

Abstract
Introduction
Matching-adjusted indirect comparisons (MAICs) were used to compare the efficacy of bimekizumab and secukinumab 150 mg and 300 mg at 52 weeks for the treatment of psoriatic arthritis (PsA) in patients who were biologic disease-modifying anti-rheumatic drug-naïve (bDMARD-naïve) or with previous inadequate response or intolerance to tumor necrosis factor inhibitors (TNFi-IR).
Methods
Relevant trials were systematically identified. Individual patient data from bimekizumab randomized controlled trials, BE OPTIMAL (N = 431) and BE COMPLETE (N = 267), were matched to aggregate data from bDMARD-naïve and TNFi-IR patient subgroups from FUTURE 2 using secukinumab 150 mg and 300 mg doses (bDMARD-naïve: N = 63/37; TNFi-IR: N = 67/33). To adjust for cross-trial differences, patients from the bimekizumab trials were re-weighted using propensity scores to match the baseline characteristics of patients in the secukinumab trials. Unanchored comparisons of recalculated bimekizumab and secukinumab 52-week non-responder imputation outcomes for 20/50/70% improvement in American College of Rheumatology score (ACR20/50/70) and minimal disease activity (MDA) index were analyzed.
Results
In patients who were bDMARD-naïve, bimekizumab had a greater likelihood of ACR70 response than secukinumab 150 mg (odds ratio [95% confidence interval] 2.39 [1.26, 4.53]; p = 0.008) and secukinumab 300 mg (2.03 [1.11, 3.72]; p = 0.021) at 52 weeks. In patients who were TNFi-IR, bimekizumab had a greater likelihood of response compared to secukinumab 150 mg for ACR20 (3.50 [1.64–7.49]; p = 0.001), ACR50 (3.32 [1.41, 7.80]; p = 0.006), ACR70 (2.95 [1.08, 8.07]; p = 0.035) and MDA (3.52 [1.38, 8.99]; p = 0.009), and a greater likelihood of response compared to secukinumab 300 mg for ACR50 (2.44 [1.06, 5.65]; p = 0.037) and MDA (2.92 [1.20, 7.09]; p = 0.018) at 52 weeks.
Conclusion
In this MAIC analysis, the efficacy of bimekizumab, as demonstrated by the likelihood of ACR20/50/70 and MDA response at 52 weeks, was greater or comparable to secukinumab 150 mg and 300 mg for patients with PsA who were bDMARD-naïve and TNFi-IR.
Trial Registration Numbers
NCT03895203, NCT03896581, NCT04009499, NCT01752634, NCT01989468, NCT02294227, NCT02404350.
Supplementary Information
The online version contains supplementary material available at 10.1007/s40744-024-00652-7.
Key Summary Points
| Why carry out this study? |
| There is currently no direct head-to-head evidence of the long-term efficacy of bimekizumab compared to other interleukin (IL)-17A inhibitors in psoriatic arthritis (PsA) |
| This study uses matching-adjusted indirect comparisons (MAICs) to compare the efficacy of bimekizumab 160 mg every 4 weeks (Q4W) and secukinumab 150 mg and 300 mg Q4W at 52 weeks for the treatment of PsA in patients who were naïve to biologic disease-modifying anti-rheumatic drugs (bDMARD-naïve) or patients who have previous inadequate response or intolerance to tumor necrosis factor (TNF) inhibitors (TNFi-IR) |
| What was learned from this study? |
| In patients who were bDMARD-naïve, bimekizumab had a greater likelihood of achieving at least a 70% improvement according to American College of Rheumatology response criteria (ACR70) outcome compared to secukinumab 150 mg and secukinumab 300 mg at 52 weeks |
| In patients who were TNFi-IR, bimekizumab had a greater likelihood of response compared to secukinumab 150 mg for ACR20, ACR70, and minimal disease activity (MDA) outcomes and a greater likelihood of response compared to secukinumab 300 mg for ACR50 and MDA outcomes at 52 weeks |
| Bimekizumab can be considered as more effective than, or at least comparable to, secukinumab in achieving long-term, positive treatment outcomes in PsA |
Introduction
Psoriatic arthritis (PsA) is a chronic, systemic disease characterized by musculoskeletal inflammation affecting up to a third of patients with psoriasis [1]. A range of biologic and targeted synthetic biological disease-modifying anti-rheumatic drugs (b/tsDMARDs) are now available for the treatment of PsA and have brought about significant clinical improvements in outcomes.
Recent focus has fallen upon the wider utility of the interleukin (IL)-17 cytokine superfamily in terms of therapeutic targeting to deliver increased efficacy in PsA, especially in treatment-resistant disease [2]. The IL-17 family includes IL-17A and IL-17F, both of which possess pro-inflammatory properties that could potentially offer synergistic therapeutic value as targets. The efficacy of IL-17A inhibition by secukinumab and ixekizumab in PsA has previously been established [2] and the FUTURE phase 3 program dataset for randomized controlled trials (RCTs) of secukinumab in the treatment of PsA has previously been published [3–7]. Bimekizumab is a humanized monoclonal immunoglobulin G1 antibody that selectively inhibits IL-17A, in addition to IL-17F, and has recently been approved in Europe for PsA. Its efficacy and safety were established in two phase 3 RCTs: BE OPTIMAL (NCT03895203) [8] in patients who were naïve to biologic disease-modifying anti-rheumatic drugs (bDMARD-naïve), and BE COMPLETE (NCT03896581) [9] in patients who had previous inadequate response or intolerance to tumor necrosis factor (TNF) inhibitors (TNFi-IR). An open-label extension (OLE) of both trials, BE VITAL (NCT04009499) [10], is also currently ongoing to assess the long-term efficacy. Bimekizumab was superior to secukinumab in plaque psoriasis in the BE RADIANT RCT (NCT03536884) [11], but as yet, there is no direct head-to-head evidence of the long-term efficacy of bimekizumab compared to other IL-17A mono-inhibitors in PsA.
When head-to-head comparisons in RCTs are unavailable, matching-adjusted indirect comparisons (MAICs) can be used to overcome limitations in assessing comparative efficacy due to insufficiently reported data (e.g., unavailable long-term placebo data in PsA) [12, 13]. A MAIC uses propensity score matching techniques to re-weight individual patient data (IPD) from one study on the basis of the summary baseline characteristics from another, enabling adjustment of differences between trials and allowing comparison between observed treatments [14]. In PsA, MAIC techniques have previously been used to compare the efficacy of secukinumab with other bDMARDs (vs adalimumab [15] and infliximab [16]). Secukinumab was therefore selected as a well-established IL-17A inhibitor for comparison to bimekizumab.
In this study, MAICs were conducted to assess the relative efficacy of bimekizumab versus secukinumab (150 mg or 300 mg doses) at 52 weeks in patients with PsA who were bDMARD-naïve or TNFi-IR. This MAIC analysis aims to provide additional long-term comparative data of bimekizumab and secukinumab following the findings of the recent network meta-analysis (NMA) up to week 24 [17].
Methods
Systematic Literature Review and Source Data
A systematic literature review (SLR) was conducted according to the Preferred Reporting Items for Systematic Reviews (PRISMA) guidelines [18] to identify relevant clinical evidence for existing bDMARD therapies in PsA published from January 1991 to December 2022. Details on SLR eligibility criteria and reasons for inclusion/exclusion were previously published [17]. This MAIC focuses on the comparison with secukinumab because of its high real-world utilization in PsA [19]. The FUTURE 2 RCT (NCT01752634) [4] for secukinumab was identified as most relevant for this MAIC analysis as it was the pivotal trial used for regulatory and Health Technology Assessment in Europe [20]. In this analysis, the efficacy of bimekizumab dosed at 160 mg every 4 weeks (Q4W) was compared to secukinumab at two dose levels, 150 mg and 300 mg Q4W. Although both secukinumab doses are common in clinical practice, the European Medicines Agency guidance recommends that the 300 mg dose be given to patients with PsA who were TNFi-IR [19–21]. Pooled data from all relevant secukinumab RCTs (FUTURE 2, FUTURE 3 [NCT01989468], FUTURE 4 [NCT02294227], and FUTURE 5 [NCT02404350]) were used for a sensitivity analysis comparison [22]. Ethical approval was obtained from the relevant institutional review boards at participating sites, and all patients provided written informed consent in accordance with local requirements. BE OPTIMAL and BE COMPLETE were conducted in accordance with Declaration of Helsinki and the International Conference on Harmonisation Guidance for Good Clinical Practice.
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors. All the results presented in this article are in aggregate form, and no personally identifiable information was used for this study.
Selection of Baseline Characteristics for Matching
Adjustment variables were selected on the basis of a review of previous MAICs in PsA [15, 16], consensus agreement with clinical experts (n = 5), and adherence to established MAIC guidelines [14]. Exploratory univariate sensitivity analyses evaluated the impact of all adjustment variables. To adjust for cross-trial differences, patients from the bimekizumab trials were re-weighted to match the baseline characteristics of the patients in FUTURE 2. Weights were determined on the basis of age, sex, methotrexate (MTX) use, Health Assessment Questionnaire Disability Index (HAQ-DI) score, percentage with psoriasis affecting ≥ 3% body surface area (BSA ≥ 3%), swollen joint count—68 joints (SJC 68), tender joint count—66 joints (TJC 66). Adjustments for race, weight, and DMARD use at baseline were excluded as they were well balanced across trials and their adjustment impact was minimal. Adjustments for dactylitis and enthesitis at baseline were excluded as the impact of the effective sample size (ESS) was assessed to be too large, leading to an unbalanced distribution of weights. For the FUTURE 2 trial, baseline characteristics were assumed to be similar for between patients randomized to secukinumab 150 mg and secukinumab 300 mg for the same study population (either bDMARD-naïve or TNFi-IR).
Adjustment of IPD to Aggregate Data and Pairwise Comparisons
The MAIC methodology as previously described by Signorovitch et al. [14] and the National Institute for Health and Care Excellence Decision Support Unit Technical Support Document 18 (NICE DSU TSD 18) was followed to create a robust population-adjusted indirect treatment comparison (ITC) [13]. All analyses were conducted with R version 3.6.2. The R program provided by the NICE DSU TSD 18 was used to implement this MAIC.
For the base case analysis in patients who were bDMARD-naïve, IPD from BE OPTIMAL was matched to summary patient data from a subgroup of bDMARD-naïve patients in FUTURE 2 for pairwise comparisons. For patients who were TNFi-IR, IPD from the bimekizumab arm of BE COMPLETE/BE VITAL and summary data from a subgroup of patients who were TNFi-IR in FUTURE 2 were used for pairwise comparisons (Fig. 1).
Fig. 1.
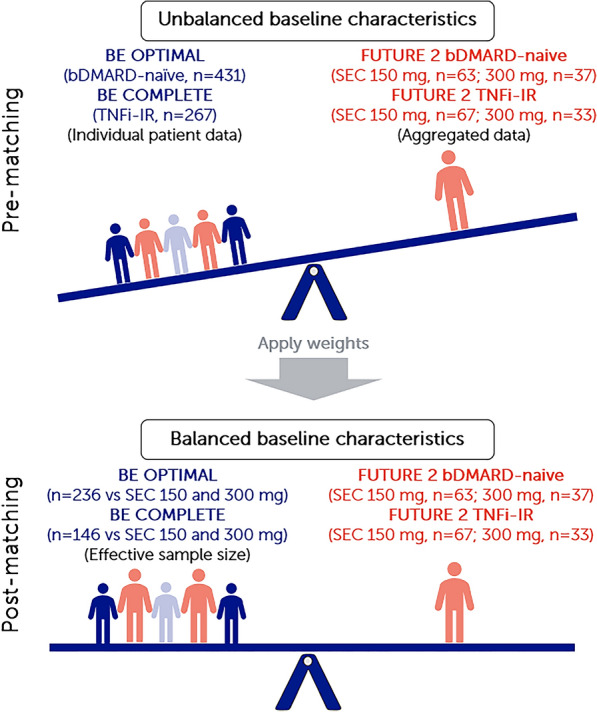
Summary of MAIC matching. Note: MAICs use IPD from trials of one treatment to match baseline aggregate statistics reported from trials of another treatment. Using propensity score weighting techniques to balance trial population characteristics, indirect comparisons can be made. Trial populations adjusted for age, sex, MTX use, HAQ-DI, BSA ≥ 3%, SJC, and TJC. bDMARD biologic disease-modifying anti-rheumatic drug-naïve, BSA body surface area, ESS effective sample size, HAQ-DI Health Assessment Questionnaire–Disability Index, IPD individual patient data, MAIC matching-adjusted indirect comparison, MTX methotrexate, Q4W every 4 weeks, Q8W every 8 weeks, SEC secukinumab, SJC swollen joint count, TJC tender joint count, TNFi-IR tumor necrosis factor inhibitor-inadequate response or intolerant
In the sensitivity analysis, IPD from BE OPTIMAL and BE COMPLETE/BE VITAL were matched to pooled patient data from FUTURE 2, 3, 4, and 5 RCTs in bDMARD-naïve and TNFi-IR patient subgroups separately, for pairwise comparisons.
Outcomes
The outcomes reported were the proportion of patients with 20/50/70% improvement in the American College of Rheumatology criteria (ACR20/50/70) and minimal disease activity (MDA, minimum 5 out of 7 domains achieved) scores. These were selected in line with the Outcomes Measures in Rheumatology (OMERACT) and the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) guidelines [23, 24]. For this MAIC analysis, week 52 data from both bimekizumab and secukinumab RCTs were used to compare outcomes as it was the longest time point at which efficacy data were available for both treatments at the time of the analysis.
Analyses of Psoriasis Area and Severity Index (PASI) scores, enthesitis resolution, dactylitis resolution, and inhibition of radiographic progression outcomes were not feasible as the baseline characteristics of the respective patient subsets were not sufficiently reported in their respective RCTs.
Reporting of Missing Data
Published outcomes included in this MAIC analysis were taken from the intent-to-treat population in all relevant trials (FUTURE 2, BE OPTIMAL, BE COMPLETE, and BE VITAL). Missing binary outcome data (ACR20/50/70 and MDA) were handled using non-responder imputation (NRI) methods.
Non-Placebo-Adjusted Outcome Comparisons
All patients randomized to placebo in the bimekizumab and secukinumab RCTs received active treatment from week 16 to 24 onwards, resulting in the absence of placebo as a common comparator in all RCTs after week 24. Non-placebo-adjusted (unanchored) outcomes at week 52 from the secukinumab 150 mg and 300 mg pooled arms of FUTURE 2 were directly compared with recalculated outcomes from the bimekizumab arms in BE OPTIMAL and BE COMPLETE/BE VITAL.
Reporting of Results
After matching, the ESS indicates the number of independent, non-weighted individuals required to give an estimate with the same precision as the weighted sample estimate and is expressed as a proportion of the original sample size (OSS) from the source trials. Recalculated outcomes were reported as adjusted response rates and the relative effects of bimekizumab versus secukinumab in different patient groups were reported as odds ratios (ORs) with 95% confidence intervals (95% CI, based on ESS). A standard value of p ≤ 0.05 was considered as the threshold for concluding statistical significance (i.e., greater/lesser likelihood or comparable at achieving an outcome response compared to secukinumab).
Results
Patient baseline values for adjusted characteristics prior to matching are provided in Table 1 for both bDMARD-naïve and TNFi-IR patient subgroups in the bimekizumab and secukinumab RCTs. Prior to matching, a greater proportion of patients in the BE OPTIMAL and BE COMPLETE/BE VITAL trials had psoriasis covering BSA ≥ 3% (TNFi-IR patients only in BE COMPLETE/BE VITAL), were receiving MTX therapy, had lower HAQ-DI scores, and had lower SJC/TJC scores compared to patients in the corresponding subgroups in FUTURE 2.
Table 1.
Baseline characteristics of patients from bimekizumab (BE OPTIMAL/BE COMPLETE/BE VITAL) and secukinumab (FUTURE 2) trials before matching
| Mean ± SD unless stated | bDMARD-naïve | TNFi-IR | ||
|---|---|---|---|---|
| BE OPTIMAL | FUTURE 2a bDMARD-naïve |
BE COMPLETE/BE VITAL | FUTURE 2a TNFi-IR |
|
| N = 431 |
N = 63 (150 mg) N = 67 (300 mg) |
N = 267 |
N = 33 (150 mg) N = 37 (300 mg) |
|
| Age, years | 49 (13) | 47 (12) | 50 (12) | 48 (12) |
| Male, % | 47 | 51 | 49 | 52 |
| MTX use, % | 59 | 49 | 45 | 40 |
| SJC (of 66 joints) | 9.0 (6.2) | 10.8 (8.6) | 9.7 (7.5) | 12.4 (9.7) |
| TJC (of 68 joints) | 16.8 (11.8) | 20.3 (14.7) | 18.4 (13.5) | 25.6 (19.1) |
| HAQ-DI score | 0.82 (0.59) | 1.2 (0.6) | 0.97 (0.59) | 1.3 (0.6) |
| BSA ≥ 3%, % | 50 | 51 | 66 | 48 |
bDMARD biologic disease-modifying anti-rheumatic drug-naïve, BSA body surface area, HAQ-DI Health Assessment Questionnaire–Disability Index, MTX methotrexate, SD standard deviation, SEC secukinumab, SJC swollen joint count, TJC tender joint count, TNFi-IR tumor necrosis factor inhibitor-inadequate response or intolerant
aFor the FUTURE 2 trial, patient baseline characteristics were assumed to be similar among patients who were bDMARD-naïve and TNFi-IR, randomized to either SEC 150 mg or 300 mg doses
Base Case Analysis—vs FUTURE 2
bDMARD-Naïve Patient Subgroup
In this MAIC analysis, patients from the bimekizumab arm of BE OPTIMAL (n = 431) were matched to a subgroup of patients who were bDMARD-naïve from FUTURE 2 (secukinumab 150 mg, n = 63; secukinumab 300 mg, n = 67). After matching, the post-matching ESS for bimekizumab was 236.15 (54.8% of the OSS) for comparison to both secukinumab 150 mg and 300 mg Q4W (Fig. 2a/c and Table S1).
Fig. 2.

Matching-adjusted odds ratio comparison of bimekizumab vs secukinumab (150 mg and 300 mg) at week 52 (NRI). a BKZ 160 mg Q4W vs SEC 150 mg Q4W in patients with PsA who were bDMARD-naïve, b BKZ 160 mg Q4W vs SEC 300 mg Q4W in patients with PsA who were bDMARD-naïve, c BKZ 160 mg Q4W vs SEC 150 mg Q4W in patients with PsA who were TNFi-IR, d BKZ 160 mg Q4W vs SEC 300 mg Q4W in patients with PsA who were TNFi-IR. *Indicates statistical significance. Figure shows a logarithmic scale. ACR American College of Rheumatology, ACR20/50/70 at least a 20/50/70% improvement according to the ACR response criteria, bDMARD biologic disease-modifying anti-rheumatic drugs, BKZ bimekizumab, CI confidence interval, ESS effective sample size, MDA minimal disease activity, NRI non-responder imputation, OR odds ratio, PsA psoriatic arthritis, pts patients, Q4W every 4 weeks, SEC secukinumab, TNFi-IR tumor necrosis factor inhibitor-inadequate response or intolerant
In patients who were bDMARD-naïve, bimekizumab had a greater likelihood of achieving ACR70 response than secukinumab 150 mg at week 52 (OR [95% CI] 2.39 [1.26, 4.53], p = 0.008) and was comparable with secukinumab 150 mg in achieving ACR20 (0.64 [0.32, 1.26], p = 0.193), ACR50 (1.20 [0.69, 2.10], p = 0.522), and MDA (1.77 [1.00, 3.15], p = 0.051) responses (Fig. 2a). Compared to secukinumab 300 mg, bimekizumab had a greater likelihood of achieving ACR70 (2.03 [1.11, 3.72], p = 0.021) response at week 52 and was comparable in achieving ACR20 (1.12 [0.62, 2.03], p = 0.704), ACR50 (1.06 [0.62, 1.83], p = 0.827), and MDA (1.53 [0.87, 2.68], p = 0.138) responses (Fig. 2c).
TNFi-IR Patient Subgroup
Patients from the bimekizumab arm of BE COMPLETE (n = 267) were matched to a subgroup of patients who were TNFi-IR from FUTURE 2 (secukinumab 150 mg, n = 37; secukinumab 300 mg, n = 33). The post-matching ESS for bimekizumab was 145.50 (54.5% of OSS) for comparison to both secukinumab 150 mg and 300 mg Q4W (Fig. 2b/d and Table S2).
In patients who were TNFi-IR, bimekizumab had a greater likelihood of response for all ACR and MDA outcomes than secukinumab 150 mg at week 52 (ACR20: 3.50 [1.64, 7.49], p = 0.001; ACR50: 3.32 [1.41, 7.80], p = 0.006; ACR70: 2.95 [1.08, 8.07], p = 0.035; MDA: 3.52 [1.38, 8.99], p = 0.009) (Fig. 2b). Compared to secukinumab 300 mg, bimekizumab had a greater likelihood of achieving ACR50 (2.44 [1.06, 5.65]; p = 0.037) and MDA (2.92 [1.20, 7.09], p = 0.018) responses at week 52 and was comparable in achieving ACR20 (1.78 [0.82, 3.87], p = 0.147) and ACR70 (2.08 [0.80, 5.37], p = 0.131) responses (Fig. 2d).
Sensitivity Analysis—vs Pooled Data from FUTURE 2–5
A sensitivity analysis was conducted using pooled data from the FUTURE 2–5 trials [22] to support the findings of the base case analysis (which used only FUTURE 2 data).
bDMARD-Naïve Patient Subgroup
Patients from the bimekizumab arm of BE OPTIMAL (n = 431) were matched to a subgroup of patients who were bDMARD-naïve from pooled analyses of FUTURE 2–5 (secukinumab 150 mg, n = 643; secukinumab 300 mg, n = 316]. The post-matching ESSs for bimekizumab were 304.81 (70.7% of OSS) and 281.93 (65.4% of OSS) for the comparison to SEC 150 mg and 300 mg Q4W, respectively (Fig. 3a/c and Table S3).
Fig. 3.

Sensitivity analysis of MAIC using pooled data from FUTURE 2–5 RCTs for secukinumab. a BKZ 160 mg Q4W vs SEC 150 mg Q4W in patients with PsA who were bDMARD-naïve, b BKZ 160 mg Q4W vs SEC 300 mg Q4W in patients with PsA who were bDMARD-naïve, c BKZ 160 mg Q4W vs SEC 150 mg Q4W in patients with PsA who were TNFi-IR, d BKZ 160 mg Q4W vs SEC 300 mg Q4W in patients with PsA who were TNFi-IR. *Indicates statistical significance. Figure shows a logarithmic scale. ACR American College of Rheumatology, ACR20/50/70 at least a 20/50/70% improvement according to the ACR response criteria, bDMARD biologic disease-modifying anti-rheumatic drugs, BKZ bimekizumab, CI confidence interval, ESS effective sample size, MAIC matching-adjusted indirect comparison, NRI non-responder imputation, OR odds ratio, pts patients, Q4W every 4 weeks, SEC secukinumab, TNFi-IR tumor necrosis factor inhibitor-inadequate response or intolerant
In patients who were bDMARD-naïve, bimekizumab had a greater likelihood of achieving ACR50 response than secukinumab 150 mg at week 52 (1.46 [1.11, 1.93], p = 0.007) and was comparable with secukinumab 150 mg in achieving ACR20 response (1.17 [0.87, 1.59], p = 0.305) (Fig. 3a). Bimekizumab was also comparable with secukinumab 300 mg in achieving ACR20 (1.02 [0.71, 1.45], p = 0.926) and ACR50 (1.19 [0.86, 1.65], p = 0.283) responses at week 52 (Fig. 3c).
TNFi-IR Patient Subgroup
Patients in the bimekizumab arm of BE COMPLETE (n = 267) were matched to a subgroup of patients who were TNFi-IR from pooled analyses of FUTURE 2–5 (secukinumab 150 mg, n = 264; secukinumab 300 mg, n = 145). The post-matching ESSs for bimekizumab were 116.71 (43.7% of OSS) and 141.99 (53.2% of OSS) for the comparison to secukinumab 150 mg and 300 mg Q4W, respectively (Fig. 3b/d and Table S4).
In patients who were TNFi-IR, bimekizumab had a greater likelihood of achieving ACR20 (2.52 [1.57, 4.03], p < 0.001) and ACR50 (2.72 [1.71, 4.32], p < 0.001) response than secukinumab 150 mg at week 52 (Fig. 3c). Bimekizumab also had a greater likelihood of achieving ACR20 (1.69 [1.04, 2.76], p = 0.034) and ACR50 (2.16 [1.32, 3.53], p = 0.002) responses than secukinumab 300 mg at week 52 (Fig. 3d).
Unadjusted and adjusted response rates and ORs for both the base case and sensitivity analyses are available from Tables S1 to S4 in the Supplementary Materials. The adjusted ORs were comparable to the unadjusted ORs for all outcomes, which provides further support for the validity of these findings.
Discussion
This study used a MAIC analysis to assess the comparative efficacy of bimekizumab 160 mg Q4W against secukinumab 150 mg or 300 mg Q4W at 52 weeks. Patients receiving bimekizumab who were bDMARD-naïve had a higher probability of achieving ACR70 response at 52 weeks compared to patients receiving secukinumab 150 mg and 300 mg doses. Bimekizumab was statistically comparable but numerically better than secukinumab 150 mg and 300 mg for all other measured outcomes (except ACR20) in bDMARD-naïve patients.
Patients treated with bimekizumab who were TNFi-IR had a higher probability of achieving any ACR response compared to those receiving secukinumab 150 mg, and a higher probability of achieving ACR50/MDA response compared to those receiving secukinumab 300 mg at 52 weeks. Bimekizumab was statistically comparable but numerically better than secukinumab 150 mg and 300 mg for all other measured outcomes in patients who were TNFi-IR. A sensitivity analysis using pooled data from FUTURE 2–5 RCTs corroborated the favorable efficacy of bimekizumab over secukinumab for ACR20 and ACR50 outcomes. These findings are also consistent with a recently published NMA in which bimekizumab ranked higher in efficacy than secukinumab on most joint outcomes at 16 to 24 weeks [17].
In PsA, IL-17A and IL-17F are considered key pro-inflammatory mediators. Their biological roles in PsA are, however, not identical and each appears to offer both overlapping and distinctive effector functions in the type 17 immune response in human systems, with the latter being considered pivotal in PsA pathogenesis [25]. The ability of a potential treatment to inhibit both IL-17A and IL-17F can provide patients with a greater inhibition of inflammation. The dual specificity of the bimekizumab antigen for IL-17A and IL-17F allows optimal binding of either isoform and prevent pro-inflammatory signaling regardless of the proportions in which the cytokines are present [26].
Study Limitations
This MAIC analysis has limitations, both intrinsic to the methodology and specific to this analysis. This MAIC analysis required the use of patient-level data of patients who were TNFi-IR from the BE VITAL OLE trial. The efficacy analyses used for BE VITAL were conducted by NRI using the total patient population that started BE COMPLETE, thereby reducing uncertainties introduced from using OLE data. All patients completing week 16 in BE COMPLETE were eligible to enroll in BE VITAL and patients receiving placebo were switched to bimekizumab. Although observed patient variables at baseline could be matched, it was not possible to control unobserved or unreported variables. There were also differences in the duration of the placebo-controlled segment between RCTs (range 16 weeks for BE OPTIMAL/BE COMPLETE to 24 weeks for FUTURE 2). For the FUTURE 2 trial, baseline characteristics had to be assumed to be similar between patients randomized to secukinumab 150 mg and secukinumab 300 mg for the same study population (either bDMARD-naïve or TNFi-IR) as patient baseline data were not stratified by different dosages. There was variation in the study designs at week 52 (dose blind [FUTURE 2–5] vs active treatment blind [BE OPTIMAL] vs open-label [BE COMPLETE/BE VITAL]), although none of the studies were placebo-controlled at that stage, meaning all patients included in this MAIC were aware that they were receiving active treatment until week 52. Analyses of PASI scores, enthesitis resolution, dactylitis resolution, and inhibition of radiographic progression were not feasible as outcomes assessed in the RCT were only based on a subset of the trial population for which baseline characteristics were not sufficiently reported. Safety outcomes could not be analyzed as the original secukinumab trial (FUTURE 2) did not provide safety data stratified by subgroups of interest (bDMARD-naïve or TNFi-IR).
Conclusion
On the basis of MAIC analysis, bimekizumab demonstrated favorable efficacy for the ACR70 outcome compared to secukinumab 150 mg and 300 mg in patients with PsA who were bDMARD-naïve. In patients with PsA who were TNFi-IR, bimekizumab demonstrated a favorable efficacy for all measured outcomes compared to secukinumab 150 mg, and for ACR50 and MDA outcomes compared to secukinumab 300 mg. The results of this analysis should be viewed in the context of the limitations for an indirect comparison, yet the use of IPD and established MAIC methodology provides comparative evidence in the absence of a confirmatory head-to-head RCT.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank the clinical investigators who provided advice on the design and implementation of this study.
Medical Writing/Editorial Assistance
The authors would like to acknowledge Heather Edens, PhD, UCB Pharma, Smyrna, GA, USA, and Costello Medical, UK for publication coordination and editorial assistance and Darryl Low, PhD, Cytel Inc, UK for medical writing and editorial assistance based on the authors’ input and direction. Support for third-party writing assistance for this article was funded by UCB Pharma.
Author Contributions
Substantial contributions to study conception and design: Damon Willems, Vanessa Taieb, Jason Eells; substantial contributions to analysis and interpretation of the data: Damon Willems, Nikos Lyris, Vanessa Taieb, Jason Eells; drafting the article or revising it critically for important intellectual content: Philip J. Mease, Richard B. Warren, Peter Nash, Jean-Marie Grouin, Damon Willems, Nikos Lyris, Vanessa Taieb, Jason Eells, Iain B. McInnes; final approval of the version of the article to be published: Philip J. Mease, Richard B. Warren, Peter Nash, Jean-Marie Grouin, Damon Willems, Nikos Lyris, Vanessa Taieb, Jason Eells, Iain B. McInnes.
Funding
This study and its publication, including the journal’s Rapid Service Fee, was sponsored by UCB Pharma. This article was based on the original studies BE OPTIMAL (NCT03895203), BE COMPLETE (NCT03896581), and BE VITAL (NCT04009499) sponsored by UCB Pharma. Support for third-party writing assistance for this article was funded by UCB Pharma and provided by Darryl Low, PhD, Cytel Inc, UK, in accordance with ISMPP Good Publication Practice (GPP 2022) guidelines [27].
Data Availability
Data from the bimekizumab clinical trials used in this analysis may be requested by qualified researchers 6 months after product approval in the USA and/or Europe, or global development is discontinued, and 18 months after trial completion. Investigators may request access to anonymized individual patient data and redacted study documents which may include: raw datasets, analysis-ready datasets, study protocol, blank case report form, annotated case report form, statistical analysis plan, dataset specifications, and clinical study report. Prior to the use of the data, proposals need to be approved by an independent review panel at www.Vivli.org and a signed data-sharing agreement will need to be executed. All documents are available in English only, for a pre-specified time, typically 12 months, on a password-protected portal.
Declarations
Conflict of Interest
Philip J. Mease: Research grants from AbbVie, Acelyrin, Amgen, BMS, Eli Lilly, Gilead, Janssen, Novartis, Pfizer, Sun Pharma and UCB Pharma; consultancy fees from AbbVie, Acelyrin, Aclaris, Alumis, Amgen, BMS, Boehringer Ingelheim, Eli Lilly, Galapagos, Gilead, GSK, Janssen, Moonlake Pharma, Novartis, Pfizer, Sun Pharma, Takeda, and UCB, and Ventyx Pharma; speakers’ bureau from AbbVie, Amgen, Eli Lilly, Janssen, Novartis, Pfizer, and UCB Pharma. Richard B. Warren: Supported by the Manchester NIHR Biomedical Research Centre; consulting fees from AbbVie, Almirall, Amgen, Arena, Astellas, Avillion, Biogen, BMS, Boehringer Ingelheim, Celgene, Eli Lilly, GSK, Janssen, LEO Pharma, Novartis, Pfizer, Sanofi, and UCB Pharma; research grants to his institution from AbbVie, Almirall, Janssen, LEO Pharma, Novartis, and UCB Pharma; and honoraria from Astellas, DiCE, GSK, and Union. Peter Nash: Research grants, clinical trials and honoraria for advice and lectures on behalf of AbbVie, Boehringer Ingelheim, BMS, Eli Lilly, Galapagos/Gilead, GSK, Janssen, Novartis, Pfizer, Samsung, Sanofi, and UCB Pharma. Jean-Marie Grouin: Consulting fees and honoraria from Acticor, Chugai, BeiGene, Inflectis, Inotrem, Ipsen, Janssen, Otsuka, SpikImm, UCB Pharma. Damon Willems, Nikos Lyris, Jason Eells, Vanessa Taieb: Employee and stockholder of UCB Pharma. Iain B. McInnes: Consulting fees and honoraria from AbbVie, AstraZeneca, BMS, Boehringer Ingelheim, Cabaletta, Causeway Therapeutics, Celgene, Evelo, Janssen, Novartis, Lilly, Moonlake, and UCB Pharma; and research support from BMS, Boehringer Ingelheim, Celgene, Janssen, Novartis, and UCB Pharma.
Ethical Approval
BE OPTIMAL and BE COMPLETE were conducted in accordance with Declaration of Helsinki and the International Conference on Harmonisation Guidance for Good Clinical Practice. Ethical approval was obtained from the relevant institutional review boards at participating sites, and all patients provided written informed consent in accordance with local requirements. This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors. All the results presented in this article are in aggregate form, and no personally identifiable information was used for this study.
Footnotes
Prior Presentation: The data in this manuscript was previously shared at the International Society for Pharmacoeconomics and Outcomes Research European Congress 2023, Copenhagen, Denmark, 12–15 November 2023 (Poster No. CO10).
References
- 1.Mease PJ, Gladman DD, Papp KA, et al. Prevalence of rheumatologist-diagnosed psoriatic arthritis in patients with psoriasis in European/North American dermatology clinics. J Am Acad Dermatol. 2013;69(5):729–735. doi: 10.1016/j.jaad.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 2.Wang EA, Suzuki E, Maverakis E, Adamopoulos IE. Targeting IL-17 in psoriatic arthritis. Eur J Rheumatol. 2017;4(4):272–277. doi: 10.5152/eurjrheum.2017.17037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mease PJ, McInnes IB, Kirkham B, et al. Secukinumab inhibition of interleukin-17A in patients with psoriatic arthritis. N Engl J Med. 2015;373(14):1329–1339. doi: 10.1056/NEJMoa1412679. [DOI] [PubMed] [Google Scholar]
- 4.Kavanaugh A, McInnes IB, Mease PJ, et al. Efficacy of subcutaneous secukinumab in patients with active psoriatic arthritis stratified by prior tumor necrosis factor inhibitor use: results from the randomized placebo-controlled FUTURE 2 study. J Rheumatol. 2016;43(9):1713–1717. doi: 10.3899/jrheum.160275. [DOI] [PubMed] [Google Scholar]
- 5.Nash P, Mease PJ, McInnes IB, et al. Efficacy and safety of secukinumab administration by autoinjector in patients with psoriatic arthritis: results from a randomized, placebo-controlled trial (FUTURE 3) Arthritis Res Ther. 2018;20(1):47. doi: 10.1186/s13075-018-1551-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kivitz AJ, Nash P, Tahir H, et al. Efficacy and safety of subcutaneous secukinumab 150 mg with or without loading regimen in psoriatic arthritis: results from the FUTURE 4 study. Rheumatol Ther. 2019;6(3):393–407. doi: 10.1007/s40744-019-0163-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Heijde D, Mease PJ, Landewe RBM, et al. Secukinumab provides sustained low rates of radiographic progression in psoriatic arthritis: 52-week results from a phase 3 study, FUTURE 5. Rheumatology (Oxf) 2020;59(6):1325–1334. doi: 10.1093/rheumatology/kez420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ritchlin CT, Coates LC, McInnes IB, et al. Bimekizumab treatment in biologic DMARD-naive patients with active psoriatic arthritis: 52-week efficacy and safety results from the phase III, randomised, placebo-controlled, active reference BE OPTIMAL study. Ann Rheum Dis. 2023;82(11):1404–14. [DOI] [PMC free article] [PubMed]
- 9.Merola JF, Landewe R, McInnes IB, et al. Bimekizumab in patients with active psoriatic arthritis and previous inadequate response or intolerance to tumour necrosis factor-alpha inhibitors: a randomised, double-blind, placebo-controlled, phase 3 trial (BE COMPLETE) Lancet. 2023;401(10370):38–48. doi: 10.1016/S0140-6736(22)02303-0. [DOI] [PubMed] [Google Scholar]
- 10.Coates LC, Landewe R, McInnes I, Mease P, Ritchlin C, Tanaka Y. Sustained efficacy and safety of bimekizumab in patients with active psoriatic arthritis and prior inadequate response to tumour necrosis factor inhibitors: results from the phase 3 BE COMPLETE study and its open-label extension up to 1 year EULAR. 2023. [DOI] [PMC free article] [PubMed]
- 11.Reich K, Warren RB, Lebwohl M, et al. Bimekizumab versus secukinumab in plaque psoriasis. N Engl J Med. 2021;385(2):142–152. doi: 10.1056/NEJMoa2102383. [DOI] [PubMed] [Google Scholar]
- 12.Dias S, Sutton AJ, Ades AE, Welton NJ. Evidence synthesis for decision making 2: a generalized linear modeling framework for pairwise and network meta-analysis of randomized controlled trials. Med Decis Mak. 2013;33(5):607–617. doi: 10.1177/0272989X12458724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. Methods for population-adjusted indirect comparisons in health technology appraisal. Med Decis Mak. 2018;38(2):200–211. doi: 10.1177/0272989X17725740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Signorovitch JE, Sikirica V, Erder MH, et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health. 2012;15(6):940–947. doi: 10.1016/j.jval.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Nash P, McInnes IB, Mease PJ, et al. Secukinumab versus adalimumab for psoriatic arthritis: comparative effectiveness up to 48 weeks using a matching-adjusted indirect comparison. Rheumatol Ther. 2018;5(1):99–122. doi: 10.1007/s40744-018-0106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strand V, McInnes I, Mease P, et al. Matching-adjusted indirect comparison: secukinumab versus infliximab in biologic-naive patients with psoriatic arthritis. J Comp Eff Res. 2019;8(7):497–510. doi: 10.2217/cer-2018-0141. [DOI] [PubMed] [Google Scholar]
- 17.Mease PJ, Gladman DD, Merola JF, et al. Comparative effectiveness of bimekizumab in psoriatic arthritis: a systematic literature review and network meta-analysis. Rheumatology. 2024. 10.1093/rheumatology/kead705. [DOI] [PMC free article] [PubMed]
- 18.Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. doi: 10.1136/bmj.n71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song J, Abé C, Banefelt J, et al. Real-world usage of biologic disease-modifying antirheumatic drugs in patients with psoriatic arthritis in Sweden. Ann Rheum Dis. 2023;82(Suppl 1):1785–1786. [Google Scholar]
- 20.EMA. Secukinumab—Summary of product characteristics: EMA; 2023. https://www.ema.europa.eu/en/documents/product-information/cosentyx-epar-product-information_en.pdf. Accessed 15 Oct 2023
- 21.Welby S, Song J, Lu C, et al. RWD174 real world treatment usage of biologic and targeted synthetic disease-modifying anti-rheumatic drugs in US patients with psoriatic arthritis: persistence, factors associated with non-persistence, and dosing patterns. Value Health. 2023;26(6):S394–S395. doi: 10.1016/j.jval.2023.03.2202. [DOI] [Google Scholar]
- 22.Orbai AM, Husni ME, Gladman DD, et al. Secukinumab efficacy on psoriatic arthritis GRAPPA-OMERACT core domains in patients with or without prior tumor necrosis factor inhibitor use: pooled analysis of four phase 3 studies. Rheumatol Ther. 2021;8(3):1223–1240. doi: 10.1007/s40744-021-00337-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tillett W, Eder L, Goel N, et al. Enhanced patient involvement and the need to revise the core set—report from the psoriatic arthritis working group at OMERACT 2014. J Rheumatol. 2015;42(11):2198–2203. doi: 10.3899/jrheum.141156. [DOI] [PubMed] [Google Scholar]
- 24.Coates LC, Soriano ER, Corp N, et al. Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA): updated treatment recommendations for psoriatic arthritis 2021. Nat Rev Rheumatol. 2022;18(8):465–479. doi: 10.1038/s41584-022-00798-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanchez-Rodriguez G, Puig L. Pathogenic role of IL-17 and therapeutic targeting of IL-17F in psoriatic arthritis and spondyloarthropathies. Int J Mol Sci. 2023;24(12):10305. [DOI] [PMC free article] [PubMed]
- 26.Tanaka Y, Shaw S. Bimekizumab for the treatment of psoriatic arthritis. Expert Rev Clin Immunol. 2024;20(2):155–68. [DOI] [PubMed]
- 27.DeTora LM, Toroser D, Sykes A, et al. Good publication practice (GPP) guidelines for company-sponsored biomedical research: 2022 update. Ann Intern Med. 2022;175(9):1298–1304. doi: 10.7326/M22-1460. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from the bimekizumab clinical trials used in this analysis may be requested by qualified researchers 6 months after product approval in the USA and/or Europe, or global development is discontinued, and 18 months after trial completion. Investigators may request access to anonymized individual patient data and redacted study documents which may include: raw datasets, analysis-ready datasets, study protocol, blank case report form, annotated case report form, statistical analysis plan, dataset specifications, and clinical study report. Prior to the use of the data, proposals need to be approved by an independent review panel at www.Vivli.org and a signed data-sharing agreement will need to be executed. All documents are available in English only, for a pre-specified time, typically 12 months, on a password-protected portal.