

Abstract
Background
The use of statin therapy in established Alzheimer's disease (AD) or vascular dementia (VaD) is a relatively unexplored area. In AD, β‐amyloid protein (Aβ) is deposited in the form of extracellular plaques and previous studies have determined Aβ generation is cholesterol dependent. Hypercholesterolaemia has also been implicated in the pathogenesis of VaD. Due to the role of statins in cholesterol reduction, it is biologically plausible they may be efficacious in the treatment of AD and VaD.
Objectives
To assess the clinical efficacy and safety of statins in the treatment of AD and VaD. To evaluate if the efficacy of statins in the treatment of AD and VaD depends on cholesterol level, ApoE genotype or cognitive level.
Search methods
We searched ALOIS, the Specialized Register of the Cochrane Dementia and Cognitive Improvement Group, The Cochrane Library, MEDLINE, EMBASE, PsycINFO, CINAHL and LILACS, as well as many trials registries and grey literature sources (20 January 2014).
Selection criteria
Double‐blind, randomised controlled trials of statins given for at least six months in people with a diagnosis of dementia.
Data collection and analysis
Two independent authors extracted and assessed data against the inclusion criteria. We pooled data where appropriate and entered them into a meta‐analysis. We used standard methodological procedures expected by The Cochrane Collaboration.
Main results
We identified four studies (1154 participants, age range 50 to 90 years). All participants had a diagnosis of probable or possible AD according to standard criteria and most participants were established on a cholinesterase inhibitor. The primary outcome in all studies was change in Alzheimer's Disease Assessment Scale ‐ cognitive subscale (ADAS‐Cog) from baseline. When we pooled data, there was no significant benefit from statin (mean difference ‐0.26, 95% confidence interval (CI) ‐1.05 to 0.52, P value = 0.51). All studies provided change in Mini Mental State Examination (MMSE) from baseline. There was no significant benefit from statins in MMSE when we pooled the data (mean difference ‐0.32, 95% CI ‐0.71 to 0.06, P value = 0.10). Three studies reported treatment‐related adverse effects. When we pooled data, there was no significant difference between statins and placebo (odds ratio 1.09, 95% CI 0.58 to 2.06, P value = 0.78). There was no significant difference in behaviour, global function or activities of daily living in the statin and placebo groups. We assessed risk of bias as low for all studies. We found no studies assessing role of statins in treatment of VaD.
Authors' conclusions
Analyses from the studies available, including two large randomised controlled trials, indicate that statins have no benefit on the primary outcome measures of ADAS‐Cog or MMSE.
Keywords: Aged; Aged, 80 and over; Humans; Middle Aged; Alzheimer Disease; Alzheimer Disease/drug therapy; Atorvastatin; Dementia; Dementia/drug therapy; Heptanoic Acids; Heptanoic Acids/therapeutic use; Hydroxymethylglutaryl‐CoA Reductase Inhibitors; Hydroxymethylglutaryl‐CoA Reductase Inhibitors/therapeutic use; Pyrroles; Pyrroles/therapeutic use; Randomized Controlled Trials as Topic; Simvastatin; Simvastatin/therapeutic use
Plain language summary
Statins for the treatment of Alzheimer's disease
Background
High levels of cholesterol (a fatty substance known as a lipid) in the blood are thought to contribute to the cause of Alzheimer's disease and vascular dementia. The statin family of medications (lovastatin, pravastatin, simvastatin and others) are powerful cholesterol‐lowering medications and are first‐line treatments for reducing cholesterol in people with, or at risk of, cardiovascular disease. There has been much interest in the possible role of statins in the treatment of dementia.
Study characteristics
Two independent authors searched scientific databases for studies in which a statin or a placebo (a pretend treatment) was given for at least six months. We included people with a probable or possible diagnosis of Alzheimer's disease according to standard clinical criteria. The findings were current to January 2014.
Key results
We identified four studies involving 1154 participants (age range 50 to 90 years). The studies used standard tests to assess the severity of Alzheimer's disease. From these trials, including two large trials, we found no evidence that statins help in the treatment of cognitive decline in dementia.
Quality of the evidence
The quality of evidence is felt to be high as two large randomised controlled trials have been included along with two smaller ones.
Summary of findings
Summary of findings for the main comparison. Statins compared with placebo for the treatment of dementia.
| Patients: patients with dementia Setting: community Intervention: statin medication Comparison: placebo | ||||
| Outcomes |
Absolute difference in means (95% CI) |
Number of participants (studies) |
GRADE Quality of evidence | Comments |
| Change in ADAS‐Cog | ‐0.26 (‐1.05 to 0.52) |
1110 (4) |
High | ‐ |
| Change in MMSE | ‐0.32 (‐0.71 to 0.06) |
1127 (4) |
High | ‐ |
| Change in CGIC | ‐0.02 (‐0.14 to 0.10) |
660 (2) |
High | ‐ |
| Change in NPI | ‐1.11 (‐2.10 to ‐0.12) |
983 (3) |
High | ‐ |
ADAS‐Cog: Alzheimer's Disease Assessment Scale ‐ cognitive subscale; CGIC: Clinical Global Impression of Change; CI: confidence interval; MMSE: Mini Mental State Examination; NPI: Neuropsychiatric Inventory.
GRADE Working Group grades of evidence:
Hign quality: Further research is very unlikely to change our confidence in the estimate of effect
Moderate quality: Further evidence is likely to have an important impact on our confidence in the estimate of effect and may change the estimate
Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate
Very low quality: We are very uncertain about the estimate
Background
Alzheimer's disease (AD) is the most common cause of dementia, accounting for 50% to 60% of all cases. Cerebrovascular disease is the second most common cause, responsible for 25% to 30% of cases and resulting in vascular dementia (VaD). AD and VaD also frequently co‐occur leading to mixed dementia. Dementia is thought to affect approximately 7% of the population older than 65 years of age and 30% of people older than 80 years of age. Other studies have quoted that 10% of people aged over 65 years are affected, rising to nearly 50% of all people aged over 85 years (Evans 1989; von Strauss 1999). Dementia is already a major public health problem and is set to become even more so due to the anticipated increase in life expectancy. As of 2013, there were an estimated 44.4 million people with dementia worldwide. This number will increase to an estimated 75.6 million in 2030 and 135.5 million in 2050 (Alzheimers Disease International 2013). Prevalence estimates from the English Cognitive Function and Ageing Study Ⅰ and Ⅱ in people aged over 65 years were 8.3% and 6.5%, respectively (Matthews 2013).
There is accumulating evidence that cholesterol may be implicated in the pathogenesis of dementia (AD and VaD) and this has led investigators to assess the possible role of lipid‐lowering agents (LLA) in the treatment of dementia. However, many questions remain unanswered. This review aimed to collate the best evidence available regarding use of statins in the treatment of dementia.
Cholesterol and Alzheimer's disease
A possible role for cholesterol in AD was based on observations of AD neuropathology among relatively young individuals with no history of dementia but with coronary heart disease (Sparks 1990). A central event in the development of AD is thought to be the abnormal processing of the cell membrane‐associated amyloid precursor protein (APP) followed by deposition of toxic β‐amyloid (Aβ) protein in the form of amyloid plaques in the extracellular space of the neocortex (Selkoe 2001). APP is a protein containing 770 amino acids. Aβ peptide is generated by the sequential cleavage of APP by beta and gamma secretase in the amyloidogenic pathway. Aβ genesis may be precluded if APP is instead cleaved first by alpha secretase within the Aβ domain, and then by gamma secretase, forming a non‐amyloidogenic fragment (Cole 2007). The non‐amyloidogenic pathway appears to be neuroprotective compared to the neurodegenerative, amyloidogenic pathway (Vetrivel 2006). Aβ occurs in two different forms, Aβ40 and Aβ42, varying in the length at the C terminus. It is the longer Aβ42 that aggregates more avidly. Early work discovered that elevated cholesterol levels led to greatly reduced levels of non‐amyloidogenic APP alpha in vitro (Bodovitz 1996). Perhaps more importantly, subsequent studies suggested that cerebral Aβ generation in vitro (Mizuno 1999; Simons 1998), and in vivo (Burns 2003; Refolo 2000; Sparks 1994), is cholesterol dependent. Cell biology investigations indicated that specialised cellular membrane microdomains rich in cholesterol and sphingolipids, termed lipid rafts, might be the link between cholesterol and amyloidogenic processing of APP. Both beta and gamma secretases are active in lipid rafts and it appears that APP processing within these lipid rafts by secretases determines the levels of Aβ production (Ehehalt 2003; Vetrivel 2004).
The ApoE ϵ4 allele is associated with sporadic AD. Meta‐analysis has shown that the ApoE ϵ4 allele increases the risk of the disease by three times in heterozygotes and by 15 times in homozygotes (Farrer 1997). It acts mainly by modifying age of onset, with each copy of the allele lowering the age at onset by almost 10 years (Corder 1993). This is significant in the context of cholesterol metabolism as ApoE acts as a cholesterol transporter in the brain. It binds directly to the Aβ peptide and influences its fibrillogenesis and clearance in vitro (Strittmatter 1993), and in vivo (Naslund 1995; Wisniewski 1995). ApoE is critically important for the formation of fibrillar Aβ in brain parenchyma in vivo (Holtzman 2000). Two genome‐wide association studies reported a significant association of AD with a locus within the clusterin (CLU) gene (Harold 2009; Lambert 2009). Functionally, clusterin has similarities to ApoE as both are major brain apolipoproteins and act as cholesterol transporters in the central nervous system. Both are also present in amyloid plaques and interact with Aβ, and regulating the conversion of Aβ into insoluble forms, co‐operating to suppress Aβ deposition and modifying Aβ clearance at the blood‐brain barrier (BBB) (van Es 2009). The amyloid cascade hypothesis states that an imbalance between production of and clearance of Aβ in the brain is the initiating event in the pathogenesis of AD, ultimately leading to neuronal degeneration and dementia (Hardy 2002), and thus theoretically links cholesterol metabolism to the development of AD.
There is also emerging evidence that genetics may play a role in responsiveness to statins. See Sabbagh 2012 for a review of findings in which new polymorphisms have been discovered that influence statin responsiveness. These have not been explored in AD and may aid in a priori identifying those most likely to benefit from treatment.
Central and peripheral cholesterol pools are separate, however, and almost all cholesterol in the brain is synthesised locally and is not transferred into plasma because of the BBB (Dietschy 2001). How serum cholesterol affects brain cholesterol has been a major question to date. Brain cholesterol content does not seem to be affected by high serum low‐density lipoprotein cholesterol (LDL‐C) or low serum high‐density lipoprotein cholesterol (HDL‐C) levels, perhaps as a result of the stability of cholesterol in myelin; however, it has not been established whether intramembranous lipid domains or intracellular cholesterol content are affected (Lane 2005). Studies in animal models have shown that diet‐induced hypercholesterolaemia increases Aβ and ApoE concentrations in the temporal and frontal cortices, but not in the cerebellum, and that these regional increases parallel the amyloid pathology observed in the AD brain (Wu 2003). Side chain‐oxidised cholesterol metabolites, such as hydroxy‐cholesterols, do cross the BBB. In the steady state in the adult brain, cholesterol clearance is facilitated by the formation and excretion of 24‐hydroxycholesterol (Lutjohann 2000). This is the major pathway for efflux of brain cholesterol and is crucial for maintenance of brain cholesterol homeostasis (Reiss 2004). 27‐Hydroxycholesterol is also found in the brain and may also provide a link between hypercholesterolaemia and AD (Heverin 2005); the contribution of the 27‐hydroxylase pathway to AD is an area in need of further exploration.
Several epidemiological studies have shown an association between high serum cholesterol levels and an increased susceptibility to AD (Jarvik 1995; Kivipelto 2002; Notkola 1998). The Notkola study was a long‐term prospective study that found elevated total serum cholesterol level was a risk factor for AD, independent of the ApoE ϵ4 allele; however, the association between AD and the ApoE ϵ4 allele became weaker after adjustment for serum total cholesterol (Notkola 1998). The authors concluded that some of the effect of the ApoE ϵ4 allele on the risk of AD might be mediated through elevated levels of total serum cholesterol. The Kivipelto study was again a prospective population‐based study that showed elevated mid‐life total cholesterol level was a risk factor for AD and was independent from risk from the ApoE ϵ4 allele and high mid‐life systolic blood pressure (Kivipelto 2002). The Jarvik study was a case‐control study, which again showed a positive association between serum cholesterol and risk of AD (Jarvik 1995). More recently, the Norwegian Counties Study, a long‐term observational study, showed dementia death was associated with increased total cholesterol levels in mid‐life (Strand 2013).
There may also be converging pathogenic mechanisms between cerebrovascular and Aβ plaque pathology ‐ cerebrovascular pathology with ischaemia resulting in upregulation of APP expression followed by Aβ deposition (Jendroska 1995). However, co‐existing pathology may occur independently of the disease process and increase the probability of exhibiting dementia in otherwise asymptomatic people (Riekse 2004).
Cholesterol and vascular dementia
VaD is the second most common form of dementia. It is characterised by both large and small vessel lesions. Subcortical ischaemic vascular disease caused by damage to tiny blood vessels that lie deep in the brain is now thought to be more prevalent than multi‐infarct dementia caused by large vessel lesions and stroke (Ballard 2000; Esiri 1997).
Sclerosis of small cerebral arteries and arterioles is considered to be responsible for the diffuse periventricular white matter abnormalities involved in the pathogenesis of subcortical VaD (Ryglewicz 2002). Risk factors for VaD are similar to risk factors for all types of vascular disease, namely hypertension, diabetes, smoking and hypercholesterolaemia (Ott 1998; Posner 2002; Stewart 1999). These factors are also important in the pathogenesis of AD (Decarli 2004); furthermore, the effects of vascular and AD pathologies are additive and in most population samples these disorders appear together (Snowdon 1997).
Plasma lipids could be associated with the risk of VaD through several mechanisms. High levels of LDL‐C and low levels of HDL‐C are established risk factors for coronary heart disease (Moroney 1999) and carotid artery atherosclerosis (Sharrett 1994). These may lead to cognitive impairment through cerebral hypoperfusion or embolism (Breteler 1994). LDL‐C may interact with ApoE to cause small vessel disease, and low levels of antioxidants known to occur in brains of people with VaD may lead to a higher susceptibility to oxidative stress and a higher grade of LDL‐C oxidation (Dantoine 2002; Paragh 2002).
One previous cross‐sectional analysis showed that the prevalence of VaD decreased with higher levels of HDL‐C and increased with higher levels of non‐HDL‐C. However, treatment with LLAs was not associated with the risk of prevalent VaD. Incidence of VaD was also calculated, again risk of VaD increased with increasing non‐HDL level but treatment with LLAs did not lower the risk of incident VaD (Reitz 2004). Other studies have found an association of VaD with decreased levels of HDL‐C (Kuriyama 1994; Muckle 1985; Zuliani 2001). The role of LDL‐C remains controversial, with some studies finding an association between increased LDL‐C and risk of VaD (Klich‐Raczka 2002; Moroney 1999; Paragh 2002), and other studies reporting a negative association (van Exel 2002; Yoshitake 1995).
Stroke is also a major risk factor for VaD. Debate continues as to whether increased cholesterol levels are a risk factor for stroke. Clinical trials indicated that statins significantly decreased stroke risk in vascular patients including patients with stroke (CTTC 2005; SPARCL 2006). The meta‐analysis carried out by the Cholesterol Treatment Trialists' Collaborators including 90,056 participants found that the use of statins caused a significant 17% proportional reduction in the incidence of first‐ever stroke of any type per 1 mmol/L LDL‐C reduction (CTTC 2005). In the secondary prevention of stroke, the Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) study showed that treatment with atorvastatin reduced the risk of recurrent cerebrovascular events in people with recent stroke or transient ischaemic attack but no history of heart disease (SPARCL 2006). By reducing the risk of stroke, statins may also act to reduce the incidence of post‐stroke dementia.
Statins
Statins are a class of drugs that inhibit 3‐hydroxy‐3‐methylglutaryl coenzyme A (HMG‐CoA) reductase. HMG‐CoA reductase is the rate‐limiting enzyme in the cascade of cellular cholesterol biosynthesis. Statins thereby reduce the formation and entry of LDL‐C into the circulation and upregulate LDL receptor activity, lowering LDL‐C and triglycerides and increasing HDL‐C. Several studies in cell culture and animals have demonstrated that treatment with cholesterol‐lowering drugs reduces the production of Aβ (Fassbender 2001; Refolo 2001; Simons 1998). It was therefore hypothesised that reduction of Aβ levels by statins may have neuroprotective effects in patients with AD (Simons 2001; Wolozin 2001). However, one study of transgenic mice found that levels of Aβ in the brains of simvastatin‐treated mice did not differ from those of untreated mice. Simvastatin treatment did lead to the reversal of learning and memory deficits and the authors hypothesised that the benefit of simvastatin may have been due to modulation of signalling pathways in memory formation (Li 2006). However, further work then demonstrated an association between antecedent statin use and neurofibrillary tangle burden at autopsy with risk for typical AD pathology reduced in statin users (Li 2007). The effects of statins on AD neuropathology are therefore not totally understood. Their possible role in the treatment of VaD includes secondary prevention of stroke and other pleiotropic effects as detailed below.
Statins are classified according to their solubility in lipids (lipophilic) or water (hydrophilic). Lipophilic statins (lovastatin, simvastatin, atorvastatin, fluvastatin) cross the BBB and penetrate cell membranes more effectively than hydrophilic statins and may be more efficient theoretically in the treatment of dementia than the hydrophilic statins (pravastatin). However, in contrast, reducing cholesterol synthesis below a critical level can induce neuronal death (Michikawa 1998), and may paradoxically make treatment with hydrophilic statins more appropriate (Sparks 2006).
Statins also have pleiotropic effects. They can improve the endothelial function of atherosclerotic vessels by decreasing endothelial 1 and angiotensin II type 1 receptors and increasing nitric oxide (Wassmann 2001). Low nitric oxide levels lead to impaired endothelial function, platelet aggregation and enhanced leukocyte adhesion to the endothelium. Statins also have antithrombotic effects as they decrease plasminogen activator levels and have anti‐inflammatory effects as they decrease adhesion molecules (Reitz 2004). They may also have the ability to reduce apoptosis and cellular death (Ruocco 2002). Many of these cholesterol‐independent effects reflect the ability of statins to block the synthesis of important isoprenoid intermediates, which serve as lipid attachments for a variety of intracellular signalling molecules (Liao 2002).
It is also possible that reduced cholesterol synthesis and concentration in the CNS caused by treatment with statins may cause neurocognitive deficits. Several investigators have therefore questioned the potential detrimental effects of lowering cholesterol on cognition (King 2003; Muldoon 2000; Wagstaff 2003; Zhang 2004). It has been shown that large doses of statins can produce substantial neurotoxicity in dogs (Berry 1988; Walsh 1996). Statins lower circulating levels of vitamin E and ubiquinone (Coenzyme Q10) and may affect the synthesis of polyunsaturated fatty acids that are integral to neuronal membranes (Palomaki 1997; Rise 1997). Researchers have speculated that low concentrations of one or more components of lipoprotein particles circulating in the bloodstream may produce subtle but measurable impairments of mental processes by influencing the supply of fat‐soluble micronutrients, specifically, vitamin E, β‐carotene and vitamin A (Muldoon 1997). However, on balance, potential benefits from statins appear to outweigh potential detrimental effects and we will assess adverse effects from statins in this review.
Statins are widely available and prescribed for treatment of dyslipidaemia and secondary prevention of cardiovascular and cerebrovascular disease. Their cost is relatively low and some have come off patent so are prescribed generically.
Statin treatment in dementia
The use of statin therapy in established AD or VaD is a relatively unexplored area. There have been a number of studies on the role of statins in the prevention of dementia but these are the focus of another Cochrane review (McGuinness 2009). Further trials have followed patients with AD and dementia; these are the focus of this review and are presented in the results section.
One post‐hoc analysis on data pooled from three double‐blind placebo‐controlled clinical trials of galantamine in AD showed no significant change in cognitive status in association with the use of statins (Winblad 2007).
One observational study in patients with AD followed for 34.8 months showed that patients treated with LLAs had a slower decline on the Mini Mental State Examination (MMSE) than patients with untreated dyslipidaemia or normolipidaemic patients. The study concluded that LLAs (including fibrates and statins) may slow cognitive decline in patients with AD and may have a neuroprotective effect, but this finding needs to be confirmed by randomised placebo‐controlled trials (Masse 2005).
Patients with mild cognitive impairment (MCI) at baseline in the observational Ginkgo Evaluation of Memory Study showed no significant cognitive benefit from treatment with LLAs including statins. This was in contrast to people without MCI at baseline who showed a reduced risk of all‐cause dementia and AD (Betterman 2012).
Other LLAs (fibrates, niacin/nicotinic acid, anion‐exchange resins) have been assessed with statins in several dementia studies. Fibrates are the main class in use other than statins. Rockwood et al. published a population‐based survey from the Canadian Study of Health and Aging (CSHA) demonstrating use of statins and other LLAs on reduced risk of AD in people younger than 80 years old (Rockwood 2002). In contrast, the UK General Practice Research Database study showed that only statins reduced the risk of dementia, other LLAs did not (Jick 2000). In a further study in patients with AD, all LLAs including statins were associated with a slower annual cognitive decline but there was no significant difference between statins and other LLAs and there was lack of statistical power to compare statins to fibrates (Masse 2005).
Fibrates do not inhibit cholesterol biosynthesis, they stimulate β‐oxidation of fatty acids and act mainly by decreasing serum triglycerides. They are only used first line in people with hypertriglyceridaemia and can be used in combination with statins in people not responding to single therapy. Fibrates also have anti‐inflammatory effects as they inhibit the production of different pro‐inflammatory molecules (Pahan 2006). However, in this review, we were primarily interested in role of statins in the treatment of dementia.
This review aimed to collate the best available evidence regarding use of statins in AD and VaD.
Statins decrease coronary events in the primary and secondary prevention of coronary heart disease significantly. The question is whether they have a significant therapeutic effect in dementia. Any intervention shown to slow the progression of dementia would have huge worldwide economic benefit.
Objectives
Primary objective
To evaluate the efficacy and safety of statins in the treatment of AD and VaD.
Secondary objective
To evaluate if the efficacy of statins in the treatment of AD and VaD depends on cholesterol level, ApoE genotype or cognitive level.
Methods
Criteria for considering studies for this review
Types of studies
Randomised, double‐blind, placebo‐controlled trials in which a statin was given for at least six months. We chose six months as we considered this to be the minimum length of time required to be on treatment to allow a disease‐modifying effect and before any cognitive benefit could be attained.
We excluded trials comparing two different statins without a placebo.
Types of participants
Patients with a diagnosis of probable or possible AD according to National Institute of Neurological and Communicative Disorders and Stroke ‐ the Alzheimer's Disease and Related Disorders Association (NINCDS‐ADRDA) criteria or acceptable equivalent.
Patients with a diagnosis of probable or possible VaD according to National Institute of Neurological Disorders and Stroke/Association International pour le Recherché at l'Enseignement en Neurosciences (NINDS/AIREN) criteria or acceptable equivalent.
We included trials with Diagnostic and Statistical Manual (DSM) III, IIIR or IV dementia, but analysed these separately from those with causal diagnoses for dementia.
Types of interventions
Any type of statin (hydrophilic and lipophilic) given in appropriate dose compared with placebo.
Types of outcome measures
Primary outcomes
Change in MMSE, Alzheimer's Disease Assessment Scale ‐ cognitive subscale (ADAS‐cog) or other accepted cognitive measure.
Secondary outcomes
Incidence and severity of adverse effects.
Change in cognitive status accounting for prior cholesterol level, ApoE genotype and cognitive level.
Patient perceived quality of life.
Change in activities of daily living (ADL).
Change in behaviour.
Search methods for identification of studies
We searched ALOIS (www.medicine.ox.ac.uk/alois) ‐ the Cochrane Dementia and Cognitive Improvement Group's Specialized Register on 20 January 2014. We used the search terms: statin OR statins OR "Hydroxymethylglutaryl‐CoA Reductase Inhibitors".
ALOIS is maintained by the Trials Search Co‐ordinator of the Cochrane Dementia and Cognitive Improvement Group and contains studies in the areas of dementia prevention, dementia treatment and cognitive enhancement in healthy people. The studies are identified from:
monthly searches of a number of major healthcare databases: MEDLINE, EMBASE, CINAHL, PsycINFO and LILACS;
monthly searches of a number of trial registers: ISRCTN; UMIN (Japan's Trial Register); the World Health Organization (WHO) portal (which covers ClinicalTrials.gov; ISRCTN; the Chinese Clinical Trials Register; the German Clinical Trials Register; the Iranian Registry of Clinical Trials and the Netherlands National Trials Register, plus others);
quarterly search of the Cochrane Central Register of Controlled Trials (CENTRAL);
six‐monthly searches of several grey literature sources: ISI Web of Knowledge Conference Proceedings; Index to Theses; Australasian Digital Theses.
To view a list of all sources searched for ALOIS see About ALOIS on the ALOIS website.
Details of the search strategies used for the retrieval of reports of trials from the healthcare databases, CENTRAL and conference proceedings can be viewed in the 'methods used in reviews' section within the editorial information about the Dementia and Cognitive Improvement Group.
We performed additional searches in many of the sources listed above to cover the timeframe from the last searches performed for ALOIS to ensure that the search for the review was as up‐to‐date and as comprehensive as possible. The search strategies used can be seen in Appendix 1.
Searches carried out in the previous version(s) of the review can be viewed in Appendix 2.
Data collection and analysis
Selection of studies
Two authors undertook the search and screening of publications (BMcG, supported by PP). Both authors agreed on and tested the MeSH terms and search strategy. Three other authors (DC, RM and RB) acted as adjudicators and reviewed the process. Authors independently selected trials for relevance against the defined inclusion criteria.
Data extraction and management
We extracted data from the published reports using a data collection form. One author (BMcG) extracted data and was by supported by a second author (RM).
Assessment of risk of bias in included studies
We assessed methodological quality of the included trials using The Cochrane Collaboration's tool for assessing risk of bias. We assessed the following domains: selection bias, performance bias, detection bias, attrition bias and reporting bias. We included trials with low risk of bias. We planned sensitivity analyses to determine inclusion/exclusion of low‐quality studies.
Measures of treatment effect
Continuous data
We extracted the mean change from baseline, the standard error of the mean change and the number of patients for each treatment group at each assessment for continuous data. Where changes from baseline were not reported, we extracted the mean, standard deviation (SD) and the number of patients for each treatment group at each time point. We also extracted available data on demographics of patients (age, gender, lipid values at baseline), statin regimen (type of statin, daily dosage, starting time, duration) and follow‐up duration. We reported 95% confidence intervals (CI).
Dichotomous data
For binary data (adverse events or not), we sought the numbers in each treatment group and the numbers experiencing the outcome of interest. We reported results as an odds ratio (OR) with 95% CI.
Dealing with missing data
For each outcome measure, we sought data on every patient assessed. To allow an intention‐to‐treat analysis, we sought the data irrespective of compliance, whether or not the patient was subsequently deemed ineligible, or otherwise excluded from treatment or follow‐up. If intention‐to‐treat data were not available in the publications, we sought "on‐treatment" or the data of those who completed the trial and indicated as such. We did not use data from titration phases prior to the randomised phase to assess safety or efficacy because patients are usually not randomised and treatments are not usually concealed.
Assessment of heterogeneity
In all cases, we presented the overall estimate from a fixed‐effect model and performed a standard Chi2 test to check for heterogeneity. We also assessed the impact of heterogeneity on the meta‐analysis using the I2 statistic. If we found significant heterogeneity, we presented a random‐effects model and reported this as the main result.
Sensitivity analysis
If heterogeneity still existed with any model, then we carried out a sensitivity analysis (excluding studies with conflicting results from the rest) thereby assessing the robustness of the results of fixed‐effect versus random‐effects models.
Results
Description of studies
Results of the search
The initial electronic searches retrieved 152 references. We included three studies in the initial meta‐analysis (ADCLT 2005;LEADe 2010;Simons 2002) (Figure 1).
1.
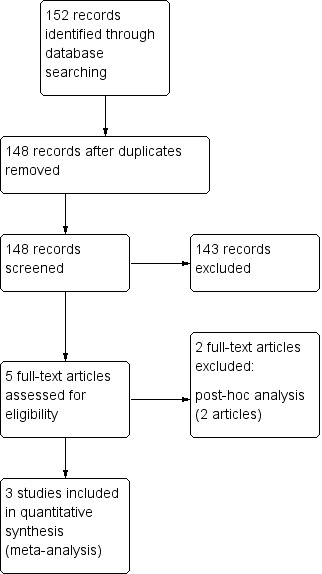
Study flow diagram initial review.
We performed two top‐up searches for this update: one in February 2013 (which yielded 105 references) and one in January 2014 (which yielded 27 references). We included one new study from the search in 2013 (Sano 2011, 406 participants) (Figure 2). We included no studies from the 2014 search.
2.
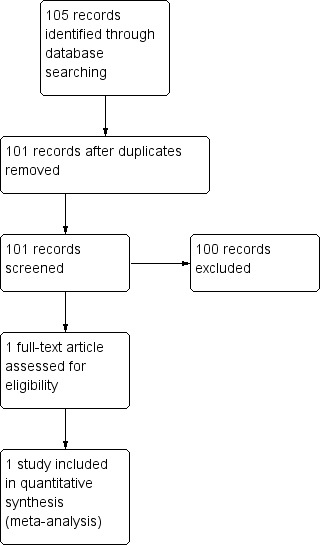
Study flow diagram update 2013 search.
Included studies
The search identified four RCTs with 1154 participants (ADCLT 2005;LEADe 2010;Sano 2011; Simons 2002). For full details, see the Characteristics of included studies table. Ages for participation ranged from 50 to 90 years with mean ages in studies of 68 to 78 years representing older adults with dementia.
ADCLT 2005 included 63 patients with a diagnosis of probable or possible AD as outlined by NINCDS‐ADRDA and Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM‐IV) criteria; people 51 years or older with mild‐to‐moderate impairment (MMSE score 12 to 28) were eligible. All but six individuals were taking cholinesterase inhibitors, three people in the atorvastatin group and three people in the placebo group. The mean age (± SD) was 78.9 ± 1.2 years in placebo group and 78.15 ± 1.3 years in atorvastatin group.
LEADe 2010 included 614 patients with a diagnosis of probable AD according to DSM‐IV and NINCDS‐ADRDA criteria and of mild‐to‐moderate severity, defined as a MMSE score of 13 to 25 at screening. Participants were 53% female, age range 50 to 90 years, mean age (± SD) 74 ± 8 years. Patients were receiving donepezil 10 mg for at least three months before randomisation and LDL‐C was 2.5 to 3.5 mmol/L for inclusion.
Sano 2011 included 406 patients with a diagnosis of mild‐to‐moderate AD according to NINCDS‐ADRDA criteria. Patients were older than 50 years, with an MMSE score of 12 to 26. Participants were 59.9% female, mean age (± SD) 75.1 ± 9.0 years in the placebo group and 58.8% female, mean age 74.0 ± 9.6 years in the intervention group. Stable use (for at least three months) of cholinesterase inhibitors and memantine was allowed. Patients were excluded if they were taking LLAs or if they had conditions requiring cholesterol‐lowering treatment according to guidelines at the time. Participants were also excluded if they had LDL‐C below 80 mg/dL (2.1 mmol/L) or triglycerides greater than 500 mg/dL (5.6 mmol/L).
Simons 2002 was primarily a study investigating whether statins alter cholesterol metabolites and reduce Aβ levels in the cerebrospinal fluid (CSF) of AD patients. Cognition was assessed as a secondary outcome. Forty‐four patients with probable AD as defined by NINCDS‐ADRDA criteria and mild‐to‐moderate severity (MMSE scores 12 to 26) were recruited. Patients were allowed to take donepezil or rivastigmine if the dose had been unchanged for three months prior to study entry and remained stable during the 26‐week study period. Mean age (± SD) was 68.5 ± 8 years in the placebo group and 68.0 ± 9 years in the simvastatin group. Participants were recruited primarily from the community.
ADCLT 2005 provided data on change in ADAS‐Cog at three‐monthly intervals for up to one year. Data were also provided on change in total cholesterol level, Clinical Global Impression of Change (CGIC) score, MMSE score, Neuropsychiatric Inventory (NPI) total score and Geriatric Depression Scale (GDS) total score between placebo and atorvastatin groups.
LEADe 2010 provided data on change in ADAS‐Cog and Alzheimer's Disease Cooperative Study‐Clinical Global Impression of Change (ADCS‐CGIC) scores between atorvastatin and placebo groups. Following randomisation, these measures were performed at three‐month intervals through month 18. Secondary outcome measures were change in NPI, Alzheimer's Disease Functional Assessment and Change Scale (ADFACS), Clinical Dementia Rating‐Sum of Boxes (CDR‐SB), MMSE and modified ADAS‐Cog. Change in total, LDL‐C and HDL‐C, and triglycerides was provided.
Sano 2011 provided data on change in ADAS‐Cog score between simvastatin and placebo groups at three, six, 12 and 18 months from baseline. Secondary outcome measures included change in ADCS‐CGIC, MMSE, dependence scale, ADCS‐ADL, NPI and three supplemental cognitive tests from the ADCS instrument protocol.
Simons 2002 provided data on change in MMSE and ADAS‐Cog score between simvastatin and placebo groups at 26 weeks.
Treatment in ADCLT 2005 consisted of atorvastatin 80 mg daily or matching placebo. Sixty‐three individuals were considered evaluable by completing the three‐month visit, 32 people received atorvastatin and 31 people received placebo. Forty‐six people completed the one‐year study, 25 received atorvastatin and 21 received placebo. Reasons for drop‐out were not provided. Atorvastatin treatment produced significant decreases in total cholesterol (40%), LDL‐C (54%), and very‐low‐density‐cholesterol (VLDL‐C) (30%) relative to placebo. ApoE genotyping was carried out on study participants. Sixty per cent of the placebo group and 62.5% of the atorvastatin group had one or more E4 allele. Change in performance among participants with screening cholesterol levels 200 mg/dL (5.2 mmol/L) or greater was compared with performance change in participants with levels less than 200 mg/dL (5.2 mmol/L). Mean change in ADAS‐Cog performance at six months was established for atorvastatin‐ and placebo‐treated participants grouped according to their ApoE genotype. Within‐group and between‐group comparisons according to presence of apolipoprotein E4 allele were performed, followed by comparisons based on dose of the apolipoprotein E4 allele.
Treatment in LEADe 2010 consisted of atorvastatin 80 mg daily or matching placebo for 72 weeks. A total of 640 patients were randomised with a modified intention‐to‐treat population of 297 in the atorvastatin group and 317 in the placebo group. Mean (± SD) prior donepezil treatment was 409 ± 407 days. Results concerning ApoE genotype were available for 511 patients, observed ApoE4 frequency was 60%. Atorvastatin treatment produced significant decreases in total cholesterol, LDL‐C (50.2%) and triglycerides but no significant change in HDL‐C. Reasons for drop‐outs were given.
Treatment in Sano 2011 consisted of simvastatin 20 mg for six weeks and 40 mg daily thereafter for the remainder of the 18‐month study. A total of 406 participants were randomised with 204 in the treatment group and 202 in the placebo group. Discontinuation reasons were given. Total cholesterol and LDL‐C levels were significantly reduced by the treatment compared with placebo; the reduction was 23% in total cholesterol and 37% in LDL‐C. HDL‐C levels were also increased with treatment by 2%. There was no difference in the use of approved anti‐dementia medications between the treatment groups during the study. Presence of the ApoE4 allele was not different in the two groups.
In Simons 2002, treatment consisted of simvastatin 40 mg daily for four weeks and 80 mg daily for the following 22 weeks. Disease duration was a mean (± SD) 2.8 ± 1.3 years in the placebo group and 2.6 ± 1.4 years in the simvastatin group. Serum LDL‐C showed few changes in the placebo group but was reduced by 52% on average in the simvastatin group. Reasons for drop‐outs were given. ApoE genotyping was not carried out.
Total adverse events were reported by LEADe 2010, Sano 2011, and Simons 2002.
Excluded studies
We excluded two studies from the analysis (Gutterman 2002;Winblad 2007). For full details, see Characteristics of excluded studies table.
Gutterman 2002 used data pooled from trials of patients treated with galantamine 24 mg daily or placebo for five to six months in randomised, double‐blind, placebo‐controlled trials. This was a post‐hoc analysis and so did not fulfil criteria for inclusion neither did it have adequate power to examine the effects of statins.
Winblad 2007 was also a post‐hoc analysis conducted on data pooled from three double‐blind, placebo‐controlled clinical trials of galantamine in patients with AD. There were four treatment groups: statin plus galantamine (42 participants), statin alone (50 participants), galantamine alone (614 participants) and neither galantamine nor statin (619 participants). As stated by the authors, it did not have sufficient power to examine the effects of statins.
Risk of bias in included studies
For full details, see 'Risk of bias' tables and Figure 3; Figure 4.
3.
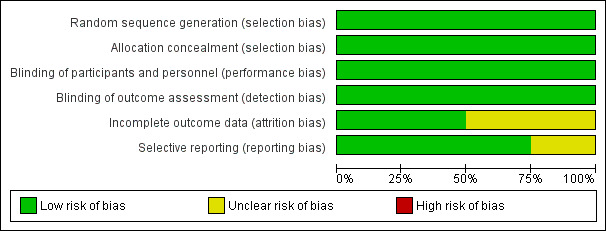
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
4.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
In ADCLT 2005, randomisation was performed in blocks of 10 using the Excel spreadsheet random‐number generator, this appeared adequate.
In LEADe 2010, 1:1 randomisation was carried out, the medication was assembled for each patient based on a randomisation code prepared by Clinical Data Operations of Pfizer Inc. This appeared satisfactory.
In Sano 2011, the randomisation sequence was generated with equal probability of assignment to drug and placebo using a random permuted block treatment assignment stratified by site. The randomisation sequence was generated by the ADCS, a consortium of US centres funded by the National Institute on Aging. This appeared satisfactory.
In Simons 2002, a randomisation list was computer generated, two copies were prepared: one was used by the packaging department of the study medication or placebo and the other was kept in a locked location until the study was completed. This appeared satisfactory.
Blinding
In ADCLT 2005, all investigators were blinded to both treatment group and cholesterol profiles after randomisation as active treatment was expected to reduce circulating cholesterol levels. Medications were supplied in bulk by the pharmaceutical company and were coded at the pharmacy. This appeared adequate.
In LEADe 2010, it is stated in the published article there was "blinding of both the investigator and the subject" and in conference proceedings "trial data remained blinded, and the authors, steering committee, and the sponsor had no information relating to study outcomes". This appeared adequate.
In Sano 2011, it is stated "scratch‐off" code‐breakers were used so that instances of unblinding would be documented; all code breakers were collected at the end of the trial. Adequacy of the blind was assessed by questionnaires completed by participants, carers, psychometrists and site investigators.
In Simons 2002, adequate blinding appears to have been carried out. "All personnel directly involved in the conduct of the study remained unaware of the treatment groups until all patients had completed the trial and all data had been retrieved". Blood results were monitored by a physician not involved in the study, this guaranteed that all investigators were kept blinded.
Incomplete outcome data
In ADCLT 2005, flow of participants through the one‐year‐long study was provided, from 63 evaluable participants, 46 attended for final assessment. Reasons for not attending were not given.
In LEADe 2010 and Sano 2011, incomplete data were addressed.
In Simons 2002, incomplete data were addressed comprehensively.
Selective reporting
There was no evidence of selective reporting.
Other potential sources of bias
We identified no other potential sources of bias.
Effects of interventions
See: Table 1
Primary outcomes
The four studies assessed change in ADAS‐Cog from baseline. We calculated the mean change and SD from the available data and entered them into a meta‐analysis.
When we combined the four studies, there was no significant difference in ADAS‐Cog between the statin group and placebo group (mean difference ‐0.26, 95% CI ‐1.05 to 0.52, P value = 0.51) (Analysis 1.1) (Figure 5). There was also considerable heterogeneity when the studies were combined (Chi2 = 7.98, degrees of freedom (df) = 3, P value = 0.05, I2 = 62%). The random‐effects model, which usually gives more weight to small studies, was therefore used to re‐pool the data; the combined results were not significant (mean difference ‐0.33, 95% CI ‐2.10 to 1.44, P value = 0.71) (Analysis 1.2) (Figure 6). We carried out a sensitivity analysis and removal of ADCLT 2005 (the study with conflicting results) resulted in a reduction of I2 to 41% with the fixed‐effect model. Again, however, there was no significant difference in ADAS‐Cog between the statin and placebo groups (P value = 0.85). As the Simons 2002 study ran for 26 weeks, we combined data from ADCLT 2005 at 24 weeks and from LEADe 2010 and Sano 2011 at 26 weeks and again there was no significant difference in ADAS‐Cog between the statin and placebo groups (mean difference ‐0.08, 95% CI ‐0.85 to 0.70). Change in ADAS‐Cog from LEADe 2010 and Sano 2011 has been provided in the tables from the various time points (Analysis 1.3; Analysis 1.4; Analysis 1.5; Analysis 1.6; Analysis 1.7). Change in modified ADAS‐Cog (13‐item, 85‐point scale, Mohs 1997) has been provided from LEADe 2010. At no time was a beneficial effect on ADAS‐Cog or modified ADAS‐Cog seen with statin treatment (Analysis 1.8).
1.1. Analysis.
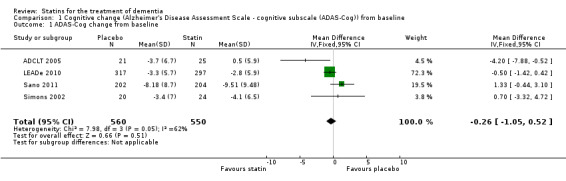
Comparison 1 Cognitive change (Alzheimer's Disease Assessment Scale ‐ cognitive subscale (ADAS‐Cog)) from baseline, Outcome 1 ADAS‐Cog change from baseline.
5.
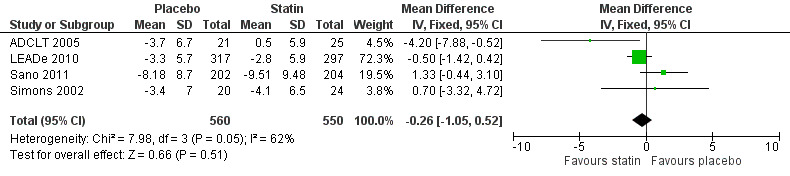
Forest plot of comparison: 1 Cognitive change (Alzheimer's Disease Assessment Scale ‐ cognitive subscale (ADAS‐Cog)) from baseline, outcome: 1.1 ADAS‐Cog change from baseline.
1.2. Analysis.
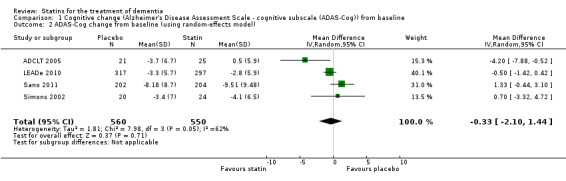
Comparison 1 Cognitive change (Alzheimer's Disease Assessment Scale ‐ cognitive subscale (ADAS‐Cog)) from baseline, Outcome 2 ADAS‐Cog change from baseline (using random‐effects model).
6.
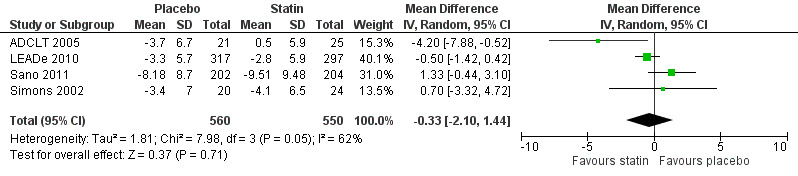
Forest plot of comparison: 1 Cognitive change (Alzheimer's Disease Assessment Scale ‐ cognitive subscale (ADAS‐Cog)) from baseline, outcome: 1.2 ADAS‐Cog change from baseline (using random‐effects model).
1.3. Analysis.
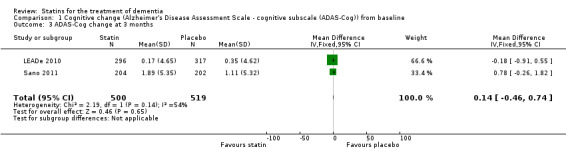
Comparison 1 Cognitive change (Alzheimer's Disease Assessment Scale ‐ cognitive subscale (ADAS‐Cog)) from baseline, Outcome 3 ADAS‐Cog change at 3 months.
1.4. Analysis.
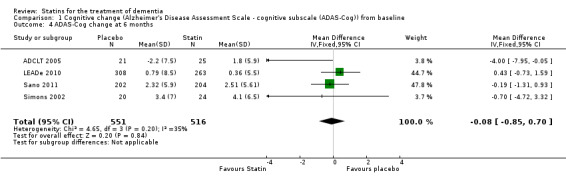
Comparison 1 Cognitive change (Alzheimer's Disease Assessment Scale ‐ cognitive subscale (ADAS‐Cog)) from baseline, Outcome 4 ADAS‐Cog change at 6 months.
1.5. Analysis.
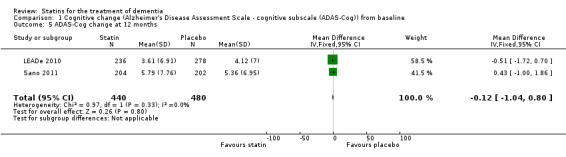
Comparison 1 Cognitive change (Alzheimer's Disease Assessment Scale ‐ cognitive subscale (ADAS‐Cog)) from baseline, Outcome 5 ADAS‐Cog change at 12 months.
1.6. Analysis.
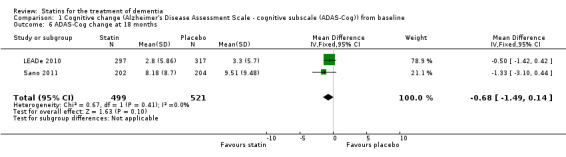
Comparison 1 Cognitive change (Alzheimer's Disease Assessment Scale ‐ cognitive subscale (ADAS‐Cog)) from baseline, Outcome 6 ADAS‐Cog change at 18 months.
1.7. Analysis.
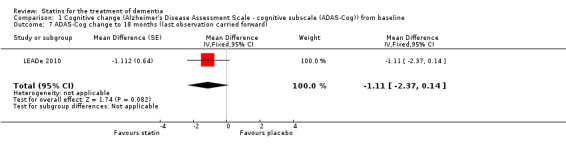
Comparison 1 Cognitive change (Alzheimer's Disease Assessment Scale ‐ cognitive subscale (ADAS‐Cog)) from baseline, Outcome 7 ADAS‐Cog change to 18 months (last observation carried forward).
1.8. Analysis.
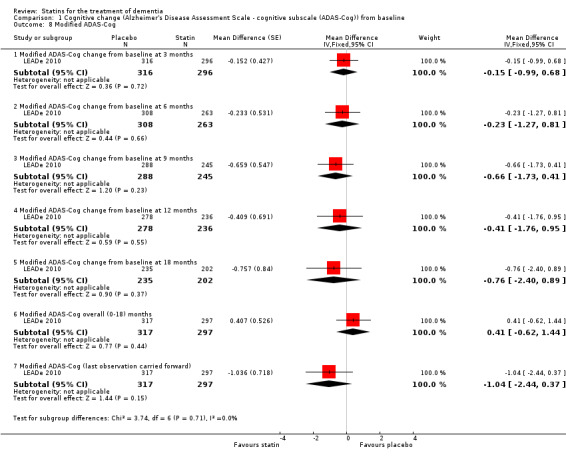
Comparison 1 Cognitive change (Alzheimer's Disease Assessment Scale ‐ cognitive subscale (ADAS‐Cog)) from baseline, Outcome 8 Modified ADAS‐Cog.
Change in MMSE was available from the four studies. When we combined data in a meta‐analysis there was no significant difference between the statin and placebo groups (mean difference ‐0.32, 95% CI ‐0.71 to 0.06, P value = 0.10) (Analysis 2.1) (Figure 7). Again there was significant heterogeneity when the studies were combined Chi2 = 15.46, df = 3, P value < 0.01, I2 = 81%) so we used the random‐effects model. There was no significant difference between statins and placebo (mean difference ‐0.88, 95% CI ‐2.04 to 0.28, P value = 0.09) (Analysis 2.2). A sensitivity analysis with Simons 2002 removed (the greatest outlier) reduced the heterogeneity (I2 statistic) to 67% with the fixed‐effect model with no effect on the outcome. Data were also compared at 24 weeks for ADCLT 2005, LEADe 2010, and Sano 2011; there was no significant difference between the statin and placebo groups (mean difference ‐0.30, 95% CI ‐0.66 to 0.05, P value = 0.09) (Analysis 2.4). Change in MMSE at different time points has been provided from LEADe 2010 and Sano 2011; at no time was there a beneficial effect from statin therapy.
2.1. Analysis.
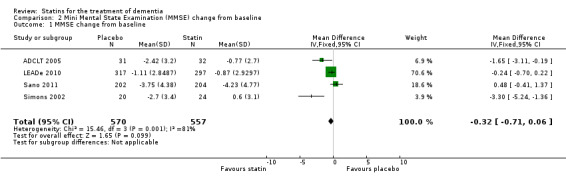
Comparison 2 Mini Mental State Examination (MMSE) change from baseline, Outcome 1 MMSE change from baseline.
7.
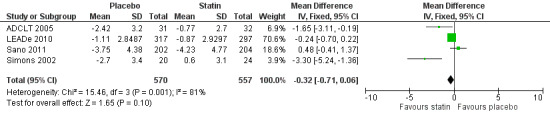
Forest plot of comparison: 2 Mini Mental State Examination (MMSE) change from baseline, outcome: 2.1 MMSE change from baseline.
2.2. Analysis.
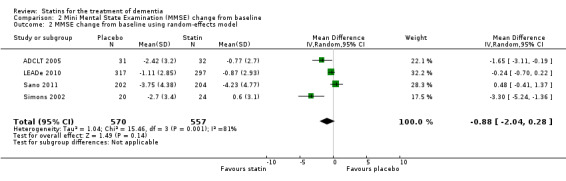
Comparison 2 Mini Mental State Examination (MMSE) change from baseline, Outcome 2 MMSE change from baseline using random‐effects model.
2.4. Analysis.
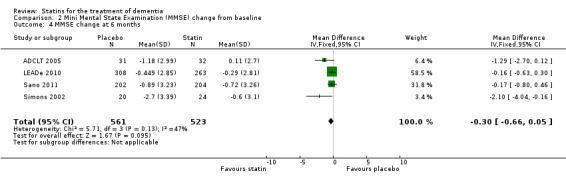
Comparison 2 Mini Mental State Examination (MMSE) change from baseline, Outcome 4 MMSE change at 6 months.
Change in ADCS‐CGIC assessing CGIC was given in two studies: ADCLT 2005 and LEADe 2010. When data from these two trials were combined in a meta‐analysis using generic inverse variance there was no significant difference between the statin and placebo groups (mean difference ‐0.02, 95% CI ‐0.14 to 0.10, P value = 0.74) (Analysis 3.1) (Figure 8). We contacted the study author for the Sano 2011 trial as ADCS‐CGIC was an outcome but we received no response.
3.1. Analysis.
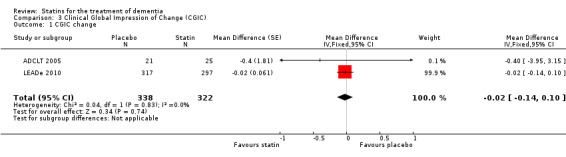
Comparison 3 Clinical Global Impression of Change (CGIC), Outcome 1 CGIC change.
8.
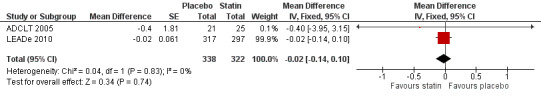
Forest plot of comparison: 3 Clinical Global Impression of Change (CGIC), outcome: 3.1 CGIC change.
Secondary outcomes
Side effects
Side effects were elicited from blood tests and from speaking to patients and carers. LEADe 2010 stated incidence of persistent elevated liver enzymes (three times upper limit of normal on two consecutive measures four to 10 days apart) in the atorvastatin group was low at 2.6% and 0% in the placebo group. There were 60 (19.1%) atorvastatin‐treated and 69 (21.2%) placebo‐treated patients who experienced serious adverse events (SAEs), six of which in the atorvastatin group and one in the placebo group were considered to be treatment related by the investigator or sponsor. The SAEs considered to be treatment related in the atorvastatin group were hepatitis, acute renal failure/rhabdomyolysis/pancreatitis, abdominal pain/nausea/chest discomfort, transaminases elevation, liver disorder and gastrointestinal haemorrhage. In Simons 2002, two patients in the simvastatin group experienced adverse events: one patient had muscle pain without elevation of creatine kinase and one patient was withdrawn because creatine kinase was elevated. No adverse effects were reported in the placebo group. In Sano 2011, placebo and treatment groups did not differ in number of participants with one or more adverse events (181/202 with placebo and 189/204 with treatment). The most commonly occurring adverse events were falls, agitation, asthenia and anxiety. There was no significant difference either between the groups in the number of participants with SAEs, number of participants with adverse events requiring hospitalisation and number of deaths. Liver enzyme elevations were noted in 2% of the treatment and 4% of the placebo groups. Data from LEADe 2010, Sano 2011, and Simons 2002 were combined and there was no significant difference between statin and placebo groups (Analysis 4.1) (Figure 9).
4.1. Analysis.
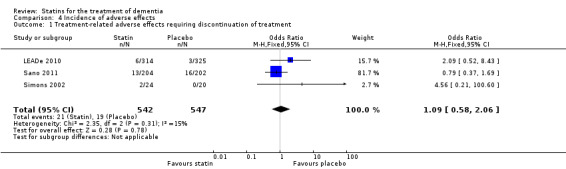
Comparison 4 Incidence of adverse effects, Outcome 1 Treatment‐related adverse effects requiring discontinuation of treatment.
9.
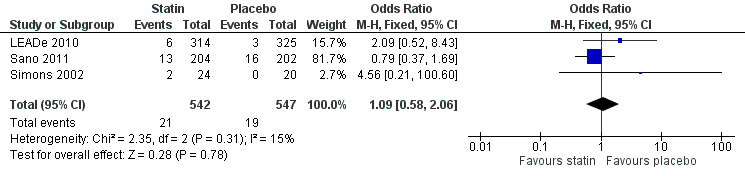
Forest plot of comparison: 4 Incidence of adverse effects, outcome: 4.1 Treatment‐related adverse effects requiring discontinuation of treatment.
Change in cognitive status accounting for prior cholesterol, ApoE genotype and cognitive level
In ADCLT 2005, among participants treated with atorvastatin, patients who had improved on the ADAS‐Cog at six months had baseline MMSE scores (mean ± SD, 21.93 ± 0.85) two points higher than patients who continued to deteriorate (19.83 ± 1.10) (P value < 0.06). Patients who improved on the ADAS‐Cog also had higher baseline cholesterol levels than patients who deteriorated (mean change (± SD) in ADAS‐Cog ‐2.14 ± 1.20 atorvastatin plus cholesterol greater than 200 mg/dL (5.2 mmol/L); 0.11 ± 0.68 atorvastatin plus cholesterol less than 200 mg/dL (5.2 mmol/L)). A significant difference was seen in ADAS‐Cog performance at six months between the atorvastatin and placebo groups in individuals with an apolipoprotein E‐4 allele (P value = 0.012) but not between the groups comprised of participants without an apolipoprotein E4 allele (P value = 0.967). (Note: there were very small numbers in all groups.)
In Sano 2011, presence of the APOE4 allele was not different in the two groups and was not associated with the rate of change in the ADAS‐Cog score. A median split was used to examine those of low and high baseline MMSE score; there was no difference in rate of ADAS‐Cog change between the simvastatin and placebo groups.
Quality of life
In LEADe 2010, there was no significant difference between the atorvastatin and placebo groups in Caregiver Burden Questionnaire and Patient Health Resources Utilization.
In Sano 2011, it is stated there was no significant difference between groups in quality of life as measured by informant or participant.
Behaviour
ADCLT 2005, LEADe 2010, and Sano 2011 provided data on NPI (information obtained from the carer). We combined data from these three studies at six (Analysis 5.1) and 12 months (Analysis 5.2) (Figure 10). There was a small but significant benefit from statins seen (mean difference at six months ‐0.84, 95% CI ‐1.64 to ‐0.05, P value = 0.04; mean difference at 12 months ‐1.11, 95% CI ‐2.10 to ‐0.12, P value = 0.03). This was no longer evident when we used the random‐effects model due to significant heterogeneity (mean difference at 12 months 1.64, 95% CI ‐3.45 to 0.18, P value = 0.11] (Analysis 5.3). In addition, if we removed ADCLT 2005 (the study with outlying results) from the analysis, the heterogeneity disappeared as did the significant treatment effect (I2 = 0%, P value = 0.13). ADCLT 2005 provided change in GDS (information obtained from the patient). Atorvastatin provided significant benefit on the GDS (P value < 0.04); there was deterioration in the placebo group and improvement in the atorvastatin group.
5.1. Analysis.
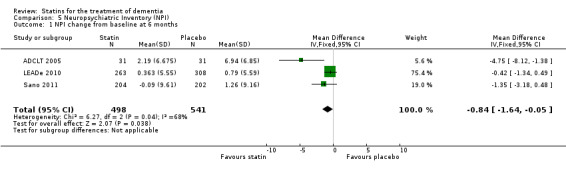
Comparison 5 Neuropsychiatric Inventory (NPI), Outcome 1 NPI change from baseline at 6 months.
5.2. Analysis.
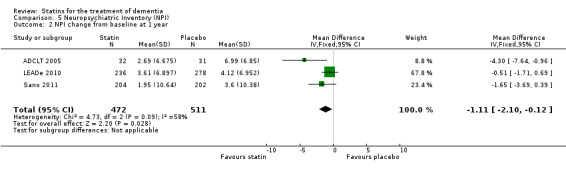
Comparison 5 Neuropsychiatric Inventory (NPI), Outcome 2 NPI change from baseline at 1 year.
10.
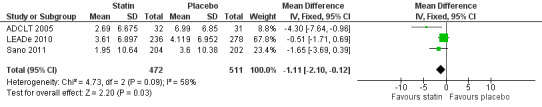
Forest plot of comparison: 5 Neuropsychiatric Inventory (NPI), outcome: 5.2 NPI change from baseline at 1 year.
5.3. Analysis.
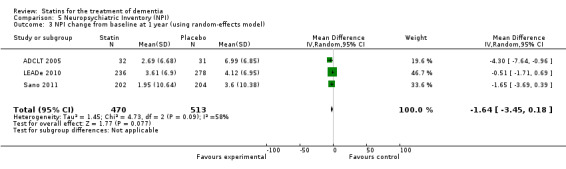
Comparison 5 Neuropsychiatric Inventory (NPI), Outcome 3 NPI change from baseline at 1 year (using random‐effects model).
Activities of daily living
ADCLT 2005 and Sano 2011 provided differences in performance on the carer‐rated Alzheimer's Disease Cooperative Study‐Activities of Daily Living (ADCS‐ADL) Inventory. In ADCLT 2005, the difference between the treatment and placebo groups did not reach significance (P value > 0.23) (no further data provided). Sano 2011 provided data on change from baseline ADCS‐ADL score. At no time was a difference seen between the placebo and treatment groups (Analysis 7.1). In LEADe 2010, there was no benefit from atorvastatin compared with placebo in the ADFACS, a measure of function (Analysis 6.4).
7.1. Analysis.
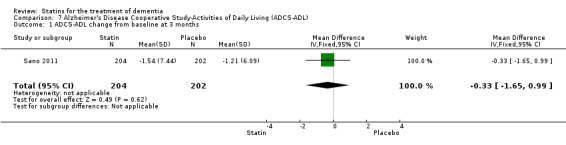
Comparison 7 Alzheimer's Disease Cooperative Study‐Activities of Daily Living (ADCS‐ADL), Outcome 1 ADCS‐ADL change from baseline at 3 months.
6.4. Analysis.
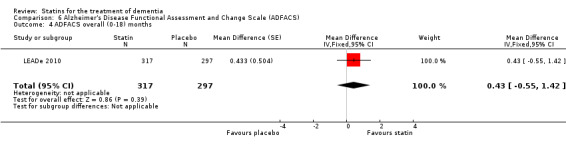
Comparison 6 Alzheimer's Disease Functional Assessment and Change Scale (ADFACS), Outcome 4 ADFACS overall (0‐18) months.
Discussion
Summary of main results
We identified four studies (ADCLT 2005; LEADe 2010; Sano 2011; Simons 2002). Mean change in ADAS‐Cog from baseline was an outcome in the four trials and there was no significant difference between the statin and the placebo groups.
Change in MMSE from baseline was also reported in all of the studies. There was no significant difference between the statin and placebo groups at any time point.
CGIC did not differ between the two groups in the two studies that recorded this measure (ADCLT 2005; LEADe 2010).
The statins were well tolerated and incidence of side effects was low. The statin group did not have a significantly higher rate of adverse effects requiring discontinuation of treatment when the data were combined.
There was some evidence from ADCLT 2005 that greater cognitive effect from atorvastatin was seen in patients with higher cholesterol at baseline, higher MMSE at baseline and in people with an apolipoprotein E4 allele present. The MMSE and ApoE effect was not seen in the larger Sano 2011 study.
There was no difference in ADL or quality of life between the two treatment groups. There was some evidence that statins provided a benefit in behaviour and mood.
The four trials included patients with AD only. We identified no trials assessing the effect of statins in the treatment of VaD.
It was not possible to assess if lipophilic statins or hydrophilic statins were more efficacious as all studies used the lipophilic statins simvastatin (Sano 2011; Simons 2002) or atorvastatin (ADCLT 2005; LEADe 2010). There appears to be a spectrum of lipophilicity within the lipophilic statins with simvastatin and cerivastatin being 2.5 to 4.0 times more lipophilic than lovastatin, fluvastatin and atorvastatin under the same physiological pH conditions (Joshi 1999). There is also debate about atorvastatin's lipophilicity and permeability across the BBB with some evidence that it crosses the BBB effectively (Kandiah 2009), and other evidence to suggest it does not (Knopp 1999). In any case, there was no convincing evidence that either atorvastatin or simvastatin were beneficial to cognition in the treatment of AD. There was no evidence that statins were detrimental to cognition.
One other comment relates to the fact that patients with elevated cholesterol levels were excluded from the large RCTs for safety and ethical reasons so the very group perhaps most likely to benefit was excluded (LEADe 2010; Sano 2011). This may be especially relevant given the ADCLT trial showed people with higher cholesterol level at baseline showed most cognitive benefit.
Overall completeness and applicability of evidence
The LEADe 2010 study was the largest with 640 patients so results from this are likely to be robust. Cognition was a primary outcome (ADAS‐Cog) along with global function (ADCS‐CGIC). Secondary outcomes included NPI, modified ADAS‐Cog, MMSE, CDR‐SB and ADFACS. Exploratory measures included a Caregiver Questionnaire and a Patient Health Resources Utilization questionnaire.
The Sano 2011 study was a large RCT with 406 participants. Cognition was a primary outcome (ADAS‐Cog) with ethnicity, baseline MMSE, ADCS‐ADL and LDL‐C meeting criteria for consideration as confounders in outcome analyses. Secondary outcomes included MMSE, Dependence Scale, ADCS‐ADL, NPI and CGIC (upon request from author). Findings from this study are considered complete and applicable.
In Simons 2002, the primary outcome was effect of statins on cholesterol metabolites and Aβ levels in the CSF of 44 patients with AD. Cognitive performance was a secondary outcome and was assessed at the beginning and end of the 26‐week study. Only 37 patients completed the study, so the impact of this study is likely to be small.
In ADCLT 2005, the primary outcomes were change in cognitive function (ADAS‐Cog) and clinical efficacy (CGIC). Secondary outcomes were change in MMSE, NPI, ADCS‐ADL and GDS, so results were applicable. However, the study was small with data available from 63 participants.
Quality of the evidence
All studies had adequate sequence generation and blinding.
Potential biases in the review process
None identified.
Agreements and disagreements with other studies or reviews
One previous systematic review assessed prevention and treatment of dementia or AD by statins (Zhou 2007). This was published before the LEADe 2010 results were available. Two studies were identified as identified in this review and there was no statistically significant difference in ADAS‐Cog between the statin and placebo groups when the trials were pooled (Simons 2002; ADCLT 2005). This is in agreement with this review.
One further review reported findings similar to this Cochrane review (Shepardson 2011). The review combined results from prevention and treatment of dementia with statins trials. Prevention of dementia with statins is the subject of another Cochrane review (McGuinness 2009). With respect to treatment trials, they identified ADCLT 2005 and LEADe 2010. They also reported negative findings for LEADe 2010 and positive findings for ADCLT 2005, but did not perform a meta‐analysis. The authors recommended future human studies take into account BBB permeabilities of the statins when analysing results, include specific analyses of the effects on LDL‐C and HDL‐C and future trials to be conducted solely in patients with mild AD, who have the best chance for disease modification.
Two more‐recent meta‐analyses found no significant benefit from statins in the primary outcome measure ADAS‐Cog in agreement with this study (Gizachew 2012; Pandey 2013). The Gizachew 2012 study assessed results from observational studies and RCTs. When they combined RCTs in a meta‐analysis, they made similar conclusions; there was no benefit from statins in treatment of dementia. The Pandey 2013 meta‐analysis included five studies and concluded no significant benefit in ADAS‐Cog from statins, they found a significant benefit on MMSE but significant heterogeneity similar to this review.
Authors' conclusions
Implications for practice.
There is no evidence to recommend statins for the treatment of Alzheimer's disease (AD) or dementia. In LEADe 2010, the first large‐scale randomised controlled trial (RCT) evaluating statins as a treatment for mild‐to‐moderate AD, the regimen of atorvastatin plus donepezil was not associated with significant benefit on clinical outcome measures over 72 weeks. Sano 2011 was a further well‐designed large RCT incorporating the regimen of simvastatin versus placebo. There again was no benefit from the statin in terms of cognition despite a lowering of cholesterol in the active treatment group. When data from these trials were pooled with two smaller‐scale studies (ADCLT 2005; Simons 2002), there was no benefit from statins seen with the primary outcome measure Alzheimer's Disease Assessment Scale ‐ cognitive subscale (ADAS‐Cog) or in Mini Mental State Examination (MMSE).
From ADCLT 2005, there was some evidence that atorvastatin treatment was more beneficial at six months in patients with AD with higher MMSE at baseline, patients with an apolipoprotein E4 allele and higher cholesterol levels at baseline. This was not replicated in Sano 2011, where there was no effect on MMSE score or ApoE genotype at baseline on the primary outcome measure.
Implications for research.
Two large‐scale RCTs and two smaller studies have assessed the effect of statins on the treatment of dementia. Combined data from the four studies and individual results from the large‐scale RCTs were negative. If further RCTs are to be carried out, lipophilic statins should be used due to their blood‐brain barrier permeability and patients with only mild cognitive impairment should be recruited.
The question of whether statins are of benefit in the treatment of vascular dementia remains unanswered and would be an avenue of research worth pursuing.
What's new
| Date | Event | Description |
|---|---|---|
| 2 July 2014 | New citation required but conclusions have not changed | Conclusions not changed |
| 20 January 2014 | New search has been performed | Two top up searches were performed for this update: one in February 2013 and one in January 2014. One new study was included from the search in 2013 ( Sano 2011, 406 participants). There were no studies for inclusion from the 2014 search |
Acknowledgements
None.
Appendices
Appendix 1. Search: February 2013 and January 2014
|
Source |
Search strategy | Hits retrieved |
| 1. ALOIS (www.medicine.ox.ac.uk/alois) [date of most recent search: 20 January 2014] | Keyword search: statin OR statins OR "Hydroxymethylglutaryl‐CoA Reductase Inhibitors" | Feb 2013: 45 Jan 2014: 0 |
| 2. MEDLINE In‐process and other non‐indexed citations and MEDLINE 1950‐present (Ovid SP) [date of most recent search: 20 January 2014] | 1. exp Dementia/ 2. Delirium/ 3. Wernicke Encephalopathy/ 4. Delirium, Dementia, Amnestic, Cognitive Disorders/ 5. dement*.mp. 6. alzheimer*.mp. 7. (Lewy* adj2 body*).mp. 8. deliri*.mp. 9. (chronic adj2 cerebrovascular).mp. 10. ("organic brain disease" or "organic brain syndrome").mp. 11. ("normal pressure hydrocephalus" and "shunt*").mp. 12. "benign senescent forgetfulness".mp. 13. (cerebr* adj2 deteriorat*).mp. 14. (cerebral* adj2 insufficient*).mp. 15. (pick* adj2 disease).mp. 16. (Creutzfeldt or jcd or cjd).mp. 17. Huntington*.mp. 18. Binswanger*.mp. 19. Korsakoff*.mp. 20. or/1‐19 21. (statin or statins).ti,ab. 22. atorvastatin.ti,ab. 23. cerivastatin.ti,ab. 24. fluvastatin.ti,ab. 25. lovastatin.ti,ab. 26. pravastatin.ti,ab. 27. simvastatin.ti,ab. 28. lipitor.ti,ab. 29. baycol.ti,ab. 30. lescol.ti,ab. 31. mevacor.ti,ab. 32. altocor.ti,ab. 33. pravachol.ti,ab. 34. lipostat.ti,ab. 35. zocor.ti,ab. 36. mevinolin.ti,ab. 37. compactin.ti,ab. 38. fluindostatin.ti,ab. 39. rosuvastatin.ti,ab. 40. Hydroxymethylglutaryl‐CoA Reductase Inhibitors/ or Lovastatin/ 41. Simvastatin/ 42. or/21‐41 43. 20 and 42 44. randomised controlled trial.pt. 45. controlled clinical trial.pt. 46. controlled clinical trial.pt. 47. randomized.ab. 48. placebo.ab. 49. drug therapy.fs. 50. randomly.ab. 51. trial.ab. 52. groups.ab. 53. or/44‐52 54. (animals not (humans and animals)).sh. 55. 53 not 54 56. 43 and 55 57. (2008* or 2009* or 2010* or 2011* or 2012*).ed. 58. 56 and 57 |
Feb 2013: 155 Jan 2014: 25 |
| 3. EMBASE 1980‐2011 week 35 (Ovid SP) [date of most recent search: 20 January 2014] |
1. exp dementia/ 2. Lewy body/ 3. delirium/ 4. Wernicke encephalopathy/ 5. cognitive defect/ 6. dement*.mp. 7. alzheimer*.mp. 8. (Lewy* adj2 body*).mp. 9. deliri*.mp. 10. (chronic adj2 cerebrovascular).mp. 11. ("organic brain disease" or "organic brain syndrome").mp. 12. "supranuclear palsy".mp. 13. ("normal pressure hydrocephalus" and "shunt*").mp. 14. "benign senescent forgetfulness".mp. 15. (cerebr* adj2 deteriorat*).mp. 16. (cerebral* adj2 insufficient*).mp. 17. (pick* adj2 disease).mp. 18. (Creutzfeldt or jcd or cjd).mp. 19. Huntington*.mp. 20. Binswanger*.mp. 21. Korsakoff*.mp. 22. CADASIL.mp. 23. or/1‐22 24. exp dementia/ 25. Lewy body/ 26. delirium/ 27. Wernicke encephalopathy/ 28. cognitive defect/ 29. dement*.mp. 30. alzheimer*.mp. 31. (Lewy* adj2 body*).mp. 32. deliri*.mp. 33. (chronic adj2 cerebrovascular).mp. 34. ("organic brain disease" or "organic brain syndrome").mp. 35. "supranuclear palsy".mp. 36. ("normal pressure hydrocephalus" and "shunt*").mp. 37. "benign senescent forgetfulness".mp. 38. (cerebr* adj2 deteriorat*).mp. 39. (cerebral* adj2 insufficient*).mp. 40. (pick* adj2 disease).mp. 41. (Creutzfeldt or jcd or cjd).mp. 42. Huntington*.mp. 43. Binswanger*.mp. 44. Korsakoff*.mp. 45. CADASIL.mp. 46. or/24‐45 47. (statin or statins).ti,ab. 48. atorvastatin.ti,ab. 49. cerivastatin.ti,ab. 50. fluvastatin.ti,ab. 51. lovastatin.ti,ab. 52. pravastatin.ti,ab. 53. simvastatin.ti,ab. 54. lipitor.ti,ab. 55. baycol.ti,ab. 56. lescol.ti,ab. 57. mevacor.ti,ab. 58. altocor.ti,ab. 59. altocor.ti,ab. 60. pravachol.ti,ab. 61. lipostat.ti,ab. 62. zocor.ti,ab. 63. mevinolin.ti,ab. 64. compactin.ti,ab. 65. fluindostatin.ti,ab. 66. rosuvastatin.ti,ab. 67. hydroxymethylglutaryl coenzyme A reductase inhibitor/ 68. simvastatin/ 69. mevinolin/ 70. fluindostatin/ 71. rosuvastatin/ 72. pravastatin/ 73. atorvastatin/ or cerivastatin/ 74. or/47‐73 75. 46 and 74 76. randomised controlled trial/ 77. controlled clinical trial/ 78. (RCT or CCT).ti,ab. 79. randomly.ab. 80. placebo.ab. 81. trial.ab. 82. randomized.ab. 83. or/76‐82 84. 75 and 83 85. (2008* or 2009* or 2010* or 2011* or 2012* or 2013*).em. 86. 84 and 85 |
Feb 2013: 222 Jan 2014: 56 |
| 4. PsycINFO 1806‐February week 1 2013 (Ovid SP) [date of most recent search: 20 January 2014] |
1. exp Dementia/ 2. exp Delirium/ 3. exp Huntingtons Disease/ 4. exp Kluver Bucy Syndrome/ 5. exp Wernickes Syndrome/ 6. exp Cognitive Impairment/ 7. dement*.mp. 8. alzheimer*.mp. 9. (Lewy* adj2 body*).mp. 10. deliri*.mp. 11. (chronic adj2 cerebrovascular).mp. 12. ("organic brain disease" or "organic brain syndrome").mp. 13. "supranuclear palsy".mp. 14. ("normal pressure hydrocephalus" and "shunt*").mp. 15. "benign senescent forgetfulness".mp. 16. (cerebr* adj2 deteriorat*).mp. 17. (cerebral* adj2 insufficient*).mp. 18. (pick* adj2 disease).mp. 19. (Creutzfeldt or jcd or cjd).mp. 20. Huntington*.mp. 21. Binswanger*.mp. 22. Korsakoff*.mp. 23. ("parkinson* disease dementia" or PDD or "parkinson* dementia").mp. 24. or/1‐23 25. (statin or statins).ti,ab. 26. atorvastatin.ti,ab. 27. cerivastatin.ti,ab. 28. fluvastatin.ti,ab. 29. lovastatin.ti,ab. 30. pravastatin.ti,ab. 31. simvastatin.ti,ab. 32. lipitor.ti,ab. 33. baycol.ti,ab. 34. lescol.ti,ab. 35. mevacor.ti,ab. 36. altocor.ti,ab. 37. pravachol.ti,ab. 38. lipostat.ti,ab. 39. zocor.ti,ab. 40. mevinolin.ti,ab. 41. compactin.ti,ab. 42. fluindostatin.ti,ab. 43. rosuvastatin.ti,ab. 44. exp Statins/ 45. randomi?ed.ab. 46. placebo.ab. 47. randomly.ab. 48. trial.ab. 49. groups.ab. 50. exp Clinical Trials/ 51. (RCT or CCT).ti,ab. 52. "double‐blind*".ti,ab. 53. or/45‐52 54. or/25‐44 55. 24 and 53 and 54 56. (2008* or 2009* or 2010* or 2011* or 2012* or 2013*).up. 57. 55 and 56 |
Feb 2013: 46 Jan 2014: 8 |
| 5. CINAHL (EBSCOhost) [date of most recent search: 20 January 2014] | S1 (MH "Dementia+") S2 (MH "Delirium") or (MH "Delirium, Dementia, Amnestic, Cognitive Disorders") S3 (MH "Wernicke's Encephalopathy") S4 TX dement* S5 TX alzheimer* S6 TX Lewy* N2 body* S7 TX deliri* S8 TX chronic N2 cerebrovascular S9 TX "organic brain disease" or "organic brain syndrome" S10 TX "normal pressure hydrocephalus" and "shunt*" S11 TX "benign senescent forgetfulness" S12 TX cerebr* N2 deteriorat* S13 TX cerebral* N2 insufficient* S14 TX pick* N2 disease S15 TX Creutzfeldt or jcd or cjd S16 TX Huntington* S17 TX Binswanger* S18 TX Korsakoff* S19 S1 or S2 or S3 or S4 or S5 or S6 or S7 or S8 or S9 or S10 or S11 or S12 or S13 or S14 or S15 or S16 or S17 or S18 S20 TX statin OR statins S21 TX atorvastatin S22 TX cerivastatin S23 TX fluvastatin S24 TX lovastatin S25 TX pravastatin S26 TX simvastatin S27 TX lipitor S28 TX baycol S29 TX lescol S30 TX mevacor S31 TX altocor S32 TX pravachol S33 TX lipostat S34 TX Zocor S35 TX mevinolin S36 TX compactin S37 TX fluindostatin S38 TX rosuvastatin S39 (MH "Statins") OR (MH "Rosuvastatin") S40 (MH "Simvastatin") S41 (MH "Atorvastatin") S42 (MH "Fluvastatin") S43 (MH "Pravastatin") S44 S20 OR S21 OR S22 OR S23 OR S24 OR S25 OR S26 OR S27 OR S28 OR S29 OR S30 OR S31 OR S33 OR S34 OR S35 OR S36 OR S37 OR S38 OR S39 OR S40 OR S41 OR S42 OR S43 S45 S19 AND S44 S46 EM 2008 S47 EM 2009 S48 EM 2010 S49 EM 2011 S50 EM 2012 S51 EM 2013 S52 S46 OR S47 OR S48 OR S49 OR S50 OR S51 S53 S45 AND S52 |
Feb 2013: 135 Jan 2014: 17 |
| 6. ISI Web of Knowledge [includes: Web of Science (1945‐present); BIOSIS Previews (1926‐present); MEDLINE (1950‐present); Journal Citation Reports] ‐ BIOSIS Previews NOT searched [date of most recent search: 20 January 2014] | Topic=(dement* OR alzheimer* OR "Lewy body*" OR DLB OR "vascular cognitive impairment*" OR FTD OF FTLD OR "cerebrovascular insufficienc*") AND Topic=(statin OR statins OR atorvastatin or cerivastatin or fluvastatin or lovastatin or pravastatin or simvastatin or lipitor or baycol or lescol or mevacor or altocor or pravachol or lipostat or zocor or mevinolin or compactin or fluindostatin or rosuvastatin OR "Hydroxymethylglutaryl‐CoA Reductase Inhibitor*") AND Topic=(random* OR placebo OR "double‐blind*" OR trial OR RCT OR CCT) AND Year Published=(2008‐2013) Timespan=All Years. Search language=English |
Feb 2013: 179 Jan 2014: 19 |
| 7. LILACS (BIREME) [date of most recent search: 20 January 2014] | dementia OR demência OR alzheimer [Words] and statin OR statins OR Hydroxymethylglutaryl‐CoA Reductase Inhibitors [Words] | Feb 2013: 8 Jan 2014: 2 |
| 8. CENTRAL (The Cochrane Library) (Issue 2 of 4, 2011) [date of most recent search: 20 January 2014] | #1 MeSH descriptor: [Dementia] explode all trees #2 MeSH descriptor: [Delirium] this term only #3 MeSH descriptor: [Wernicke Encephalopathy] this term only #4 MeSH descriptor: [Delirium, Dementia, Amnestic, Cognitive Disorders] this term only #5 dement* #6 alzheimer* #7 "Lewy* body*" #8 deliri* #9 "chronic cerebrovascular" #10 "organic brain disease" or "organic brain syndrome" #11 "normal pressure hydrocephalus" and "shunt*" #12 "benign senescent forgetfulness" #13 "cerebr* deteriorat*" #14 "cerebral* insufficient*" #15 "pick* disease" #16 Creutzfeldt or jcd or cjd #17 Huntington* #18 Binswanger* #19 Korsakoff* #20 #1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 or #9 or #10 or #11 or #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19 #21 statin or statins #22 atorvastatin or cerivastatin or fluvastatin or lovastatin or pravastatin or simvastatin or lipitor or baycol or lescol or mevacor or altocor or pravachol or lipostat or zocor or mevinolin or compactin or fluindostatin or rosuvastatin #23 #21 or #22 #24 MeSH descriptor: [Hydroxymethylglutaryl‐CoA Reductase Inhibitors] explode all trees #25 #23 or #24 #26 #25 and #20 #27 #26 from 2008 to 2013 |
Feb 2013: 98 Jan 2014: |
| 9. Clinicaltrials.gov (www.clinicaltrials.gov) [date of most recent search: 20 January 2014] | Interventional Studies | dementia OR alzheimer OR alzheimers OR Lewy OR vascular cognitive impairment | statin OR statins OR Hydroxymethylglutaryl‐CoA Reductase Inhibitors | received from 01/01/2008 to 02/13/2013 | Feb 2013: 10 Jan 2014: 1 |
| 10. ICTRP Search Portal (apps.who.int/trialsearch) [includes: Australian New Zealand Clinical Trials Registry; ClinicalTrilas.gov; ISRCTN; Chinese Clinical Trial Registry; Clinical Trials Registry ‐ India; Clinical Research Information Service – Republic of Korea; German Clinical Trials Register; Iranian Registry of Clinical Trials; Japan Primary Registries Network; Pan African Clinical Trial Registry; Sri Lanka Clinical Trials Registry; The Netherlands National Trial Register] [date of most recent search: 20 January 2014] | Interventional Studies | dementia OR alzheimer OR alzheimers OR Lewy OR vascular cognitive impairment | statin OR statins OR Hydroxymethylglutaryl‐CoA Reductase Inhibitors | received from 01/01/2008 to 02/13/2013 [Status: ALL] | Feb 2013: 80 Jan 2014: 0 |
| TOTAL before de‐duplication | Feb 2013: 915 Jan 2014: 132 |
|
| TOTAL after de‐dupe and first‐assess | Feb 2013: 110 Jan 2014: 27 |
|
Appendix 2. Search: October 2008
| Database/source | Search strategy | Notes |
| Specialized Register | statin* OR lipophilic OR hydrophilic | |
| The Cochrane Library | 1. statin*.tiabkw. 2. hydroxymethylglutaryl‐CoA Reductase Inhibitors/ (all subheadings) 3. 1 OR 2 4. Alzheimer‐disease/ all subheadings 5. exp dementia‐vascular/ all subheadings 6. creutzfeldt‐jakob‐syndrome/ all subheadings 7. kluver‐bucy‐syndrome/ all subheadings 8. lewy‐body‐disease/ all subheadings 9. pick‐disease‐of‐the‐brain/ all subheadings 10. Huntington‐disease/ all subheadings 11. delirium/ all subheading 12. wernicke‐encephalopathy/ all subheadings 13. (dement$ OR Alzheimer$).tiab. 14. (Lewy$ AND body$).tiab. 15. ((cognit$ OR memor$ OR mental) and (decline$ OR impair$ OR los$ OR deteriorate$)).tiab. 16. (chronic AND cerebrovascular).tiab. 17. ((organic brain syndrome) OR (organic brain disease)).tiab. 18. ((cerebr$ AND deteriorate$) OR (cerebr$ AND insufficien$)).tiab. 19. ((pick$ and disease) or (Creutzfeldt or JCD or CJD) or Huntington$ or Binswanger$ or Korsakoff$).tiab. 20. 4 OR 5 OR 6 OR 7 OR 8 OR 9 OR 10 OR 11 OR 12 OR 13 OR 14 OR 15 OR 16 OR 17 OR 18 OR 19 21. 20 AND 4 22. limit 21 to (randomised controlled trial).pt. | |
| MEDLINE (Ovid SP) | 1. (statin$ OR lipophilic OR hydrophilic).mp. 2. (lovastatin OR simvastatin OR cervistatin OR atorvastatin OR rosuvastatin OR pravastatin OR fluvastatin).mp. 3. Hydroxymethylglutaryl‐CoA Reductase Inhibitors/ all subheadings 4. Alzheimer‐disease/ all subheadings 5. exp dementia‐vascular/ all subheadings 6. creutzfeldt‐jakob‐syndrome/ all subheadings 7. kluver‐bucy‐syndrome/ all subheadings 8. lewy‐body‐disease/ all subheadings 9. pick‐disease‐of‐the‐brain/ all subheadings 10. Huntington‐disease/ all subheadings 11. delirium/ all subheading 12. wernicke‐encephalopathy/ all subheadings 13. (dement$ OR Alzheimer$).mp. 14. (Lewy$ AND body$).mp. 15. ((cognit$ OR memor$ OR mental) and (decline$ OR impair$ OR los$ OR deteriorate$)).mp. 16. (chronic AND cerebrovascular).mp. 17. ((organic brain syndrome) OR (organic brain disease)).mp. 18. ((cerebr$ AND deteriorate$) OR (cerebr$ AND insufficien$)).mp. 19. ((pick$ and disease) or (Creutzfeldt or JCD or CJD) or Huntington$ or Binswanger$ or Korsakoff$).mp. 20. 1 OR 2 OR 3 21. 4 OR 5 OR 6 OR 7 OR 8 OR 9 OR 10 OR 11 OR 12 OR 13 OR 14 OR 15 OR 16 OR 17 OR 18 OR 19 22. 20 AND 21 23. randomized controlled trial.pt. 24. controlled clinical trial.pt. 25. randomized.ab. 26. placebo.ab. 27. drug therapy.fs. 28. randomly.ab. 29. trial.ab. 30. groups.ab. 31. 23 OR 24 OR 25 OR 26 OR 27 OR 28 OR 29 OR 30 32. humans.sh. 33. 31 AND 32 34. 22 AND 33 | |
| EMBASE (Ovid SP) | 1. (statin$ OR lipophilic OR hydrophilic).mp. 2. (lovastatin OR simvastatin OR cervistatin OR atorvastatin OR rosuvastatin OR pravastatin OR fluvastatin).mp. 3. Hydroxymethylglutaryl‐CoA Reductase Inhibitors/ all subheadings 4. Alzheimer‐disease/ all subheadings 5. exp dementia‐vascular/ all subheadings 6. creutzfeldt‐jakob‐syndrome/ all subheadings 7. kluver‐bucy‐syndrome/ all subheadings 8. lewy‐body/ all subheadings 9. pick‐presenile‐dementia/ all subheadings 10. Huntington‐chorea/ all subheadings 11. delirium/ all subheading 12. wernicke‐encephalopathy/ all subheadings 13. (dement$ OR Alzheimer$).mp. 14. (Lewy$ AND body$).mp. 15. ((cognit$ OR memor$ OR mental) and (decline$ OR impair$ OR los$ OR deteriorate$)).mp. 16. (chronic AND cerebrovascular).mp. 17. ((organic brain syndrome) OR (organic brain disease)).mp. 18. ((cerebr$ AND deteriorate$) OR (cerebr$ AND insufficien$)).mp. 19. ((pick$ and disease) or (Creutzfeldt or JCD or CJD) or Huntington$ or Binswanger$ or Korsakoff$).mp. 20. 1 OR 2 OR 3 21. 4 OR 5 OR 6 OR 7 OR 8 OR 9 OR 10 OR 11 OR 12 OR 13 OR 14 OR 15 OR 16 OR 17 OR 18 OR 19 22. 20 AND 21 23. randomized controlled trial.pt. 24. controlled clinical trial.pt. 25. randomized.ab. 26. placebo.ab. 27. drug therapy.fs. 28. randomly.ab. 29. trial.ab. 30. groups.ab. 31. 23 OR 24 OR 25 OR 26 OR 27 OR 28 OR 29 OR 30 32. humans.sh. 33. 31 AND 32 34. 22 AND 33 | |
| CINAHL (Ovid SP) | 1. (statin$ OR lipophilic OR hydrophilic).mp. 2. (lovastatin OR simvastatin OR cervistatin OR atorvastatin OR rosuvastatin OR pravastatin OR fluvastatin).mp. 3. Hydroxymethylglutaryl‐CoA Reductase Inhibitors/ all subheadings 4. Alzheimer‐disease/ all subheadings 5. exp dementia‐vascular/ all subheadings 6. creutzfeldt‐jakob‐syndrome/ all subheadings 7. kluver‐bucy‐syndrome/ all subheadings 8. lewy‐body‐disease/ all subheadings 9. pick‐disease‐of‐the‐brain/ all subheadings 10. Huntington‐disease/ all subheadings 11. delirium/ all subheading 12. wernicke‐encephalopathy/ all subheadings 13. (dement$ OR Alzheimer$).mp. 14. (Lewy$ AND body$).mp. 15. ((cognit$ OR memor$ OR mental) and (decline$ OR impair$ OR los$ OR deteriorate$)).mp. 16. (chronic AND cerebrovascular).mp. 17. ((organic brain syndrome) OR (organic brain disease)).mp. 18. ((cerebr$ AND deteriorate$) OR (cerebr$ AND insufficien$)).mp. 19. ((pick$ and disease) or (Creutzfeldt or JCD or CJD) or Huntington$ or Binswanger$ or Korsakoff$).mp. 20. 1 OR 2 OR 3 21. 4 OR 5 OR 6 OR 7 OR 8 OR 9 OR 10 OR 11 OR 12 OR 13 OR 14 OR 15 OR 16 OR 17 OR 18 OR 19 22. 20 AND 21 23. randomized controlled trial.pt. 24. controlled clinical trial.pt. 25. randomized.ab. 26. placebo.ab. 27. drug therapy.fs. 28. randomly.ab. 29. trial.ab. 30. groups.ab. 31. 23 OR 24 OR 25 OR 26 OR 27 OR 28 OR 29 OR 30 32. humans.sh. 33. 31 AND 32 34. 22 AND 33 | |
| PsycINFO (Ovid SP) | 1. (statin$ OR lipophilic OR hydrophilic).mp. 2. (lovastatin OR simvastatin OR cervistatin OR atorvastatin OR rosuvastatin OR pravastatin OR fluvastatin).mp. 3. Hydroxymethylglutaryl‐CoA Reductase Inhibitors/ all subheadings 4. Alzheimer‐disease/ all subheadings 5. exp dementia‐vascular/ all subheadings 6. creutzfeldt‐jakob‐syndrome/ all subheadings 7. kluver‐bucy‐syndrome/ all subheadings 8. lewy‐body‐disease/ all subheadings 9. pick‐disease‐of‐the‐brain/ all subheadings 10. Huntington‐disease/ all subheadings 11. delirium/ all subheading 12. wernicke‐encephalopathy/ all subheadings 13. (dement$ OR Alzheimer$).mp. 14. (Lewy$ AND body$).mp. 15. ((cognit$ OR memor$ OR mental) and (decline$ OR impair$ OR los$ OR deteriorate$)).mp. 16. (chronic AND cerebrovascular).mp. 17. ((organic brain syndrome) OR (organic brain disease)).mp. 18. ((cerebr$ AND deteriorate$) OR (cerebr$ AND insufficien$)).mp. 19. ((pick$ and disease) or (Creutzfeldt or JCD or CJD) or Huntington$ or Binswanger$ or Korsakoff$).mp. 20. 1 OR 2 OR 3 21. 4 OR 5 OR 6 OR 7 OR 8 OR 9 OR 10 OR 11 OR 12 OR 13 OR 14 OR 15 OR 16 OR 17 OR 18 OR 19 22. 20 AND 21 23. randomized controlled trial.pt. 24. controlled clinical trial.pt. 25. randomized.ab. 26. placebo.ab. 27. drug therapy.fs. 28. randomly.ab. 29. trial.ab. 30. groups.ab. 31. 23 OR 24 OR 25 OR 26 OR 27 OR 28 OR 29 OR 30 32. humans.sh. 33. 31 AND 32 34. 22 AND 33 | |
| LILACS | (statin* OR lipophilic OR hydrophilic) AND (alzheimer$ OR dementia) |
Data and analyses
Comparison 1. Cognitive change (Alzheimer's Disease Assessment Scale ‐ cognitive subscale (ADAS‐Cog)) from baseline.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ADAS‐Cog change from baseline | 4 | 1110 | Mean Difference (IV, Fixed, 95% CI) | ‐0.26 [‐1.05, 0.52] |
| 2 ADAS‐Cog change from baseline (using random‐effects model) | 4 | 1110 | Mean Difference (IV, Random, 95% CI) | ‐0.33 [‐2.10, 1.44] |
| 3 ADAS‐Cog change at 3 months | 2 | 1019 | Mean Difference (IV, Fixed, 95% CI) | 0.14 [‐0.46, 0.74] |
| 4 ADAS‐Cog change at 6 months | 4 | 1067 | Mean Difference (IV, Fixed, 95% CI) | ‐0.08 [‐0.85, 0.70] |
| 5 ADAS‐Cog change at 12 months | 2 | 920 | Mean Difference (IV, Fixed, 95% CI) | ‐0.12 [‐1.04, 0.80] |
| 6 ADAS‐Cog change at 18 months | 2 | 1020 | Mean Difference (IV, Fixed, 95% CI) | ‐0.68 [‐1.49, 0.14] |
| 7 ADAS‐Cog change to 18 months (last observation carried forward) | 1 | Mean Difference (Fixed, 95% CI) | ‐1.11 [‐2.37, 0.14] | |
| 8 Modified ADAS‐Cog | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 8.1 Modified ADAS‐Cog change from baseline at 3 months | 1 | 612 | Mean Difference (Fixed, 95% CI) | ‐0.15 [‐0.99, 0.68] |
| 8.2 Modified ADAS‐Cog change from baseline at 6 months | 1 | 571 | Mean Difference (Fixed, 95% CI) | ‐0.23 [‐1.27, 0.81] |
| 8.3 Modified ADAS‐Cog change from baseline at 9 months | 1 | 533 | Mean Difference (Fixed, 95% CI) | ‐0.66 [‐1.73, 0.41] |
| 8.4 Modified ADAS‐Cog change from baseline at 12 months | 1 | 514 | Mean Difference (Fixed, 95% CI) | ‐0.41 [‐1.76, 0.95] |
| 8.5 Modified ADAS‐Cog change from baseline at 18 months | 1 | 437 | Mean Difference (Fixed, 95% CI) | ‐0.76 [‐2.40, 0.89] |
| 8.6 Modified ADAS‐Cog overall (0‐18) months | 1 | 614 | Mean Difference (Fixed, 95% CI) | 0.41 [‐0.62, 1.44] |
| 8.7 Modified ADAS‐Cog (last observation carried forward) | 1 | 614 | Mean Difference (Fixed, 95% CI) | ‐1.04 [‐2.44, 0.37] |
Comparison 2. Mini Mental State Examination (MMSE) change from baseline.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 MMSE change from baseline | 4 | 1127 | Mean Difference (IV, Fixed, 95% CI) | ‐0.32 [‐0.71, 0.06] |
| 2 MMSE change from baseline using random‐effects model | 4 | 1127 | Mean Difference (IV, Random, 95% CI) | ‐0.88 [‐2.04, 0.28] |
| 3 MMSE change at 3 months | 2 | 1017 | Mean Difference (IV, Fixed, 95% CI) | 0.04 [‐0.31, 0.38] |
| 4 MMSE change at 6 months | 4 | 1084 | Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐0.66, 0.05] |
| 5 MMSE change at 12 months | 4 | 1027 | Mean Difference (IV, Fixed, 95% CI) | ‐0.46 [‐0.90, ‐0.03] |
| 6 MMSE change at 18 months | 2 | 845 | Mean Difference (IV, Fixed, 95% CI) | ‐0.14 [‐0.68, 0.41] |
2.3. Analysis.
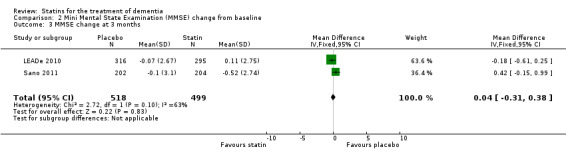
Comparison 2 Mini Mental State Examination (MMSE) change from baseline, Outcome 3 MMSE change at 3 months.
2.5. Analysis.
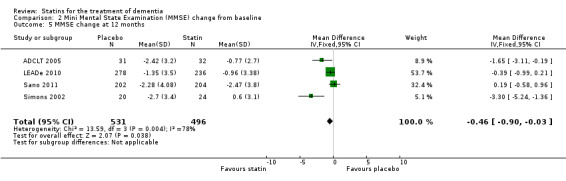
Comparison 2 Mini Mental State Examination (MMSE) change from baseline, Outcome 5 MMSE change at 12 months.
2.6. Analysis.
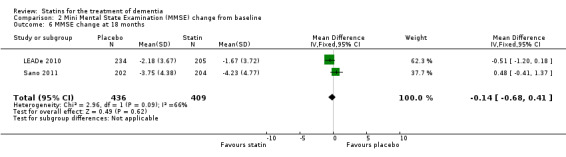
Comparison 2 Mini Mental State Examination (MMSE) change from baseline, Outcome 6 MMSE change at 18 months.
Comparison 3. Clinical Global Impression of Change (CGIC).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 CGIC change | 2 | 660 | Mean Difference (Fixed, 95% CI) | ‐0.02 [‐0.14, 0.10] |
| 2 CGIC change at 3 months | 1 | 603 | Mean Difference (Fixed, 95% CI) | 0.01 [‐0.10, 0.12] |
| 3 CGIC change at 6 months | 1 | 564 | Mean Difference (Fixed, 95% CI) | 0.03 [‐0.11, 0.18] |
| 4 CGIC change at 9 months | 1 | 527 | Mean Difference (Fixed, 95% CI) | ‐0.02 [‐0.18, 0.14] |
| 5 CGIC change at 12 months | 1 | 505 | Mean Difference (Fixed, 95% CI) | ‐0.03 [‐0.20, 0.13] |
| 6 CGIC change at 15 months | 1 | 486 | Mean Difference (Fixed, 95% CI) | 0.02 [‐0.16, 0.20] |
| 7 CGIC change at 18 months | 1 | 435 | Mean Difference (Fixed, 95% CI) | 0.12 [‐0.07, 0.31] |
| 8 CGIC (last observation carried forward) | 1 | 614 | Mean Difference (Fixed, 95% CI) | 0.16 [‐0.01, 0.33] |
3.2. Analysis.
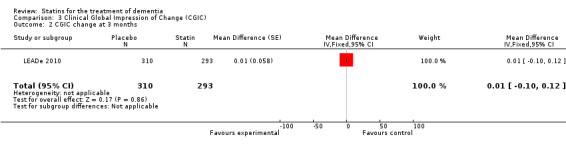
Comparison 3 Clinical Global Impression of Change (CGIC), Outcome 2 CGIC change at 3 months.
3.3. Analysis.
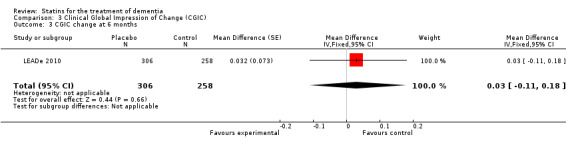
Comparison 3 Clinical Global Impression of Change (CGIC), Outcome 3 CGIC change at 6 months.
3.4. Analysis.
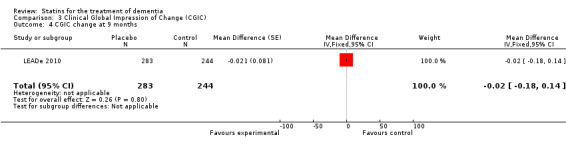
Comparison 3 Clinical Global Impression of Change (CGIC), Outcome 4 CGIC change at 9 months.
3.5. Analysis.
Comparison 3 Clinical Global Impression of Change (CGIC), Outcome 5 CGIC change at 12 months.
3.6. Analysis.
Comparison 3 Clinical Global Impression of Change (CGIC), Outcome 6 CGIC change at 15 months.
3.7. Analysis.
Comparison 3 Clinical Global Impression of Change (CGIC), Outcome 7 CGIC change at 18 months.
3.8. Analysis.
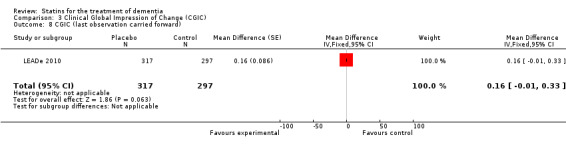
Comparison 3 Clinical Global Impression of Change (CGIC), Outcome 8 CGIC (last observation carried forward).
Comparison 4. Incidence of adverse effects.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Treatment‐related adverse effects requiring discontinuation of treatment | 3 | 1089 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.58, 2.06] |
Comparison 5. Neuropsychiatric Inventory (NPI).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 NPI change from baseline at 6 months | 3 | 1039 | Mean Difference (IV, Fixed, 95% CI) | ‐0.84 [‐1.64, ‐0.05] |
| 2 NPI change from baseline at 1 year | 3 | 983 | Mean Difference (IV, Fixed, 95% CI) | ‐1.11 [‐2.10, ‐0.12] |
| 3 NPI change from baseline at 1 year (using random‐effects model) | 3 | 983 | Mean Difference (IV, Random, 95% CI) | ‐1.64 [‐3.45, 0.18] |
Comparison 6. Alzheimer's Disease Functional Assessment and Change Scale (ADFACS).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ADFACS change from baseline at 6 months | 1 | 596 | Mean Difference (Fixed, 95% CI) | 0.45 [‐0.36, 1.26] |
| 2 ADFACS change from baseline at 12 months | 1 | 512 | Mean Difference (Fixed, 95% CI) | 0.4 [‐0.74, 1.54] |
| 3 ADFACS change from baseline at 18 months | 1 | 470 | Mean Difference (Fixed, 95% CI) | 0.45 [‐0.99, 1.89] |
| 4 ADFACS overall (0‐18) months | 1 | 614 | Mean Difference (Fixed, 95% CI) | 0.43 [‐0.55, 1.42] |
| 5 ADFACS (last observation carried forward) | 1 | 614 | Mean Difference (Fixed, 95% CI) | 0.04 [‐1.24, 1.32] |
6.1. Analysis.
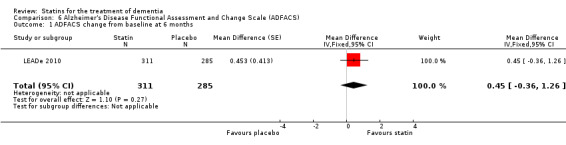
Comparison 6 Alzheimer's Disease Functional Assessment and Change Scale (ADFACS), Outcome 1 ADFACS change from baseline at 6 months.
6.2. Analysis.
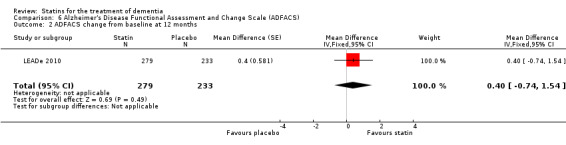
Comparison 6 Alzheimer's Disease Functional Assessment and Change Scale (ADFACS), Outcome 2 ADFACS change from baseline at 12 months.
6.3. Analysis.
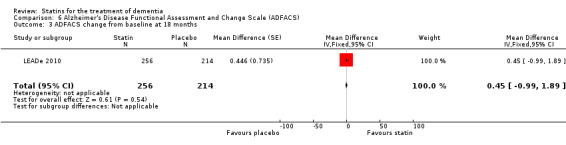
Comparison 6 Alzheimer's Disease Functional Assessment and Change Scale (ADFACS), Outcome 3 ADFACS change from baseline at 18 months.
6.5. Analysis.
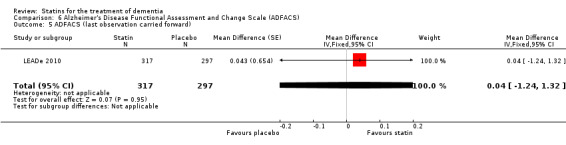
Comparison 6 Alzheimer's Disease Functional Assessment and Change Scale (ADFACS), Outcome 5 ADFACS (last observation carried forward).
Comparison 7. Alzheimer's Disease Cooperative Study‐Activities of Daily Living (ADCS‐ADL).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ADCS‐ADL change from baseline at 3 months | 1 | 406 | Mean Difference (IV, Fixed, 95% CI) | ‐0.33 [‐1.65, 0.99] |
| 2 ADCS‐ADL change from baseline at 6 months | 1 | 406 | Mean Difference (IV, Fixed, 95% CI) | 0.29 [‐1.33, 1.91] |
| 3 ADCS‐ADL change from baseline at 12 months | 1 | 406 | Mean Difference (IV, Fixed, 95% CI) | ‐1.24 [‐3.30, 0.82] |
| 4 ADCS‐ADL change from baseline at 18 months | 1 | 406 | Mean Difference (IV, Fixed, 95% CI) | ‐0.85 [‐3.50, 1.80] |
7.2. Analysis.
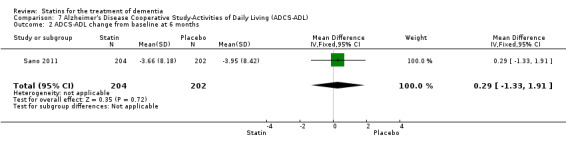
Comparison 7 Alzheimer's Disease Cooperative Study‐Activities of Daily Living (ADCS‐ADL), Outcome 2 ADCS‐ADL change from baseline at 6 months.
7.3. Analysis.
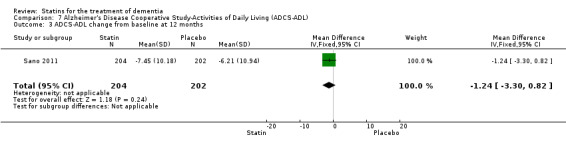
Comparison 7 Alzheimer's Disease Cooperative Study‐Activities of Daily Living (ADCS‐ADL), Outcome 3 ADCS‐ADL change from baseline at 12 months.
7.4. Analysis.
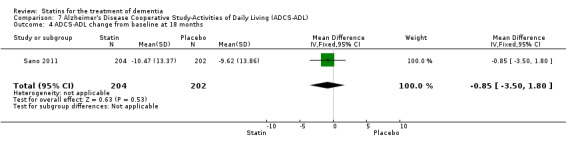
Comparison 7 Alzheimer's Disease Cooperative Study‐Activities of Daily Living (ADCS‐ADL), Outcome 4 ADCS‐ADL change from baseline at 18 months.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
ADCLT 2005.
| Methods | Randomised controlled trial | |
| Participants | 63 participants (32 intervention, 31 control) with probable or possible AD (NINCDS‐ADRDA and DSM‐IV guidelines), 9th grade education or equivalent, speak English fluently and of good general health as evidenced by physical, neurological and clinical laboratory examination. Aged ≥ 51 years, mean (± SD) 78.9 ± 1.2 years in placebo group, 78.15 ± 1.3 years in atorvastatin group. MMSE 12‐28, score ≤ 4 on the modified Hachinski scale and ≤ 20 on the GDS. Individuals were allowed to continue stable dose use of cholinesterase inhibitor and medications treating non‐excluded medical conditions. Patients recruited from a single site in the USA Duration of study: 1 year Mean (± SD) total cholesterol at entry was 208.00 ± 6.41 mg/dL (5.39 ± 0.17 mmol/L) in the placebo group and 207.97 ± 5.98 mg/dL (5.39 ± 0.15 mmol/L) in the atorvastatin group, mean (± SD) LDL‐C 122.22 ± 6.19 mg/dL (3.16 ± 0.16 mmol/L) and 124.47 ± 5.92 mg/dL (3.22 ± 0.15 mmol/L), mean (± SD) VLDL‐C 26.65 ± 2.26 mg/dL in the placebo group and 27.84 ± 2.13 mg/dL in the atorvastatin group (to convert total cholesterol from mg/dL to mmol/L, multiply by 0.0259; LDL‐C mg/dL to mmol/L, multiply by 0.02586) |
|
| Interventions | Intervention: atorvastatin 80 mg daily Control: matching placebo |
|
| Outcomes | Primary outcomes: change in ADAS‐Cog and CGIC Secondary outcomes: change in MMSE, NPI Caregiver Distress Scale, GDS, ADCS‐ADL |
|
| Notes | Participants were excluded with a neurological or psychiatric disease other than AD, including suspected Parkinson's disease or dementia with Lewy bodies, significant systemic illness, organ failure, myocardial infarction, cardiac or thromboembolic vascular disease, major depression according to DSM‐IV criteria, current anticholinergic use. Individuals with a history of head injury; significant liver disease or elevated transaminase levels, or both; allergy to statin medication or screen cholesterol levels < 2.3 mmol/L were also excluded. No participant was using memantine or allowed to initiate cholinesterase inhibitor use after trial entrance and continue participation | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed in blocks of 10 (5 active medication and 5 placebo) using an Excel spreadsheet random number generator Comment: probably done |
| Allocation concealment (selection bias) | Low risk | The sequence was inspected to ensure there was no duplication of random sequences and that there were 5 individuals assigned to each treatment group Comment: probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All investigators were blinded to both treatment groups and cholesterol profiles after randomisation. Only the physician safety monitor, who was not involved in any other aspect of the trial, viewed quarterly cholesterol levels to ensure patient safety. Bottles of study medication were coded at the pharmacy. Atorvastatin calcium tablets, 40 mg, and identical placebo tablets were supplied in bulk by Pfizer Pharmaceuticals, Inc. Bottles of study medication were coded at the pharmacy according to the randomisation schedule, transmitted to the clinic and thereafter provided to the participant/carer Comment: probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double blind Comment: probably done |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Flow table provided detailing flow of participants through the study. 29 active treatment and 27 placebo patients evaluated at visit 2, 26 active treatment and 22 placebo evaluated at visit 3 and 25 active treatment and 21 placebo patients evaluated at visit 4. Reasons for drop‐out not given |
| Selective reporting (reporting bias) | Low risk | Change in cognitive scales as detailed in methods section provided |
LEADe 2010.
| Methods | Randomised, multicentre, parallel‐group, placebo‐controlled, double‐blind study with a double‐blind randomised withdrawal phase | |
| Participants | 640 patients with a diagnosis of probable AD (DSM‐IV and NINCDS‐ADRDA criteria) and of mild‐to‐moderate severity (MMSE 13‐25 at screening). Aged 50‐90 years, mean (± SD) 74 ± 8 years. A CT or MRI brain scan consistent with the diagnosis of probable AD and without other significant co‐morbid abnormalities was required within the previous 12 months. People with diabetes mellitus who had stable blood sugars with diet or treatment with antidiabetic drugs were permitted to enter the study if they had haemoglobin A1c levels of < 10% and fasting serum glucose levels of < 9.4 mmol/L and LDL‐C values 2.5‐3.5 mmol/L. At entry all other participants had to have LDL‐C levels of 2.5‐5.0 mmol/L and must not have required treatment for dyslipidaemia with any lipid‐lowering drug in the opinion of the investigator Duration of study: 72‐week trial period followed by 8‐week atorvastatin withdrawal phase Mean (± SD) total cholesterol at study entry was 5.8 ± 0.8 mmol/L in the groups combined, mean (± SD) LDL‐C was 3.7 ± 0.7 mmol/L, mean (± SD) HDL‐C 1.6 ± 0.5 mmol/L, mean (± SD) VLDL‐C 0.47 ± 0.38 mmol/L and mean (± SD) triglycerides 1.5 ± 0.7 mmol/L 53% women, 47% men Patients recruited from Australia, Austria, Canada, Denmark, Germany, South Africa, Spain, Sweden, the UK and the USA. 96% white |
|
| Interventions | Patients already receiving donepezil 10 mg for at least 3 months before screening Intervention group: atorvastatin 80 mg daily Control group: matching placebo |
|
| Outcomes | Primary outcomes: change in ADAS‐Cog and ADCS‐CGIC Secondary outcomes: change in behaviour (NPI), general cognitive status (MMSE), overall dementia severity (CDR‐SB), activities of daily living (measured by ADFACS Additional analyses on effect of statin on cholesterol/lipid components (ApoB, ApoE, serum total cholesterol, serum LDL‐C, serum VLDL‐C, triglycerides and HDL‐C) Caregiver Burden and Patient Healthcare Resource Utilisation Questionnaire Rate of change in MRI whole brain and hippocampal volumes |
|
| Notes | Participants were excluded if they were taking any medications that affect lipid metabolism or cholinesterase activity other than donepezil within 3 months of screening, or if they had known hypersensitivity to HMG‐CoA reductase inhibitors. Participants were also excluded if they were also experiencing any clinically significant or unstable medical condition including dermatological, haematological, pulmonary, cardiovascular, renal, hepatic, gastrointestinal, genitourinary, endocrine or neurological disease (other than AD). Participants were discouraged from taking any putative cognitive enhancer (e.g. gingko biloba, high‐dose vitamin E, non‐steroidal anti‐inflammatory drugs), but in people who were taking them, the dose must have been stable 3 months before randomisation and throughout the study. Participants with medical conditions that could change the bioavailability or metabolism of the study medication or affect the results of the study and people with a current primary psychiatric disorder other than AD, and who within the previous 5 years met DSM‐IV criteria for drug or alcohol abuse were also excluded from the study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | 1:1 randomisation carried out Comment: probably done |
| Allocation concealment (selection bias) | Low risk | The medication was assembled for each patient based on a randomisation code prepared by Clinical Data Operations of Pfizer Inc Comment: probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participants and personnel stated Comment: probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double blind Comment: probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Trial profile illustrated in a figure with reasons for not completing trial. 1088 people selected for screening, 368 people excluded (319 did not meet entry criteria, 21 no longer willing to participate, 19 other, 8 adverse events, 1 protocol violation) 640 people underwent randomisation: 326 participants assigned to placebo, 325 treated, 245 completed (75.2%), 80 discontinued (24.5%). 314 participants assigned to atorvastatin, 314 treated, 207 completed (65.9%), 107 discontinued (34.1%) 317 participants on placebo analysed for efficacy by modified intention to treat, 325 analysed for safety 297 participants on atorvastatin analysed for efficacy by modified intention to treat, 314 analysed for safety |
| Selective reporting (reporting bias) | Low risk | All cognitive outcomes stated in methods were reported on in both the paper and online supplementary information |
Sano 2011.
| Methods | Randomised, placebo‐controlled, double‐blind trial. Multicentre trial (45 sites) conducted by the ADCS, a consortium of US centres funded by the National Institute of Aging | |
| Participants | Individuals with probable AD according to NINCDS‐ADRDA criteria Inclusion criteria included aged > 50 years and MMSE score 12‐26. Mean (± SD) age 74.6 ± 9.3 years. Stable use (for at least 3 months) of cholinesterase inhibitors and memantine was allowed Duration of study: 18 months Mean (± SD) total cholesterol in the groups combined at study entry was 211.9 ± 30.8 mg/dL, mean (± SD) LDL‐C 126.0 ± 25.1 mg/dL, mean (± SD) HDL‐C 60.9 ± 16.4 mg/dL. 59.4% women |
|
| Interventions | Intervention group: simvastatin 20 mg for 6 weeks and simvastatin 40 mg thereafter for the remainder of the 18‐month study Placebo group: matching placebo |
|
| Outcomes | Primary outcome: change in ADAS‐Cog score Secondary outcomes: ADCG‐CGIC, MMSE, Dependence Scale, ADCS‐ADL, NPI 3 supplemental cognitive tests from the ADCS instrument protocol Quality of life ADCS Resource Use Instrument Adverse events |
|
| Notes | Individuals were excluded if they had other neurological or psychiatric diagnosis that could interfere with cognitive function, if they were taking lipid‐lowering drugs or if they had conditions requiring cholesterol‐lowering treatment as defined by Adult Treatment Guidelines at that time. They were also excluded if they had LDL‐C < 80 mg/dL or triglycerides > 500 mg/dL, if they had recently taken drugs with significant central anticholinergic effects, sedatives, anti‐parkinsonian medications or any investigational treatment for AD. Other excluded medications were those that are specifically contraindicated with simvastatin as well as those that could interact with CYP 3A4 to either increase or decrease levels of simvastatin | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation carried out using a random permuted block treatment assignment stratified by site with equal probability of assignment to drug and placebo. Randomisation sequence generated by the ADCS data centre Comment: probably done |
| Allocation concealment (selection bias) | Low risk | Randomisation sequence generated by the ADCS data centre Comment: probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Scratch‐off" code breakers were used so that instances of unblinding would be documented: all code breakers were collected at the end of the trial. Adequacy of the blinding was assessed by questionnaires completed by participants, carers, psychometrists and site investigators Comment: probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double blind Comment: probably done |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Flow of participants through the study detailed in a figure. 685 subjects screened, 406 randomised, 279 not randomised as did not meet criteria. 204 participants in treatment group, 15 lost to follow‐up before month 18, 145 completed month 18 on medication, 3 completed month 18 off medication. 202 participants in placebo group, 10 participants lost to follow‐up before month 18, 152 completed month 18 on medication, 4 completed month 18 off medication. Predominant reasons for early discontinuation were side effects (16 in placebo arm, 13 in treatment arm) and study partner's unwillingness or inability to continue (7 in placebo arm, 22 in treatment arm). There were no statistically significant differences in baseline characteristics between study participants who discontinued early and study completers (data not published, available by request from authors) |
| Selective reporting (reporting bias) | Unclear risk | Most cognitive and functional outcomes reported. CGIC data available upon request but not provided on contacting study author |
Simons 2002.
| Methods | Randomised, placebo‐controlled, double‐blind trial | |
| Participants | Patients eligible if they fulfilled a diagnosis of probable AD according to NINCDS‐ADRDA criteria. MMSE score 12‐26, then patients subgrouped into mild (MMSE 21‐26) and moderate (MMSE 12‐20) AD. Age (± SD) 68.5 ± 8 years in placebo group, 68.0 ± 9 years in simvastatin group. All patients had a CT scan to rule out vascular encephalopathy as a cause of dementia. Patients were allowed to take donepezil or rivastigmine if the dose had been unchanged for the last 3 months before study entry and remained stable during the 26‐week study period. 44 patients randomised. 47% women in placebo group; 63% women in intervention group Duration of study: 26 weeks Patients recruited from Germany Mean (± SD) serum LDL‐C 134 ± 32 mg/dL in the placebo group and 137 ± 42 mg/dL in the simvastatin group |
|
| Interventions | Intervention: simvastatin up to 80 mg daily Control: matching placebo |
|
| Outcomes | Those analysed in review: change in MMSE and ADAS‐Cog score Those not analysed in review: CSF Aβ40, Aβ42, lathosterol, cholesterol, 24S‐hydroxycholesterol |
|
| Notes | Participants were excluded if they had a Hachinski score > 3 and a continuous intake of anti‐inflammatory drugs | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A randomisation list was computer generated Comment: probably done |
| Allocation concealment (selection bias) | Low risk | 2 copies of the randomisation list were prepared: 1 was used by the packaging department of the study medication or placebo, 1 was kept in a locked location until the study was completed Comment: probably done |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | All personnel directly involved in the conduct of the study remained unaware of the treatment groups until all patients had completed the trial and all data had been retrieved. Serum blood results were controlled monthly by a physician who was otherwise not involved in the study; this guaranteed that all the investigators were kept blinded Comment: probably done |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double blind Comment: probably done |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Trial profile illustrated in a figure with reasons for not completing trial. 3 participants withdrew consent from placebo group and 17 completed treatment. 1 withdrew consent from simvastatin group, 1 was deemed non‐compliant because serum LDL‐C level decreased to < 10%, 1 withdrew due to muscle pain without elevation of creatine kinase, 1 was withdrawn as creatine kinase was elevated and 20 completed treatment |
| Selective reporting (reporting bias) | Low risk | Cognitive outcomes specified in methods reported on |
AD: Alzheimer's disease; ADAS‐Cog: Alzheimer's Disease Assessment Scale ‐ cognitive subscale; ADCS: Alzheimer's Disease Cooperative Study; ADCS‐ADL: Alzheimer's Disease Cooperative Study‐Activities of Daily Living Inventory; ADCS‐CGIC: Alzheimer's Disease Cooperative Study‐Clinical Global Impression of Change; ADFACS: Alzheimer's Disease Functional Assessment and Change Scale; ApoB: apolipoprotein B; ApoE: apolipoprotein E; CDR‐SB: Clinical Dementia Rating‐Sum of Boxes; CGIC: Clinical Global Impression of Change; CT: computed tomography; DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition; GDS: Geriatric Depression Scale; HDL‐C: high‐density lipoprotein cholesterol; HMG‐CoA: 3‐hydroxy‐3‐methylglutaryl coenzyme A; LDL‐C: low‐density lipoprotein cholesterol; MMSE: Mini Mental State Examination; MRI: magnetic resonance imaging; NINCDS‐ADRDA: National Institute of Neurological and Communicative Disorders and Stroke‐the Alzheimer's Disease and Related Disorders Association; NPI: Neuropsychiatric Inventory; SD: standard deviation; VLDL‐C: very‐low‐density lipoprotein cholesterol.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Gutterman 2002 | Post‐hoc analysis from randomised placebo‐controlled clinical trials of galantamine in patients with AD. Not sufficiently powered according to authors to examine the effects of statins on cognition. Using last observation carried forward, mean ADAS‐Cog change from baseline was measured. Of 1311 patients, 8.8% were taking statins. There were 4 treatment groups: placebo (598 participants), galantamine alone (598 participants), statin alone (60 participants) and galantamine plus statin (55 participants). While galantamine use was associated with a significant change in mean ADAS‐Cog from baseline (P value < 0.001), statin use (P value = 0.195) or the interaction of galantamine with statins (P value = 0.372) were not. The conclusion was the use of statins did not lead to significant improvement of cognitive function among patients with AD either alone or in combination with galantamine. Longer periods of statin treatment were advised by authors |
| Winblad 2007 | Post‐hoc analysis from randomised placebo‐controlled clinical trials of galantamine in patients with AD. Not sufficiently powered according to authors to establish a 2‐point difference in ADAS‐Cog/11 scale over the study period, which would be clinically significant. Results based on a non‐random sample of pooled participants with regards to statin use, resulting in baseline differences between the statin and non‐statin treatment groups with respect to age and gender. Administration of statins was heterogeneous with respect to specific statin, dose and treatment duration. There were 4 treatment groups: statin plus galantamine (42 participants), statin alone (50 participants), galantamine alone (614 participants) and neither galantamine nor statin (619 participants). While galantamine was associated with a significant beneficial effect on cognitive status (P value < 0.001), there was no association seen with use of statins (P value = 0.083). There was no significant effect on cognition with use of statin plus galantamine (P value = 0.183) |
AD: Alzheimer's disease; ADAS‐Cog: Alzheimer's Disease Assessment Scale ‐ cognitive subscale.
Contributions of authors
BMcG: all correspondence, drafting of review versions, search for trials, obtaining copies of trial reports, selection of trials for inclusion/exclusion, extraction of data, entry of data and interpretation of data analyses.
RM: statistical analysis and interpretation of data analyses.
DC: drafting of reviews and interpretation of data analyses.
RB: drafting of review versions and interpretation of data analyses.
APP: all correspondence, drafting of review versions, selection of trials for inclusion/exclusion and interpretation of data analyses.
Declarations of interest
Peter Passmore: investigator in LEADe study received grants from Pfizer, MSD, BMS and Novartis and honoraria from Pfizer, MSD, Novartis and Astra Zeneca.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
ADCLT 2005 {published data only (unpublished sought but not used)}
- Sparks DL, Connor DJ, Sabbagh MN, Petersen RB, Lopez J, Browne P. Circulating cholesterol levels, apolipoprotein E genotype and dementia severity influence the benefit of atorvastatin treatment in Alzheimer's disease: results of the Alzheimer's Disease Cholesterol‐Lowering Treatment (ADCLT) trial. Acta Neurologica Scandinavica 2006;114(Suppl 185):3‐7. [DOI] [PubMed] [Google Scholar]
- Sparks DL, Sabbagh M, Connor D, Soares H, Lopez J, Stankovic G, et al. Statin therapy in Alzheimer's disease. Acta Neurologica Scandinavica 2006;114(Suppl 185):78‐86. [DOI] [PubMed] [Google Scholar]
- Sparks DL, Sabbagh MN, Connor DJ, Lopez J, Launer LJ, Browne P, et al. Atorvastatin for the treatment of mild to moderate Alzheimer disease. Archives of Neurology 2005;62:753‐7. [DOI] [PubMed] [Google Scholar]
LEADe 2010 {published and unpublished data}
- Feldman H, Jones RW, Kivipelto M, Sparks L, Doody R, Waters D, et al. The LEADe study: a randomized, controlled trial investigating the effect of atorvastatin on cognitive and global function in patients with mild‐to‐moderate Alzheimer's disease receiving background therapy of donepezil. Neurology 2008;71:153‐6. [Google Scholar]
- Feldman HH, Doody RS, Kivipelto M, Sparks DL, Waters DD, Jones RW, et al. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease. LEADe. Neurology 2010;74:956‐64. [DOI] [PubMed] [Google Scholar]
- Jones RW, Kivipelto M, Feldman H, Sparks L, Doody R, Waters DD, et al. The Atorvastatin/Donepezil in Alzheimer's Disease Study (LEADe): design and baseline characteristics. Alzheimer's & Dementia 2008;4:145‐53. [DOI] [PubMed] [Google Scholar]
Sano 2011 {published data only}
- Sano M, Bell KL, Galasko D, Galvin JE, Thomas RG, Dyck CH, et al. A randomized, double‐blind, placebo‐controlled trial of simvastatin to treat Alzheimer disease. Neurology 2011;77:556‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Simons 2002 {published data only}
- Simons M, Schwarzler F, Lutjohann D, Bergmann K, Beyreuther K, Dichgans J, et al. Treatment with simvastatin in normocholesterolaemic patients with Alzheimer's disease: a 26‐week randomized, placebo‐controlled, double‐blind trial. Annals of Neurology 2002;52:346‐50. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Gutterman 2002 {published data only}
- Gutterman E, Markowitz JS, Lilienfeld S. Cognitive maintenance of Alzheimer's disease patients in pooled randomized, placebo‐controlled clinical trials of galantamine: the effect of statins. International Journal of Neuropsychopharmacology 2002;5(Suppl 1):S92‐3. [Google Scholar]
Winblad 2007 {published data only}
- Winbald B, Jelic V, Kershaw P, Amatniek J. Effects of statins on cognitive function in patients with Alzheimer's disease in galantamine clinical trials. Drugs Aging 2007;24:57‐61. [DOI] [PubMed] [Google Scholar]
Additional references
Alzheimers Disease International 2013
- Alzheimers Disease International. Dementia statistics. www.alz.co.uk/research/statistics (accessed 16 June 2014).
Ballard 2000
- Ballard C, McKeith I, O'Brien J, Kalaria R, Jaros E, Ince P, et al. Neuropathological substrates of dementia and depression in vascular dementia, with a particular focus on cases with small infarct volumes. Dementia, Geriatric and Cognitive Disorders 2000;11(2):59‐65. [DOI] [PubMed] [Google Scholar]
Berry 1988
- Berry PH, MacDonald JS, Alberts AW, Molon‐Noblot S, Chen JS, Lo CY, et al. Brain and optic system pathology in hypocholesterolemic dogs treated with a competitive inhibitor of 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase. American Journal of Pathology 1988;132(3):427‐43. [PMC free article] [PubMed] [Google Scholar]
Betterman 2012
- Betterman K, Arnold AM, Williamson J, Rapp S, Sink K, Toole JF, et al. Statins, risk of dementia, and cognitive function: secondary analysis of the ginkgo evaluation of memory study. Journal of Stroke and Cerebrovascular Disease 2012;6:436‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bodovitz 1996
- Bodovitz S, Klein WL. Cholesterol modulates alpha‐secretase cleavage of amyloid precursor protein. Journal of Biological Chemistry 1996;271(8):4436‐40. [DOI] [PubMed] [Google Scholar]
Breteler 1994
- Breteler MM, Swieten JC, Bots ML, Grobbee DE, Claus JJ, Hout JH, et al. Cerebral white matter lesions, vascular risk factors, and cognitive function in a population‐based study: the Rotterdam Study. Neurology 1994;44(7):1246‐52. [DOI] [PubMed] [Google Scholar]
Burns 2003
- Burns MP, Noble WJ, Olm V, Gaynor K, Casey E, LaFrancois J, et al. Co‐localization of cholesterol, apolipoprotein E and fibrillar Abeta in amyloid plaques. Brain Research. Molecular Brain Research 2003;110(1):119‐25. [DOI] [PubMed] [Google Scholar]
Cole 2007
- Cole SL, Vassar R. The Alzheimer's disease beta‐secretase enzyme, BACE1. Molecular Neurodegeneration 2007;2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Corder 1993
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993;261(5123):921‐3. [DOI] [PubMed] [Google Scholar]
CTTC 2005
- Cholesterol Treatment Trialists' Collaborators (CTTC). Efficacy and safety of cholesterol‐lowering treatment: prospective meta‐analysis of data from 90,056 participants in 14 randomized trials of statins. Lancet 2005;366:1267. [DOI] [PubMed] [Google Scholar]
Dantoine 2002
- Dantoine TF, Debord J, Merle L, Lacroix‐Ramiandrisoa H, Bourzeix L, Charmes JP. Paraoxonase 1 activity: a new vascular marker of dementia?. Annals of the New York Academic Science 2002;977:96‐101. [DOI] [PubMed] [Google Scholar]
Decarli 2004
- Decarli C. Vascular factors in dementia: an overview. Journal of Neurological Science 2004;226(1‐2):19‐23. [DOI] [PubMed] [Google Scholar]
Dietschy 2001
- Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Current Opinions in Lipidology 2001;12(2):105‐12. [DOI] [PubMed] [Google Scholar]
Ehehalt 2003
- Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer beta‐amyloid precursor protein depends on lipid rafts. Journal of Cell Biology 2003;160(1):113‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Esiri 1997
- Esiri MM, Wilcock GK, Morris JH. Neuropathological assessment of the lesions of significance in vascular dementia. Journal of Neurology, Neurosurgery and Psychiatry 1997;63(6):749‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Evans 1989
- Evans DA, Funkenstein HH, Albert MS, Scherr PA, Cook NR, Chown MJ, et al. Prevalence of Alzheimer's disease in a community population of older persons. Higher than previously reported. JAMA 1989;262(18):2551‐6. [PubMed] [Google Scholar]
Farrer 1997
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997;278(16):1349‐56. [PubMed] [Google Scholar]
Fassbender 2001
- Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, et al. Simvastatin strongly reduces levels of Alzheimer's disease beta ‐amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proceedings of the National Academy of Science USA 2001;98(10):5856‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gizachew 2012
- Gizachew S, Makonnen E. The role of statins in Alzheimer's disease: a meta‐analysis. African Journal of Neurological Sciences 2012;2:2002‐12. [Google Scholar]
Hardy 2002
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297(5580):353‐6. [DOI] [PubMed] [Google Scholar]
Harold 2009
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome‐wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nature Genetics 2009;41(10):1088‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Heverin 2005
- Heverin M, Meaney S, Lutjohann D, Diczfalusy U, Wahren J, Bjorkhem I. Crossing the barrier: net flux of 27‐hydroxycholesterol into the human brain. Journal of Lipid Research 2005;46(5):1047‐52. [DOI] [PubMed] [Google Scholar]
Holtzman 2000
- Holtzman DM, Fagan AM, Mackey B, Tenkova T, Sartorius L, Paul SM, et al. Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer's disease model. Annals of Neurology 2000;47(6):739‐47. [PubMed] [Google Scholar]
Jarvik 1995
- Jarvik GP, Wijsman EM, Kukull WA, Schellenberg GD, Yu C, Larson EB. Interactions of apolipoprotein E genotype, total cholesterol level, age, and sex in prediction of Alzheimer's disease: a case‐control study. Neurology 1995;45(6):1092‐6. [DOI] [PubMed] [Google Scholar]
Jendroska 1995
- Jendroska K, Poewe W, Daniel SE, Pluess J, Iwerssen‐Schmidt H, Paulsen J, et al. Ischemic stress induces deposition of amyloid beta immunoreactivity in human brain. Acta Neuropathology 1995;90(5):461‐6. [DOI] [PubMed] [Google Scholar]
Jick 2000
- Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet 2000;356:1627‐31. [DOI] [PubMed] [Google Scholar]
Joshi 1999
- Joshi HN, Fakes MG, Serajuddin ATM. Differentiation of 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase inhibitors by their relative lipophilicity. Pharmacy and Pharmacology Communications 1999;5:269‐71. [Google Scholar]
Kandiah 2009
- Kandiah N, Feldman HH. Therapeutic potential of statins in Alzheimer's disease. Journal of the Neurological Sciences 2009;283(1‐2):230‐4. [DOI] [PubMed] [Google Scholar]
King 2003
- King DS, Wilburn AJ, Wofford MR, Harrell TK, Lindley BJ, Jones DW. Cognitive impairment associated with atorvastatin and simvastatin. Pharmacotherapy 2003;23(12):1663‐7. [DOI] [PubMed] [Google Scholar]
Kivipelto 2002
- Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K, et al. Apolipoprotein E epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late‐life Alzheimer disease. Annals of Internal Medicine 2002;137(3):149‐55. [DOI] [PubMed] [Google Scholar]
Klich‐Raczka 2002
- Klich‐Raczka A, Necki M, Wizner B, Baron T, Adamkiewicz‐Piejko A, Gryglewska B, et al. Vascular dementia and systemic changes. Przeglad Lekarski 2002;59(4‐5):269‐71. [PubMed] [Google Scholar]
Knopp 1999
- Knopp RH. Drug treatment of lipid disorders. New England Journal of Medicine 1999;341(7):498‐511. [DOI] [PubMed] [Google Scholar]
Kuriyama 1994
- Kuriyama M, Takahashi K, Yamano T, Hokezu Y, Togo S, Osame M, et al. Low levels of serum apolipoprotein A I and A II in senile dementia. Japanese Journal of Psychiatry and Neurology 1994;48(3):589‐93. [DOI] [PubMed] [Google Scholar]
Lambert 2009
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nature Genetics 2009;41(10):1094‐9. [DOI] [PubMed] [Google Scholar]
Lane 2005
- Lane RM, Farlow MR. Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer's disease. Journal of Lipid Research 2005;46(5):949‐68. [DOI] [PubMed] [Google Scholar]
Li 2006
- Li L, Cao D, Kim H, Lester R, Fukuchi K‐I. Simvastatin enhances learning and memory independent of amyloid load in mice. Annals of Neurology 2006;60:729‐39. [DOI] [PubMed] [Google Scholar]
Li 2007
- Li G, Larson EB, Sonnen JA, Shofer JB, Petrie EC, Schantz A, et al. Statin therapy is associated with reduced neuropathologic changes of Alzheimer disease. Neurology 2007;69(9):878‐85. [DOI] [PubMed] [Google Scholar]
Liao 2002
- Liao JK. Isoprenoids as mediators of the biological effects of statins. Journal of Clinical Investigation 2002;110(3):285‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lutjohann 2000
- Lutjohann D, Papassotiropoulos A, Bjorkhem I, Locatelli S, Bagli M, Oehring RD, et al. Plasma 24S‐hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. Journal of Lipid Research 2000;41(2):195‐8. [PubMed] [Google Scholar]
Masse 2005
- Masse I, Bordet R, Deplanque D, Al Khedr A, Richard F, Libersa C, et al. Lipid lowering agents are associated with a slower cognitive decline in Alzheimer's disease. Journal of Neurology, Neurosurgery and Psychiatry 2005;76(12):1624‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Matthews 2013
- Matthews FE, Arthur A, Barnes LE, Bond J, Jagger C, Robinson L, et al. A two‐decade comparison of prevalence of dementia in individuals aged 65 years and older from three geographical areas of England: results of the Cognitive Function and Ageing Study I and II. Lancet 2013;382:1405‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
McGuinness 2009
- McGuinness B, Craig D, Bullock R, Passmore P. Statins for the prevention of dementia. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD003160.pub2] [DOI] [PubMed] [Google Scholar]
Michikawa 1998
- Michikawa M, Yanagisawa K. Apolipoprotein E4 induces neuronal cell death under conditions of suppressed de novo cholesterol synthesis. Journal of Neuroscience Research 1998;54(1):58‐67. [DOI] [PubMed] [Google Scholar]
Mizuno 1999
- Mizuno T, Nakata M, Naiki H, Michikawa M, Wang R, Haass C, et al. Cholesterol‐dependent generation of a seeding amyloid beta‐protein in cell culture. Journal of Biological Chemistry 1999;274(21):15110‐14. [DOI] [PubMed] [Google Scholar]
Mohs 1997
- Mohs RC, Knopman D, Petersen RC, Ferris SH, Ernesto C, Grundman M, et al. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer's Disease Assessment Scale that broaden its scope: The Alzheimer's Disease Cooperative Study. Alzheimer Disease and Associated Disorders 1997;11 Suppl 2:S13‐21. [PubMed] [Google Scholar]
Moroney 1999
- Moroney JT, Tang MX, Berglund L, Small S, Merchant C, Bell K, et al. Low‐density lipoprotein cholesterol and the risk of dementia with stroke. JAMA 1999;282(3):254‐60. [DOI] [PubMed] [Google Scholar]
Muckle 1985
- Muckle TJ, Roy JR. High‐density lipoprotein cholesterol in differential diagnosis of senile dementia. Lancet 1985;1(8439):1191‐3. [DOI] [PubMed] [Google Scholar]
Muldoon 1997
- Muldoon MF, Ryan CM, Matthews KA, Manuck SB. Serum cholesterol and intellectual performance. Psychosomatic Medicine 1997;59(4):382‐7. [DOI] [PubMed] [Google Scholar]
Muldoon 2000
- Muldoon MF, Barger SD, Ryan CM, Flory JD, Lehoczky JP, Matthews KA, et al. Effects of lovastatin on cognitive function and psychological well‐being. American Journal of Medicine 2000;108(7):538‐46. [DOI] [PubMed] [Google Scholar]
Naslund 1995
- Naslund J, Thyberg J, Tjernberg LO, Wernstedt C, Karlstrom AR, Bogdanovic N, et al. Characterization of stable complexes involving apolipoprotein E and the amyloid beta peptide in Alzheimer's disease brain. Neuron 1995;15(1):219‐28. [DOI] [PubMed] [Google Scholar]
Notkola 1998
- Notkola IL, Sulkava R, Pekkanen J, Erkinjuntti T, Ehnholm C, Kivinen P, et al. Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer's disease. Neuroepidemiology 1998;17(1):14‐20. [DOI] [PubMed] [Google Scholar]
Ott 1998
- Ott A, Slooter AJ, Hofman A, Harskamp F, Witteman JC, Broeckhoven C, et al. Smoking and risk of dementia and Alzheimer's disease in a population‐based cohort study: the Rotterdam Study. Lancet 1998;351(9119):1840‐3. [DOI] [PubMed] [Google Scholar]
Pahan 2006
- Pahan K. Lipid‐lowering drugs. Cellular and Molecular Life Sciences 2006;63(10):1165‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Palomaki 1997
- Palomaki A, Malminiemi K, Metsa‐Ketela T. Enhanced oxidizability of ubiquinol and alpha‐tocopherol during lovastatin treatment. FEBS Letters 1997;410(2‐3):254‐8. [DOI] [PubMed] [Google Scholar]
Pandey 2013
- Pandey RD, Gupta PP, Jha D, Kumar S. Role of statins in Alzheimer's disease: a retrospective meta‐analysis for commonly investigated clinical parameters in RCTs. International Journal of Neuroscience 2013;123:521‐5. [DOI] [PubMed] [Google Scholar]
Paragh 2002
- Paragh G, Balla P, Katona E, Seres I, Egerhazi A, Degrell I. Serum paraoxonase activity changes in patients with Alzheimer's disease and vascular dementia. European Archives of Psychiatry and Clinical Neuroscience 2002;252(2):63‐7. [DOI] [PubMed] [Google Scholar]
Posner 2002
- Posner HB, Tang MX, Luchsinger J, Lantigua R, Stern Y, Mayeux R. The relationship of hypertension in the elderly to AD, vascular dementia, and cognitive function. Neurology 2002;58(8):1175‐81. [DOI] [PubMed] [Google Scholar]
Refolo 2000
- Refolo LM, Malester B, LaFrancois J, Bryant‐Thomas T, Wang R, Tint GS, et al. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiological Disorders 2000;7(4):321‐31. [DOI] [PubMed] [Google Scholar]
Refolo 2001
- Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas‐Bryant T, et al. A cholesterol‐lowering drug reduces beta‐amyloid pathology in a transgenic mouse model of Alzheimer's disease. Neurobiological Disorders 2001;8(5):890‐9. [DOI] [PubMed] [Google Scholar]
Reiss 2004
- Reiss AB, Siller KA, Rahman MM, Chan ES, Ghiso J, Leon MJ. Cholesterol in neurologic disorders of the elderly: stroke and Alzheimer's disease. Neurobiology of Aging 2004;25(8):977‐89. [DOI] [PubMed] [Google Scholar]
Reitz 2004
- Reitz C, Tang MX, Luchsinger J, Mayeux R. Relation of plasma lipids to Alzheimer disease and vascular dementia. Archives of Neurology 2004;61(5):705‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Riekse 2004
- Riekse RG, Leverenz JB, McCormick W, Bowen JD, Teri L, Nochlin D, et al. Effect of vascular lesions on cognition in Alzheimer's disease: a community‐based study. Journal of the American Geriatrics Society 2004;52(9):1442‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rise 1997
- Rise P, Colombo C, Galli C. Effects of simvastatin on the metabolism of polyunsaturated fatty acids and on glycerolipid, cholesterol, and de novo lipid synthesis in THP‐1 cells. Journal of Lipid Research 1997;38(7):1299‐307. [PubMed] [Google Scholar]
Rockwood 2002
- Rockwood K, Kirkland S, Hogan DB, MacKnight C, Merry H, Verreault R, et al. Use of lipid‐lowering agents, indication bias, and the risk of dementia in community‐dwelling elderly people. Archives of Neurology 2002;59(2):223‐7. [DOI] [PubMed] [Google Scholar]
Ruocco 2002
- Ruocco A, Postiglione A, Santillo M, Seru R, Avvedimento EV, Cuda G, et al. New possible role of statins in age‐related diseases. Journal of the American Geriatrics Society 2002;50(12):2099‐100. [DOI] [PubMed] [Google Scholar]
Ryglewicz 2002
- Ryglewicz D, Rodo M, Kunicki PK, Bednarska‐Makaruk M, Graban A, Lojkowska W, et al. Plasma antioxidant activity and vascular dementia. Journal of Neurological Science 2002;203‐204:195‐7. [DOI] [PubMed] [Google Scholar]
Sabbagh 2012
- Sabbagh MN, Sparks DL. Statins to treat Alzheimer's disease: an incomplete story. Expert Reviews in Neurotherapeutics 2012;12:27‐30. [DOI] [PubMed] [Google Scholar]
Selkoe 2001
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiological Review 2001;81(2):741‐66. [DOI] [PubMed] [Google Scholar]
Sharrett 1994
- Sharrett AR, Patsch W, Sorlie PD, Heiss G, Bond MG, Davis CE. Associations of lipoprotein cholesterols, apolipoproteins A‐I and B, and triglycerides with carotid atherosclerosis and coronary heart disease. The Atherosclerosis Risk in Communities (ARIC) Study. Arteriosclerosis Thrombosis 1994;14(7):1098‐104. [DOI] [PubMed] [Google Scholar]
Shepardson 2011
- Shepardson NE, Shankar GM, Selkoe DJ. Cholesterol level and statin use in Alzheimer disease. II. Review of human trials and recommendations. Archives in Neurology 2011;68(11):1385‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Simons 1998
- Simons M, Keller P, Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta‐amyloid in hippocampal neurons. Proceedings of the National Academy of Science of the USA 1998;95(11):6460‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Simons 2001
- Simons M, Keller P, Dichgans J, Schulz JB. Cholesterol and Alzheimer's disease: is there a link?. Neurology 2001;57(6):1089‐93. [DOI] [PubMed] [Google Scholar]
Snowdon 1997
- Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer's disease. The Nun Study. JAMA 1997;277(10):813‐17. [PubMed] [Google Scholar]
SPARCL 2006
- SPARCL Investigators. High‐dose atorvastatin after stroke or transient ischaemic attack. New England Journal of Medicine 2006;355:549‐59. [DOI] [PubMed] [Google Scholar]
Sparks 1990
- Sparks DL, Hunsaker JC, 3rd, Scheff SW, Kryscio RJ, Henson JL, Markesbery WR. Cortical senile plaques in coronary artery disease, aging and Alzheimer's disease. Neurobiology of Aging 1990;11(6):601‐7. [DOI] [PubMed] [Google Scholar]
Sparks 1994
- Sparks DL, Scheff SW, Hunsaker JC, 3rd, Liu H, Landers T, Gross DR. Induction of Alzheimer‐like beta‐amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Experimental Neurology 1994;126(1):88‐94. [DOI] [PubMed] [Google Scholar]
Sparks 2006
- Sparks DL, Connor DJ, Sabbagh MN, Petersen RB, Lopez J, Browne P. Circulating cholesterol levels, apolipoprotein E genotype and dementia severity influence the benefit of atorvastatin treatment in Alzheimer's disease: results of the Alzheimer's Disease Cholesterol‐Lowering Treatment (ADCLT) trial. Acta Neurology Scandinavia Supplement 2006;185:3‐7. [DOI] [PubMed] [Google Scholar]
Stewart 1999
- Stewart R, Liolitsa D. Type 2 diabetes mellitus, cognitive impairment and dementia. Diabetes Medicine 1999;16(2):93‐112. [DOI] [PubMed] [Google Scholar]
Strand 2013
- Strand BH, Langballe EM, Hjellvik M, Handal M, Naess O, Knudsen GP, et al. Midlife vascular risk factors and their association with dementia deaths: results from a Norwegian prospective study followed up for 35 years. Journal of the Neurological Sciences 2013;324:124‐30. [DOI] [PubMed] [Google Scholar]
Strittmatter 1993
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak‐Vance M, et al. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform‐specific effects and implications for late‐onset Alzheimer disease. Proceedings of the National Academy of Sciences of the USA 1993;90(17):8098‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
van Es 2009
- Es MA, Berg LH. Alzheimer's disease beyond APOE. Nature Genetics 2009;41(10):1047‐8. [DOI] [PubMed] [Google Scholar]
van Exel 2002
- Exel E, Craen AJ, Gussekloo J, Houx P, Bootsma‐van der Wiel A, Macfarlane PW, et al. Association between high‐density lipoprotein and cognitive impairment in the oldest old. Annals of Neurology 2002;51(6):716‐21. [DOI] [PubMed] [Google Scholar]
Vetrivel 2004
- Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, et al. Association of gamma‐secretase with lipid rafts in post‐Golgi and endosome membranes. Journal of Biological Chemistry 2004;279(43):44945‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vetrivel 2006
- Vetrivel KS, Thinakaran G. Amyloidogenic processing of beta‐amyloid precursor protein in intracellular compartments. Neurology 2006;66(2 Suppl 1):S69‐73. [DOI] [PubMed] [Google Scholar]
von Strauss 1999
- Strauss E, Viitanen M, Ronchi D, Winblad B, Fratiglioni L. Aging and the occurrence of dementia: findings from a population‐based cohort with a large sample of nonagenarians. Archives of Neurology 1999;56(5):587‐92. [DOI] [PubMed] [Google Scholar]
Wagstaff 2003
- Wagstaff LR, Mitton MW, Arvik BM, Doraiswamy PM. Statin‐associated memory loss: analysis of 60 case reports and review of the literature. Pharmacotherapy 2003;23(7):871‐80. [DOI] [PubMed] [Google Scholar]
Walsh 1996
- Walsh KM, Albassam MA, Clarke DE. Subchronic toxicity of atorvastatin, a hydroxymethylglutaryl‐coenzyme A reductase inhibitor, in beagle dogs. Toxicology Pathology 1996;24(4):468‐76. [DOI] [PubMed] [Google Scholar]
Wassmann 2001
- Wassmann S, Laufs U, Baumer AT, Muller K, Ahlbory K, Linz W, et al. HMG‐CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension 2001;37(6):1450‐7. [DOI] [PubMed] [Google Scholar]
Wisniewski 1995
- Wisniewski T, Lalowski M, Golabek A, Vogel T, Frangione B. Is Alzheimer's disease an apolipoprotein E amyloidosis?. Lancet 1995;345(8955):956‐8. [DOI] [PubMed] [Google Scholar]
Wolozin 2001
- Wolozin B. A fluid connection: cholesterol and Abeta. Proceedings of the National Academy of Science USA 2001;98(10):5371‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wu 2003
- Wu CW, Liao PC, Lin C, Kuo CJ, Chen ST, Chen HI, et al. Brain region‐dependent increases in beta‐amyloid and apolipoprotein E levels in hypercholesterolemic rabbits. Journal of Neural Transmission 2003;110(6):641‐9. [DOI] [PubMed] [Google Scholar]
Yoshitake 1995
- Yoshitake T, Kiyohara Y, Kato I, Ohmura T, Iwamoto H, Nakayama K, et al. Incidence and risk factors of vascular dementia and Alzheimer's disease in a defined elderly Japanese population: the Hisayama Study. Neurology 1995;45(6):1161‐8. [DOI] [PubMed] [Google Scholar]
Zhang 2004
- Zhang J, Muldoon MF, McKeown RE. Serum cholesterol concentrations are associated with visuomotor speed in men: findings from the third National Health and Nutrition Examination Survey, 1988‐1994. American Journal of Clinical Nutrition 2004;80(2):291‐8. [DOI] [PubMed] [Google Scholar]
Zhou 2007
- Zhou B, Teramukai S, Fukushima M. Prevention and treatment of dementia or Alzheimer's disease by statins: a meta‐analysis. Dementia and Geriatric Cognitive Disorders 2007;23(3):194‐201. [DOI] [PubMed] [Google Scholar]
Zuliani 2001
- Zuliani G, Ble' A, Zanca R, Munari MR, Zurlo A, Vavalle C, et al. Lipoprotein profile in older patients with vascular dementia and Alzheimer's disease. BMC Geriatrics 2001;1:5. [DOI] [PMC free article] [PubMed] [Google Scholar]