

Summary
Differentiation of human pluripotent stem cells (hPSCs) into subtype-specific neurons holds substantial potential for disease modeling in vitro. For successful differentiation, a detailed understanding of the transcriptional networks regulating cell fate decisions is critical. The heterochronic nature of neurodevelopment, during which distinct cells in the brain and during in vitro differentiation acquire their fates in an unsynchronized manner, hinders pooled transcriptional comparisons. One approach is to “translate” chronologic time into linear developmental and maturational time. Simple binary promotor-driven fluorescent proteins (FPs) to pool similar cells are unable to achieve this goal, due to asynchronous promotor onset in individual cells. We tested five fluorescent timer (FT) molecules expressed from the endogenous paired box 6 (PAX6) promoter in 293T and human hPSCs. Each of these FT systems faithfully reported chronologic time in 293T cells, but none of the FT constructs followed the same fluorescence kinetics in human neural progenitor cells.
Subject areas: Natural sciences, Biological sciences, Biochemistry, Molecular biology, Neuroscience, Cellular neuroscience, Cell biology
Graphical abstract
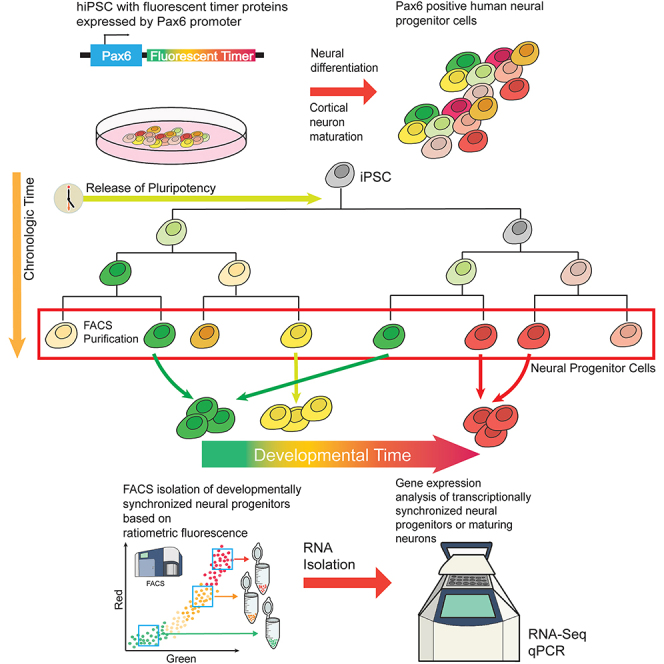
Highlights
-
•
Five iPSC lines expressing fluorescent timers (FT) from the PAX6 promoter
-
•
FT designs to isolate developmentally synchronized cortical progenitor populations
-
•
Sorting of iPSCs by ratiometric fluorescence during neural differentiation
-
•
No tested FT faithfully reports developmental time in human neural differentiation
Natural sciences; Biological sciences; Biochemistry; Molecular biology; Neuroscience; Cellular neuroscience; Cell biology
Introduction
Differentiation of human pluripotent stem cells (hPSCs) into distinct neuronal populations holds substantial potential for disease modeling in vitro.1,2,3,4,5 This could enable both elucidation of pathobiological mechanisms and screening of potential therapeutic agents. However, detailed understanding of the transcriptional networks and their dynamic programs regulating and controlling endogenous cell fate decisions is critical for successful differentiation of hPSCs into subtype- and disease-specific neurons for disease modeling in vitro. Understanding of human-specific versions and subtleties of these networks and dynamics with hPSCs is very limited compared with major fundamental understanding of parallel neuron subtype differentiation in mice that has been deeply investigated over the past two decades.6,7,8,9
One major roadblock to this understanding and thus molecular manipulation for more refined and precise subtype-specific differentiation of hPSCs is the heterochronic nature of neurodevelopment, during which distinct cells and cell types in the brain and during in vitro differentiation mature and acquire their fates in an unsynchronized manner, hindering pooled transcriptional comparisons.7,10,11 Thus, to optimally understand and potentially control the sequential steps of cell fate acquisition and neuronal maturation, it is important to identify and analyze cells that share the same developmental stage. One potential approach would be to re-organize and conceptually translate chronologic time into linear developmental and maturational time.
Single cell RNA sequencing (scRNA-seq) is used extensively to investigate cellular and developmental heterogeneity during neuronal differentiation in vitro12 and in vivo.13 However, scRNA-seq typically only captures a relatively small part of the transcriptome due to limited sequencing depth, thus typically only allowing classification based on relatively highly expressed transcripts, and likely not capturing diverse and progressive cell states in a population of cells. One potential solution is to purify many cells at the same developmental—thus transcriptional—stage, enabling much deeper sequencing at sequential development stages to elucidate dynamics and regulation of gene expression.
One promising approach toward this goal is the widely used approach of genetically encoded fluorescent protein (FP) reporters expressed from cell-fate determining and cell type-specific promoters, typically used for real-time observation of cell states.14,15,16,17 However, such FPs are binary, so individual cells within a pool of cells that all express a given FP might have begun expressing that FP at substantially different times, thus not allowing synchronization and analysis of more transcriptionally homogeneous cells based on promoter onset. Further, even cells expressing the same marker from the same promoter at a time when assessed can still differ in other aspects, such as epigenetic landscape, metabolic state, or expression of other genes. Therefore, it is important to know not just whether a cell expresses a cell-type marker, but also to cluster cells based on the timing and initiation of a distinguishing promoter activity. This would offer potential to optimally analyze dynamics and regulation of transcriptional state.
To circumvent these challenges, specific FP variants with changing fluorescence spectra and intensities over relatively short and reproducible time periods—fluorescent timers (FTs)—can be used as a form of molecular clock.18,19,20,21,22,23 These FTs display time-dependent spectral conversion from one color to another during maturation, e.g., green to red or blue to red. In theory, this might allow determination of timing of promoter activation based on ratiometric fluorescence of the two colors.
Alternatively, two spectrally distinct FPs with distinct maturation constants—e.g., a fast-maturing green FP and a slow-maturing red FP—might be used as ratiometric molecular clocks without such limits of intensity.1,24 Both FPs might be expressed from the same promoter, and the distinct and reproducible maturation times of the green and red FPs might theoretically enable reliable ratiometric measurement and determination of timing of promoter onset. FTs expressed from one or more endogenous, fate-determining promoters, in combination with fluorescence-activated cell sorting (FACS),25,26 might theoretically enable purification of cells synchronized based on their ratiometric fluorescence. Recently, an approach of this type has been used to study enteroendocrine progenitor cells in the intestinal epithelium of mice.27 It is not known whether FPs and/or FTs also might be used to investigate transcriptional dynamics of fate decision and maturation of hPSCs toward human neural progenitors and neurons in vitro. This could “translate” chronological time to developmental-maturational time, thus might enable reliable analysis of substantially more transcriptionally homogeneous cell states.
In the work presented here, we generated and tested a range of knock-in hPSC lines that express five distinct FPs/FTs from the endogenous paired box 6 (PAX6) promoter,28 highly expressed by dorsal cerebral cortex (cortical) progenitors of all cortical projection neurons. PAX6 expression indicates one of the first transition points in cortical neuron development, when pluripotent stem cells exit their pluripotent stage and become committed toward neuronal differentiation. Importantly, PAX6 protein expression in neural progenitor cells is expected to be heterogeneous, since the starting hPSCs are all slightly different, exiting their pluripotency state over a range of time. Together, these characteristics make PAX6 an attractive promoter to test alternative FT systems. We differentiated each of these engineered hPSC lines toward cortical progenitor identity, and validated that each hPSC line differentiated into populations of progenitors with high levels of PAX6 and FT expression. We report here the negative results that none of the tested FP/FT constructs were successful in identification of distinct progenitor populations based on ratiometric fluorescence, thus unsuccessful toward isolating distinct, developmentally synchronized cortical progenitors.
Results
SlowFT expression in 293T cell and neural progenitor cells
In a first set of experiments, designed to assess dynamics of one FT within cultured mammalian cells, we generated stable knock-in 293T cells expressing the monomeric, blue to red maturing, FT SlowFT19 from the doxycycline (Dox) inducible Tet-ON promoter.
We induced SlowFT expression with a 24 h pulse of Dox, and measured fluorescence changes at specific times after induction using flow cytometry. We first detected a strong increase of blue fluorescence 6 h post-induction. After 24 h, we detected double-positive blue and red cells exhibiting increased fluorescence intensity over time, with red fluorescence highest in cells that also exhibited the highest blue fluorescence, indicating proper maturation of the FT (Figure S1A). We also observed in 293T cells expressing SlowFT that fluorescence levels of both blue and red variants of SlowFT are lower than classical single FPs such as EGFP or mCherry. These experiments demonstrate that SlowFT is expressed and properly matures in human cells, thus can be used to track promoter onset based on ratiometric blue to red fluorescence measurements.
The transcription factor (TF) PAX6 is highly expressed by the early human neuroectoderm in vivo and in vitro, and represses pluripotency genes during differentiation from hPSC into dorsal neural progenitor cells, making the endogenous PAX6 promoter an especially useful candidate as the first fate-determining promoter to target with an FT.28 We used the CRISPR-Cas929 system to integrate SlowFT downstream of the endogenous PAX6 gene using a P2A splice acceptor site30 (Figure S2A). This leaves the endogenous PAX6 gene intact, and results in SlowFT expression levels comparable to endogenous PAX6 RNA, thus appropriately reporting PAX6 promoter onset (Figure S2B). We differentiated PAX6-SlowFT iPSC clones into PAX6-expressing dorsal neural progenitor cells,31 and analyzed their fluorescence daily from the start of neuronal differentiation at day 0 up to day 8, using flow cytometry. Uninduced PAX6-SlowFT iPSC did not exhibit detectable blue or red fluorescence. We started to detect a strong increase of blue fluorescence from day 2 onward. However, contrary to our results with 293T cells, we did not detect an increase of red fluorescence over the same period to day 8 (Figure S1B).
Because PAX6 expression is virtually absent on day 2 in this in vitro neuronal differentiation system, we wondered if the strong increase of blue fluorescence might be simply evolving autofluorescence during neuronal differentiation, so we analyzed wild-type (WT) iPSC and WT progenitor cells at day 8 (both lacking any FT construct). Indeed, we found that WT cells showed a strong blue shift, indicating that the increase of blue fluorescence from day 2 onward in PAX6-SlowFT engineered cells is autofluorescence, and not fluorescence of SlowFT protein (Figure S1C). This was further confirmed by confocal imaging of PAX6-SlowFT neural progenitor cells on day 12, revealing strong PAX6 protein expression, but no detectable blue or red fluorescence (Figure S1D). Finally, we tested whether SlowFT mRNA is present in PAX6-SlowFT progenitor cells by RT-PCR on WT and PAX-SlowFT iPSC and progenitor cells at day 8. We found that PAX6 was highly expressed in WT and PAX6-SlowFT progenitors, and we detected SlowFT only in PAX6-SlowFT progenitors, indicating that SlowFT RNA is expressed by PAX6-SlowFT neural progenitors, and increases together with PAX6 mRNA (Figure S1E). While we detected strongly increased SlowFT mRNA, we did not detect SlowFT fluorescence, indicating either sub-detection limit intensity or potentially incorrect folding and maturation of SlowFT in human neural progenitors. Additionally, the strong increase of blue autofluorescence detected during WT neural differentiation could further mask low-intensity SlowFT fluorescence. Together, these results demonstrate that SlowFT is not suitable for tracking PAX6 promoter activation in neural progenitor cells.
TandemFP expression in 293T and neural progenitor cells
Monomeric FTs like SlowFT have the advantage that they are small and therefore easy to incorporate into expression systems, but they can suffer from low intensity when expressed from endogenous promoters. Alternatively, a combination of spectrally distinct, fast maturing and slow maturing, FPs can be used as a FT pair (dual FT). In this approach, the useful range of the dual FT is determined by the difference in maturation times of the two FPs.1 When expressed from an endogenous promoter (e.g., PAX6 and Fezf232), ratiometric fluorescence measurements between the fast and slow maturing FP theoretically might be used to time promoter activation. This approach would enable the use of bright fluorescence molecules, and any spectral combination is theoretically possible if their maturation times are significantly different, thus potentially making this system more flexible compared to a monomeric FT, and more suitable in systems in which more than one FT is expressed sequentially from distinct promoters.
To test the feasibility of such a dual FT approach to purify progenitors with synchronized developmental/transcriptional time based on PAX6 promoter activation, we generated a new FT (TandemFP) consisting of a fusion of fast-maturing green fluorescent protein sfGFP33 and slow-maturing red fluorescent protein DsRed234 (Figure S2B). We chose this pair for three central reasons: (1) because DsRed2 matures 28-times slower than sfGFP, theoretically offering a long enough time window to measure promoter activation based on ratiometric fluorescence; (2) because of the improved folding kinetics of sfGFP when fused to other proteins; and (3) because of sfGFP’s superior brightness compared to regular GFP variants.33
We generated 293T cells with Dox-inducible TandemFP, and pulse-induced them to test whether TandemFP faithfully reports promoter activation. Six hours after Dox induction, we detected strongly increased green fluorescence with further increases over time. At 24 h, two distinct populations emerged—one GFP-only and one GFP/DsRed2-double positive. At 24 h, almost all cells are GFP positive and do not further increase their green fluorescence intensity. At 72 h, only GFP/DsRed double-positive cells were detected. (Figures 1A and S3A). These results demonstrate that the fluorescence changes observed follow reported maturation times for these FPs, and that ratiometric green-to-red measurements can be used to time promoter activation—something not possible using a single fluorophore alone.
Figure 1.

Distinct dynamics of TandemFP expression by 293T cells and human PGP-1 iPSC-derived neural progenitor cells
(A) FACS plots of 293T cell expressing TandemFP from a Tet-ON promoter. Expression was induced by adding Dox to the cell culture medium. Cells were harvested and analyzed after 4 periods of culture—6, 24, 48, and 72 h. Green fluorescence increases strongly by 6 h after induction, with most cells displaying both green and red fluorescence after 72 h.
(B) FACS plots of neural progenitor cells derived from PGP-1 iPSCs expressing TandemFP from the endogenous PAX6 promoter. PGP1-PAX6-tandemFP iPSCs were induced at day (D) 0, and neural progenitors were harvested and analyzed daily. Green fluorescence is detected by D5, however no red fluorescence is detected by D8.
To investigate whether TandemFP successfully times PAX6 promoter onset in human neural progenitor cells, we integrated TandemFP into the 3′ end of the PAX6 gene (Figures S2A and S2B). After confirming correct heterozygous TandemFP integration Figure S2C), we differentiated these PAX6-TandemFP iPSCs into neural progenitor cells, and longitudinally measured their fluorescence using flow cytometry. Based on our successful TandemFP experiments using 293T cells, we expected to first detect “GFP-only” progenitors, followed by GFP/DsRed2 double-positive progenitors, then a further increase of the DsRed2 signal.
Similar to PGP1 WT iPSCs, which do not exhibit fluorescence in their stem- and neural progenitor cell stage, we did not detect any fluorescence in PAX6-TandemFP iPSCs or neural progenitor cells until day 3 of differentiation. GFP-positive progenitors appeared from day 4 onward, with GFP fluorescence levels increasing until day 8. However, we did not detect GFP/DsRed2 double-positive progenitors (Figures 1B, and Figure S3D).
These unanticipated results raised questions of whether the TandemFP fusion protein is correctly expressed and folded, or whether it does not properly mature in human neural progenitor cells. We investigated these questions by performing qPCR and western blot (WB) analysis in WT and PAX6-TandemFP expressing progenitor cells. We designed three qPCR primer sets that either target the sfGFP region, the DsRed2 region, or the middle region, covering both the sfGFP and the DsRed2 fusion region of the TandemFP mRNA. 12 days after induction, we detected strong upregulation, with all three primer sets in TandemFP progenitors, demonstrating that the TandemFP mRNA is fully expressed in neural progenitors. We further detected strong upregulation of PAX6 mRNA in both WT and TandemFP neural progenitors, indicating that the knock-in of TandemFP into the PAX6 locus did not alter its endogenous expression pattern (Figure S3B). Next, we investigated whether TandemFP mRNA is translated into a full-length fusion protein by performing WB against the N-terminal part of TandemFP using an antibody against GFP. Again, we found a strong upregulation of the PAX6 protein in both WT and TandemFP progenitors at day 8 of differentiation. When probing the same samples with the GFP antibody, we detected a strong band correlating with the molecular weight of TandemFP in the PAX6-TandemFP neural progenitors, indicating correct translation of the TandemFP DsRed2::sfGFP fusion protein. Interestingly we also detected a weaker band correlating with the MW of GFP (Figure S3C).
These results demonstrate that full length TandemFP is expressed and translated in human neural progenitor cells, and that its expression correlates with PAX6 mRNA and protein levels. However, we did not detect endogenous DsRed2 signal from the TandemFP fusion protein, indicating either issues with correct folding and maturation of the fusion protein or low DsRed fluorescence signal levels in progenitors, resulting in TandemFP not being suitable as an FT in human neural progenitor cells.
Design and refinement of MolTimer dual fluorescent protein timer constructs
To address these suboptimal results in our next dual FP design, we aimed to increase brightness by using different FPs and adding a NLS35 to further increase signal by concentrating the FPs to the nucleus. We also separated the individual FPs with a P2A/T2A30 site, resulting in a single polycistronic mRNA that is translated into two separate FPs that are expected to fold and mature independently of each other. Recently, a very similar FT (Chrono)27 was successfully used to investigate enteroendocrine progenitor cells in the intestinal epithelium in mice. The Chrono design added a degron to the fast-maturing green fluorescent protein, thus increasing the effective time range of the FT. We based our next FT on a similar design, with some modifications. First, we changed dTomato36 to mScarlet,37 which is roughly twice as bright as dTomato, with a slower maturation time, theoretically making it superior to dTomato in an FT construct. Second, we used a codon-optimized version of mNeonGreen (hmNeonGreen)38 to facilitate translation in human cells (Figure S2B). We chose NeonGreen over sfGFP in this construct because NeonGreen is substantially brighter than GFP or EGFP, and faster maturing than sfGFP, theoretically making NeonGreen superior in an FT construct compared with other GFP variants.38
We used this FT construct (MolTimer1.0) to generate heterogeneous knock-in iPSCs that express MolTimer1.0 from the endogenous PAX6 promoter (Figure S2B and S2C). We differentiated these PAX6-MolTimer1.0 iPSCs into neural progenitor cells, and longitudinally assayed MolTimer1.0 fluorescence in PFA-fixed cells using flow cytometry. Uninduced PAX6-MolTimer1.0 iPSCs did not exhibit fluorescence. From day 4 on, we detected a small population of red, mScarlet-positive cells that increased in both number and fluorescence intensity until day 10. They also exhibited weak, hmNeonGreen fluorescence. However, contrary to published maturation times that predicted faster maturation for mNeonGreen compared to mScarlet, we did not detect hmNeonGreen-only cells (Figures 2A, S4B, and S4C). These results were confirmed by longitudinal confocal imaging of endogenous mScarlet and hmNeonGreen fluorescence in PFA fixed PAX6-MolTimer1.0 iPSCs and neural progenitor cells, in which we detected a linear increase of fluorescence for both FPs, but no hmNeonGreen-only cells (Figure 2B). These results were not due to an incomplete expression of MolTimer1.0 mRNA, because we detected both mScarlet and the hmNeonGreen mRNA sequences (Figure 2C).
Figure 2.

MolTimer1.0 is expressed by human iPSC-derived neural progenitor cells with appropriate multi-day dynamics
(A) FACS plots of human iPSC-derived neural progenitor cells expressing MolTimer1.0 from the endogenous PAX6 promoter. PGP1-PAX6-MolTimer1.0 iPSCs were induced at day (D) 0, and neural progenitors were harvested and analyzed daily. Green and red double-positive cells start to appear from D6 onward, with increasing fluorescence intensity until D10.
(B) Z-projections of confocal imaging stacks of endogenous MolTimer1.0 expression in PGP1-PAX6-MolTimer1.0 neural progenitor cells from D1 to D10. Green and red fluorescence is detected from D6 onward, increasing in intensity until D10.
(C) qPCR analysis of hmNeonGreen, mScarlet, and PAX6 reveals increased mRNA-level expression of both fluorophores together with increased PAX6 expression.
To maintain experimental conditions as directly comparable as possible, and to avoid inherent minor day-to-day variability of the flow cytometer instrument, flow cytometric analysis and confocal imaging was initially performed on PFA-fixed cells. However, PFA fixation can impact fluorescence intensity of certain FPs, and might make a low hmNeonGreen signal undetectable. Therefore, we repeated the longitudinal fluorescence analysis in live cells, and limited analysis to the times just before we detected a fluorescence signal (day 3) to the times when cells were double-positive (day 6). We started the differentiation of all iPSCs on the same day, to avoid variability in the starting cell population, and we analyzed neural progenitor cells on consecutive days, using the same flow cytometer with identical settings. Similar to results in PFA-fixed progenitor cells, we first detected a small population of red, mScarlet-expressing cells at day 4 that increased in fluorescence intensity and number. Again, we detected a slight increase in hmNeonGreen fluorescence in the most highly mScarlet-expressing cells. However, we did not detect single fluorescence, hmNeonGreen-only cells (Figure S5A). These results both indicate that fixation did not quench or negatively impact the hmNeonGreen signal in our time course experiment using PFA-fixed cells (Figures 2A, S4B, and S4C) and further confirm that published maturation times for mScarlet and mNeonGreen, in heterologous expression systems, do not fully reflect maturation times in human neural progenitor cells in vitro.
The differentiation protocol we used includes constant dual SMAD inhibition (SMADi) to produce a homogeneous, PAX6-positive, neural progenitor population. Since we did not detect hmNeonGreen-only cells in the PAX6-MolTimer1.0 progenitor population, we tested whether a more heterogeneous progenitor pool might enhance identification of hmNeonGreen-only cells.
To achieve a more heterogeneous progenitor pool, we pulse treated PAX6-MolTimer1.0 iPSCs with dual SMAD inhibitors for 24 h (D0–D1) and removed the small molecules thereafter. We hypothesized that this short pulse would either lead to no neural induction or to a heterogeneous population of which some cells become neural progenitors and some remain in their undifferentiated pluripotent state. We introduced an additional level of heterogeneity by mixing neural progenitors with matched but uninduced iPSCs prior to flow cytometric analysis. We chose edited but uninduced iPSCs over a simpler WT iPSC control to better control for potential PAX6 promoter leakiness and/or autofluorescence. In these experiments, PAX6-MolTimer1.0 progenitors were either pulse treated or continuously treated with dual SMAD inhibitors, then mixed with PAX6-MolTimer1.0 iPSCs (Figure S5E, 1/20 ratio, progenitors/iPSCs) prior to analysis. Surprisingly, we found that a single, 24 h pulse of dual SMAD inhibition led to MolTimer1.0 expression levels comparable to continuous dual SMAD inhibition, and not to the expected higher degree of heterogeneity (Figure S5B). However, our mixing approach succeeded; mixing of PAX6-MolTimer1.0 progenitors with PAX6-MolTimer iPSCs prior to analysis resulted in a heterogeneous population, with only a small distinct fluorescent population in a large non-fluorescent background population. This was particularly apparent on days 5–6, when only a small population displayed hmNeonGreen and mScarlet fluorescence, with most cells not exhibiting any detectable fluorescence (Figure S5C). While these mixing experiments made it easier to identify MolTimer1.0 expressing progenitors, we still did not identify hmNeonGreen-only cells with these experiments.
The MolTimer1.0 design was based on Chrono,27 but we employed mScarlet instead of dTomato, which has a different maturation time, fluorescence intensity, and fluorescence spectrum, thus might have potentially masked hmNeonGreen fluorescence. Therefore, we generated a heterozygous PAX6-Chrono iPSC reporter line (Figure S2C) to test whether Chrono, expressed from the endogenous PAX6 promoter might be utilized to isolate time resolved human neural progenitor cells. We differentiated these PAX6-Chrono iPSCs into neural progenitors using pulsed dual SMAD inhibition, and analyzed live cells from day 3–7 either as a progenitor population or mixed with PAX6-Chrono iPSC prior to analysis. Similar to our observations with MolTimer1.0-expressing progenitors, a small population of mNeonGreen and dTomato double-positive cells was first detected from day 4, increasing in number and fluorescence intensity over time. However, we did not detect mNeonGreen-only cells (Figure S6). These results demonstrate that MolTimer1.0 and Chrono expressed from the PAX6 promoter do not function as molecular timers in human neural progenitor cells, since we could not capture cells that were single-positive for the fast-maturing mNeonGreen.
MolTimer1.0 and Chrono both contain a destabilized mNeonGreen to theoretically increase the time period over which the FT can report promoter onset. However, we hypothesized that this might also lead to faster degradation of mNeonGreen, making it undetectable. Additionally, mNeonGreen might theoretically mature more slowly in human neural progenitors, leading to its incompatibility with mScarlet or dTomato.
Therefore, we simplified the FT design by removing the degron, and changed hmNeonGreen to sfGFP (MolTimer2.0). We chose mScarlet and sfGFP as the FP pair because both displayed strong expression in neural progenitors (Figures 1B, and 2A) and because their maturation times are substantially different. We targeted the endogenous PAX6 promoter to generate heterozygous PAX6-MolTimer2.0 iPSCs (Figures S2B and S2C), differentiated those iPSCs into neural progenitors, and longitudinally assayed their fluorescence in PFA fixed cells using fluorescence cytometry and confocal microscopy.
As with all the prior PAX6 reporter lines, we did not detect fluorescence until day 4. From day 4 on, we detected an increase of both sfGFP and mScarlet fluorescence, with both number of detected cells and their intensities increasing roughly linearly until day 10, when all cells showed high level red and green fluorescence. Again, we did not detect sfGFP-only cells with this construct (Figures 3A and S7C). These results were confirmed by confocal microscopy, with no detectable fluorescence until day 4, then a roughly linear increase of both FPs until day 10 (Figure 3B). qPCR for sfGFP and mScarlet confirmed that the full MolTimer2.0 polycistronic mRNA was present in progenitors, and that its expression increased together with PAX6 during progenitor differentiation. These results exclude simple problems with expression of the MolTimer2.0 construct (Figure 3C). To again investigate whether there might be negative effects of PFA fixation on the fluorescence signal, we repeated this analysis with live progenitor cells, and followed their fluorescence from day 3 to day 6. Again, we did not detect any sfGFP-only MolTimer2.0-expressing neural progenitors (Figure S8A).
Figure 3.

MolTimer2.0 is also expressed by human iPSC-derived neural progenitor cells with appropriate multi-day dynamics
(A) FACS plots of human iPSC-derived neural progenitor cells expressing MolTimer2.0 from the endogenous PAX6 promoter. PGP1-PAX6-MolTimer2.0 iPSCs were induced at D0, and neural progenitors were harvested and analyzed daily. Green and red double-positive cells start to appear from D6 onward, with increasing fluorescence intensity until D10.
(B) Z-projections of confocal imaging stacks of endogenous MolTimer2.0 expression in PGP1-PAX6-MolTimer2.0 neural progenitor cells from D1 to D10. Green and red fluorescence is detected from D6 onward, increasing in intensity until D10.
(C) qPCR analysis of sfGFP, mScarlet, and PAX6 reveals increasing mRNA-level expression of both fluorophore mRNAs together with increased PAX6 expression. Data are represented as mean ± SD.
Previous experiments found that pulsed induction with dual SMAD inhibitors strongly induces MolTimer1.0 expression in neural progenitors (Figure S5B). To again generate a heterogeneous progenitor population, we employed pulsed dual SMAD induction of PAX6-MolTimer2.0 iPSC followed by mixing of these induced MolTimer2.0 expressing progenitors with un-induced MolTimer2.0 iPSC (Figure S8E). Interestingly, we detected a small sfGFP-only population emerging on day 4 that transitioned into a sfGFP+ and mScarlet+ double-positive population on day 5 and 6 (Figure S8C).
Since an sfGFP-only population was also visible on day 5, we asked whether these cells might represent an early, immature progenitor population that just started to express PAX6 and is distinct from slightly more mature double-positive progenitors. We tested this hypothesis by FACS isolation and subsequent qPCR analysis for PAX6 and sfGFP expression on day 5. We pulse-differentiated PAX6-MolTimer2.0 iPSC into neural progenitors, mixed them with un-induced PAX6-MolTimer2.0 iPSC (1/20 ratio, progenitors/iPSCs) and isolated four distinct populations based on ratiometric green and red fluorescence (Figures 4A and 4B). One population consisted of non-fluorescent cells, expected to contain mostly PAX6-negative iPSCs. As a second population, we collected weakly GFP+ cells, expected to consist of progenitors that just started to express MolTimer2.0, with low PAX6 expression levels. The third population consisted of sfGFP/mScarlet double-positive cells, expected to contain more mature progenitors with moderate PAX6 levels. The fourth population consisted of high level expressing sfGFP/mScarlet double-positive cells, expected to contain the most mature, highly PAX6-expressing progenitor population (Figure 4C). Of note, we detected roughly equally high levels of PAX6 and sfGFP expression in the sfGFP/mScarlet double-positive populations. In contrast to our expectations, we did not detect PAX6 or sfGFP expression in the sfGFP-only population, indicating that this population might mainly consist of iPSC, and not a distinct, early PAX6-expressing progenitor population (Figure 4D).
Figure 4.

FACS isolation and qPCR of MolTimer2.0 expressing neural progenitor cells
(A) Schematic of the mixing and FACS isolation of MolTimer2.0 expressing neural progenitor cells. PGP1-PAX6-MolTimer2.0 neural progenitors were mixed with undifferentiated PGP1-PAX6-MolTimer2.0 iPSC prior to FACS analysis.
(B) FACS plots of PGP1-PAX6-MolTimer2.0 iPSC (left), D5 PGP1-PAX6-MolTimer2.0 neural progenitor cells (middle), and the 1:20 mix of D5 PGP1-PAX6-MolTimer2.0 progenitor cells and undifferentiated PGP1-PAX6-MolTimer2.0 iPSCs used for isolation and qPCR analysis (right).
(C) Gating strategy to isolate four distinct populations of MolTimer2.0 expressing neural progenitor cells: non-fluorescent GFP−/mScarlet−, green-only GFP+/mScarlet−, dual fluorescent GFP+/mScarlet+, and high level dual fluorescent GFP+/mScarlet+.
(D) qPCR analysis of PAX6 and sfGFP expression. PAX6 and sfGFP are only detected in the low level GFP+/mScarlet+ and high level GFP+/mScarlet+ populations. Data are represented as mean ± SD.
Discussion
We investigated the feasibility of multiple, progressively refined FT proteins to report “developmental time” after onset of expression of a developmentally indicative TF, as opposed to chronologic time. In particular, we tested two new designs, MolTimer1.0 and MolTimer2.0, and the Chrono construct published in 2019 by Gehart et al.,27 during neural differentiation of human iPSCs into neural progenitor cells. Ideally, multiple, spectrally distinct FT proteins would be expressed from the promoter of such a fate-determining TF. The sequential expression of those FTs, and their ratiometric use in FACS, would operate in a sense like a chronometer, enabling isolation of distinct, time resolved cellular populations during differentiation of human pluripotent stem cells into subtype-specific neurons or other cell types.
As a first step toward this goal of a sequential FT protein expression system, we engineered five human iPSC lines to express molecularly distinct FTs from the endogenous PAX6 promoter. These FTs ranged from a single FT that changes color over time, SlowFT, to constructs of various levels of complexity with distinct green and red fluorescent proteins, including the recently published Chrono.27 We found that four out of these five FT constructs lead to strong and readily detectable fluorescence upon neuronal induction, with their expression levels increasing with PAX6 expression. In these four FT constructs, we combined a fast-maturing green fluorescent protein with a slow-maturing red fluorescent protein. We predicted that the difference in maturation times would enable ratiometric green-to-red fluorescence measurements, thus isolation of distinct neural progenitor cells. In contrast to these predictions and results in pilot HEK 293T cells, we found that fluorescence of both fluorophores increased in tandem in a roughly linear fashion when expressed from the PAX6 promoter. In contrast, 293T cells first exhibited green fluorescence, then slowly became double-positive, faithfully timing promoter activation as we predicted with these rationally designed constructs.
Our results raise the question of why the FT protein constructs faithfully time promoter onset in 293T cells, or with Chrono in mouse enteroendocrine progenitor cells, but not in human iPSC-derived neural progenitor cells. First, expression levels are different between 293T and neural progenitor cells. When the PAX6 promoter becomes active, initial FT protein levels are low, so green fluorescence is likely below detection threshold. However, on average, the PAX6 promoter is one of the strongest promoters in neural progenitor cells.39 Even with the relatively strong expression of the PAX6 promoter, we detected much higher fluorescence when we heterotopically expressed FT proteins in 293T cells. A second possible explanation is sampling frequency. We assayed cells every 24 h, and might have missed a very short phase when only the fast-maturing FP was detectable in maturing neural progenitor cells. However, in 293T cells, we detected the first green cells 6 h after promoter activation, and could still detect GFP-only cells 24 h later, making this explanation less likely.
Potentially more likely, fluorescent proteins might mature differently in distinct cell types. Except for DsRed2, we always detected both green and red fluorescence in neural progenitor cells, indicating correct processing of both proteins. However, it remains unclear how efficiently FT proteins mature in neural progenitor cells after they are expressed and translated. We carefully selected fluorescent protein pairs based on their maturation times and selected spectrally distinct variants with large differences in their known maturation times. However, those values might be quite different in previously unstudied neural progenitor cells compared to published values derived in pure protein extracts, heterotopic expression systems, or E. coli.40
Further, it is known that distinct promoters have different activation and expression kinetics.41 Human neurogenesis in vivo and in vitro is a lengthy process that occurs over weeks and months compared to days in mice. The PAX6 promoter is one of the first promoters activated when human iPSC exit their pluripotent state and become committed toward neural fate. During this process, PAX6 slowly increases in neural progenitor cells over a long period of time, which also leads to slow, steady increase of FT expression in dividing neural progenitors. This is quite different from pulsed and transient FT expression in 293T cells or in mouse enteroendocrine cells that differentiate from a common progenitor into mature cells in 24 h. In a pulsed system, a limited amount of an FT is made relatively synchronously, then matures, thus operating effectively as a developmental “chronometer” reporting promoter activation. Our experiments indicate that, in striking and limiting contrast, FT expression from the PAX6 promoter in neural progenitor cells slowly increases. Thus, it is likely that each individual cell contains a mixture of an individual FT at a range of protein maturation stages. This might likely explain our experimental results of lack of detectable ratiometric distinction between two FT proteins after early, possibly weak fluorescence following PAX6 promoter activation.
There are also differences between expression systems. Early phases of in vitro neural differentiation from iPSCs appear largely synchronized, with most cells expressing PAX6 at similar levels. This would likely make it difficult to identify subtly developmentally distinct cells based on small fluorescence differences between cells, and heterogeneous cell cycle states could further complicate analysis of dividing progenitors. This is in sharp contrast to the mouse enteroendocrine system, in which only a small population starts to express a promoter, thus FT protein, at distinct times, thus enabling isolation of time-resolved cells from a largely dark, non-FT-expressing background. Though we predicted this difference between mouse and human systems, and designed our FT constructs and experiments to overcome this by mixing FT-expressing neural progenitors with matched iPSCs prior to analysis, this strategy was not sufficient. While these design steps made it possible to identify FT-expressing cells, we were unsuccessful in differentially isolating cells that had just started to express PAX6 vs. cells that were developmentally more mature. These results reinforce the presence of quite homogeneous and developmentally synchronized neural progenitor pool. Therefore, a different promoter (e.g., SOX2, BCL11B, and SATB2) that is active only in a select population of cells, ideally only transiently, might perform better and overcome this limitation. We opted for the PAX6 promoter because it enabled us to test and iterate a range of FT designs early in differentiation, which provides critical early differentiation “trajectory” information not available using a late onset neuronal differentiation promoter such as BCL11B (CTIP2), which peaks in expression at around day 80.39
Taken together, our sequential design and testing of a range of five knock-in hPSC lines that express five distinct single and dual FT proteins from the endogenous paired box 6 (PAX6) promoter demonstrates that none of the tested FP/FT constructs were successful in identification and isolation of distinct progenitor populations in vitro based on ratiometric fluorescence and FACS. Such isolation of distinct, developmentally synchronized cortical progenitors in vitro would enable direct investigation of developmental trajectories of transcriptional regulation. While these dual FP or FT systems faithfully reported chronologic time when expressed from a strong inducible promoter in 293T cells, none of the tested FP/FT constructs followed the same fluorescence kinetics in developing human neural progenitor cells. Additional experiments expressing FTs in neural progenitor cells from a doxycycline-inducible promoter, rather than from a fate-specifying promoter, might further clarify if the differences we report between 293T cells and human neural progenitor cells are due to cell type differences, expression levels, expression kinetics, or differences in protein folding and maturation of the FT when expressed in specific cell types and promoter systems.
Our comparison to 293T cells, and the use of a published FT, demonstrates that FT proteins need to be carefully selected and tested for specific cell types and differentiation systems. This work highlights unique and often surprising expression kinetics and regulation in specific cell types differentiating from hPSCs.
Limitations of the study
There are three broad categories of limitations of this study. First, we employed PGP1 human induced pluripotent stem cells (hiPSCs) as a widely used hiPSC line. Though broadly studied and known to be quite competent in neuronal differentiation, PGP1 cells might differ from some alternative iPSC or ES cell lines such as H1 or H9 (though we do not expect substantially different expression kinetics and regulation of fluorescent proteins between distinct hiPSC or ES lines). Second, while we assessed fluorescence changes at 24 h and over several days thereafter, based on the multi-day kinetics of each FP, we cannot exclude the unlikely theoretical possibility that there might exist a very short phase during the first 24 h when only the fast-maturing FP is detected in neural progenitor cells, Third, we expressed our FT timer constructs from only the PAX6 promoter. It is possible that a different promoter, e.g., with a more heterogenous and pulsed expression profile, might enable isolation of developmentally distinct cells based on ratiometric fluorescence measurements. Thus, the limitations reported might not fully generalize to all hiPSC lines and promoters at some very specific time.
In our experiments using 293T cells, we induced FT expression with one doxycycline concentration that rapidly leads to strong induction of the FT. A gradual doxycycline increase might potentially mimic a slower and more heterogeneous FT increase, with cells containing a mixture of FT at various ranges of protein concentration, similar to expression in neural progenitor cells. Such experiments might further elucidate whether a mixture of FTs at varying maturation states hinders identification of the fast-maturing FP. Further, higher or lower doxycycline concentrations might lead to different FT performances in 293T cells, but we did not investigate this possibility.
These experiments employ the doxycycline-inducible FT system in 293T cells, but not in other cell types. Thus, it remains possible that expressing FTs in neural progenitor cells from a tetracycline-on system might further elucidate whether FT systems are able to report chronologic time in neural progenitors, at least when ectopically expressed and at high levels.
Another theoretical variable is the sequence order of the FPs in the MolTimer1.0, 2.0, and Chrono constructs. FPs were linked with a P2A/T2A site that reliably results in equal expression levels of both proteins, in this case both FP proteins. However, there is the theoretical possibility that the FP placed upstream in the expression construct is, for some atypical reason, expressed at higher levels compared to the downstream FP. In our constructs, the red FP was placed upstream of the green FP. This might theoretically lead to a higher concentration of the red vs. the green FP, which theoretically might hinder identification of the green FP. This would be quite atypical for dual constructs linked by a P2A/T2A site. Future experiments could potentially reverse the order to test for such unexpected effects.
Finally, we assessed SlowFT expression using RT-PCR but not by WB. Further characterization with antibodies against SlowFT might potentially elucidate whether SlowFT was not detected due to low expression levels or maturation issues that might lead to rapid degradation in neural progenitors.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-β-Actin 1/10000 | Sigma Aldrich | Cat# A5441; RRID: AB_476744 |
| Rabbit anti-GFP 1/1000 | Thermo Fisher | Cat# A-11122; RRID: AB_221569 |
| Mouse anti-DSRed2 1/200 | Santa Cruz Biotechnology | Cat# sc-101526; RRID: AB_1562589 |
| Mouse anti-PAX6 1/500 | Thermo Fisher | Cat# 14-9914-95; RRID: AB_2865518 |
| Mouse anti-RPL22 1/1000 | Santa Cruz Biotechnology | Cat# sc-373993; RRID: AB_10918294 |
| Rabbit anti-mNeonGreen Tag 1/1000 | CellSignalling Technologies | Cat# 53061; RRID: AB_2799426 |
| Rabbit anti-RFP 1/1000 | Rockland | Cat# 600-401-379, RRID:AB_2209751 |
| Chemicals, peptides, and recombinant proteins | ||
| mTeSR+ medium | StemCell Technologies | 100-0276 |
| Penicillin/Streptomycin solution | Thermo Fisher | 15140122 |
| Geltrex LDEV-Free hESC-Qualified | Thermo Fisher | A1413302 |
| DMEM medium | Thermo Fisher | 10566016 |
| FBS | Thermo Fisher | 10438026 |
| Lipofectamin3000 | Thermo Fisher | L3000015 |
| Puromycin Dihydrochloride | Thermo Fisher | A1113803 |
| Trypsin-EDTA | Thermo Fisher | 25200056 |
| DMSO | Santa Cruz Biotechnology | sc-358801 |
| Doxycyline | Sigma Aldrich | D9891 |
| CloneR | StemCell Technologies | 5888 |
| Accutase | Innovative Cell Technologies | AT104 |
| Y-27632 | StemCell Technologies | 72302 |
| SB43152 | Seleckchem | S1067 |
| Dorsomorphin | StemCell Technologies | 72102 |
| DMEM:F12 | Thermo Fisher | 10565018 |
| Neurobasal medium | Thermo Fisher | 21103049 |
| Insulin | Sigma Aldrich | I9278 |
| 2-mercaptoethanol | Thermo Fisher | 31350010 |
| Non-essential amino acids | Thermo Fisher | 11140050 |
| Sodium Pyruvate | Sigma Aldrich | S8636 |
| N-2 supplement | Thermo Fisher | 17502048 |
| B-27 supplement | Thermo Fisher | 17504044 |
| GlutaMax | Thermo Fisher | 35050061 |
| Kapa HiFi HotStart ReadyMix | Roche | 7958935001 |
| GeneRuler 1kb ladder | Thermo Fisher | SM0311 |
| SuperScript IV First-Strand Synthesis system | ThermoFisher | 18091050 |
| the PowerUp SYBR Green Master Mix | Thermo Fisher | A25741 |
| Cell Lysis buffer | Thermo Fisher | 78442 |
| 8 well μ-Slides | Ibidi | 80826 |
| Paraformaldehyde | Thermo Fisher | 28906 |
| Triton-X100 | Sigma Aldrich | X-100 |
| 4′,6-Diamidino-2-Phenylindole | Thermo Fisher | D1306 |
| SYTOX Blue Dead Cell Stain | Thermo Fisher | S34857 |
| Goat serum | Thermo Fisher | 16210072 |
| Critical commercial assays | ||
| QuickExtract DNA Extraction Solution | Lucigen | QE09050 |
| AllPrep DNA/RNA Mini Kit | Qiagen | 80004 |
| RNeasy Plus Micro kit | Qiagen | 74034 |
| Experimental models: Cell lines | ||
| Human PGP1 iPSC | G.Church lab | Coriell GM23338 |
| 293T cells | ATCC | CRL-3216 |
| Software and algorithms | ||
| Fiji | Schindelin 2012, Nat Methods | https://fiji.sc/ |
| Prism | GraphPad | https://www.graphpad.com/features |
| FlowJo | BD | https://www.flowjo.com |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jeffrey D. Macklis (jeffrey_macklis@harvard.edu).
Materials availability
Cell lines used in this study are available from their respective repositories (see experimental model and subject details).
Data and code availability
This study did not generate/analyze novel transcriptomic or proteomic data or any code in this study.
Experimental model and subject details
All cell lines were maintained at 37°C at 5% CO2 in a humidified incubator.
We selected the widely employed adult-derived human induced pluripotent stem cell line PGP1 (Personal Genome Project 1) as the target iPSC line rather than H1/H9 ES cell lines because PGP1 is so widely used in the neuronal differentiation field, making it a relative “consensus” line for disease and mechanistic modeling. PGP1 was obtained from the laboratory of G.Church,42 and iPSCs were grown in mTeSR+ medium (StemCell Technologies, 100-0276) supplemented with penicillin/streptomycin (Pen/Strep, 1% vol/vol, ThermoFisher, 15140122) on Geltrex-coated coated cell culture dishes (ThermoFisher A1413302).
293T cells were obtained from ATCC (CRL-3216) and grown in DMEM (ThermoFisher, 10566016) supplemented with FBS (10% vol/vol, ThermoFisher, 10438026) and Pen/Strep (1% vol/vol).
Generation of doxycycline (DOX) inducible, knock-in fluorescent timer (FT) 293T cells
DOX-inducible 293T-FT cells were generated using the piggyBack transposon system. The FT construct was cloned into a piggyBack transposon targeting vector containing a tetracycline- inducible expression cassette and a puromycin selection cassette flanked by piggyBac transposase specific inverted terminal repeats. 293T cells were grown to 60% confluency in one well of a 6 well plate, and co-transfected with 1ug transposon targeting plasmid and 1ug transposase plasmid using Lipofectamin3000 (ThermoFisher, L3000015) following the manufacturer’s protocol. Medium was changed 24h later, and cells were incubated for additional 48h. After 48h, puromycin (1ug/ml, ThermoFisher, A1113803) was added to the medium. 48h later, cells were harvested with Trypsin-EDTA (ThermoFisher, 25200056) to generate a single cell solution, and single clones were collected into 96 well plates using FACS for genotyping. Positive clones were frozen in complete DMEM medium supplemented with DMSO (20% vol/vol, SantaCruz, sc-358801).
DOX induction of FT in 293T cells
DOX inducible FT 293T cells were grown in complete medium to reach 50% confluency. The FT was induced by adding doxycycline (500ng/ml, Sigma, D9891) to the complete medium. 24h later, medium was changed to fresh medium without doxycycline. Cells were harvested with trypsin-EDTA and prepared for FACS analysis.
Generation of PAX6-fluorescent timer lines
We used CRISPR/Cas9 editing to integrate one of a series of iterative FT constructs downstream of the endogenous PAX6 promoter. The donor plasmid contained (from 5′ to 3′) the 3′end of the PAX6 gene without the stop codon, a P2A peptide, and one of this set of iterative FT constructs (“SlowFT”, “TandemFP”, “MolTimer 1.0”, “MolTimer 2.0”, “Chrono”), and the DNA sequence directly downstream of the Pax6 stop codon. TandemFP consists of a fusion of DsRed2 and sfGFP; MolTimer 1.0 consists of NLS-mScarlet-P2A-Ubiquitin-Degron-humanNeonGreen-NLS and MolTimer 2.0 consists of NLS-mScarlet-T2A-sfGFP-NLS. The Chrono plasmid27 was generously provided by Professor Hans Clevers, University Utrecht). The Cas9-guideRNA plasmid pSpCas9(BB)-2A-GFP (Addgene, 48138) was obtained from Addgene, and the Pax6 guideRNA (5′ GGCCAGTATTGAGACATATC 3′) was cloned downstream of the human U6 promoter.
To integrate one of a set of FT constructs downstream of the Pax6 promoter, iPSCs were first disassociated with Accutase (Innovative Cell Technologies, AT104) into a single cell suspension. Nucleofection was performed with an Amaxa 4D-Nucleofector System (Lonza) using the P3 Primary Cell nucleofector kit (Lonza, V4XP-3012). 8x105 cells were resuspended in 100μl P3 solution, combined with 3mg donor plasmid and 2mg Cas9-guideRNA plasmid, and nucleofected using the CA-137 program. After nucleofection, cells were plated on Geltrex-coated 10cm plates in mTeSR+ supplemented with CloneR (StemCell Technologies, 05888). A full medium change was performed 48h post nucleofection with fresh mTesR+ supplemented with CloneR. Four days after nucleofection, the medium was changed to fresh mTesR+ containing 0.3ug/ml puromycin. Medium changes were performed daily from day 4 onwards. On day 6 the medium was changed to fresh mTeSR+ without puromycin, and cells were grown until clones were manually picked into 96 well plates for genotyping. Genomic DNA was isolated with the QuickExtract DNA Extraction Solution (Lucigen, QE09050) or the AllPrep DNA/RNA Mini Kit (Qiagen, 80004). Correct PAX6 integration of the appropriate FT construct was confirmed by PCR. PCR was performed using Kapa HiFi HotStart ReadyMix (Roche, 07958935001) and primers specified in Table S1. PCR products were run on a 1.5% agarose gel, alongside a GeneRuler 1kb ladder (ThermoFisher, SM0311). The full FT cassette was Sanger sequenced to confirm correct genomic integration into the 3′ end of the Pax6 gene, and clones were banked in mFreSR freezing medium (StemCell Technologies, 05854).
Cortical differentiation
Neuronal induction was performed following a cortical induction protocol.43 IPSCs were disassociated with Accutase (Innovative Cell Technologies, AT104) to generate a single cell suspension, and 2.8x105 cells/cm2 were plated on Geltrex-coated plates in mTesR+ supplemented with ROCK inhibitor Y-27632 (10uM, StemCell Technologies, 72302). Neuronal induction was started 24h later (day 0) by changing mTesR+ medium to neuronal induction medium consisting of neuronal maintenance medium (NMM) supplemented with SB43152 (10μM, Selleckchem, S1067) and Dorsomorphin (DM, 10μM, Stemcell Technologies, 72102). The medium was changed daily until day 12. For pulsing experiments DM and SB43152 was omitted from the NMM from day one onwards.
NMM consisted of DMEM:F12 (50% vol/vol, ThermoFisher, 10565018), Neurobasal medium (50% vol/vol ThermoFisher, 21103049), insulin (0.03% vol/vol, Sigma, I9278), 2-mercaptoethanol (0.1% vol/vol, ThermoFisher, 31350010), non-essential amino acids (0.5% vol/vol, ThermoFischer, 11140050), Sodium Pyruvate (0.5% vol/vol Sigma, S8636), Pen/Strep (0.5% vol/vol, ThermoFisher, 15140122), N-2 supplement (0.5% vol/vol, ThermoFisher, 17502048), B-27 supplement (1% vol/vol ThermoFisher, 17504044) and Glutamax (0.5% vol/vol ThermoFisher, 35050061).
qPCR analysis
Cells were harvested with Accutase and washed twice with PBS. RNA was isolated using the RNeasy Plus Micro kit (Quiagen, 74034), and cDNA was synthesized with the SuperScript IV First-Strand Synthesis system (ThermoFisher, 18091050) following the manufacturer’s protocols. PCR targets were amplified with the PowerUp SYBR Green Master Mix (ThermoFisher, A25741) using the primer sequences in the “qPCRPrimer” supplementary table.
Western blot analysis
Cells were harvested with Accutase and washed twice with PBS. Whole cell protein extraction was performed by lysing the cell pellet with Cell Lysis buffer (ThermoFisher, FNN0011) supplemented with protease inhibitor cocktail (ThermoFisher, 78442) following the manufacturer’s protocol. Western blot analysis was performed with the following antibodies: β-actin (1:10,000 Sigma, A5441), GFP (1:1,000, ThermoFisher, A11122), DsRed2 (1:200 SantaCruz, sc-101526), PAX6 (1:500 ThermoFisher, 14-9914-95), RPL22 (1:1,000 SantaCruz, sc-373993), mNeonGreen Tag (1:1,000 CellSignallingTechnologies, 53061), RFP (1:1,000, Rockland 600-401-379).
Confocal imaging
Cells were differentiated on 8 well μ-Slides (Ibidi, 80826). Cells were washed twice with PBS, and fixed with PFA (4% w/vol, ThermoFisher, 28906) for 10min at room temperature (RT). PFA was removed, and cells were washed twice with PBS and stored in PBS at 4°C. On the imaging day, the PBS was removed, and cells were permeabilized by incubation with PBS containing Triton-X100 (0.3% vol/vol, Sigma Aldrich X-100) for 5 min at RT. Nuclei were stained with 4′,6-Diamidino-2-Phenylindole (DAPI, 5μg/ml, DAPI, ThermoFisher D1306) in PBS for 5 min at RT, followed by a PBS wash. Cells were imaged on an inverted Zeiss LSM 880 confocal microscope. All images were processed with Fiji.44
Immunofluorescence labeling
Cells were washed twice with PBS, and fixed with PFA (4% w/vol, ThermoFisher, 28906) for 10min at room temperature (RT). PFA was removed, and cells were washed three times with PBS, incubated for 15min at RT with PBS + 0.1% Triton-X, and blocked with PBS + 5% goat serum for 1h at RT. Cells were incubated with primary PAX6 (1:100 ThermoFisher, 14-9914-95) antibody in PBS + 5% goat serum overnight at 4°C. On the next day, cells were washed three times with PBS and incubated with Alexa-dye conjugated secondary antibodies (ThermoFisher) diluted 1:500 in PBS + 5% goat serum at RT form 1h. Cells were washed 3 times with PBS and TO-PRO3 (TO-PRO-3 Iodide (642/661), ThermoFisher T3605) was added to the second wash step.
Live cell imaging
Cells were differentiated on 8 well μ-Slides and imaged for 24h at a 20min time interval on a Zeiss Cell Discoverer7 live-cell-imaging system.
Flow cytometry analysis
For fixed FACS analysis, cells were harvested with Accutase to generate a single cell suspension and washed twice with PBS. Cells were incubated for 5min with PFA (4% w/vol), washed twice with PBS, and stored at 4°C. Cells were filtered through a 35μm cell strainer (Corning, 352235) prior to analysis. For live cell FACS analysis, cells were harvested with Accutase, washed twice with PBS, and filtered through a 35μm cell strainer. For live cell FACS mixing experiments, induced FT timer cells were mixed with edited but uninduced FT-PGP1 iPSCs instead of WT PGP1 iPSCs to rigorously control for potential endogenous autofluorescence and/or PAX6 promoter leakiness. Cells were kept on ice and incubated with SYTOX Blue Dead Cell Stain (ThermoFisher, S34857) prior to FACS analysis. All samples were analyzed on a LSRII (BD Bioscience) or FACSymphony A5 (BD Bioscience), cytometer and plots were prepared using FlowJo (BD Bioscience). We performed each experiment twice, both with PFA fixed and with live cells and live imaging. Since results replicated and were essentially the same twice, we moved to further iterations based on negative results.
Acknowledgments
We thank Holly McKee and Karen Wang for their excellent technical support; members of the Macklis laboratory for scientific discussions and helpful suggestions; Joyce LaVecchio and Nema Kheradmand of the HSCRB-HSCI Flow Cytometry Core; the Harvard Center for Biological Imaging for infrastructure and support. This work was supported by the following grants to J.D.M.: Paul G. Allen Frontiers Group—Allen Distinguished Investigator award #11855; National Institutes of Health OD DP1 NS106665; additional infrastructure support from NIH grantsNS104055, NS045523, and NS049553; and the Max and Anne Wien Professor of Life Sciences fund.
Author contributions
M.P., S.S., and J.D.M conceived the overall project and experiments; M.P. and S.S. designed initial experiments; M.P. and J.D.M. designed later experiments; J.H. performed the PCR genotyping experiments and M.P. performed all other experiments; M.P. and J.D.M. analyzed and interpreted the data, integrated the findings, and wrote and edited the manuscript. All authors contributed to discussions and manuscript editing.
Declaration of interests
The authors declare no competing interests.
Published: May 7, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109911.
Supplemental information
References
- 1.Barry J.D., Donà E., Gilmour D., Huber W. TimerQuant: a modelling approach to tandem fluorescent timer design and data interpretation for measuring protein turnover in embryos. Development. 2016;143:174–179. doi: 10.1242/dev.125971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelley K.W., Pașca S.P. Human brain organogenesis: Toward a cellular understanding of development and disease. Cell. 2022;185:42–61. doi: 10.1016/j.cell.2021.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Penney J., Ralvenius W.T., Tsai L.H. Modeling Alzheimer's disease with iPSC-derived brain cells. Mol. Psychiatry. 2020;25:148–167. doi: 10.1038/s41380-019-0468-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sances S., Bruijn L.I., Chandran S., Eggan K., Ho R., Klim J.R., Livesey M.R., Lowry E., Macklis J.D., Rushton D., et al. Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat. Neurosci. 2016;19:542–553. doi: 10.1038/nn.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang X., Hu D., Shang Y., Qi X. Using induced pluripotent stem cell neuronal models to study neurodegenerative diseases. Biochim. Biophys. Acta, Mol. Basis Dis. 2020;1866 doi: 10.1016/j.bbadis.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ozkan A., MacDonald J.L., Fame R.M., Itoh Y., Peter M., Durak O., Macklis J.D. In: Patterning and Cell Type Specification in the Developing CNS and PNS. Second Edition) Rubenstein J., Rakic P., Chen B., Kwan K.Y., editors. Academic Press; 2020. Chapter 19 - Specification of cortical projection neurons: transcriptional mechanisms; pp. 427–459. [DOI] [Google Scholar]
- 7.Greig L.C., Woodworth M.B., Galazo M.J., Padmanabhan H., Macklis J.D. Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci. 2013;14:755–769. doi: 10.1038/nrn3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Catela C., Shin M.M., Dasen J.S. Assembly and function of spinal circuits for motor control. Annu. Rev. Cell Dev. Biol. 2015;31:669–698. doi: 10.1146/annurev-cellbio-100814-125155. [DOI] [PubMed] [Google Scholar]
- 9.Kepecs A., Fishell G. Interneuron cell types are fit to function. Nature. 2014;505:318–326. doi: 10.1038/nature12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Libe-Philippot B., Vanderhaeghen P. Cellular and Molecular Mechanisms Linking Human Cortical Development and Evolution. Annu. Rev. Genet. 2021;55:555–581. doi: 10.1146/annurev-genet-071719-020705. [DOI] [PubMed] [Google Scholar]
- 11.Close J.L., Yao Z., Levi B.P., Miller J.A., Bakken T.E., Menon V., Ting J.T., Wall A., Krostag A.R., Thomsen E.R., et al. Single-Cell Profiling of an In Vitro Model of Human Interneuron Development Reveals Temporal Dynamics of Cell Type Production and Maturation. Neuron. 2017;93:1035–1048.e5. doi: 10.1016/j.neuron.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uzquiano A., Kedaigle A.J., Pigoni M., Paulsen B., Adiconis X., Kim K., Faits T., Nagaraja S., Antón-Bolaños N., Gerhardinger C., et al. Proper acquisition of cell class identity in organoids allows definition of fate specification programs of the human cerebral cortex. Cell. 2022;185:3770–3788.e27. doi: 10.1016/j.cell.2022.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trevino A.E., Muller F., Andersen J., Sundaram L., Kathiria A., Shcherbina A., Farh K., Chang H.Y., Pasca A.M., Kundaje A., et al. Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell. 2021;184:5053–5069.e5023. doi: 10.1016/j.cell.2021.07.039. [DOI] [PubMed] [Google Scholar]
- 14.Ozair M.Z., Kirst C., van den Berg B.L., Ruzo A., Rito T., Brivanlou A.H. hPSC Modeling Reveals that Fate Selection of Cortical Deep Projection Neurons Occurs in the Subplate. Cell Stem Cell. 2018;23:60–73.e6. doi: 10.1016/j.stem.2018.05.024. [DOI] [PubMed] [Google Scholar]
- 15.Nehme R., Zuccaro E., Ghosh S.D., Li C., Sherwood J.L., Pietilainen O., Barrett L.E., Limone F., Worringer K.A., Kommineni S., et al. Combining NGN2 Programming with Developmental Patterning Generates Human Excitatory Neurons with NMDAR-Mediated Synaptic Transmission. Cell Rep. 2018;23:2509–2523. doi: 10.1016/j.celrep.2018.04.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maroof A.M., Keros S., Tyson J.A., Ying S.W., Ganat Y.M., Merkle F.T., Liu B., Goulburn A., Stanley E.G., Elefanty A.G., et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kmet M., Guo C., Edmondson C., Chen B. Directed differentiation of human embryonic stem cells into corticofugal neurons uncovers heterogeneous Fezf2-expressing subpopulations. PLoS One. 2013;8 doi: 10.1371/journal.pone.0067292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terskikh A., Fradkov A., Ermakova G., Zaraisky A., Tan P., Kajava A.V., Zhao X., Lukyanov S., Matz M., Kim S., et al. Fluorescent timer": protein that changes color with time. Science. 2000;290:1585–1588. doi: 10.1126/science.290.5496.1585. [DOI] [PubMed] [Google Scholar]
- 19.Subach F.V., Subach O.M., Gundorov I.S., Morozova K.S., Piatkevich K.D., Cuervo A.M., Verkhusha V.V. Monomeric fluorescent timers that change color from blue to red report on cellular trafficking. Nat. Chem. Biol. 2009;5:118–126. doi: 10.1038/nchembio.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyatsuka T., Matsuoka T.A., Sasaki S., Kubo F., Shimomura I., Watada H., German M.S., Hara M. Chronological analysis with fluorescent timer reveals unique features of newly generated beta-cells. Diabetes. 2014;63:3388–3393. doi: 10.2337/db13-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyatsuka T., Li Z., German M.S. Chronology of islet differentiation revealed by temporal cell labeling. Diabetes. 2009;58:1863–1868. doi: 10.2337/db09-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knop M., Edgar B.A. Tracking protein turnover and degradation by microscopy: photo-switchable versus time-encoded fluorescent proteins. Open Biol. 2014;4 doi: 10.1098/rsob.140002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bending D., Prieto Martín P., Paduraru A., Ducker C., Marzaganov E., Laviron M., Kitano S., Miyachi H., Crompton T., Ono M. A timer for analyzing temporally dynamic changes in transcription during differentiation in vivo. J. Cell Biol. 2018;217:2931–2950. doi: 10.1083/jcb.201711048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khmelinskii A., Keller P.J., Bartosik A., Meurer M., Barry J.D., Mardin B.R., Kaufmann A., Trautmann S., Wachsmuth M., Pereira G., et al. Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nat. Biotechnol. 2012;30:708–714. doi: 10.1038/nbt.2281. [DOI] [PubMed] [Google Scholar]
- 25.Poulopoulos A., Murphy A.J., Ozkan A., Davis P., Hatch J., Kirchner R., Macklis J.D. Subcellular transcriptomes and proteomes of developing axon projections in the cerebral cortex. Nature. 2019;565:356–360. doi: 10.1038/s41586-018-0847-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Catapano L.A., Arnold M.W., Perez F.A., Macklis J.D. Specific neurotrophic factors support the survival of cortical projection neurons at distinct stages of development. J. Neurosci. 2001;21:8863–8872. doi: 10.1523/JNEUROSCI.21-22-08863.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gehart H., van Es J.H., Hamer K., Beumer J., Kretzschmar K., Dekkers J.F., Rios A., Clevers H. Identification of Enteroendocrine Regulators by Real-Time Single-Cell Differentiation Mapping. Cell. 2019;176:1158–1173.e16. doi: 10.1016/j.cell.2018.12.029. [DOI] [PubMed] [Google Scholar]
- 28.Zhang X., Huang C.T., Chen J., Pankratz M.T., Xi J., Li J., Yang Y., Lavaute T.M., Li X.J., Ayala M., et al. Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell. 2010;7:90–100. doi: 10.1016/j.stem.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Z., Chen O., Wall J.B.J., Zheng M., Zhou Y., Wang L., Vaseghi H.R., Qian L., Liu J. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017;7:2193. doi: 10.1038/s41598-017-02460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi Y., Kirwan P., Smith J., Robinson H.P.C., Livesey F.J. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 2012;15:477–486. doi: 10.1038/nn.3041. S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Molyneaux B.J., Arlotta P., Hirata T., Hibi M., Macklis J.D. Fezl is required for the birth and specification of corticospinal motor neurons. Neuron. 2005;47:817–831. doi: 10.1016/j.neuron.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 33.Pedelacq J.D., Cabantous S., Tran T., Terwilliger T.C., Waldo G.S. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 34.Bevis B.J., Glick B.S. Rapidly maturing variants of the Discosoma red fluorescent protein (DsRed) Nat. Biotechnol. 2002;20:83–87. doi: 10.1038/nbt0102-83. [DOI] [PubMed] [Google Scholar]
- 35.Kalderon D., Roberts B.L., Richardson W.D., Smith A.E. A short amino acid sequence able to specify nuclear location. Cell. 1984;39:499–509. doi: 10.1016/0092-8674(84)90457-4. [DOI] [PubMed] [Google Scholar]
- 36.Shaner N.C., Campbell R.E., Steinbach P.A., Giepmans B.N.G., Palmer A.E., Tsien R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 37.Bindels D.S., Haarbosch L., van Weeren L., Postma M., Wiese K.E., Mastop M., Aumonier S., Gotthard G., Royant A., Hink M.A., Gadella T.W.J., Jr. mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat. Methods. 2017;14:53–56. doi: 10.1038/nmeth.4074. [DOI] [PubMed] [Google Scholar]
- 38.Tanida-Miyake E., Koike M., Uchiyama Y., Tanida I. Optimization of mNeonGreen for Homo sapiens increases its fluorescent intensity in mammalian cells. PLoS One. 2018;13 doi: 10.1371/journal.pone.0191108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van de Leemput J., Boles N.C., Kiehl T.R., Corneo B., Lederman P., Menon V., Lee C., Martinez R.A., Levi B.P., Thompson C.L., et al. CORTECON: a temporal transcriptome analysis of in vitro human cerebral cortex development from human embryonic stem cells. Neuron. 2014;83:51–68. doi: 10.1016/j.neuron.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 40.Balleza E., Kim J.M., Cluzel P. Systematic characterization of maturation time of fluorescent proteins in living cells. Nat. Methods. 2018;15:47–51. doi: 10.1038/nmeth.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molyneaux B.J., Goff L.A., Brettler A.C., Chen H.H., Hrvatin S., Rinn J.L., Arlotta P. DeCoN: genome-wide analysis of in vivo transcriptional dynamics during pyramidal neuron fate selection in neocortex. Neuron. 2015;85:275–288. doi: 10.1016/j.neuron.2014.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Church G.M. The personal genome project. Mol. Syst. Biol. 2005;1:2005.0030. doi: 10.1038/msb4100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi Y., Kirwan P., Livesey F.J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 2012;7:1836–1846. doi: 10.1038/nprot.2012.116. [DOI] [PubMed] [Google Scholar]
- 44.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze novel transcriptomic or proteomic data or any code in this study.