

SUMMARY
Little is known of the brain mechanisms that mediate sex-specific autism symptoms. Here, we demonstrate that deletion of the autism spectrum disorder (ASD)-risk gene, Pten, in neocortical pyramidal neurons (NSEPten knockout [KO]) results in robust cortical circuit hyperexcitability selectively in female mice observed as prolonged spontaneous persistent activity states. Circuit hyperexcitability in females is mediated by metabotropic glutamate receptor 5 (mGluR5) and estrogen receptor α (ERα) signaling to mitogen-activated protein kinases (Erk1/2) and de novo protein synthesis. Pten KO layer 5 neurons have a female-specific increase in mGluR5 and mGluR5-dependent protein synthesis. Furthermore, mGluR5-ERα complexes are generally elevated in female cortices, and genetic reduction of ERα rescues enhanced circuit excitability, protein synthesis, and neuron size selectively in NSEPten KO females. Female NSEPten KO mice display deficits in sensory processing and social behaviors as well as mGluR5-dependent seizures. These results reveal mechanisms by which sex and a high-confidence ASD-risk gene interact to affect brain function and behavior.
Graphical abstract
In brief
Molinaro et al. report enhanced cortical circuit excitability, selectively in females, in mouse models of Pten deletion. Hyperexcitability is driven by mGluR5 and ERα signaling to ERK and protein synthesis. Enhanced mGluR5-ERα interactions and enhanced mGluR5-dependent protein synthesis in Pten KO neurons may drive female-specific circuit effects.
INTRODUCTION
Autism spectrum disorder (ASD) is diagnosed four times more often in males than in females.1 However, increasing evidence indicates that ASD is expressed differently in males and females on a behavioral level and affects brain function and connectivity differently. Because of these sex differences, it has been suggested that ASD is not as well detected in females with current ASD diagnostic tests and is more prevalent in females than previously recognized.2–7 Some symptoms and co-morbidities, such as deficits in sensory processing and epilepsy, are more severe or common in females with autism.8–11 There is little known of the sex-dependent brain mechanisms that interact with ASD-linked genes and how ASD genes affect female brain physiology and behaviors differently than in males. One prominent ASD gene associated with sex-dependent interactions, at least in cancer, is the Phosphatase and tensin homolog deleted on chromosome 10 (PTEN), a lipid phosphatase that dephosphorylates phosphatidylinositol triphosphate and inactivates phosphatidylinositol 3-kinase (PI3K) and its downstream signaling to Akt and mammalian target of rapamycin (mTOR).12,13 PTEN was first discovered as a tumor suppressor and subsequently linked to ASD.14 Individuals with germline PTEN mutations display variable and complex phenotypes including macrocephaly, autism, intellectual disability (ID), and epilepsy that are collectively termed PTEN hamartoma syndrome (PTHS).12,15–18 Importantly, mutations in PTEN or other suppressors of the PI3K/Akt/mTOR pathway are the most common causes of breast cancers; cancers that are ~100-fold more prevalent in women than in men.19 This sex bias is because 70%–80% of breast cancers express estrogen receptor α (ERα) and depend on estrogen signaling for growth.20 Mutations in PTEN or other genes that lead to hyperactivation of Akt/mTOR in breast cancer cells result in enhanced ERα function.21–23 Thus, combination therapies that inhibit ERα and PI3K/mTOR are most effective for breast cancer.23
Although there is no bias toward females in ASD with germline PTEN mutations, as there is with breast cancer, the rate of ASD in females with PTEN mutations is high (47%) and similar to that of males (66%).17 A mouse model of PTHS, a germline heterozygous deletion of Pten, displays female-specific deficits in social behavior.18,24,25 Similarly, human relevant Pten mutation in mice, leading to a cytoplasmic predominant expression of PTEN (Ptenm3m4), results in a male-selective enhancement in sociability.26 The sex-dependent effects of PTEN on brain physiology are unknown, nor are the molecular mechanisms that lead to sex-dependent behavior deficits with PTEN deletion. ERα is expressed in brain, including cortical neurons, where it has important regulatory functions on different processes such as cognition, anxiety, and addiction.27,28 Many of the effects of estrogen on neurons and behavior are mediated by ERα and rely on activity of metabotropic glutamate receptor 5 (mGluR5), a Gq-coupled glutamate receptor and candidate therapeutic target in ASD. If or how PTEN deletion in neurons affects ERα or mGluR5 function or whether these signaling pathways contribute to brain phenotypes in PTEN loss-of-function models is also unknown.
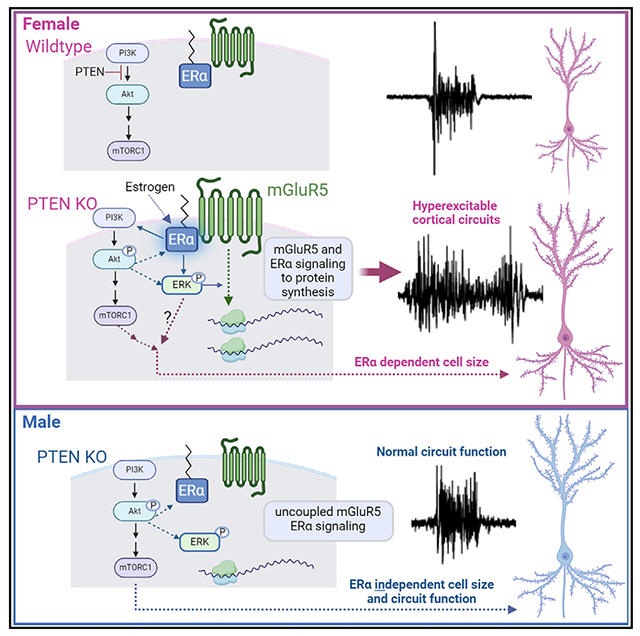
To explore the effects of Pten deletion on brain function, we studied an established mouse model of PTEN deletion with ASD-relevant behaviors, NSEPten knockout (KO).29 NSEPten KO mice have a mosaic deletion of PTEN, primarily in the excitatory neurons in cortical layers 3–5 (L3–L5). To assess overall cortical circuit function, we measured spontaneous persistent activity states, or UP states, in acute slices of the somatosensory cortex. UP states are driven by the activity of local cortical circuits and are an overall readout of the balance of excitation and inhibition. UP states also reflect the state of maturation of cortical circuits.30,31 We have shown that UP-state duration is prolonged in another mouse model of ASD, the Fmr1 KO model of fragile X syndrome, reflecting a circuit hyperexcitability. Surprisingly, we observed prolonged UP states in female, but not in male NSEPten KO mice in contrast to our previous work in male Fmr1 KO mice. Prolonged UP states in female NSEPten KO are mediated by enhanced signaling of mGluR5 and ERα and required de novo protein synthesis. We find that Pten deletion in NSE-cre expressing neurons results in enhanced mGluR5 levels, mGluR5- and ERα-driven protein synthesis, and mGluR5-dependent seizures, in females but not in males. Notably, genetic reduction of ERα in the same neurons corrects circuit hyperexcitability, enhanced protein synthesis, and cell size, again selectively in females. The prolonged cortical circuit activity we observe in slices from female NSEPten KO is associated with a female-selective deficit in temporal processing of sensory stimuli as measured with electroencephalography (EEG) recordings in vivo as well as deficits in social behavior. Female-specific cortical circuit hyperexcitability and mGluR5-dependent seizures are also observed in a mouse model of PTHS. Our results suggest a model in which deletion of PTEN results in a female-specific enhancement of mGluR5-ERα function that drives cortical hyperexcitability through ERK and de novo protein synthesis. These results may suggest distinct therapeutic strategies for males and females with PTEN mutations and highlight the importance of studying ASD-risk genes in both male and female brain physiology.
RESULTS
Hyperexcitable sensory neocortical circuits in female mouse model of Pten deletion
To determine the effects of Pten deletion on cortical circuit function, we utilized a previously characterized mouse in which Pten is deleted with Neuron-specific enolase (NSE) promoter-driven cre (NSEPten KO). In brain, NSE-Cre is expressed postnatally (~postnatal day 2 [P2]) in discrete mature neuronal populations in L3–L5 as well as in neurons in the CA3 region and dentate gyrus of the hippocampus.29 In cortex, NSE-Cre is most abundantly expressed in sensory areas, and the vast majority of neurons are excitatory based on γ-aminobutyric acid staining (>99%). Importantly, NSEPten KO mice show ASD-relevant behaviors such as deficits in social behavior, learning and memory, and sensory hypersensitivity, and thus may be a good model for the study of ASD-related circuit alterations with PTEN deletion.29 To assess sensory cortical circuit function, we performed extra-cellular multi-unit recordings of spontaneous persistent activity, or UP states, in acute slices of somatosensory barrel neocortex from P18–P25 male and female mice (Figure 1A). Mice were either NSE-Cre(+)/Ptenfl/fl or NSE-Cre(−) Ptenfl/fl or Ptenfl/+ littermates, indicated in the figures as Cre(+) or Cre(−), respectively (Figure 1B). UP states are spontaneous, oscillatory (0.5–1 Hz), synchronized firing of neocortical neuron networks driven by recurrent excitatory and inhibitory synaptic circuits and provide a readout of the intact functioning of neocortical circuits.32,33 The duration of UP states measured in L4, was increased by 56% in female slices compared to Cre(−) controls (Figure 1C). In contrast, slices from Cre(+) males had normal UP-state duration. There was no difference in UP-state duration between male and female Cre(−) mice. Similar results were obtained when recordings were performed in L2/3 or L5 (Figure 1C). UP states were longer in Cre(+) females, but not in males, as compared to Cre(−) controls, and there was a significant interaction of sex and genotype on UP-state duration in both L2/3 (two-way ANOVA; F(1, 73) = 5.555; p < 0.05) and L5 (F(1, 71) = 7.332; p < 0.01). There was no effect of sex or genotype on the amplitude or frequency of UP states in any layer (Table S1). Because similar results were obtained in all layers, subsequent experiments were focused in L4.
Figure 1. Female-specific cortical circuit hyperexcitability in acute slices of NSEPten KO mice.
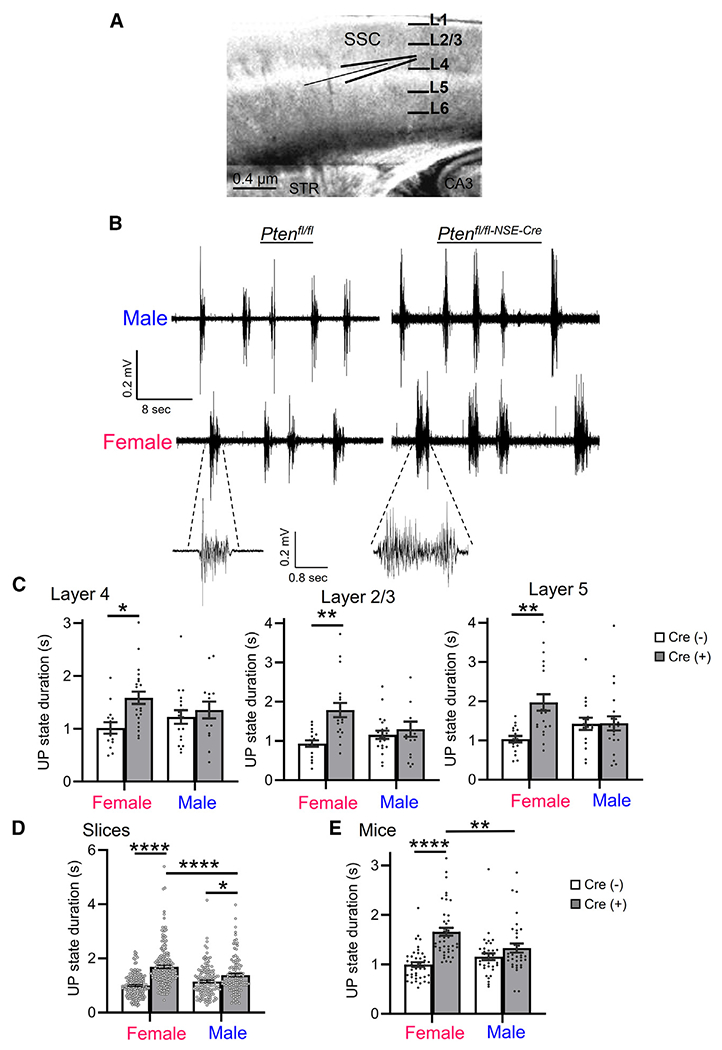
(A) Schematic of thalamocortical slice of somatosensory cortex and extracellular multi-unit recording in layer 4 (L4).
(B) Representative spontaneous, persistent activity states, or UP states from extracellular recordings in L4 of male and female Ptenfl/fl Cre(−) and Ptenfl/fl-NSE-Cre Cre(+) somatosensory cortex.
(C) Group average UP-state duration in male and female Cre(−) and Cre(+) mice in L4, L2/3, and L5. Each black dot is an individual slice from 4–6 mice/sex/genotype.
(D and E) UP-state duration reported as average per slice (D) or per mouse (E). Each black dot is an individual slice (D) or mouse (E) from 35–45 mice/sex/genotype.
Data are presented as mean ± SEM; two-way ANOVA with Šídák’s test for multiple comparisons. *p < 0.05, **p < 0.01, ****p < 0.0001.
In some experiments, we did observe increases in UP-state duration in male Cre(+) mice as compared to male Cre(−) mice. Combining data from vehicle or untreated slices across experiments, we observed a small (~20%) genotypic increase in UP-state duration in males (Cre(−): 1,145 ± 45 ms, n = 148 slices; Cre(+): 1,383 ± 62 ms, n = 114; p < 0.05) (Figure 1D). However, the genotypic increase in UP-state duration in females was large, ~70% (Cre(−): 997 ± 31 ms, n = 175; female Cre(+): 1,689 ± 58 ms, n = 176; p < 0.001)and durations were longer than in Cre(+) males (p < 0.001). No difference in UP-state duration was observed between Cre(−) males and females. This resulted in a significant interaction of sex × genotype on UP-state duration (F(1, 609) = 20.47; p < 0.0001). Averaging UP-state duration from all slices per mouse yielded similar results (Figure 1E) and a significant sex × genotype interaction (F(1, 153) = 11.58; p < 0.001).
The female-specific effects on UP-state duration were not attributable to sex-dependent differences in deletion of PTEN in NSEPten KO mice as measured with fluorescent immunohistochemistry or western blotting. Reflecting the expression pattern of NSE-Cre, we observed a mosaic deletion of PTEN in L5 neurons as indicated by NeuN labeling that was similar in males and females (female Cre(+): 63% ± 2% of NeuN+ L5 neurons, n = 14 slices/3 mice; male Cre(+): 64% ± 2%, n = 10 slices/3 mice; Figures S1A and S1B). Total PTEN levels were quantified in L5-enriched cortical lysates, which revealed similar decreases of PTEN in male and female Cre(+) mice (female Cre(+); 76% ± 5%, n = 10 mice; male Cre(+): 72% ± 6% of same-sex Cre(−), n = 6 mice; Figures S1C and S1D). Taken together these results suggest sex-specific circuit hyperexcitability in female Pten-deleted cortex.
Pharmacological antagonism of mGluR5 rescues UP states in female Cre(+) mice
Genetic or pharmacological reduction of mGluR5 activity rescues prolonged UP states in Fmr1 KO mice.34 To test whether mGluR5 activity mediates the longer UP states in female Cre(+) mice, we examined the effects of a specific mGluR5-negative allosteric modulator, 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP). Slices from Cre(+) mice or Cre(−) controls of both sexes were pre-treated with vehicle or MTEP (3 μM). We confirmed a robust increase in UP-state duration in female Cre(+) mice (218% of Cre(−) controls), which was reduced to normal levels with MTEP treatment. UP-state duration in Cre(−) females was unaffected by MTEP, and there was a significant interaction of MTEP with genotype (F(1, 110) = 12.05; p < 0.01; Figures 2A and 2B). In males, there was a small increase in UP-state duration in Cre(+) mice (139% of Cre(−) controls; Figure 2B) that was unaffected by MTEP. These results suggest a sex-specific gain of function of mGluR5 in female Cre(+) mice that drives circuit hyperexcitability.
Figure 2. Prolonged UP states in female NSEPten KO mice are rescued by an mGluR5-negative allosteric modulator and are associated with increased mGluR5 in cortical layer 5.
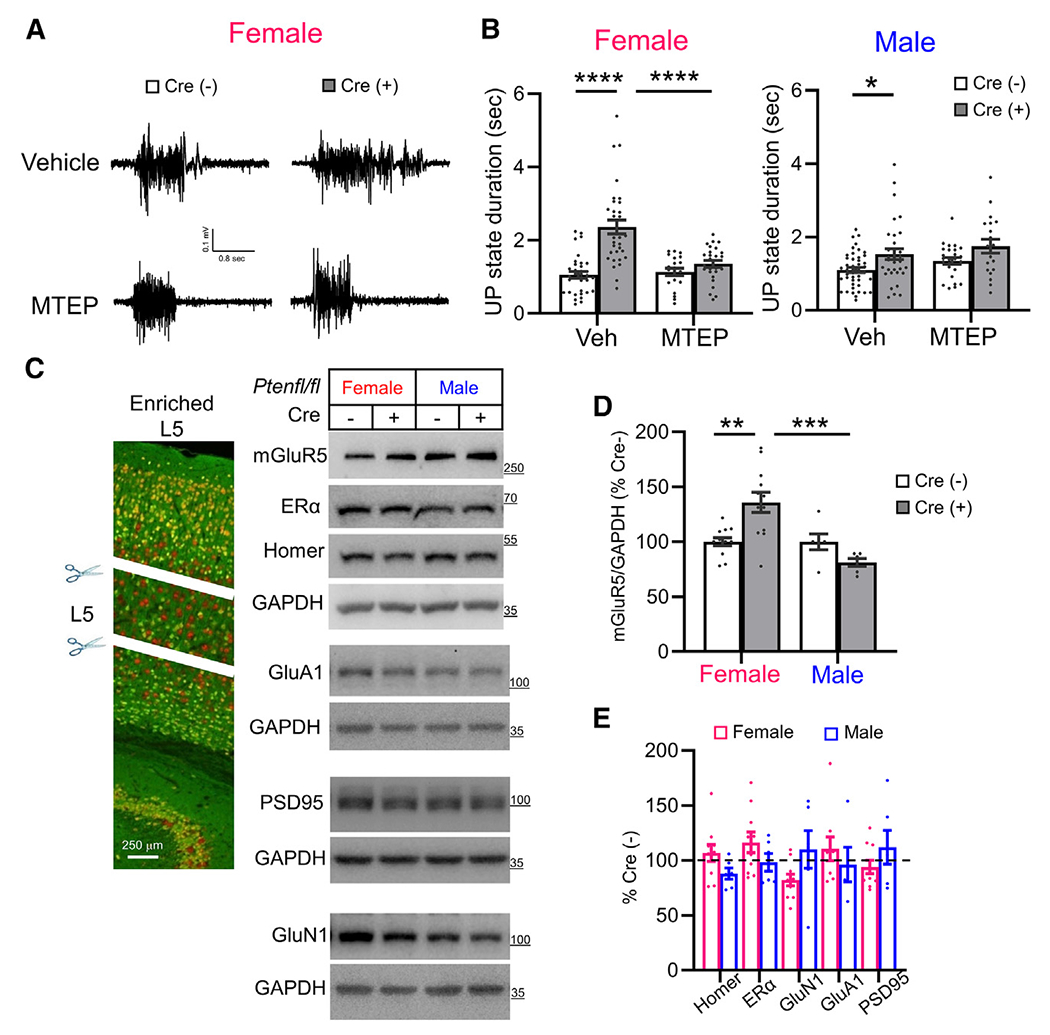
(A and B) The selective mGluR5 negative allosteric modulator, MTEP (3 μM; 2 h), corrected UP-state duration only in female Cre(+) mice without affecting UP states in Cre(−) mice. (A) Representative UP states from female Cre(−) and Cre(+) slices. (B) Group data with n = 25–45 slices from 4–8 mice/genotype/sex.
(C and D) Representative western blots (C) and Quantified group data (D) show that mGluR5 levels are elevated in L5 (microdissected from slices of NSEPten KO mice) of female, but not of male, Cre(+) mice as compared to Cre(−) same-sex littermates. mGluR5, Homer, and ERα were immunoblotted on the same blot.
(E) Quantification of mGluR5 interacting proteins, Homer and ERα, AMPA or NMDA receptors, or PSD-95 revealed no changes in L5 of Cre(+) mice of either sex.
Each black dot in (D) and (E) represents an individual mouse
Data are presented as mean ± SEM; two-way ANOVA with Šídák’s test for multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.0005, ****p < 0.0001.
mGluR5 expression is selectively enhanced in female Cre(+) mice
To determine whether mGluR5 levels were altered with Pten deletion, we performed western blotting of lysates from somatosensory cortical slices of NSEPten KO mice whereby we attempted to enrich for Pten KO neurons by microdissection of lower cortical layers, including L5. Western blots of cortical L5-enriched lysates revealed a small but significant (35%) increase in mGluR5 in Cre(+) female mice as compared to Cre(−) controls. mGluR5 protein levels were unaffected in male Cre(+) mice, and there was a significant interaction of sex × genotype (F(1, 32) = 12.80; p < 0.01; Figures 2C and 2D). The increase of mGluR5 in Cre(+) female lysates was not associated with changes in protein levels for other glutamate receptors (GluN1 or GluA1) or excitatory synaptic proteins (PSD-95 or Homer) (Figure 2E). These data suggest a female-specific upregulation of mGluR5 protein in PTEN KO neurons. In Fmr1 KO mice, mGluR5 is less associated with its binding partner Homer, a scaffolding protein that regulates its localization to synapses, signaling to effectors and cortical circuit excitability.35,36 To determine whether mGluR5 is dissociated from Homer in NSEPten KO mice, we performed co-immunoprecipitation (coIP) of Homer and mGluR5 from lysates of either whole or L5-enriched cortical lysates from male and female mice. In contrast to Fmr1 KO, we were not able to detect differences in mGluR5 coIP with Homer in Cre(+) mice as compared to their same-sex Cre(−) controls in either whole or L5-enriched cortical lysates (Figures S2A and S2B). This suggests other mechanisms of mGluR5 dysfunction in NSEPten KO females or that we are unable to detect changes with a bulk coIP method because of the mosaic nature of Pten deletion in the NSEPten KO mice.
Antagonism or genetic reduction of ERα in L5 neurons rescues prolonged UP states in female NSEPten KO mice
A large body of evidence indicates that mGluR5 functionally interacts with ERα specifically in the female brain. mGluR5 mediates the rapid non-genomic effect of ERα on multiple neuronal functions including synaptic transmission, cognition, and feeding behavior, among others.37–40 Furthermore, mutations in PTEN in breast cancer cells result in enhanced ERα function through phosphorylation of ERα by either by Akt or S6K.21–23 To determine whether ERα activation contributes to circuit dysfunction in female Cre(+) somatosensory cortex, we used two structurally distinct antagonists of ERα, methyl-piperidino-pyrazole (MPP; 1 μM)41,42 or GNE-149.43 In females, MPP or GNE-149 rescued the increase in UP-state duration in Cre(+) mice and had no effect on UP states in Cre(−) mice Figures 3A and 3B). There was also a significant interaction of genotype with MPP (F(1, 79) = 8.291; p < 0.01) or GNE-149 (F(1, 65) = 8.682; p = 0.01) on UP-state duration. Neither MPP nor GNE-149 affected UP-state duration in slices from Cre(+) male mice (Figure S3A). These results suggest that, like mGluR5, there is a female-selective signaling of ERα in Cre(+) mice that promotes UP-state duration. To confirm the role of ERα in UP states using a genetic strategy and determine whether increased ERα function occurred in the NSE-Cre(+) cortical neurons with Pten deletion, we obtained floxed Esr1 mice and bred them to create mice with reduced ERα levels in NSE-Cre(+) and Pten-deleted neurons (NSE-Cre/Ptenfl/fl/Esr1fl/+) and measured UP states in these mice in comparison to NSE-Cre/Ptenfl/fl and Cre(−) littermates. In females, UP-state duration in slices from NSE-Cre/Ptenfl/fl/Esr1fl/+ mice was reduced compared to NSE-Cre/Ptenfl/fl littermates and equal to durations measured in Cre(−) controls (Figures 3C and 3D). In contrast, UP-state duration in male Cre(+) mice was unaffected by genetic reduction of ERα (Figure 3D). Heterozygosity of ERα in cortical neurons without PTEN deletion (NSE-Cre/Pten+/+/Esr1fl/+) had no effect on UP-state duration in females (Figure S3B). These results suggest that ERα is either overactive and/or dysfunctional in Pten-deleted NSE-Cre(+) neurons, which promotes cortical circuit hyperexcitability in females but not in males. This also suggests a cell-autonomous regulation of ERα by PTEN in NSE-Cre(+) neurons that contributes to circuit dysfunction.
Figure 3. Estrogen receptor α is required in L5 neurons for prolonged UP states and is more associated with mGluR5 in females.
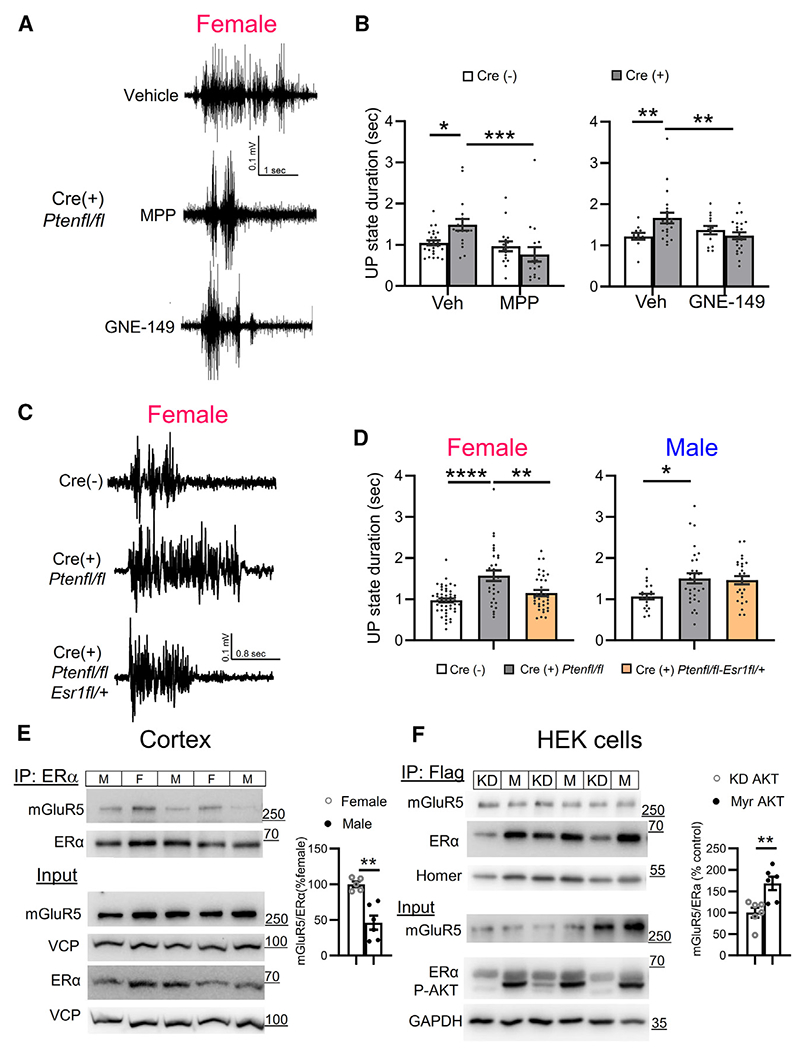
(A and B) Representative UP states (A) and group data (B) show that two ERα-selective antagonists (MPP and GNE, 1 μM; 1.5–2 h) reduce UP-state duration in slices from Cre(+) female mice. n = 16–32 slices from 4–8 mice/genotype.
(C) Representative UP states from female NSE-Cre/Ptenfl/fl(Cre(+)) and Cre(−) mice and NSE-Cre/Ptenfl/fl/Esr1fl/+, with a genetic reduction of ERα in NSE-Cre(+) neurons.
(D) Genetic reduction of ERα expression reduces UP-state duration in female (left), but not in male (right), Cre(+) mice. n = 20–40 slices from 3–8 mice/sex/genotype.
(E) Left: representative western blots (left) and quantified group data (right) show that cortical ERα-mGluR5 physical interactions (coIP) are enhanced in female as compared to male wild-type mice (n = 5–6).
(F) Active AKT increased association of ERα and mGluR5 in HEK cells. Representative western blots (left) and quantification (right) show coIP of FLAG-mGluR5 and HA-ERα from lysates of HEK-293T cells co-expressing either constitutively active AKT (myrAKT; M) or a kinase dead (KD) AKT. n = 6.
Data are presented as mean ± SEM; two-way ANOVA with Šídák’s test for multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.0005, ****p < 0.0001.
mGluR5-ERα complexes are enhanced in female cortex and in response to active Akt
ERα mediates many non-genomic effects of estradiol by physically or functionally interacting with mGluR1 and mGluR5 in brain regions such as the striatum and hippocampus.39,44 To determine whether mGluR5 physically interacts with ERα in cortex and whether this is regulated by sex, we performed coIP experiments and observed an association of mGluR5 with ERα in cortical lysates that was enhanced, by ~2-fold in female wild-type (WT) mice as compared to males (Figures 3E and S3C). Total mGluR5 and ERα levels were not different between sexes. In subcellular fractionation experiments, we observed that mGluR5 was present in both nuclear and non-nuclear fractions from cortical lysates and that ERα was primarily non-nuclear, as previously shown45,46 (Figure S3D). This suggests that ERα and mGluR5 interact in a non-nuclear complex in cortex. To determine whether mGluR5-ERα interaction was affected by Pten, we performed a coIP of ERα and mGluR5 from total cortical lysates of Cre(+) and Cre(−) male and female mice (Figures S3E and S3F). We observed a non-significant main effect of genotype (F(1, 20) = 4.251; p = 0.056) on mGluR5-ERα interaction, with a tendency to increase in Cre(+) mice, but did not observe sex-specific effects. The mosaic nature of the NSE-Cre(+) deletion of PTEN in cortical neurons likely prohibits us from detecting molecular changes in whole cortical homogenates. We attempted to co-immunoprecipitate mGluR5- ERα from microdissected cortical slices to enrich for PTEN KO neurons but encountered technical challenges. To overcome challenges of working with endogenous ERα, we expressed ERα together with mGluR5 and Homer2 in HEK293 cells. As we observed in the brain, mGluR5 and ERα interacted in a complex (Figure S3G). We observed that co-expression of Homer2 increased mGluR5-ERα interaction (Figure S3H), so we co-expressed Homer2 in this assay. To mimic the effect of Pten knockdown, we overexpressed a constitutively active form of AKT (myrAKT) or a kinase-dead form of AKT (KD-Akt; K179M).47 myr-AKT increased the association of ERα and mGluR5 (Figure 3F). These results predict that PTEN deletion and enhanced Akt activity increase mGluR5 association with ERα.
Acute inhibition of ERK activation or protein synthesis corrects UP-state duration in female NSE-Cre PTEN KO mice
Candidate signaling pathways downstream of mGluR5 that may regulate circuit excitability include mTOR complex 1 (mTORC1), the mitogen-activated kinase ERK, and de novo protein synthesis.48–55 To test the role of these candidate pathways in prolonged UP states in female Cre(+) mice, slices were pretreated with either an inhibitor of mTORC1 (rapamycin; 200 nM), or the upstream activating kinase of ERK, MEK, U0126 (20 μM), or one of two different protein synthesis inhibitors (anisomycin [20 μM] or cycloheximide [60 μM]; Figures 4A and 4B). Surprisingly, rapamycin pre-treatment (200 nM, 2 h) had no effect on UP-state duration in female Cre(−) or Cre(+) mice and failed to correct the prolonged UP states (Figures S4A and S4B). U0126 reduced and corrected UP-state duration in Cre(+) females compared to that observed in Cre(−) females (Figures 4A and 4B). There was no effect of U0126 on UP-state duration in Cre(−) females. Similarly, anisomycin and cycloheximide both corrected UP-state duration in Cre(+) females and had no effect on UP states in Cre(−) controls (Figures 4A–4D). Thus, there was a significant interaction of inhibitor with genotype on UP-state duration for both anisomycin (F(1, 57) = 5.236; p < 0.05) and cycloheximide (F(1, 55) = 8.317; p < 0.01). In males, although there was no effect of genotype on UP-state duration, there was a main effect of U0126 to reduce UP-state duration (F(1, 51) = 6.076; p < 0.05), and this was significant in Cre(+) but not in Cre(−) mice (Figure S4C). Similarly, there were main effects of anisomycin (F(1, 76) = 15.01; p < 0.001) and cycloheximide (F(1, 41) = 8.443; p < 0.01) to reduce UP-state duration in males, but this reached statistical significance only in Cre(+) and not in Cre(−), males (Figures S4D and S4E). Although the UP-state durations are not robustly increased in male Cre(+) mice, the UP states are sensitive to ERK and protein synthesis inhibitors. Taken together, our results suggest that ERK signaling and protein synthesis downstream signaling pathways from mGluR5 and ERα promote female-selective cortical hyperexcitability in Cre(+) mice.
Figure 4. Inhibitors of ERK activation and de novo protein synthesis correct prolonged UP states in female NSEPten KO mice.
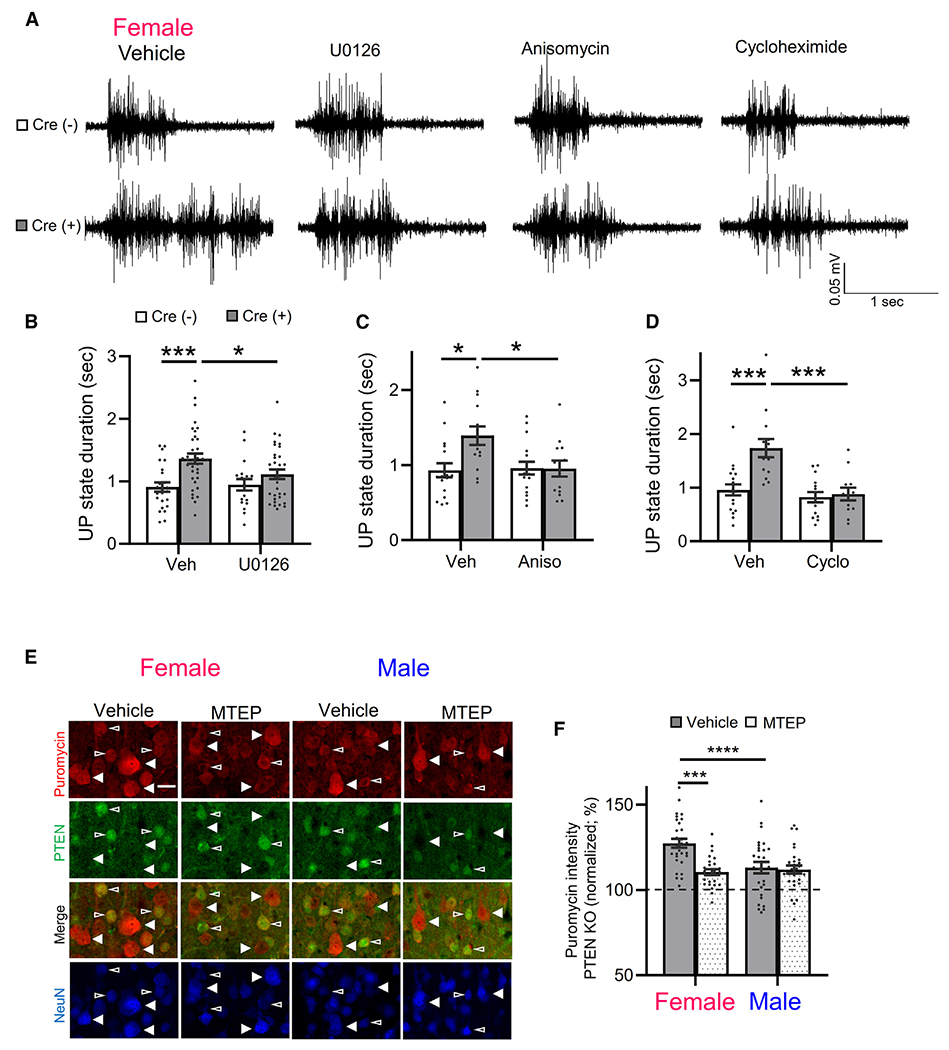
(A) Representative UP states from female Cre(−) and Cre(+) mice pre-incubated in vehicle, U126 (20 μM, 2 h), anisomycin (20 μM, 1 h) or cycloheximide (60 μM, 1 h).
(B–D) Group averages of UP-state duration in inhibitors of ERK activation or protein synthesis in female Cre(+) and Cre(−) mice. n = 12–30 slices from 3–6 mice/genotype/condition.
(E) Representative images of puromycin immunolabeling (red) of L5 neurons in cortical sections from male and female Cre(+) mice, pre-treated with vehicle or MTEP (3 μM, 2 h). NeuN (blue) and PTEN (green) immunolabeling identify PTEN KO (filled arrows) and neighboring PTEN+ neurons (open arrows). Scale bar, 30 μm.
(F) Quantified group data of puromycin fluorescent intensity of PTEN KO neurons normalized to neighboring PTEN+ neurons. n = 26–30 sections from 4 mice/sex.
Data are presented as mean ± SEM; two-way ANOVA with Šídák’s test for multiple comparisons. *p < 0.05, ***p < 0.0005, ****p < 0.0001.
From these results, we hypothesize that there is an enhanced mGluR5 signaling to protein synthesis in female Cre(+) mice that promotes cortical circuit excitability. To test this hypothesis, we measured bulk protein synthesis in male and female cortical neurons with and without Pten deletion using surface sensing of translation (SUnSET) in the presence and absence of MTEP. SUnSET measures de novo protein translation by incorporation of a tRNA analog, puromycin.56 Puromycin incorporation into newly synthesized protein is detected with a puromycin antibody and fluorescence immunohistochemistry (IHC).57 Cortical slices from Cre(+) male and female were acutely prepared as for UP-state recordings, incubated in puromycin (40 min) with or without MTEP (3 μM), and processed for IHC. Neurons were identified using an antibody for NeuN or NeuroTrace (Nissl), and NSE-Cre(+), Pten-deleted (KO) L5 neurons were identified with a PTEN antibody. The mosaic nature of NSE-Cre expression allowed us to compare puromycin incorporation in the soma of neighboring Cre(−) or “WT” neurons, identified by positive staining for PTEN, and Cre(+) or “KO” neurons that were negative for PTEN in the same section. Pre-incubation of slices in anisomycin strongly reduced puromycin labeling, indicating that it is reflecting de novo protein synthesis (Figure S4F). Analysis of puromycin fluorescence intensity revealed that PTEN KO L5 neurons in females have enhanced puromycin intensity as compared to neighboring WT neurons (127% ± 3% of WT; n = 30 sections/4 mice), and this increase was reduced by MTEP treatment (113% ± 3%, n = 27/4 mice; Figures 4E and 4F). Using a Cre-reporter (TdTomato) mice, to identify Cre(+) neurons, we determined that puromycin intensity is not elevated in female NSE-Cre(+) L5 neurons without Pten deletion (Figures S4G and S4H). Therefore, the increase in protein synthesis in PTEN KO neurons is a result of Pten deletion and not an artifact of NSE-Cre expression. In male mice, protein synthesis, as measured by puromycin intensity, was elevated in PTEN KO L5 neurons (110% ± 2% of neighboring WT, n = 26; p < 0.0001; one-sample t test), but this increase was less than observed in female PTEN KO neurons (p < 0.0001; šídák’s multiple comparison) and unaffected by MTEP (112% ± 2%; n = 29). As a result, there was a significant interaction of MTEP and sex on puromycin intensity in PTEN KO neurons (F(1, 108) = 9.293; p < 0.01; Figure 4F). These results indicate that mGluR5 activity drives protein synthesis in PTEN KO neurons but does so only in females. Because inhibition of ERK signaling with U0126 rescued UP-state duration in female Cre(+) mice, but rapamycin did not, we tested whether U0126 or rapamycin reduced protein synthesis in PTEN KO neurons in female mice (Figures S4I and S4J). Surprisingly, we observed that both U0126 and rapamycin pre-treatment of slices reduced puromycin incorporation into PTEN KO neurons to a similar degree. These results suggest that inhibition of bulk protein synthesis does not correlate with rescue of the circuit hyperexcitability and suggest that ERK-dependent translation of specific transcripts in PTEN KO neurons drives circuit excitability.
Genetic reduction of ERα levels reduces protein synthesis and cell size in PTEN KO L5 neurons from female mice
Estrogen and ERα promote mRNA translation in neurons and in cancer cells.58–60 To determine whether ERα heterozygosity reduced protein synthesis in PTEN KO neurons, we measured puromycin incorporation in neighboring WT (PTEN+) and KO (PTEN−) L5 neurons in the cortices of female NSE-Cre/Ptenfl/fl and NSE-Cre/Ptenfl/fl/Esr1fl/+ mice (Figures 5A and 5B). PTEN KO L5 neurons had a 129% ± 3% increase in puromycin intensity as compared to neighboring WT (Cre(−)) neurons (n = 17 sections/3 mice). ERα heterozygosity in PTEN KO neurons reduced puromycin intensity to 118% ± 4% of WT (n = 17; p < 0.05), suggesting that ERα contributes to the enhanced protein synthesis in female PTEN KO neurons. To determine whether ERα contributed to other cellular phenotypes of PTEN KO neurons in a sex-specific manner, we measured the size of their soma.29 The soma size of PTEN KO L5 neurons was ~40% larger than neighboring WT neurons in both males and females (Figures 5C–5F). ERα heterozygosity reduced the soma size of PTEN KO neurons in females but had no effect in males (Figures 5C–5F). PTEN deletion results in activation of the mTORC1-ribosomal S6 kinase (p70S6K) pathway, observed by phosphorylation (P) of ribosomal protein S6 (P-S6; Ser235/23629). This pathway may contribute to cell size and enhanced protein synthesis.61–63 In female mice, we observed enhanced P-S6 in PTEN KO L5 neurons in comparison to neighboring WT neurons (Figures S5A and S5B). ERα heterozygosity reduced P-S6 in PTEN KO neurons. However, the percent increase in P-S6 in PTEN KO neurons as normalized to neighboring WT neurons is unchanged by ERα deletion, in contrast to protein synthesis. This may be because of the small, trending decrease in P-S6 in Cre(−) neurons. These results indicate that ERα contributes to the enhanced protein synthesis and cell size of PTEN KO neurons in females. Activation of P-S6 may be contributing to ERα-driven protein synthesis in PTEN KO neurons, but there are likely other signaling pathways that play a role.
Figure 5. ERα promotes protein synthesis and soma size in Pten KO L5 neurons in female mice.
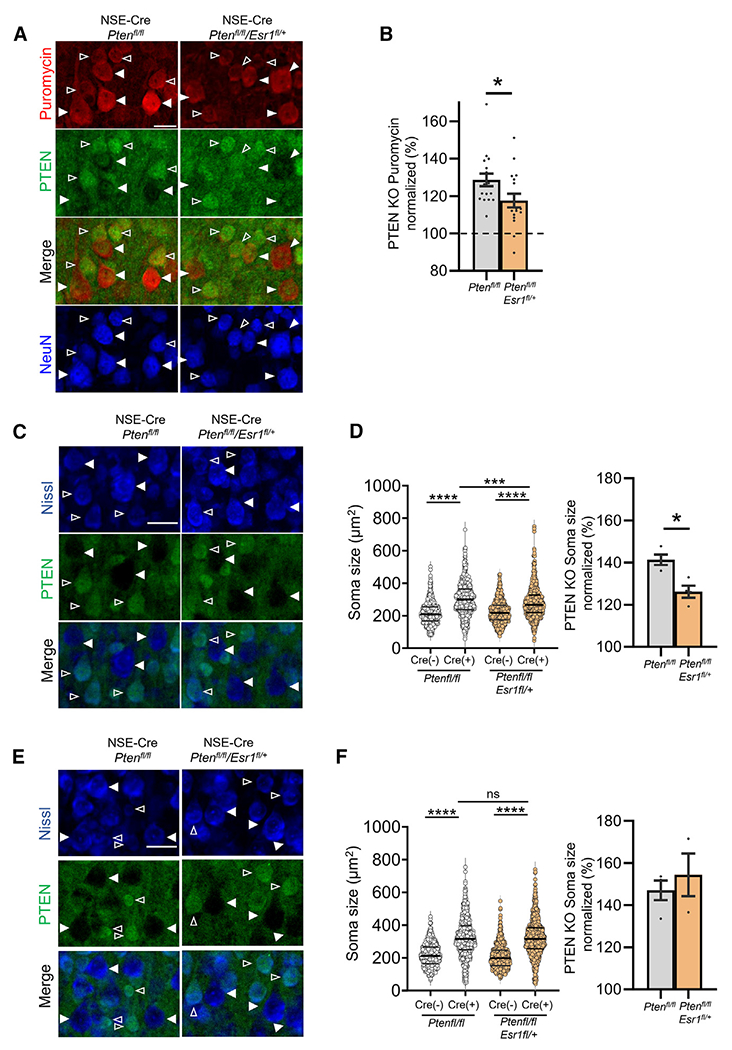
(A and B) Representative images of puromycin immunolabeling (red) of L5 neurons in cortical sections from female NSE-Cre/Ptenfl/fl or NSE-Cre/Ptenfl/fl/Esr1fl/+ mice (A), and quantified group data of PTEN KO L5 neurons normalized to neighboring PTEN+ neurons within each genotype. n = 17 sections from 3 mice/genotype; t test .Scale bar, 50 μm.
(C and E) Representative images of Nissl staining (blue) of L5 neurons in cortical sections from female (C) and male (E) NSE-Cre/Ptenfl/fl or NSE-Cre/Ptenfl/fl/Esr1fl/+ mice. PTEN (green) immunolabeling identifies PTEN KO (filled arrows) and neighboring PTEN+ neurons (open arrows). Scale bar, 40 μm.
(D and F) Raw (left) and normalized (right) mouse averages show an increase in soma size of PTEN KO L5 neurons that is reduced by genetic reduction of Esr1 in females (D) but not in males (F). n = 20–21 sections from 3–4 mice/sex.
Data are presented as mean ± SEM; two-way ANOVA with Šídák’s test for multiple comparisons. *p < 0.05, ***p < 0.0005, ****p < 0.0001. ns, not significant.
Estrogen activates ERK via ERα in neurons, and ERK can regulate mRNA translation through phosphorylation of eIF4E, 4E-binding protein, and other factors.59,64,65 To determine whether ERK signaling is selectively elevated in female PTEN KO neurons and whether this relies on mGluR5 and/or ERα, we performed IHC for phosphorylated (P) ERK (Thr202/Tyr204) in L5 neurons in sections from Cre(+) male and female mice. P-ERK levels were elevated by ~40% in Cre(+) neurons, as compared to neighboring Cre(−) neurons in L5, in both males and females (one-sample Wilcoxon test; Figures 5C–5F). Surprisingly, P-ERK levels were similarly elevated in Cre(+) neurons in slices pre-treated with MTEP from either female or male mice. In contrast, antagonism of ERα with GNE-149 prevented P-ERK increases in female, but not in male, Cre(+) neurons. These results suggest that ERα and mGluR5 may signal through distinct pathways in female PTEN KO neurons to promote protein synthesis and cortical circuit hyperexcitability.
Sex-specific alterations in cortical temporal processing of sensory stimuli
The prolonged cortical UP states in young female NSE-Cre PTEN KO mice suggests altered timing of cortical circuits that may impact temporal processing of sensory stimuli. High-fidelity temporal processing of sound is necessary for speech and language function66–68 and is affected in individuals with ASD.69–72 Individuals with ASD show deficits in detection of sound duration, onset and offset, and rapid changes in spectrotemporal properties.73–75 Impaired temporal gap detection threshold in children are associated with lower phonological processing scores.70 To determine whether there are sex-specific alterations in temporal processing, particularly with rapid gaps in sounds, we performed epidural EEG recordings on the auditory cortex (AC) and frontal cortex (FC) in young (P21–P23) NSEPten KO mice. To address temporal acuity, we used a gap-in-noise auditory steady-state response (gap-ASSR) paradigm.76 The gap-ASSR elicits a steady-state response by presenting short gaps in continuous noise at 40 Hz. Using short gap widths and shallow-amplitude modulation depths (~75%), we compared the limits of temporal processing of cortical circuits across experimental groups. To measure the consistency of cortical responses to the gap-ASSR across trials, we calculated the inter-trial phase clustering (ITPC) at 40 Hz. We also measured ITPC during the ASSR without any gaps and termed this “baseline ITPC.” Both male and female Cre(+) mice showed deficits in ITPC of the gap-ASSR measured in the AC and FC with a brief, 4–6 ms, gap (Figures 6A and 6B). However, in females, the ITPC during the gap-ASSR was not greater than baseline ITPC or when no gaps were present. Male Cre(+) mice had a greater ITPC during the gap-ASSR as compared to baseline. These results suggest that female Cre(+) mice are not consistently able to detect brief gaps in sound, and the cortical responses are similar to noise. Male Cre(+) mice can detect the brief gaps in sound, but their responses are not as consistent as those of Cre(−) male mice. These results suggest deficits in temporal processing of sensory stimuli in both male and female Cre(+) mice, and these deficits are more severe in females.
Figure 6. Deficits in temporal processing of sensory stimuli and mGluR5-dependent seizures female NSEPten KO mice.
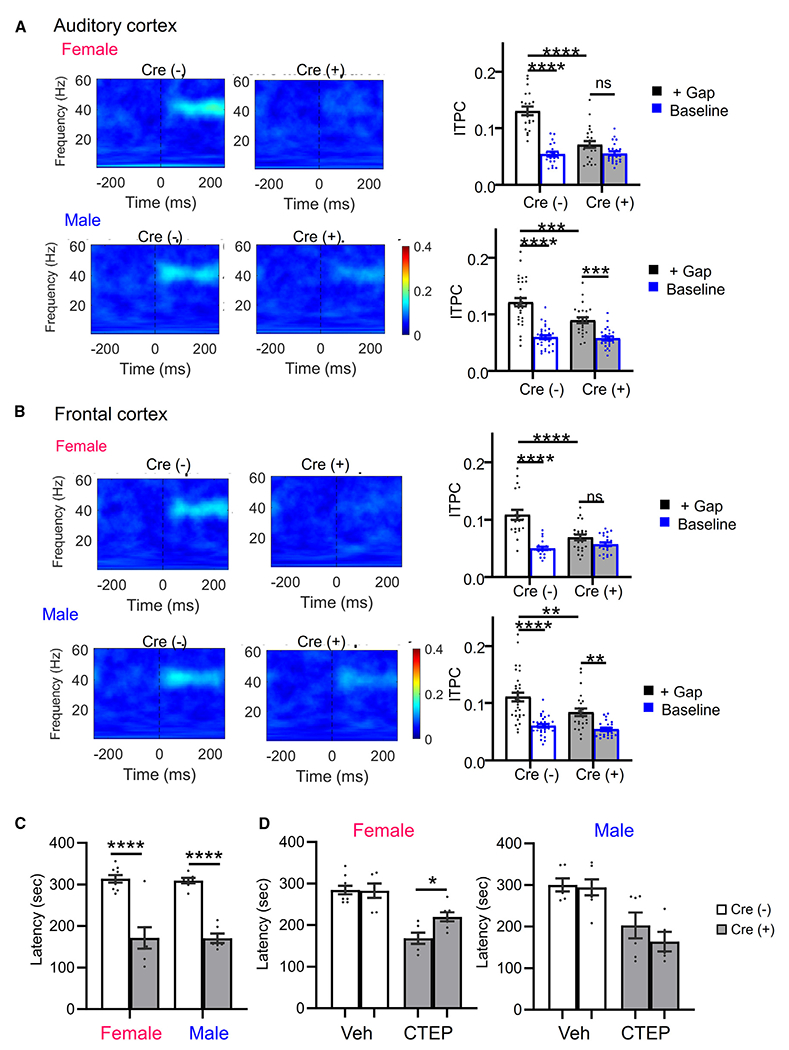
(A and B) (Left) Average ITPC heatmaps of P21 female and male Cre(−) and Cre(+) mice from the auditory cortex (A) or frontal cortex (B). Heatmaps show the average ITPC produced across presentations of the 6 ms/75% modulated gap. (Right) Both male and female Cre(+) mice show ITPC deficits when presented with gap interrupted noise (+Gap). Cre(+) females’ ITPC during the gap-ASSR was not greater than baseline levels (unmodulated noise). Male Cre(+) mice show increased ITPC during the gap-ASSR as compared to baseline.
(C) Latency to myoclonic twitch in response to flurothyl in 6- to 8-week-old male and female Cre(+) and Cre(−) mice.
(D) mGluR5 negative allosteric modulator CTEP increases latency to myoclonic twitch in female (left) but not male (right) Cre(+) mice. n = 6–9 mice/sex/genotype/treatment.
Data are presented as mean ± SEM; two-way ANOVA with Šídák’s test for multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.0005, ****p < 0.0001. ns, not significant.
Seizure susceptibility in female NSE-Cre PTEN KO mice
Our slice results suggest an mGluR5-dependent hyperexcitability of cortical circuits in female NSE-Cre PTEN KO mice. To determine whether there is sex-specific circuit hyperexcitability in vivo, we measured susceptibility to seizures induced with the volatile convulsant flurothyl. The latency to onset to distinct seizure behaviors, myoclonic twitch or tonic-clonic extension, provides a reliable index of seizure threshold in naive mice, and flurothyl seizures are highly penetrant regardless of genetic background.77,78 At 3 weeks of age, similar to the UP-state experiments, neither male nor female Cre(+) mice had an increased susceptibility to flurothyl-induced seizures, indicating that the cortical excitability we observe in female Cre(+) acute slices is not sufficient to affect seizure latency (latency to myoclonic twitch) in female Cre(−): 221.4 ± 14.2 s, n = 7; female Cre(+): 214.2 ± 24.7, n = 5; male Cre(−): 207.5 ± 6.9, n = 4; male Cre(+): 192 ± 3.2, n = 3). However, at 6–8 weeks of age, both male and female Cre(+) mice had reduced latency to myoclonic twitch and tonic-clonic seizure in response to a single flurothyl exposure (Figure 6C). Because hyperexcitability of cortical circuits in female Cre(+) mice is dependent on mGluR5, we tested whether seizure susceptibility was also dependent on mGluR5 by administering the brain penetrant mGluR5 negative allosteric modulator, 2-chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-1H-imidazol-4-yl)ethynyl)pyridine (CTEP; 2 mg/kg intraperitoneally [i.p.]), prior to flurothyl exposure. CTEP increased the latency to myoclonic twitch in female but not in male Cre(+) mice (Figure 6D). CTEP did not affect seizure onset latency in male or female Cre(−) mice. These results indicate that both male and female NSE-Cre PTEN KO mice develop increased susceptibility to seizures as they age, but this is dependent on mGluR5 only in females.
Female-specific behavioral alterations in NSE-Cre PTEN KO mice
Previous work demonstrated ASD-relevant behavioral alterations in NSE-Cre PTEN KO mice such as social interaction deficits, but any sex dependence of these deficits was not reported.29 Female mice with a germline heterozygous deletion of Pten (Pten+/−) display deficits in social preference,24,79,80 and we hypothesized that NSEPten KO mice would exhibit sex-dependent behavioral deficits. To test this hypothesis, we performed a battery of behavioral tests in Cre(+) and Cre(−) male and female mice. In previous work, NSEPten KO mice exhibited deficits in social interaction, enhanced responses to sensory stimuli, anxiety-like behaviors, seizures, and decreased learning, which are features associated with ASD.29 Consistent with this previous work, Cre(+) mice displayed a deficit in the three-chamber social interaction test, but this was only observed in females (Figure S6A). Female Cre(−) mice spent more time sniffing a novel mouse in one chamber as compared to an object in another chamber, whereas female Cre(+) mice sniffed the mouse and object similarly, which resulted in a significant interaction of genotype × chamber (F(1, 42) = 7.340; p < 0.01). In contrast, male Cre(+) mice performed similarly to male Cre(−) littermates and sniffed a novel mouse more than an object (genotype × chamber: F(1, 44) = 0.01; not significant). Female Cre(+) mice also displayed an increase in locomotor activity, observed in dim light (females; genotype main effect: F(1, 31) = 5.874; p < 0.05; Figure S6B). However, in a brightly lit open field, both male and female Cre(+) mice were hyperlocomotive as assessed by total distance traveled, and there was no sex or genotype effect on time spent in the center of open field, a measure of anxiety (Figure S6C). In another measure of anxiety, the dark-light box, both male and female Cre(+) mice spent decreased time in the light side of the box (Figure S6D). These results suggest a tendency for increased anxiety in both male and female Cre(+) mice. To assess potential sex dependence in a learning task, we assayed delay cue and context-dependent fear, or threat conditioning. Female Cre(+) mice displayed enhanced cue-induced fear, as measured by percent of time freezing (Figure S6E). Baseline freezing and context-induced freezing were normal. Male Cre(+) had normal freezing both at baseline and in response to cue or context. These results suggest female-specific effects of NSE-Cre-mediated Pten deletion in mice on social preference, locomotion, and fear similar to other Pten-deletion models.25
Female-specific circuit excitability in a mouse model of PTEN hamartoma syndrome
To determine whether there were sex-dependent alterations in cortical excitability in a mouse model with construct validity for individuals with PTEN mutations, we examined UP states in acute slices of somatosensory cortex from mice with germline haploinsufficiency of Pten (Pten+/−). In contrast to NSEPten KO mice, we did not observe altered UP-state duration in slices from juvenile female, or male, Pten+/− mice (P18–P25; Figure S7A). However, in young adult mice (6–8 weeks), we observed a female-specific cortical circuit hyperexcitability in Pten+/− mice (Figures 7A and 7B) measured as an increase in the number of UP states and total time spent in an UP state (Figures 7A and 7B). In Pten+/− males, UP-state duration was reduced, and the number of UP states and total time spent in an UP state was unchanged. There was a significant interaction of genotype × sex for both UP-state number (F(1, 193) = 5.041; p < 0.05) and time in UP state (F(1, 193) = 7.671; p < 0.01). Surprisingly, WT females had fewer UP states and less time in an UP state when compared to WT males. These results reveal female-selective increase in excitability of cortical circuits with germline haploinsufficiency of Pten, a human-disease-relevant model of loss-of-function mutations in PTEN.
Figure 7. Female-selective circuit excitability and mGluR5-dependent seizure severity in a mouse model of PTEN Hamartoma syndrome.
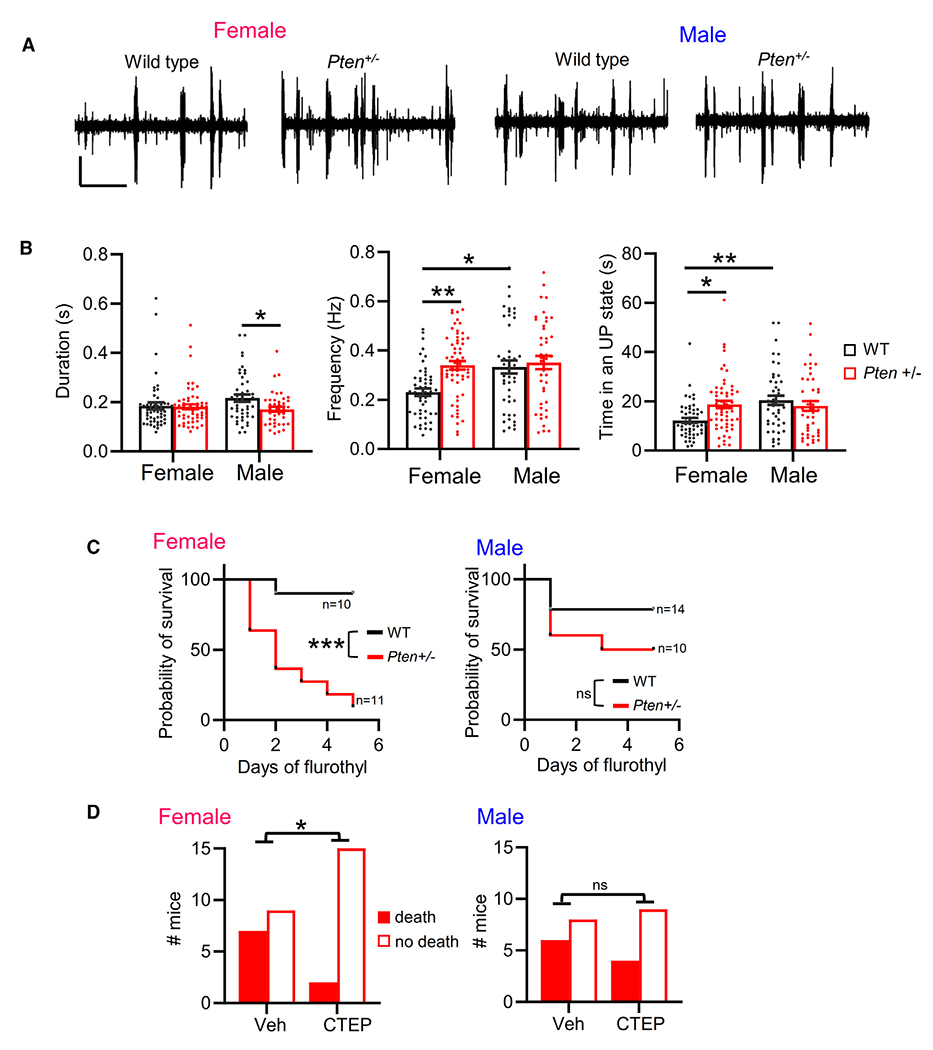
(A) Representative UP states from 6- to 8-week-old male and female Pten+/− mice. Scale bars, 100 μV/7.5 s.
(B) Group averages of UP-state duration, frequency, and total time in an UP state in female and male Pten+/− slices. n = 43–58 slices from 8–12 mice/sex/genotype.
(C) Survival curve of female and male Pten+/− mice (P90–P100) with repeated daily flurothyl exposure. Mantel-Cox test. n = 10–14 mice/sex/genotype.
(D) Survival of female (left) or male (right) Pten+/− mice on first day of flurothyl treatment with or without pre-treatment with CTEP. n = 13–17 mice/sex/treatment condition. Chi-square test.
In (B), data are presented as mean ± SEM; two-way ANOVA with Šídák’s test for multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.0005. ns, not significant.
We next tested whether there was sex-dependent seizure susceptibility in Pten+/− mice in response to flurothyl treatment. We observed a main effect of Pten+/− to reduce the latency for the onset of myoclonic twitch and tonic-clonic seizures but did not observe a female-specific reduction in seizure latency (Figure S7B). However, in a repeated flurothyl treatment paradigm (daily for 5 days), female Pten+/− mice had an increased probability of death during flurothyl treatment as compared to WT females (Figure 7C). Male Pten+/− mice died at a rate similar to that of WT males. To determine whether the flurothyl-induced death was mediated by mGluR5, we pre-treated mice daily with CTEP (i.p.) 90 min prior to flurothyl. CTEP treatment resulted in greater survival of Pten+/− females on the first day of flurothyl exposure and over the course of a 3-day repeated exposure (Figures 7D and S7C). In contrast, CTEP did not affect the survival of male Pten+/− mice. CTEP did not affect the latency to myoclonic twitch or tonic-clonic seizure in male or female Pten+/− mice and had no effect on survival of WT mice with repeated flurothyl (Figures S7C and S7D). These results reveal a female-specific, mGluR5-dependent seizure severity in a mouse model of human PTEN haploinsufficiency.
DISCUSSION
Understanding how ASD genes affect and interact with sex-specific brain functions and signaling pathways will be key to developing more personalized therapeutics for individuals. Pten mutations have a strong female bias for causing breast cancer, but the sex-specific roles of PTEN in the brain are relatively unknown. Here, we identify a female-specific hyperexcitability of cortical circuits and an mGluR5-dependent seizure severity in mice with postnatal deletion of Pten in L5 neurons or with germline heterozygous deletion of Pten. Using the NSEPten KO mouse model, we identified a female-specific, cell-autonomous effect of Pten deletion in L5 neurons that results in enhanced signaling of mGluR5-ERα to protein synthesis and drives hyperexcitability and abnormal timing of developing cortical circuits. This abnormal timing of cortical circuit correlates with reduced temporal processing of sensory stimuli in young female NSEPten KO mice, as measured by the gap-ASSR. Young adult NSEPten KO displayed female-specific ASD-relevant behaviors, such as reduced social interaction, hyperactivity, and enhanced fear learning. Our results reveal a molecular mechanism for the sex-specific effects of loss of function of an ASD-risk gene on neuronal and circuit function and suggest distinct pharmacological strategies, based on sex, for individuals with PTEN mutations and perhaps other genetic causes of ASD.
Female-selective activity of mGluR5 signaling drives excitability in PTEN KO cortical neurons
Our results suggest that either enhanced or altered signaling of mGluR5 and ERα underlies the female-specific effects of Pten deletion. Prolonged UP states, enhanced protein synthesis, and seizure susceptibility in NSEPten KO mice were dependent on mGluR5 in females but not in males. Antagonism or genetic reduction of ERα corrected UP states and protein synthesis in female but not in male NSEPten KO mice. Previous work in rodents has shown that estrogen signals through mGluR5 selectively in female brains, and this contributes to sex-dependent effects of estrogen in drug addiction, learning and memory, and other behaviors.40,81 Consistent with these studies, we observe enhanced mGluR5-ERα complexes in female cortex. In addition, Pten deletion results in a female-specific enhancement of mGluR5 levels in L5-enriched cortical lysates. Together, these results point to enhanced or altered mGluR5 and ERα function or signaling in female PTEN KO neurons, which drives protein synthesis. In further support of this idea, genetic reduction in ERα reduced protein synthesis and cell size in L5 PTEN KO neurons in a cell-autonomous fashion. Thus, our results suggest direct effects of PTEN on ERα function and its coupling with mGluR5 within L5 neurons. Hyperactivation of Akt in breast cancer cells results in phosphorylation and constitutive activity of ERα that further drives activation of PI3K/mTOR and tumor growth.21–23,64 Pten deletion in neurons may result in similar alterations in ERα phosphorylation and activity.
Dysfunction of mGluR5, ERK signaling, and protein synthesis: Convergence of ASD-risk genes
Like female NSEPten KO, prolonged UP states and other measures of hyperexcitability in male Fmr1 KO mice are dependent on mGluR5,34 ERK (Figure S4E),54 and de novo protein synthesis55 (G.M. and K.M.H., unpublished data). Acute antagonism of ERK activation and de novo protein synthesis, but, surprisingly, not mTORC1, corrected long UP states in female NSEPten KO slices. Although rapamycin reduced protein synthesis in PTEN KO neurons, it did not correct circuit excitability, in contrast to the inhibitor of ERK activation. Similarly, in Fmr1 KO, inhibition of ERK, but not mTORC1, has been implicated in circuit hyperexcitability and seizures.53,54,82 This suggests that an ERK-dependent signaling pathway(s) promotes translation of specific mRNA transcripts that lead to circuit hyperexcitability with loss of function of either Fmr1 or Pten. Although inhibitors of ERK suppressed both long UP states and protein synthesis rates in female PTEN KO neurons, P-ERK levels were similarly elevated in both male and female PTEN KO L5 neurons and unaffected by MTEP. This contrasts with the female-selective increases in protein synthesis in PTEN KO L5 neurons that are mGluR5 driven. Instead, our results suggest that ERα promotes ERK signaling in female but not in male PTEN KO neurons. Therefore, mGluR5 and ERα may stimulate distinct signaling pathways to protein synthesis in PTEN KO neurons. Genetic reduction of ERα did not correct P-S6 levels in PTEN KO neurons and may be driven by mGluR5. Additional work is needed to determine whether there are altered mGluR5-ERα complexes in Pten-deleted neurons and their signaling pathways to protein synthesis.
In Fmr1 KO mice, mGluR5 is less associated with its scaffolding protein Homer.35,83 Restoration of mGluR5-Homer interactions in Fmr1 KO mice corrects prolonged UP states, seizures, and some behaviors.36,84 In NSEPten KO mice, we were unable to detect changes in mGluR5-Homer in either sex, which may have been because of the mosaic nature of Pten deletion. Alternatively, mGluR5 may be activated in PTEN KO neurons through its interaction with a hyperactive ERα. However, in both models, dysfunctional mGluR5 drives elevated mRNA translation and cortical circuit hyperexcitability. Deletion of Tsc2, another ASD-risk gene and suppressor of mTORC1 activation, results in mGluR5-dependent hyperexcitability or seizures in mice. The sex dependence of this effect was neither tested49 nor evident.85 As in breast cancer, one may expect dysregulation of ERα in neurons with loss-of-function mutations in suppressors of PI3K and sex-dependent effects on brain function and behavior.
Developmental regulation of PTEN phenotypes and the role of estrogen
The sex-specific, ERα-dependent cortical circuit dysfunction in NSEPten KO was observed in young pre-pubertal mice (P18–P25), when circulating testosterone and estrogen levels are low.86 Our results with MPP suggest that estrogen is necessary to drive ERα activity and long UP states. If so, brain synthesized estrogen may mediate ERα activation in this context. Observing sex-specific effects of Pten-deletion in young mice, before puberty, also suggests that PTEN interacts with mechanisms of sexual differentiation of the brain, a process that occurs during the first few postnatal days in rodents.86 Hyperexcitability of cortical circuits was also observed in female Pten+/− but not until the young adult stage (6–8 weeks). This may be a consequence of the heterozygous expression of Pten that maintains circuit function in developing cortex. Although reductions in seizure latency were not sex dependent in NSEPten KO mice, seizures were sensitive to CTEP only in females. We did observe enhanced seizure severity in female Pten+/− mice as compared to males, which we measured as an increased probability of death. This was also selectively prevented in females by CTEP. These results are consistent with a female-specific, enhanced function of mGluR5 that leads to circuit hyperexcitability with Pten deletion. Epilepsy is more common in females with autism as compared to males,87,88 and brain synthesized estrogen promotes seizures.89 This suggests that there may be roles for estrogen and/or mGluR5-ERα signaling in epilepsy in females with autism. In this study, we did not control for estrus cycle in phenotypes in adult female Pten+/− mice and would predict that estrus cycle and circulating estrogen would modulate these phenotypes.
Sex-specific deficits in temporal sensory processing in young NSEPten KO mice
UP states reflect the balance of excitation and inhibition in cortical circuits and provide a measure of dynamic circuit function.30,32 By measuring UP states, we revealed altered function of developing cortical circuits in the form of prolonged circuit activity as well as an overall increase in circuit activity in females with Pten deletion. The contribution of this prolonged circuit activity and hyperexcitability to in vivo cortical function is unknown but may contribute to the reduced ability of cortical circuits to rapidly respond and synchronize to changing sensory stimuli, as revealed here by the reduced ability of female NSEPten KO mice to consistently respond to brief gaps in a steady-state sound. Our previous work in the Fmr1 KO mouse model identified prolonged duration of cortical UP states. Because Fmr1 is X-linked, we only tested males in these studies. Similar to NSEPten KO mice, cortical circuits of juvenile Fmr1 KO mice have a reduced ability to synchronize to spectrotemporally dynamic sounds at 40–80 Hz.90 These results suggest similar mechanisms of cortical circuit dysfunction in developing NSEPten KO and Fmr1 KO that results in prolonged circuit activity and deficits in the temporal processing of sensory stimuli. In male NSEPten KO mice, we observed smaller and less reliable increases in cortical UP-state duration and smaller deficits in the gap-ASSR. These results indicate that effects of PTEN deletion are not exclusive to females but are more robust in females as compared to males. Responses to narrow gaps in sounds are commonly used measures of auditory acuity that show prolonged maturation in children and predict phonological aspects of speech.70 These data suggest that observed sex-dependent differences in gap processing may underlie abnormal speech recognition in females to a greater extent than in males with PTEN mutations. Girls with ASD have more severe deficits in sensory processing as compared to boys, particularly in the auditory and somatosensory domains.8 Studies of sensory circuits and their development across sexes in ASD mouse models may contribute to understanding these deficits and developing therapies.
Deficits in female-specific behaviors in mouse models of PTEN deletion
Increasing evidence indicates that there are sex-specific effects on social behavior in multiple mouse models of PTEN loss of function.25 Most notably, female, but not male, Pten+/− mice have deficits in social preference.18,24,25 Interestingly, male Ptenm3m4 mutant mice, expressing a cytoplasmic predominant form of PTEN, exhibit enhanced sociability.26 Consistent with these studies, we demonstrate a female-specific deficit in social interaction in NSEPten KO mice. Pten+/− mice have female-specific alterations in fear conditioning, as we observe in the NSEPten KO mice.25 We observed a female-specific hyperlocomotion under dim light but not in the open field, where both males and females were hyperlocomotive. Modulators of mGluR5, either positive or negative, affect and correct behaviors in adult mouse ASD models caused by loss-of-function mutation in suppressors of mTOR or mRNA translation91–93 and inbred strains,94 but sex-specific effects were not reported in these studies. Similarly, the role of brain ERα in sex-specific ASD-relevant behaviors has not been reported in any mouse models. Modulation of mGluR5 and/or ERα may have sex specificity on behavioral phenotypes in mouse models of Pten deletion and other ASD models.
It is increasingly recognized that ASD manifests differently in females and males. For example, as discussed, females with autism are more likely to have epilepsy and more severe problems in sensory processing in some modalities. Here we demonstrate female-selective hyperexcitability of sensory circuits, enhanced severity of seizures, and female-specific pharmacology of circuit function with loss of function of a high-impact ASD-risk gene. Importantly, we reveal roles for brain estrogen receptors in ASD-relevant phenotypes and suggest that ASD-risk genes that regulate the PI3K pathway, as in cancer, also regulate ERα in the brain and may give rise to sex-specific alterations in brain function and behavior. Our results also predict sex-specific efficacy in therapeutic strategies for individuals with PTEN mutations and, perhaps more generally, in ASD.
Limitations of the study
Whether the sex-specific effects of Pten deletion we observe in mice will be similar in human neurons or patients with PTEN mutations is unknown. Furthermore, we also performed most experiments with homozygous Pten deletion in neurons, which we think is necessary to observe robust and reproducible results in mice. Although we observed some similarities with heterozygous deletion of Pten in mice, whether the same effects on cortical circuit function and mGluR5 and ERα function will be observed in human neuron Pten heterozygous deletion or from patients is unknown.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kimberly M Huber (kimberly.huber@utsouthwestern.edu).
Materials availability
Plasmids generated in this study are available and will be shared by the lead contact upon request.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
Custom Labview software has been deposited to Zenodo and the access link is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Monoclonal Anti-PTEN (Clone 138G6) | Cell Signaling Technology | Cat# 9559; RRID:AB_390810 |
| Rabbit anti-Phospho-S6 Ribosomal Protein (Ser235/236)-Alexa Fluor® 647 | Cell Signaling Technology | Cat# 4851: RRID:AB_10695457 |
| Mouse anti-Puromycin Antibody (clone 12D10) | Millipore | Cat# MABE343: RRID:AB_2566826 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP® Rabbit mAb | Cell Signaling Technology | Cat# 4370: RRID:AB_2315112 |
| Mouse anti-NeuN | Millipore | Cat# MAB377: RRID:AB_2298772 |
| Goat Anti-Rabbit IgG Antibody (H + L), Biotinylated (BA-1000-1.5) | Vector Laboratories | Cat# BA-1000; RRID:AB_2313606 |
| Streptavidin, Alexa Fluor™ 594 conjugate | Thermo Fisher Scientific | Cat# S11227 |
| Guinea Pig anti-NeuN | Synaptic Systems | Cat# 266 004; RRID:AB_2619988 |
| Rabbit anti-Estrogen Receptor alpha (clone 60C) | Millipore | Cat# 04–820; RRID:AB_1587018 |
| Rabbit anti-Metabotropic Glutamate Receptor 5 (mGluR5) | Millipore | Cat# AB5675; RRID:AB_2295173 |
| Goat anti-Homer (E–18) | Santa Cruz Biotechnology | Cat# sc-8921; RRID:AB_648368 |
| Mouse monoclonal anti-Homer (D3) | Santa Cruz Biotechnology | Cat# sc-17842; RRID:AB_627742 |
| Mouse monoclonal anti-PSD-95 (clone 7E3) | Santa Cruz Biotechnology | Cat# sc-32290;RRID:AB_628114 |
| Mouse monoclonal anti-GluA1/GluR1 glutamate receptor | UC Davis/NIH NeuroMab Facility | Cat# N355/1; RRID:AB_2877405 |
| Mouse monoclonal anti-GluN1 glutamate receptor | UC Davis/NIH NeuroMab Facility | Cat# N308/48; RRID:AB_2877408 |
| Mouse monoclonal anti-Glyceraldehyde-3-PDH (GAPDH) | Millipore | Cat# MAB374; RRID:AB_2107445 |
| Mouse monoclonal anti-Actin (clone C4) | Millipore | Cat# MAB1501; RRID:AB_2223041 |
| Rabbit polyclonal anti-VCP | Cell Signaling Technology | Cat# 2648; RRID:AB_2214632 |
| Mouse monoclonal anti-Lamin A/C | Thermo Fisher Scientific | Cat# 14-9688-80; RRID:AB_2572956 |
| Mouse monoclonal ANTI-FLAG® M2 | Sigma-Aldrich | Cat# F1804; RRID:AB_262044 |
| Rabbit IgG, monoclonal [EPR25A] Antibody | Abcam | Cat# ab172730; RRID:AB_2687931 |
| Normal mouse IgG | Santa Cruz Biotechnology | Cat# sc-2025; RRID: AB_737182) |
| Fluorescein (FITC)-AffiniPure Goat Anti-Rabbit IgG (H + L) | Jackson ImmunoResearch Labs | Cat# 111-095-144; RRID: AB_2337978 |
| Peroxidase AffiniPure Goat Anti-Rabbit IgG (H + L) | Jackson ImmunoResearch Labs | Cat# 111-035-144; RRID: AB_2307391 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG (H + L) | Jackson ImmunoResearch Labs | Cat# 115-035-146; RRID: AB_2307392 |
| Goat anti-Rabbit IgG Fc, Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | Thermo Fisher Scientific | Cat# A78957; RRID:AB_2925780) |
| Rabbit TrueBlot®: Anti-Rabbit IgG HRP | Rockland Immunochemicals | Cat# 18-8816-31; RRID:AB_2610847 |
| Goat anti-Guinea Pig IgG (H + L) Secondary Antibody, Alexa Fluor™ 647 | Thermo Fisher Scientific | Cat# A-21450; RRID:AB_141882 |
| Goat anti-Mouse IgG (H + L) Secondary Antibody, Texas Red-X | Thermo Fisher Scientific | Cat# T-6390; RRID:AB_2556778 |
| NeuroTrace™ 435/455 blue fluorescent Nissl stain | Thermo Fisher Scientific | Cat# N21479 |
| Chemicals, peptides, and recombinant proteins | ||
| Xylazine | Dechra | N/A |
| GNE-149 | MCE MedChemExpress | Cat# HY-145341; CAS: 1953132-75-6 |
| MTEP hydrochloride | Tocris Biosciences | Cat# 2921; CAS: 1186195-60-7 |
| Ketamine | UTSW Medical Center Veterinary Drug service | N/A |
| MPP dihydrochloride | Tocris Biosciences | Cat# 1991; CAS: 911295-24-4 |
| U0126 | Tocris Biosciences | Cat# 1144; CAS: 109511-58-2 |
| Rapamycin | Tocris Biosciences | Cat# 1292; CAS: 53123-88-9 |
| Cycloheximide | Tocris Biosciences | Cat# 0970; CAS: 66-81-9 |
| Puromycin dihydrochloride from Streptomyces alboniger powder | Sigma-Aldrich | P8833; CAS: 58-58-2 |
| Anisomycin from Streptomyces griseolus | Sigma-Aldrich | A9789; CAS: 22862-76-6 |
| Lipofectamine™ 2000 Transfection Reagent | Thermo Fisher Scientific | Cat# 11668019 |
| Bis(2,2,2-trifluoroethyl) ether | Sigma-Aldrich | Cat# 287571; CAS Number: 333-36-8 |
| CTEP | Sigma-Aldrich | Cat# SML2306; CAS Number: 871362-31-1 |
| Gibco™ GlutaMAX™ Supplement | Thermo Fisher Scientific | Cat#35050061 |
| Gibco™ Antibiotic-Antimycotic (100X) | Thermo Fisher Scientific | Cat#15240096 |
| Invitrogen™ Dynabeads™ Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat#10003D |
| Invitrogen™ Dynabeads™ Protein A for Immunoprecipitation | Thermo Fisher Scientific | Cat#10001D |
| 2x Laemmli Sample Buffer | Bio-Rad | Cat #1610737 |
| Buprenorphine | Sigma-Aldrich | Cat #B9275 |
| TWEEN® 80 | Sigma-Aldrich | Cat #: P1754 |
| Critical commercial assays | ||
| Streptavidin/Biotin Blocking Kit | Vector Laboratories | Cat# SP-2002 |
| Pierce™ BCA Protein Assay Kits | Thermo Fisher Scientific | Cat# 23225 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | Thermo Fisher Scientific | Cat#34577 |
| Experimental models: Cell lines | ||
| HEK293 | ATCC | CRL-1573 |
| Experimental models: Organisms/strains | ||
| Mouse: Congenic NSE-Cre mice | Kwon et al.95 | N/A |
| Mouse: B6.129S4-Ptentm1Hwu/J | The Jackson Laboratory | JAX: 006440; RRID: IMSR_JAX:006440 |
| Mouse: B6(Cg)-Esr1tm4.1Ksk/J | The Jackson Laboratory | JAX: 032173 RRID: IMSR_JAX:032173 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | The Jackson Laboratory | JAX: 007914 RRID:IMSR_JAX:007914 |
| Mouse: B6.C-Tg(CMV-cre)1Cgn/J | The Jackson Laboratory | JAX: 006054 RRID:IMSR_JAX:006054 |
| Mouse: Pten heterozygous mice | This study | N/A |
| Recombinant DNA | ||
| N-terminal, flag-tagged mGluR5a | Bhave et al.96 | N/A |
| C-terminal HA-tagged ERα | This paper | N/A |
| Myc-tagged Homer2 | Guo et al.84 | N/A |
| Esr1 (tGFP-tagged) - Mouse estrogen receptor 1 (alpha) (Esr1) | Origene | Cat# MG227304 |
| 901 pLNCX myr HA Akt1 | Ramaswamy et al.47 | Addgene Plasmid #9005 |
| 902 pLNCX myr HA Akt1 K179M | Ramaswamy et al.47 | Addgene Plasmid #9006 |
| pcDNA™3.1 (+) Mammalian Expression Vector | Thermo Fisher Scientific | Cat#: V79020 |
| Software and algorithms | ||
| ImageJ/FiJI | NIH Schindelin et al.97 | https://imagej.net/software/fiji/ |
| GraphPad Prism 9 | GraphPad Prism 9 | https://www.graphpad.com/ |
| Custom Labview software | This study | https://doi.org/10.5281/zenodo.10819515 |
| ChemiDoc Imaging System | Bio-Rad | Cat# 12003153 |
| Image Lab Software | Bio-Rad | Cat# 12012931 |
| EthoVision XT | Noldus | https://www.noldus.com/ethovision-xt?utm_term=&utm_campaign=&utm_source=adwords&utm_medium=ppc&hsa_acc=5401040478&hsa_cam=20436101206&hsa_grp=&hsa_ad=&hsa_src=x&hsa_tgt=&hsa_kw=&hsa_mt=&hsa_net=adwords&hsa_ver=3&gad_source=1&gclid=EAIaIQobChMI28O82oT3hAMVSCetBh2B7ATHEAAYASAAEgJEAvD_BwE |
| Med Associates software | Med Associates Inc., St. Albans | https://med-associates.com/ |
| Other | ||
| Vibratome Leica VT 1200S | Leica | N/A |
| Interface recording chamber | Harvard Instruments | BSC-HT |
| SCREW 00-96 X 1/16(1.6 MM) (for AC and FC electrodes) | P1 technologies | 8L0X3905201F |
| EEG cable | Tucker Davis Technologies | N/A |
| RA4LI/RA4PA headstage/pre-amp | Tucker Davis Technologies | N/A |
| RZ6 multi-I/O processor | Tucker Davis Technologies | N/A |
| OpenEx (TDT) | Tucker Davis Technologies | N/A |
| Zeiss LSM 710 laser-scanning confocal microscope | Zeiss | N/A |
| PVC box (45 × 45 × 30 cm) | UTSW Medical center -Rodent Behavior Core Facility | N/A |
| 0.5 MΩ Tungsten Microelectrodes | FHC | N/A |
| Vibratome Leica VT 1000S | Leica | N/A |
| Scrambled shock generator | Med Associates Inc., St. Albans | N/A |
| Foredom dental drill | Foredom | N/A |
| Dental cement | PANAVIA™ SA Cement Universal | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Mice were group housed (5 maximum) in non-environmentally enriched cages with unrestricted food and water access and a 12 h light-dark cycle. Teklad Global 16% Protein Rodent Diet was used. This diet does not contain alfalfa or soybean meal, thus minimizing the occurrence of natural phytoestrogens. Room temperature was maintained at 21 ± 2°C. Animal husbandry was carried out by UT Southwestern Medical Center technical staff. The animal use protocols used in this manuscript were approved by the UT Southwestern Institutional Animal Care and Use Committee and approved by the Institutional Animal Care and Use Committee at the University of California, Riverside. Congenic NSE-Cre mice95 on the C57BL6 background were originally obtained from Dr. Luis F Parada (Memorial Sloan Kettering Cancer Center, New York, NY) and were crossed with floxed Pten (Strain #:006440). Where indicated, we also utilized the floxed Esr1 (Strain #:032173) and Ai14 Cre-reporter mice (Strain #007914) all obtained from Jackson Laboratories (Bar Harbor, ME). Germline Pten+/− mice were generated by crossing CMV-Cre (B6.C-Tg(CMV-cre)1Cgn/J) (JAX stock number: 006054) with floxed Pten (Strain #:006440). F1 with the Pten mutation was then bred on the C57BL/6 J background to obtain WT and Pten+/− mice. Unless otherwise specified, all experiments were done in male and female mice 19 to 22 days old. UP state were also recorded from Pten+/− mice of both sexes when 6–9 weeks old. Flurothyl exposure was performed in male and female Pten Cre(−) and Cre(+) at 3 weeks and 6 weeks of age. Pten+/− mice of both sexes were exposed to Flurothyl when 13–15 weeks old. Behavioral tests were done in male and female Pten Cre(−) and Cre(+) when they were 9–12 weeks old.
Cultured cell lines
The HEK293 (ATCC# CRL-1573) cell line was used for heterologous expression. Cells were grown at 37°C, 5% CO2 in 1X DMEM + Glutamax (GIBCO) containing 10% Fetal Bovine Serum and 1X Antibiotic-Antimycotic (GIBCO). Cells used in experiments were passaged a maximum of twenty times. Plasmid constructs were transfected or co-transfected into HEK293 using Lipofectamine 2000 reagent (ThermoFisher #11668019) based on manufacturer’s instructions.
METHOD DETAILS
Plasmids production
The N-terminal, flag-tagged mGluR5a was gifted from Dr. Robert Gereau (Washington University, St. Louis, MO).96 The C-terminal HA-tagged ERα was constructed from the purchased ERα cDNA expression clone (Origene #MG227304) into BamHI and EcoRI sites of pcDNA3 vector using PCR amplification. Myc-tagged Homer2 was previously described.84 The myr_HA_Akt1 and myr_-HA_Akt1_K179M were gifts from Dr. William Sellers (Addgene plasmids #9005 and #9006 respectively) and previously described.47 All constructs were verified by sequencing.
Slice preparation and electrophysiology
Cortical slices were acutely prepared from male and female mice of P18-60 age. In brief, mice were deeply anesthetized with Ketamine/Xylazine (20 mg/kg xylazine and 150 mg/kg ketamine) and decapitated. The brain was transferred into ice-cold dissection buffer containing (in mM): 87 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 7 MgCl2, 0.5 CaCl2, 20 d-glucose, 75 sucrose, and 1.3 ascorbic acid aerating with 95% O2-5% CO2. Thalamocortical slices, 400 μm, were made on an angled block,98 using a vibratome Leica VT 1200S. Following cutting, slices were transferred to an interface recording chamber (Harvard Instruments) and allowed to recover for 1 h in nominal “low activity” artificial CSF (ACSF) at 32°C containing (in mM): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 MgCl2, 2 CaCl2, and 25 d-glucose. Slices were then perfused with a modified “high activity” ACSF that mimics physiological ionic concentrations in vivo, which contained (in mM): 126 NaCl, 5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1 MgCl2, 1 CaCl2, and 25 d-glucose. Slices remained in “high activity” ACSF for 45 min prior to UP state recordings.34 Drugs were prepared fresh daily in vehicle and were added in low and high activity ACSF or as indicated.
Spontaneously generated UP states were recorded extracellularly for 5 min/slice, using 0.5 MΩ tungsten microelectrodes (FHC) placed in layer 4 of primary somatosensory cortex (S1) or layers 2/3 or 5, as indicated. Recordings were amplified 10,000-fold, sampled at 2.5 kHz, and filtered on-line between 500 Hz and 3 kHz. All measurements were analyzed offline using custom Labview software. For visualization and analysis of UP states, traces were offset to zero, rectified, and low-pass filtered with a 0.2 Hz cutoff frequency. Using these processed traces, the threshold for detection was set at 15× the RMS noise. An event was defined as an UP state if its amplitude remained above the threshold for at least 200 ms. The end of the UP state was determined when the amplitude decreased below threshold for >600 ms. Two events occurring within 600 ms of one another were grouped into a single UP state. UP state amplitude was defined based on the filtered/rectified traces and was unitless since it was normalized to the detection threshold. This amplitude may be considered a coarse indicator of the underlying firing rates of neuronal populations. Direct measures of firing rates were not possible because individual spikes could not be isolated except during the quiet periods (the DOWN states). Data are represented by the mean ± SEM. Significant differences were determined using t tests, one-way ANOVA, two-way ANOVA, where appropriate (all performed with GraphPad Prism). Repeated-measures ANOVA was also used when appropriate. Šídák post hoc tests were performed following ANOVAs.
mGluR5 Co-Immunoprecipitation in vivo
Neocortices from P21 male and female NSEPten KO and WT mice were lysed in co-immunoprecipitation (CoIP) buffer (50 mM Tris, pH 7.4, 120 mM NaCl, 1 mM EDTA, 1% Triton X-100 containing Protease and Phosphatase inhibitor mixture), and further homogenized using brief (5–10 s) pulses of sonication until lysates were clear. After homogenization, lysates were rotated for 30 min at 4°C followed by centrifugation at 10,000 X g for 10 min at 4°C. Then, 10% of the lysate was set aside to be used as an input control and the rest was incubatedovernight at 4°C with 7 μL of ERα antibody or 1.4 μg of Homer antibody (Santa Cruz Biotechnology, D-3). The same isotype IgG was used as control for the specificity of the ERα (Abcam #ab172730) and Homer D3 antibodies. 25 μL of Dynabeads Protein A or G slurry (Invitrogen) were washed in co-immunoprecipitation buffer and added to the samples for 3 h at 4°C with gentle rotation and the beads were then washed with CoIP buffer. Immunoprecipitated proteins were eluted with 2x SDS Sample Buffer (Bio-Rad). Eluted immunoprecipitated proteins were run in parallel with input lysate. Western blotting was performed with antibodies against Homer (Santa Cruz, E–18, 1:5000), mGluR5 (Millipore, 1/20000), and ERα (Millipore, 1:1000). The TrueBlot secondary Ab (18-8816-31 Rockland, (1:5000) was used as secondary for ERα detection. For analysis of each experiment, the CoIP band intensity values were normalized by the intensity of the corresponding IP protein band. (e.g., mGluR5/ERα or mGluR5/Homer).
Co-Immunoprecipitations in HEK cells
Plasmid constructs were transfected or co-transfected into HEK293 using Lipofectamine 2000 reagent. 48 h post-transfection, cells were lysed in CoIP buffer (50 mm Tris, pH 7.4, 120 mm NaCl, 1 mM EDTA, 1% Triton X-100 containing protease and phosphase inhibitor mixture), and insoluble debris was removed by centrifugation (10 min, 10,000 g). The cleared lysate was incubated with an anti-Flag antibody (Sigma-Aldrich, F1804, 7 μg) or same IgG isotype control, while rotating, overnight at 4°C and then for 3–4 h with Dynabeads G beads. Bound proteins were eluted with sample buffer and loaded on gradient SDS-PAGE gels and processed for western blot as described above. Input lysate and CoIP complexes were blotted with anti-mGluR5 (Millipore, 1:50000 for input and 1:200000 for IP), anti-ERα antibodies (Millipore, 1:50000 for Input and 1:200000 for IP) and anti-Homer (Santa Cruz E18, 1:10000 for both IP and Input). GAPDH (Millipore, 1:300000) was used a loading control for input samples.
Subcellular fractionation
Postnatal day 21 mouse cortices were processed as described99 by mincing tissue on ice with a razor blade into 1 mm cubes. Tissue cubes were resuspended in 10 volumes of Buffer “A” medium containing 2.0 mM MgCl2, 25 mM KCl, 10 mM HEPES (pH 7.5), and protease inhibitors (Complete Tablets; Roche Applied Science, Indianapolis, IN). After incubating for 10 min on ice, cells were homogenized in a Wheaton glass homogenizer using 15 strokes with a “B” pestle. The homogenate was filtered through 3 layers of sterile gauze and centrifuged at 1000 × g for 10 min. The nuclear pellet was resuspended in 3 mL of Buffer N containing 0.25 M sucrose in buffer A. Resuspended nuclei were layered over 2 mL of medium containing 1.1 M sucrose in buffer A, and then recentrifuged at 1000 × g. This step was repeated twice. The final nuclear pellet was resuspended in SDS sample buffer. The supernatant from the first pellet was concurrently further fractionated by centrifugation at 35,000 × g for 40 min. This second supernatant represented soluble, cytoplasmic proteins whereas the high-speed pellet contained plasma membrane proteins. Aliquots from each fraction were used for gel electrophoresis. Protein concentrations were determined using the BCA protein assay kit (Pierce).
Western blotting
Western blotting was performed on lysates from somatosensory cortex, or microdissected lower cortical layers (to enrich for Pten KO neurons), subcellular fractions from P21 mouse cortices, and eluted immunoprecipitated proteins. With the exclusion of the immunoprecipitated protein, samples were lysed in lysis buffer containing 50 mM Tris, pH 7.4, 120 mM NaCl, 1 mM EDTA and 1% Triton X-100, containing Protease Inhibitor Mixture, (Sigma P8340); and phosphatase inhibitor mixture 2 and 3, (Sigma P5726 and P0040). Samples were further homogenized using brief (5–10 s) pulses of sonication until lysates were clear. Lysates were then centrifuged at 10,000 g for 10 min at 4°C, to remove insoluble material. Protein concentration was determined by the BCA protein assay kit (Pierce). To equalize protein concentrations, samples were diluted with lysis buffer and 4x SDS-Sample buffer to a final concentration of 1–3 μg/μL. Equal amount of protein (5–15 μg) in an equal volume were loaded for each sample on 8–10% Tris-glycine polyacrylamide gels; the gels were run using Tris-glycine SDS running buffer at constant voltage and then transferred to a PVDF membrane. Membranes were cut at appropriate molecular weights to allow immunoblotting of the same membrane for multiple proteins. After blocking with 5% milk in 1 × TBS, 0.05% Tween 20 for 1 h, membranes were incubated with the following primary antibodies in blocking buffer overnight at 4°C: anti-mGluR5 (Millipore, 1:50000 to 1:200000), anti-ERα antibodies (Millipore, 1:1000), anti-Homer (Santa Cruz E18, 1:5000), anti-GluA1/GluR1 (UC Davis/NIH NeuroMab Facility, 1:100000), anti-GluN1 (UC Davis/NIH NeuroMab Facility, 1:50000) and anti-PSD95 (Santa Cruz Biotechnology, 1:100000). Following incubation in primary antibody, blots were incubated with HRP-conjugated secondary antibodies (Jackson ImmunoResearch Labs, 1:5000) in 5% milk in 1× TBS, 0.05% Tween 20 for 1 h at room temperature. Membranes were washed and developed using the SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific). Signal was acquired using Chemidoc MP (BioRad) and analyzed using ImageLab (BioRad). Protein bands were normalized using GAPDH (Millipore, 1:3000000), VCP (Cell Signaling Technology, 1:25000) or Actin (Millipore, 1:1000000) as housekeeping proteins. The details of the primary antibodies used, and species they were raised in, are provided in the key resources table.
Surface sensing of translation (SUnSET) in brain slice
To measure protein synthesis in L5 of S1 somatosensory cortex, we used the immunohistochemical version of SUnSET.56 Acute slices were prepared as for UP states and recovered for 2 h (1 h in low activity ACSF and then 1 h in high activity ACSF) in a submerged chamber with continuous aeration with 95% O2/5% CO2. Puromycin (2.0 μg/μL) (P8833, Sigma-Aldrich Inc.) was then added to ACSF for 40 min. MTEP (3 μM), rapamycin (200 nM) or U0126 (20 μM) were present during the 2 h recovery time and during the puromycin incubation. For anisomycin (20 μM) control experiment, slices were incubated with the protein synthesis inhibitor for 60 min before puromycin incubation. At the end of puromycin incubation, slices were washed in cold ACSF and then fixed for 3 h in 4% PFA, rinsed in PBS and stored at 4°C. Slices were then re-sectioned (50 μm thickness) using a vibratome (Leica VT 1000 S) and stored in anti-freezing solution (30% Glycerol, 30% Ethylene Glycol, 40% PBS) at −20°C until subjected to IHC.
Immunohistochemistry (IHC)
For PTEN, Phospho-S6 IHC and soma size measurement (Figure 5), mice were anesthetized with Ketamine/xylazine and transcardially perfused with PBS followed by 4% paraformaldehyde (PFA) in 0.01 M phosphate buffer. Brains were then post-fixed overnight in 4% PFA. After postfixation, brains were washed with PBS and sectioned into 50 μm thick coronal sections using the vibratome. For P-ERK, and Puromycin staining after SUnSET, post-fixed slices were washed in 0.01 M PBS and re-sectioned into 50 μm thick sections containing the somatosensory cortex. Sections containing the somatosensory cortex were permeabilized with 0.3% Triton X-100 for 30 min, blocked with 0.1% Triton X-100, 5% normal goat serum, and then incubated for 24 to 48 h at 4°C with the following primary antibodies: rabbit anti-PTEN (Cell Signaling, 1; 100), rabbit anti-Phospho-S6 Ribosomal Protein (Ser235/236) Alexa Fluor 647 (Cell Signaling, 1:500), anti-puromycin (Millipore, 1:500), anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (Cell Signaling, 1:50), anti-NeuN (Millipore, 1:1000 for Pten positive cell counting in Figure S1A), anti-NeuN (Synaptic System, 1:1000). After the incubation in primary antibodies, sections were washed in PBS 0.04% Triton X-100 at room temperature (3 × 10 min). Following the last wash, slices were incubated in appropriate secondary antibodies for 2 h at room temperature, followed by 3 × 10 min washes in PBS-Triton X-100 at room temperature. Secondary antibodies used were the following: goat Anti-Rabbit IgG fluorescein (FITC, 1:500), goat anti-Mouse Texas Red-X (Invitrogen, Catalog # T-6390, 1:500), Goat anti-Guinea Pig IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 (Invitrogen, Cat. N.: Cat # A-21450, 1; 500). NeuroTrace 435/455 Blue Fluorescent Nissl Stain (Invitrogen, 1:400) was also used to label neurons and for soma size analysis and was incubated along with secondary antibodies. For P-S6 staining, PTEN stained sections (primary + secondary and Neurotrace) were incubated with the anti P-S6 antibody Alexa Fluor 647 Conjugate overnight at 4°C. Following wash with PBS 0.04% Triton X-100, sections were mounted using Aqua-Poly/Mount (Polysciences) on glass slides and imaged. For ERK phosphorylation, slices were permeabilized and blocked as for the other staining and the Streptavidin/Biotin blocking Kit (Vector Laboratories) was used as directed by the manufacturer. Sections were incubated with the primary antibodies for Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (for 48 h at 4°C). After that, the sections were washed and incubated first with a Biotinylated Goat Anti-Rabbit IgG Antibody (over-night at 4°C, Vector Laboratories), and then with Alexa Fluor 594 streptavidin (3 h – room temperature, Thermo Fisher). After extensive washing, slices were incubated again with a rabbit antibody raised against PTEN over-night at 4°C (Cell Signaling). After washing, the sections were then incubated with a Goat anti-rabbit IgG conjugated with Alexa Fluor 647 (Thermo Fisher Scientific) and NeuroTrace 435/455 blue fluorescent Nissl stain (Thermo Fisher Scientific) to identify neuron’s soma. This combination of reagents prevents nonspecific binding and overlap between the P-ERK and the PTEN staining.
Confocal microscopy and image quantification
Fluorescence images (1024 × 1024 pixels, 8 bit) were taken from L5 of S1 cortex using a Zeiss LSM 710 laser-scanning confocal microscope with a 20× objective, 1× zoom, 2 μm z-step and a resolution of 2.41 pixels/μm. For all quantitative comparison experiments, the same microscope and acquisition settings were used for each image and samples were processed in batches to include matched control and experimental samples. All images were processed using Fiji software (https://imagej.net/software/fiji/). For image analysis, the investigators were blind to the treatment condition.
To quantify puromycin, P-S6 and P-ERK fluorescence intensity and soma size, background was subtracted using the “Subtract background” function on Fiji (rolling ball radius: 50 pixel). After background was subtracted from each image, regions of interest (ROIs) were manually drawn around neuronal bodies in the puromycin channel (red channel), P-S6 channels (ultrared channel) and Neurotrace channel (blue channel) of each image with the “Freehand” tool and were then used to measure area and fluorescence mean gray value in the Puromycin, P-S6, P-ERK, Neurotrace or NeuN channels. Non neuronal cells were excluded from the analysis based on the NeuN (for the Puromycin IHC) or Nissl staining. Cre(+) or Cre(−) neurons were then identified and separated based on PTEN immunofluorescence (green channel). The fluorescence intensity and area of 30–60 Cre(−) and (+) neurons was measured in each section for Puromycin, P-S6, P-ERK and soma size evaluations, which was then averaged for each section and the mean value of the fluorescence intensity of Cre(+) was expressed as % of the Cre(−) neurons for each channel. 5-8 sections were analyzed from 4 mice for each condition/treatment. n for these data represent the # of sections. To quantify PTEN (+) and PTEN (−) neurons in Figure S1, NeuN+ neurons were counted in Fiji by an author blinded to the sex within L5 cortical region and then cells subsequently defined as PTEN-positive or negative.
Flurothyl induced seizures
Flurothyl (bis(2,2,2-trifluoroethyl) ether, Sigma-Aldrich) seizure experiments were performed as described.100 Each mouse was placed in a 2 L glass chamber inside of a chemical fume hood and allowed to habituate for 1 min before the top of the chamber was closed. 10% flurothyl in 95% ethanol was then infused at a rate of 200 μL/min onto a disk of filter paper (Whatman, Grade 1) suspended at the top of the chamber. Mice exhibit various stages of increasing seizure severity in response to flurothyl exposure, including myoclonic seizure (sudden involuntary jerk/shock-like movements involving the face, trunk, and/or limbs) and generalized seizure (also known as clonic-forebrain seizures that are characterized by clonus of the face and limbs, loss of postural control, rearing, and falling). Generalized seizures can immediately progress into brain stem seizures manifested by tonic extension of the limbs. Upon emergence of a generalized seizure, the lid of the chamber was immediately removed, allowing for rapid dissipation of the flurothyl vapors and exposure of the mouse to fresh air. Mice were then returned to their home cage following recovery from behavioral seizures. One mouse at a time was tested in the flurothyl chamber, which was recharged with a fresh filter paper, cleaned using water, and thoroughly dried between subjects. In the same experiments, flurothyl exposures were repeated once daily over five consecutive days. Mouse behavior during each flurothyl exposure was video-recorded and viewed by investigators (blind to genotype) who determined latency to the onset of myoclonic and generalized seizures.
The mGluR5 specific negative allosteric modulator CTEP 2-(chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-1H-imidazol-4-yl)ethynyl)pyridine) was prepared fresh daily as a microsuspension in vehicle (0.9% NaCl, 0.3% Tween-80) and administered by intraperitoneal (i.p.) injection 90 min prior to flurothyl exposure.
Behavioral tests
Behavior tests were performed during the light phase of the light/dark cycle. The battery of tests was performed in the same order for all mice (Locomotor Activity-Open Field-Dark Light Box-Three-chamber social approach-Fear conditioning) and designed to minimize carryover effects.101 Mice were moved into the testing area at least 1 h prior to testing. Manual scoring for social recognition and fear conditioning (with a stopwatch) was performed by a trained observer blind to sex and genotype.
Locomotor activity
To assess locomotor activity, mice were placed in a new home cage with minimal bedding for 2 h and horizontal activity was monitored using photobeams linked to computer data acquisition software (San Diego Instruments) under red light condition.
Open field test
The open field test was used to evaluate the voluntary exploratory behavior, and anxiety of experimental animals to a new and different environment.102 The light intensity in the open field test was 60 lux. At the beginning of the test, the mice were placed in a PVC box (45 × 45 × 30 cm) at the same starting position and an overhead camera and tracking software (Noldus Ethovision software version 12.5) was used to monitor the mouse movement for 10 min. Total distance and time traveled in different compartment of the chamber (center, non-periphery, periphery) were measured. The chamber was cleaned after each trial. All behavioral results were analyzed by investigators blind to genotypes.
Light–dark (L/D) test
The apparatus comprised of a box divided into two separate compartments. The light chamber was painted white, while the other chamber was entirely black and enclosed. The L/D chambers were separated by a sliding door. After 2 min of habituation in the dark chamber, the door to the light chamber was opened and mice were allowed to explore for 10 min. Behavioral parameters such as latency time to leave the dark chamber and total time spent in the light and dark chamber were recorded.102
Three chamber sociability test
Social preference of a mouse was evaluated in a three-chamber choice task.103 The sociability test was used to evaluate a mouse’s preference for a social object over a non-social object. All tests were conducted in a white opaque acrylic three-chambered apparatus. The apparatus was divided into three chambers connected by doorways under white light conditions. During the habituation phase, the test subject was acclimated for 10 min in the center chamber, followed by another 10 min exploring the entire apparatus. Then, the social interaction test was conducted. The test subject was first confined to the center chamber with both doorways closed. An empty wire cage was then placed in one side chamber and an identical wire cage containing an unfamiliar, sex-matched mouse (stranger #1) was placed in the other side chamber. The doorways were then re-opened, and the test subject was allowed to freely explore the testing chambers for 10 min. Time spent directly interacting with the novel animal and novel object (sniffing time) was scored with a stopwatch by an examiner who was blind to genotype. Animals were tested between 10 and 14 weeks of age. Light at the center of the three-chambered apparatus was 60 lux for all experiments.
Fear conditioning
Fear conditioning was measured in boxes equipped with a metal grid floor connected to a scrambled shock generator (Med Associates Inc., St. Albans) as described.102 For training, mice were individually placed in the chamber. After 2 min, the mice received 3 tone-shock pairings (30 s white noise, 80 dB, co-terminated with a 2 s, 0.5 mA footshock, 1 min intertrial interval). The following day, memory of the context was measured by placing the mice into the same chambers and freezing was measured automatically by the Med Associates software for 5 min. Forty-eight hours after training, memory for the white noise cue was measured by placing the mice in a box with altered floors and walls, different lighting, and a vanilla smell. Freezing was measured for 3 min, then the noise cue was turned on for an additional 3 min and freezing was measured.
EEG recordings
Mice underwent epidural electrode implant surgery at P18-20. Mice were anesthetized using i.p. injections of 80/20 mg/kg of ketamine/xylazine and were monitored closely throughout the procedure by toe pinch reflex every 10–15 min. ETHIQA-XR (1-shot buprenorphine, 3.25 mg/kg body weight) was administered (s.c.) prior to surgery to ensure an adequate analgesic level was present post-surgically. Following the removal of fur and skin, and sterilization (alcohol and iodine wipes) of the scalp, an incision was made to expose the scalp. A Foredom dental drill was utilized to drill ~1mm diameter holes in the skull over the right auditory cortex (AC), right frontal cortex (FC), and left occipital cortex. The screw positions were determined using skull landmarks and coordinates previously reported104 and were based on single unit mapping.105 The wires extending from three-channel posts were wrapped around 1 mm screws and screwed into holes. Dental cement was applied around the screws, on the base of the post, and exposed skull, to secure the implant. Mice were placed on a heating pad until fully awake and were allowed 48–72 h for recovery before EEG recordings were made. Mice were housed individually following surgery. All EEG recordings were obtained from awake and freely moving mice. EEG recordings were performed at P20-23. Recordings were obtained from the AC and FC electrodes, using the occipital screw as reference. Mice were placed in an arena where they could move freely during the recording. The arena was inside a Faraday cage placed on a vibration isolation table in a sound-insulated and anechoic booth (Gretch-Ken, OR). Mice were briefly anesthetized with isoflurane and attached to an EEG cable. The EEG recording set-up has been previously reported.76 Briefly, the attached cable was connected via a commutator to a TDT (Tucker Davis Technologies, FL) RA4LI/RA4PA headstage/pre-amp, which was connected to a TDT RZ6 multi-I/O processor. OpenEx (TDT) was used to simultaneously record EEG signals and operate the LED light used to synchronize the video and waveform data. TTL pulses marked stimulus onsets on a separate channel in the collected EEG data. The EEG signals were recorded at a sampling rate of 24 kHz and down-sampled to 1024 Hz for analysis. All raw EEG recordings were visually examined prior to analysis for artifacts, including loss of signal or signs of clipping, but none were seen.
Gap-ASSR
The stimulus used to assess auditory temporal processing is termed the ‘40 Hz gaps in noise-ASSR’ (auditory steady state response, henceforth, ‘gap-ASSR’)76 and contains alternating 250 ms segments of noise and gap interrupted noise presented at 75 dB SPL. The gaps are placed 25 ms apart, resulting in a repetition rate of 40 Hz. The 40 Hz modulation rate produces the strongest ASSR signal across temporal and frontal cortex and may reflect the resonance frequency of the underlying neural circuits.106,107 The gap width and modulation depth are chosen at random for each gap-in-noise segment. For data shown in Figure 6, gaps included 4 or 6 ms and modulation depths of 75%. To measure the ability of the cortex to consistently respond to the gaps in noise, inter-trial phase clustering (ITPC) at 40 Hz was measured.108 The EEG trace was transformed using a dynamic complex Morlet wavelet transform. The trials corresponding to each parametric pair (gap duration + modulation depth) were grouped together. The ITPC was calculated for each time-frequency point as the average vector for each of the phase unit vectors recorded across trials (trial count >100 trials per parametric pair). The ITPC values at 40Hz were averaged to extract the mean ITPC for the parametric pairs in the AC and FC. To ensure that the calculated ITPC was not due to spurious or transient phase locking, we measured ‘baseline’ ITPC. Baseline ITPC was calculated during the periods of unmodulated noise (no gaps) and compared to the ITPC generated during periods of modulated noise.
QUANTIFICATION AND STATISTICAL ANALYSIS
Whenever possible, data was analyzed by an investigator blind to the genotype or sex of mice. Results in figures are graphically represented as mean ± SEM. Statistics were performed in GraphPad Prism. Normal distribution was tested by the D’Agostino & Pearson test. For 2-group comparison, we used a student unpaired 2-tailed t test, or a Mann-Whitney nonparametric test. For multiple group comparison, data were analyzed using ordinary 1-way ANOVA or Kruskal-Wallis (nonparametric) or two-way ANOVA where appropriate. Significance was determined using post-hoc multiple comparisons testing for ANOVA analyses. The sample size, statistical test, and p value are denoted in results and figure legends.
Supplementary Material
Highlights.
Sex and a high-confidence ASD-risk gene, Pten, interact to affect brain function and behavior
Deletion of Pten causes robust hyperexcitability of neocortical circuits in female mice
Circuit hyperexcitability is driven by mGluR5 and ERα signaling to ERK and protein synthesis
Female Pten KO neurons have enhanced mGluR5 and ERα-dependent protein synthesis
ACKNOWLEDGMENTS
This work was supported by NIH grant R37NS114516 to K.M.H. We would like to thank Dr. Luis Parada for providing the NSE-Cre mouse line and Courtney Saqueton and Christopher Williams for technical assistance.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114056.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Maenner MJ, Warren Z, Williams AR, Amoakohene E, Bakian AV, Bilder DA, Durkin MS, Fitzgerald RT, Furnier SM, Hughes MM, et al. (2023). Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years — Autism and Developmental Disabilities Monitoring Network. MMWR. Surveill. Summ 72, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bölte S, Neufeld J, Marschik PB, Williams ZJ, Gallagher L, and Lai MC (2023). Sex and gender in neurodevelopmental conditions. Nat. Rev. Neurol 19, 136–159. 10.1038/s41582-023-00774-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans SC, Boan AD, Bradley C, and Carpenter LA (2019). Sex/Gender Differences in Screening for Autism Spectrum Disorder: Implications for Evidence-Based Assessment. J. Clin. Child Adolesc. Psychol 53, 840–854. 10.1080/15374416.2018.1437734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hull L, Petrides KV, and Mandy W (2020). The Female Autism Phenotype and Camouflaging: a Narrative Review. Rev. J. Autism Dev. Disord 7, 306–317. 10.1007/s40489-020-00197-9. [DOI] [Google Scholar]
- 5.Frazier TW, Georgiades S, Bishop SL, and Hardan AY (2014). Behavioral and cognitive characteristics of females and males with autism in the Simons Simplex Collection. J. Am. Acad. Child Adolesc. Psychiatry 53, 329–340.e403. 10.1016/j.jaac.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loomes R, Hull L, and Mandy WPL (2017). What Is the Male-to-Female Ratio in Autism Spectrum Disorder? A Systematic Review and Meta-Analysis. J. Am. Acad. Child Adolesc. Psychiatry 56, 466–474. 10.1016/j.jaac.2017.03.013. [DOI] [PubMed] [Google Scholar]
- 7.Lawrence KE, Hernandez LM, Fuster E, Padgaonkar NT, Patterson G, Jung J, Okada NJ, Lowe JK, Hoekstra JN, Jack A, et al. (2022). Impact of autism genetic risk on brain connectivity: a mechanism for the female protective effect. Brain 145, 378–387. 10.1093/brain/awab204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osório JMA, Rodríguez-Herreros B, Richetin S, Junod V, Romascano D, Pittet V, Chabane N, Jequier Gygax M, and Maillard AM (2021). Sex differences in sensory processing in children with autism spectrum disorder. Autism Res. 14, 2412–2423. 10.1002/aur.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amiet C, Gourfinkel-An I, Bouzamondo A, Tordjman S, Baulac M, Lechat P, Mottron L, and Cohen D (2008). Epilepsy in autism is associated with intellectual disability and gender: evidence from a meta-analysis. Biol. Psychiatr 64, 577–582. 10.1016/j.biopsych.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 10.Jokiranta E, Sourander A, Suominen A, Timonen-Soivio L, Brown AS, and Sillanpää M (2014). Epilepsy Among Children and Adolescents with Autism Spectrum Disorders: A Population-Based Study. J. Autism Dev. Disord 44, 2547–2557. 10.1007/s10803-014-2126-6. [DOI] [PubMed] [Google Scholar]
- 11.Besag FMC, and Vasey MJ (2021). Seizures and Epilepsy in Autism Spectrum Disorder. Psychiatr. Clin 44, 51–68. 10.1016/j.psc.2020.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Tilot AK, Frazier TW 2nd, and Eng C. (2015). Balancing Proliferation and Connectivity in PTEN-associated Autism Spectrum Disorder. Neurotherapeutics 12, 609–619. 10.1007/s13311-015-0356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varga EA, Pastore M, Prior T, Herman GE, and McBride KL (2009). The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet. Med 11, 111–117. 10.1097/GIM.0b013e31818fd762. [DOI] [PubMed] [Google Scholar]
- 14.McBride KL, Varga EA, Pastore MT, Prior TW, Manickam K, Atkin JF, and Herman GE (2010). Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 3, 137–141. 10.1002/aur.132. [DOI] [PubMed] [Google Scholar]
- 15.O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, et al. (2012). Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. 10.1126/sci-ence.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou J, and Parada LF (2012). PTEN signaling in autism spectrum disorders. Curr. Opin. Neurobiol 22, 873–879. 10.1016/j.conb.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Busch RM, Srivastava S, Hogue O, Frazier TW, Klaas P, Hardan A, Martinez-Agosto JA, Sahin M, and Eng C; Developmental Synaptopathies Consortium (2019). Neurobehavioral phenotype of autism spectrum disorder associated with germline heterozygous mutations in PTEN. Transl. Psychiatry 9, 253. 10.1038/s41398-019-0588-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clipperton-Allen AE, and Page DT (2014). Pten haploinsufficient mice show broad brain overgrowth but selective impairments in autism-relevant behavioral tests. Hum. Mol. Genet 23, 3490–3505. 10.1093/hmg/ddu057. [DOI] [PubMed] [Google Scholar]
- 19.Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, and Nakshatri H (2001). Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J. Biol. Chem 276, 9817–9824. 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- 20.Vilgelm A, Lian Z, Wang H, Beauparlant SL, Klein-Szanto A, Ellenson LH, and Di Cristofano A (2006). Akt-mediated phosphorylation and activation of estrogen receptor alpha is required for endometrial neoplastic transformation in Pten+/− mice. Cancer Res. 66, 3375–3380. 10.1158/0008-5472.CAN-05-4019. [DOI] [PubMed] [Google Scholar]
- 21.Yamnik RL, Digilova A, Davis DC, Brodt ZN, Murphy CJ, and Holz MK (2009). S6 kinase 1 regulates estrogen receptor alpha in control of breast cancer cell proliferation. J. Biol. Chem 284, 6361–6369. 10.1074/jbc.M807532200. [DOI] [PubMed] [Google Scholar]
- 22.Ishida N, Baba M, Hatanaka Y, Hagio K, Okada H, Hatanaka KC, Togashi K, Matsuno Y, and Yamashita H (2018). PIK3CA mutation, reduced AKT serine 473 phosphorylation, and increased ERalpha serine 167 phosphorylation are positive prognostic indicators in postmenopausal estrogen receptor-positive early breast cancer. Oncotarget 9, 17711–17724. 10.18632/oncotarget.24845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciruelos Gil EM (2014). Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer. Cancer Treat Rev. 40, 862–871. 10.1016/j.ctrv.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 24.Page DT, Kuti OJ, Prestia C, and Sur M (2009). Haploinsufficiency for Pten and Serotonin transporter cooperatively influences brain size and social behavior. Proc. Natl. Acad. Sci. USA 106, 1989–1994. 10.1073/pnas.0804428106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clipperton-Allen AE, and Page DT (2020). Connecting Genotype with Behavioral Phenotype in Mouse Models of Autism Associated with PTEN Mutations. Cold Spring Harb. Perspect. Med 10, a037010. 10.1101/cshperspect.a037010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tilot AK, Gaugler MK, Yu Q, Romigh T, Yu W, Miller RH, Frazier TW 2nd, and Eng C (2014). Germline disruption of Pten localization causes enhanced sex-dependent social motivation and increased glial production. Hum. Mol. Genet 23, 3212–3227. 10.1093/hmg/ddu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Do Rego JL, Seong JY, Burel D, Leprince J, Luu-The V, Tsutsui K, Tonon MC, Pelletier G, and Vaudry H (2009). Neurosteroid biosynthesis: enzymatic pathways and neuroendocrine regulation by neurotransmitters and neuropeptides. Front. Neuroendocrinol 30, 259–301. 10.1016/j.yfrne.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Becker JB, and Chartoff E (2019). Sex differences in neural mechanisms mediating reward and addiction. Neuropsychopharmacology 44, 166–183. 10.1038/s41386-018-0125-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, and Parada LF (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron 50, 377–388. 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haider B, and McCormick DA (2009). Rapid neocortical dynamics: cellular and network mechanisms. Neuron 62, 171–189. 10.1016/j.neuron.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rigas P, Adamos DA, Sigalas C, Tsakanikas P, Laskaris NA, and Skaliora I (2015). Spontaneous Up states in vitro: a single-metric index of the functional maturation and regional differentiation of the cerebral cortex. Front. Neural Circ 9, 59. 10.3389/fncir.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanchez-Vives MV, and McCormick DA (2000). Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat. Neurosci 3, 1027–1034. [DOI] [PubMed] [Google Scholar]
- 33.Rigas P, and Castro-Alamancos MA (2007). Thalamocortical Up States: Differential Effects of Intrinsic and Extrinsic Cortical Inputs on Persistent Activity. J. Neurosci 27, 4261–4272. 10.1523/jneurosci.0003-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hays SA, Huber KM, and Gibson JR (2011). Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J. Neurosci 31, 14223–14234. 10.1523/JNEUROSCI.3157-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giuffrida R, Musumeci S, D’Antoni S, Bonaccorso CM, Giuffrida-Stella AM, Oostra BA, and Catania MV (2005). A reduced number of metabotropic glutamate subtype 5 receptors are associated with constitutive homer proteins in a mouse model of fragile X syndrome. J. Neurosci 25, 8908–8916. 10.1523/JNEUROSCI.0932-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ronesi JA, Collins KA, Hays SA, Tsai NP, Guo W, Birnbaum SG, Hu JH, Worley PF, Gibson JR, and Huber KM (2012). Disrupted Homer scaffolds mediate abnormal mGluR5 function in a mouse model of fragile X syndrome. Nat. Neurosci 15, 431–440, S1. 10.1038/nn.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boulware MI, Weick JP, Becklund BR, Kuo SP, Groth RD, and Mermelstein PG (2005). Estradiol activates group I and II metabotropic glutamate receptor signaling, leading to opposing influences on cAMP response element-binding protein. J. Neurosci 25, 5066–5078. 10.1523/JNEUROSCI.1427-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fagan MP, Ameroso D, Meng A, Rock A, Maguire J, and Rios M (2020). Essential and sex-specific effects of mGluR5 in ventromedial hypothalamus regulating estrogen signaling and glucose balance. Proc. Natl. Acad. Sci. USA 117, 19566–19577. 10.1073/pnas.2011228117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martinez LA, Peterson BM, Meisel RL, and Mermelstein PG (2014). Estradiol facilitation of cocaine-induced locomotor sensitization in female rats requires activation of mGluR5. Behav. Brain Res 271, 39–42. 10.1016/j.bbr.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tonn Eisinger KR, Larson EB, Boulware MI, Thomas MJ, and Mermelstein PG (2018). Membrane estrogen receptor signaling impacts the reward circuitry of the female brain to influence motivated behaviors. Steroids 133, 53–59. 10.1016/j.steroids.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun J, Huang YR, Harrington WR, Sheng S, Katzenellenbogen JA, and Katzenellenbogen BS (2002). Antagonists selective for estrogen receptor alpha. Endocrinology 143, 941–947. 10.1210/endo.143.3.8704. [DOI] [PubMed] [Google Scholar]
- 42.Harrington WR, Sheng S, Barnett DH, Petz LN, Katzenellenbogen JA, and Katzenellenbogen BS (2003). Activities of estrogen receptor alpha- and beta-selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepression. Mol. Cell. Endocrinol 206, 13–22. 10.1016/s0303-7207(03)00255-7. [DOI] [PubMed] [Google Scholar]
- 43.Liang J, Blake R, Chang J, Friedman LS, Goodacre S, Hartman S, Ingalla ER, Kiefer JR, Kleinheinz T, Labadie S, et al. (2020). Discovery of GNE-149 as a Full Antagonist and Efficient Degrader of Estrogen Receptor alpha for ER+ Breast Cancer. ACS Med. Chem. Lett 11, 1342–1347. 10.1021/acsmedchemlett.0c00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tabatadze N, Huang G, May RM, Jain A, and Woolley CS (2015). Sex Differences in Molecular Signaling at Inhibitory Synapses in the Hippocampus. J. Neurosci 35, 11252–11265. 10.1523/JNEUROSCI.1067-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sergin I, Jong YJI, Harmon SK, Kumar V, and O’Malley KL (2017). Sequences within the C Terminus of the Metabotropic Glutamate Receptor 5 (mGluR5) Are Responsible for Inner Nuclear Membrane Localization. J. Biol. Chem 292, 3637–3655. 10.1074/jbc.M116.757724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tabatadze N, Smejkalova T, and Woolley CS (2013). Distribution and posttranslational modification of synaptic ERalpha in the adult female rat hippocampus. Endocrinology 154, 819–830. 10.1210/en.2012-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, and Sellers WR (1999). Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. USA 96, 2110–2115. 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Campanac E, Gasselin C, Baude A, Rama S, Ankri N, and Debanne D(2013). Enhanced intrinsic excitability in basket cells maintains excitatory-inhibitory balance in hippocampal circuits. Neuron 77, 712–722. 10.1016/j.neuron.2012.12.020. [DOI] [PubMed] [Google Scholar]
- 49.Potter WB, Basu T, O’Riordan KJ, Kirchner A, Rutecki P, Burger C, and Roopra A (2013). Reduced juvenile long-term depression in tuberous sclerosis complex is mitigated in adults by compensatory recruitment of mGluR5 and Erk signaling. PLoS Biol. 11, e1001627. 10.1371/journal.pbio.1001627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Merlin LR, Bergold PJ, and Wong RK (1998). Requirement of protein synthesis for group I mGluR-mediated induction of epileptiform discharges. J. Neurophysiol 80, 989–993. 10.1152/jn.1998.80.2.989. [DOI] [PubMed] [Google Scholar]
- 51.Zhao W, Bianchi R, Wang M, and Wong RKS (2004). Extracellular signal-regulated kinase 1/2 is required for the induction of group I metabotropic glutamate receptor-mediated epileptiform discharges. J. Neurosci 24, 76–84. 10.1523/jneurosci.4515-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhong J, Chuang SC, Bianchi R, Zhao W, Lee H, Fenton AA, Wong RKS, and Tiedge H (2009). BC1 regulation of metabotropic glutamate receptor-mediated neuronal excitability. J. Neurosci 29, 9977–9986. 10.1523/jneurosci.3893-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Osterweil EK, Krueger DD, Reinhold K, and Bear MF (2010). Hypersensitivity to mGluR5 and ERK1/2 Leads to Excessive Protein Synthesis in the Hippocampus of a Mouse Model of Fragile X Syndrome. J. Neurosci 30, 15616–15627. 10.1523/JNEUROSCI.3888-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Osterweil EK, Chuang SC, Chubykin AA, Sidorov M, Bianchi R, Wong RKS, and Bear MF (2013). Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77, 243–250. 10.1016/j.neuron.2012.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stoppel DC, McCamphill PK, Senter RK, Heynen AJ, and Bear MF (2021). mGluR5 Negative Modulators for Fragile X: Treatment Resistance and Persistence. Front. Psychiatr 12, 718953. 10.3389/fpsyt.2021.718953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmidt EK, Clavarino G, Ceppi M, and Pierre P (2009). SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275–277. 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- 57.Bergeman J, and Huot MÉ (2017). Quantitative Immunofluorescence to Measure Global Localized Translation. J. Vis. Exp 55909. 10.3791/55909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sarkar SN, Smith LT, Logan SM, and Simpkins JW (2010). Estrogen-induced activation of extracellular signal-regulated kinase signaling triggers dendritic resident mRNA translation. Neuroscience 170, 1080–1085. 10.1016/j.neuroscience.2010.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Briz V, and Baudry M (2014). Estrogen Regulates Protein Synthesis and Actin Polymerization in Hippocampal Neurons through Different Molecular Mechanisms. Front. Endocrinol 5, 22. 10.3389/fendo.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xie J, Kusnadi EP, Furic L, and Selth LA (2021). Regulation of mRNA Translation by Hormone Receptors in Breast and Prostate Cancer. Cancers 13, 3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bohlen J, Roiuk M, and Teleman AA (2021). Phosphorylation of ribosomal protein S6 differentially affects mRNA translation based on ORF length. Nucleic Acids Res. 49, 13062–13074. 10.1093/nar/gkab1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meyuhas O, and Dreazen A (2009). Ribosomal protein S6 kinase from TOP mRNAs to cell size. Prog. Mol. Biol. Transl. Sci 90, 109–153. 10.1016/S1877-1173(09)90003-5. [DOI] [PubMed] [Google Scholar]
- 63.Biever A, Valjent E, and Puighermanal E (2015). Ribosomal Protein S6 Phosphorylation in the Nervous System: From Regulation to Function. Front. Mol. Neurosci 8, 75. 10.3389/fnmol.2015.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khatpe AS, Adebayo AK, Herodotou CA, Kumar B, and Nakshatri H (2021). Nexus between PI3K/AKT and Estrogen Receptor Signaling in Breast Cancer. Cancers 13, 369. 10.3390/cancers13030369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mahoney SJ, Dempsey JM, and Blenis J (2009). Chapter 2 Cell Signaling in Protein Synthesis: Ribosome Biogenesis and Translation Initiation and Elongation. In Progress in Molecular Biology and Translational Science (Academic Press; ), pp. 53–107. 10.1016/S1877-1173(09)90002-3. [DOI] [PubMed] [Google Scholar]
- 66.Michalewski HJ, Starr A, Nguyen TT, Kong YY, and Zeng FG (2005). Auditory temporal processes in normal-hearing individuals and in patients with auditory neuropathy. Clin. Neurophysiol 116, 669–680. 10.1016/j.clinph.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 67.Tallal P, Merzenich MM, Miller S, and Jenkins W (1998). Language learning impairments: integrating basic science, technology, and remediation. Exp. Brain Res 123, 210–219. 10.1007/s002210050563. [DOI] [PubMed] [Google Scholar]
- 68.Trehub SE, and Henderson JL (1996). Temporal resolution in infancy and subsequent language development. J. Speech Hear. Res 39, 1315–1320. 10.1044/jshr.3906.1315. [DOI] [PubMed] [Google Scholar]
- 69.Abbeduto L, Brady N, and Kover ST (2007). Language development and fragile X syndrome: profiles, syndrome-specificity, and within-syndrome differences. Ment. Retard. Dev. Disabil. Res. Rev 13, 36–46. 10.1002/mrdd.20142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Foss-Feig JH, Schauder KB, Key AP, Wallace MT, and Stone WL (2017). Audition-specific temporal processing deficits associated with language function in children with autism spectrum disorder. Autism Res. 10, 1845–1856. 10.1002/aur.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jeste SS, and Nelson CA 3rd. (2009). Event related potentials in the understanding of autism spectrum disorders: an analytical review. J. Autism Dev. Disord 39, 495–510. 10.1007/s10803-008-0652-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tager-Flusberg H, and Caronna E (2007). Language disorders: autism and other pervasive developmental disorders. Pediatr. Clin 54, 469–481. 10.1016/j.pcl.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 73.Kwakye LD, Foss-Feig JH, Cascio CJ, Stone WL, and Wallace MT (2011). Altered auditory and multisensory temporal processing in autism spectrum disorders. Front. Integr. Neurosci 4, 129. 10.3389/fnint.2010.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lepistö T, Silokallio S, Nieminen-von Wendt T, Alku P, Näätänen R, and Kujala T (2006). Auditory perception and attention as reflected by the brain event-related potentials in children with Asperger syndrome. Clin. Neurophysiol 117, 2161–2171. 10.1016/j.clinph.2006.06.709. [DOI] [PubMed] [Google Scholar]
- 75.Oram Cardy JE, Flagg EJ, Roberts W, Brian J, and Roberts TPL (2005). Magnetoencephalography identifies rapid temporal processing deficit in autism and language impairment. Neuroreport 16, 329–332. 10.1097/00001756-200503150-00005. [DOI] [PubMed] [Google Scholar]
- 76.Rumschlag JA, and Razak KA (2021). Age-related changes in event related potentials, steady state responses and temporal processing in the auditory cortex of mice with severe or mild hearing loss. Hear. Res 412, 108380. 10.1016/j.heares.2021.108380. [DOI] [PubMed] [Google Scholar]
- 77.Krasowski MD (2000). Differential modulatory actions of the volatile convulsant flurothyl and its anesthetic isomer at inhibitory ligand-gated ion channels. Neuropharmacology 39, 1168–1183. 10.1016/s0028-3908(99)00221-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kadiyala SB, Papandrea D, Herron BJ, and Ferland RJ (2014). Segregation of seizure traits in C57 black mouse substrains using the repeated-flurothyl model. PLoS One 9, e90506. 10.1371/journal.pone.0090506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Séjourné J, Llaneza D, Kuti OJ, and Page DT (2015). Social Behavioral Deficits Coincide with the Onset of Seizure Susceptibility in Mice Lacking Serotonin Receptor 2c. PLoS One 10, e0136494. 10.1371/journal.pone.0136494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang W-C, Chen Y, and Page DT (2016). Hyperconnectivity of prefrontal cortex to amygdala projections in a mouse model of macrocephaly/autism syndrome. Nat. Commun 7, 13421. 10.1038/ncomms13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tonn Eisinger KR, Gross KS, Head BP, and Mermelstein PG (2018). Interactions between estrogen receptors and metabotropic glutamate receptors and their impact on drug addiction in females. Horm. Behav 104, 130–137. 10.1016/j.yhbeh.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saré RM, Song A, Loutaev I, Cook A, Maita I, Lemons A, Sheeler C, and Smith CB (2017). Negative Effects of Chronic Rapamycin Treatment on Behavior in a Mouse Model of Fragile X Syndrome. Front. Mol. Neurosci 10, 452. 10.3389/fnmol.2017.00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ango F, Prézeau L, Muller T, Tu JC, Xiao B, Worley PF, Pin JP, Bockaert J, and Fagni L (2001). Agonist-independent activation of metabotropic glutamate receptors by the intracellular protein Homer. Nature 411, 962–965. [DOI] [PubMed] [Google Scholar]
- 84.Guo W, Ceolin L, Collins KA, Perroy J, and Huber KM (2015). Elevated CaMKIIalpha and Hyperphosphorylation of Homer Mediate Circuit Dysfunction in a Fragile X Syndrome Mouse Model. Cell Rep. 13, 2297–2311. 10.1016/j.celrep.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kelly E, Schaeffer SM, Dhamne SC, Lipton JO, Lindemann L, Honer M, Jaeschke G, Super CE, Lammers SH, Modi ME, et al. (2018). mGluR5 Modulation of Behavioral and Epileptic Phenotypes in a Mouse Model of Tuberous Sclerosis Complex. Neuropsychopharmacology 43, 1457–1465. 10.1038/npp.2017.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schwarz JM (2016). Chapter 10 - Sex and the Developing Brain. In Sex Differences in the Central Nervous System, Shansky RM, ed. (Academic Press; ), pp. 221–245. 10.1016/B978-0-12-802114-9.00010-X. [DOI] [Google Scholar]
- 87.Lax-Pericall MT, Bird V, and Taylor E (2019). Gender and psychiatric disorders in children with epilepsy. A meta-analysis. Epilepsy Behav. 94, 144–150. 10.1016/j.yebeh.2019.02.014. [DOI] [PubMed] [Google Scholar]
- 88.Blackmon K, Bluvstein J, MacAllister WS, Avallone J, Misajon J, Hedlund J, Goldberg R, Bojko A, Mitra N, Giridharan R, et al. (2016). Treatment Resistant Epilepsy in Autism Spectrum Disorder: Increased Risk for Females. Autism Res. 9, 311–320. 10.1002/aur.1514. [DOI] [PubMed] [Google Scholar]
- 89.Sato SM, and Woolley CS (2016). Acute inhibition of neurosteroid estrogen synthesis suppresses status epilepticus in an animal model. Elife 5, e12917. 10.7554/eLife.12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pirbhoy PS, Rais M, Lovelace JW, Woodard W, Razak KA, Binder DK, and Ethell IM (2020). Acute pharmacological inhibition of matrix metalloproteinase-9 activity during development restores perineuronal net formation and normalizes auditory processing in Fmr1 KO mice. J. Neurochem 155, 538–558. 10.1111/jnc.15037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aguilar-Valles A, Matta-Camacho E, Khoutorsky A, Gkogkas C, Nader K, Lacaille JC, and Sonenberg N (2015). Inhibition of Group I Metabotropic Glutamate Receptors Reverses Autistic-Like Phenotypes Caused by Deficiency of the Translation Repressor eIF4E Binding Protein 2. J. Neurosci 35, 11125–11132. 10.1523/jneurosci.4615-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.de Esch CEF, van den Berg WE, Buijsen RAM, Jaafar IA, Nieuwenhuizen-Bakker IM, Gasparini F, Kushner SA, and Willemsen R (2015). Fragile X mice have robust mGluR5-dependent alterations of social behaviour in the Automated Tube Test. Neurobiol. Dis 75, 31–39. 10.1016/j.nbd.2014.12.021. [DOI] [PubMed] [Google Scholar]
- 93.Auerbach BD, Osterweil EK, and Bear MF (2011). Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–68. 10.1038/nature10658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Silverman JL, Smith DG, Rizzo SJS, Karras MN, Turner SM, Tolu SS, Bryce DK, Smith DL, Fonseca K, Ring RH, and Crawley JN (2012). Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci. Transl. Med 4, 131ra51. 10.1126/sci-translmed.3003501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kwon CH, Zhou J, Li Y, Kim KW, Hensley LL, Baker SJ, and Parada LF (2006). Neuron-specific enolase-cre mouse line with cre activity in specific neuronal populations. Genesis 44, 130–135. 10.1002/gene.20197. [DOI] [PubMed] [Google Scholar]
- 96.Bhave G, Nadin BM, Brasier DJ, Glauner KS, Shah RD, Heinemann SF, Karim F, and Gereau RW 4th. (2003). Membrane topology of a metabotropic glutamate receptor. J. Biol. Chem 278, 30294–30301. 10.1074/jbc.M303258200. [DOI] [PubMed] [Google Scholar]
- 97.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Agmon A, and Connors BW (1991). Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience 41, 365–379. 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
- 99.O’Malley KL, Jong YJI, Gonchar Y, Burkhalter A, and Romano C (2003). Activation of metabotropic glutamate receptor mGlu5 on nuclear membranes mediates intranuclear Ca2+ changes in heterologous cell types and neurons. J. Biol. Chem 278, 28210–28219. 10.1074/jbc.M300792200. [DOI] [PubMed] [Google Scholar]
- 100.Ferland RJ (2017). The Repeated Flurothyl Seizure Model in Mice. Bio. Protoc 7, e2309. 10.21769/BioProtoc.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cnops V, Iyer VR, Parathy N, Wong P, and Dawe GS (2022). Test, rinse, repeat: A review of carryover effects in rodent behavioral assays. Neurosci. Biobehav. Rev 135, 104560. 10.1016/j.neubiorev.2022.104560. [DOI] [PubMed] [Google Scholar]
- 102.Celen C, Chuang JC, Luo X, Nijem N, Walker AK, Chen F, Zhang S, Chung AS, Nguyen LH, Nassour I, et al. (2017). Arid1b haploinsufficient mice reveal neuropsychiatric phenotypes and reversible causes of growth impairment. Elife 6, e25730. 10.7554/eLife.25730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jaramillo TC, Liu S, Pettersen A, Birnbaum SG, and Powell CM (2014). Autism-related neuroligin-3 mutation alters social behavior and spatial learning. Autism Res. 7, 264–272. 10.1002/aur.1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lovelace JW, Ethell IM, Binder DK, and Razak KA (2020). Minocycline Treatment Reverses Sound Evoked EEG Abnormalities in a Mouse Model of Fragile X Syndrome. Front. Neurosci 14, 771. 10.3389/fnins.2020.00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Trujillo M, Measor K, Carrasco MM, and Razak KA (2011). Selectivity for the rate of frequency-modulated sweeps in the mouse auditory cortex. J. Neurophysiol 106, 2825–2837. 10.1152/jn.00480.2011. [DOI] [PubMed] [Google Scholar]
- 106.Llinás RR, Grace AA, and Yarom Y (1991). In vitro neurons in mammalian cortical layer 4 exhibit intrinsic oscillatory activity in the 10- to 50-Hz frequency range. Proc. Natl. Acad. Sci. USA 88, 897–901. 10.1073/pnas.88.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hwang E, Brown RE, Kocsis B, Kim T, McKenna JT, McNally JM, Han HB, and Choi JH (2019). Optogenetic stimulation of basal forebrain parvalbumin neurons modulates the cortical topography of auditory steady-state responses. Brain Struct. Funct 224, 1505–1518. 10.1007/s00429-019-01845-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cohen MX (2014). Analyzing Neural Time Series Data: Theory and Practice (The MIT Press; ). 10.7551/mitpress/9609.001.0001. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
Custom Labview software has been deposited to Zenodo and the access link is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Monoclonal Anti-PTEN (Clone 138G6) | Cell Signaling Technology | Cat# 9559; RRID:AB_390810 |
| Rabbit anti-Phospho-S6 Ribosomal Protein (Ser235/236)-Alexa Fluor® 647 | Cell Signaling Technology | Cat# 4851: RRID:AB_10695457 |
| Mouse anti-Puromycin Antibody (clone 12D10) | Millipore | Cat# MABE343: RRID:AB_2566826 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP® Rabbit mAb | Cell Signaling Technology | Cat# 4370: RRID:AB_2315112 |
| Mouse anti-NeuN | Millipore | Cat# MAB377: RRID:AB_2298772 |
| Goat Anti-Rabbit IgG Antibody (H + L), Biotinylated (BA-1000-1.5) | Vector Laboratories | Cat# BA-1000; RRID:AB_2313606 |
| Streptavidin, Alexa Fluor™ 594 conjugate | Thermo Fisher Scientific | Cat# S11227 |
| Guinea Pig anti-NeuN | Synaptic Systems | Cat# 266 004; RRID:AB_2619988 |
| Rabbit anti-Estrogen Receptor alpha (clone 60C) | Millipore | Cat# 04–820; RRID:AB_1587018 |
| Rabbit anti-Metabotropic Glutamate Receptor 5 (mGluR5) | Millipore | Cat# AB5675; RRID:AB_2295173 |
| Goat anti-Homer (E–18) | Santa Cruz Biotechnology | Cat# sc-8921; RRID:AB_648368 |
| Mouse monoclonal anti-Homer (D3) | Santa Cruz Biotechnology | Cat# sc-17842; RRID:AB_627742 |
| Mouse monoclonal anti-PSD-95 (clone 7E3) | Santa Cruz Biotechnology | Cat# sc-32290;RRID:AB_628114 |
| Mouse monoclonal anti-GluA1/GluR1 glutamate receptor | UC Davis/NIH NeuroMab Facility | Cat# N355/1; RRID:AB_2877405 |
| Mouse monoclonal anti-GluN1 glutamate receptor | UC Davis/NIH NeuroMab Facility | Cat# N308/48; RRID:AB_2877408 |
| Mouse monoclonal anti-Glyceraldehyde-3-PDH (GAPDH) | Millipore | Cat# MAB374; RRID:AB_2107445 |
| Mouse monoclonal anti-Actin (clone C4) | Millipore | Cat# MAB1501; RRID:AB_2223041 |
| Rabbit polyclonal anti-VCP | Cell Signaling Technology | Cat# 2648; RRID:AB_2214632 |
| Mouse monoclonal anti-Lamin A/C | Thermo Fisher Scientific | Cat# 14-9688-80; RRID:AB_2572956 |
| Mouse monoclonal ANTI-FLAG® M2 | Sigma-Aldrich | Cat# F1804; RRID:AB_262044 |
| Rabbit IgG, monoclonal [EPR25A] Antibody | Abcam | Cat# ab172730; RRID:AB_2687931 |
| Normal mouse IgG | Santa Cruz Biotechnology | Cat# sc-2025; RRID: AB_737182) |
| Fluorescein (FITC)-AffiniPure Goat Anti-Rabbit IgG (H + L) | Jackson ImmunoResearch Labs | Cat# 111-095-144; RRID: AB_2337978 |
| Peroxidase AffiniPure Goat Anti-Rabbit IgG (H + L) | Jackson ImmunoResearch Labs | Cat# 111-035-144; RRID: AB_2307391 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG (H + L) | Jackson ImmunoResearch Labs | Cat# 115-035-146; RRID: AB_2307392 |
| Goat anti-Rabbit IgG Fc, Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | Thermo Fisher Scientific | Cat# A78957; RRID:AB_2925780) |
| Rabbit TrueBlot®: Anti-Rabbit IgG HRP | Rockland Immunochemicals | Cat# 18-8816-31; RRID:AB_2610847 |
| Goat anti-Guinea Pig IgG (H + L) Secondary Antibody, Alexa Fluor™ 647 | Thermo Fisher Scientific | Cat# A-21450; RRID:AB_141882 |
| Goat anti-Mouse IgG (H + L) Secondary Antibody, Texas Red-X | Thermo Fisher Scientific | Cat# T-6390; RRID:AB_2556778 |
| NeuroTrace™ 435/455 blue fluorescent Nissl stain | Thermo Fisher Scientific | Cat# N21479 |
| Chemicals, peptides, and recombinant proteins | ||
| Xylazine | Dechra | N/A |
| GNE-149 | MCE MedChemExpress | Cat# HY-145341; CAS: 1953132-75-6 |
| MTEP hydrochloride | Tocris Biosciences | Cat# 2921; CAS: 1186195-60-7 |
| Ketamine | UTSW Medical Center Veterinary Drug service | N/A |
| MPP dihydrochloride | Tocris Biosciences | Cat# 1991; CAS: 911295-24-4 |
| U0126 | Tocris Biosciences | Cat# 1144; CAS: 109511-58-2 |
| Rapamycin | Tocris Biosciences | Cat# 1292; CAS: 53123-88-9 |
| Cycloheximide | Tocris Biosciences | Cat# 0970; CAS: 66-81-9 |
| Puromycin dihydrochloride from Streptomyces alboniger powder | Sigma-Aldrich | P8833; CAS: 58-58-2 |
| Anisomycin from Streptomyces griseolus | Sigma-Aldrich | A9789; CAS: 22862-76-6 |
| Lipofectamine™ 2000 Transfection Reagent | Thermo Fisher Scientific | Cat# 11668019 |
| Bis(2,2,2-trifluoroethyl) ether | Sigma-Aldrich | Cat# 287571; CAS Number: 333-36-8 |
| CTEP | Sigma-Aldrich | Cat# SML2306; CAS Number: 871362-31-1 |
| Gibco™ GlutaMAX™ Supplement | Thermo Fisher Scientific | Cat#35050061 |
| Gibco™ Antibiotic-Antimycotic (100X) | Thermo Fisher Scientific | Cat#15240096 |
| Invitrogen™ Dynabeads™ Protein G for Immunoprecipitation | Thermo Fisher Scientific | Cat#10003D |
| Invitrogen™ Dynabeads™ Protein A for Immunoprecipitation | Thermo Fisher Scientific | Cat#10001D |
| 2x Laemmli Sample Buffer | Bio-Rad | Cat #1610737 |
| Buprenorphine | Sigma-Aldrich | Cat #B9275 |
| TWEEN® 80 | Sigma-Aldrich | Cat #: P1754 |
| Critical commercial assays | ||
| Streptavidin/Biotin Blocking Kit | Vector Laboratories | Cat# SP-2002 |
| Pierce™ BCA Protein Assay Kits | Thermo Fisher Scientific | Cat# 23225 |
| SuperSignal™ West Pico PLUS Chemiluminescent Substrate | Thermo Fisher Scientific | Cat#34577 |
| Experimental models: Cell lines | ||
| HEK293 | ATCC | CRL-1573 |
| Experimental models: Organisms/strains | ||
| Mouse: Congenic NSE-Cre mice | Kwon et al.95 | N/A |
| Mouse: B6.129S4-Ptentm1Hwu/J | The Jackson Laboratory | JAX: 006440; RRID: IMSR_JAX:006440 |
| Mouse: B6(Cg)-Esr1tm4.1Ksk/J | The Jackson Laboratory | JAX: 032173 RRID: IMSR_JAX:032173 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | The Jackson Laboratory | JAX: 007914 RRID:IMSR_JAX:007914 |
| Mouse: B6.C-Tg(CMV-cre)1Cgn/J | The Jackson Laboratory | JAX: 006054 RRID:IMSR_JAX:006054 |
| Mouse: Pten heterozygous mice | This study | N/A |
| Recombinant DNA | ||
| N-terminal, flag-tagged mGluR5a | Bhave et al.96 | N/A |
| C-terminal HA-tagged ERα | This paper | N/A |
| Myc-tagged Homer2 | Guo et al.84 | N/A |
| Esr1 (tGFP-tagged) - Mouse estrogen receptor 1 (alpha) (Esr1) | Origene | Cat# MG227304 |
| 901 pLNCX myr HA Akt1 | Ramaswamy et al.47 | Addgene Plasmid #9005 |
| 902 pLNCX myr HA Akt1 K179M | Ramaswamy et al.47 | Addgene Plasmid #9006 |
| pcDNA™3.1 (+) Mammalian Expression Vector | Thermo Fisher Scientific | Cat#: V79020 |
| Software and algorithms | ||
| ImageJ/FiJI | NIH Schindelin et al.97 | https://imagej.net/software/fiji/ |
| GraphPad Prism 9 | GraphPad Prism 9 | https://www.graphpad.com/ |
| Custom Labview software | This study | https://doi.org/10.5281/zenodo.10819515 |
| ChemiDoc Imaging System | Bio-Rad | Cat# 12003153 |
| Image Lab Software | Bio-Rad | Cat# 12012931 |
| EthoVision XT | Noldus | https://www.noldus.com/ethovision-xt?utm_term=&utm_campaign=&utm_source=adwords&utm_medium=ppc&hsa_acc=5401040478&hsa_cam=20436101206&hsa_grp=&hsa_ad=&hsa_src=x&hsa_tgt=&hsa_kw=&hsa_mt=&hsa_net=adwords&hsa_ver=3&gad_source=1&gclid=EAIaIQobChMI28O82oT3hAMVSCetBh2B7ATHEAAYASAAEgJEAvD_BwE |
| Med Associates software | Med Associates Inc., St. Albans | https://med-associates.com/ |
| Other | ||
| Vibratome Leica VT 1200S | Leica | N/A |
| Interface recording chamber | Harvard Instruments | BSC-HT |
| SCREW 00-96 X 1/16(1.6 MM) (for AC and FC electrodes) | P1 technologies | 8L0X3905201F |
| EEG cable | Tucker Davis Technologies | N/A |
| RA4LI/RA4PA headstage/pre-amp | Tucker Davis Technologies | N/A |
| RZ6 multi-I/O processor | Tucker Davis Technologies | N/A |
| OpenEx (TDT) | Tucker Davis Technologies | N/A |
| Zeiss LSM 710 laser-scanning confocal microscope | Zeiss | N/A |
| PVC box (45 × 45 × 30 cm) | UTSW Medical center -Rodent Behavior Core Facility | N/A |
| 0.5 MΩ Tungsten Microelectrodes | FHC | N/A |
| Vibratome Leica VT 1000S | Leica | N/A |
| Scrambled shock generator | Med Associates Inc., St. Albans | N/A |
| Foredom dental drill | Foredom | N/A |
| Dental cement | PANAVIA™ SA Cement Universal | N/A |