

Abstract
Cerebrovascular diseases (CVDs) are a major threat to global health. Elucidation of the molecular mechanisms underlying the pathology of CVDs is critical for the development of efficacious preventative and therapeutic approaches. Accumulating studies have highlighted the significance of ubiquitin‐modifying enzymes (UMEs) in the regulation of CVDs. UMEs are a group of enzymes that orchestrate ubiquitination, a post‐translational modification tightly involved in CVDs. Functionally, UMEs regulate multiple pathological processes in ischemic and hemorrhagic stroke, moyamoya disease, and atherosclerosis. Considering the important roles of UMEs in CVDs, they may become novel druggable targets for these diseases. Besides, techniques applying UMEs, such as proteolysis‐targeting chimera and deubiquitinase‐targeting chimera, may also revolutionize the therapy of CVDs in the future.
Keywords: cerebrovascular disease, deubiquitinating enzyme, disease mechanism, therapeutic target, ubiquitinating enzyme, ubiquitination
Ubiquitin‐modifying enzyme (UMEs), comprising E1, E2, E3 ubiquitinating enzymes and deubiquitinating enzymes, orchestrate ubiquitination and thereby critically regulate the pathophysiology of cerebrovascular diseases (CVDs).
Alteration in the abundance or activity of UMEs affects the outcome of CVDs.
UME‐targeting therapy and therapeutic techniques applying UMEs may be beneficial for the treatment of CVDs.
1. INTRODUCTION
As the “central processing unit” of the body, the brain needs a relatively large amount of energy to maintain its sophisticated functionality. Although its weight accounts for only 2% of the body weight, the brain consumes about 20% of the human body's oxygen and glucose supply. 1 , 2 Unlike other major energy consumers of the body, such as the liver, the brain has barely any reservation of energy materials. Therefore, the cerebrovascular system is the only important source of glucose and oxygen for the brain. Cerebral blood flow, which is mainly supplied by internal carotid arteries and vertebral arteries, accounts for up to 20% of total cardiac output. 3 , 4 Given the importance of the cerebrovascular system in brain energy supply, interruption of the cerebral blood supply leads to termination of cerebral electrical activity and irreversible brain damage within minutes. Chronic cerebrovascular diseases (CVDs), such as moyamoya disease (MMD) and intracranial atherosclerotic disease, cause insufficient blood supply to the brain, inciting symptoms including headache, visual disturbance, paresthesia, motor nerve dysfunction, mental abnormality, and cognitive impairment. 5 , 6 , 7 , 8 , 9 These chronic CVDs and other factors, for example, hypertension, can also trigger life‐threatening acute cerebrovascular accidents such as ischemic and hemorrhagic stroke. 5 , 6 , 8 CVDs have become the main cause of disability and mortality in both developing and developed countries. 10
Accumulative studies have shown that the pathological processes of CVDs are closely regulated by ubiquitination, a post‐translational modification (PTM) that is widely involved in signal transduction, inflammatory responses, metabolism, and cell fate determination. 11 , 12 , 13 , 14 , 15 , 16 , 17 In the process of ubiquitination, ubiquitin, a 76‐amino acid small protein, is covalently conjugated to a lysine residue of the substrate protein under the sequential catalysis of ubiquitin‐activating enzymes (E1s), ubiquitin‐conjugating enzymes (E2s), and ubiquitin ligases (E3s) 18 (Figure 1A). First, an E1 hydrolyzes ATP and forms a thioester bond between the sulfhydryl group of its active cysteine and the carboxyl group of the ubiquitin C‐terminal glycine. Second, the activated ubiquitin is attached to an active cysteine of the E2 through a new E2‐ubiquitin thioester bond. Then, the E2‐ubiquitin complex interacts with an E3, which recognizes the protein substrate to mediate the final ubiquitin transfer. 18 E3s containing the really interesting new gene (RING) domain catalyze the direct transfer of ubiquitin from the E2 to the substrate. In contrast, E3s belonging to the RING‐between‐RING (RBR) and homologous to the E6AP carboxyl terminus (HECT) subfamilies form a ubiquitin‐E3 thioester intermediate before transferring the ubiquitin to the substrate 18 , 19 (Figure 1A). According to the type of ubiquitin attachment, ubiquitination can be classified as polyubiquitination, monoubiquitination, and multi‐monoubiquitination (Figure 1A). In polyubiquitin chains, ubiquitin monomers are linked together via isopeptide bonds between the C‐terminal glycine carboxyl group of one ubiquitin and the amino group of the N‐terminal methionine residue (M1) or any one of the internal lysine residues (K6, K11, K27, K29, K33, K48, K63) in the next ubiquitin. 20 Of note, polyubiquitin chains can be added to substrates by “sequential addition” or “en bloc transfer.” In the sequential addition, individual ubiquitin monomers are transferred stepwise to the end of a growing polyubiquitin chain. By contrast, in the en bloc mechanism, a pre‐assembled polyubiquitin chain is transferred as a whole to a substrate. 21 Functionally, ubiquitination controls the localization, activity, stability, or binding partners of protein substrates. As a reversible PTM, ubiquitination is antagonized by deubiquitinating enzymes (DUBs), which are divided into seven subfamilies based on sequence and domain conservation: ubiquitin‐specific proteases (USPs), ovarian tumor proteases (OTUs), ubiquitin C‑terminal hydrolases (UCHs), Machado–Josephin domain‐containing proteases (MJDs), JAB1/MPN/MOV34 family (JAMMs), motif interacting with ubiquitin‐containing novel DUB family (MINDYs), and zinc finger with UFM1‐specific peptidase domain protein (ZUFSP/ZUP1). 22 , 23 Mostly, DUBs directly remove ubiquitin molecules from protein substrates (Figure 1B). However, OTUB1, a DUB of the OTU subfamily, possesses a non‐canonical function that enables it to obstruct the ubiquitination of protein targets by blocking ubiquitin transfer from E2s 24 , 25 , 26 (Figure 1C). Of note, the removed ubiquitin molecules from substrates will be recycled for new ubiquitination processes. Besides, DUBs can also enrich the free ubiquitin pool by cleaving ubiquitin precursors encoded by UBA52, RPS27A, UBB, and UBC to release free ubiquitin molecules 27 (Figure 1D).
FIGURE 1.
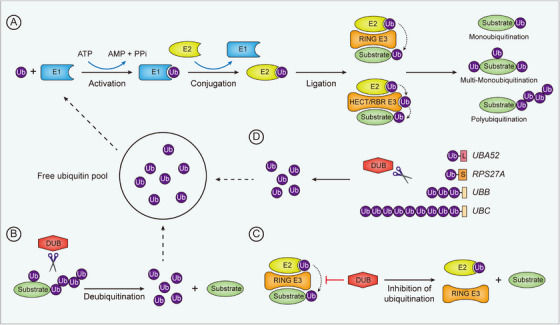
Overview of the process of ubiquitination and deubiquitination. (A) Ubiquitination is an enzymatic cascade catalyzed sequentially by E1s, E2s, and E3s. Ubiquitin is first activated by an E1 and subsequently transferred to an E2. An E3 mediates the final attachment of the ubiquitin molecule to a protein substrate. In contrast to HECT and RBR E3s, which receive ubiquitin molecules from E2s before transferring to substrates, RING E3s mediate the transfer of ubiquitins directly from E2s to substrates. (B,C) Ubiquitination is inhibited by DUBs, which either directly remove ubiquitins from substrates (B) or block the ubiquitination process. (D) DUBs cleave ubiquitin precursor proteins to release free ubiquitin molecules, which are further used for ubiquitination.
Ubiquitin‐modifying enzymes (UMEs) comprising E1s, E2s, E3s, and DUBs orchestrate the diverse and exquisite ubiquitination of target proteins to precisely control their cellular localization, protein interaction, enzymatic activity, and degradation, thereby affecting the initiation and/or outcome of brain diseases. For example, loss‐of‐function mutations in the TRIM37 gene, which encodes the E3 ligase TRIM37, cause the Mulibrey (muscle–liver–brain–eye) nanism. 28 , 29 In the scope of CVDs, the gene encoding the E3 ligase RNF213 has been identified as the most prominent susceptibility gene for MMD. 30 , 31 , 32 , 33 , 34 In this review, we elaborate on the functional roles and molecular mechanisms of UMEs in CVDs and discuss the possibility of exploring UMEs as therapeutic targets for these diseases.
1.1. UMEs regulate cerebral injury after ischemic stroke
Stroke is the second leading cause of death and disability worldwide. Around the world, there are more than 12 million new stroke cases per year and approximately 100 million people are living with the aftermath of a stroke. 10 Stroke is caused by infarction or rupture of a blood vessel in the brain, and accordingly can be classified into ischemic stroke and hemorrhagic stroke, with the former accounting for nearly 80% of all stroke cases. 35 , 36 Thrombolytic therapy with tissue plasminogen activator (tPA) and mechanical thrombectomy are two common ways to restore blood supply after ischemic stroke. 37 However, blood reperfusion can cause secondary brain damage known as cerebral ischemia/reperfusion (CI/R) injury. Multiple mechanisms, such as calcium overload, excitotoxicity, mitochondrial damage, oxidative stress, blood‐brain barrier (BBB) disruption, and inflammation, are involved in CI/R injury, culminating in neuronal cell death and neurological deficits. 38 The middle cerebral artery occlusion (MCAO) model, an animal model phenocopying key features of human ischemic stroke, is widely used to study the pathophysiology and treatment of ischemic stroke. With the MCAO model, regulatory functions of UMEs in ischemic stroke injury have been revealed. Recent studies have shown that UMEs affect the injury in ischemic stroke predominantly by regulating neuronal death, axonal function, neuroinflammation, BBB integrity, and mitochondrial dysfunction.
1.1.1. UMEs regulate neuronal death after ischemic stroke
Neuronal cell death is the basic pathophysiology of stroke, and the ischemic insult sequentially induces two major forms of programmed cell death, that is, necroptosis and apoptosis 39 (Figure 2). Necroptosis is a regulated necrosis that is rapidly induced in neurons shortly after cerebral ischemia. 39 In response to necroptosis‐inducing stimuli, receptor‐interacting protein kinase 1 (RIPK1) undergoes a conformational change and recruits RIPK3 to form a RIPK1/RIPK3 oligomer. In this kinase complex, RIPK3 is activated and then phosphorylates the effector molecule mixed lineage kinase domain‐like protein (MLKL). 40 Phosphorylated MLKL forms oligomers that translocate to the plasma membrane. At the membrane, MLKL undergoes conformational changes, leading to rapid breakage of the cell membrane and cell death. 41 , 42 Due to membrane permeabilization, necroptotic cells leak intracellular contents comprising damage‐associated molecular patterns (DAMPs), which further activate innate immune responses to incite inflammation 43 (Figure 2). Inhibition of necroptosis has been shown to be neuroprotective in mice subjected to MCAO. 44 , 45 Necroptosis is inhibited by E3 ligases TRAF2, CHIP, and Triad3, and they can mitigate cerebral ischemic injury by attenuating necroptosis and neuroinflammation. 46 , 47 , 48 Consistently, viral vector‐mediated overexpression of CHIP has been shown to prevent neuronal cell death after cerebral ischemia. 49
FIGURE 2.
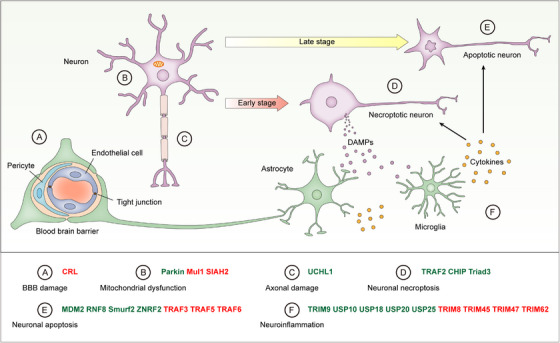
The role of UMEs in ischemic stroke. Ischemic stroke induces BBB damage, mitochondrial dysfunction, axonal damage, and neuronal death. Shortly after cerebral ischemia/reperfusion, neurons undergo necroptosis, leading to the release of DAMPs including S100A8/A9 and HMGB1. These DAMPs stimulate adjacent astrocytes and microglia to produce pro‐inflammatory cytokines. Neuroinflammation in turn promotes neuronal necroptosis in a positive feedback loop and instigates neuronal apoptosis in the late stage. UMEs influence ischemic stroke injury by regulating BBB damage (A), mitochondrial dysfunction (B), axonal damage (C), neuronal necroptosis (D), neuronal apoptosis (E), and neuroinflammation (F). UMEs inhibiting ischemic stroke injury are in green and UMEs promoting ischemic stroke injury are in red.
In mice, MCAO‐induced neuronal cell death undergoes the transition from necroptosis to apoptosis over time, and apoptosis becomes the main type of neuronal cell death following the initial necroptosis 39 (Figure 2). Ischemia‐induced neuronal apoptosis is inhibited by UMEs such as MDM2, RNF8, Smurf2 and ZNRF2, and enhanced by TRAF3, TRAF5, and TRAF6. 11 , 50 , 51 , 52 , 53 , 54 , 55 Cerebral ischemia induces the upregulation of MDM2, an E3 ligase that negatively regulates p53 via both repressing p53 target gene transcription and ubiquitinating p53 for degradation. 50 Consistent with studies showing that p53 promotes stroke‐induced apoptosis and affects functional recovery after stroke, 56 , 57 the single‐nucleotide polymorphism of the MDM2 gene (SNP309T > G), which enhances MDM2 expression, is associated with better functional outcomes in patients with ischemic or hemorrhagic stroke. 50 RNF8 is an E3 ligase that is involved in DNA damage repair via histone ubiquitination, and ablation of RNF8 leads to DNA damage accumulation and neuronal apoptosis. 58 , 59 In mice subjected to MCAO, RNF8 plays a neuroprotective role by inducing the ubiquitination and degradation of HDAC2, which enhances oxygen‐glucose deprivation (OGD)‐induced neuronal apoptosis via regulating GSK3β activation. 51 Smurf2 is another E3 ligase that can inhibit neuronal apoptosis induced by cerebral ischemia and OGD, and overexpression of Smurf2 reduces brain injury in mice subjected to MCAO. 11 Smurf2 ubiquitinates Yin Yang 1 (YY1) for proteasome‐dependent degradation, thereby suppressing apoptosis via inactivating the apoptosis‐inducing YY1/HIF1α/DDIT4 axis. 11 CI/R‐induced neuronal apoptosis can also be inhibited by the E3 ligase ZNRF2, which inhibits apoptosis by preventing excessive autophagy, and overexpression of ZNRF2 attenuates cerebral injury in rats after MCAO. 52 In sharp contrast to the aforementioned apoptosis‐inhibiting E3 ligases, several E3 ligases of the tumor necrosis factor receptor‐associated factor (TRAF) family, including TRAF3/5/6 can enhance CI/R‐induced neuronal apoptosis. 53 , 54 , 55 For example, TRAF6 potentiates CI/R‐induced neuronal apoptosis by K63 ubiquitinating and activating Rac1. 54
1.1.2. UME regulates axonal function after ischemic stroke
In addition to grey matter, white matter can also be injured by ischemic stroke. 60 , 61 UCHL1 is a neuron‐specific DUB that is essential for axonal function. 14 After cerebral ischemia, UCHL1 is deactivated by reactive lipids, which bind to the C152 residue of UCHL1, leading to an impaired ubiquitin‐proteasome pathway. However, the UCHL1 C152A mutant preserves the ubiquitin hydrolase activity in the presence of reactive lipids. 62 As compared with wild‐type controls, the UCHL1 C152A knock‐in mice show decreased accumulation of ubiquitinated proteins and axonal injury after MCAO, suggesting that UCHL1 plays a critical role in maintaining axonal function after ischemic stroke. 14
1.1.3. UMEs regulate neuroinflammation after ischemic stroke
Neuroinflammation is an indispensable component of the pathological machinery in ischemic stroke. 63 Shortly after ischemic stroke, DAMPs such as S100A8/A9 and HMGB1 are released from necroptotic cells. These DAMPs are recognized by microglia and astrocytes, two innate immune cell populations in the brain, through pattern recognition receptors, resulting in the production of pro‐inflammatory cytokines and chemokines 39 , 64 (Figure 2). The post‐stroke neuroinflammation is driven by various pro‐inflammatory signaling pathways, in particular the nuclear factor‐kappa B (NF‐κB) pathway, which is tightly regulated by ubiquitination and UMEs. 63 , 65 , 66 , 67 , 68 Noteworthy, in response to pro‐inflammatory stimuli, polyubiquitin chains catalyzed by UMEs provide large scaffolds to induce multi‐protein structures comprising IκB kinases (IKKs), which serve as an upstream organizing center regulating NF‐κB activation. 69 , 70 As such, multiple UMEs have been shown to influence ischemic stroke injury by regulating neuroinflammation (Figure 3). Ischemic stroke‐induced neuroinflammation has been shown to be promoted by TRIM8, 71 TRIM45, 72 TRIM47 73 and TRIM62, 74 and inhibited by TRIM9, 13 USP10, 75 USP18, 76 USP20, 77 and USP25. 78 For example, after CI/R injury, microglia‐mediated neuroinflammation and neurological deficit are enhanced by the E3 ligase TRIM45. 72 After OGD/R, TRIM45 catalyzes K63‐specific polyubiquitination on TAB2, which is crucial for the phosphorylation of TAK1 and the subsequent activation of NF‐κB signaling. Moreover, microglia‐specific knockdown of TRIM45 significantly mitigates neurological deficit following CI/R injury in mice. 72 In sharp contrast to TRIM45, the DUB USP25 inhibits CI/R‐induced K63 ubiquitination of TAB2 in microglia. 78 In both mice and humans, microglial expression of USP25 is upregulated in the ischemic penumbra. 78 USP25 physically interacts with TAB2 through the UIM2 domain and cleaves K63 polyubiquitin chains on TAB2. In mice, ablation of USP25 significantly exacerbated MCAO‐induced cerebral deficits by enhancing neuroinflammation. 78 Ubiquitination and degradation of IκBα, the inhibitor that retains NF‐κB in the cytoplasm in resting cells, is essential for the activation of NF‐κB signaling. Of note, the ubiquitination of IκBα is induced by an E3 ligase complex comprising β‐TrCP. 79 As a counter‐regulating mechanism, the degradation of IκBα is inhibited by TRIM9, which competes with IκBα for β‐TrCP interaction and thereby inhibits the ubiquitination of IκBα. 80 Upon ischemic stroke, TRIM9 inhibits NF‐κB‐mediated neuroinflammation by stabilizing IκBα, resulting in alleviated cerebral damage. 13
FIGURE 3.
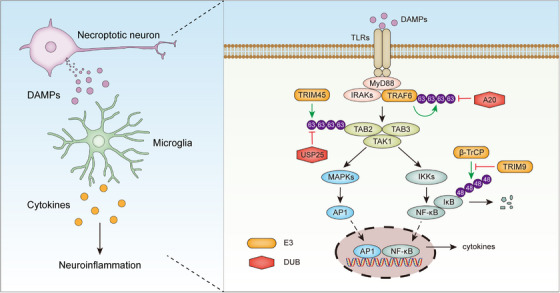
UMEs regulate neuroinflammatory signal transduction after ischemic stroke. DAMPs released from necroptotic neurons induce the production of pro‐inflammatory cytokines in glial cells mainly by activating NF‐κB and MAPK signaling pathways. E3s and DUBs tightly control the activity of these signaling pathways, thereby affecting neuroinflammation and ischemic stroke outcomes.
1.1.4. UMEs regulate BBB integrity after ischemic stroke
BBB disruption, characterized by loss of BBB junctional proteins and enhanced permeability, is another pathological process associated with cerebral ischemic stroke. Disrupted BBB subsequently leads to cerebral edema and neuronal cell death. 81 After ischemic stroke, BBB damage is promoted by the E3 ubiquitin ligase CRL, which induces the degradation of the protective protein neurofibromatosis 1 (NF1). 82 The activity of CRL is inhibited by the small‐molecular inhibitor MLN4924, and treatment of mice with MLN4924 ameliorates ischemic brain injury by inducing the accumulation of NF1. 82 In addition to neuronal apoptosis, BBB disruption is also mitigated in TRAF5 knockout mice after CI/R, indicating a role of TRAF5 in regulating BBB damage. 53
1.1.5. UMEs regulate mitochondrial dysfunction after ischemic stroke
Mitochondrial dysfunction is a key mechanism contributing to brain injury in ischemic stroke. 83 Mul1 is a mitochondrial membrane protein with dual E3 ligase functions in both ubiquitination and sumoylation, a ubiquitination‐like PTM. 84 , 85 Mul1 is upregulated in the rat brain after MCAO, and it aggravates mitochondrial dysfunction by regulating the protein abundance of the mitochondrial fission protein Drp1 and the mitochondrial fusion protein Mfn2 through sumoylation and ubiquitination, respectively. 84 Knockdown of Mul1 ameliorates MCAO‐induced brain injury by restoring protein abundance of Drp1 and Mfn2. 84 In response to OGD, the E3 ligase SIAH2 is activated in neurons, and it induces the ubiquitination and degradation of mitochondrial NCX3, a protein essential for mitochondrial integrity and neuronal survival during hypoxia. As compared with control neurons, SIAH2‐deficient neurons show improved mitochondrial function under OGD conditions due to elevated NCX3 levels. 86 Another study demonstrated that SIAH2 could also aggravate ischemia‐induced mitochondrial damage by inducing the ubiquitination and proteasomal degradation of AKAP121, a mitochondrial scaffold protein essential for mitochondria activity. 87 Therefore, the two studies jointly show that SIAH2 contributes to mitochondrial damage upon ischemic stress. 86 , 87 Selective mitochondrial autophagy, known as mitophagy, serves as a key mechanism in clearing damaged mitochondria and it is activated in ischemic brains. 88 Upon ischemic injury, the E3 ligase Parkin is recruited to the damaged mitochondria and ubiquitinates mitochondrial membrane proteins to trigger mitophagy. 89 In mice, Parkin‐mediated mitophagy has been shown to be a key protective mechanism in CI/R injury. 88 , 90
In aggregate, these studies show that, after ischemic stroke, UMEs impinge on cerebral injury by regulating a broad range of biological activities, implying that UMEs may serve as potential therapeutic targets for ischemic stroke.
1.2. UMEs regulate cerebral injury after hemorrhagic stroke
Nearly 20% of stroke cases are hemorrhagic, with intracerebral hemorrhage (ICH) accounting for about 10−15%. 91 , 92 ICH causes neuronal cell death and neurological deficits by hematoma‐associated mechanical damage and secondary injury mechanisms such as oxidative stress, mitochondrial dysfunction, neuronal excitotoxicity, calcium overload, neuroinflammation, and free radical production. 93 UMEs have been shown to affect the outcome of ICH by regulating many of these pathological processes. 94 , 95 , 96 , 97 , 98 The DUB A20, encoded by the TNFAIP3 gene, serves as a brake on inflammatory responses via suppressing multiple pro‐inflammatory signaling pathways, such as NF‐κB and JAK‐STAT signaling 66 , 99 (Figure 3). Mutations in or close to the TNFAIP3 gene are associated with various autoimmune diseases including systemic lupus erythematosus, rheumatoid arthritis, multiple sclerosis, and colitis. 66 , 100 , 101 ICH‐induced inflammatory injury is also inhibited by A20, and overexpression of A20 ameliorates brain damage after ICH. 94 Moreover, in humans, A20 mRNA levels in peripheral blood mononuclear cells are negatively correlated with neurological deficits after ICH, indicating that A20 is a key suppressor for ICH injury. 94 Mitochondrial dysfunction and oxidative stress are inhibited by PGC‐1α and enhanced by RNF34, an E3 ligase inducing the ubiquitination‐mediated degradation of PGC‐1α. 95 , 102 Overexpression of RNF34 exacerbates ICH‐induced brain injury by promoting PGC‐1α protein degradation and increasing oxidative stress and mitochondrial dysfunction. 95 Necroptosis is an important mechanism causing brain injury after ICH and it can be regulated by CHIP, which ubiquitinates the key component of necroptosis, RIPK3, for lysosomal degradation. 103 , 104 , 105 Overexpression of CHIP inhibits neuronal necroptosis and neuroinflammation in rats after ICH, resulting in reduced hemorrhagic lesions. Concordantly, CHIP deficiency leads to aggravated brain injury after ICH. 96 In addition to necroptosis, apoptosis is also induced by ICH, and it can be enhanced by the DUBs USP4 and USP11. 97 , 98
Subarachnoid hemorrhage (SAH) is another subtype of hemorrhagic stroke, accounting for nearly 5−10% of acute stroke. 92 , 106 The E3 ligase RNF216, also known as Triad3A, has been shown to modulate synaptic plasticity in glutamatergic neurons by inducing the ubiquitination and degradation of Arc. 107 Upon SAH, RNF216 increases oxyhemoglobin‐induced intracellular Ca2+ accumulation in neurons by restraining the Arc‐AMPAR pathway, thereby promoting cytotoxicity and neuronal apoptosis. Moreover, the downregulation of RNF216 ameliorates brain injury following SAH. 108 In addition to RNF216, neuronal apoptosis induced by SAH is also enhanced by the E3 ligase TRAF3, which enhances SAH‐induced NF‐κB and MAPK signaling by activating TAK1. 109 Inflammation is a key pathological process contributing to early brain injury (EBI) following SAH. After experimental SAH, microglia upregulate the expression of Peli1, an E3 ligase that positively regulates neuroinflammation by promoting c‐IAP2 ubiquitination and downstream inflammatory signaling in microglia. 65 , 110 Consistently, the knockdown of Peli1 reduces neuroinflammation and improves neurological outcomes during EBI after SAH. 110
1.3. UMEs regulate moyamoya disease (MMD)
MMD is an idiopathic cerebral vasculopathy characterized by progressive narrowing of the intracranial portion of the internal carotid artery and its main branches including the middle cerebral and anterior cerebral arteries. 7 , 111 In MMD, a hazy network of collateral arteries named moyamoya vessels develops around the occlusive region to compensate for the blood flow. MMD usually causes cerebral ischemia in pediatric and adult patients, but half of adult patients can also develop intracranial bleeding. 112 Therefore, MMD poses a key risk factor for both hemorrhagic and ischemic stroke. 111 , 112
The annual incidence of MMD is as high as 0.5–1.5/100 000 in East Asian countries including China, Japan, and Korea, but as low as 0.1/100 000 in other parts of the world. 7 The difference in MMD prevalence is largely due to genetic susceptibility factors in East Asian populations. Indeed, RNF213, which encodes the E3 ligase RNF213, was identified as the principal susceptibility gene for MMD. 7 , 30 , 31 The heterozygous p.Arg4810Lys variant of RNF213 has been identified as a founder mutant present in East Asian MMD patients. 30 , 32 , 33 , 34 Around 1.5% of the population of South Korea and Japan carry this variant, but it is rarely found in Caucasians, which may explain, at least partly, the nearly tenfold higher frequency of MMD in East Asian countries than in other regions.
RNF213 is the biggest E3 ligase in the human proteome with a mass of 591 kD, consisting of an N‐terminal stalk, a dynein‐like ATPase core, and a C‐terminal multidomain E3 module. 113 Since most of the pathological MMD variants map to the E3 module of RNF213, these MMD‐related variants may disturb the E3 ligase activity of RNF213. 113 Consistently, a recent study found that proteins encoded by MMD‐associated RNF213 variants, including the most prevalent Arg4810Lys variant, had reduced ubiquitination activity, suggesting that decreased E3 ligase activity of RNF213 contributes to the pathogenesis of MMD. 114 A recent study found that ablation of RNF213 disrupted the barrier function of human cerebral endothelium in vitro, which could be a potential pathogenic mechanism causing MMD. 15 However, the exact biological functions and molecular mechanisms of RNF213 in MMD remain largely unclear. In the future, studies on macromolecular interactions, conformational dynamics, and biochemical functions of RNF213 may reveal the role and mechanism of action of this E3 in MMD and accelerate the development of RNF213‐targeting therapies for MMD.
1.4. UMEs regulate atherosclerosis
Atherosclerosis is a chronic vascular disease resulting from the complex interplay between lipid metabolism and immune responses. Besides, atherosclerosis can also contribute to the development of other diseases of the circulation system, such as coronary artery disease, peripheral artery disease, and stroke. 6 , 115 , 116 The chronic build‐up of atherosclerotic plaques in the sub‐endothelial intimal layer of medium‐ and large‐sized arteries causes stenosis and restricts blood flow to critical organs, particularly the brain. In addition, rupture of the atherosclerotic plaque leads to acute thrombo‐occlusive events, including ischemic stroke. 117 The p.Arg4810Lys variant of RNF213, which represents the most prevalent genetic abnormality in East Asian MMD patients, is also closely associated with intracranial atherosclerosis and ischemic stroke. 118 , 119 , 120 , 121 Besides, this RNF213 variant predisposes patients with symptomatic intracranial atherosclerosis to stroke recurrence. 122
Atherosclerosis is a cholesterol‐related disease caused by the deposition of lipoproteins, especially low‐density lipoproteins (LDLs), in the intimal space of arteries (Figure 4). In the intima, LDLs are oxidized by free radicals to form oxidized LDLs (OxLDLs), which can be taken up primarily by macrophages through scavenger receptors (SRs). 123 , 124 Macrophages and vascular smooth muscle cells (VSMCs) engulfing excessive OxLDLs differentiate into foam cells, and the accumulation of foam cells contributes to the development of atherosclerotic fatty streaks and plaques. 125 , 126 In addition to driving the transition of macrophages and VSMCs to foam cells, OxLDLs as a group of metabolism‐associated molecular patterns (MAMPs) can also promote atherosclerosis by triggering inflammation, which is a key pathological process underlying the pathogenesis and progression of atherosclerosis 123 (Figures 4 and 5). UMEs have emerged as key regulators of atherosclerosis and they affect the onset and progression of atherosclerosis by modulating endothelial cell function, foam cell formation, and vascular inflammation.
FIGURE 4.
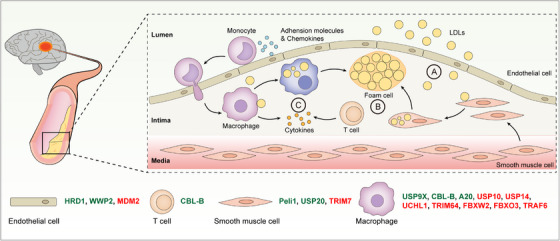
The role of UMEs in atherosclerosis. Atherosclerosis is a primary risk factor for stroke. During the initial stages of atherosclerosis, LDLs are transported across dysfunctional endothelial cells to the sub‐endothelial space of arteries (A). LDLs in the artery intima are engulfed by macrophages through scavenger receptors. After ingesting overdose LDLs, macrophages laden with lipids become foam cells. Apart from macrophages, smooth muscle cells can also become foam cells after ingesting LDLs (B). Accumulation of foam cells further leads to the formation of atherosclerotic plaques. Besides, plaque formation is strongly promoted by pro‐inflammatory cytokines produced by macrophages and T cells (C). UMEs can influence the pathogenesis and development of atherosclerosis by regulating various cell populations including endothelial cells, T cells, macrophages, and smooth muscle cells. UMEs inhibiting atherosclerosis are in green and UMEs promoting atherosclerosis are in red.
FIGURE 5.
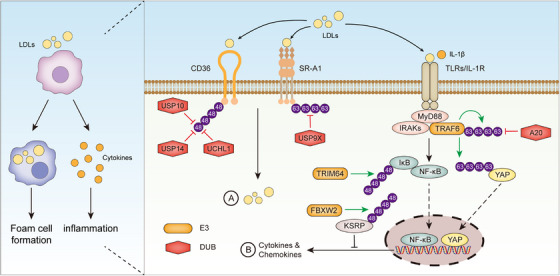
UMEs regulate macrophage functions in atherosclerosis. Macrophages are a key cell population promoting atherosclerosis. On the one hand, macrophages ingest LDLs to become foam cells (A). On the other hand, macrophages produce pro‐inflammatory cytokines in response to LDLs (B). The two processes are closely controlled by UMEs.
1.4.1. UMEs regulate endothelial cell function in atherosclerosis
The atherosclerotic process begins with the accumulation of LDLs in the sub‐endothelial space of arteries 127 (Figure 4A). Endothelial cell dysfunction, such as altered permeability and apoptosis, is the critical initial step in atherogenesis, and this process is tightly regulated by E3 ligases HRD1, MDM2, and WWP2. 128 , 129 , 130 The binding of OxLDLs with lectin‐like oxidized LDL receptor‐1 (LOX‐1), the specific scavenger receptor for OxLDLs on endothelial cells, induces endothelial dysfunction and OxLDL uptake. 131 LOX‐1 can be ubiquitinated by the E3 ligase HRD1 for degradation. 128 HRD1 expression is downregulated in human atherosclerotic intima and its overexpression attenuates OxLDL‐induced apoptosis of endothelial cells by reducing LOX‐1 abundance, indicating that decreased HRD1 expression induces endothelial dysfunction in atherosclerosis. 128 Oxidative stress is a primary driving factor in endothelial dysfunction. The E3 ligase MDM2 promotes OxLDL‐induced mitochondrial damage and oxidative stress in endothelial cells. 129 MDM2 induces the UPS‐dependent degradation of retinoid X receptor beta (RXRβ), a protein that plays a protective role in endothelial cells upon OxLDL stimulation. In LDLr−/− mice, pharmacological inhibition of MDM2 increases the protein abundance of RXRβ in the aorta and decreases the formation of atherosclerotic lesions. 129 In sharp contrast, the E3 ligase WWP2 can inhibit OxLDL‐induced endothelial cell injury by antagonizing oxidative stress. Mechanistically, WWP2 ubiquitinates PDCD4 for degradation, thereby activating the antioxidant HO‐1 pathway in endothelial cells. In ApoE−/− mice, overexpression of WWP2 ameliorates atherosclerosis by reducing oxidative stress and inflammation. 130
1.4.2. UMEs regulate foam cell formation in atherosclerosis
Foam cell formation is a hallmark of atherosclerosis (Figure 4B). A majority of foam cells are derived from macrophages, which ingest OxLDLs through scavenger receptors SR‐A1 and SR‐B2 (CD36) 124 , 132 (Figure 5A). Upon binding with OxLDLs, SR‐A1 is K63 polyubiquitinated at the K27 residue, and this PTM facilitates SR‐A1 internalization, OxLDL uptake, and foam cell formation. 133 The K63‐linked polyubiquitination of SR‐A1 is counter‐regulated by the DUB USP9X (Figure 5A). Pharmacological or genetic inhibition of USP9X increases OxLDL‐induced SR‐A1 ubiquitination and internalization in macrophages. Furthermore, disrupting the interaction between SR‐A1 and USP9X with a cell‐penetrating peptide exacerbates atherosclerosis by increasing foam cell formation, showing that USP9X is an important beneficial regulator of atherosclerosis. 133 The other key scavenger receptor CD36 can also be ubiquitinated, and the ubiquitination of CD36 leads to its proteasomal degradation. 134 , 135 Of note, the ubiquitination and degradation of CD36 is inhibited by DUBs including USP10, USP14, and UCHL1 136 , 137 , 138 (Figure 5A). Inhibition of USP10, USP14 or UCHL1 reduces CD36 protein abundance in macrophages and thereby diminishes OxLDL‐induced foam cell formation. 136 , 137 , 138 Apart from macrophages, another important source of foam cells is VSMCs. The E3 ligase TRIM7 promotes the proliferation and migration of VSMCs in atherosclerosis, and the downregulation of TRIM7 alleviates atherosclerosis in ApoE−/− mice. 139 On the contrary, the E3 ligase Peli1 inhibits atherosclerosis progression by reducing inflammation and the transition of VSMCs to foam cells. 140
1.4.3. UMEs regulate inflammation in atherosclerosis
Atherosclerosis is characterized by continuous low‐grade inflammation in the artery wall, and inflammation accelerates plaque expansion and destabilization 141 , 142 (Figure 4C). Macrophages are the predominant source of pro‐inflammatory molecules in atherosclerosis and macrophage‐mediated inflammatory responses are regulated by UMEs including A20, TRIM64, FBXW2, FBXO3, TRAF6 143 , 144 , 145 , 146 , 147 (Figure 5B). A20, a special UME with both DUB and E3 ligase activities, is an NF‐κB inhibitor and critically regulates inflammatory responses in various diseases. 99 , 148 , 149 A20 was found to play a protective role in atherosclerosis by suppressing the expression of NF‐κB target genes including cytokines and adhesion molecules. 143 As compared with control ApoE−/− mice, atherosclerotic lesions are increased in A20‐haploinsufficient mice and decreased in A20‐overexpressing mice. 143 In contrast, OxLDL‐induced NF‐κB‐dependent inflammation in macrophages is promoted by the E3 ligase TRIM64, which enhances IκBα degradation by ubiquitinating IκBα at the K67 residue. 144 The E3 ligase FBXW2 is an F‐box protein and acts as a substrate‐binding component of the E3 ligase complex termed Skp1‐Cullin‐F‐box protein (SCF) complex. 145 FBXW2 is upregulated in macrophages in atherosclerotic plaques. FBXW2 enhances the production of pro‐inflammatory factors by mediating the ubiquitination and degradation of KSRP, an RNA‐binding protein that negatively regulates the synthesis of a subset of cytokines and chemokines. Consistently, myeloid cell‐specific ablation of FBXW2 mitigates atherosclerosis in mice, accompanied by reduced expression of pro‐inflammatory factors in atherosclerotic lesions. 145 Another F‐box protein of the SCF complex, FBXO3, can also promote atherosclerosis by enhancing inflammation. 146 FBXO3 is predominantly expressed in macrophages in human carotid atherosclerotic plaques, and FBXO3 depletion in macrophages diminishes OxLDL‐induced inflammatory responses. Intriguingly, individuals carrying a hypo‐functioning FBXO3 variant are less susceptible to atherosclerosis. 146 YAP is an essential signaling molecule of the Hippo pathway and it was recently shown to exacerbate atherosclerosis by promoting chemokine production in macrophages. 147 YAP expression is upregulated in macrophages in mouse and human atherosclerotic lesions, and myeloid cell‐specific YAP overexpression aggravates atherosclerosis in mice. Upon stimulation with IL‐1β, a key pro‐inflammatory cytokine involved in atherosclerosis, YAP is K63 ubiquitinated by the E3 ligase TRAF6 at the K252 residue, leading to its protein stabilization and nuclear translocation. This study found that IL‐1β enhanced YAP‐mediated chemokine production in macrophages by activating TRAF6, highlighting a pivotal role of TRAF6 in the inflammation‐driven progression of atherosclerosis. 147
Akin to macrophages, T cells are a dominant immune cell type in atherosclerotic plaques and mediate the inflammatory responses underlying atherosclerosis 150 , 151 (Figure 4C). In both human and mouse atherosclerotic plaques, the E3 ligase CBL‐B is mainly expressed in infiltrating macrophages and T cells. 152 CBL‐B deficiency exacerbates vascular inflammation in mice by increasing the abundance and cytotoxicity of CD8+ T cells as well as macrophage activation, resulting in aggravated atherosclerosis. 152 In addition to macrophages and T cells, VSMCs can also contribute to inflammation in atherosclerosis. The DUB USP20 has been shown to inhibit IL‐1‐ and TNF‐evoked inflammatory responses in VSMCs by deubiquitinating RIPK1. In vivo, specific overexpression of USP20 in VSMCs significantly reduces vascular inflammation and ameliorates atherosclerosis. 153
Collectively, these reports demonstrate that UMEs regulate various key aspects in the pathogenesis and progression of atherosclerosis. Therefore, enhancing the beneficial functions and/or inhibiting the detrimental functions of UMEs may impede the progression of atherosclerosis, preventing the occurrence of more severe CVDs such as stroke.
1.5. UMEs as therapeutic targets and tools
Considering that UMEs serve as versatile and critical regulators in CVDs, potent and specific UME inhibitors/agonists may become efficacious drugs for the prevention and treatment of CVDs. For example, the USP14 inhibitor IU1 has been shown to attenuate neurological deficits caused by ischemic stroke. 154 , 155 Since inhibition of USP14 diminishes foam cell formation, IU1 may also ameliorate atherosclerosis. 137 Of note, compared with E1, E2, and E3 ubiquitinating enzymes, DUBs are more favorable targets for the development of small‐molecule inhibitors. 156 , 157 In the past two decades, DUBs have emerged as novel drug targets for cancer and immune disorders. 158 In the foreseen future, UME inhibitors, particularly DUB inhibitors, may also enrich the therapeutic armamentarium for CVDs. Notably, in the NF‐κB signaling, which critically regulates inflammation and cell death in multiple CVDs, stimulus‐specific polyubiquitin scaffolds provide the docking sites for key upstream signaling molecules including IKKs. 69 , 70 In light of this, compared with the conventional “target‐centric” inhibitors that inhibit single UMEs, “network‐centric” inhibitors, which inhibit ubiquitin‐mediated assembly of signaling complexes, may be more specific and effective. 159 In addition, given that UMEs tightly control the abundance, location, and activation of key proteins involved in CVDs, UMEs can also be applied to treat CVDs by precisely inhibiting detrimental proteins and enhancing beneficial proteins. Indeed, techniques based on UMEs, such as deubiquitinase‐targeting chimera (DUBTAC) and proteolysis‐targeting chimera (PROTAC), are gaining increasing attention as innovative therapeutic methods. 160 , 161 , 162 Therefore, UMEs may become novel drug targets and therapeutic tools, opening up new possibilities for the prevention and treatment of CVDs.
2. CONCLUSION AND PERSPECTIVE
CVDs are a leading cause of disability and death in both developing and developed countries. In 2020, CVDs caused 7.08 million deaths worldwide, surging from 6.6 million deaths in 2019. 116 , 163 Recent studies have elucidated the pivotal roles of UMEs in CVDs, shedding light on the mechanism and therapy of these medical emergencies. Despite these advances, several critical aspects concerning UMEs in CVDs remain to be strengthened. First, more effects are needed to delineate the disease linkage of UMEs with CVDs. Inflammation is of particular importance in the progression of CVDs. Some UMEs, such as Pellino, are impactful regulators of inflammatory signaling, but their roles in CVDs remain largely unknown. Besides, no UME has been found to regulate cerebral small vessel diseases to date. It is intriguing and meaningful to identify new CVD‐regulating UMEs. Second, the function of some UMEs in CVDs has yet to be clarified. Although RNF213 has been established as an essential protein in MMD, its exact function in MMD remains unclear. In the future, the in‐depth investigation of new biochemical functions, interacting partners, and substrates of RNF213 may unravel the pathogenic mechanisms of MMD and inspire new therapies for MMD. Third, the clinical relevance of UMEs with human CVDs should be confirmed. Given that animal models cannot fully recapitulate human diseases and most studies have explored the function of UMEs in CVDs using animal models, these findings cannot be simply extrapolated to clinical situations. Comprehensive studies involving clinical research or humanized mice are more favorable for concluding the function of UMEs in CVDs. Fourth, the research and development of therapeutic approaches and drugs for CVDs based on UMEs should be accelerated. Despite recent advances, the study on UME inhibitors/agonists and PROTAC/DUBTAC is still in its infancy. Further studies in this burgeoning field may improve or even revolutionize the treatment for CVDs.
AUTHOR CONTRIBUTIONS
Conceptualization: Xu Wang. Data collection and analysis: Jingyong Huang, Zhenhu Zhu, and Xu Wang. Writing‐original draft: Jingyong Huang, Zhenhu Zhu, and Xu Wang. Writing‐review and editing: Jingyong Huang, Zhenhu Zhu, Dirk Schlüter, Kate Lykke Lambertsen, Weihong Song, and Xu Wang. Funding acquisition: Weihong Song and Xu Wang.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
ETHICAL APPROVAL
Not applicable.
ACKNOWLEDGEMENTS
This work was supported by grants from the Natural Science Foundation of Zhejiang Province (LZ24H090003 to Xu Wang) and the National Natural Science Foundation of China (82150710557 and 82293642 to Weihong Song; 81971143 and 81900496 to Xu Wang).
Huang J, Zhu Z, Schlüter D, Lambertsen KL, Song W, Wang X. Ubiquitous regulation of cerebrovascular diseases by ubiquitin‐modifying enzymes. Clin Transl Med. 2024;14:e1719. 10.1002/ctm2.1719
Contributor Information
Weihong Song, Email: weihong@wmu.edu.cn.
Xu Wang, Email: sunrim@163.com.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Mergenthaler P, Lindauer U, Dienel GA, Meisel A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013;36(10):587‐597. doi: 10.1016/j.tins.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: Focus on astrocyte‐neuron metabolic cooperation. Cell Metab. 2011;14(6):724‐738. doi: 10.1016/j.cmet.2011.08.016 [DOI] [PubMed] [Google Scholar]
- 3. Xing CY, Tarumi T, Liu J, et al. Distribution of cardiac output to the brain across the adult lifespan. J Cereb Blood Flow Metab. 2017;37(8):2848‐2856. doi: 10.1177/0271678x16676826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Claassen J, Thijssen DHJ, Panerai RB, Faraci FM. Regulation of cerebral blood flow in humans: physiology and clinical implications of autoregulation. Physiol Rev. 2021;101(4):1487‐1559. doi: 10.1152/physrev.00022.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gutierrez J, Khasiyev F, Liu M, et al. Determinants and outcomes of asymptomatic intracranial atherosclerotic stenosis. J Am Coll Cardiol. 2021;78(6):562‐571. doi: 10.1016/j.jacc.2021.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Banerjee C, Chimowitz MI. Stroke caused by atherosclerosis of the major intracranial arteries. Circ Res. 2017;120(3):502‐513. doi: 10.1161/CIRCRESAHA.116.308441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ihara M, Yamamoto Y, Hattori Y, et al. Moyamoya disease: diagnosis and interventions. Lancet Neurol. 2022;21(8):747‐758. doi: 10.1016/S1474-4422(22)00165-X [DOI] [PubMed] [Google Scholar]
- 8. Asselman C, Hemelsoet D, Eggermont D, Dermaut B, Impens F. Moyamoya disease emerging as an immune‐related angiopathy. Trends Mol Med. 2022;28(11):939‐950. doi: 10.1016/j.molmed.2022.08.009 [DOI] [PubMed] [Google Scholar]
- 9. Tekle WG, Hassan AE. Intracranial Atherosclerotic disease: current concepts in medical and surgical management. Neurology. 2021;97(20):S145‐S157. doi: 10.1212/WNL.0000000000012805 Suppl 2 [DOI] [PubMed] [Google Scholar]
- 10. Owolabi MO, Thrift AG, Mahal A, et al. Primary stroke prevention worldwide: translating evidence into action. Lancet Public Health. 2022;7(1):e74‐e85. doi: 10.1016/S2468-2667(21)00230-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu H, Sun S, Liu B. Smurf2 exerts neuroprotective effects on cerebral ischemic injury. J Biol Chem. 2021;297(2):100537. doi: 10.1016/j.jbc.2021.100537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu YL, Chou RH, Shyu WC, et al. Smurf2‐mediated degradation of EZH2 enhances neuron differentiation and improves functional recovery after ischaemic stroke. EMBO Mol Med. 2013;5(4):531‐547. doi: 10.1002/emmm.201201783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zeng J, Wang Y, Luo Z, et al. TRIM9‐Mediated resolution of neuroinflammation confers neuroprotection upon ischemic stroke in mice. Cell Rep. 2019;27(2):549‐560. doi: 10.1016/j.celrep.2018.12.055 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu H, Povysheva N, Rose ME, et al. Role of UCHL1 in axonal injury and functional recovery after cerebral ischemia. Proc Natl Acad Sci U S A. 2019;116(10):4643‐4650. doi: 10.1073/pnas.1821282116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roy V, Ross JP, Pepin R, et al. Moyamoya disease susceptibility gene RNF213 regulates endothelial barrier function. Stroke. 2022;53(4):1263‐1275. doi: 10.1161/STROKEAHA.120.032691 [DOI] [PubMed] [Google Scholar]
- 16. Ndoja A, Reja R, Lee SH, et al. Ubiquitin ligase COP1 suppresses neuroinflammation by degrading c/EBPbeta in microglia. Cell. 2020;182(5):1156‐1169. doi: 10.1016/j.cell.2020.07.011 e12 [DOI] [PubMed] [Google Scholar]
- 17. Heger K, Wickliffe KE, Ndoja A, et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature. 2018;559(7712):120‐124. doi: 10.1038/s41586-018-0256-2 [DOI] [PubMed] [Google Scholar]
- 18. Zheng N, Shabek N. Ubiquitin ligases: Structure, function, and regulation. Annu Rev Biochem. 2017;86:129‐157. doi: 10.1146/annurev-biochem-060815-014922 [DOI] [PubMed] [Google Scholar]
- 19. Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol. 2014;21(4):301‐307. doi: 10.1038/nsmb.2780 [DOI] [PubMed] [Google Scholar]
- 20. Harrigan JA, Jacq X, Martin NM, Jackson SP. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat Rev Drug Discovery. 2018;17(1):57‐78. doi: 10.1038/nrd.2017.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deol KK, Lorenz S, Strieter ER. Enzymatic logic of ubiquitin chain assembly. Front Physiol. 2019;10:835. doi: 10.3389/fphys.2019.00835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Clague MJ, Urbe S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol. 2019;20(6):338‐352. doi: 10.1038/s41580-019-0099-1 [DOI] [PubMed] [Google Scholar]
- 23. Liu B, Ruan J, Chen M, et al. Deubiquitinating enzymes (DUBs): Decipher underlying basis of neurodegenerative diseases. Mol Psychiatry. 2022;27(1):259‐268. doi: 10.1038/s41380-021-01233-8 [DOI] [PubMed] [Google Scholar]
- 24. Juang YC, Landry MC, Sanches M, et al. OTUB1 co‐opts Lys48‐linked ubiquitin recognition to suppress E2 enzyme function. Mol Cell. 2012;45(3):384‐397. doi: 10.1016/j.molcel.2012.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wiener R, Zhang X, Wang T, Wolberger C. The mechanism of OTUB1‐mediated inhibition of ubiquitination. Nature. 2012;483(7391):618‐622. doi: 10.1038/nature10911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nakada S, Tai I, Panier S, et al. Non‐canonical inhibition of DNA damage‐dependent ubiquitination by OTUB1. Nature. 2010;466(7309):941‐946. doi: 10.1038/nature09297 [DOI] [PubMed] [Google Scholar]
- 27. Sheng X, Xia Z, Yang H, Hu R. The ubiquitin codes in cellular stress responses. Protein Cell. 2023;15(3):157‐190. doi: 10.1093/procel/pwad045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gu W, Zhang J, Li Q, et al. The TRIM37 variants in Mulibrey nanism patients paralyze follicular helper T cell differentiation. Cell Discovery. 2023;9(1):82. doi: 10.1038/s41421-023-00561-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brigant B, Demont Y, Ouled‐Haddou H, et al. TRIM37 is highly expressed during mitosis in CHON‐002 chondrocytes cell line and is regulated by miR‐223. Bone. 2020;137:115393. doi: 10.1016/j.bone.2020.115393 [DOI] [PubMed] [Google Scholar]
- 30. Liu W, Morito D, Takashima S, et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS One. 2011;6(7):e22542. doi: 10.1371/journal.pone.0022542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kamada F, Aoki Y, Narisawa A, et al. A genome‐wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet. 2011;56(1):34‐40. doi: 10.1038/jhg.2010.132 [DOI] [PubMed] [Google Scholar]
- 32. Liu W, Hitomi T, Kobayashi H, Harada KH, Koizumi A. Distribution of moyamoya disease susceptibility polymorphism p.R4810K in RNF213 in East and Southeast Asian populations. Neurol Med Chir. 2012;52(5):299‐303. doi: 10.2176/nmc.52.299 [DOI] [PubMed] [Google Scholar]
- 33. Jang MA, Shin S, Yoon JH, Ki CS. Frequency of the moyamoya‐related RNF213 p.Arg4810Lys variant in 1,516 Korean individuals. BMC Med Genet. 2015;16:109. doi: 10.1186/s12881-015-0252-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xue Y, Zeng C, Ge P, et al. Association of RNF213 variants with periventricular anastomosis in moyamoya disease. Stroke. 2022;53(9):2906‐2916. doi: 10.1161/STROKEAHA.121.038066 [DOI] [PubMed] [Google Scholar]
- 35. Collaborators GBDS . Global, regional, and national burden of stroke, 1990–2016: A systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2019;18(5):439‐458. doi: 10.1016/S1474-4422(19)30034-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hankey GJ. Stroke. Lancet. 2017;389(10069):641‐654. doi: 10.1016/S0140-6736(16)30962-X [DOI] [PubMed] [Google Scholar]
- 37. Prabhakaran S, Ruff I, Bernstein RA. Acute stroke intervention: a systematic review. JAMA. 2015;313(14):1451‐1462. doi: 10.1001/jama.2015.3058 [DOI] [PubMed] [Google Scholar]
- 38. Bavarsad K, Barreto GE, Hadjzadeh MA, Sahebkar A. Protective effects of curcumin against ischemia‐reperfusion injury in the nervous system. Mol Neurobiol. 2019;56(2):1391‐1404. doi: 10.1007/s12035-018-1169-7 [DOI] [PubMed] [Google Scholar]
- 39. Naito MG, Xu D, Amin P, et al. Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke. Proc Natl Acad Sci U S A. 2020;117(9):4959‐4970. doi: 10.1073/pnas.1916427117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Green DR. The coming decade of cell death research: five riddles. Cell. 2019;177(5):1094‐1107. doi: 10.1016/j.cell.2019.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Galluzzi L, Kepp O, Chan FK, Kroemer G. Necroptosis: mechanisms and relevance to disease. Annu Rev Pathol. 2017;12:103‐130. doi: 10.1146/annurev-pathol-052016-100247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yuan J, Amin P, Ofengeim D. Necroptosis and RIPK1‐mediated neuroinflammation in CNS diseases. Nat Rev Neurosci. 2019;20(1):19‐33. doi: 10.1038/s41583-018-0093-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311‐320. doi: 10.1038/nature14191 [DOI] [PubMed] [Google Scholar]
- 44. Deng XX, Li SS, Sun FY. Necrostatin‐1 Prevents necroptosis in brains after ischemic stroke via inhibition of RIPK1‐mediated RIPK3/MLKL signaling. Aging Dis. 2019;10(4):807‐817. doi: 10.14336/AD.2018.0728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112‐119. doi: 10.1038/nchembio711 [DOI] [PubMed] [Google Scholar]
- 46. Li J, Zhang J, Zhang Y, et al. TRAF2 protects against cerebral ischemia‐induced brain injury by suppressing necroptosis. Cell Death Dis. 2019;10(5):328. doi: 10.1038/s41419-019-1558-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yao D, Zhang S, Hu Z, et al. CHIP ameliorates cerebral ischemia‐reperfusion injury by attenuating necroptosis and inflammation. Aging (Albany NY). 2021;13(23):25564‐25577. doi: 10.18632/aging.203774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yuan Z, Yi‐Yun S, Hai‐Yan Y. Triad3A displays a critical role in suppression of cerebral ischemic/reperfusion (I/R) injury by regulating necroptosis. Biomed Pharmacother. 2020;128:110045. doi: 10.1016/j.biopha.2020.110045 [DOI] [PubMed] [Google Scholar]
- 49. Cabral‐Miranda F, Nicoloso‐Simoes E, Adao‐Novaes J, et al. rAAV8‐733‐Mediated gene transfer of CHIP/Stub‐1 prevents hippocampal neuronal death in experimental brain ischemia. Mol Ther. 2017;25(2):392‐400. doi: 10.1016/j.ymthe.2016.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rodriguez C, Ramos‐Araque ME, Dominguez‐Martinez M, et al. Single‐nucleotide polymorphism 309T>G in the MDM2 promoter determines functional outcome after stroke. Stroke. 2018;49(10):2437‐2444. doi: 10.1161/STROKEAHA.118.022529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhu X, Li J, You D, Xiao Y, Huang Z, Yu W. Neuroprotective effect of E3 ubiquitin ligase RNF8 against ischemic stroke via HDAC2 stability reduction and reelin‐dependent GSK3beta inhibition. Mol Neurobiol. 2022;59(8):4776‐4790. doi: 10.1007/s12035-022-02880-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gu C, Yang J, Luo Y, et al. ZNRF2 attenuates focal cerebral ischemia/reperfusion injury in rats by inhibiting mTORC1‐mediated autophagy. Exp Neurol. 2021;342:113759. doi: 10.1016/j.expneurol.2021.113759 [DOI] [PubMed] [Google Scholar]
- 53. Wang L, Lu Y, Guan H, et al. Tumor necrosis factor receptor‐associated factor 5 is an essential mediator of ischemic brain infarction. J Neurochem. 2013;126(3):400‐414. doi: 10.1111/jnc.12207 [DOI] [PubMed] [Google Scholar]
- 54. Li T, Qin JJ, Yang X, et al. The Ubiquitin E3 Ligase TRAF6 exacerbates ischemic stroke by ubiquitinating and activating Rac1. J Neurosci. 2017;37(50):12123‐12140. doi: 10.1523/JNEUROSCI.1751-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gong J, Li ZZ, Guo S, et al. Neuron‐Specific tumor necrosis factor receptor‐associated factor 3 is a central regulator of neuronal death in acute ischemic stroke. Hypertension. 2015;66(3):604‐616. doi: 10.1161/HYPERTENSIONAHA.115.05430 [DOI] [PubMed] [Google Scholar]
- 56. Gomez‐Sanchez JC, Delgado‐Esteban M, Rodriguez‐Hernandez I, et al. The human Tp53 Arg72Pro polymorphism explains different functional prognosis in stroke. J Exp Med. 2011;208(3):429‐437. doi: 10.1084/jem.20101523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rodriguez C, Sobrino T, Agulla J, et al. Neovascularization and functional recovery after intracerebral hemorrhage is conditioned by the Tp53 Arg72Pro single‐nucleotide polymorphism. Cell Death Differ. 2017;24(1):144‐154. doi: 10.1038/cdd.2016.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ouyang S, Song Y, Tian Y, Chen Y, Yu X, Wang D. RNF8 deficiency results in neurodegeneration in mice. Neurobiol Aging. 2015;36(10):2850‐2860. doi: 10.1016/j.neurobiolaging.2015.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mailand N, Bekker‐Jensen S, Faustrup H, et al. RNF8 ubiquitylates histones at DNA double‐strand breaks and promotes assembly of repair proteins. Cell. 2007;131(5):887‐900. doi: 10.1016/j.cell.2007.09.040 [DOI] [PubMed] [Google Scholar]
- 60. Wang Y, Liu G, Hong D, Chen F, Ji X, Cao G. White matter injury in ischemic stroke. Prog Neurobiol. 2016;141:45‐60. doi: 10.1016/j.pneurobio.2016.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stetler RA, Gao Y, Leak RK, et al. APE1/Ref‐1 facilitates recovery of gray and white matter and neurological function after mild stroke injury. Proc Natl Acad Sci U S A. 2016;113(25):E3558‐E3567. doi: 10.1073/pnas.1606226113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu H, Li W, Rose ME, et al. The point mutation UCH‐L1 C152A protects primary neurons against cyclopentenone prostaglandin‐induced cytotoxicity: implications for post‐ischemic neuronal injury. Cell Death Dis. 2015;6(11):e1966. doi: 10.1038/cddis.2015.323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shichita T, Ooboshi H, Yoshimura A. Neuroimmune mechanisms and therapies mediating post‐ischaemic brain injury and repair. Nat Rev Neurosci. 2023;24(5):299‐312. doi: 10.1038/s41583-023-00690-0 [DOI] [PubMed] [Google Scholar]
- 64. Heckmann BL, Tummers B, Green DR. Crashing the computer: apoptosis vs. necroptosis in neuroinflammation. Cell Death Differ. 2019;26(1):41‐52. doi: 10.1038/s41418-018-0195-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xiao Y, Jin J, Chang M, et al. Peli1 promotes microglia‐mediated CNS inflammation by regulating Traf3 degradation. Nat Med. 2013;19(5):595‐602. doi: 10.1038/nm.3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang X, Deckert M, Xuan NT, et al. Astrocytic A20 ameliorates experimental autoimmune encephalomyelitis by inhibiting NF‐kappaB‐ and STAT1‐dependent chemokine production in astrocytes. Acta Neuropathol. 2013;126(5):711‐724. doi: 10.1007/s00401-013-1183-9 [DOI] [PubMed] [Google Scholar]
- 67. Mulas F, Wang X, Song S, et al. The deubiquitinase OTUB1 augments NF‐kappaB‐dependent immune responses in dendritic cells in infection and inflammation by stabilizing UBC13. Cell Mol Immunol. 2021;18(6):1512‐1527. doi: 10.1038/s41423-020-0362-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang X, Mulas F, Yi W, et al. OTUB1 inhibits CNS autoimmunity by preventing IFN‐gamma‐induced hyperactivation of astrocytes. EMBO J. 2019;38(10):e100947. doi: 10.15252/embj.2018100947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cruz JA, Mokashi CS, Kowalczyk GJ, et al. A variable‐gain stochastic pooling motif mediates information transfer from receptor assemblies into NF‐kappaB. Sci Adv. 2021;7(30):eabi9410. doi: 10.1126/sciadv.abi9410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tarantino N, Tinevez JY, Crowell EF, et al. TNF and IL‐1 exhibit distinct ubiquitin requirements for inducing NEMO‐IKK supramolecular structures. J Cell Biol. 2014;204(2):231‐245. doi: 10.1083/jcb.201307172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bai X, Zhang YL, Liu LN. Inhibition of TRIM8 restrains ischaemia‐reperfusion‐mediated cerebral injury by regulation of NF‐kappaB activation associated inflammation and apoptosis. Exp Cell Res. 2020;388(2):111818. doi: 10.1016/j.yexcr.2020.111818 [DOI] [PubMed] [Google Scholar]
- 72. Xia Q, Zhan G, Mao M, Zhao Y, Li X. TRIM45 causes neuronal damage by aggravating microglia‐mediated neuroinflammation upon cerebral ischemia and reperfusion injury. Exp Mol Med. 2022;54(2):180‐193. doi: 10.1038/s12276-022-00734-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hao MQ, Xie LJ, Leng W, Xue RW. Trim47 is a critical regulator of cerebral ischemia‐reperfusion injury through regulating apoptosis and inflammation. Biochem Biophys Res Commun. 2019;515(4):651‐657. doi: 10.1016/j.bbrc.2019.05.065 [DOI] [PubMed] [Google Scholar]
- 74. Liu X, Lei Q. TRIM62 knockout protects against cerebral ischemic injury in mice by suppressing NLRP3‐regulated neuroinflammation. Biochem Biophys Res Commun. 2020;529(2):140‐147. doi: 10.1016/j.bbrc.2020.06.014 [DOI] [PubMed] [Google Scholar]
- 75. Wang L, Wu D, Xu Z. USP10 protects against cerebral ischemia injury by suppressing inflammation and apoptosis through the inhibition of TAK1 signaling. Biochem Biophys Res Commun. 2019;516(4):1272‐1278. doi: 10.1016/j.bbrc.2019.06.042 [DOI] [PubMed] [Google Scholar]
- 76. Xiang J, Zhang X, Fu J, Wang H, Zhao Y. USP18 overexpression protects against focal cerebral ischemia injury in mice by suppressing microglial activation. Neuroscience. 2019;419:121‐128. doi: 10.1016/j.neuroscience.2019.09.001 [DOI] [PubMed] [Google Scholar]
- 77. Pan R, Xie Y, Fang W, Liu Y, Zhang Y. USP20 mitigates ischemic stroke in mice by suppressing neuroinflammation and neuron death via regulating PTEN signal. Int Immunopharmacol. 2022;103:107840. doi: 10.1016/j.intimp.2021.107840 [DOI] [PubMed] [Google Scholar]
- 78. Li Z, Liu B, Lambertsen KL, et al. USP25 inhibits neuroinflammatory responses after cerebral ischemic stroke by deubiquitinating TAB2. Adv Sci. 2023;10(28):e2301641. doi: 10.1002/advs.202301641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Frescas D, Pagano M. Deregulated proteolysis by the F‐box proteins SKP2 and beta‐TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8(6):438‐449. doi: 10.1038/nrc2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Shi M, Cho H, Inn KS, et al. Negative regulation of NF‐kappaB activity by brain‐specific TRIpartite Motif protein 9. Nat Commun. 2014;5:4820. doi: 10.1038/ncomms5820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Spitzer D, Guerit S, Puetz T, et al. Profiling the neurovascular unit unveils detrimental effects of osteopontin on the blood‐brain barrier in acute ischemic stroke. Acta Neuropathol. 2022;144(2):305‐337. doi: 10.1007/s00401-022-02452-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yu H, Luo H, Chang L, et al. The NEDD8‐activating enzyme inhibitor MLN4924 reduces ischemic brain injury in mice. Proc Natl Acad Sci U S A. 2022;119(6):e2111896119. doi: 10.1073/pnas.2111896119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Narne P, Pandey V, Phanithi PB. Interplay between mitochondrial metabolism and oxidative stress in ischemic stroke: an epigenetic connection. Mol Cell Neurosci. 2017;82:176‐194. doi: 10.1016/j.mcn.2017.05.008 [DOI] [PubMed] [Google Scholar]
- 84. Ren KD, Liu WN, Tian J, et al. Mitochondrial E3 ubiquitin ligase 1 promotes brain injury by disturbing mitochondrial dynamics in a rat model of ischemic stroke. Eur J Pharmacol. 2019;861:172617. doi: 10.1016/j.ejphar.2019.172617 [DOI] [PubMed] [Google Scholar]
- 85. Prudent J, Zunino R, Sugiura A, Mattie S, Shore GC, McBride HM. MAPL SUMOylation of Drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol Cell. 2015;59(6):941‐955. doi: 10.1016/j.molcel.2015.08.001 [DOI] [PubMed] [Google Scholar]
- 86. Sisalli MJ, Ianniello G, Savoia C, Cuomo O, Annunziato L, Scorziello A. Knocking‐out the Siah2 E3 ubiquitin ligase prevents mitochondrial NCX3 degradation, regulates mitochondrial fission and fusion, and restores mitochondrial function in hypoxic neurons. Cell Commun Signal. 2020;18(1):42. doi: 10.1186/s12964-020-0529-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Carlucci A, Adornetto A, Scorziello A, et al. Proteolysis of AKAP121 regulates mitochondrial activity during cellular hypoxia and brain ischaemia. EMBO J. 2008;27(7):1073‐1084. doi: 10.1038/emboj.2008.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Zhang X, Yan H, Yuan Y, et al. Cerebral ischemia‐reperfusion‐induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy. 2013;9(9):1321‐1333. doi: 10.4161/auto.25132 [DOI] [PubMed] [Google Scholar]
- 89. Geisler S, Holmstrom KM, Skujat D, et al. PINK1/Parkin‐mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119‐131. doi: 10.1038/ncb2012 [DOI] [PubMed] [Google Scholar]
- 90. Zhang X, Yuan Y, Jiang L, et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2‐dependent mitophagy. Autophagy. 2014;10(10):1801‐1813. doi: 10.4161/auto.32136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Feigin VL, Lawes CM, Bennett DA, Barker‐Collo SL, Parag V. Worldwide stroke incidence and early case fatality reported in 56 population‐based studies: A systematic review. Lancet Neurol. 2009;8(4):355‐369. doi: 10.1016/S1474-4422(09)70025-0 [DOI] [PubMed] [Google Scholar]
- 92. Claassen J, Park S. Spontaneous subarachnoid haemorrhage. Lancet. 2022;400(10355):846‐862. doi: 10.1016/S0140-6736(22)00938-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke. 2011;42(6):1781‐1786. doi: 10.1161/STROKEAHA.110.596718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Meng Z, Zhao T, Zhou K, et al. A20 Ameliorates intracerebral hemorrhage‐induced inflammatory injury by regulating TRAF6 polyubiquitination. J Immunol. 2017;198(2):820‐831. doi: 10.4049/jimmunol.1600334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Qu X, Wang N, Chen W, Qi M, Xue Y, Cheng W. RNF34 overexpression exacerbates neurological deficits and brain injury in a mouse model of intracerebral hemorrhage by potentiating mitochondrial dysfunction‐mediated oxidative stress. Sci Rep. 2019;9(1):16296. doi: 10.1038/s41598-019-52494-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhang S, Hu ZW, Luo HY, et al. AAV/BBB‐mediated gene transfer of CHIP attenuates brain injury following experimental intracerebral hemorrhage. Transl Stroke Res. 2020;11(2):296‐309. doi: 10.1007/s12975-019-00715-w [DOI] [PubMed] [Google Scholar]
- 97. Liu C, Liu C, Liu H, et al. Increased expression of ubiquitin‐specific protease 4 participates in neuronal apoptosis after intracerebral hemorrhage in adult rats. Cell Mol Neurobiol. 2017;37(3):427‐435. doi: 10.1007/s10571-016-0375-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Xu Z, Li X, Chen J, et al. USP11, Deubiquitinating enzyme, associated with neuronal apoptosis following intracerebral hemorrhage. J Mol Neurosci. 2016;58(1):16‐27. doi: 10.1007/s12031-015-0644-0 [DOI] [PubMed] [Google Scholar]
- 99. Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol. 2012;12(11):774‐785. doi: 10.1038/nri3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ruan J, Schluter D, Naumann M, Waisman A, Wang X. Ubiquitin‐modifying enzymes as regulators of colitis. Trends Mol Med. 2022;28(4):304‐318. doi: 10.1016/j.molmed.2022.01.006 [DOI] [PubMed] [Google Scholar]
- 101. Razani B, Whang MI, Kim FS, et al. Non‐catalytic ubiquitin binding by A20 prevents psoriatic arthritis‐like disease and inflammation. Nat Immunol. 2020;21(4):422‐433. doi: 10.1038/s41590-020-0634-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. St‐Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC‐1 transcriptional coactivators. Cell. 2006;127(2):397‐408. doi: 10.1016/j.cell.2006.09.024 [DOI] [PubMed] [Google Scholar]
- 103. Chu X, Wu X, Feng H, et al. Coupling between interleukin‐1R1 and necrosome complex involves in hemin‐induced neuronal necroptosis after intracranial hemorrhage. Stroke. 2018;49(10):2473‐2482. doi: 10.1161/STROKEAHA.117.019253 [DOI] [PubMed] [Google Scholar]
- 104. Shen H, Liu C, Zhang D, et al. Role for RIP1 in mediating necroptosis in experimental intracerebral hemorrhage model both in vivo and in vitro. Cell Death Dis. 2017;8(3):e2641. doi: 10.1038/cddis.2017.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Seo J, Lee EW, Sung H, et al. CHIP controls necroptosis through ubiquitylation‐ and lysosome‐dependent degradation of RIPK3. Nat Cell Biol. 2016;18(3):291‐302. doi: 10.1038/ncb3314 [DOI] [PubMed] [Google Scholar]
- 106. Chen S, Feng H, Sherchan P, et al. Controversies and evolving new mechanisms in subarachnoid hemorrhage. Prog Neurobiol. 2014;115:64‐91. doi: 10.1016/j.pneurobio.2013.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Mabb AM, Je HS, Wall MJ, et al. Triad3A regulates synaptic strength by ubiquitination of arc. Neuron. 2014;82(6):1299‐1316. doi: 10.1016/j.neuron.2014.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chen T, Zhu J, Wang YH. RNF216 mediates neuronal injury following experimental subarachnoid hemorrhage through the Arc/Arg3.1‐AMPAR pathway. FASEB J. 2020;34(11):15080‐15092. doi: 10.1096/fj.201903151RRRR [DOI] [PubMed] [Google Scholar]
- 109. Zhou Y, Tao T, Liu G, et al. TRAF3 mediates neuronal apoptosis in early brain injury following subarachnoid hemorrhage via targeting TAK1‐dependent MAPKs and NF‐kappaB pathways. Cell Death Dis. 2021;12(1):10. doi: 10.1038/s41419-020-03278-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Huang XP, Peng JH, Pang JW, et al. Peli1 Contributions in microglial activation, neuroinflammatory responses and neurological deficits following experimental subarachnoid hemorrhage. Front Mol Neurosci. 2017;10:398. doi: 10.3389/fnmol.2017.00398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Scott RM, Smith ER. Moyamoya disease and moyamoya syndrome. N Engl J Med. 2009;360(12):1226‐1237. doi: 10.1056/NEJMra0804622 [DOI] [PubMed] [Google Scholar]
- 112. Kuroda S, Houkin K. Moyamoya disease: Current concepts and future perspectives. Lancet Neurol. 2008;7(11):1056‐1066. doi: 10.1016/S1474-4422(08)70240-0 [DOI] [PubMed] [Google Scholar]
- 113. Ahel J, Lehner A, Vogel A, et al. Moyamoya disease factor RNF213 is a giant E3 ligase with a dynein‐like core and a distinct ubiquitin‐transfer mechanism. eLife. 2020;9:e56185. doi: 10.7554/eLife.56185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bhardwaj A, Banh RS, Zhang W, Sidhu SS, Neel BG. MMD‐associated RNF213 SNPs encode dominant‐negative alleles that globally impair ubiquitylation. Life Sci Alliance. 2022;5(5):e202000807. doi: 10.26508/lsa.202000807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. 2019;124(2):315‐327. doi: 10.1161/CIRCRESAHA.118.313591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Tsao CW, Aday AW, Almarzooq ZI, et al. Heart disease and stroke statistics‐2022 update: A report from the American Heart Association. Circulation. 2022;145(8):e153‐e639. doi: 10.1161/CIR.0000000000001052 [DOI] [PubMed] [Google Scholar]
- 117. Qureshi AI, Caplan LR. Intracranial atherosclerosis. Lancet. 2014;383(9921):984‐998. doi: 10.1016/S0140-6736(13)61088-0 [DOI] [PubMed] [Google Scholar]
- 118. Bang OY, Chung JW, Cha J, et al. A polymorphism in RNF213 is a susceptibility gene for intracranial atherosclerosis. PLoS One. 2016;11(6):e0156607. doi: 10.1371/journal.pone.0156607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Okazaki S, Morimoto T, Kamatani Y, et al. Moyamoya disease susceptibility variant RNF213 p.R4810K Increases the risk of ischemic stroke attributable to large‐artery atherosclerosis. Circulation. 2019;139(2):295‐298. doi: 10.1161/CIRCULATIONAHA.118.038439 [DOI] [PubMed] [Google Scholar]
- 120. Kamimura T, Okazaki S, Morimoto T, et al. Prevalence of RNF213 p.R4810K variant in early‐onset stroke with intracranial arterial stenosis. Stroke. 2019;50(6):1561‐1563. doi: 10.1161/STROKEAHA.118.024712 [DOI] [PubMed] [Google Scholar]
- 121. Miyawaki S, Imai H, Shimizu M, et al. Genetic variant RNF213 c.14576G>A in various phenotypes of intracranial major artery stenosis/occlusion. Stroke. 2013;44(10):2894‐2897. doi: 10.1161/STROKEAHA.113.002477 [DOI] [PubMed] [Google Scholar]
- 122. Kim HJ, Choi EH, Chung JW, et al. Role of the RNF213 variant in vascular outcomes in patients with intracranial atherosclerosis. J Am Heart Assoc. 2021;10(1):e017660. doi: 10.1161/JAHA.120.017660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wang X, Wang Y, Antony V, Sun H, Liang G. Metabolism‐associated molecular patterns (MAMPs). Trends Endocrinol Metab. 2020;31(10):712‐724. doi: 10.1016/j.tem.2020.07.001 [DOI] [PubMed] [Google Scholar]
- 124. Kunjathoor VV, Febbraio M, Podrez EA, et al. Scavenger receptors class A‐I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. 2002;277(51):49982‐49988. doi: 10.1074/jbc.M209649200 [DOI] [PubMed] [Google Scholar]
- 125. Chinetti‐Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015;12(1):10‐17. doi: 10.1038/nrcardio.2014.173 [DOI] [PubMed] [Google Scholar]
- 126. Shankman LS, Gomez D, Cherepanova OA, et al. KLF4‐dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21(6):628‐637. doi: 10.1038/nm.3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Skalen K, Gustafsson M, Rydberg EK, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417(6890):750‐754. doi: 10.1038/nature00804 [DOI] [PubMed] [Google Scholar]
- 128. Li Q, Xuan W, Jia Z, et al. HRD1 prevents atherosclerosis‐mediated endothelial cell apoptosis by promoting LOX‐1 degradation. Cell Cycle. 2020;19(12):1466‐1477. doi: 10.1080/15384101.2020.1754561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zeng Y, Cao J, Li CX, Wang CY, Wu RM, Xu XL. MDM2‐mediated ubiquitination of RXRbeta contributes to mitochondrial damage and related inflammation in atherosclerosis. Int J Mol Sci. 2022;23(10):5766. doi: 10.3390/ijms23105766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Wang X, Ma L, Zhang S, Song Q, He X, Wang J. WWP2 ameliorates oxidative stress and inflammation in atherosclerotic mice through regulation of PDCD4/HO‐1 pathway. Acta Biochim Biophys Sin. 2022;54(8):1057‐1067. doi: 10.3724/abbs.2022091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Akhmedov A, Sawamura T, Chen CH, Kraler S, Vdovenko D, Luscher TF. Lectin‐like oxidized low‐density lipoprotein receptor‐1 (LOX‐1): a crucial driver of atherosclerotic cardiovascular disease. Eur Heart J. 2021;42(18):1797‐1807. doi: 10.1093/eurheartj/ehaa770 [DOI] [PubMed] [Google Scholar]
- 132. Rahaman SO, Lennon DJ, Febbraio M, Podrez EA, Hazen SL, Silverstein RL. A CD36‐dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006;4(3):211‐221. doi: 10.1016/j.cmet.2006.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wang B, Tang X, Yao L, et al. Disruption of USP9X in macrophages promotes foam cell formation and atherosclerosis. J Clin Invest. 2022;132(10):e154217. doi: 10.1172/JCI154217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Tran TT, Poirier H, Clement L, et al. Luminal lipid regulates CD36 levels and downstream signaling to stimulate chylomicron synthesis. J Biol Chem. 2011;286(28):25201‐25210. doi: 10.1074/jbc.M111.233551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Srikanthan S, Li W, Silverstein RL, McIntyre TM. Exosome poly‐ubiquitin inhibits platelet activation, downregulates CD36 and inhibits pro‐atherothombotic cellular functions. J Thromb Haemost. 2014;12(11):1906‐1917. doi: 10.1111/jth.12712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Xia X, Hu T, He J, et al. USP10 deletion inhibits macrophage‐derived foam cell formation and cellular‐oxidized low density lipoprotein uptake by promoting the degradation of CD36. Aging. 2020;12(22):22892‐22905. doi: 10.18632/aging.104003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Zhang F, Xia X, Chai R, et al. Inhibition of USP14 suppresses the formation of foam cell by promoting CD36 degradation. J Cell Mol Med. 2020;24(6):3292‐3302. doi: 10.1111/jcmm.15002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Xia X, Xu Q, Liu M, et al. Deubiquitination of CD36 by UCHL1 promotes foam cell formation. Cell Death Dis. 2020;11(8):636. doi: 10.1038/s41419-020-02888-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Ji R, Gu Y, Zhang J, et al. TRIM7 promotes proliferation and migration of vascular smooth muscle cells in atherosclerosis through activating c‐Jun/AP‐1. IUBMB Life. 2020;72(2):247‐258. doi: 10.1002/iub.2181 [DOI] [PubMed] [Google Scholar]
- 140. Burger F, Baptista D, Roth A, Brandt KJ, Miteva K. The E3 ubiquitin ligase peli1 deficiency promotes atherosclerosis progression. Cells. 2022;11(13):11132014. doi: 10.3390/cells11132014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Soehnlein O, Libby P. Targeting inflammation in atherosclerosis—from experimental insights to the clinic. Nat Rev Drug Discovery. 2021;20(8):589‐610. doi: 10.1038/s41573-021-00198-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Roy P, Orecchioni M, Ley K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat Rev Immunol. 2022;22(4):251‐265. doi: 10.1038/s41577-021-00584-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Wolfrum S, Teupser D, Tan M, Chen KY, Breslow JL. The protective effect of A20 on atherosclerosis in apolipoprotein E‐deficient mice is associated with reduced expression of NF‐kappaB target genes. Proc Natl Acad Sci U S A. 2007;104(47):18601‐18606. doi: 10.1073/pnas.0709011104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Zhu C, Chen W, Cui H, et al. TRIM64 promotes ox‐LDL‐induced foam cell formation, pyroptosis, and inflammation in THP‐1‐derived macrophages by activating a feedback loop with NF‐kappaB via IkappaBalpha ubiquitination. Cell Biol Toxicol. 2023;39(3):607‐620. doi: 10.1007/s10565-022-09768-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Wang C, Xu W, Chao Y, Liang M, Zhang F, Huang K. E3 ligase FBXW2 is a new therapeutic target in obesity and atherosclerosis. Adv Sci. 2020;7(20):2001800. doi: 10.1002/advs.202001800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Chandra D, Londino J, Alexander S, et al. The SCFFBXO3 ubiquitin E3 ligase regulates inflammation in atherosclerosis. J Mol Cell Cardiol. 2019;126:50‐59. doi: 10.1016/j.yjmcc.2018.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Liu M, Yan M, Lv H, et al. Macrophage K63‐linked ubiquitination of YAP promotes its nuclear localization and exacerbates atherosclerosis. Cell Rep.2020;32(5):107990. doi: 10.1016/j.celrep.2020.107990 [DOI] [PubMed] [Google Scholar]
- 148. Wertz IE, O'Rourke KM, Zhou H, et al. De‐ubiquitination and ubiquitin ligase domains of A20 downregulate NF‐kappaB signalling. Nature. 2004;430(7000):694‐699. doi: 10.1038/nature02794 [DOI] [PubMed] [Google Scholar]
- 149. Martens A, Priem D, Hoste E, et al. Two distinct ubiquitin‐binding motifs in A20 mediate its anti‐inflammatory and cell‐protective activities. Nat Immunol. 2020;21(4):381‐387. doi: 10.1038/s41590-020-0621-9 [DOI] [PubMed] [Google Scholar]
- 150. Kyaw T, Winship A, Tay C, et al. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in apoE‐deficient mice. Circulation. 2013;127(9):1028‐1039. doi: 10.1161/CIRCULATIONAHA.112.001347 [DOI] [PubMed] [Google Scholar]
- 151. Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. 2020;17(7):387‐401. doi: 10.1038/s41569-020-0352-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Seijkens TTP, Poels K, Meiler S, et al. Deficiency of the T cell regulator Casitas B‐cell lymphoma‐B aggravates atherosclerosis by inducing CD8+ T cell‐mediated macrophage death. Eur Heart J. 2019;40(4):372‐382. doi: 10.1093/eurheartj/ehy714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Jean‐Charles PY, Wu JH, Zhang L, et al. USP20 (ubiquitin‐specific protease 20) inhibits TNF (tumor necrosis factor)‐triggered smooth muscle cell inflammation and attenuates atherosclerosis. Arterioscler, Thromb, Vasc Biol. 2018;38(10):2295‐2305. doi: 10.1161/ATVBAHA.118.311071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Min JW, Lu L, Freeling JL, Martin DS, Wang H. USP14 inhibitor attenuates cerebral ischemia/reperfusion‐induced neuronal injury in mice. J Neurochem. 2017;140(5):826‐833. doi: 10.1111/jnc.13941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Hou W, Yao J, Liu J, et al. USP14 inhibition promotes recovery by protecting BBB integrity and attenuating neuroinflammation in MCAO mice. CNS Neurosci Ther. 2023;29(11):3612‐3623. doi: 10.1111/cns.14292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Lange SM, Armstrong LA, Kulathu Y. Deubiquitinases: from mechanisms to their inhibition by small molecules. Mol Cell. 2022;82(1):15‐29. doi: 10.1016/j.molcel.2021.10.027 [DOI] [PubMed] [Google Scholar]
- 157. Chan WC, Liu X, Magin RS, et al. Accelerating inhibitor discovery for deubiquitinating enzymes. Nat Commun. 2023;14(1):686. doi: 10.1038/s41467-023-36246-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Ren J, Yu P, Liu S, et al. Deubiquitylating Enzymes in Cancer and Immunity. Adv Sci. 2023;10(36):e2303807. doi: 10.1002/advs.202303807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Pabon NA, Zhang Q, Cruz JA, Schipper DL, Camacho CJ, Lee REC. A network‐centric approach to drugging TNF‐induced NF‐kappaB signaling. Nat Commun. 2019;10(1):860. doi: 10.1038/s41467-019-08802-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Farrell K, Jarome TJ. Is PROTAC technology really a game changer for central nervous system drug discovery? Expert Opin Drug Discovery. 2021;16(8):833‐840. doi: 10.1080/17460441.2021.1915979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Bekes M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discovery. 2022;21(3):181‐200. doi: 10.1038/s41573-021-00371-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Henning NJ, Boike L, Spradlin JN, et al. Deubiquitinase‐targeting chimeras for targeted protein stabilization. Nat Chem Biol. 2022;18(4):412‐421. doi: 10.1038/s41589-022-00971-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Virani SS, Alonso A, Aparicio HJ, et al. Heart disease and stroke statistics‐2021 update: A report from the American Heart Association. Circulation. 2021;143(8):e254‐e743. doi: 10.1161/CIR.0000000000000950 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.