of response at week 100 in patients who had responded at week 52 across several measures; indeed, most patients who achieved ACR20/50/70, PASI 90, MDA, and/or clinically meaningful improvement in patient’s report of pain at week 52 maintained their response at week 100.
Risankizumab was generally well tolerated, and no new safety signals were identified; the safety profile was similar to what has been previously reported [21]. Of the six treatment-emergent patient deaths reported, none were assessed by the investigators as being related to the study drug. The KEEPsAKE 1 study was conducted during the COVID-19 pandemic, and the long-term safety data indicate that COVID-19-related TEAEs increased from 0.4 E/100 PY at 24 weeks to 8.0 E/100 PY at 100 weeks. Global cases of COVID-19 at the week 24 data cutoff date in October 2020 were estimated at 83.51 cases per million people, but by the week 100 data cutoff date in March 2022 (when many countries had relaxed COVID-19 restrictions, and the more contagious Omicron variant had emerged), there were substantially more infections with cases estimated at 1157.70 per million people at the peak in January 2022 [22]. No cases of active tuberculosis were reported.
In addition, reports of major adverse cardiovascular events, serious infections, opportunistic infections, herpes zoster, and serious hypersensitivity remained stable. Overall, treatment with risankizumab had a favorable benefit–risk profile supported by multiple studies [11, 12, 14, 15].
The relatively homogenous population enrolled in this trial may be a potential limitation that could affect the generalizability of these results. This study also included open-label administration, which may bias efficacy and safety. To mitigate this potential bias, missing efficacy results including patient-reported outcomes were imputed using nonresponder imputation.
Conclusions
The week 100 results of the ongoing KEEPsAKE 1 trial demonstrate that treatment with risankizumab provides a durable improvement in the signs and symptoms of PsA in patients who are csDMARD-IR. Risankizumab is generally well tolerated with a long-term stable safety profile. The KEEPsAKE 1 study remains ongoing for continued efficacy and safety analysis.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
AbbVie and the authors thank all study investigators for their contributions and the patients who participated in this study
Medical Writing and Editorial Assistance.
Medical writing support was provided by Abegale Templar, PhD, and Ray Beck, Jr, PhD, of JB Ashtin and Trisha Rettig, PhD, of AbbVie, and funded by AbbVie.
Author Contributions
LEK, MK, KP, LM, DW, KC, RL, HP, LD, AMS, MC, BP, and FB had access to relevant data and participated in the development, review, approval, and decision to submit this manuscript for publication. No honoraria or payments were made for authorship. In addition, AMS and BP contributed to the conception and design of the study. LEK, MK, KP, LM, DW, and FB were involved in the data acquisition. LEK, KP, DW, HP, AMS, MC, BP, and FB contributed to the data interpretation. MC also helped to conduct the statistical analyses.
Funding
AbbViefunded this study and participated in the study design, research, data collection, analysis, and interpretation of data, reviewing, and approval of this manuscript. The Rapid Service Fee of this manuscript was funded by AbbVie.
Data Availability
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized individual and trial-level data (analysis data sets) and other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the United States and Europe and after acceptance of this manuscript for publication. These data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://vivli.org/ourmember/abbvie/, then select “Home.”
Declarations
Conflict of Interest
Lars Erik Kristensen has received honoraria or fees for serving as a speaker or consultant from AbbVie, Amgen, Biogen, Bristol Myers Squibb, Gilead, Janssen, Lilly, Merck, Novartis, Pfizer, and UCB. He has also received IIT research grants from Lilly, Novartis, Pfizer, and UCB. Mauro Keiserman has served as a speaker for AbbVie, Bristol Myers Squibb, Celgene, Eli Lilly, Janssen, Novartis, Pfizer, Roche, and UCB; was a consultant for AbbVie, Bristol Myers Squibb, GlaxoSmithKline, MSD, Biogen, Sanofi, and Novartis; and receives grant/research support from AbbVie, Bristol Myers Squibb, GlaxoSmithKline, MSD, Biogen, Sanofi, and Novartis. Kim Papp has received research funds from AbbVie, Aceleryn, Amgen, Arcutis, Bausch Health, Baxalta, Boehringer Ingelheim, Bristol Myers Squibb, Coherus, Dermavant, Forward Pharma, Galderma, Incyte, Janssen, LEO Pharma, Lilly, Novartis, Ortho Dermatologics, Pfizer, Sanofi Genzyme, Sun Pharma, and UCB. He is a consultant for AbbVie, Aceleryn, Amgen, Arcutis, Astellas, Bausch Health, Boehringer Ingelheim, Bristol Myers Squibb, Coherus, Dermavant, Forward Pharma, Galderma, Incyte, Janssen, LEO Pharma, Lilly, Meiji Seika Pharma, Merck, Mitsubishi Tanabe Pharma, Novartis, Pfizer, Sandoz, Sanofi Genzyme, Stiefel, Sun Pharma, Takeda, and UCB. He is a speaker for AbbVie, Amgen, Arcutis, Bausch Health, Boehringer Ingelheim, Bristol Myers Squibb, Dermavant, Incyte, Janssen, LEO Pharma, Lilly, Novartis, Pfizer, Sandoz, Sanofi Genzyme, Sun Pharma, and UCB. He is a committee member for PSOLAR (Psoriasis Longitudinal Assessment and Registry) and PURE (registry of patients with moderate-to-severe chronic plaque psoriasis in Latin America and Canada). Leslie McCasland has received fees for serving on an advisory board from Lilly. Douglas White has received honoraria or fees for serving on advisory boards or as a speaker or consultant from AbbVie. Kyle Carter, Ralph Lippe, Huzefa Photowala, Leonidas Drogaris, Michael Chen, and Byron Padilla are full-time employees of AbbVie, and may hold AbbVie stock or stock options. Ahmed M. Soliman is a full-time employee of AbbVie, may hold AbbVie stock or stock options, and is a coinventor on AbbVie patents. Frank Behrens has received research grants, honoraria, or fees for serving as a consultant or speaker from AbbVie, Amgen, Affibody, Acelyrin, BMS, Boehringer Ingelheim, Chugai, Celltrion, Galapagos, Genzyme, Gilead, GlaxoSmithKline, Janssen, Lilly, Merck, MoonLake, Novartis, Pfizer, Roche, Sandoz and Sanofi.
Ethical Approval
The study protocol, informed consent forms, and recruitment materials were approved by the independent ethics committee or institutional review board at each study site before patient enrollment began (Supplementary Table 1). The studies were conducted in accordance with the International Conference for Harmonisation guidelines, applicable regulations, and the Declaration of Helsinki. All patients provided written informed consent before entering the study.
Footnotes
Prior Presentation: The 100-week analyses of KEEPsAKE 1 were previously presented at ACR Convergence 2022, November 10–14, Philadelphia, PA (abstract 2145), and the 31st EADV Congress, 7–10 September 2022, Milan, Italy, and online (presentation D3T01.1D).
References
- 1.Ogdie A, Weiss P. The epidemiology of psoriatic arthritis. Rheum Dis Clin North Am. 2015;41:545–568. doi: 10.1016/j.rdc.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mease PJ, Gladman DD, Papp KA, et al. Prevalence of rheumatologist-diagnosed psoriatic arthritis in patients with psoriasis in European/North American dermatology clinics. J Am Acad Dermatol. 2013;69:729–735. doi: 10.1016/j.jaad.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 3.Kristensen LE, Jorgensen TS, Christensen R, et al. Societal costs and patients’ experience of health inequities before and after diagnosis of psoriatic arthritis: a Danish cohort study. Ann Rheum Dis. 2017;76:1495–1501. doi: 10.1136/annrheumdis-2016-210579. [DOI] [PubMed] [Google Scholar]
- 4.Gladman DD, Antoni C, Mease P, Clegg DO, Nash P. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis. 2005;64:14–17. doi: 10.1136/ard.2004.032482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gottlieb AB, Merola JF. Axial psoriatic arthritis: an update for dermatologists. J Am Acad Dermatol. 2021;84:92–101. doi: 10.1016/j.jaad.2020.05.089. [DOI] [PubMed] [Google Scholar]
- 6.Gossec L, Baraliakos X, Kerschbaumer A, et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 update. Ann Rheum Dis. 2020;79:700–712. doi: 10.1136/annrheumdis-2020-217159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coates LC, Cook R, Lee KA, Chandran V, Gladman DD. Frequency, predictors, and prognosis of sustained minimal disease activity in an observational psoriatic arthritis cohort. Arthritis Care Res (Hoboken) 2010;62:970–976. doi: 10.1002/acr.20162. [DOI] [PubMed] [Google Scholar]
- 8.Theander E, Husmark T, Alenius GM, et al. Early psoriatic arthritis: short symptom duration, male gender and preserved physical functioning at presentation predict favourable outcome at 5-year follow-up. Results from the Swedish Early Psoriatic Arthritis Register (SwePsA) Ann Rheum Dis. 2014;73:407–413. doi: 10.1136/annrheumdis-2012-201972. [DOI] [PubMed] [Google Scholar]
- 9.Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376:1551–1560. doi: 10.1056/NEJMoa1607017. [DOI] [PubMed] [Google Scholar]
- 10.Singh S, Kroe-Barrett RR, Canada KA, et al. Selective targeting of the IL23 pathway: generation and characterization of a novel high-affinity humanized anti-IL23A antibody. MAbs. 2015;7:778–791. doi: 10.1080/19420862.2015.1032491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kristensen LE, Keiserman M, Papp K, et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 24-week results from the randomised, double-blind, phase 3 KEEPsAKE 1 trial. Ann Rheum Dis. 2022;81:225–231. doi: 10.1136/annrheumdis-2021-221019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Östor A, Van den Bosch F, Papp K, et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 24-week results from the randomised, double-blind, phase 3 KEEPsAKE 2 trial. Ann Rheum Dis. 2022;81:351–358. doi: 10.1136/annrheumdis-2021-221048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kristensen LE, Soliman AM, Papp K, et al. Effects of risankizumab on nail psoriasis in patients with active psoriatic arthritis: results from KEEPsAKE 1. J Eur Acad Dermatol Venereol. 2022;36:e389–e392. doi: 10.1111/jdv.17931. [DOI] [PubMed] [Google Scholar]
- 14.Kristensen LE, Keiserman M, Papp K, et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 52-week results from the KEEPsAKE 1 study. Rheumatology (Oxford) 2023;62:2113–2121. doi: 10.1093/rheumatology/keac607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Östor A, Van den Bosch F, Papp K, et al. Efficacy and safety of risankizumab for active psoriatic arthritis: 52-week results from the KEEPsAKE 2 study. Rheumatology (Oxford) 2023;62:2122–2129. doi: 10.1093/rheumatology/keac605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heijde DVD, Sharp J, Wassenberg S, Gladman DD. Psoriatic arthritis imaging: a review of scoring methods. Ann Rheum Dis. 2005;64:ii61–ii64. doi: 10.1136/ard.2004.030809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cassell SE, Bieber JD, Rich P, et al. The modified Nail Psoriasis Severity Index: validation of an instrument to assess psoriatic nail involvement in patients with psoriatic arthritis. J Rheumatol. 2007;34:123–129. [PubMed] [Google Scholar]
- 18.Hudgens S, Sundaram M, Williams D. Evaluation of a novel clinician reported outcome in nail psoriasis. Value Health. 2016;19:A127. doi: 10.1016/j.jval.2016.03.519. [DOI] [Google Scholar]
- 19.Mease PJ, Woolley JM, Bitman B, Wang BC, Globe DR, Singh A. Minimally important difference of Health Assessment Questionnaire in psoriatic arthritis: relating thresholds of improvement in functional ability to patient-rated importance and satisfaction. J Rheumatol. 2011;38:2461–2465. doi: 10.3899/jrheum.110546. [DOI] [PubMed] [Google Scholar]
- 20.Coates LC, FitzGerald O, Merola JF, et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis/Outcome Measures in Rheumatology consensus-based recommendations and research agenda for use of composite measures and treatment targets in psoriatic arthritis. Arthritis Rheumatol. 2018;70:345–355. doi: 10.1002/art.40391. [DOI] [PubMed] [Google Scholar]
- 21.Gordon KB, Lebwohl M, Papp KA, et al. Long-term safety of risankizumab from 17 clinical trials in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2022;186:466–475. doi: 10.1111/bjd.20818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agyapon-Ntra K, McSharry PE. A global analysis of the effectiveness of policy responses to COVID-19. Sci Rep. 2023;13:5629. doi: 10.1038/s41598-023-31709-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized individual and trial-level data (analysis data sets) and other information (e.g., protocols, clinical study reports, or analysis plans), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent, scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time after approval in the United States and Europe and after acceptance of this manuscript for publication. These data will be accessible for 12 months, with possible extensions considered. For more information on the process or to submit a request, visit the following link: https://vivli.org/ourmember/abbvie/, then select “Home.”