

Abstract
Genome stability is governed by chromatin structural dynamics, which modify DNA accessibility under the influence of intra- and inter-nucleosomal contacts, histone post-translational modifications (PTMs) and variations, besides the activity of ATP-dependent chromatin remodelers. These are the main ways by which chromatin dynamics are regulated and connected to nuclear processes, which when dysregulated can frequently be associated with most malignancies. Recently, functional crosstalk between histone modifications and chromatin remodeling has emerged as a critical regulatory method of transcriptional regulation during cell destiny choice. Therefore, improving therapeutic outcomes for patients by focusing on epigenetic targets dysregulated in malignancies should help prevent cancer cells from developing resistance to anticancer treatments. For this reason, SET domain bifurcated histone lysine methyltransferase 1 (SETDB1) has gained a lot of attention recently as a cancer target. SETDB1 is a histone lysine methyltransferase that plays an important role in marking euchromatic and heterochromatic regions. Hence, it promotes the silencing of tumor suppressor genes and contributes to carcinogenesis. Some studies revealed that SETDB1 was overexpressed in various human cancer types, which enhanced tumor growth and metastasis. Thus, SETDB1 appears to be an attractive epigenetic target for new cancer treatments. In this review, we have discussed the effects of its overexpression on the progression of tumors and the development of inhibitor drugs that specifically target this enzyme.
The SETDB1 protein's structure and primary function are succinctly described in this review article. It also gives light on potential inhibitory mechanisms that, in the future, can be employed as a cancer target.
1. Introduction
Cancer remains one of the deadliest diseases in the modern era.1,2 Although several drugs and treatments have been developed so far, enabling the targeting of multiple cancer hallmarks, the success of cancer treatment is limited by the development of chemoresistance, the poor effectiveness against metastatic disease and general toxicity.3
Indeed, drug resistance remains a huge challenge in cancer treatment. It is considered a complex process where cancer cells tend to survive the treatment due to either a genetic predisposition or acquired modifications driven by the continuous administration of a specific treatment.4–8 Different mechanisms can lead to the latter, such as drug inactivation, multi-drug resistance, suppression of apoptosis, prevention of cell death, alterations in drug metabolism, modifications in drug targets, enhanced DNA repair, and target gene amplification.9–11 Recent reports have linked drug resistance to the ability of cancer cells to easily adapt to their environment through what is known as epigenetic machinery, a non-genetic resistance mechanism.6,9,11,12
Despite sharing the same genetic make-up, epigenetic reprogramming refers to the widespread modification of epigenetic markers that results in a change in phenotypes. In other words, an individual's gene behavior is not solely determined by their genetic makeup; it can also be influenced by changes to the way their genes are expressed through the epigenetic machinery.13–17 Additionally, it provides a framework for comprehending individual distinctions and the uniqueness of certain cells, tissues, or organs.18,19 Small-interfering RNAs, DNA methylation, histone modifications and nucleosome positioning are the main categories of interconnected mechanisms that can affect the gene stability, DNA folding, chromatin compaction, and ultimately nuclear organization and gene expression.20–23 Epigenetic reprogramming has emerged as a major player in cancer resistance, linked to transcriptional plasticity and heterogeneity in tumor cell populations, meaning that tumor cells can transform, adapt, and change their profile towards a persistent state.7,9–11 Those mechanisms could be linked to dysregulation of the expression of genes associated with the silencing of tumor suppressors, the activation of oncogenic pathways or the overexpression of oncogenes.4–7 In 2018, Yue et al. conducted a study in which they demonstrated that miR-483-3p, a non-coding RNA that has been thoroughly investigated as a potential biomarker and therapeutic target for numerous cancers, increases the susceptibility of gefitinib-resistant non-small cell lung cancer (NSCLC) to the medication by inhibiting the growth of the resistant cells and promoting apoptosis, in addition to preventing the migration, invasion, and metastasis of gefitinib-resistant NSCLC cells.24,25 The researchers started by identifying miR-483-3p as the unique miRNAs that contributed to acquired resistance. To achieve this, they created an in vitro model using NSCLC cell lines that were susceptible to gefitinib. With this information in hand, they used miRNA microarrays to perform genome-wide miRNA chip analyzes in parent cell lines that were sensitive to gefitinib and those that were resistant to it. They identified miR-483-3p as the ideal candidate because they saw that it was one of the miRNAs that were downregulated the most in gefitinib resistance (GR) cells.24 qPCR was used to validate the result.24
To confirm that miR-483-3p downregulation is associated with acquired resistance to gefitinib, researchers examined the survival of cells after gefitinib therapy in those that had miR-483-3p expression that had been altered by transiently transfecting miR-483-3p mimic or inhibitor into NSCLC cells.24 As anticipated, they observed a sharp decline in the viability of the miR-483-3p mimic, whereas the viability of the parental cells was only marginally increased by the miR-483-3p inhibitor in the presence and absence of gefitinib. This finding suggests that miR-483-3p silencing contributed to acquired resistance while miR-483-3p replenishment increased gefitinib sensitivity in vitro.24 Result that was later, validated by means of the in vivo acquired resistance model.24 Moreover, the team showed that miR-483-3p influences the levels of markers associated with the epithelial-to-mesenchymal transition (EMT).24 The team found that miR-483-3p was upregulated in HCC827GR cells, which hindered the EMT phenotype and altered cell motility, invasion, and metastasis. Most importantly, when resistant tumors were injected with a miR-483-3p mimic as opposed to a negative control, EMT was reversed, confirming the role of miR-483-3p on improved gefitinib sensitivity.24 Mechanistically, this study has demonstrated that miR-483-3p can downregulate integrin β3 level by targeting its 3′-UTR directly, in vivo and in vitro.
Other studies, including the one by Yue et al., investigate the use of miRNA to regulate EMT markets on gefitinib-resistant NSCLC cells.24
This enduring mechanism could be related to dysregulation of the expression of genes associated with the silencing of tumor suppressor genes or the induced overexpression of oncogenes.15,16,26–28 Most importantly, those genes could also be regulated by the activity of specific enzymes related to the epigenetic mechanisms in cells, which play a crucial role in the occurrence and development of tumors.3,7,9
SETDB1 is a lysine methyltransferase (KMT) that is responsible for catalyzing the di- and trimethylation of histone 3 on lysine 9 (H3K9) and has been associated with a poor prognosis in cancer patients.29,30 Several studies have reported the overexpression of SETDB1 in the progression of various types of cancer including lung,31 liver,32,33 colon29,34 and breast cancer.35,36
In this review, we highlight the major roles of SETDB1 activity in cancer development and progression, along with current studies of inhibitors of this protein. Our aim is to explore the role of medicinal chemistry in the discovery and development of novel small compounds that can bind to and modulate SETDB1 as part of integrative therapies.
2. KMTs SET-domain family and SETDB1
Histone proteins, which make up nucleosomes, the major constituents of chromatin, have positively charged amino-terminal tails that are exposed on the exterior of nucleosomes.
Numerous post-translational covalent modifications, such as acetylation, phosphorylation, ubiquitination, sumoylation, and methylation are observed to occur within these histone tails. Acetylation and phosphorylation are two reversible, dynamic changes that are frequently related to the inducible control of the expression of specific genes. Heterochromatin organization is fixed in a heritable way by histone methylation, which tends to be more permanent and appears to be involved in the cellular memory of the transcriptional status.18,19
Histone methyltransferases (HMTs), are responsible for the biochemical transfer of a methyl group from the S-adenosyl-l-methionine (SAM) cosubstrate to arginine or lysine residues on the histone tails of histones 3 and 4 (H3 & H4). Following along, KMTs are exclusively in charge of mono-, di-, and trimethylating the ε nitrogen atom of lysine residues in substrates that are histones and other proteins. Mutations, genetic translocations, and altered gene expression involving these KMTs are frequently reported in cancer, developmental abnormalities, and other diseases. The KMTs are responsible for the mono-, di-, and trimethylation of the ε-nitrogen of lysine residues side chains.4,37,38
There are two domains with annotated lysine methyltransferase activity. The SET domain, named for three Drosophila melanogaster proteins originally recognized as containing the domain, the suppressor of variegation 3-9 (SUV39), the enhancer of zeste and the enhancer of trithorax. And the seven-beta-strand (7βS) domain, which is found on enzymes ranging from the histone lysine methyltransferases (HKMT) hDOT1L to DNA methyltransferases.39
The first class of KMTs consists of about 55 proteins that contain the SET domain. These proteins have a variable I-SET insert, flanked by pre- and post-SET regions, and a common evolutionary conserved SET domain that is about 130 amino acid residues long and responsible for their catalytic activity.4,37–40
Sequence similarities found within SET domains and neighboring sequences, as well as other structural characteristics like the existence of additional clearly defined protein domains, are used to group SET-domain enzymes into a variety of families. The SUV39 family is the primary factor in the methylation of histone 3 lysine 9 (H3K9) in humans and has been most well described. The six distinct proteins Euchromatic histone lysine methyltransferase 1 (EHMT1/GLP), euchromatic histone lysine methyltransferase 2 (EHMT2/G9a), suppressor of variegation 3–9 (Drosophila) homolog 1 histone lysine methyltransferase (SUV39H1), suppressor of variegation 3–9 (Drosophila) homolog 2 histone lysine methyltransferase (SUV39H2), SETDB1 and SET domain bifurcated histone lysine methyltransferase 2 (SETDB2) make up the SUV39 family.41
With a conserved SET domain and variable I-SET insert, flanked by pre- and post-SET regions, and canonical features like a pseudo-knot next to the catalytic site, distinct cosubstrate and substrate binding areas meeting at the site of methyl transfer, and a narrow substrate lysine docking channel, the SUV39 structures adopt a typical fold, similar to the SET-domain. They also possess Cys-rich modules, which bind the three structural zinc ions necessary for the proper folding of the SET domain's core and, consequently, for its catalytic activity. The N-SET region, which encircles the core SET domain and is situated N-terminal to the pre-SET, is another feature shared by the four architectures.42 Similar to other HKMTs, the H3K9 side chain inserts itself in a groove created by the I-SET and post-SET domains and interacts extensively with the enzyme through interactions with its side chains and backbone.41–43
In a mutagenesis screen to detect proteins involved in inhibiting position effect variegation or heterochromatin development, the SUV39 was originally identified in Drosophila melanogaster. It is the founder member of the superfamily of methyltransferases that contain SET domains. It has been demonstrated that the SUV39H1 and SUV39H2 human homologues of Drosophila SUV39 are expressed in a tissue-specific way. The substrate specificity of G9a and GLP was shown to be comparable to SUV39H's introduction of H3K9me2 (di-methylation) on chromatin. In euchromatic regions of the genome, SETDB1 introduces H3K9me2/3 (di-/tri-methylation). It has also been shown to perform a dynamic role in chromatin modification and gene regulation by establishing H3K4me3/H3K9me3 and H3K14Ac/H3K9me3 post-translational modifications by identifying these previous modifications and modulating these bivalent nucleosome states through the activity of its triple TUDOR domain (TTD).41 A closely related paralogue of SETDB1, SETDB2 likewise presents H3K9 methylation activity in a tissue-specific manner. Differently from the rest of the SUV39 family, the SET domain in SETDB1/2 has been divided into two domains by the introduction of the so-called I-SET domain, giving rise to the names SETDB1 and SETDB2.41
3. SETDB1 structure
SETDB1 was first identified on human chromosome 1q21 and encodes a 143 kDa protein that is composed of 1291 amino acids.44 SETDB1 is the newest-characterized member of the family of histone methyltransferases containing a SET-domain. The SET-domain proteins play an important role in many chromatin remodeling events including transcriptional silencing, heterochromatin formation and genomic imprinting.40,45,46
SETDB1 comprises a group of proteins that specifically target H3K9, which raises significant attention as an important target in cancer development and cell reprogramming.30 It has been demonstrated that SETDB1 is able to change the expression of important genes involved in several biological processes, which include the development and silencing of endogenous retroviruses (ERV), neural diseases and cancer.30,40,45
SETDB1 structure includes an N-terminal portion which contains a TTD, a methyl-DNA binding domain (MBD), and C-terminus composed of a pre-SET, a SET and a post-SET domains, as illustrated below (Fig. 1).42,43
Fig. 1. Representation of the structural motifs within the SETDB1 protein sequence. In the image the domains are being represented by different colors.

The MBD has long been known for its role in DNA methylation readout, being involved in the recruitment of chromatin remodelers, histone deacetylases, methylases and other factors, associating gene repression with methylated DNA. In SETDB1, the MBD assists on the heterochromatin formation and transcriptional repression, though the action of two conserved arginines (R619 and R636) that are involved in the recognition of CpG islands within DNA, contributing to the specific interaction between the enzyme and methylated DNA, and is responsible for the coupling of DNA methylation with H3K9 trimethylation through an interplay with DNA Methyltransferase 3 (DNMT3), thus inducing gene silencing.44,46–49
The SET-domain topology, which is linked by a pseudo-knot topology, is composed of the pre-SET, SET-I, and post-SET subdomains. These subdomains are primarily found in enzymes that belong to the SUV39 family, including SETDB1.42,43,50,51
This unusual pseudo-knot is formed by the C-terminal segment of the SET domain passing through a loop formed by a preceding stretch of the sequence. The C-terminal segment and the loop contain the two most-conserved sequence motifs in the SET domains consisting of ELxF/YDY and NHxCxPN (where x is any amino acid), respectively.51,52
The majority of the SET-domain proteins crystal structures currently available reveal that the conserved SET domain has a unique structural fold. The SET domain is folded into a number of tiny sheets that are packed next to one another and around a structure resembling a knot.51,52 It has also been shown that the pre-SET and post-SET, two non-contiguous regions, are considered the core SET domain that is structurally conserved and includes residues critical for the methyltransferase activity.53
Pre-SET domain is characterized by nine invariant cysteine residues that are grouped into two segments of five and four cysteines, in complex with three zinc(ii) cations. The cysteinates are clustered together in a triangular arrangement, where each zinc ion is coordinated by four anionic cysteine residues in a tetrahedral manner, with each pair of zinc ions sharing one common cysteine, therefore forming an equilateral triangular cluster, Zn3Cys9.52,54,55 This zinc cluster is buried and plays a key structural role.
Interestingly, SETDB1 presents a 347-amino-acid insertion resulting in a bifurcated SET domain. It is observed that the presence of this particular insertion is evolutionarily conserved from the human protein to lower eukaryotes, including Caenorhabditis elegans and D. melanogaster, indicating that the SET domain may have functionally distinct domains.56 However, compared to other SET proteins, SETDB1 has the largest length of I-SET.56–58 Although some recent studies have linked the ubiquitination at lysine 867, located in the I-SET domain of the SETDB1 protein, as an important player in the methyltransferase activity of the protein, Schultz et al. states that this unique insertion appears to have no direct participation in the catalytic activity of SETDB1, and instead may be associated with the regulation of the enzyme's activity through conformational changes induced by post translational modifications (PTMs), such as the aforementioned ubiquitination (Fig. 1).56
The post-SET region is folded into an unusual knot-like structure that brings together the two most-conserved sequence motifs of the SET domain (NHxCxPN and ELxF/YDY) (Fig. 2) to establish an active site in close proximity to the peptide–substrate-binding cleft and the pocket where the methyl donor binds the enzyme.42,51,52 According to Zhang et al., these two highly conserved segments participate in intramolecular side chain-main chain interactions, hydrophobic structural packing, and the formation of the active site, which is situated right adjacent to the peptide binding cleft and SAM binding pocket.55
Fig. 2. The specifics of how two tyrosine residues of 3 SET-domain containing protein (GLP, SUV39H2 and G9a) interact with H3K9Me peptide. The structures were obtained from the PDB under the respective codes 3HNA (a), 2R3A (b) and 2O8J (c).

Moreover, the post-SET region, just like all proteins of the SET-family, includes the three conserved cysteine residues CxCx4C, which are fundamental for the histone lysine methyltransferase catalytic activity. The three post-SET domain cysteines coordinate the catalytic zinc ion tetrahedrally together with a Cys residue from the SET signature motif (NHxCxPN) in the pseudo knot region, near the active site, forming a narrow channel that will accommodate the side chain of the target lysine residue (Fig. 3).55,59–61
Fig. 3. Sequence alignment of the methyltransferase domain of some members of H3K9 HKMTs. Highlights represent conserved regions of the sequence. Colors red, blue, magenta and green represent small and hydrophobic, acidic, basic and hydroxyl or thiol or amine residues, respectively. Large inserts present in SETDB1 sequences (875 to 1202) are not shown for clarity.

Many studies have stated that specific conserved residues, particularly those found on the post-SET domain, are crucial to the catalytic activity of SETDB1, even though the precise catalytic mechanism is still unclear.
Schultz et al. and Yang et al. have observed that the SETDB1 H3-methylase activity is compromised by single amino acid substitutions at the highly conserved cysteines within the catalytic domain. Furthermore, it is suggested that a tyrosine in the post-SET domain within the conserved motif ELxF/YDY (TYR1265) and, specifically, the hydroxyl group of tyrosine 832 contribute to the orientation of the substrate and cosubstrate in the active site through hydrophobic- and hydrogen-bonds, favoring the transfer of the methyl from the SAM to the substrate's lysine residue. These two tyrosine residues are of particular importance for catalysis, according to studies of the SET-domain family of proteins (Fig. 2).42,56,62–64
It is believed that the SET domain folds into a small hydrophobic channel approximating the cosubstrate SAM and the peptide substrate. While the methylated lysine is buried in the channel at the active site of the enzyme, the cosubstrate binds in a separate cleft on the opposite surface of the SET domain.51,62,63 By creating hydrogen bonds with the main-chain atoms of residues within the conserved NHxCxPN motif of the pseudo-knot and GxG motif of the N-terminal region, the cosubstrate SAM bound to the SET domain adopts a U-shaped conformation, in which its methyl group faces the ε-nitrogen of the substrate lysine residue.51,62,63
It has been proposed that, mechanistically, the departing methyl of the cosubstrate is located near the de-protonated ε-nitrogen of the substrate lysine at the bottom of the lysine channel. It is currently accepted by the scientific community that the methyl group transfer from the SAM to the ε-amino moiety of the target lysine residue most likely occurs by a direct bimolecular nucleophilic substitution (SN2), even if the precise catalytic mechanism between the ternary complexes of SET-domain enzymes and their substrates and cosubstrate is yet unknown.51,62,65
It is believed that the hydroxyl group of a catalytic tyrosine residue, present in the C-terminal region of the enzymes, and nearby main chain carbonyl oxygens contribute to increase the electrophilicity of the leaving methyl group of the SAM (Fig. 4).51,62
Fig. 4. Putative catalytic mechanism of the SET-domain enzymes. Catalytic tyrosine residues side chains are shown in red, while the substrate lysine side chain is shown in blue. The cosubstrate SAM is shown in black. Relevant lone electron pairs are represented by double dots on atoms and relevant hydrogen bonds are shown as dashed lines. Electron pair movement is represented by curved solid arrows.

The mechanism is intimately related to the hydrogen bonds established in the SET domain active site, however given that only a deprotonated substrate lysine bearing a free lone pair of electrons is capable of the nucleophilic attack on the SAM methyl group, there are two main theories that may account for this phenomenon.51,62
First, it is thought that the aforementioned Tyr residue can function as a base to deprotonate the lysine substrate before methyl transfer, or at least donate some electron density to it, as to stabilize its positive charge, allowing the N–H bond electron pair to be more densely located at the nitrogen atom, making it more nucleophilic towards the electrophilic methyl group of the cosubstrate SAM, even if the precise process is still unknown (Fig. 4). Although tyrosine residues often do not carry out this function due to their relatively high pKa, owing to the nature of the enzyme pocket micro-environment, the pKa of the tyrosine residues may be sufficiently lowered to allow for proton extraction.51,62,65 Furthermore, the Lys side chain ρ-ammonium cation and the SAM sulfonium cation, two positively charged ligand moieties, are closely juxtaposed in a deeply buried position in the productive Michaelis complex, which could further favor the deprotonation of the substrate Lys residue.51,62
An alternative for the target lysine deprotonation is that it may lose a proton to the solvent due to the SET domain flexible C-flanking sequence exposing the substrate lysine to the solvent media.51
Parallel to this, a different Tyr residue that surrounds the lysine ε-nitrogen aligns its lone electron pair with the scissile methyl–sulfur bond by forming a hydrogen bond with the substrate lysine. The lysine is then methylated and S-adenosyl-l-homocysteine (SAH) is released as a consequence of the Lys lone pair of electrons nucleophilic attack upon the antibonding orbital of the sigma bond between the methyl group and the sulfur atom of the cosubstrate (Fig. 4).51,62,65
The TTD, whose function has just lately been clarified through crystallography, was revealed to bind a novel bivalent chromatin state comprised of H3K9me1/2 and H3K14ac, which it subsequently facilitates the H3K9 trimethylation.46–48 The co-crystal structure of TTD and H3K9, elucidated by Mader et al., indicates two interconnected pockets within the protein that are important for the recognition of the di-methylation and acetylation of the histone peptide. The first, a small pocket, is where the dimethyl lysine binds, while the second relatively larger and deeper pocket is engaged by the acetyl lysine (Fig. 5).66
Fig. 5. Mader et al. (2019) revealed the crystal structure of SETDB1-TTD in complex with the H3K9me2K14ac peptide (PDB 6BHD). The Kac and Kme pocket pockets are colored yellow and orange, respectively.

The TTD is thus involved in the protein–protein recognition process and various studies have demonstrated that the three TTD subdomains are crucial for the formation of important protein complexes that regulate transcriptional activity via chromatin modifications, such as histone deacetylase 1/2 (HDAC1/2) and Kruppel-associated box zinc finger proteins associated protein-1 (KRAB-ZFP-KAP-1).56,67,68
According to Blackburn, the mouse SETDB1 gene promoter region has a high GC content and contains binding regions for GATA-binding factor 1 (GATA-1), nuclear transcription factor Y (NF-Y) and specificity protein-1 (Sp-1).69 This fact could prove to be of great importance in the repurposing of some well-known epigenetic and chemotherapeutic drugs.29,70–73
4. SETDB1 physiologic role
As previously mentioned, SETDB1 is essential for the H3K9 trimethylation-mediated silencing of genes and retrotransposons, but little is known about the regulation of this enzyme's activity. According to several studies, the nuclear activating transcription factor 7 interacting protein (ATF7IP), a known binding partner of SETDB1, can control the enzymatic activities of SETDB1.37,74,75
ATF7IP apparently regulates SETDB1 retention inside the nucleus by blocking its nuclear export by tying to the N-terminal region of SETDB1, which contains the nuclear export signal motifs, and also by increasing its nuclear import. The ubiquitinated, more active form of SETDB1 is increased by nuclear localization.74,75
Recent research has also shed light on a previously unknown area where SETDB1 is tightly linked and unaffected by repressive histone marks.76 According to Warrier et al., SETDB1 co-bound with CTCF and cohesin in a region that is in close proximity to genes and devoid of any known repressive histone marks, suggests that they serve entirely different purposes from that of the well-known activities of SETDB1, such as the regulator of lineage-specific genes and ERVs.76 The team claims that following SETDB1 ChIP-Seq data processing, a unique area known as DiSCs was found, where a strong binding between SETDB1 and cohesin was revealed.76 More significantly, the co-occupancy of SETDB1 and cohesin at these locations suggested their interdependency, and it was shown that cohesin enrichment at these sites was strongly dependent on SETDB1 binding.76 The study also demonstrated that the reduction of ATF7IP led to the depletion of SMC1A and the whole cohesin complex, revealing a relationship between SETDB1 and cohesin complex.76
Another important repressive complex that has been extensively reported is the SETDB1/KAP1/ KRAB-Zfp. The Krüppel-associated box (KRAB) domain-containing zinc-finger proteins (KRAB-Zfp), scaffold protein that can assemble large repressive epigenetic machinery, was reported to be able to associate with both SETDB1 and Heterochromatin protein 1α (HP1αa).74
Through its plant homeodomain (PHD) and bromodomain, associated protein 1 (KAP1) interacts with KRAB-Zfp directly and recruits the histone deacetylase complex, nucleosome remodeling deacetylase complex (NuRD), heterochromatin protein 1 (HP1), and SETDB1. The KAP1 PHD domain enhances the binding of SETDB1 and serves as an intramolecular E3 ligase for sumoylation of the neighboring bromodomain.37,56 The SETDB1/KAP1/KRAB-Zfp complex favors the creation of a microenvironment of heterochromatin at euchromatic sites together with all of the accompanying proteins, which helps mammalian cells silence some genes and exhibit position-effect variegation.37,56
In addition to its effect on gene silencing, the SETDB1/KAP1 complex has also been linked to genomic stability by preventing homologous recombination among the several zinc finger domains containing proteins (ZNFs). Despite the fact that the SETDB1/KAP1 complex is well known for playing a crucial part in the silencing of ERVs, ZNFs have evolved alongside waves of ERVs and are thought to be an adaptive defense against viral invasion. Therefore, it is conceivable that the SETDB1/KAP1 complex served two roles during evolution, one being the suppression of ERV to ensure genomic stability and the other being the restriction of ZNF gene expansion to allow for a certain level of retrotransposon activity to bring new genomic diversity.37
The interaction between the N-terminal domain of SETDB1 and the PHD domain of DNA methyltransferase 3A (DNMT3A) is another gene transcription suppression mechanism that has been extensively researched. Studies have shown that SETDB1 specifically interacts with DNMT3s but not DNA methyltransferase 1 (DNMT1) in vitro and in vivo.37,47 The researchers additionally found a connection between SETDB1 and DNMT3B3, a DNA methyltransferase 3 beta (DNMT3B) splicing variant devoid of methylase activity, indicating that this protein participates in both DNA methylase activity-dependent and -independent processes. The work showed direct communication between DNMT3A and SETDB1, as well as the self-replicating epigenetic cycle in which SETDB1 methylates H3-K9 at areas of methylated DNA and enlists DNMT3A to support DNA methylation maintenance. It's also possible that SETDB1 is involved in de novo epigenetic silencing, which involves DNA methylation and histone H3-K9 trimethylation, in addition to retaining heritable marks. This interaction also highlights the importance of SETDB1 as a pivot on the de novo DNA methylation of the Ras association domain family protein1 isoform A (RASSF1A) promoter. The SETDB1-HDAC1 complex is initially attracted to the unmethylated RASSF1A promoter in this process, resulting in the gene inhibition. Through its direct contact with SETDB1, DNMT3A is then recruited to the promoter region and starts de novo DNA methylation. Similar mechanism was observed in studies on NSCLC cells that showed a crosstalk of SETDB1-DNMT3A on Forkhead box A2 protein (FOXA2) silencing.77
5. SETDB1: function in cancer context
SETDB1 plays a key role in the silencing of tumor suppressor genes. This KMT is a potential target in multiple signaling pathways of carcinogenesis.31,32,34,78,79 Several studies have reported that the overexpression of SETDB1 and its activity are related to aggressive tumor phenotypes,80 such as in breast,35,81,82 colon,83,84 liver32,33,85 and lung cancers,78,86–89 as well as in myeloid leukemia.90,91 Furthermore, lung cancer studies have shown that the upregulation of the protein was vital for its rapid growth and proliferation.92 Similarly, the overexpression of SETDB1 has also been described as necessary for the survival of more than 70% of acute myeloid leukemia (AML) cell lines.48 According to Monaghan et al. (2019), the knockdown of SETDB1 induced apoptosis in AML cells through important immune response activators. Hence, SETDB1 may also be associated with immune system evasion, one of the hallmarks of cancer.48,93–95 Most studies characterize SETDB1 as an oncogenic agent, showing its impact on the expression of important tumor suppressor genes, such as apolipoprotein E (APOE), tumor protein P53 (TP53) and homeobox A protein (HoxA).93,94
While SETDB1 is strongly linked to tumorigenesis and a poor prognosis, it functions differently in cancer invasion depending on the kind of cell, either as an activator or a suppressor.96–100 For example, in comparison to wild-type A549 cells, SETDB1 knockout (KO) was able to cause an increase of migratory cells in lung cancer.96 Na et al. suggest that this may be explained by the close relationship between SETDB1 and the functional localization and activity of E-cadherin and β-catenin, two complexes that are essential for preserving epithelial integrity. Disrupting this complex has an impact on a cell's adhesive repertoire as well.96,101 Wu et al. previously published a similar result, demonstrating that SETDB1 suppresses the EMT. Mechanistically, SETDB1 forms a complex with SMAD2/3 to decrease annexin A2 (ANXA2) transcription, a crucial molecular marker of cell invasion and metastasis that is inversely linked with E-cadherin expression.102–104 The same process was also observed in breast cancer.35,98
However, research on hepatocellular carcinoma (HCC) and gastric cancer (GC) revealed that SETDB1 facilitated proliferation and metastasis.33,85,99 In GC, Sp1 activity is regulated by SETDB1 overexpression. Sp1 activity has been demonstrated to be stabilized via the Wnt signaling pathway in a β-catenin manner and linked to the up-regulation of EMT, which is directly related to invasion and metastasis.101,105 Moreover, SETDB1 demonstrated a favorable effect on cell invasion, migration, and proliferation in HCC by a quite distinct mechanism. T-lymphoma invasion and metastasis-inducing protein-1 (Tiam1) and SETDB1 interact to produce a complex that is responsible for the oncogenic properties of SETDB1.33
In conclusion, the data suggests SETDB1 activity has a considerable impact in the pathophysiology of these types of cancer, however there is some disagreement over how it relates to cancer invasion and migration.
Additionally, more than its epigenetic silencing function, SETDB1 may also directly activate or even contribute to the stability of proteins with important oncogenic roles, such as the protein kinase B (AKT) enzyme or the mutated form of the peptide P53, through their methylation (Table 1).32,106–108 Although SETDB1 can methylate TP53 gene promoter,79 this protein may also interact with P53 directly and catalyze P53K370 di-methylation;32 both mechanisms are related to carcinogenesis in colon and liver cancer, respectively. Another example besides the epigenetic silencing function of SETDB1 is SETDB1-mediated AKT K64 methylation, which has been shown to be upregulated and correlated with AKT hyperactivation in lung cancer.108 Therefore, the following sections explain the proto-oncogene function of SETDB1 that has so far been identified.
Impact of SETDB1 on important cancer signaling pathway.
| Cell type | Target | Regulation | Effect | Oncogenic role | Publication year | Ref. |
|---|---|---|---|---|---|---|
| Lung cancer | P53 | Decrease expression | Stimulate WNT-β-catenin pathway activation and diminished P53 expression, maintaining cell self-renewal and stemness | Activation | 2015 | 109 |
| Lung cancer | Akt, β-catenin, E-cadherin, STAT3 | Downregulate expression of E-cadherin and b-catenin along with the upregulation of STAT3 and Akt expression | Increase migration and invasion via complex regulations on E-cadherin, b-catenin, STAT3, and Akt | Activation | 2020 | 110 |
| Lung cancer | WNT/β-catenin | Stimulates its activation | Stimulate WNT-β-catenin pathway activation and diminished P53 expression, maintaining cell self-renewal and stemness | Activation | 2015 | 109 |
| Breast cancer | SMAD7 | Decrease expression | Promote cell proliferation and metastasis by down-regulation of SMAD7 | Activation | 2019 | 35 |
| Breast cancer | c-MYC | Enhance c-MYC and CCND1 mRNA translation | Promote cell cycle progression, and provides a growth/self-renewal advantage to breast cancer cells | Activation | 2018 | 36 |
| Breast cancer | ΔNp63α | Interacts with ΔNp63α and co-repress P63 | Stimulate cell growth and self-renewal | Activation | 2016 | 81 |
| Colon cancer | AKT | Protein methylation | Promote cell proliferation, survival, and cancer progression through AKT activation | Activation | 2019 | 106 |
| Colorectal cancer | STAT1 | Increase expression | Induce STAT1-CCND1/CDK6 activation leading to an increase in cell proliferation | Activation | 2020 | 111 |
| Colorectal cancer | p21 | Silencing | Promote cell proliferation, migration, and invasion by inhibition cell-cycle arrest | Activation | 2020 | 34 |
| Colorectal cancer | WNT/β-catenin | Epigenetic markers | Inactivate the activity of the transcription factor PPARγ and therefore maintaining cell progression and cell stemness | Activation | 2012 | 83 |
| Epithelial cancer | AKT | Protein methylation | Promote cell proliferation, survival, and cancer progression through AKT activation | Activation | 2019 | 106 |
| Fibroblast | AKT | Protein methylation | Promote cell proliferation, survival, and cancer progression through AKT activation | Activation | 2019 | 108 |
| Gastric cancer | ERG genes | Increase expression of cyclin D1 (CCND1) and matrix metalloproteinase 9 (MMP9) | Interact with ERG to promote the transcription CCND1 and MMP9 plays key roles in gastric carcinogenesis and metastasis | Activation | 2021 | 112 |
| Leukemia | Six1, Dock1, Hoxa9, MEIS1 | Decrease expression | Suppresses cells growth, self-renewal and induce cell differentiation | Inactivation | 2020 | 91 |
| Liver cancer | H3K9 markers | Epigenetic markers | Activation of important oncogenic marker as well as silence of important tumor suppressor | Activation | 2016 | 85 |
| Liver cancer | P53-wild | Silencing | Decrease expression of P53 and increase gene instability and tumor progression | Activation | 2015 | 32 |
| Liver cancer | P53-mutant | Stability | Increase mutant P53 stability and increase tumor growth and proliferation | Activation | 2015 | 32 |
| Lung cancer | SMAD2/3 | Form a complex with SMAD to repress TGF-B metastatic pathway | Suppress proliferation and invasion | Inactivation | 2014 | 97 |
| Melanoma | AKT | Protein methylation | Promote cell proliferation, survival, and cancer progression through AKT activation | Activation | 2019 | 106 |
| Ovarian cancer | ||||||
| Melanoma | HOX | Decrease expression | Increase tumor growth and progression | Activation | 2011 | 113 |
5.1. SETDB1 and AKT
AKT is a serine/threonine kinase known for being one of the most important intracellular signaling hubs for integrating diverse extracellular signals into control of cell cycle progression, cell metabolic regulation, genome stability, transcription, and protein synthesis.114 As a result, dysregulation of AKT signaling is associated with severe illnesses, particularly cancer, in which AKT hyperactivity is a major biomarker of tumor severity. Intense ongoing efforts have uncovered a myriad of AKT PTMs that influence its activity, with two activating phosphorylation events by the pyruvate dehydrogenase kinase 1 (PDK1) and mammalian target of rapamycin (mTOR) kinases being the most prominent. Apart from phosphorylation, novel and promising PTMs were also recently discovered.
The discovery of SETDB1 as a novel AKT interacting partner was made by Gao et al. in 2007.115 According to the author, SETDB1 may regulate the nuclear activity of AKT1 by a non-substrate interaction. The interaction occurs in the TUDOR domain, therefore suggesting that binding to AKT1 may not affect the methyltransferase activity of SETDB1, since the SET domain is responsible for the methyltransferase activity.115 Moreover, they observed that SETDB1 may recruit AKT to the nucleus where it would participate in the regulation of nuclear activity, which is crucial for tumorigenesis.
Furthermore, the data suggests that SETDB1 not only interacts with and recruits AKT but also plays a critical role in the activation of this kinase via SETDB1-mediated lysine methylation.106,108 In response to growth factor stimulation, SETDB1 causes AKT K64 methylation, which is essential for AKT ubiquitination and activation. Based on the mechanistic study performed by the researchers on K64 methylation, AKT is recognized by Jumonji domain-containing protein 2A (JMJD2A) resulting in the formation of the complex with E3 ligases and promoting AKT K63-linked ubiquitination and cell membrane recruitment. Once recruited to the cell membrane, AKT is finally activated by PDK1-mediated phosphorylation on Thr308 (Fig. 6).108
Fig. 6. Scheme of the SETDB1-mediated Akt K64 methylation mechanism. The image represents the working model of SETDB1-mediated Akt K64 methylation in growth-factor-mediated Akt ubiquitination, cell membrane recruitment and activation which was shown in the study by Wang et al. (2019).108.

On the other hand, Guo et al. describes a different mechanism by which SETDB1 may assist AKT activation. In accordance with earlier research on the conformational shift of AKT, SETDB1 can interact with the PHD of AKT to permit methylation at the linker-region. Upon methylation, AKT can interact with its upstream kinase PDK1. In this mechanism, JMJD2A acts as an antagonist of SETDB1 activity via demethylation. As demonstrated by Wang and colleagues, SETDB1 was able to enhance AKT ubiquitination and the subsequent phosphatidylinositol 3,4,5-trisphosphate (PIP3) binding and membrane translocation, which is a crucial step in AKT activation.
It is important to note that both studies found that SETDB1 is crucial for the activation of AKT and, more critically, that its function extends beyond the suppression of gene transcription to include interactions with particular cytoplasmic proteins like AKT and active signaling pathways activity.
5.2. SETDB1 and P53
As previously mentioned, SETDB1 regulates the expression of a number of critical genes, and also directly influences the activity of several cellular effectors, including TP53, that are involved in normal cell function. Often referred to as “the guardian of the genome,” the tumor suppressor P53 regulates the cellular stress response and abnormal cellular physiology by either triggering a repair response in damaged cells or eliminating them through programmed cell death. This helps maintain cell homeostasis.116 Mutant TP53 frequently acquires new, innate carcinogenic functions in addition to losing its capacity to regulate tumor growth. This process is referred to as “gain-of-function” (GOF), and it contributes to genomic instability, P63 and P73 inactivation, aberrant gene transcription, anti-apoptosis activity, and increased tumor cell invasion and migration.32,117 Thus, the relationship between SETDB1 and TP53 is demonstrated in multiple researches pertaining to this oncogenic pathway as a novel mechanism that controls the proliferation of cancer cells.32,78,79,118
SETDB1 was shown to regulate TP53 stability through the di-methylation of residue K370 in the protein.32 The study used two cancer cell models, with GOF oncogenic TP53 mutations and wild-type TP53 cells. The group demonstrated that cells carrying GOF-mutant TP53 have a higher affinity for SETDB1 methylation of P53 compared to wild-type cells, resulting in a more stable protein, and therefore conferring cell growth advantages (Fig. 7). The mechanism by which cells respond differently is still unclear and needs to be investigated further.32
Fig. 7. Mechanistic scheme of SETDB1 and P53 interaction. The scheme showed that GOF TP53 mutations are relatively stable and often oncogenic. Moreover, this stability could be enhanced through interaction with the SETDB1 through di-methylation at K370, resulting in inhibition of the TP53-MDM2 complex formation (Fei et al., 2015).32.

In addition, a cohort analysis using colorectal cancer (CRC) samples revealed that SETDB1 was overexpressed in most CRC samples and that this overexpression was linked to TP53 gene promoter methylation, which was strongly related with a poor prognosis for CRC patients.79 This study further confirms the oncogenic effect of SETDB1 and its essential role in the proliferation and migration of CRC cells.
SETDB1 appeared to contribute to the development of pancreatic ductal adenocarcinoma (PDAC) by inhibiting the expression of TP53 genes, through binding into TP53 promoter regions and regulating cell apoptosis.118 The investigative team demonstrated that SETDB1 loss resulted in a massive pancreatic atrophy and the increased apoptosis of inflamed cells concomitant with the upregulation of TP53 expression as well as other apoptotic genes such as APAF1 and NOXA. Most importantly, this study demonstrated that SETDB1 deletion protected against PDAC formation by inducing P53-mediated apoptosis.
5.3. SETDB1 and the WNT pathway
The WNT/β-catenin signaling pathway is essential for controlling stem cell renewal as well as proliferation and differentiation during development. Because of these characteristics, it is a promising candidate for researchers looking for new druggable targets in the treatment of cancer, a disease in which Wnt/β-catenin signaling is frequently involved.119–121
On that basis, studies have demonstrated that SETDB1 controls Wingless-type MMTV integration site family, member 1 (WNT1) and β-catenin via epigenetic changes. First demonstrated in CRC, SETDB1 was shown to suppress the WNT pathway via the NOTCH pathway, therefore regulating WNT/β-catenin target genes in CRC cells. According to Kim et al., SETDB1 was found to be phosphorylated by NLK, a downstream target of Notch receptor 1 (NOTCH1) and a known inhibitor of the WNT signaling pathway. It leads to the formation of a corepressor complex that will modify histone status, such as an increased level of H3K9 methylation, on Apcmin tumors, and more specifically on WNT/β-catenin promoter regions, silencing the expression of this tumor suppressor.83
In NSCLC, SETDB1 was shown to be upregulated in over 72% of the samples. Intriguingly, SETDB1 resulted in the positive activation of the WNT/β-catenin pathway and diminished TP53 expression, resulting in an enhanced NSCLC growth and proliferation rate. Results showed that SETDB1 is essential for WNT pathway activation, consequently playing an oncogenic role in NSCLC cells (Fig. 8).109
Fig. 8. Schematic illustration of the proposed signaling interaction of SETDB1 and the Wnt pathway.

Recently, it has been showed that SETDB1 activation of WNT1 and β-catenin increases the proliferation, invasion, and migration of NSCLC by downregulating the expression of LINC00476, which is a long non-coding RNA involved in the tumorigenesis of NSCLC.122
5.4. SETDB1 and MAPK pathway
The relationship between SETDB1 and the mitogen-activated protein kinase (MAPK) signaling pathway was reported during anticancer treatment, more specifically its impact on human melanoma cells metastasis.89,110 Unexpectedly, various studies demonstrated that doxorubicin-treated, taxol-treated and siSETDB1-transfection A549 cells showed decreased expression of SETDB1 along with increased levels of FBJ murine osteosarcoma viral oncogene homolog B (FosB), which according to the group, indicated the possible negative role of SETDB1 during anticancer drug treatment.89,110 Later, the same group of researchers demonstrated that the mechanism linked to this reverse relationship between the expression of SETDB1 and FosB during anticancer drug treatment was mediated by extracellular signal-regulated kinase 2 (ERK2) activation.89
Notably, SETDB1 KO in mouse embryonic fibroblasts cells showed increased activity of the downstream MAPK transcription factors, activator protein 1 (AP1).123 Additionally, a recent study demonstrated that the inhibition of SETDB1 may sensitize cells to MAPK inhibitor-based therapies in A375 melanoma cells.70
5.5. SETDB1 and immunotherapy
Due to the fact that immunotherapies utilize the immune system capabilities and may be able to prevent escape-resistance mechanisms within tumor cells, they have received a great deal of attention when it comes to new therapy targets and combinatorial therapy. When immunotherapy is combined with conventional cytotoxic medicines, immunogenic cell death is enhanced, according to promising results.124–126
Despite its complexity, experts are currently working to put each piece of data together to build a more complete picture. Understanding the key actors in the immune system is crucial to start with.127–130
In the conventional classification, the immune system is split into two parts, the innate and adaptive systems, which work together to respond to foreign antigens or to distinguish between one's own body and that of another. However, as new technologies are developing, this idea is disproved because we now know that the immune system interacts not only with other immune components but also with commensal microorganisms and other critical systems like the endocrine and nervous systems. With this, the scientific community could begin to comprehend the intricacy of the immune system and control of the immune response in both health and sickness.128,130
Regarding cancer, immune surveillance consists of the mechanisms of recognition and elimination of any mutant cells by the lymphocytes, which act as sentinels that would recognize transformed cells. During tumor progression, cancer cells show low immunogenicity and resistance to immune effector cells, therefore, expanding and escaping immune control, being able to even modify the immune system by a mechanism known as immune editing.127,128
The key to immune surveillance is the production of tumor antigens, which are proteins produced by tumor cells that may be utilized to diagnose mutant cells, as targets for targeted immunotherapy once the disease has advanced, or even as targets for cancer vaccines.130
Despite these indicators, one way for tumor cells to escape being recognized by the immune system is through antigenic alteration. These escape mechanisms are crucial to immunotherapy because they aid in the creation of plans for using this knowledge to increase the effectiveness of cancer immunotherapy.128,130
However, there are many possible immunosuppressive mechanisms that can also vary throughout patients and tumors, which make things more complex. Within a population of tumor cells there are numerous mechanisms of immunosuppression and escaping such as downregulation of tumor antigens in cells, upregulation of immune checkpoints and immunosuppressive tumor microenvironment.128–131
Given the intricacy and significance of the immune response in cancer survival, some research has looked at the influence and function of SETDB1 in this scenario.80,95,132–134 For example, Griffin et al. demonstrates SETDB1 as an immune escaping mediator and immune sensitivity modulator of cancer cells. More notably, the study demonstrated that, in patients with advanced renal cell carcinoma, SETDB1 amplification or overexpression also predicted poor prognosis in response to programmed death 1 (PD1) blockage but not mTOR inhibition. It is significant to highlight the significance of PD1, a cell surface receptor that acts as a T-cell checkpoint and is crucial in controlling T-cell fatigue. Overexpressed PD1 is prevalent in tumor cells, and when it is activated, downstream signaling pathways are also activated, which slows T cell activation and aids the immune system escape mechanism.135 Additionally, mTOR has recently been shown to control T-cell exhaustion, which has significant implications for cancer therapy that include PD1 blocking.136 Based on their findings that SETDB1 can decrease tumor-intrinsic immunity by repressing transposable elements (TEs) and immune response-related genes, the team believes that SETDB1 may serve as an epigenetic checkpoint. By suppressing transposable elements (TEs) with potential for gene regulation, the data suggests that SETDB1 buffers the epigenetic memory in these areas, therefore acting as a key player on the mechanism of genome evolution.93 Just like presented by Griffin et al., Pan et al. went on to show that SETDB1 loss could greatly increase endogenous retrovirus activation and be followed by type I interferon induction, making murine tumors susceptible to radiotherapy reliant on type I interferons and cytotoxic T cells.93,137
The impact of SETDB1 in immune system response also was demonstrated by Lin et al., which showed that loss of SETDB1-TRIM28 complex can upregulate CD247 expression and increase the infiltration of effector CD8+ T cells through activation of the GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) pathway and thus synergizes with immune checkpoint blockade.132,138 In order to increase programmed cell death 1 ligand 1 (PDL1) expression, the team used a CRISPR-Cas9 screen and IFN treatment on ID8 mouse ovarian cells before identifying three gene hits that had a high expression of PDL1 sgRNAs.132
The experiments showed that PDL1 levels were highest in AT-rich interaction domain 1A (ARID1A), tripartite motif containing 28 (TRIM28), and lysine demethylase 1A (KDM1A) knock-down cells. These genes are also known to be negatively associated with CD274, which is the gene responsible for encoding the PDL1 immune checkpoint. The group wants to concentrate on TRIM28 since its involvement in controlling PDL1 expression and antitumor immunity has not been well investigated. As SETDB1 is the catalytic subunit of the HUSH complex, which includes TRIM28 as a component, the KO of this gene was also examined. As a result, PDL1 expression increased in ID8 Setdb1 KO cells both at the basal level and in response to the type I IFN stimulation. Results which were confirmed by microarray expression of SETDB1–TRIM28 and PD-L1 in tumor samples of 131 cases of human high-grade serous ovarian cancer (HGSOC). Furthermore, the team demonstrated that SETDB1 and TRIM28 both negatively correlated with tumor-infiltrating CD8+ T cells and activated CD8+ T-cells, as demonstrated in granzyme B+/CD8+ T-cells. However, no correlation was seen with immune suppressive populations like regulatory T cells, leading the team to the conclusion that SETDB1–TRIM28 suppresses PDL1 expression and prevents the infiltration of effector CD8+ T-cells. The activation of the cyclic GMP–AMP synthase (cGAS)–stimulator of interferon genes (STING) innate immune response pathway (cGAS–STING) ability to simultaneously upregulate PDL1 expression and boost immune cell infiltration via micronuclei-induced interferon-stimulated genes (ISGs), such Ccl5 and Cxcl10, is the mechanism behind the impact of SETDB1–TRIM28 complex loss.132
Along with this research, Hu et al. demonstrated that Atf7ip and Setdb1, its interacting partner, expression inversely correlated with antigen processing and presentation pathways, interferon signaling, and T-cell infiltration and cytotoxicity in human cancers, results that complement previous studies performed by Griffin et al.93,95 In this study, the group demonstrated significant upregulation in the expression of ERV derived antigens, endogenous retroviruses, upon ATF7IP and SETDB1 disruption and subsequent antitumor immune responses in immunocompetent hosts. Moreover, the group observed increased T-cell infiltration and activation as a result of upregulated antigen production and presentation, which results in improved antitumor immune responses, in addition to a slower growth of tumors in immunocompetent hosts lacking ATF7IP and SETDB1 genes.95
It was later shown that lysine demethylase 5B (KDM5B)- an H3K4 demethylase- expression decreased the levels of T-cell markers, antigen presentation, and cytokines expression, including two crucial effector cytokine genes, the IFNG (encoding IFN-γ) and TNF-α, as well as the T-cell chemoattractant genes CXCL9 and CXCL10, that may be connected to immune checkpoint blockade resistance. In this study, the research team demonstrated that KDM5B inhibits anti-tumor immunity through retroelement epigenetic silencing, where they showed that deletion of Kdm5b in the immunogenic YUMMER1.7 mouse melanoma model, lead to increased levels of T cells, particularly CD8+ T cells, and an increase in tumor cell death. It was demonstrated to be connected to KDM5B's function in RNA- and DNA-sensing pathways, which are essential for the type-I interferon pathway's activation. Plus, bidirectional transcripts from retroelements, such as long terminal repeat (LTR)-containing endogenous retroviruses (ERVs) and non-LTR elements, like long interspersed nuclear elements (LINEs), were detected in greater amounts in YUMMER1.7 cells following the loss of KDM5B, according to strand-specific RNA-seq analysis. Remarkably, low amounts of H3K9me3 at the retroelement loci are associated with this decline, suggesting that it is independent of its demethylase activity. KDM5B deletion decreased H3K9me3 levels at the retroelement loci, whereas H3K27me3 levels stayed the same. Furthermore, whereas the deletion of G9a and Suv39h1 had no influence on the expression of retroelements and interferon-stimulated genes (ISGs), the removal of Setdb1 significantly increased the induction of endogenous retroelement MMVL30. Moreover, in Kdm5b−/− cells, SETDB1 binding to those retroelements loci diminished and was partially restored upon reintroduction of either wild-type KDM5B or catalytically inactive KDM5B. The team conducted ChIP followed by sequencing (ChIP–seq) analyses of YUMMER1.7 cells to determine the cause of this phenomena. The results indicated that SETDB1 and H3K9me3 were more abundant within KDM5B-binding peaks in wild-type cells as compared to Kdm5b−/− cells, and that there were only minor changes in the levels of H3K4me3 and H3K4me2 following Kdm5b loss. In addition, a noteworthy positive correlation has been seen between the peaks of KDM5B and SETDB1 as well as between KDM5B and H3K9me3, indicating a potential interaction between KDM5B and SETDB.134
These works allow us to propose SETDB1 as a key participant in the tumor-cell immune escape process and a potential therapeutic target.
5.6. SET-domain family inhibition by small molecules
By adding methyl groups to histones, the epigenetic modulator SETDB1 inhibits the transcription of its target genes. This process, as previously stated, occurs at the SET domain of proteins in the SET-domain family, where the donor methyl group of the SAM cosubstrate is close to the deprotonated nitrogen of the substrate lysine at the bottom of the lysine channel. The cosubstrate and substrate peptide bind into two distinct pockets that join to create the catalytic site, giving rise to two potential drug design approaches: competitive peptide binding or competitive cosubstrate inhibition.65 Despite the fact that both the substrate and cosubstrate binding sites are expected to be druggable, the majority of co-crystallized effective and selective SET domain inhibitors made public in the Protein Data Bank (PDB) compete with the substrate lysine residue. The protein–ligand complexes G9a–UNC0638 (PDB 3RJW),139 SMYD2–AZ505 (PDB 3S7B),140 and SET7-inhibitor (PDB 4E47) are examples.
As previously stated, it is crucial to comprehend the known inhibitors of the family, since the SET-domain enzymes not only have a structurally conserved trait but also contain comparable residues that are essential for their catalytic activity. It is also important to note that the SET domain composition and the areas around it affect substrate selectivity.
The insert area between the N-SET and C-SET region can partially explain the selectivity of the SET family of enzymes. However, since SET domain proteins (such as SUV39H1 and SETDB1), which have similar selectivity profiles, can have remarkably varied insert regions, it is doubtful that SET-I is exclusively responsible for substrate selectivity. Since it can alter the catalytically competent conformation, which is attained by subsequently closing the post-SET domain on the substrate upon binding, it is necessary to take into account additional factors that influence ligand selectivity in these proteins, such as the SET domain composition itself and that of the SET flanking regions.42,54,55,59,60 This phenomenon may explain the selectivity profile of UNC0638, an inhibitor that specifically inhibits the H3K9 KMTs G9a and GLP but not the H3K4 KMT SETD7, the H4K20 KMT SETD8, or even the H3K9 KMT SUV39H2.139,141,142
In terms of H3K9 methylation agents, only selective inhibitors of SUV39H1, G9a, and GLP have been reported.
5.7. SET-domain family small molecule inhibitors
In 2005, researchers revealed the first SUV39H1/H2 selective inhibitor. With an IC50 of 0.8 μM and a SAM-competitive behavior, chaetocin was noted as one of the most effective inhibitors. Follow-up research, however, revealed that this inhibition was non-specific and time-dependent, rendering chaetocin ineffective as a selective chemical probe for histone lysine methyltransferases.139,142,147–150
The first G9a and GLP specific small-molecule inhibitor was first described by Kubicek et al. in 2007.143 A noteworthy advancement in the field of protein lysine methyltransferases (PKMT) inhibitors was the identification of two hits using high-throughput screening (HTS) of over 125 000 previously selected compounds, compound 1 and compound 2, shown in Table 2,143 chemical structure shown in Table 2.
SET-domain small molecule inhibitors explored in this review.
| Compound number in this review | Compound name in original study | Compound chemical structure | IC50 | PubChem CID | Ref. |
|---|---|---|---|---|---|
| 1 | BIX-01338 |

|
G9a: 4700 nM | 56676725 | 143 |
| 2 | BIX-01294 |

|
G9a: 1900 nM | 25150857 | 143 |
| GLP: 700 nM | |||||
| 3 | UNC0224 |

|
G9a: 15 nM | 44251522 | 144 |
| 4 | UNC0638 |

|
G9a: <15 nM | 46224516 | 139 |
| GLP: 19 nM | |||||
| 5 | UNC0642 |

|
G9a: <2.5 nM | 53315878 | 145 |
| 6 | UNC0321 |

|
G9a: 9 nM | 46901937 | 142 |
| 7 | A-366 |

|
G9a: 3.3 nM | 76285486 | 146 |
To maximize the enzymatic activity and protein expression for the HTS experiment, a recombinant GLP–G9a fusion was employed. A preselected list of 125 000 compounds was used in the HTS after the Boehringer Ingelheim chemical compound library was filtered using structural and chemical prediction in the first stage. Seven verified hits corresponding to four distinct molecular backbones were acquired and subsequently examined on the dimethylating enzyme G9a KMT, the trimethylating enzyme SUV39H1(H320R) mutant, and the arginine-methyltransferase PRMT1. Out of the seven hits, compound 1 demonstrated a broad effect on all examined enzymes and compound 2 demonstrated a strong selectivity towards G9a. Mass spectrometry was used to examine this data in more detail and examine the methylation profile (mono-, di-, and trimethylation states). The extremely selective small molecule 2 was shown to affect G9a, and only at higher concentrations did it also affect the closely related GLP/Eu-KMT1.143,151In vitro, compound 2 exhibits an IC50 of 1.7 μM and is noncompetitive with the cosubstrate SAM, according to the Kubicek et al. experiments. Those results were later explored by Chang et al., which further confirmed the inhibitory effect of 2 on GLP and G9a (with IC50 of 1.9 μM for G9a and 0.7 μM for GLP). It also demonstrated that the concentration used by Kubicek et al. was the result of an oversaturating reaction, which led to nearly all of the substrate being converted to the non physiologically relevant product H3K9me3.152
The GLP SET domain in association with compound 2 and SAH was crystallized, and the complex structure elucidated by Chang et al. This structure demonstrates that the inhibitor interacts with the substrate binding groove of GLP (Fig. 9).152 In the structure, Chang et al. showed that the diazepane ring, quinazoline ring, and related dimethoxy moieties that make up the compound 2 are the main sites of interaction between the enzyme and the inhibitor.144 The fact that four aspartates—Asp1131 and Asp1135, Asp1140, and Asp1145—specific to G9a and GLP surround compound 2 is more significant and accounts for its high selectivity toward GLP. These aspartates all demonstrated a close and strong interaction that is similar to the bound conformation of the histone peptide substrate.144
Fig. 9. GLP SET domain structure showing the two separate pockets for SAH and compound 2 interaction (PDB: 3FPD) (a). (b), An illustration of the main interactions between GLP and compound 2 was generated using the Biovia Discovery Studio Visualizer.

Followed by compound 2 discovery, using 2,4-diaminoquinazoline core of 2 as a starting scaffold in a structure–activity relationship (SAR) study, Liu et al. presented compound 3 (Table 2), a selective G9a and GLP inhibitor.142 Compound 3 was less toxic than 2, but it was also less potent. Liu et al. begin by examining the side chains found in the 7-dimethylaminopropoxy, following the advice given in the Chang et al. study discussion. To boost the already established potency, the benzyl moiety of the molecule was tackled first. The second adjustment would be to substitute different cyclic and acyclic amines for the seven-membered diazepane ring, as suggested by Chang's study. This would create a new point of interaction with the binding groove.142,152
The group also suggests adding a 7-aminoalkoxy side chain to the quinazoline template in order to take advantage of this nearby lysine binding channel. This will allow the lysine binding channel to be used for new interactions while the rest of the molecule maintains its interactions with the peptide binding groove.
The result was the synthesis of compound 3, which combines the best 2- and 4-amino moieties and has a 7-dimethylaminopropoxy chain. According to the ITC assay, the novel molecule was almost five times as effective than compound 2.142 A high-resolution X-ray crystal structure of the G9a in combination with the inhibitor was reported in this work (Fig. 10).
Fig. 10. The X-ray co-crystal structure of G9a and the inhibitor compound 3 (PDB code: 3K5K). The inhibitor is shown as a stick model with the carbon atoms in salmon (a). (b) Biovia Discovery Studio Visualizer was used to illustrate the significant interactions between G9a and compound 3.

In the same study, the 7-dimethylaminopropoxy side chain of 3 was optimized, resulting in the discovery of compound 6 (Table 2).
Due to the scaffold’s poor cell permeability, the team used the previously identified structure–activity relationship to create a new class of inhibitors that had improved cell permeability while retaining the same potency.139,153 Among those potential inhibitors, compound 4, viewed Table 2, a substrate-competitive inhibitor of G9a (IC50 < 15 nM, Ki = 3 nM) and the closely related GLP (IC50 = 19 nM), demonstrated significant on-target actions in cells with minimal cell damage.153
Using the fluorescence-based S-adenosyl-l-homocysteine hydrolase (SAHH)-coupled assay, which tracks the conversion of the cosubstrate, SAM, to the cosubstrate product, SAH, compound 4 demonstrated an IC50 of 19 nM for GLP and an IC50 less than 15 nM for G9a. The endoproteinase-coupled microfluidic capillary electrophoresis (MCE) experiment validated this.139 The mechanism of action (MOA) was also explored showing that 4 was competitive with the peptide substrate (Ki = 3.0 ± 0.05 nM) but noncompetitive with the cosubstrate SAM. H3K9me2 antibody cell immunofluorescence or in-cell western test was used to evaluate the cellular potency of compound 4, showing that 4 reduced global H3K9me2 levels in MDA-MB-231 cells, a human breast carcinoma cell line, with significantly greater potency (IC50 = 81 ± 9 nM) than 2 (IC50 = 500 ± 43 nM). In addition, 4 displayed considerably reduced cytotoxicity (EC50 = 11 000 ± 710 nM) compared to 2 (EC50 = 2700 ± 76 nM) in MDA-MB-231 cells.139 Even while compound 4 was useful as a chemical probe based on cells, its pharmacokinetic (PK) qualities in animals were not good.
Conversely, compound 5, the first in vivo chemical probe of G9a and GLP structures, as illustrated in Table 2, is a selective inhibitor of G9a that preferentially decreases H3K9me2 and possesses enhanced in vivo pharmacokinetic (PK) properties, qualifying it for use in animal studies. It also possesses excellent selectivity, low cell toxicity, and good in vitro and cellular potency, retaining high in vitro potency for G9a and GLP (IC50 < 2.5 nM) and was >20 000-fold selective for G9a/GLP over other methyltransferases (e.g., MLL1, SETD7, SETD8, SETDB1, PRMT3, PRMT5, SMYD2, SMYD3, SUV39H2, SUV420H1, SUV420H2, DOT1L, and DNMT1).145
According to MOA compound 5 study, it is noncompetitive with the cosubstrate SAM and competitive with the peptide substrate, similar to compound 4 (Ki = 3.7 ± 1 nM). Furthermore, 5 toxicity was accessed in MDA-MB-231 cells, resulting in a high potency (IC50 < 150 nM) in reducing cellular levels of H3K9me2 and low cell toxicity (EC50 > 3000 nM), although far similar to the one observed in compound 4.145
Furthermore, in vivo PK characteristics in male Swiss Albino mice were evaluated, revealing that 5 had a relatively low brain penetration with a brain/plasma ratio of 0.33 and a plasma Cmax that was over ten times greater than compound 4.145
Pappano et al. propose a novel peptide-competitive histone methyltransferase inhibitor, compound 7 (Table 2), an indole core-based compound that specifically inhibits G9a and the closely related GLP (EHMT1) but no other histone methyltransferases. Despite having comparable biological activity on H3K9me2 methylation to other known G9a/GLP small molecule inhibitors, 7 demonstrated noticeably fewer harmful effects on the proliferation of tumor cell types. The selectivity profile of compound 7 has also aided researchers in discovering G9a key role in leukemia cells differentiation.154 Because of its low toxicity, long-term G9a inhibition was made possible, demonstrating the role of G9a on the epigenetic mechanisms responsible for suppressing the differentiation of these tumors and inducing similar effects to the ones seen in earlier studies upon G9a KO.155
6. SETDB1 gene modulation by small molecules
Despite extensive study on SET-domain inhibition, none of the inhibitors have been well outlined in light of SETDB1 so far. Paclitaxel, mithramycin, and 3-deazaneplanocin A (DZNep) are a few possible non-specific inhibitors that have been suggested (Fig. 11).70,71,73
Fig. 11. Structures of the studied non-specific SETDB1 inhibitors: mithramycin A, DZNep and paclitaxel.

Mithramycin A is a DNA/RNA polymerase inhibitor, DNA-binding transcriptional inhibitor, antibiotic and the first drug that was reported to be capable of inhibiting SETDB1 gene expression. According to Ryu and colleagues, SETDB1 expression depends on the activity of the transcriptional activators Sp1 and Sp3. These are inhibited by mithramycin, which is a clinically approved guanosine–cytosine-rich DNA binding antitumor antibiotic that interferes with the binding of Sp-family of transcription factors to DNA.73 The same hypothesis was further confirmed in a recent study using malignant melanoma cells (Fig. 12).70
Fig. 12. Mechanism of action of SETDB1 genetic inhibitors. In the figure we can see that each compound works in a different way to inhibit the expression of SETDB1, but that all of the compounds work on the mRNA expression of SETDB1.

Another drug that had been reported to interfere in SETDB1 transcription was the nucleotide analogue DZNep, which is an epigenetic anticancer drug that indirectly suppresses SAM-dependent cellular methylation by inhibiting SAHH.71 According to the researchers, although the mechanism by which DZNep decreases SETDB1 transcription is not clear, they believe that it may be linked to the formation of a repressive complex in the promoter region of the SETDB1 gene (Fig. 12).
As an alternate SETDB1 inhibitor, paclitaxel has also been studied. It is known as an antitumor drug that acts as a natural antimicrotubule agent, paclitaxel was shown to significantly decrease SETDB1 transcription, through a mechanism of action linked to the increase in P53 protein levels which appeared to directly bind to the distant part of the SETDB1 promoter region. In the study, the group assumed that paclitaxel-induced P53 epigenetically represses SETDB1 gene expression by forming a repressive complex with SUV39H1 (Fig. 12).72
7. Rational drug discovery: computer-aided drug design (CADD)
Drug discovery and development is a multidisciplinary process that is exceedingly difficult, expensive, and takes a very long time to complete. Modern drug design techniques built on an understanding of disease pathophysiology, the research of biochemical pathways, and the identification of molecular targets are crucial for optimizing this process.156–158
For the discovery and development of novel medications, the use of computational techniques has produced useful information and made the whole process more efficient. In medicinal chemistry, the development of bioactive molecules benefits greatly from the coordinated application of computational and experimental methods, and the use of computational models is essential to reduce costs and time in the creation of lead compounds with optimal pharmacodynamic and pharmacokinetic properties.157
Prototype structures are modified rationally during the optimization step according to the various pharmacophoric contributions and their affinities and specificities for the chosen target.158
Among those techniques, two should be highlighted, they are: structure based drug design (SBDD) and ligand based drug design (LBDD).157,159,160
The use of drug design based on the structure of the receptor (SBDD) is only viable when dealing with a situation in which the structures of the target or ligand–receptor complexes have been already elucidated, so that methodologies such as molecular docking-based virtual screening and structure-based pharmacophore modeling can be employed.157–161
In contrast, when a set of ligands with known activity exists, it is possible to apply methods that investigate the traits and features of a number of candidate ligands using the previously defined bioactive molecules as reference. A ligand-based pharmacophoric model is used to design novel compounds, or screen existing ones, that could interact favorably with the target by using this well-known molecule or set of molecules to select the chemical motifs and spatial configuration that a ligand must possess in order to successfully bind to and elicit a biological activity upon the target, this strategy is known as LBDD.157–159,162
It is crucial to emphasize that, depending on the data the researchers have available, the combined use of SBDD and LBDD methods may often create beneficial information. The complementarity between the results of both strategies leads to more robust inferences.162
8. Drug design in SETDB1
Motivated by the need for selective and safe inhibitors, some efforts had been made to use computational tools to design new chemical entities as alternatives to cancer chemotherapy having the SETDB1 enzyme as a biomolecular target. The strategies of CADD have an important role in the modern drug design pipeline to search for new drug candidates, to improve the pharmacokinetic and pharmacodynamic properties of known compounds and to understand the mechanism of action of existing drugs or compounds.163–165
Research has investigated the protein's catalytic region by creating a computationally predicted human structure for this segment of the SETDB1 protein.166,167 A process known as docking-based virtual screening was used in the computationally built structure to find putative small peptide-competitive inhibitors by searching through databases. Park and colleagues employed a structure-based approach, using a 3D protein structure to assess the binding of the ligands that passed the cutoff from the previous phase by molecular docking, in addition to a ligand-based pharmacophore to screen against a collection of databases.
Five potential hit candidates (10, 11, 12, 13 and 14) were identified using computational analysis that showed a potential inhibitory effect upon H3K9me3. Based on binding analysis, chemicals 10 and 12 were chosen as the final hits (Table 3). Surface plasmon resonance (SPR) experiments were carried out in vitro in order to examine the direct interaction of compounds 10 and 12 with the recombinant SETDB1/ESET protein. SPR kinetic data showed that compounds 10 and 12 could bind to the SETDB1/ESET protein with Kd values of 3.26 ± 1.71 and 0.232 ± 0.146 μM, respectively (Table 3).
SETDB1 inhibitors explored in this review.
| Compound number in this review | Compound name in original study | Compound chemical structure | K d | IC50 | Compound IUPAC name | PubChem CID | Ref. |
|---|---|---|---|---|---|---|---|
| 10 | VH01 |

|
3.26 μM | — | 7-Chloro-2-[3-(dimethylamino)propyl]-1-(3-ethoxyphenyl)-1,2-dihydrochromeno[2,3-c]pyrrole-3,9-dione | 4551170 | 167 |
| 11 | VH03 |

|
— | — | N-(3,4-Dimethoxyphenyl)-3-(4-oxo-3,4,5,6,7,8-hexahydrobenzo[4,5]thieno[2,3-d]pyrimidin-2-yl)propanamide | — | 167 |
| 12 | VH06 |

|
0.232 μM | — | N-[2-(Diethylamino)ethyl]-2-[[5,7-dimethyl-6-[(2-methylbenzyl)-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl]thio]acetamide | 22422875 | 167 |
| 13 | VH08 |
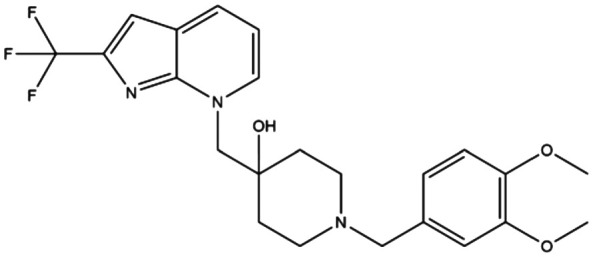
|
— | — | 1-[(3,4-Dimethoxybenzyl)-4-[[2-(trifluoromethyl)-7H-pyrrolo[2,3-b]pyridin-7-yl]methyl]piperidin-4-ol | — | 167 |
| 14 | VH21 |

|
— | — | N-[(3,4-Dimethoxyphenethyl]-3-[4-oxo-2-(pyridin-3-ylmethyl)thio)quinazolin-3(4H)]propanamide | — | 167 |
| 15 | APQ |
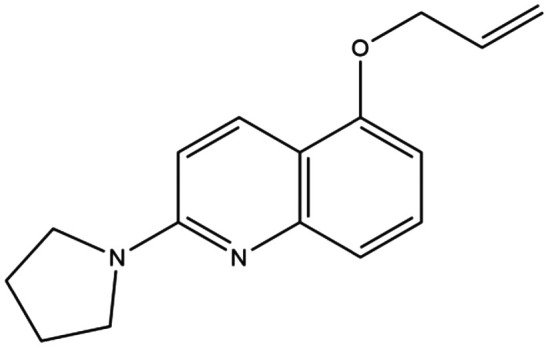
|
— | 65 μM | 5-Allyloxy-2-(pyrrolidin-1-yl)quinolone | — | 166 |
| 16 | Cpd1 |

|
4.4 μM | — | 3,5-Dimethyl-2-[[(3R,5R)-1-methyl-5-phenylpiperidin-3-yl]amino]pyrrolo[3,2-d]pyrimidin-4-one | 155535148 | 170 |
| 17 | C. 12 |
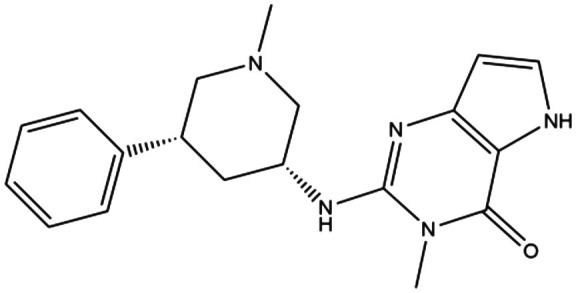
|
1.6 μM | — | 3,5-Dimethyl-2-{[(3R,5R)-1-methyl-5-phenylpiperidin-3-yl]amino}-3,5-dihydro-4H-pyrrolo[3,2- d]pyrimidin-4-one | — | 170 |
| 18 | C. 45 |

|
0.21 μM | — | 2-{[(3R,5R)-5-(4-(Benzyloxy)phenyl)-1-methylpiperidin-3-yl]amino}-3-methyl-3,5-dihydro-4H-pyrrolo[3,2-d]pyrimidin-4-one | — | 170 |
| 19 | C. R,R-59 |

|
0.08 μM | H3K9me2: 750 μM | 3-Allyl-2-{[(3R,5R)-5-(4-(benzyloxy)pheny)-1-methylpiperidin-3-yl]amino}-3,5-dihydro-4H-pyrrolo[3,2-d]pyrimidin-4-one | — | 170 |
| H3K9me3: 930 μM | |||||||
| 20 | 1 |

|
— | — | (S)-N-(tert-Butyl)-1,2,3,4- tetrahydroisoquinoline-3-carboxamide | 688344 | 66 |
| 21 | 2 |

|
— | — | 1-Methyl-4H,6H-benzo[e][1,2,4]triazolo[3,4-c][1,4]oxazepine | — | 66 |
Later, based on this study, experiments were performed which identified compound 15 as a novel SETDB1 inhibitor (Table 3).166 Combining in silico and in vitro cell-based assays, the group showed that 15 blocked the connecting pore of lysine binding site and SAM binding pocket on the catalytic domain of the SETDB1 protein, thus inhibiting the methylation process.166 To confirm the mechanism of inhibition of 15 on SETDB1, the team first carried out a docking investigation. The Gold suite-5.2 program was used to dock the 15 within the modeled enzyme structure.167 According to the docking experiments, the 15 molecule binds with a PLP score of 70.27 in the connecting pore between the lysine binding site and the SAM binding pocket.
In this study, it was also demonstrated that the two tyrosine residues, Tyr1265 and Tyr832, which have been extensively discussed in the literature on the SET-domain family of proteins, interact with Quinoline ring intersects with pi–pi-T-shaped interaction. These residues are crucial in the interaction between SET proteins and their substrates.
Subsequent to this outcome, in vitro investigations were conducted utilizing the STHdh striatal cell line, a widely used cell culture model for examining the cell-level elements of the pathogenesis of Huntington's disease (HD).168,169 To further assess the impact of 15 on SETDB1, the team used Western blot analysis. The findings demonstrated a dose-dependently significant reduction in H3K9me3 levels and SETDB1 protein levels in STHdhQ111/111 cells HD, the mutant-type HD cells.166
The team also carried out a chemo-luminescence test to assess APQ's inhibitory effects on the SETDB1 enzyme. SETDB1 activity decreased in a dose-dependent manner, despite the fact that 15's IC50 value is just 65 μM. Crucially, the MTT test, which was employed to ascertain whether 15 was cytotoxic to STHdhQ7/7 cells—the wild-type STHdh striatal cells—showed no decline in cell viability. This implies that 15 could lower SETDB1 expression levels, which would lower H3K9me3 markers without impairing the viability of normal cells, thus having a selective suppressing effect on cancer cells.166
The team conducted a network analysis to determine the effect of 15 on additional potential routes. Over 68 different YAC128 mice epigenomes were affected by 15 in terms of the occupancy of H3K9me3 in promoter regions. The network showed close connections between the epigenome involved in chromatin modification (Suv39h1, Mecp2, KDM5c, etc.), transcription control (Elk1, Foxp4, Hoxa6, Dhx9, etc.), metabolic process (Acsl4, Fbp2, Nat2, etc.), and signal transduction pathways (Cnksr2, Dusp9, Rab9, etc.). In particular, SUV39H1, which significantly decreased in YAC128 animals and was comparable to the level in WT mice.166
Recently, other studies have deciphered the importance of the TUDOR domain in the SETDB1 protein as a target of computational approaches.5,66 Just like the SET-domain in SETDB1, the TUDOR domain plays an important role in the epigenetic alterations resulting from this protein. It was shown that TUDOR domain I was not only responsible for the localization of this protein with the histones but also assisted important proteins like DNMT2 and DNMT3 in their epigenetic alteration of the DNA.46
SETDB1 presents two important adjacent pockets, a smaller one that interacts with the methylated lysine and forms the active site of the enzyme together with the SAM pocket, and a bigger one that recognizes the acetylated lysine residue were discovered in the crystal structure of the TUDOR domain, as was discussed in chapter 3.66 After the secondary pocket was identified, fragment-based screening techniques were used to find small-molecule ligands that could potentially bind the acetyl–lysine pocket of the SETDB1–TUDOR domain. These led to the identification of compounds 20 and 21, as shown in Table 3. These findings may subsequently provide the basis for additional research into novel small compounds with higher binding affinities and specificities.66
In order to search for SETDB1–TTD inhibitors, Guo and colleagues later conducted a screening against an internal chemical library that contained roughly 5000 compounds that the team had produced. The researchers identified compound 16 (Table 3), as a viable SETDB1-TTD inhibitor. Compound 16, which has a thermal shift (ΔTm) of 1.588 C in the DSF assay and a Kd value of 4.4 ± 1.7 μM in the isothermal titration calorimetry (ITC) assay.170
Additionally, the group was able to recognize the region between TUDOR 2 and TUDOR 3 as the binding domain of compound 16 based on the crystal structure of TTD in complex with compound 16.170
This study identifies three potential compound 16 chemical modifications that may be explored in order to increase the protein binding affinity, still in accordance with the cocrystal structure.170
The first moiety identified is the methyl group on the pyrrole nitrogen because of E386 and pyrrole nitrogen (–NH) form a hydrogen bonding interaction that might be enhanced by replacing the 5-position methyl group with hydrogen. Indeed, this analogue resulted in a novel compound 17 (Table 3), with ΔTm and Kd values that were significantly higher than those of compound 16, which had values of 5.168 °C and 1.6 ± 0.22 μM, respectively. More significantly, compound 17 continues to bind to TTD in the same position as 16 and maintains the crucial interactions between 16 and TTD.170
The second moiety that may be changed is the phenyl ring. The study team notes that this ring can be enlarged with substituents to improve its fit and binding to the pocket produced by the hydrophobic residues Thr210, Thr212, and Trp275 in Tudor 1 and Tudor 2. One of the byproducts of this alteration, compound 18 (Table 3), has a para-benzyloxy group. With ΔTm and Kd values of 8.158 °C and 0.21 ± 0.13 μM, respectively, this chemical showed high potency.170
The reason for this outcome is that compound 18 not only occupies the same binding site with 16 and 17 in the same pose, but it is also clear that Trp275's indole ring rotates 238 counterclockwise, positioning it parallel to the benzene ring of benzyloxy, forming an F-type π–π stack interaction and also improves its steric interactions.170
And finally the third moiety is the 3-methyl group of pyrrolo[3,2-d]pyrimidin-4-one, which has a sizable empty space in the binding pocket surrounding the methyl group. In this step, five novel compounds were produced, one of them, compound 19 (Table 3), which has a propenyl group, showed enhanced efficacy in the ITC experiment with a Kd value of 0.08 ± 0.045 μM.
It's interesting to note that 19 shares the same binding location as 16, 17, and 18, and shows a similar binding pose in the enzyme pocket. The essential interactions between compound 18 and TTD are all still present. Important to highlight, 19's C C double bond of propenyl comes into contact with Tyr268's phenolic hydroxyl at distances of 2.995 (C1–O) and 2.822 (C2–O), suggesting that a σ*–π interaction is established, which explains why 19 shows higher affinity than the other compounds.170
The research team finds 19 binding sites to be groomed between TD2 and TD3, which suggests that 19 may prevent or at least interfere with the binding of H3 protein. Additionally, in order to assess the protein–protein interaction between H3K9me2/K14ac and TTD in the presence of the inhibitor 19, the homogeneous time-resolved fluorescence (HTRF) experiment was conducted. The TTD—H3K9me2/K14ac and TTD—H3K9me3/K14ac interactions were shown to have IC50 values of 0.75 mM and 0.93 mM for 19, respectively.
The group performed a fluorescence recovery after photobleaching (FRAP) assay to further explore the activity of 19 in living cells. They carried out this through the use of enhanced green fluorescent protein (eGFP) tagged SETDB1–TTD (wild-type) and Y268A mutant plasmids, a specific mutation that loses the ability to bind H3 endogenous binders. Both the 19 treatment group and the Y268A mutant group had much lower t1/2 values, suggesting that the majority of the protein is unbound. All of the experimental findings support cellular target engagement of 19. Consequently, the research culminated in a potent and selective cell-active SETDB1–TUDOR inhibitor.170
Based on the crystallographic structure of the enzyme–inhibitor complex, the new compound 19 is shown to overlap the H3 binding site, therefore competing with the protein for its binding domain (Fig. 13).
Fig. 13. Cartoon representation of TTD domain. A) Crystal structure of the TTD–H3 fragment peptide (PDB 6BHD), B) crystal structure of the TTD–compound 19 complex (PDB 7CJT) C) An alignment between structures of the TTD–compound 19 complex (PDB entry 7CJT) and TTD–H3 fragment peptide (PDB ID 6BHD). Image was generated employing the software Pymol using the structure described in Guo et al. study.

Although there has been significant progress in developing more potent and selective inhibitors of the SETDB1 protein, no study has fully mapped out the protein structure. To enable the development of more effective SETDB1 modulators, additional research into its three-dimensional structure, catalytic site folding and function, ligand binding modes, and inhibitory mechanisms must be carried out.
9. Conclusion
Cancer remains one of the deadliest diseases worldwide and the development of resistance to anti-cancer treatment has been shown to be one of the core issues in the survival of patients. Multiple physiological mechanisms have been linked to the development of therapy-resistant malignancies, including the accumulation of epigenetic alterations, which makes modulating its effectors a promising strategy to reverse cancer progression and re-sensitize tumors to chemotherapeutic agents.5 On top of that, depending on the physiological complexity of the cancer in question, linked to its heterogeneity and plasticity, epigenetic alterations influence multiple aspects in tumor progression, like the expression of oncogenes, silencing of tumor suppressor genes and alterations in signal transduction pathways, therefore, are key determinants on cancer initiation, growth, invasion, metastasis, immune evasion, and drug resistance.171
Although being extensively studied, epigenetic therapy still has some challenges that need to be addressed and developed upon.4–7,172 One of the most important challenges faced by epi-drugs and epigenetic therapy strategies is their selectivity, since most epigenetic events are ubiquitously distributed across normal and cancer cells, making it difficult to specifically target only the tumorigenic cells.9,171,173 In addition, epi-drugs have an off-target nature, and more specifically, there are some unwanted epigenetic modifications that may inhibit important cell anticancer defense mechanisms. Therefore, it is important and highly necessary to establish a safety profile for their applicability in clinical trials.171,172 Aside from all of these challenges, the effectiveness of epi-drugs alone or administered with other chemotherapy regimens in clinical trials is indisputable, showing great improvement in patient outcomes.173,174
In this review, we explored SETDB1 implication in the development of many tumors. The repression of important genes by H3K9me3 silencing has made SETDB1 activity a very promising target against the increase in tumor proliferation and drug resistance.
According to the studies discussed here, SETDB1 is a novel epigenome-modifying target that needs further investigation for the potential development of new therapies in cancer treatment. Moreover, the mechanisms by which its epigenetic alteration might impact cancer progression and sensitivity needs to be better researched, as well as the development of SETDB1-specific inhibitors, not to mention the elucidation of the tridimensional structure of this enzyme.
Abbreviationdefinition
- AKT
Protein kinase B
- AML
Acute myeloid leukemia
- ANXA2
Annexin A2
- AP1
Activator protein 1
- Apc
Adenomatous polyposis coli
- APOE
Apolipoprotein E
- APQ
5-Allyloxy-2-(pyrrolidin-1-yl) quinoline
- ARID1A
AT-rich interaction domain 1A
- ATF7IP
Activating transcription factor 7 interacting protein
- c-MYC
MYC proto-oncogene
- Ccl5
C–C motif chemokine ligand 5
- CCND1
Cyclin D1
- CD274
Cluster of differentiation 274
- CDK6
Cyclin dependent kinase 6
- cDNAs
Complementary DNA
- cGAS
Cyclic GMP–AMP synthase
- Cpd1
3,5-Dimethyl-2-[(3R,5R)-1-methyl-5-phenylpi-peridin-3-yl]amino
- CRC
Colorectal cancer
- CRISPR-Cas9
Clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9
- CXCL10
C-X-C motif chemokine ligand 10
- CXCL9
C-X-C motif chemokine ligand 9
- DNA
Deoxyribonucleic acid
- DNMT1
DNA methyltransferase 1
- DNMT3
DNA methyltransferase 3
- DNMT3A
DNA methyltransferase 3 alpha
- DNMT3B
DNA methyltransferase 3 beta
- Dock 1
Dedicator of cytokinesis 1
- DSF
Differential scanning fluorimetry
- DZNep
3-Deazaneplanocin A
- eGFP
Enhanced green fluorescent protein
- EGFR
Epidermal growth factor receptor
- EMT
Epithelial-to-mesenchymal transition
- Erk
Extracellular signal-regulated kinases
- ERK2
Extracellular signal-regulated kinase 2
- ERV
Endogenous retroviruses
- FosB
FBJ murine osteosarcoma viral oncogene homolog B
- FOXA2
Forkhead box A2 protein
- FRAP
Fluorescence recovery after photobleaching
- G9a
Euchromatic histone lysine methyltransferase 2
- GATA-1
GATA-binding factor 1
- GC
Gastric cancer
- GLP
Euchromatic histone lysine methyltransferase 1
- GOF
Gain of function
- H3K9
Histone 3, lysine 9
- HCC
Hepatocellular carcinoma
- HD
Huntington's disease
- HDAC1
Histone deacetylase 1
- HDAC1/2
Histone deacetylase 1/2
- HMT
Histone methyltransferase
- HoxA
Gene encode homeobox A
- Hoxa9
Homeobox A9
- HP1αa
Heterochromatin protein 1α
- HUSH
Human silencing hub
- IC50
Inhibition concentration of 50%
- IFNG
Interferon gamma
- ISGs
Interferon-stimulated genes
- ITC
Isothermal titration calorimetry assay
- JMJD2A
Jumonji domain-containing protein 2A
- KAP-1
Associated protein-1
- KD
Dissociation constant value
- KDM1A
Lysine demethylase 1A
- KDM5B
Lysine demethylase 5B
- KMTs
Lysine methyltransferases
- KO
Knock out
- KRAB
Kruppel-associated box
- LINC00476
Long intergenic non-protein coding RNA 476
- MAPK
Mitogen-activated protein kinase
- MCE
Microfluidic capillary electrophoresis
- MEIS1
Meis homeobox 1
- MMP9
Matrix metalloproteinase 9
- mTOR
Mammalian target of rapamycin
- NF-Y
Nuclear transcription factor Y
- NLK
Nemo like kinase
- NOTCH1
Notch receptor 1
- NSCLC
Non-small cell lung cancer
- NuRD
Nucleosome remodeling deacetylase
- P21
Cyclin-dependent kinase inhibitor 1A
- P53
Tumor protein P53
- P63
Tumor protein P63
- PD1
Programmed death 1
- PDAC
Pancreatic ductal adenocarcinoma
- PDK1
Pyruvate dehydrogenase kinase 1
- PDL1
Programmed cell death 1 ligand 1
- PHD
Plant homeodomain containing
- PIP3
Phosphatidylinositol 3,4,5-trisphosphate
- PPARγ
Peroxisome proliferator-activated receptor gamma
- PTMs
Post-translational modifications
- RASSF1A
Ras association domain family protein1 isoform A
- RNA
Ribonucleic acid
- SAH
S-Adenosyl-l-homocysteine
- SAHH
S-Adenosyl-l-homocysteine hydrolase
- SAM
S-Adenosyl-l-methionine
- SETDB1
SET domain bifurcated histone lysine methyltransferase 1
- SETDB2
SET domain bifurcated histone lysine methyltransferase 2
- sgRNAs
single guide RNA
- siSETDB1
Small interfering RNA SETDB1
- Six1
SIX homeobox 1
- SMAD7
SMAD family member 7
- SP-1
Specificity protein-1
- SPR
Surface plasmon resonance
- STAT1
Signal transducer and activator of transcription 1
- STAT3
Signal transducer and activator of transcription 3
- STING
Stimulator of interferon genes
- SUV39
Suppressor of Variegation 3-9
- SUV39H1
Suppressor of variegation 3-9 (Drosophila) homolog 1
- SUV39H2
Suppressor of variegation 3-9 (Drosophila) homolog 2
- TEs
Transposable elements
- Tiam1
T-lymphoma invasion and metastasis-inducing protein-1
- TKI
Tyrosine kinase inhibitor
- TNF
Tumor necrosis factor
- TP53
Gene encode tumor protein P53
- TRIM28
Tripartite motif containing 28
- WNT
Wingless-type MMTV integration site family
- WNT1
Wingless-type MMTV integration site family, member 1
- ZFP
Zinc finger proteins
- ZNFs
Zinc finger domains
- ΔNp63α
N-terminally truncated isoform of p63
Conflicts of interest
There is no conflict of interest to declare.
Supplementary Material
Acknowledgments
The authors would like to thank FAPESP (The São Paulo Research Foundation), CNPq (Brazilian National Council for Scientific and Technological Development) and CAPES (Coordination for the Improvement of Higher Education Personnel) for financial support and fellowship.
References
- Nat. Cancer. 2020;1:1–2. doi: 10.1038/s43018-019-0023-9. [DOI] [PubMed] [Google Scholar]; , https://www.nature.com/articles/s43018-019-0023-9#citeas
- Laconi E. Marongiu F. DeGregori J. Br. J. Cancer. 2020;122:943–952. doi: 10.1038/s41416-019-0721-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.-H. Qu Q. Qi T.-T. Teng X.-Q. Zhu H.-H. Wang J.-J. Lu Q. Qu J. J. Exp. Clin. Cancer Res. 2021;40:174. doi: 10.1186/s13046-021-01974-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feoli A. Viviano M. Cipriano A. Milite C. Castellano S. Sbardella G. RSC Chem. Biol. 2022;3:359–406. doi: 10.1039/D1CB00196E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L. Lee Y.-T. Zhou Y. Huang Y. Semin. Cancer Biol. 2022;83:487–502. doi: 10.1016/j.semcancer.2020.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S. Jiang J. Lu Y. Nice E. C. Huang C. Zhang J. He W. Signal Transduction Targeted Ther. 2020;5:1–36. doi: 10.1038/s41392-019-0089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quagliano A. Gopalakrishnapillai A. Barwe S. P. Front. Oncol. 2020;10 doi: 10.3389/fonc.2020.00992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan R. B. A. Brennan P. E. RSC Chem. Biol. 2021;2:759–795. doi: 10.1039/D1CB00016K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz M. H. Ahmad A. Cancer Drug Resist. 2020;3:113–116. doi: 10.20517/cdr.2020.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansoori B. Mohammadi A. Davudian S. Shirjang S. Baradaran B. Adv. Pharm. Bull. 2017;7:339. doi: 10.15171/apb.2017.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasan N. Baselga J. Hyman D. M. Nature. 2019;575:299–309. doi: 10.1038/s41586-019-1730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N. Ma T. Yu B. Signal Transduction Targeted Ther. 2023;8:1–24. doi: 10.1038/s41586-022-05669-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M. A. Kouzarides T. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- Lazo P. A. Cancers. 2022;14:4050. doi: 10.3390/cancers14164050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. A. Marcum R. D. He Y. J. Mol. Biol. 2021;433:166929. doi: 10.1016/j.jmb.2021.166929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geis F. K. Goff S. P. Viruses. 2020;12:884. doi: 10.3390/v12080884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. Liu H. Liu J. Qi J. Wei B. Yang J. Liang H. Chen Y. Chen J. Wu Y. Guo L. Zhu J. Zhao X. Peng T. Zhang Y. Chen S. Li X. Li D. Wang T. Pei D. Nat. Genet. 2013;45:34–42. doi: 10.1038/ng.2491. [DOI] [PubMed] [Google Scholar]
- Egger G. Liang G. Aparicio A. Jones P. A. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- Rajpathak S. N., Biradar V. S. and Deobagkar D. D., in Medical Epigenetics, Academic Press, Tollefsbol, 2021, vol. 29, pp. 903–918 [Google Scholar]
- Cantone I. Fisher A. G. Nat. Struct. Mol. Biol. 2013;20:282–289. doi: 10.1038/nsmb.2489. [DOI] [PubMed] [Google Scholar]
- Hajkova P. Philos. Trans. R. Soc., B. 2011;366:2266–2273. doi: 10.1098/rstb.2011.0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W. Dean W. Walter J. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- Reik W. Walter J. Nat. Rev. Genet. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- Yue J. Lv D. Wang C. Li L. Zhao Q. Chen H. Xu L. Oncogene. 2018;37:4300–4312. doi: 10.1038/s41388-018-0276-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X. Chen L. Liu L. Niu X. Front. Oncol. 2019;9 doi: 10.3389/fonc.2019.01044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter C. J. Saito A. Hasegawa Y. Tanaka Y. Nagpal M. Perez G. Alway E. Espeso-Gil S. Fayyad T. Ratner C. Dincer A. Gupta A. Devi L. Pappas J. G. Lalonde F. M. Butman J. A. Han J. C. Akbarian S. Kamiya A. Nat. Commun. 2019;10:4112. doi: 10.1038/s41467-019-12013-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs P. Ding D. Bergmaier P. Lamp B. Schlagheck C. Finkernagel F. Nist A. Stiewe T. Mermoud J. E. Nat. Commun. 2019;10:1335. doi: 10.1038/s41467-019-09078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase K. A. Sharma R. P. Int. J. Neuropsychopharmacol. 2013;16:1129–1138. doi: 10.1017/S1461145712001101. [DOI] [PubMed] [Google Scholar]
- Hou Z. Sun L. Xu F. Hu F. Lan J. Song D. Feng Y. Wang J. Luo X. Hu J. Wang G. Cancer Lett. 2020;487:63–73. doi: 10.1016/j.canlet.2020.05.029. [DOI] [PubMed] [Google Scholar]
- Torrano J. Al Emran A. Hammerlindl H. Schaider H. Clin. Epigenet. 2019;11:43. doi: 10.1186/s13148-019-0644-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y.-K. Min B. Front. Genet. 2020;11:573515. doi: 10.3389/fgene.2020.573515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei Q. Shang K. Zhang J. Chuai S. Kong D. Zhou T. Fu S. Liang Y. Li C. Chen Z. Zhao Y. Yu Z. Huang Z. Hu M. Ying H. Chen Z. Zhang Y. Xing F. Zhu J. Xu H. Zhao K. Lu C. Atadja P. Xiao Z.-X. Li E. Shou J. Nat. Commun. 2015;6:8651. doi: 10.1038/ncomms9651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Huang J. Li Q. Chen K. Liang Y. Zhan Z. Ye F. Ni W. Chen L. Ding Y. BMC Cancer. 2018;18:539. doi: 10.1186/s12885-018-4464-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao N. Yu Y. Zhu H. Chen M. Chen P. Zhuo M. Mao Y. Li L. Zhao Q. Wu M. Ye M. Cell Death Dis. 2020;11:1–13. doi: 10.1038/s41419-019-2182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu T. Y. Kim K. Kim S.-K. Oh J.-H. Min J.-K. Jung C.-R. Son M.-Y. Kim D.-S. Cho H.-S. BMB Rep. 2019;52:139–144. doi: 10.5483/BMBRep.2019.52.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J.-F. Sun Q.-Y. Ding L.-W. Chien W. Liu X.-Y. Mayakonda A. Jiang Y.-Y. Loh X.-Y. Ran X.-B. Doan N. B. Castor B. Chia D. Said J. W. Tan K. T. Yang H. Fu X.-Y. Lin D.-C. Koeffler H. P. J. Pathol. 2018;246:89–102. doi: 10.1002/path.5126. [DOI] [PubMed] [Google Scholar]
- Yuan L. Sun B. Xu L. Chen L. Ou W. Int. J. Mol. Sci. 2021;22:7416. doi: 10.3390/ijms22147416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelraheem E. Thair B. Varela R. F. Jockmann E. Popadić D. Hailes H. C. Ward J. M. Iribarren A. M. Lewkowicz E. S. Andexer J. N. Hagedoorn P.-L. Hanefeld U. ChemBioChem. 2022;23:e202200212. doi: 10.1002/cbic.202200212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husmann D. Gozani O. Nat. Struct. Mol. Biol. 2019;26:880–889. doi: 10.1038/s41594-019-0298-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninova M. Fejes Tóth K. Aravin A. A. Development. 2019;146:dev181180. doi: 10.1242/dev.181180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padeken J. Methot S. P. Gasser S. M. Nat. Rev. Mol. Cell Biol. 2022;23:623–640. doi: 10.1038/s41580-022-00483-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H. Min J. Lunin V. V. Antoshenko T. Dombrovski L. Zeng H. Allali-Hassani A. Campagna-Slater V. Vedadi M. Arrowsmith C. H. Plotnikov A. N. Schapira M. PLoS One. 2010;5:e8570. doi: 10.1371/journal.pone.0008570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markouli M. Strepkos D. Piperi C. Life. 2021;11:817. doi: 10.3390/life11080817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto K. Kawamata N. Uchihara Y. Okubo M. Fujimoto R. Gotoh E. Kakinouchi K. Mizohata E. Hino N. Okada Y. Mochizuki Y. Tanaka T. Hamakubo T. Sakai J. Kodama T. Inoue T. Tachibana K. Doi T. PLoS One. 2016;11:e0165766. doi: 10.1371/journal.pone.0165766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L. Fang J. Mol. Cell. 2016;62:958–966. doi: 10.1016/j.molcel.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y. Sun D. Jakovcevski M. Jiang Y. Transl. Psychiatry. 2020;10:1–8. doi: 10.1038/s41398-019-0665-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Rauch T. Chen Z.-X. Szabó P. E. Riggs A. D. Pfeifer G. P. J. Biol. Chem. 2006;281:19489–19500. doi: 10.1074/jbc.M513249200. [DOI] [PubMed] [Google Scholar]
- Monaghan L. Massett M. E. Bunschoten R. P. Hoose A. Pirvan P.-A. Liskamp R. M. J. Jørgensen H. G. Huang X. Front. Oncol. 2019;9:705. doi: 10.3389/fonc.2019.00705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X. Ma W. Solov'yov I. A. Chipot C. Schulten K. Nucleic Acids Res. 2012;40:2747–2758. doi: 10.1093/nar/gkr1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X. Collins R. E. Zhang X. Annu. Rev. Biophys. Biomol. Struct. 2005;34:267–294. doi: 10.1146/annurev.biophys.34.040204.144452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian C. Zhou M.-M. Cell. Mol. Life Sci. 2006;63:2755–2763. doi: 10.1007/s00018-006-6274-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon S. C. Zhang X. Trievel R. C. Cheng X. Genome Biol. 2005;6:227. doi: 10.1186/gb-2005-6-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S. Eisenhaber F. O'Carroll D. Strahl B. D. Sun Z. W. Schmid M. Opravil S. Mechtler K. Ponting C. P. Allis C. D. Jenuwein T. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- Wilson J. R. Jing C. Walker P. A. Martin S. R. Howell S. A. Blackburn G. M. Gamblin S. J. Xiao B. Cell. 2002;111:105–115. doi: 10.1016/S0092-8674(02)00964-9. [DOI] [PubMed] [Google Scholar]
- Zhang X. Tamaru H. Khan S. I. Horton J. R. Keefe L. J. Selker E. U. Cheng X. Cell. 2002;111:117–127. doi: 10.1016/S0092-8674(02)00999-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D. C. Ayyanathan K. Negorev D. Maul G. G. Rauscher F. J. Genes Dev. 2002;16:919–932. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harte P. J. Wu W. Carrasquillo M. M. Matera A. G. Cytogenet. Cell Genet. 1999;84:83–86. doi: 10.1159/000015220. [DOI] [PubMed] [Google Scholar]
- Marmorstein R. Trends Biochem. Sci. 2003;28:59–62. doi: 10.1016/S0968-0004(03)00007-0. [DOI] [PubMed] [Google Scholar]
- Min J. Zhang X. Cheng X. Grewal S. I. S. Xu R.-M. Nat. Struct. Biol. 2002;9:828–832. doi: 10.1038/nsb860. [DOI] [PubMed] [Google Scholar]
- Southall S. M. Wong P.-S. Odho Z. Roe S. M. Wilson J. R. Mol. Cell. 2009;33:181–191. doi: 10.1016/j.molcel.2008.12.029. [DOI] [PubMed] [Google Scholar]
- Zhang X. Yang Z. Khan S. I. Horton J. R. Tamaru H. Selker E. U. Cheng X. Mol. Cell. 2003;12:177–185. doi: 10.1016/S1097-2765(03)00224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trievel R. C. Beach B. M. Dirk L. M. A. Houtz R. L. Hurley J. H. Cell. 2002;111:91–103. doi: 10.1016/S0092-8674(02)01000-0. [DOI] [PubMed] [Google Scholar]
- Williamson K. Schneider V. Jordan R. A. Mueller J. E. Pozzi M. H. Bryk M. PLoS One. 2013;8:e57974. doi: 10.1371/journal.pone.0057974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L. Xia L. Wu D. Y. Wang H. Chansky H. A. Schubach W. H. Hickstein D. D. Zhang Y. Oncogene. 2002;21:148–152. doi: 10.1038/sj.onc.1204998. [DOI] [PubMed] [Google Scholar]
- Schapira M. Curr. Chem. Genomics. 2011;5:85–94. doi: 10.2174/1875397301005010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader P. Mendoza-Sanchez R. Iqbal A. Dong A. Dobrovetsky E. Corless V. B. Liew S. K. Houliston S. R. De Freitas R. F. Smil D. Sena C. C. D. Kennedy S. Diaz D. B. Wu H. Dombrovski L. Allali-Hassani A. Min J. Schapira M. Vedadi M. Brown P. J. Santhakumar V. Yudin A. K. Arrowsmith C. H. Bioorg. Med. Chem. 2019;27:3866–3878. doi: 10.1016/j.bmc.2019.07.020. [DOI] [PubMed] [Google Scholar]
- Jurkowska R. Z. Qin S. Kungulovski G. Tempel W. Liu Y. Bashtrykov P. Stiefelmaier J. Jurkowski T. P. Kudithipudi S. Weirich S. Tamas R. Wu H. Dombrovski L. Loppnau P. Reinhardt R. Min J. Jeltsch A. Nat. Commun. 2017;8:2057. doi: 10.1038/s41467-017-02259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenneville S. Turelli P. Bojkowska K. Raclot C. Offner S. Kapopoulou A. Trono D. Cell Rep. 2012;2:766–773. doi: 10.1016/j.celrep.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn M. L. Chansky H. A. Zielinska-Kwiatkowska A. Matsui Y. Yang L. Biochim. Biophys. Acta. 2003;1629:8–14. doi: 10.1016/S0167-4781(03)00155-6. [DOI] [PubMed] [Google Scholar]
- Federico A. Steinfass T. Larribère L. Novak D. Morís F. Núñez L.-E. Umansky V. Utikal J. Mol. Ther.--Oncolytics. 2020;18:83–99. doi: 10.1016/j.omto.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-K. Kim K.-C. Biochem. Biophys. Res. Commun. 2013;438:647–652. doi: 10.1016/j.bbrc.2013.07.128. [DOI] [PubMed] [Google Scholar]
- Noh H.-J. Kim K.-A. Kim K.-C. Biochem. Biophys. Res. Commun. 2014;446:43–48. doi: 10.1016/j.bbrc.2014.02.053. [DOI] [PubMed] [Google Scholar]
- Ryu H. Lee J. Hagerty S. W. Soh B. Y. McAlpin S. E. Cormier K. A. Smith K. M. Ferrante R. J. Proc. Natl. Acad. Sci. U. S. A. 2006;103:19176–19181. doi: 10.1073/pnas.0606373103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. An W. Cao R. Xia L. Erdjument-Bromage H. Chatton B. Tempst P. Roeder R. G. Zhang Y. Mol. Cell. 2003;12:475–487. doi: 10.1016/j.molcel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Tsusaka T. Shimura C. Shinkai Y. EMBO Rep. 2019;20:e48297. doi: 10.15252/embr.201948297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrier T. El Farran C. Zeng Y. Ho B. S. Q. Bao Q. Zheng Z. H. Bi X. Ng H. H. Ong D. S. T. Chu J. J. H. Sanyal A. Fullwood M. J. Collins J. J. Li H. Xu J. Loh Y.-H. Nucleic Acids Res. 2022;50:7326–7349. doi: 10.1093/nar/gkac531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueshima S. Fang J. Oncogene. 2022;41:3370–3380. doi: 10.1038/s41388-022-02345-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B. Wang J. Wang J. Wang H. Gu X. Tang L. Feng X. Biosci. Rep. 2018;38:BSR20180678. doi: 10.1042/BSR20180678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K. Zhang F. Ding J. Liang Y. Zhan Z. Zhan Y. Chen L.-H. Ding Y. J. Cancer. 2017;8:3318–3330. doi: 10.7150/jca.20482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J. Chen X. Chen M. Hou J. BioMed Res. Int. 2022;2022:3307873. doi: 10.1155/2022/3307873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regina C. Compagnone M. Peschiaroli A. Lena A. Annicchiarico-Petruzzelli M. Piro M. C. Melino G. Candi E. Onco Targets Ther. 2016;7:28836–28848. doi: 10.18632/oncotarget.7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W. Su Y. Hou C. Chen L. Zhou D. Ren K. Zhou Z. Zhang R. Liu X. Oncol. Rep. 2019;41:1284–1292. doi: 10.3892/or.2018.6871. [DOI] [PubMed] [Google Scholar]
- Kim H.-A. Koo B.-K. Cho J.-H. Kim Y.-Y. Seong J. Chang H. J. Oh Y. M. Stange D. E. Park J.-G. Hwang D. Kong Y.-Y. J. Clin. Invest. 2012;122:3248–3259. doi: 10.1172/JCI61216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. Lee C. Hwang C. Y. Kim D. Han Y. Hong S. N. Kim S.-H. Cho K.-H. Mol. Cancer Res. 2020;18:118–129. doi: 10.1158/1541-7786.MCR-19-0450. [DOI] [PubMed] [Google Scholar]
- Wong C.-M. Wei L. Law C.-T. Ho D. W.-H. Tsang F. H.-C. Au S. L.-K. Sze K. M.-F. Lee J. M.-F. Wong C. C.-L. Ng I. O.-L. Hepatology. 2016;63:474–487. doi: 10.1002/hep.28304. [DOI] [PubMed] [Google Scholar]
- Cruz-Tapias P. Zakharova V. Perez-Fernandez O. M. Mantilla W. RamÍRez-Clavijo S. Ait-Si-Ali S. Cancers. 2019;11:1134. doi: 10.3390/cancers11081134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafuente-Sanchis A. Zúñiga Á. Galbis J. M. Cremades A. Estors M. Martínez-Hernández N. J. Carretero J. Clin. Transl. Oncol. 2016;18:798–804. doi: 10.1007/s12094-015-1440-6. [DOI] [PubMed] [Google Scholar]
- Liu S. Li B. Xu J. Hu S. Zhan N. Wang H. Gao C. Li J. Xu X. Front. Cell. Dev. Biol. 2020;8:213. doi: 10.3389/fcell.2020.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na H.-H. Kim K.-C. Biochem. Biophys. Res. Commun. 2018;495:512–518. doi: 10.1016/j.bbrc.2017.10.176. [DOI] [PubMed] [Google Scholar]
- Cho S. Park J. S. Kang Y.-K. J. Biol. Chem. 2011;286:41115–41124. doi: 10.1074/jbc.M111.248534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropa J. Saha N. Hu H. Peterson L. F. Talpaz M. Muntean A. G. Haematologica. 2020;105:2273–2285. doi: 10.3324/haematol.2019.223883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Paredes M. Martinez de Paz A. Simó-Riudalbas L. Sayols S. Moutinho C. Moran S. Villanueva A. Vázquez-Cedeira M. Lazo P. A. Carneiro F. Moura C. S. Vieira J. Teixeira M. R. Esteller M. Oncogene. 2014;33:2807–2813. doi: 10.1038/onc.2013.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin G. K. Wu J. Iracheta-Vellve A. Patti J. C. Hsu J. Davis T. Dele-Oni D. Du P. P. Halawi A. G. Ishizuka J. J. Kim S. Y. Klaeger S. Knudsen N. H. Miller B. C. Nguyen T. H. Olander K. E. Papanastasiou M. Rachimi S. Robitschek E. J. Schneider E. M. Yeary M. D. Zimmer M. D. Jaffe J. D. Carr S. A. Doench J. G. Haining W. N. Yates K. B. Manguso R. T. Bernstein B. E. Nature. 2021;595:309–314. doi: 10.1038/s41586-021-03520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harjes U. Nat. Rev. Cancer. 2021;21:412–412. doi: 10.1038/s41568-021-00373-x. [DOI] [PubMed] [Google Scholar]
- Hu H. Khodadadi-Jamayran A. Dolgalev I. Cho H. Badri S. Chiriboga L. A. Zeck B. Cancer Immunol. Res. 2021;9:1298–1315. doi: 10.1158/2326-6066.CIR-21-0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na H.-H. Moon S. Kim K.-C. Biochem. Biophys. Res. Commun. 2020;533:486–492. doi: 10.1016/j.bbrc.2020.09.026. [DOI] [PubMed] [Google Scholar]
- Wu P.-C. Lu J.-W. Yang J.-Y. Lin I.-H. Ou D.-L. Lin Y.-H. Chou K.-H. Huang W.-F. Wang W.-P. Huang Y.-L. Hsu C. Lin L.-I. Lin Y.-M. Shen C.-K. J. Tzeng T.-Y. Cancer Res. 2014;74:7333–7343. doi: 10.1158/0008-5472.CAN-13-3572. [DOI] [PubMed] [Google Scholar]
- Smad3-mediated recruitment of the methyltransferase SETDB1/ESET controls Snail1 expression and epithelial–mesenchymal transition, https://www.embopress.org/doi/epdf/10.15252/embr.201744250?src=getftr, (accessed December 1, 2023) [DOI] [PMC free article] [PubMed]
- Fan Y. Yang L. Ren Y. Wu Y. Li L. Li L. J. Gastric Cancer. 2022;22:319–338. doi: 10.5230/jgc.2022.22.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyropoulou A. Gargalionis A. Dalagiorgou G. Adamopoulos C. Papavassiliou K. A. Lea R. W. Piperi C. Papavassiliou A. G. NeuroMol. Med. 2014;16:70–82. doi: 10.1007/s12017-013-8254-x. [DOI] [PubMed] [Google Scholar]
- Kim W. K. Kwon Y. Jang M. Park M. Kim J. Cho S. Jang D. G. Lee W.-B. Jung S. H. Choi H. J. Min B. S. Il Kim T. Hong S. P. Paik Y.-K. Kim H. Sci. Rep. 2019;9:18440. doi: 10.1038/s41598-019-54890-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P.-C. Lu J.-W. Yang J.-Y. Lin I.-H. Ou D.-L. Lin Y.-H. Chou K.-H. Huang W.-F. Wang W.-P. Huang Y.-L. Hsu C. Lin L.-I. Lin Y.-M. Shen C.-K. J. Tzeng T.-Y. Cancer Res. 2014;74:7333–7343. doi: 10.1158/0008-5472.CAN-13-3572. [DOI] [PubMed] [Google Scholar]
- Rocha M. R. Barcellos-de-Souza P. Sousa-Squiavinato A. C. M. Fernandes P. V. De Oliveira I. M. Boroni M. Morgado-Diaz J. A. Sci. Rep. 2018;8:11285. doi: 10.1038/s41598-018-29703-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. Yuan J. Zhang J. Tian R. Ji W. Zhou Y. Yang Y. Song W. Zhang F. Niu R. Onco Targets Ther. 2015;6:30975–30992. doi: 10.18632/oncotarget.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir R. Sharma A. Pradhan S. J. Galande S. Mol. Cell. Biol. 2018;38:e00188-18. doi: 10.1128/MCB.00188-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. Dai X. Laurent B. Zheng N. Gan W. Zhang J. Guo A. Yuan M. Liu P. Asara J. M. Toker A. Shi Y. Pandolfi P. P. Wei W. Nat. Cell Biol. 2019;21:226–237. doi: 10.1038/s41556-018-0261-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. Wei W. Cell Cycle. 2019;18:917–922. doi: 10.1080/15384101.2019.1609832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G. Long J. Gao Y. Zhang W. Han F. Xu C. Sun L. Yang S.-C. Lan J. Hou Z. Cai Z. Jin G. Hsu C.-C. Wang Y.-H. Hu J. Chen T.-Y. Li H. Lee M. G. Lin H.-K. Nat. Cell Biol. 2019;21:214–225. doi: 10.1038/s41556-018-0266-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q.-Y. Ding L.-W. Xiao J.-F. Chien W. Lim S.-L. Hattori N. Goodglick L. Chia D. Mah V. Alavi M. Kim S. R. Doan N. B. Said J. W. Loh X.-Y. Xu L. Liu L.-Z. Yang H. Hayano T. Shi S. Xie D. Lin D.-C. Koeffler H. P. J. Pathol. 2015;235:559–570. doi: 10.1002/path.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na H.-H. Noh H.-J. Cheong H.-M. Kang Y. Kim K.-C. BMB Rep. 2016;49:238–243. doi: 10.5483/BMBRep.2016.49.4.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L. Ye F. Li Y.-Y. Zhan Y.-Z. Liu Y. Yan H.-M. Fang Y. Xie Y.-W. Zhang F.-J. Chen L.-H. Ding Y. Chen K.-L. Carcinogenesis. 2020;41:678–688. doi: 10.1093/carcin/bgz131. [DOI] [PubMed] [Google Scholar]
- Shang W. Wang Y. Liang X. Li T. Shao W. Liu F. Cui X. Wang Y. Lv L. Chai L. Qu L. Zheng L. Jia J. J. Pathol. 2021;253:148–159. doi: 10.1002/path.5568. [DOI] [PubMed] [Google Scholar]
- Ceol C. J. Houvras Y. Jane-Valbuena J. Bilodeau S. Orlando D. A. Battisti V. Fritsch L. Lin W. M. Hollmann T. J. Ferré F. Bourque C. Burke C. J. Turner L. Uong A. Johnson L. A. Beroukhim R. Mermel C. H. Loda M. Ait-Si-Ali S. Garraway L. A. Young R. A. Zon L. I. Nature. 2011;471:513–517. doi: 10.1038/nature09806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y. Shi X. Sheng K. Han G. Li W. Zhao Q. Jiang B. Feng J. Li J. Gu Y. Mol. Med. Rep. 2019;19:783–791. doi: 10.3892/mmr.2018.9713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H. Yu Z. Bi D. Jiang L. Cui Y. Sun J. Ma R. Mol. Cell. Biochem. 2007;305:35–44. doi: 10.1007/s11010-007-9525-3. [DOI] [PubMed] [Google Scholar]
- Olovnikov I. A. Kravchenko J. E. Chumakov P. M. Semin. Cancer Biol. 2009;19:32–41. doi: 10.1016/j.semcancer.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. Zhang T. Su W. Dou Z. Zhao D. Jin X. Lei H. Wang J. Xie X. Cheng B. Li Q. Zhang H. Di C. Cell Death Dis. 2022;13:1–14. doi: 10.1038/s41419-022-05408-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa S. Fukuda A. Matsumoto Y. Hanyu Y. Sono M. Fukunaga Y. Masuda T. Araki O. Nagao M. Yoshikawa T. Goto N. Hiramatsu Y. Tsuda M. Maruno T. Nakanishi Y. Hussein M. S. Tsuruyama T. Takaori K. Uemoto S. Seno H. Gastroenterology. 2020;159:682–696.e13. doi: 10.1053/j.gastro.2020.04.047. [DOI] [PubMed] [Google Scholar]
- Chiarini F. Paganelli F. Martelli A. M. Evangelisti C. Int. J. Mol. Sci. 2020;21:1098. doi: 10.3390/ijms21031098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danieau G. Morice S. Rédini F. Verrecchia F. Royer B. B.-L. Int. J. Mol. Sci. 2019;20:3751. doi: 10.3390/ijms20153751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Wang X. J. Hematol. Oncol. 2020;13:165. doi: 10.1186/s13045-020-00990-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G. Fu X. Wang W. Chen X. Liu S. Cao P. Zhao S. Radiat. Res. 2021;195:275–283. doi: 10.1667/RADE-20-00105.1. [DOI] [PubMed] [Google Scholar]
- Kato M. Takemoto K. Shinkai Y. Nat. Commun. 2018;9:1683. doi: 10.1038/s41467-018-04132-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson N. M. Simon M. C. Curr. Biol. 2020;30:R921–R925. doi: 10.1016/j.cub.2020.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro N. AACN Adv. Crit. Care. 2019;30:113–125. doi: 10.4037/aacnacc2019415. [DOI] [PubMed] [Google Scholar]
- Wang Z. Wang B. Cao X. Signal Transduction Targeted Ther. 2021;6:1–3. doi: 10.1038/s41392-020-00451-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arneth B. Medicina. 2019;56:15. doi: 10.3390/medicina56010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Morales S. Aranda-Uribe I. S. Pérez-Amado C. J. Ramírez-Bello J. Hidalgo-Miranda A. Front. Immunol. 2021;12:737340. doi: 10.3389/fimmu.2021.737340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers W. H., Rabinowich H. and Herberman R. B., in Holland-Frei Cancer Medicine, BC Decker, 6th edn, 2003 [Google Scholar]
- Toor S. M. Sasidharan Nair V. Decock J. Elkord E. Semin. Cancer Biol. 2020;65:1–12. doi: 10.1016/j.semcancer.2019.06.021. [DOI] [PubMed] [Google Scholar]
- Varadé J. Magadán S. González-Fernández Á. Cell. Mol. Immunol. 2021;18:805–828. doi: 10.1038/s41423-020-00530-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. Guo D. Liu H. Zhou W. Wang C. Müller I. Kossenkov A. V. Drapkin R. Bitler B. G. Helin K. Zhang R. Cancer Immunol. Res. 2021;9:1413–1424. doi: 10.1158/2326-6066.CIR-21-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J. Wang W. Zhu J. Zhuang Y. Qi C. Cai Z. Yan W. Lu W. Shang A. J. Immunol. Res. 2022;2022:4012920. doi: 10.1155/2022/4012920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.-M. Cai W. L. Liu X. Thakral D. Luo J. Chan L. H. McGeary M. K. Song E. Blenman K. R. M. Micevic G. Jessel S. Zhang Y. Yin M. Booth C. J. Jilaveanu L. B. Damsky W. Sznol M. Kluger H. M. Iwasaki A. Bosenberg M. W. Yan Q. Nature. 2021;598:682–687. doi: 10.1038/s41586-021-03994-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun D. A. Hou Y. Bakouny Z. Ficial M. Sant' Angelo M. Forman J. Ross-Macdonald P. Berger A. C. Jegede O. A. Elagina L. Steinharter J. Sun M. Wind-Rotolo M. Pignon J.-C. Cherniack A. D. Lichtenstein L. Neuberg D. Catalano P. Freeman G. J. Sharpe A. H. McDermott D. F. Van Allen E. M. Signoretti S. Wu C. J. Shukla S. A. Choueiri T. K. Nat. Med. 2020;26:909–918. doi: 10.1038/s41591-020-0839-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando S. Perkins C. M. Sajiki Y. Chastain C. Valanparambil R. M. Wieland A. Hudson W. H. Hashimoto M. Ramalingam S. S. Freeman G. J. Ahmed R. Araki K. J. Clin. Invest. 2023;133:e160025. doi: 10.1172/JCI160025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D. Bao X. Hu M. Jiao M. Li F. Li C.-Y. Cancer Res. 2022;82:2748–2760. doi: 10.1158/0008-5472.CAN-21-3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio F. P. Trombetta D. Rossi A. Sparaneo A. Castellana S. Muscarella L. A. Ther. Adv. Med. Oncol. 2018;10:1758835918815598. doi: 10.1177/1758835918815598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedadi M. Barsyte-Lovejoy D. Liu F. Rival-Gervier S. Allali-Hassani A. Labrie V. Wigle T. J. Dimaggio P. A. Wasney G. A. Siarheyeva A. Dong A. Tempel W. Wang S.-C. Chen X. Chau I. Mangano T. J. Huang X.-P. Simpson C. D. Pattenden S. G. Norris J. L. Kireev D. B. Tripathy A. Edwards A. Roth B. L. Janzen W. P. Garcia B. A. Petronis A. Ellis J. Brown P. J. Frye S. V. Arrowsmith C. H. Jin J. Nat. Chem. Biol. 2011;7:566–574. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson A. D. Larsen N. A. Howard T. Pollard H. Green I. Grande C. Cheung T. Garcia-Arenas R. Cowen S. Wu J. Godin R. Chen H. Keen N. Structure. 1993;2011(19):1262–1273. doi: 10.1016/j.str.2011.06.011. [DOI] [PubMed] [Google Scholar]
- Chandar Charles M. R. Li M.-C. Hsieh H.-P. Coumar M. S. ACS Omega. 2021;6:6100–6111. doi: 10.1021/acsomega.0c04710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F. Chen X. Allali-Hassani A. Quinn A. M. Wigle T. J. Wasney G. A. Dong A. Senisterra G. Chau I. Siarheyeva A. Norris J. L. Kireev D. B. Jadhav A. Herold J. M. Janzen W. P. Arrowsmith C. H. Frye S. V. Brown P. J. Simeonov A. Vedadi M. Jin J. J. Med. Chem. 2010;53:5844–5857. doi: 10.1021/jm100478y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek S. O'Sullivan R. J. August E. M. Hickey E. R. Zhang Q. Teodoro M. L. Rea S. Mechtler K. Kowalski J. A. Homon C. A. Kelly T. A. Jenuwein T. Mol. Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Liu F. Chen X. Allali-Hassani A. Quinn A. M. Wasney G. A. Dong A. Barsyte D. Kozieradzki I. Senisterra G. Chau I. Siarheyeva A. Kireev D. B. Jadhav A. Herold J. M. Frye S. V. Arrowsmith C. H. Brown P. J. Simeonov A. Vedadi M. Jin J. J. Med. Chem. 2009;52:7950–7953. doi: 10.1021/jm901543m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F. Barsyte-Lovejoy D. Li F. Xiong Y. Korboukh V. Huang X.-P. Allali-Hassani A. Janzen W. P. Roth B. L. Frye S. V. Arrowsmith C. H. Brown P. J. Vedadi M. Jin J. J. Med. Chem. 2013;56:8931–8942. doi: 10.1021/jm401480r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweis R. F. Pliushchev M. Brown P. J. Guo J. Li F. Maag D. Petros A. M. Soni N. B. Tse C. Vedadi M. Michaelides M. R. Chiang G. G. Pappano W. N. ACS Med. Chem. Lett. 2014;5:205–209. doi: 10.1021/ml400496h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherblanc F. L. Chapman K. L. Brown R. Fuchter M. J. Nat. Chem. Biol. 2013;9:136–137. doi: 10.1038/nchembio.1187. [DOI] [PubMed] [Google Scholar]
- Fujishiro S. Dodo K. Iwasa E. Teng Y. Sohtome Y. Hamashima Y. Ito A. Yoshida M. Sodeoka M. Bioorg. Med. Chem. Lett. 2013;23:733–736. doi: 10.1016/j.bmcl.2012.11.087. [DOI] [PubMed] [Google Scholar]
- Greiner D. Bonaldi T. Eskeland R. Roemer E. Imhof A. Nat. Chem. Biol. 2013;9:137–137. doi: 10.1038/nchembio.1188. [DOI] [PubMed] [Google Scholar]
- Iwasa E. Hamashima Y. Fujishiro S. Higuchi E. Ito A. Yoshida M. Sodeoka M. J. Am. Chem. Soc. 2010;132:4078–4079. doi: 10.1021/ja101280p. [DOI] [PubMed] [Google Scholar]
- Charles M. R. C. Dhayalan A. Hsieh H.-P. Coumar M. S. Future Med. Chem. 2019;11:993–1014. doi: 10.4155/fmc-2018-0396. [DOI] [PubMed] [Google Scholar]
- Chang Y. Zhang X. Horton J. R. Upadhyay A. K. Spannhoff A. Liu J. Snyder J. P. Bedford M. T. Cheng X. Nat. Struct. Mol. Biol. 2009;16:312–317. doi: 10.1038/nsmb.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F. Barsyte-Lovejoy D. Allali-Hassani A. He Y. Herold J. M. Chen X. Yates C. M. Frye S. V. Brown P. J. Huang J. Vedadi M. Arrowsmith C. H. Jin J. J. Med. Chem. 2011;54:6139–6150. doi: 10.1021/jm200903z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappano W. N. Guo J. He Y. Ferguson D. Jagadeeswaran S. Osterling D. J. Gao W. Spence J. K. Pliushchev M. Sweis R. F. Buchanan F. G. Michaelides M. R. Shoemaker A. R. Tse C. Chiang G. G. PLoS One. 2015;10:e0131716. doi: 10.1371/journal.pone.0131716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnertz B. Pabst C. Su L. Miller M. Liu F. Yi L. Zhang R. Krosl J. Yung E. Kirschner J. Rosten P. Underhill T. M. Jin J. Hébert J. Sauvageau G. Humphries R. K. Rossi F. M. Genes Dev. 2014;28:317–327. doi: 10.1101/gad.236794.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guido R. V. C. Andricopulo A. D. Oliva G. Estudos Avançados. 2010;24:81–98. doi: 10.1590/S0103-40142010000300006. [DOI] [Google Scholar]
- Lima E. J. C. Gomes R. A. Fornari E. Emery F. da S. Trossini G. H. G. Mini-Rev. Med. Chem. 2021;21:2227–2248. doi: 10.2174/1389557521666210226145328. [DOI] [PubMed] [Google Scholar]
- Lima L. Quím. Nova. 2007;30:1456–1468. doi: 10.1590/S0100-40422007000600015. [DOI] [Google Scholar]
- Ferraz W. R. Gomes R. A. Novaes A. L. S. Goulart Trossini G. H. Future Med. Chem. 2020;12:1815–1828. doi: 10.4155/fmc-2020-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V., Wakode S. and Kumar H., in Chemoinformatics and Bioinformatics in the Pharmaceutical Sciences, ed. N. Sharma, H. Ojha, P. K. Raghav and R. K. Goyal, Academic Press, 2021, pp. 27–53 [Google Scholar]
- Ajjarapu S. M., Tiwari A., Ramteke P. W., Singh D. B. and Kumar S., in Bioinformatics, ed. D. B. Singh and R. K. Pathak, Academic Press, 2022, pp. 233–252 [Google Scholar]
- Vázquez J. López M. Gibert E. Herrero E. Luque F. J. Molecules. 2020;25:4723. doi: 10.3390/molecules25204723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiriiri G. K. Njogu P. M. Mwangi A. N. Future J. Pharm. Sci. 2020;6:27. doi: 10.1186/s43094-020-00047-9. [DOI] [Google Scholar]
- Veselovsky A. V. Ivanov A. S. Curr. Drug Targets Infect. Disord. 2003;3:33–40. doi: 10.2174/1568005033342145. [DOI] [PubMed] [Google Scholar]
- Yu W. MacKerell A. D. Methods Mol. Biol. 2017;1520:85–106. doi: 10.1007/978-1-4939-6634-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang Y. J. Hyeon S. J. Kim Y. Lim S. Lee M. Y. Kim J. Londhe A. M. Gotina L. Kim Y. Pae A. N. Cho Y. S. Seong J. Seo H. Kim Y. K. Choo H. Ryu H. Min S.-J. J. Enzyme Inhib. Med. Chem. 2021;36:856–868. doi: 10.1080/14756366.2021.1900160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I. Hwang Y. J. Kim T. Viswanath A. N. I. Londhe A. M. Jung S. Y. Sim K. M. Min S.-J. Lee J. E. Seong J. Kim Y. K. No K. T. Ryu H. Pae A. N. J. Comput.-Aided Mol. Des. 2017;31:877–889. doi: 10.1007/s10822-017-0052-3. [DOI] [PubMed] [Google Scholar]
- Trettel F. Rigamonti D. Hilditch-Maguire P. Wheeler V. C. Sharp A. H. Persichetti F. Cattaneo E. MacDonald M. E. Hum. Mol. Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Singer E. Walter C. Weber J. J. Krahl A.-C. Mau-Holzmann U. A. Rischert N. Riess O. Clemensson L. E. Nguyen H. P. Sci. Rep. 2017;7:16880. doi: 10.1038/s41598-017-17275-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y. Mao X. Xiong L. Xia A. You J. Lin G. Wu C. Huang L. Wang Y. Yang S. Angew. Chem., Int. Ed. 2021;60:8760–8765. doi: 10.1002/anie.202017200. [DOI] [PubMed] [Google Scholar]
- Cheng Y. He C. Wang M. Ma X. Mo F. Yang S. Han J. Wei X. Signal Transduction Targeted Ther. 2019;4:1–39. doi: 10.1038/s41392-018-0034-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y. Chan Y.-T. Tan H.-Y. Li S. Wang N. Feng Y. Mol. Cancer. 2020;19:79. doi: 10.1186/s12943-020-01197-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S. De Carvalho D. D. FEBS J. 2022;289:1214–1239. doi: 10.1111/febs.15750. [DOI] [PubMed] [Google Scholar]
- Morel D. Jeffery D. Aspeslagh S. Almouzni G. Postel-Vinay S. Nat. Rev. Clin. Oncol. 2020;17:91–107. doi: 10.1038/s41571-019-0267-4. [DOI] [PubMed] [Google Scholar]