

Abstract
We present a case of primary angiosarcoma, an exceedingly rare malignant breast lesion, in a 21-year-old female patient with unilateral breast enlargement. Primary angiosarcoma is an aggressive form of breast cancer with variable presentation and limited clinical experience due to the rarity of the disease. Despite an initial diagnostic challenge, this report showcases the importance of a systematic diagnostic approach and multidisciplinary management in the context of uncommon breast lesions in young patients. Our patient underwent a mastectomy followed by breast reconstruction, achieving favourable cosmetic outcomes. This case serves as a contribution to our understanding of the diagnostic considerations surrounding angiosarcoma of the breast in a young patient.
Keywords: Angiosarcoma, PASH, mastectomy, breast tumour
Introduction
Angiosarcoma represents an exceptionally rare form of malignant breast lesion which is present in around 0.05% of the patients diagnosed to have breast malignancy. 1
This is a malignant and aggressive tumour arising from the vascular endothelium, with rapid growth and infiltration into the local issues. Angiosarcoma is also associated with poor prognosis. 2
Breast angiosarcoma was first described by Borman in 1907. 3 Breast angiosarcoma is described according to the aetiology as primary (de novo) and secondary. The median age for primary breast angiosarcoma (PAS) is 40 years and 70 years for secondary breast angiosarcoma (SAS). The risk factors for angiosarcomas include radiation history, long-standing lymphoedema, carcinogens and some familial syndromes.4–6
Management of angiosarcoma mainly depends on the stage of the disease, histologic grade and tumour size. At present, there is limited knowledge and no evidence-based guidelines regarding the treatment of angiosarcoma. Wide surgical resection along with other modes of therapy such as chemoradiotherapy appears to be the commonly used treatment option. 7
Case presentation
A 21-year-old female presented with an 8-month history of progressively increasing lump in the right breast accompanied by breast heaviness. She did not complain of any history of trauma or nipple discharge and had no history of breast disease. There was no family history suggestive of breast malignancy.
On clinical examination, a large, ill-defined, non-tender mass involving the entire right breast was noted which was also close to the nipple-areolar complex (Figure 1). The left breast and axillae appeared unremarkable.
Figure 1.
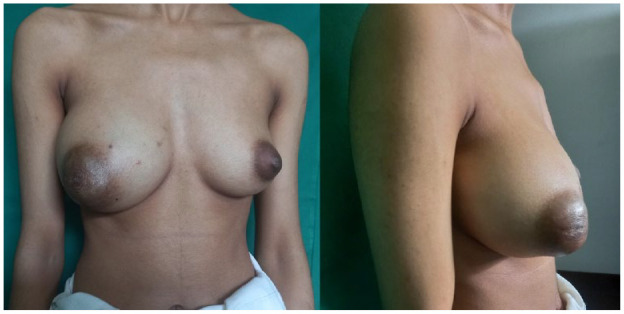
Right breast enlargement with distortion of the nipple-areolar complex.
Ultrasonography (USS) of the breast demonstrated diffuse right breast enlargement with an ill-defined mixed echogenic lesion in the retro-areolar region (22 mm × 12 mm). No enlarged lymph nodes were observed, and the left breast and axilla appeared normal. Magnetic resonance imaging (MRI) scan of the breasts showed an ill-defined mass lesion in the right breast mainly involving central and medial quadrants measuring 6.0(T) × 7.7(AP) × 9.0 cm (CC) with heterogeneous signals in it. The medial aspect of the tumour was solid and showed T1W low signals, T2W fat at iso signals and avid contrast enhancement. These areas showed diffusion restriction and rapid washout in the kinetic curve. Prominent internal mammary vessels were noted adjacent to the medial margin of the lesion. A 2.6 × 3.2 × 2.4 cm multi-loculated cystic area was noted within the lesion medial to the nipple. The heterogeneous enhancing solid component of the lesion is in the retro areolar region with skin infiltration (5 cm diameter). A breast neoplasm such as angiosarcoma was the likely diagnosis on MRI interpretation (Figure 2). To rule out metastasis, a contrast-enhanced computed tomography of the chest and abdomen was performed, yielding negative findings.
Figure 2.
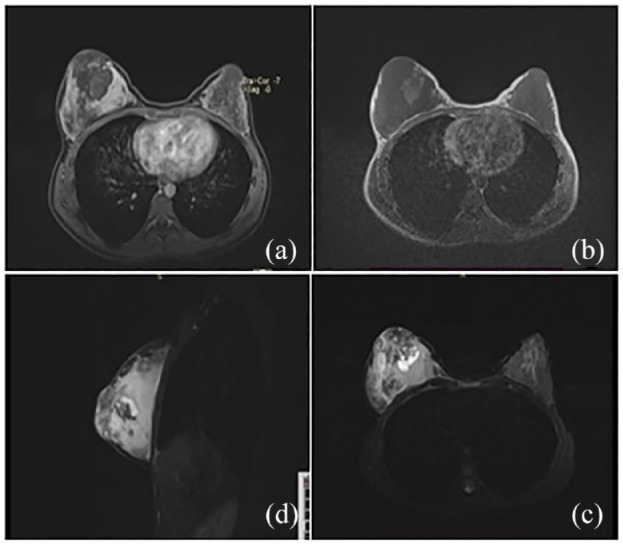
Magnetic resonance imaging images of the tumour. (a) Post-contrast enhancement in the posterior aspect of the tumour with retro areolar non-enhancing area with some serpiginous enhancement in the periphery. (b) T1-weighted view – Heterogenous central low and peripheral high-intensity signal. (c) T2-weighted view – Diffuse enhancement, the high-intensity signal in the peripheral area and heterogeneous signal in the retro areolar region. (d) TIRM sagittal – High signal lesion diffusely involving right breast with central necrotic area in a medial quadrant, infiltrating into the subcutaneous tissue.
Multiple core biopsies were obtained from different sites within the lesion. The histopathological analysis revealed a vascular lesion characterized by complex anastomosing spaces and a dense collagenous keloid-like stroma. These spaces were mostly empty and the lining cells were devoid of nuclear atypia or mitoses negating a malignant diagnosis. These features suggested a pseudoangiomatous stromal hyperplasia (PASH), offering no evidence of a definite malignancy. 8
The patient was extensively discussed at the multidisciplinary meeting. Since there was a discrepancy in the radiological and pathological diagnosis, the decision was made to carry out a mastectomy with or without reconstruction following a discussion with the patient. The diagnosis, the discrepancies, possible outcomes and pros and cons of reconstruction were discussed in detail with the patient. The patient requested an immediate reconstruction. Following this, a decision was made to proceed with skin-sparing mastectomy with reconstruction using a latissimus dorsi muscle flap and implant which yielded satisfactory cosmetic results.
The macroscopy of the right mastectomy specimen revealed a tumour mass occupying the entire specimen and measuring 100 × 90 × 30 mm. Microscopy revealed predominantly bland endothelial lined vascular channels. The erythroblast transformation-specific related gene was positive. Twenty percent of the tumour was solid with pleomorphic cells. Ki67 proliferative index was performed which was 50%. It was diagnosed as a well-differentiated primary angiosarcoma of the breast. All margins were free of tumour/R0 resection (Figure 3).
Figure 3.
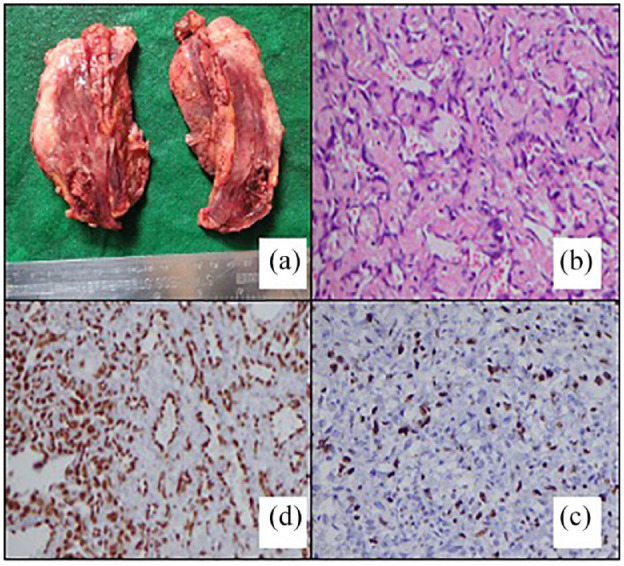
Macroscopic and microscopic images of the tumour. (a) Macroscopically pink solid tumour mass. (b) Vascular channels with bland endothelial cells. (H &E X200). (c) Strong nuclear staining of the endothelium (IHC-ERG X200). (d) Ki 67 proliferative index of 50% (IHC Ki 67X200).
The patient was re-discussed at the multidisciplinary meeting and considered for radical mastectomy and chemo-radiotherapy. The patient declined further treatment and is on close follow-up. The patient was counselled regarding the possibility of recurrence, the need for close follow-up and the necessity for radical surgery if recurs. She is being followed up in the breast clinic 3 monthly with clinical assessment and ultrasonographic imaging. Post op 6 month, the patient is disease free and is continuing the follow-up plan.
Discussion
Angiosarcoma is an aggressive tumour described to be originating from lymphatic or vascular endothelial cells. PAS of the breast is rarer than SAS and has no known risk factors. It is more likely to occur in younger women with dense breasts. SAS usually develops in women who have been previously treated for breast cancer with radiation therapy. It typically develops 6–10 years after radiation treatment. Another risk factor for SAS is chronic lymphedema which can occur after breast cancer treatment. This is also known as Stewart-Trevis syndrome. Other well-known risk factors include environmental carcinogens such as vinyl chloride and arsenic. SAS usually arises in the dermal and subcutaneous layers of the skin and may not necessarily involve the parenchyma.9–12,20
PAS typically occurs in younger females with a median age of 40 years. However, our patient was 21 years of age and is one of the youngest patients reported in the literature.
PAS often presents as a large mass that arises within the breast parenchyma typically without any skin changes. They can also present as rapidly growing tumours. By contrast, SAS usually presents as a painless bruising. Examination findings in our patient are comparable to the reported literature on PAS.13,14 The tumour size of our patient was 10 cm × 9 cm which is quite large compared to the 6 cm median size reported in past studies. 15
Thrombocytopenia and haemorrhagic manifestations (Kasabach–Merritt syndrome) are sometimes noted in large tumours. Although our patient had a significantly large tumour, she did not develop bleeding manifestations. 16
Imaging for angiosarcoma with mammogram or ultrasound imaging do not usually depict any specific or pathognomic findings. MRI is the most helpful imaging modality that can show typical malignant signs (hyperintensity on T2 images and a rapid initial intense phase followed by washout). 17
In our patient, the USS was non-diagnostic and revealed only a vascular lesion. However, the MRI was more indicative of a malignant lesion such as angiosarcoma as the images showed unilateral right breast enlargement with an avidly heterogeneously enhancing breast mass lesion involving central and medial quadrants, infiltrating the areolar skin. The lesion contained multi-loculated vascular components with haemorrhagic and necrotic areas, strongly suggesting a malignant pathology.
As the core biopsy was not representative of solid areas and the bland endothelial cells mimicked a benign lesion the diagnosis of PASH was suggested. The heterogeneity within the tumour was a pitfall in this patient and lesions of vascular proliferation in the breast warrant the exclusion of primary angiosarcoma by performing multiple cores and Ki-67 index. ERG is a sensitive marker of endothelial differentiation and is expressed in vascular tumours, including angiosarcomas as seen in our patient. 18 Other markers such as C myc, AE1 were not performed as other variants were not suspected in the diagnosis.
This discordance in radiology and histology is commonly noted in PAS and is reported in several studies. 19
With hardly any prospective studies and minimal retrospective data, optimal treatment of PAS is purely based on experience and expert judgement.
At present, PAS and SAS management are almost similar, with surgical excision being the most common primary management. Most of the current literature shows that PAS has been commonly managed with mastectomy over breast conservation. However, the optimum surgical management remains unclear, due to the rarity of the disease and with lack of long-term outcome data comparing wide excision versus mastectomy. 20
Most studies show that an incomplete excision is strongly associated with both local relapse and poor survival. Therefore, the principle of surgery should be R0 resection with optimal margins. 21 There is no conclusive data on the axillary staging or surgery. Axillary nodal metastasis is also not common in angiosarcoma as metastases are primarily due to haematogenous spread. Therefore, the need for axillary lymph node staging is not clearly recommended. 22
There is no international consensus or guideline about the role of chemotherapy and radiotherapy in the current literature. The available limited literature shows promising results with improvement in overall survival with the use of chemotherapy. Electrochemotherapy is also described as an effective treatment for angiosarcoma with cutaneous involvement which is mainly seen in SAS. 23
There is also no clear consensus regarding radiotherapy. Some studies have demonstrated a reduction in local recurrence with radiotherapy in large tumours. Therefore, radiotherapy could be considered as adjuvant treatment in selected cases. 24
The overall prognosis of PAS is poor compared to invasive breast cancer. Studies show 5-year overall survival rate to be less than 40%.21,25
Tumour size and grade are considered the important prognostic factors for both PAS in regard to both overall survival and disease-free survival. Studies show that larger tumour size and higher grade are associated with an increased risk of local recurrence and reduced overall survival. 26
Although clear margins were achieved in our patient, we offered the patient radical mastectomy and adjuvant treatment with chemotherapy. However, the patient declined further treatment and is currently being followed up.
Conclusion
PAS is a rare malignancy; therefore, most of the existing literature is in the form of case reports or single institutional experiences with no standard guidelines. Optimal surgical resection and wide margins remain to be the most agreed upon approach.
We report a case of managing a young female with PAS and the diagnostic and management challenges faced during the treatment. We underline the importance of the multidisciplinary approach and shared consensus on the management of PAS which would directly benefit the patient. Further studies are needed to define the best treatment options for this rare tumour especially because it affects the younger female population.
Acknowledgments
None declared.
Footnotes
Author contributions: All authors contributed to the concept, material preparation and drafting of the manuscript. The first draft of the manuscript was written by B.W. and J.S. and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval: Our institution does not require ethical approval for reporting individual cases or case series.
Informed consent: Written informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
ORCID iDs: Jeewantha Senavirathna  https://orcid.org/0000-0003-0471-9218
https://orcid.org/0000-0003-0471-9218
Malith Nandasena
https://orcid.org/0000-0001-5188-9848
References
- 1. Park J, Kang BJ, Kim SH, et al. A case report of breast angiosarcoma in a young woman. Investig Magn Reson Imaging 2021; 25(2): 135. [Google Scholar]
- 2. Liberman L, Dershaw DD, Kaufman RJ, et al. Angiosarcoma of the breast. Radiology 1992; 183(3): 649–654. [DOI] [PubMed] [Google Scholar]
- 3. Borrmann R. Metastasenbildung bei histologisch gutartigen Geschwulsten (Fall von metastasierendem Angiome.). Beitr. z. Pathol. Anat. Allg. Pathol 1906; 40: 372–392. https://acsjournals.onlinelibrary.wiley.com/doi/pdf/10.1002/1097-0142(19800715)46:2%3C368::AID-CNCR2820460226%3E3.0.CO;2-E [Google Scholar]
- 4. Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res 2019; 9(11): 2303. [PMC free article] [PubMed] [Google Scholar]
- 5. Salm R. The nature of the so-called postmastectomy lymphangiosarcoma. J Pathol Bacteriol 1963; 85: 445–456. [DOI] [PubMed] [Google Scholar]
- 6. Conti M, Morciano F, Rossati C, et al. Angiosarcoma of the breast: overview of current data and multimodal imaging findings. J Imaging 2023; 9(5): 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bordoni D, Bolletta E, Falco G, et al. Primary angiosarcoma of the breast. Int J Surg Case Rep 2016; 20(Suppl): 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Steininger R, Bouton M, Marsh J. Masson’s tumor of the breast: rare differential for new or recurrent breast cancer—Case report, pathology, and review of the literature. Breast J 2020; 26(4): 752–754. [DOI] [PubMed] [Google Scholar]
- 9. Young RJ, Brown NJ, Reed MW, et al. Angiosarcoma. Lancet Oncol 2010; 11: 983–991. [DOI] [PubMed] [Google Scholar]
- 10. Boughey JC, Hoskin TL, Cheville AL, et al. Risk factors associated with breast lymphedema. Ann Surg Oncol 2014; 21(4): 1202–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bonito FJP, de Almeida Cerejeira D, Dahlstedt-Ferreira C, et al. Radiation-induced angiosarcoma of the breast: a review. Breast J 2020; 26(3): 458–463. [DOI] [PubMed] [Google Scholar]
- 12. Parisi S, Gambardella C, Iovino F, et al. Post-irradiation breast angiosarcoma: all the possible treatments and electrochemotherapy. Case report and literature review. J Clin Med 2024; 13(2): 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bordoni D, Bolletta E, Falco G, et al. Primary angiosarcoma of the breast. Int J Surg Case Rep 2016; 20(Suppl): 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ginter PS, McIntire PJ, Shin SJ. Vascular tumours of the breast: a comprehensive review with focus on diagnostic challenges encountered in the core biopsy setting. Pathology 2017; 49(2): 197–214. [DOI] [PubMed] [Google Scholar]
- 15. Abdou Y, Elkhanany A, Attwood K, et al. Primary and secondary breast angiosarcoma: single center report and a meta-analysis. Breast Cancer Res Treat 2019; 178: 523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bernathova M, Jaschke W, Pechlahner C, et al. Primary angiosarcoma of the breast associated Kasabach-Merritt syndrome during pregnancy. Breast 2006; 15(2): 255–258. [DOI] [PubMed] [Google Scholar]
- 17. Bousquet G, Confavreux C, Magné N, et al. Outcome and prognostic factors in breast sarcoma: a multicenter study from the rare cancer network. Radiother Oncol 2007; 85(3): 355–361. [DOI] [PubMed] [Google Scholar]
- 18. Sullivan HC, Edgar MA, Cohen C, et al. The utility of ERG, CD31 and CD34 in the cytological diagnosis of angiosarcoma: an analysis of 25 cases. J Clin Pathol 2015; 68(1): 44–50. [DOI] [PubMed] [Google Scholar]
- 19. Varghese B, Deshpande P, Dixit S, Koppiker CB, Jalnapurkar N. Primary angiosarcoma of the breast: a case report. J Radiol Case Rep 2019; 13(2): 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arora TK, Terracina KP, Soong J, et al. Primary and secondary angiosarcoma of the breast. Gland Surg 2014; 3(1): 28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yin M, Wang W, Drabick JJ, et al. Prognosis and treatment of non-metastatic primary and secondary breast angiosarcoma: a comparative study. BMC Cancer 2017; 17(1): 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Adem C, Reynolds C, Ingle JN, et al. Primary breast sarcoma: clinicopathologic series from the Mayo Clinic and review of the literature. Br J Cancer 2004; 91(2): 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Benevento R, Carafa F, Di Nardo D, et al. An-giosarcoma of the breast: a new therapeutic approach? Int. J. Surg. Case Rep 2015; 13: 30–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ghareeb ER, Bhargava R, Vargo JA, et al. Primary and radiation-induced breast angiosarcoma: clinicopathologic predictors. Am J Clin Oncol 2016; 39(5): 463–467. [DOI] [PubMed] [Google Scholar]
- 25. Wang XY, Jakowski J, Tawfik OW, et al. Angiosarcoma of the breast: a clinicopathologic analysis of cases from the last 10 years. Ann Diagn Pathol 2009; 13: 147–150. [DOI] [PubMed] [Google Scholar]
- 26. Kim YJ, Ryu JM, Lee SK, et al. Primary angiosarcoma of the breast: a single-center retrospective study in Korea. Curr Oncol 2022; 29(5): 3272–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]