

Summary
Cancer is a multi-faceted disease with intricate relationships between mutagenic processes, alterations in cellular signaling, and the tissue microenvironment. To date, these processes have been largely studied in isolation. A systematic understanding of how they interact and influence each other is lacking. Here, we present a framework for systematically characterizing the interaction between pairs of mutational signatures and between signatures and signaling pathway alterations. We applied this framework to large-scale data from TCGA and PCAWG and identified multiple positive and negative interactions, both cross֊tissue and tissue֊specific, that provide new insights into the molecular routes observed in tumorigenesis and their respective drivers. This framework allows for a more fine-grained dissection of common and distinct etiology of mutational signatures. We further identified several interactions with both positive and negative impacts on patient survival, demonstrating their clinical relevance and potential for improving personalized cancer care.
Subject areas: Biological sciences, Bioinformatics, Cancer systems biology
Graphical abstract
Highlights
-
•
A framework defining and assessing interactions of mutational signatures in cancers
-
•
Multiple interactions among mutational signatures and with signaling pathways
-
•
Evaluation of the impact of detected interactions on patient survival
-
•
A resource for mutational signature interactions in PCAWG and TCGA
Biological sciences; Bioinformatics; Cancer systems biology
Introduction
Cancer genomes reflect the history of endogenous and exogenous genomic stressors from the zygote through organismal development to disease initiation and progression until sample collection. Endogenous stressors that create DNA damage during normal cellular functioning include transcription1 and replication,2 deamination of methylated cytosines,3,4 oxidative stress, and in some cases deficiencies in DNA damage sensing and repair machineries.5,6 Exogenous threats to DNA integrity range from radiation7,8 to numerous cytotoxic or genotoxic agents such as those involved in tobacco smoke,9,10 food production,11 or environmental toxins.12
In the past ten years, mutational signatures revolutionized the understanding of genomic stressors. Signatures of single-base substitutions (SBS), indels, double base substitutions, and copy-number variations have been compiled, and some of the etiologies have been identified.13,14,15 SBS signatures, defined by patterns of mutations in a trinucleotide context, are particularly powerful tools due to comparatively clearer readouts and ongoing success in annotations. These signatures exhibit characteristic probability distributions over mutation types, and many of them have been linked to specific mutagenic processes.13,14,15 Hence, our study is based on SBS signatures as defined and compiled in the COSMIC database.16
Cancer genomes, findings based on in vivo, in vitro, and organoid models, have identified mutational signatures of environmental carcinogens,17 deficient DNA repair pathways,6,18,19 endogenous mutagenic processes,20,21 as well as combinations of genotoxic agents and deficient DNA repair.22 These studies have mapped the effects of processes involving proteins and exposures directly affecting DNA. Yet, other variables such as impaired signaling pathways additionally affect and can be impacted by mutational processes through their crosstalk with DNA damage sensing and repair.23,24 Furthermore, signature activities resulting from mutagenic processes might preferentially lead to the accumulation of driver mutations.25
The clinical value of mutational signatures as biomarkers for therapy response, prognosis, and therapy contraindication has been recently reviewed.26 Several synthetic lethal interactions within DNA damage and repair pathways as well as their associated signatures have been identified as biomarkers for targeted therapies, highlighting the clinical value of these interactions. Exploiting the interaction between homologous recombination deficiency (HRD) and PARP, a gene involved in DNA repair, has led to successful targeted therapy.27 Hence, a more comprehensive analysis of signature interactions could uncover novel targetable interactions.
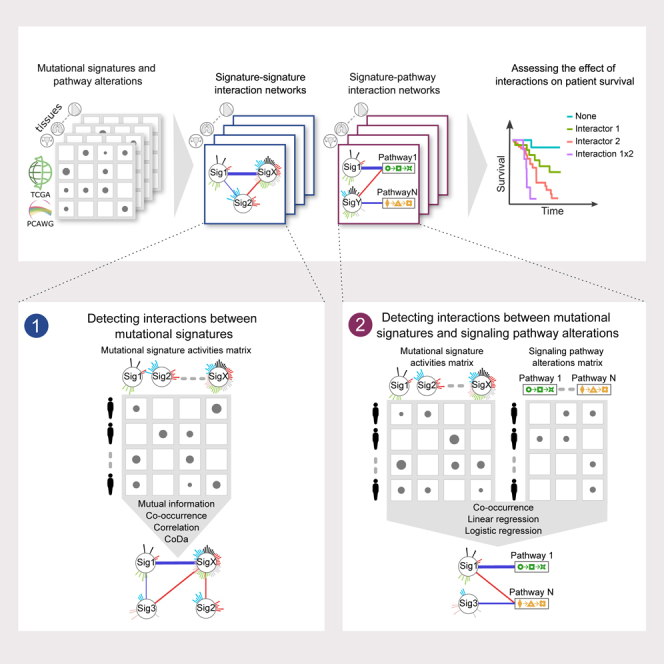
While the landscape of individual DNA damage, repair, and oncogenic signaling pathways has been extensively studied, a systematic analysis of their interactions, coexpression and correlation patterns, and the resulting mutational landscape is still missing. Identifying these interaction patterns can shed light on concurrent and consecutive events and molecular processes that underlie cancer initiation and progression, as well as offer potentially targetable interactions. Here, we developed a framework to systematically explore the patterns of interactions between mutational signatures (signature-signature) and between mutational signatures and signaling pathways (signature-pathway) (Figure 1A). We applied this framework to observational data from The Cancer Genome Atlas (TCGA) and the Pan-Cancer Analysis of Whole Genomes (PCAWG).13,14,28 Finally, we evaluated the clinical impact of these interactions based on their effect on patient survival.
Figure 1.

Overview of the study and input data
(A) Schematic demonstration of interactions between mutational signatures, pathway alterations, and immune state.
(B) Number of samples in PCAWG across cancer types.
(C) Number of active mutational signatures in PCAWG across cancer types. Colors in (B) and (C) correspond to individual cancer types with >25 samples. See also Figure S1.
Results
We obtained mutational signatures extracted from the TCGA and PCAWG datasets.14 After filtering samples with low reconstruction accuracy (<0.85), a low number of total mutations (<90), and excluding tissues with less than 25 samples, we retained 2,591 (out of 2,780) and 3,547 (out of 9,493) samples, for 25 and 15 tissues in PCAWG and TCGA, respectively (Figures 1B and S1A). Since we were interested in interactions between mutational processes rather than individual signatures, we aggregated all signatures with a common etiology as annotated in the COSMIC16 database (v3.2, Table S1). The number of active signatures varied depending on the tissue and cancer type, as well as individual sample exposures; however, more than 77% of all samples in both datasets had three to four active aggregated signatures (Figures 1C and S1B). We also note that the number of signatures scaled linearly with the number of available samples for a particular cancer type (Figure S1C), hence, future analyses with larger sample sizes should recover rare signature interactions.
A framework for signature-signature interaction analysis
The dynamics of mutational signature accumulation in cancer genomes depend on the signature etiology and are incompletely understood. Clock-like signatures have been shown to accumulate linearly over time,29,30 APOBEC-related signatures in episodic bursts31,32 and signatures associated with environmental exposures accumulate over the periods of active exposure. As a result, the interactions between signatures may be linear or nonlinear in nature, depending on their specific combination. To overcome the limitations of individual metrics in capturing different linear or nonlinear aspects of interaction, we used four complementary metrics (Figure S1D): (1) first, we quantified the cooccurrence between mutational signature activities treated as a binary function using Fisher’s exact test. (2) We quantified the bias corrected mutual information (BCMI) to assess potentially nonlinear and nonmonotonic associations based on jackknife bias correction.33 (3) Next, we used the Spearman coefficient to assess rank correlations. (4) Finally, we used a CoDa (compositional data) approach to detect interactions between mutational process activities. Mutational signature activities are not independent, and many mutation types in the 96-trinucleotide channel are shared among several signatures. Furthermore, cosine similarities between signature pairs are greater than 0.5 in ∼11% of all comparisons, indicating high intermixing34 potential between several signature pairs including SBS1 and SBS6, SBS3 and SBS40, SBS8 and SBS4, etc. High cosine similarity between signature pairs not only means impaired algorithmic decoupling but also that the number of mutations attributed to each signature will depend on mutation counts attributed to all the other signatures with high cosine similarity. This suggests that mutational signature attribution for individual signatures depends on the attribution to similar signatures. Hence, mutational signatures are compositional and must be treated through the lens of compositional data analysis. To evaluate whether the activities of these signatures change in accordance with each other, we devised an interaction metric based on calculating symmetric pivot coordinates35 followed by Pearson’s correlation. Importantly, the CoDa metric can only be applied to samples where both signatures are present. We calculated all signature-interaction metrics through bootstrapping (Figure S1E). Further, we set the significance threshold at 5% or 95% percentile of the null model for each interacting signature-signature pair calculated by signature shuffling for each tissue independently (STAR Methods).
All applied metrics may result in either positive or negative interactions, generally indicating either synergistic or antagonistic relationships, respectively. Depending on the precise signature pair, either direction may have several interpretations. Positive interactions can indicate that the respective signatures have a common initiation mechanism. This is clearly observed for signatures with shared etiology or split-signatures, such as SBS7a-d and SBS10a-d, and may extend to signatures with still unknown but linked etiologies. Alternatively, one mutational signature could instigate another in the course of disease progression. For example, a large-scale study of clonal evolution in cancers related to environmental exposures revealed that early clonal mutations are caused by the exposure, whereas subsequent mutations occurring during tumor progression arise from downstream endogenous processes, e.g., cell division or disruption of DNA repair pathways.36 Similarly, negative interactions may reflect different underlying mechanisms. Some could be indicative of synthetic lethality, where concurrent mutations in different pathways result in cell death, as in the case of homologous recombination combined with PARP or POLQ mutations. Alternatively, negative interactions may be a result of independent driver events.
Mutational signature interaction networks reveal complex relationships between mutagenic processes
We calculated signature interaction metrics separately for each tissue in the PCAWG and TCGA datasets. Significant signature-signature interaction metrics are reported for both datasets across tissues in Tables S2 and S3. The results can be summarized by signature-signature interaction networks, allowing to assess interaction patterns within a given tissue, and how different metrics capture different aspects of interactions. We aggregated all metrics for a given signature pair by selecting interactions that are supported by at least two metrics and classified them into positive and negative interactions. In PCAWG, we identified 88 positive and 50 negative interactions across 25 tissues ranging from 1 interaction in biliary adenocarcinoma to 18 in B cell non-Hodgkin lymphoma (Table S4). In TCGA, we identified 50 positive and 47 negative interactions (Table S4). We summarized all detected interactions in Figures 2A and S2A. The network reveals several basic principles. Firstly, we observe an interconnected core of clock-like signature SBS1, clock-like signature SBS5, and SBS40 that are abundantly active across tissues. These interactions are likely by-products of basic cellular repair and cellular metabolic processes that are shared across tissues, found in cancer and normal tissues, and constitute the collective signature of cellular aging.37 SBS1 and SBS5 are both so-called clock-like signatures accumulating over a lifetime that have been linked to stem-cell divisions and environmental exposures, respectively.29 Secondly, endogenous processes are densely connected both with clock-like signatures and with other endogenous signatures. Notably, signatures of APOBEC mutagenesis or DNA repair pathways such as homologous recombination can be active across multiple tissues. The high interconnectedness of DDR pathways potentially facilitates these interactions between signatures of endogenous processes.
Figure 2.

Signature-signature interactions in PCAWG
(A) Network summary of all detected signature-signature interactions in PCAWG. CL stands for clock-like.
(B) Heatmap of signature activities in pancreatic adenocarcinoma.
(C) Signature-signature interaction network for samples in (B).
(D) Summary of common interactions in PCAWG across tissues. Positive (red) and negative (blue) interactions are summarized separately, and the numbers indicate the number of tissues in which this interaction was detected. We filtered the displayed signatures to have at least four interactions. The infographic at the bottom right side demonstrates how common signatures across tissues were selected.
(E) Network of interactions found both in PCAWG and TCGA. The icons on the edges indicate the tissue where the interaction is observed. See also Figure S2.
In Figure 2, we present examples of the signature activities for pancreatic adenocarcinoma as a heatmap (Figure 2B) and extracted signature-signature interactions (Figure 2C). While some interactions are supported by only one or two metrics, others are supported by three or four. Notably, metrics such as CoDa appear to capture unique aspects of the interactions. Looking at these networks allows to spot the intricacies of signature interactions. In Figure 2C, we observe a positive interaction between APOBEC and HRD, clock-like signature SBS1 and SBS40, while the latter negatively interacts with ROS and HRD. These interactions reflect the complex structure of how stressors like oxidative stress, genomic instability, APOBEC mutagenesis, aging, and unknown endogenous mutagenic processes act in unique combinations in different patient groups.
Cross-tissue signature-signature interactions reveal principles behind signature manifestation
We binned the detected interactions into three main categories based on their prevalence: common interactions across multiple tissues (>10), intermediate interactions appearing in 2–4 tissues, and interactions unique to a specific tissue.
Figure 2D shows the interactions aggregated across all tissues for PCAWG (Figure S2B for TCGA). Three interactions were highly abundant—a positive interaction between clock-like signatures SBS1 and SBS5, SBS1 and SBS40, and a negative interaction between SBS5 and SBS40, reflecting the signatures of cellular aging discussed in the earlier section. Notably, SBS5 has also been linked to naturally occurring environmental/external processes, supported by the association with oxidative processes and nucleotide excision repair pathway.38
Most of the consensus interactions found in 2–4 tissues involve exposures and cellular processes that correspond to localized perturbations. For example, in PCAWG the exposure to reactive oxygen species (ROS, signature SBS18) varies greatly across tissues. Consequently, interactions of the ROS signature with other signatures are restricted to tissues where this kind of damage is more common. Specifically, ROS is positively interacting with SBS5 in pancreatic (Panc_AdenoCA), prostate (Prost_AdenoCA), and stomach (Stomach_AdenoCA) adenocarcinomas and negatively interacting with SBS40 in medulloblastoma (CNS_Medullo), ovarian (Ovary_AdenoCA), pancreatic (Panc_AdenoCA), and prostate (Prost_AdenoCA) adenocarcinomas. Further, interactions between signatures caused by homologous recombination deficiency (HRD) and the signature associated with APOBEC family of cytidine deaminases (APOBEC) have been identified in three tissues—breast, esophageal, and pancreatic adenocarcinomas. This interaction recapitulates recent results demonstrating that dysregulated APOBEC promotes genomic instability and DNA damage, thereby further activating homologous recombination,38,39,40,41 hence demonstrating the potential of our framework to detect valid interactions. In this category, we also notice that some interactions have opposing directionality across tissues. Specifically, clock-like SBS1 and APOBEC signatures interact positively in pancreatic endocrine tumors and melanoma, while they interact negatively in breast adenocarcinoma. We notice that APOBEC mutation levels are relatively high in breast adenocarcinoma (interquartile range IQR = [212, 1204], mean = 2257), while, in the two other tissues, the mean mutation count is under 200 mutations (IQR = 0). We hypothesize that higher levels of APOBEC mutagenesis might interfere with cell cycle and consequently with accumulation of clock-like SBS1 signature, supported by recent work showing how increased APOBEC activity can impair cell-cycle progression.42
In TCGA samples, we identified a negative interaction between APOBEC and MMR signatures in breast, stomach, and uterine adenocarcinomas (Figure 3D). The closer inspection of samples has revealed that these signatures are mutually exclusive, potentially indicating two alternative mutagenic processes in these tissues.
Figure 3.

Signature-pathway interactions in PCAWG
(A) Summary of pathway alterations in PCAWG across tissues.
(B) Summary of signature-pathway interactions across tissues in PCAWG. Positive (red) and negative (blue) interactions are summarized separately, and the numbers indicate the number of tissues in which this interaction has been detected.
(C) Bipartite graph of signature-pathway interactions in PCAWG.
(D) Heatmap of logged signature activities in stomach adenocarcinoma (Stomach_Adeno) in PCAWG with annotated pathway alterations and two classes C1 and C2 with high and low levels of mismatch repair deficiency (dMMR) signature.
(E) Signature-pathway cooccurrence and exclusivity in Stomach_AdenoCA.
(F) Common signature-pathway interactions in TCGA and PCAWG. Icons on the edges indicate the tissue in which the interaction was detected in both datasets.
Finally, the unique interactions are linked to highly tissue-specific exposures. In particular, the UV signature and its interactions are only found in melanoma.
We compared detected signature interactions between PCAWG and TCGA cohorts (Figure 2E). Of note, the two cohorts have vastly different numbers of samples sequenced per tissue, which may compromise the detection power for some interactions. Nonetheless, we identified 27 interactions that were shared between the two datasets with matching tissue and directionality. The most common interaction partner detected in both datasets was SBS5, which is positively linked to signatures of both environmental (tobacco smoking or UV signatures) and endogenous origin (mismatch repair deficiency—dMMR, clock-like signature SBS1 linked to cell division). Thus, SBS5 might be a by-product of a wide array of mutagenic processes irrespective of their origin.
We detected an unexpected negative interaction between SBS38 and UV signature. Previously, SBS38 has been speculated to be caused by indirect effect of UV light; however, our results suggest that SBS38 signature is UV independent.14
Mutational signatures and pathway alterations
Cancer mutations agglomerate in a small set of important signaling pathways.43 We probed the interactions between mutational signatures and signaling pathways using signatures extracted from TCGA and PCAWG and pathway alteration status of 10 pathways that were previously shown to be frequently aberrant in cancers: cell cycle, HIPPO, MYC, NOTCH, NRF2, PI-3-Kinase/Akt, RTK-RAS, TGFβ signaling, TP53, and β-catenin/WNT. We first harmonized the two datasets by mapping the driver mutations in the PCAWG dataset44 within these pathways.
The occurrence of pathway alterations in PCAWG was concordant with TCGA43 across tissues (Figures 3A and S3A). We identified samples with both signature and driver mutation information available, and limited our analysis to tissues with at least 30 samples, resulting in 1,471 samples from 14 tissues in PCAWG and 3,132 samples from 12 tissues in TCGA.
As in the case of signature-signature interactions, there can be diverse modes of interactions between signatures and oncogenic pathways that cannot be captured by a single metric. We therefore used two complementary strategies. First, we assessed the co-occurrence or exclusivity patterns of signatures and pathways by treating them as binary features (i.e., present or absent). Secondly, we applied regression models between signature activities and the number of driver events in pathways to have a more detailed quantification of the interaction. Significant interactions for all interaction metrics are provided in Tables S5 and S6.
Signature-pathway cooccurrence uncovers diverse routes of tumorigenesis
We assessed the co-occurrence between signatures and pathways by creating contingency tables for individual tissues over signature-pathway occurrence and applied Fisher’s exact test. The results are summarized in Figures 3B and S3B, and lists of tissues for each interaction are provided in Table S7 and summarized in bipartite networks in Figures 3C and S3D.
First, we note that certain signature-pathway interactions exhibited opposing directionality in different tissues. For example, in PCAWG, the homologous recombination signature and the TP53 pathway interact positively in breast adenocarcinoma and negatively in pancreatic neuroendocrine tumors. Similarly, in TCGA, PI3K pathway alterations and the homologous recombination signature interact positively in esophageal adenocarcinoma and negatively in breast cancer. These opposing modes of interaction between signatures and pathways potentially indicate alternative routes of disease initiation and progression. Specifically, our results indicate that in HRD-driven breast cancer, tumorigenesis is more commonly mediated through TP53 (supported by both TCGA and PCAWG), cell cycle, and MYC alterations and less often through mutations in the PI3K and TGF-beta pathways (Figures S3E and S3F). Conversely, in esophageal adenocarcinoma, homologous recombination deficiency seems to favor PI3K mutations, indicating a potential synergy between these pathways in the development of this cancer type.
Next, a closer inspection of samples highlights how different mutational signature compositions favor different pathway routes in the disease progression in a tissue-specific manner. Specifically, in TCGA, we observe an association of mismatch repair deficiency (dMMR) signatures with multiple pathways, namely, WNT, TGF-beta, PI3K, NOTCH, and HIPPO (Figure S3B), while we observed negative interactions between TP53 and dMMR in two tissues. We will contextualize these observations for stomach adenocarcinoma (Figures 3D and 3E). Hierarchical clustering of all samples based on mutational signatures reveals two major clusters. In addition to clock-like SBS1 and SBS5 signatures, the first cluster (C1) carries high levels of dMMR signature, while the second cluster (C2) carries a combination of SBS40, SBS17, and APOBEC and is predominantly characterized by the absence of the MMR signature. This clustering further translates to different levels of pathway alterations in TP53, PI3K, HIPPO, WNT, NOTCH, and TGF-beta between clusters C1 and C2 (Figure 3E). Thus, our approach shows that signature-pathway interactions can distinguish molecularly distinct sub-groups.
We observed a similar pattern in melanoma samples (Figures S3G and S3H), where clustering based on mutational signatures revealed clusters with differential driver mutations. Specifically, samples carrying high levels of UV mutational signatures were characterized with high loads of RTK-RAS mutations (Figure S3C). Samples characterized by low levels of UV signatures, on the other hand, carried a combination of SBS38, APOBEC, and SBS40 (Figure 2C). These samples were characterized by higher occurrence of HIPPO and NOTCH pathway alterations and lower levels of PI3K and RTK-RAS pathways compared to the UV-high cluster. Indeed, previously reported molecular characterization of acral melanoma has revealed copy-number alterations in the YAP1 gene, a key player in the HIPPO pathway.45 Further, YAP1 overexpression has been mechanistically linked to increased nuclear rupture in an acral melanoma mouse model.46
These examples in stomach adenocarcinoma and melanoma demonstrate how the tumorigenic processes, manifested through mutational signatures, preferentially lead to distinct driver mutations that will consequently steer the cancer progression.
Finally, we evaluated the consistency of the detected interactions between the two datasets (Figure 3F). Notably, only four tissues were sufficiently sequenced and characterized on the pathway level (Liver_HCC, Breast_AdenoCA, Eso_AdenoCA, Skin_Melanoma) in both datasets. In two tissues, breast, and skin, the interactions were detected in both TCGA and PCAWG. In melanoma, we identified a positive interaction between the RTK-RAS pathway and the UV signature, as expected, as well as between HIPPO and SBS38. Meanwhile, SBS38 and UV are mutually exclusive, hence we speculate that HIPPO is one of the driving pathways among the UV-low melanoma samples. In breast adenocarcinoma, we identified co-occurrences between the homologous recombination deficiency (HRD) signature and TP53, MYC, and cell-cycle pathway alterations as discussed before (Figures S3E and S3F). This suggests a potential synergistic interaction between HRD and these pathway alterations or that HRD biases the tumorigenesis toward acquisition of mutations in these driver pathways throughout disease progression.
Exploring the causal relationship between mutational processes and pathway alterations
Next, we assessed the interaction between signatures and pathways by fitting regression models. Compared to contingency table analysis, regression models not only consider the signatures as binary (presence/absence) but also account for their intensity. For common signatures, regression models are thus more potent tools to assess interactions. The interaction between mutational signatures and pathway alterations can be viewed as a causal relationship, where either the mutational process induces a driver mutation or the pathway alteration triggers cellular processes, which, in turn, activate mutational processes. A prominent example of this type of causal interaction is the initiation of melanoma due to an increased exposure to UV that induces driver mutations in the RTK-RAS pathway. We evaluated the validity of our approach by fitting a regression model between the number of driver mutations in the RTK-RAS pathway and the log-transformed number of UV signatures in melanoma samples (Figure S3C). We recovered a clear monotonic increase for both variables.
Since for most of the interacting pairs the causal directionality is not clear, we fit both logistic and linear regression models to signature-pathway interactions and summarized the results in Figure S4. In TCGA, both regression models indicate strong interactions between clock-like signatures SBS1 and SBS5 with nearly all pathway alterations (Figures 4A and S4A). This finding recapitulates our current understanding of age as the most important cancer risk factor.47 In PCAWG, we had limited power to verify these observations due to fewer sample counts. Nonetheless, both regression models supported the interaction between clock-like signatures and TP53, NOTCH, and cell-cycle pathways across tissues, with a more prominent interaction for the clock-like signature SBS1. The linear regression model in PCAWG results in a positive interaction between clock-like signature SBS1 and TP53 driver mutations in five tissues, chronic lymphocytic leukemia (Lymph_CLL), breast adenocarcinoma (Breast_AdenoCA), esophageal adenocarcinoma (Eso_AdenoCA), pancreatic adenocarcinoma (Panc_AdenoCA), and prostate adenocarcinoma (Prost_AdenoCA); therefore, we focused on this relationship. For all these cancer types, the disease incidence dramatically increases among aged patients, with pancreatic cancer peaking at around 85–89 years,48 prostate cancer 75–79 years,49 and CLL 85–89 years50 according to Cancer Research UK. This suggests that ageing-related mutational signatures might be prominent causes of driver accumulation in TP53 in these cancers. To test this hypothesis, we inspected all the mutations in the six genes of the TP53 pathway in pancreatic adenocarcinoma, since we had most samples for this tissue. We identified an increase of C>T mutations in the CpG context among TP53 pathway genes compared to the total mutations identified in these samples (Figure 4B, hypergeometric test p = 0.002). Of note, C>T mutations in the CpG context correspond to clock-like signature SBS1. This observation suggests that TP53 mutations in this cancer type are largely originating from spontaneous mutations arising from cell division.
Figure 4.

Linear regression of signature-pathway interactions in TCGA
(A) Summary of the regression model of predicting signature activities from pathway alteration counts in TCGA. Positive (red) and negative (blue) interactions are summarized separately, and the numbers indicate the number of tissues in which the interaction has been detected.
(B) Mutation types in TP53 pathway vs. background mutations in pancreatic adenocarcinoma. The p value was calculated using the hypergeometric test. The asterisk indicates significance—p < 0.05. See also Figure S4.
Overall, our results recapitulate expected relationships such as that between UV and RTK-RAS pathway, uncover a causal link between TP53 and clock-like signature SBS1, as well as several other interactions that remain to be mechanistically understood.
Signature interactions and patient survival
To assess the impact of interaction on patient survival, we fit a range of Cox regression models (Figure 1A). We focused on two main classes of models while in both cases the interactors were treated as binary variables. In the first class, the regression model was constructed from the two interactors and an interaction term. This class of models allowed to determine the impact of individual interactors as well as their epistatic effect on survival in a unified fashion (see STAR Methods). However, in practice, we observed that accurate estimation of regression coefficients required a high sample size, and estimates were obtained from only a few well-sampled tissues. In the second class of models, we classified the samples depending on whether they had either of the interactors, both or none, and fit a Cox regression model using the sample class as a regression parameter. While this approach cannot definitively expose all interactions with synergistic or antagonistic relationships, it provides a stringent filter for further case-by-case inspection and is applicable for tissues with lower sample sizes. Notably, for both classes of models, we iteratively added the age of diagnosis and log-transformed tumor mutational burden (TMB). In cases where models were fit to multiple cancer types, the cancer type was also provided as a covariate. Further, all the fitted models underwent a pairwise comparison using the partial likelihood ratio (PLR) test for non-nested models or the likelihood ratio (LR) test for nested models,51 and the best models were reported.
To benchmark our results, we first combined and summarized the impact of age and TMB on survival across tissues and datasets (Figures 5A and 5B). As expected, age was linked to increased mortality (log hazard ratio log(HR) > 0) in a range of tissues (11 out of 23). TMB, on the other hand, had no effect on survival for 17 out of 23 tissues, a positive effect for five tissues (log(HR) < 0), and a negative effect in one tissue (log(HR) > 0). This dual effect of TMB on overall survival has been observed previously.52
Figure 5.

The effect of signature-signature and signature-pathway interactions on patient survival
(A and B) Summary of survival effect of age at diagnosis and tumor mutational burden (TMB) across samples in TCGA and PCAWG. The letters to the left indicate the dataset. The error bars in A,B, C, D, F correspond to the 95% confidence interval of the HR estimate.
(C and D) The log hazard ratio of signature-signature interactions with a significant effect on survival in PCAWG and TCGA.
(E) Survival curves of pancreatic, prostate, ovarian adenocarcinomas, and medulloblastoma with SBS40 signature, ROS signature (SBS18), a combination, or none, adjusted for age at diagnosis, cancer type, and log-transformed tumor mutational burden.
(F) The log hazard ratio of signature-pathway interactions with a significant effect on survival in TCGA. See also Figure S5.
We now proceed to discuss some of the significant survival effects among signature-signature and signature-pathway interactions identified in PCAWG and TCGA datasets (Figures 2D, S2B, S3B, and S3B). All the models and model comparison results for interactions with moderate or significant effects on patient survival (p-value < 0.1) are reported in Tables S8, S9, S10, S11, S12, S13, S14, and S15.
Impact of signature-signature interactions on patient survival
We first investigated the best models for survival across model classes and covariates. Among these models, only two of the tested signature-signature interactions in PCAWG data showed a significant effect on survival (Figure 5C). Both interactions were detected in hepatocellular carcinoma and involved the signature SBS40. First, in the positive interaction between clock-like signature SBS1 and SBS40, patients who carried only SBS40 were marked with significantly compromised survival, while carrying both signatures rescued the phenotype. On the contrary, the negative interaction between SBS29 and SBS40 significantly exacerbated the phenotype (hazard ratio [HR] = 6.42, 95% confidence interval [CI] = [1.22, 33.83]). Notably, in both examples the number of samples in some sample groups was low, hence, more targeted investigation would be required to validate these observations.
In TCGA, we identified four interactions in two tissues, where the best model reported a significant effect among carriers of both signatures (Figure 5D). In colorectal adenocarcinoma, the positive interaction between MMR and SBS5 had a significantly improving effect on survival (HR = 0.037, 95% CI = [0.007, 0.2]), while both signatures individually were linked to worse survival compared to the reference (MMR only HR = 13 with 95% CI = [3.01, 56.13], SBS5 only HR = 2.74, 95% CI = [1.05, 7.16]). Complementary to this observation, the negative interaction between MMR and SBS40 had a worse effect on survival (HR = 24.54, 95% CI = [3.9, 154.58]), while patients carrying MMR alone had a significantly better survival (HR = 0.1248 with 95% CI = [0.02, 0.7208]). These divergent effects of SBS40 and SBS5 on patient survival in combination with MMR signature suggest that these signatures might originate at different stages of cancer progression. Recent work on the evolutionary history of cancers supports this hypothesis, showing that mutations attributed to SBS5 tend to be more clonal, while SBS40 mutations tend to be more subclonal36 (Figure S6A).
Further, in melanoma samples, the positive interaction between UV and clock-like SBS1 signatures had a significantly ameliorating effect (HR = 0.14, 95% CI = [0.036, 0.54]), while SBS1 signature alone was linked with worse survival than the reference of non-carries (HR = 5.88, HR = [1.72, 20.11]). SBS1 signature is caused by spontaneous deamination of methylated cytosines and has been linked to cell division.3 This effectively explains the correlation of this signature with patient age14,29 and suggests an association with tumor stage, potentially explaining its impact on patient survival observed in our analysis.53,54
Next, to explore the potential interactions in tissues with fewer samples, we relaxed the model-selection criteria and focused on models that classified samples into carriers of either of the signatures or both and used the non-carriers as reference. The addition of age and TMB to the regression model as covariates was selected using the PLR and LR tests (see STAR Methods). We further performed a case-by-case inspection to detect potentially interesting interactions (Figures S5A and S5C).
In PCAWG, for the negative interaction between SBS40 and ROS signatures, our models showed that carriers of both signatures had worse survival in pancreatic adenocarcinoma. This negative interaction has been detected in four tissues in PCAWG—pancreatic, prostate, and ovarian adenocarcinomas and medulloblastoma. Since the number of samples with both signatures was low in tissues individually, we pooled samples from different tissues and fit a range of Cox regression models, while performing a model selection as described earlier. In these models, we provided the cancer type as a covariate. The pooled analysis reveals an HR of the synergistic interaction between SBS40 and ROS signatures to be HR = 2.62 (95% CI = [1.29, 5.29]). Our model shows that in these tissues ROS alone is linked to poor survival outcome (HR = 1.59, 95% CI = [1.15, 2.19]), which is amplified by signature SBS40 (Figures 5E and S5B). The effect of ROS in pancreatic adenocarcinoma is dual—low levels of ROS can induce proliferation in the early stages of the tumor, while higher levels can promote metastasis.55
Impact of signature-pathway interactions on patient survival
Next, we tested the effect of interacting signature-pathway pairs on survival by performing model fitting and selection as described earlier.
Our results recapitulate several survival effects of both mutational signatures and pathway alterations. As expected, in some cancer types signatures and pathway alterations alone had an impact on patient survival. One such example was the mutual exclusivity between TP53 pathway mutation and PolH mutational signature, which recapitulates the complex landscape of CLL, where individually both TP53 and IGHV mutations (PolH-linked marker) are important prognostic markers.56 TP53 and IGHV mutations are linked to worse and better outcomes, respectively, also detected in our regression model, although we had no samples carrying both markers.
In PCAWG, survival models that could account for synergistic effects did not report significant interactions, while in TCGA, we had four pairs with a significant impact on survival in three tissues (Figure 5F).
The negative interaction between APOBEC signature and NOTCH pathway had a deteriorating effect on survival (HR = 10.92, 95% CI = [1.33, 89.9]), similar to the negative interaction between SBS40 and NOTCH (HR = 14.67, 95% CI = [1.5, 143.42]). Notably, the samples with APOBEC and SBS40 signatures form a cluster, and our results demonstrate that in this cluster samples that carry alterations in the NOTCH pathway have markedly worse survival.
In breast adenocarcinoma, the positive interaction between HRD signature and TP53 pathway had a favorable impact over the background of either HRD signature or TP53 pathway mutation carries (HR = 0.06, 95% CI = [0.004, 0.79]), while HRD signature carriers alone had significantly worse survival (HR = 15.26, 95% CI = [1.54, 151.29]). Notably, this result agrees with a recent study reporting that TP53 mutation in the background of germline BRCA2 mutation has a beneficial effect on survival.57
Similarly, in esophageal adenocarcinoma, the negative interaction between SBS17 signature and PI3K pathway alteration enhanced survival on the background of either interactors (HR = 0.27, 95% CI = [0.075, 0.94]).
Further, the positive interaction between MMR signature and HIPPO pathway in the uterine adenocarcinoma had a deteriorating impact on survival (HR = 14.29, 95% CI = [0.96, 212.61], corresponding to a p-value of 0.053).
We then relaxed the model selection criteria and focused on regression models that classified the samples into carriers of either of the interactors, both, or none. The survival times were compared between these groups with the reference of non-carriers. These models revealed more potentially interesting interactions that were missed in the more complex models due to the low sample size.
In PCAWG, despite relatively low sample numbers, we detected two positive interactions in two tissues, all impacting survival unfavorably (Figure S5D). The interaction between ROS signature and MYC pathway was linked to overall worse survival in medulloblastoma (CNS_Medullo). Since both ROS and MYC were linked to worse survival already individually, their interaction potentially has only an additive effect on survival. In prostate adenocarcinoma (Prost_AdenoCA), the interaction between ROS and TP53 was also linked to worse survival. The prevalence of TP53 mutations in metastatic prostate adenocarcinoma58 increases dramatically compared to primary tumors, and the role of concurrently acquired ROS mutational signature remains to be understood in the context of metastasis.
In TCGA, we identified three interactions in two tissues where the carriers of both interactors had significant survival differences compared to non-carriers, including the interactions between the NOTCH signaling pathway and APOBEC or SBS40 signatures in uterine adenocarcinoma discussed earlier (Figure S5E). The negative interaction between APOBEC and TGF-beta in cervical cancer indicates a mildly positive effect on survival among the carriers of mutations in TGF-beta pathway; however, the number of samples carrying none of the interactors was very low (three samples) for an accurate estimate.
Discussion
A complete understanding of how tumors emerge and reshape their environment will require the identification of principles underlying the interplay between mutational signatures, signaling pathways, metabolic states, and tissue remodeling. Our work opens a conceptual and methodological framework for interrogating these interactions and could serve as a resource for hypothesis generation. To showcase the potential of our framework, we applied it to thousands of cancer samples across tissues in TCGA and PCAWG datasets and identified numerous interactions. We have implemented our methodology in an R package and made all results available through a web application (https://ahakobyan.shinyapps.io/mutsigapp/).
Our results showed that some signatures imply a preference for specific driver mutations throughout tumorigenesis, advancing our understanding of the causal relationship between pathways and signatures. We recapitulated the case of melanoma, where UV-exposed tumors tend to have mutations in the RTK-RAS pathway. We observed a similar phenomenon in tissues with a mismatch repair deficiency (MMR) signature. Specifically, in stomach adenocarcinoma, MMR-high samples had higher levels of NOTCH, WNT, TGF-beta, HIPPO, and PI3K and lower levels of TP53 alterations than MMR-low tumors.
We investigated the effect of all detected interactions on patient survival and identified several interactions with significant impact. Specifically, we identified a negative interaction between the ROS and SBS40 mutational signatures that had a detrimental effect on survival across tissues. We also found that the MMR signature and HIPPO pathway had a negative synergistic effect on survival in uterine adenocarcinomas, highlighting a potential avenue for treatment. Furthermore, our analysis revealed that SBS40 has distinct interaction preferences compared to SBS5. These signatures have unknown etiologies and due to high cosine similarity, they have been speculated to be an effect of intermixing. Our results from the survival analysis suggest that SBS40 has a behavior distinct from SBS5 and is potentially activated at a later stage of tumor progression, underscoring the need for a better understanding of its etiology and context.
Limitations of the study
We acknowledge several limitations in our current approach and envision some strategies to address these constraints: (1) differential numbers of samples available for tissues across datasets; (2) TCGA and PCAWG provide whole exome and genome resolution, respectively, thus the activity of signatures could not be inferred with the same accuracy; (3) spatial and temporal characteristics, as well as clonality were not taken into account; (4) we only considered SBS signatures; (5) we used signatures extracted by Alexandrov et al.; however, the final extraction results can vary depending on the extraction method.
Applying the framework to large, uniform, and high-quality datasets, as well as accounting for the spatial, temporal, and clonal structure of the data would be a way forward for more detailed understanding, which may lead to the identification of novel tumor vulnerabilities and the subsequent development of targeted interventions.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Mutational signatures and somatic variant calls from PCAWG and TCGA | ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium14,28 | https://dcc.icgc.org/releases/PCAWG |
| Software and algorithms | ||
| R (4.1.2) | R core team59 | https://www.R-project.org/ |
| R package tidyverse (2.0.0) | Wickham et al.60 | https://www.tidyverse.org/ |
| R package ggplot2 (3.4.4) | Wickham H.61 | https://ggplot2.tidyverse.org/ |
| R package mpmi (0.43.2.1) | Pardy et al.33 | https://CRAN.R-project.org/package=mpmi |
| R package MutationalPatterns (3.4.1) | Blokzijl et al.34 | https://bioconductor.org/packages/release/bioc/html/MutationalPatterns.html |
| R package mutsigintsKit | This paper | https://zenodo.org/doi/10.5281/zenodo.10818464 |
| R code mutsigints | This paper | https://zenodo.org/doi/10.5281/zenodo.10818459 |
| R code mutsigapp | This paper | https://github.com/annahyan/mutsigapp |
| Shiny app mutsigapp | This paper | https://ahakobyan.shinyapps.io/mutsigapp/ |
| R package pheatmap (1.0.12) | N/A | https://CRAN.R-project.org/package=pheatmap |
| R package rms (6.3.0) | Harrell Jr FE.62 | https://CRAN.R-project.org/package=rms |
| R package robCompositions (2.4.1) | Templ et al.63 | https://CRAN.R-project.org/package=robCompositions |
| R package survminer (0.4.9) | Kassambara et al. 64 | https://CRAN.R-project.org/package=survminer |
| R package survival (3.4.0) | Therneau T.65 | https://CRAN.R-project.org/package=survival |
| R package nonnestcox | Implementation of Fine et al.51 | https://github.com/thomashielscher/nonnestcox |
Resource availability
Lead contact
Further information and requests for resources should be directed to Jörg Menche (joerg.menche@univie.ac.at).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
This paper analyzes existing, publicly available data. We used the mutational signatures extracted by the PCAWG Mutational Signatures Working Group from PCAWG and TCGA datasets14 available at https://dcc.icgc.org/releases/PCAWG.
-
•
All original code for analysis is available at: https://github.com/menchelab/mutsigints. The toolkit for the analysis has been compiled into a package called mutsigintsKit and is available at: https://github.com/menchelab/mutsigintsKit. Analysis results can be explored with a web app at: https://ahakobyan.shinyapps.io/mutsigapp/, with the source code available at: https://github.com/annahyan/mutsigapp. The DOI’s are listed in the Key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Method details
Signature-signature interaction metrics
Correlation metric based on compositional data analysis
Mutational signatures are compositional by design, resulting from the procedure of their extraction. Each mutation in a sample is being assigned to a signature and the sum of mutations assigned to all mutational signatures is constrained by the number of total mutations in a sample. The data, which is constrained by a constant sum, is by design compositional and lies on a simplex (Figure S1A). This means that data that is described by N parameters lies on an N‒1 dimensional simplex. Thus, classical statistical tools are not applicable and can give rise to spurious correlations of ratios first noted by Pearson.66 These issues have been addressed through the framework of compositional data (CoDa) analysis, which provides tools for data transformation into a space, where relations between individual features can be assessed.35
Assuming we have a compositional vector:
To transform the data from dimensional simplex to N‒1 dimensional orthonormal space, isometric log-ratio (ILR) coordinates are used, also called pivot coordinates:
The first element in ILR coordinates defines the relative dominance of the first component with respect to the geometric mean of all the other components:
For mutational signatures this will accordingly determine the relative dominance of the first signature with respect to other signatures. We are, however, interested in the relation between two such components. The interpretability of the first component in ILR coordinates allows for the construction of symmetric pivot coordinates,35 in order to assess the relative dominance of two components with respect to all the other components in a symmetric manner. The first two elements of pivot coordinates , which are symmetric with respect to the first two components are calculated with
and
These coordinates can then be used to calculate correlation metrics, which will show whether the dominance of selected mutational signatures is correlated. The function to calculate symmetric pivot coordinates was adapted from the robCompositions63 package in R and is available as part of the mutsigintsKit package.
Bias-corrected mutual information: We calculated bias-corrected mutual information using the R package mpmi (0.43.2).33
Bootstrapped calculation of interaction metrics
Accurate assessment of mutational signature interactions depends on the number of available samples, the incidence of mutational signatures, and the number of mutations these signatures contribute. We assessed signature interactions across available tissues and datasets and identified several properties in the data that could confound the analysis. First, (i) tissue specificity of signatures is highly variable. Signatures linked to environmental exposures are highly specific to tissues subjected to those agents, (e.g. UV signatures SBS7a-d in skin, tobacco smoking signature SBS4 in lungs). Other signatures, such as those associated with endogenous DNA damage and repair processes, are less selective and can be active in multiple tissues. For example, signature SBS6 is associated with DNA mismatch repair deficiency and is active across multiple tissues. Similarly, the ageing-associated mutational signature SBS1 is omnipresent across tissues. Further, (ii) the incidence of a signature in the tissue is highly patient-specific, and the signature composition of samples within tissues is linked to and contributes to disease heterogeneity (Figures 1C and S1B). (iii) The number of mutations generated by each signature type is highly variable both between signatures and between tissues for a given signature. (iv) Signature incidence varies among different samples of the same tissue. (v) The number of samples sequenced for each tissue type is variable, yet critical for a comprehensive understanding of active signatures and their activities. Indeed, we found that the number of signatures detected in a tissue depends on the number of available samples (Figure S1C). All of these factors hinder the uniform assessment of signature interactions. To reduce sampling bias, we therefore performed bootstrapped calculations of signature interactions for all metrics (Figure S2B). Specifically, for each tissue we sampled 90% of the samples with replacement for 100 iterations and calculated the interaction metrics. Further, we assumed the interaction was present if the metric was non-zero for at least 50% of the sampling iterations. Otherwise, an interaction was considered absent. Non-zero interaction metrics were averaged.
Null models for interaction metrics
We computed all the metrics for each tissue and applied the p-value threshold of 0.05 after the Benjamini-Hochberg correction.
The null models for individual metrics were defined by 1000 random shufflings and repeated calculations of each metric in each tissue. The permutations were performed for each signature individually, hence the combinations of active signatures and signatures and pathways in permuted samples were randomized each time. After we identified the empirical distribution of metric values generated from shuffled mutational matrices, we set the 0.95 quantile values as the significance threshold for each pair of interacting signatures. Additionally, for the mutual information, we applied a hard cutoff of 0.2.
Interaction metric aggregation
In signature-signature analysis, the application of different metrics resulted in multiple values for each interaction. All the interactions supported with less than two metrics were discarded. All the used metrics could provide directionality except for mutual information. The final interaction direction was defined after summarizing the metric values of all the other interactions and taking the sign. This approach could be ambiguous in cases when the metrics imply different interaction types. However, in practice, the interaction directions were highly concordant, and discordant interactions were ignored.
Signature-pathway interactions
We obtained the curate list of recurrently mutated signaling pathways as well as the pathway alterations across TCGA samples from Sanchez-Vega et al,43 while we obtained the PCAWG driver mutations across samples from the ICGC Data Portal (https://dcc.icgc.org/releases/PCAWG). PCAWG driver mutations were mapped to 10 oncogenic signaling pathways. For this analysis, we filtered out samples with available mutational signatures as well as pathway alteration data. Further, we filtered out all the tissues with less than 30 samples for further analysis.
We performed the co-occurrence analysis between mutational signatures and pathway alterations by creating a contingency table for each signature-pathway pair and performed Fisher’s exact test with a p-value threshold of 0.05. Linear and logistic regressions were run with R functions lm and glm. Furthermore, we created a null model for each interaction type and signature-pathway interacting pair by performing 1000 tissue shufflings for each tissue individually and set the 5% and 95% percentile of each signature-pathway interaction as an additional cutoff.
Survival analysis
All the analyses were run in the R (4.1.2) language59 and RStudio environment. Survival analysis was performed using the survival package (3.2-13)65 and survival curves were produced using survminer package (0.4.9).64 Adjusted survival curves were produced by ggadjustedcurves function.
We used Cox regression model, which links the hazard function ⅄(t) to covariates through an exponential function. The hazard function models survival-time or time-to-death.
represents the baseline survival and is the regression coefficient for the covariate .
To detect the effect of interacting signatures we have extensively tested different regression models with different interaction modes and a range of covariates.
To assess the interaction effect we tested two different classes of models:
-
1.
To test the epistatic effect of a pair of interactors (signature-signature or signature-pathway) on survival we built a model by providing the presence of individual interactors as well as their co-occurrence as individual regression terms:
where represent the coefficients for first interactor, second interactor, and their interaction term, while ’s are the coefficients for the covariates.
This model allows to differentiate between additive vs epistatic effects of interactors, however, in practice, the accurate estimation requires a relatively high sample size.
-
2.
In the second approach, for interaction between interactor s1 and interactor s2 we classified the samples into 4 groups depending on interactor activities - None, s1, s2, s1_and_s2.
where is the status of the sample and is the respective coefficient. Notably, this model treats samples carrying both signatures as a separate category and each is estimated separately. The sample set that carries none of the mutations serves as a reference. This model does not allow to account for individual effects of interactors in the interaction, yet, it allows to select the tissues where the samples carrying both interactors have significantly different survival outcomes even for small sample sizes, and can be useful for manual selection and inspection of individual tissues.
Acknowledgments
The results published here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. This work was supported by the Vienna Science and Technology Fund (WWTF) through project VRG15-005 (to J.M.). M.M. was supported by a DOC fellowship of the Austrian Academy of Sciences (25757, awarded to M.M.). We thank all the members of the Menche lab and the Loizou lab for their valuable inputs throughout this project.
Author contributions
Conceptualization, A.H., J.M., L.V., and J.H.; methodology, A.H., L.V., J.H., and J.M.; investigation, A.H. and N.V.; writing—original draft, A.H.; writing—review & editing, A.H., M.M., L.V., J.H., J.I.L., and J.M.; funding acquisition, J.M.; resources, J.M. and J.I.L.; supervision, J.M. and J.I.L.
Declaration of interests
J.I.L. is now an employee of AstraZeneca. The authors declare no other competing interests.
Published: May 2, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109873.
Supplemental information
Interaction metric values along with empirical p values are provided. For non-significant interactions (p >0.05) p values are inflated to 1.
Interaction metric values along with empirical p values are provided. For non-significant interactions (p > 0.05) p values are inflated to 1.
For each interaction, we present all tissues where the interaction has been observed. Negative and positive interactions across datasets are presented separately.
Interaction metric values along with empirical p values are provided. For non-significant interactions (p > 0.05) p values are inflated to 1.
Interaction metric values along with empirical p values are provided. For non-significant interactions (p > 0.05) p values are inflated to 1.
For each interaction, we present all tissues where the interaction has been observed. Negative and positive interactions across datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p-value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
References
- 1.Kim N., Jinks-Robertson S. Transcription as a source of genome instability. Nat. Rev. Genet. 2012;13:204–214. doi: 10.1038/nrg3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCulloch S.D., Kunkel T.A. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell Res. 2008;18:148–161. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duncan B.K., Miller J.H. Mutagenic deamination of cytosine residues in DNA. Nature. 1980;287:560–561. doi: 10.1038/287560a0. [DOI] [PubMed] [Google Scholar]
- 4.Wijesinghe P., Bhagwat A.S. Efficient deamination of 5-methylcytosines in DNA by human APOBEC3A, but not by AID or APOBEC3G. Nucleic Acids Res. 2012;40:9206–9217. doi: 10.1093/nar/gks685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haradhvala N.J., Kim J., Maruvka Y.E., Polak P., Rosebrock D., Livitz D., Hess J.M., Leshchiner I., Kamburov A., Mouw K.W., et al. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat. Commun. 2018;9:1746. doi: 10.1038/s41467-018-04002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meier B., Volkova N.V., Hong Y., Schofield P., Campbell P.J., Gerstung M., Gartner A. Mutational signatures of DNA mismatch repair deficiency in C. elegans and human cancers. Genome Res. 2018;28:666–675. doi: 10.1101/gr.226845.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pfeifer G.P., You Y.-H., Besaratinia A. Mutations induced by ultraviolet light. Mutat. Res. 2005;571:19–31. doi: 10.1016/j.mrfmmm.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 8.Parplys A.C., Petermann E., Petersen C., Dikomey E., Borgmann K. DNA damage by X-rays and their impact on replication processes. Radiother. Oncol. 2012;102:466–471. doi: 10.1016/j.radonc.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 9.Denissenko M.F., Pao A., Tang M., Pfeifer G.P. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science. 1996;274:430–432. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- 10.Kucab J.E., van Steeg H., Luijten M., Schmeiser H.H., White P.A., Phillips D.H., Arlt V.M. TP53 mutations induced by BPDE in Xpa-WT and Xpa-Null human TP53 knock-in (Hupki) mouse embryo fibroblasts. Mutat. Res. 2015;773:48–62. doi: 10.1016/j.mrfmmm.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldman R., Shields P.G. Food mutagens. J. Nutr. 2003;133:965S–973S. doi: 10.1093/jn/133.3.965S. [DOI] [PubMed] [Google Scholar]
- 12.Yang M. A current global view of environmental and occupational cancers. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2011;29:223–249. doi: 10.1080/10590501.2011.601848. [DOI] [PubMed] [Google Scholar]
- 13.Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A.J.R., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.-L., et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexandrov L.B., Kim J., Haradhvala N.J., Huang M.N., Tian Ng A.W., Wu Y., Boot A., Covington K.R., Gordenin D.A., Bergstrom E.N., et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578:94–101. doi: 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steele C.D., Abbasi A., Islam S.M.A., Bowes A.L., Khandekar A., Haase K., Hames-Fathi S., Ajayi D., Verfaillie A., Dhami P., et al. Signatures of copy number alterations in human cancer. Nature. 2022;606:984–991. doi: 10.1038/s41586-022-04738-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E., et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kucab J.E., Zou X., Morganella S., Joel M., Nanda A.S., Nagy E., Gomez C., Degasperi A., Harris R., Jackson S.P., et al. A Compendium of Mutational Signatures of Environmental Agents. Cell. 2019;177:821–836.e16. doi: 10.1016/j.cell.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zou X., Owusu M., Harris R., Jackson S.P., Loizou J.I., Nik-Zainal S. Validating the concept of mutational signatures with isogenic cell models. Nat. Commun. 2018;9:1744. doi: 10.1038/s41467-018-04052-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jager M., Blokzijl F., Kuijk E., Bertl J., Vougioukalaki M., Janssen R., Besselink N., Boymans S., de Ligt J., Pedersen J.S., et al. Deficiency of nucleotide excision repair is associated with mutational signature observed in cancer. Genome Res. 2019;29:1067–1077. doi: 10.1101/gr.246223.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hwang T., Reh S., Dunbayev Y., Zhong Y., Takata Y., Shen J., McBride K.M., Murnane J.P., Bhak J., Lee S., et al. Defining the mutation signatures of DNA polymerase θ in cancer genomes. NAR Cancer. 2020;2 doi: 10.1093/narcan/zcaa017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reijns M.A.M., Parry D.A., Williams T.C., Nadeu F., Hindshaw R.L., Rios Szwed D.O., Nicholson M.D., Carroll P., Boyle S., Royo R., et al. Signatures of TOP1 transcription-associated mutagenesis in cancer and germline. Nature. 2022;602:623–631. doi: 10.1038/s41586-022-04403-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Volkova N.V., Meier B., González-Huici V., Bertolini S., Gonzalez S., Voeringer H., Abascal F., Martincorena I., Campbell P.J., Gartner A., Gerstung M. Mutational signatures are jointly shaped by DNA damage and repair. Nat. Commun. 2020;11 doi: 10.1038/s41467-020-15912-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X.-P., Liu F., Wang W. Two-phase dynamics of p53 in the DNA damage response. Proc. Natl. Acad. Sci. USA. 2011;108:8990–8995. doi: 10.1073/pnas.1100600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cáceres-Gutiérrez R.E., Alfaro-Mora Y., Andonegui M.A., Díaz-Chávez J., Herrera L.A. The Influence of Oncogenic RAS on Chemotherapy and Radiotherapy Resistance Through DNA Repair Pathways. Front. Cell Dev. Biol. 2022;10 doi: 10.3389/fcell.2022.751367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Temko D., Tomlinson I.P.M., Severini S., Schuster-Böckler B., Graham T.A. The effects of mutational processes and selection on driver mutations across cancer types. Nat. Commun. 2018;9:1857. doi: 10.1038/s41467-018-04208-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brady S.W., Gout A.M., Zhang J. Therapeutic and prognostic insights from the analysis of cancer mutational signatures. Trends Genet. 2022;38:194–208. doi: 10.1016/j.tig.2021.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chopra N., Tovey H., Pearson A., Cutts R., Toms C., Proszek P., Hubank M., Dowsett M., Dodson A., Daley F., et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020;11:2662. doi: 10.1038/s41467-020-16142-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93. doi: 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexandrov L.B., Jones P.H., Wedge D.C., Sale J.E., Campbell P.J., Nik-Zainal S., Stratton M.R. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015;47:1402–1407. doi: 10.1038/ng.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li C.H., Haider S., Boutros P.C. Age influences on the molecular presentation of tumours. Nat. Commun. 2022;13:208. doi: 10.1038/s41467-021-27889-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petljak M., Alexandrov L.B., Brammeld J.S., Price S., Wedge D.C., Grossmann S., Dawson K.J., Ju Y.S., Iorio F., Tubio J.M.C., et al. Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell. 2019;176:1282–1294.e20. doi: 10.1016/j.cell.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roper N., Gao S., Maity T.K., Banday A.R., Zhang X., Venugopalan A., Cultraro C.M., Patidar R., Sindiri S., Brown A.-L., et al. APOBEC Mutagenesis and Copy-Number Alterations Are Drivers of Proteogenomic Tumor Evolution and Heterogeneity in Metastatic Thoracic Tumors. Cell Rep. 2019;26:2651–2666.e6. doi: 10.1016/j.celrep.2019.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pardy C., Galbraith S., Wilson S.R. Integrative exploration of large high-dimensional datasets. Ann. Appl. Stat. 2018;12:178–199. doi: 10.1214/17-AOAS1055. [DOI] [Google Scholar]
- 34.Blokzijl F., Janssen R., van Boxtel R., Cuppen E. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018;10:33. doi: 10.1186/s13073-018-0539-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Filzmoser P., Hron K., Templ M. Springer International Publishing; 2018. Applied Compositional Data Analysis: With Worked Examples in R 1st Ed. [DOI] [Google Scholar]
- 36.Gerstung M., Jolly C., Leshchiner I., Dentro S.C., Gonzalez S., Rosebrock D., Mitchell T.J., Rubanova Y., Anur P., Yu K., et al. The evolutionary history of 2,658 cancers. Nature. 2020;578:122–128. doi: 10.1038/s41586-019-1907-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee-Six H., Olafsson S., Ellis P., Osborne R.J., Sanders M.A., Moore L., Georgakopoulos N., Torrente F., Noorani A., Goddard M., et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature. 2019;574:532–537. doi: 10.1038/s41586-019-1672-7. [DOI] [PubMed] [Google Scholar]
- 38.Kim Y.-A., Wojtowicz D., Sarto Basso R., Sason I., Robinson W., Hochbaum D.S., Leiserson M.D.M., Sharan R., Vadin F., Przytycka T.M. Network-based approaches elucidate differences within APOBEC and clock-like signatures in breast cancer. Genome Med. 2020;12:52. doi: 10.1186/s13073-020-00745-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Talluri S., Samur M.K., Buon L., Kumar S., Potluri L.B., Shi J., Prabhala R.H., Shammas M.A., Munshi N.C. Dysregulated APOBEC3G causes DNA damage and promotes genomic instability in multiple myeloma. Blood Cancer J. 2021;11:166. doi: 10.1038/s41408-021-00554-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farmanbar A., Firouzi S., Kneller R., Khiabanian H. Mutational signatures reveal ternary relationships between homologous recombination repair, APOBEC, and mismatch repair in gynecological cancers. J. Transl. Med. 2022;20:65. doi: 10.1186/s12967-022-03259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jakobsdottir G.M., Brewer D.S., Cooper C., Green C., Wedge D.C. APOBEC3 mutational signatures are associated with extensive and diverse genomic instability across multiple tumour types. BMC Biol. 2022;20:117. doi: 10.1186/s12915-022-01316-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCann J.L., Klein M.M., Leland E.M., Law E.K., Brown W.L., Salamango D.J., Harris R.S. The DNA deaminase APOBEC3B interacts with the cell-cycle protein CDK4 and disrupts CDK4-mediated nuclear import of Cyclin D1. J. Biol. Chem. 2019;294:12099–12111. doi: 10.1074/jbc.RA119.008443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanchez-Vega F., Mina M., Armenia J., Chatila W.K., Luna A., La K.C., Dimitriadoy S., Liu D.L., Kantheti H.S., Saghafinia S., et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018;173:321–337.e10. doi: 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sabarinathan R., Pich O., Martincorena I., Rubio-Perez C., Juul M., Wala J., Schumacher S., Shapira O., Sidiropoulos N., Waszak S.M., et al. The whole-genome panorama of cancer drivers. bioRxiv. 2017;190330 doi: 10.1101/190330. [DOI] [Google Scholar]
- 45.Shi Q., Liu L., Chen J., Zhang W., Guo W., Wang X., Wang H., Guo S., Yue Q., Ma J., et al. Integrative Genomic Profiling Uncovers Therapeutic Targets of Acral Melanoma in Asian Populations. Clin. Cancer Res. 2022;28:2690–2703. doi: 10.1158/1078-0432.CCR-21-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seo J., Kim H., Min K.I., Kim C., Kwon Y., Zheng Z., Kim Y., Park H.-S., Ju Y.S., Roh M.R., et al. Weight-bearing activity impairs nuclear membrane and genome integrity via YAP activation in plantar melanoma. Nat. Commun. 2022;13:2214. doi: 10.1038/s41467-022-29925-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Risk Factors: Age. National Cancer Institute; 2015. https://www.cancer.gov/about-cancer/causes-prevention/risk/age [Google Scholar]
- 48.Pancreatic Cancer Incidence Statistics. Cancer Research UK; 2015. https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/pancreatic-cancer/incidence [Google Scholar]
- 49.Prostate Cancer Incidence Statistics. Cancer Research UK; 2015. https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/prostate-cancer/incidence [Google Scholar]
- 50.Chronic Lymphocytic Leukaemia (CLL) Statistics. Cancer Research UK; 2015. https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/leukaemia-cll [Google Scholar]
- 51.Fine J.P. Comparing nonnested Cox models. Biometrika. 2002;89:635–648. doi: 10.1093/biomet/89.3.635. [DOI] [Google Scholar]
- 52.Ding H., Zhao J., Zhang Y., Wang G., Cai S., Qiu F. Tumor mutational burden and prognosis across pan-cancers. Ann. Oncol. 2018;29:viii16–viii17. doi: 10.1093/annonc/mdy269.055. [DOI] [Google Scholar]
- 53.Wan J.C.M., Stephens D., Luo L., White J.R., Stewart C.M., Rousseau B., Tsui D.W.Y., Diaz L.A., Jr. Genome-wide mutational signatures in low-coverage whole genome sequencing of cell-free DNA. Nat. Commun. 2022;13:4953. doi: 10.1038/s41467-022-32598-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abbas S., Pich O., Devonshire G., Zamani S.A., Katz-Summercorn A., Killcoyne S., Cheah C., Nutzinger B., Grehan N., Lopez-Bigas N., et al. Mutational signature dynamics shaping the evolution of oesophageal adenocarcinoma. Nat. Commun. 2023;14:4239. doi: 10.1038/s41467-023-39957-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang L., Li J., Zong L., Chen X., Chen K., Jiang Z., Nan L., Li X., Li W., Shan T., et al. Reactive Oxygen Species and Targeted Therapy for Pancreatic Cancer. Oxid. Med. Cell. Longev. 2016;2016 doi: 10.1155/2016/1616781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zenz T., Eichhorst B., Busch R., Denzel T., Häbe S., Winkler D., Bühler A., Edelmann J., Bergmann M., Hopfinger G., et al. TP53 mutation and survival in chronic lymphocytic leukemia. J. Clin. Oncol. 2010;28:4473–4479. doi: 10.1200/JCO.2009.27.8762. [DOI] [PubMed] [Google Scholar]
- 57.Kim J., Jeong K., Jun H., Kim K., Bae J.M., Song M.G., Yi H., Park S., Woo G.-U., Lee D.-W., et al. Mutations of TP53 and genes related to homologous recombination repair in breast cancer with germline BRCA1/2 mutations. Hum. Genomics. 2023;17:2. doi: 10.1186/s40246-022-00447-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abeshouse A., Ahn J., Akbani R., Ally A., Amin S., Andry C.d., Annala M., Aprikian A., Armenia J., Arora A., et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.R Core Team . Preprint at R Foundation for Statistical Computing; 2021. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 60.Wickham H., Averick M., Bryan J., Chang W., McGowan L., François R., Grolemund G., Hayes A., Henry L., Hester J., et al. Welcome to the tidyverse. J. Open Source Softw. 2019;4:1686. doi: 10.21105/joss.01686. [DOI] [Google Scholar]
- 61.Wilkinson L. Ggplot2: Elegant graphics for data analysis by WICKHAM, H. Biometrics. 2011;67:678–679. doi: 10.1111/j.1541-0420.2011.01616.x. [DOI] [Google Scholar]
- 62.Harrell F., Jr. 2023. Rms: Regression Modeling Strategies. [Google Scholar]
- 63.Templ M., Hron K., Filzmoser P. In: Compositional Data Analysis Monographs on Statistics and Applied Probability. Pawlowsky-Glahn V., Buccianti A., editors. John Wiley & Sons, Ltd; 2011. robCompositions: An R-package for Robust Statistical Analysis of Compositional Data; pp. 341–355. [DOI] [Google Scholar]
- 64.Kassambara A., Kosinski M., Biecek P. survminer: Drawing Survival Curves using “ggplot2.” R package. 2021. [Google Scholar]
- 65.Therneau T.M. 2022. A Package for Survival Analysis in R. [Google Scholar]
- 66.Pearson K. Mathematical contributions to the theory of evolution.—On a form of spurious correlation which may arise when indices are used in the measurement of organs. Proc. R. Soc. Lond. 1897;60:489–498. doi: 10.1098/rspl.1896.0076. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Interaction metric values along with empirical p values are provided. For non-significant interactions (p >0.05) p values are inflated to 1.
Interaction metric values along with empirical p values are provided. For non-significant interactions (p > 0.05) p values are inflated to 1.
For each interaction, we present all tissues where the interaction has been observed. Negative and positive interactions across datasets are presented separately.
Interaction metric values along with empirical p values are provided. For non-significant interactions (p > 0.05) p values are inflated to 1.
Interaction metric values along with empirical p values are provided. For non-significant interactions (p > 0.05) p values are inflated to 1.
For each interaction, we present all tissues where the interaction has been observed. Negative and positive interactions across datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p-value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Here we present tested survival models and fitted coefficients for all interactions where the p value of the interaction term in the best fitted model was <0.1. The results for signature-signature and signature-pathway interactions, for positive and negative interactions, and the two datasets are presented separately.
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. We used the mutational signatures extracted by the PCAWG Mutational Signatures Working Group from PCAWG and TCGA datasets14 available at https://dcc.icgc.org/releases/PCAWG.
-
•
All original code for analysis is available at: https://github.com/menchelab/mutsigints. The toolkit for the analysis has been compiled into a package called mutsigintsKit and is available at: https://github.com/menchelab/mutsigintsKit. Analysis results can be explored with a web app at: https://ahakobyan.shinyapps.io/mutsigapp/, with the source code available at: https://github.com/annahyan/mutsigapp. The DOI’s are listed in the Key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.