

Abstract
The fragile WWOX gene, encompassing the chromosomal fragile site FRA16D, is frequently altered in human cancers. While vulnerable to DNA damage itself, recent evidence has shown that the WWOX protein is essential for proper DNA damage response (DDR). Furthermore, the gene product, WWOX, has been associated with multiple protein networks, highlighting its critical functions in normal cell homeostasis. Targeted deletion of Wwox in murine models suggests its in vivo requirement for proper growth, metabolism, and survival. Recent molecular and biochemical analyses of WWOX functions highlighted its role in modulating aerobic glycolysis and genomic stability. Cumulatively, we propose that the gene product of FRA16D, WWOX, is a functionally essential protein that is required for cell homeostasis and that its deletion has important consequences that contribute to the neoplastic process. This review discusses the essential role of WWOX in tumor suppression and genomic stability and how its alteration contributes to cancer transformation.
Keywords: Common fragile site, FRA16D, Genomic instability, Tumor suppressor, WWOX, ATM, HIF1
Introduction
Fourteen years ago, three research groups reported the cloning and mapping of WWOX (WW domain-containing oxidoreductase). The Aldaz group [1] followed by the Richards group [2] demonstrated that WWOX (also known as FOR) spans a chromosome region at 16q involved in cancer. This observation was then followed by cloning the murine WOX1 sequence by the Chang group [3]. The protein product, WWOX, contains two WW domains at its N-terminus and a central domain homologous to the short-chain dehydrogenase/reductase (SDR) family. Through its WW1 domain, WWOX binds with proline-tyrosine (PY) motifs-containing proteins and acts as an adapter protein regulating transactivation and localization (reviewed in [4]). Early evidence demonstrated that overexpression of WWOX in WWOX-negative cancer cells promotes apoptosis and suppresses tumorigenicity (reviewed in [5, 6]). Characterization of Wwox mouse strains revealed its essential role for proper growth, survival, steroidogenesis, and metabolism. Wwox-deficient mice die within 3–4 weeks with metabolic defects [7–10], precluding adult tumor analysis. Nevertheless, analysis of heterozygous mice revealed increased incidence of tumor development [7, 11, 12]. New conditional mouse models have recently been established that can be used to study specific in vivo roles of WWOX in development and tumorigenesis [13, 14].
The WWOX gene spans the common fragile site FRA16D, a genomic region that is involved in chromosome translocation in multiple myeloma and in homo and hemizygous deletions in cancer and cancer-derived cell lines [2]. Common fragile sites (CFS) have been defined cytogenetically as gaps or breaks on metaphase chromosomes in cells treated with DNA replication inhibitors, such as aphidicolin [15, 16]. CFS are preferential targets of replication stress in preneoplastic lesions [17] and emerging evidence suggests that they represent early warning sensors for DNA damage [18–20]. Both genetic and epigenetic factors are thought to regulate the fragility of CFS [21, 22] (see also B. Kerem and M. Debatisse chapters in this issue). Recent profiling studies of CFS provide evidence that the fragility of CFS is tissue specific [23–25]. Altogether, these observations suggest complex regulation of CFS.
Chromosomal instability, including structural and numerical changes, is the most common type of genomic instability in non-inherited human cancers [26]. Defects in DNA replication, impaired checkpoint responses, and oxidative stress contribute to chromosomal instability during all stages of neoplastic progression. Importantly, CFS correlate with chromosomal breakpoints in tumors and are considered preferential hot spots for chromosomal instability [27, 28]. While the FRA16D is highly susceptible to DNA damage, its product, the WWOX protein, behaves as a tumor suppressor. The mechanism of tumor suppression of WWOX involves apoptosis [29, 30], modulation of the extracellular matrix [31], and modulation of cell bioenergetics [32, 33]. Strikingly, recent evidence also revealed that the WWOX gene product functions as an upstream component of the DDR and is essential for proper activation of the DNA damage checkpoint-signaling pathway [34]. The findings that gene products of CFSs, such as WWOX (this review) and FHIT (see K. Huebner chapter in this issue), have driving roles in carcinogenesis argue against CFS being inert structures that are passenger events in cancer development. This review will discuss the emerging tumor suppressor functions of the WWOX protein and its implication for neoplastic progression.
Alteration of WWOX in cancer
Aberrant expression of WWOX in cancer is a common event (reviewed in [35]). Various reports have associated WWOX loss or low expression with numerous types of cancer, including breast [36, 37], prostate [38], gastric [39], lung [40, 41], and pancreatic [42, 43] carcinomas. WWOX aberrant expression was also reported in osteosarcomas [44, 45] as well as in hematopoietic malignancies [46, 47]. WWOX’s alteration in these cancers is mainly due to genomic modifications as a result of chromosomal deletions and translocations [35]. Additional mechanisms include hypermethylation of the regulatory element [48] and protein degradation [49]. Initial studies to investigate WWOX expression in cancer focused on RT-PCR analysis and revealed the presence of many forms of WWOX truncated variants in clinical samples [50, 51]. These forms exhibited deleted exons 5, 6, 7, and 8, the later corresponding to the core of FRA16D. These data were initially interpreted as random genomic deletions, however emerging evidence suggests that other mechanisms could be involved. For example, in one study, it was demonstrated that the splicing factor hnRNP A2/B1 regulates tumor suppressor genes splicing, including WWOX, in glioblastoma [52]. Although the presence of these aberrant transcripts or isoforms was evident in multiple cancer types, truncated protein expression in these cancers was rarely detected.
The protein expression of WWOX was also studied in a broad spectrum of cancers using immunohistochemistry. In most cases, the absence or reduction of cytoplasmic WWOX was associated with advanced stages of cancers [35]. However, some reports also documented increased levels of WWOX, suggesting a complex regulation of WWOX in cancer [53, 54]. Whether this increase in WWOX level is of any advantage to the neoplastic process is unlikely, but it remains to be determined. This latter phenomena could also be an artifact resulting from the expression of truncated forms or cancer-specific isoforms that are detected by immunohistochemistry but not detected by immunoblotting. Alternatively, WWOX expression, if retained, could be induced in certain contexts or as a result of specific stimuli during the neoplastic process.
A recent comprehensive analysis of somatic copy number alterations in a large sample of cancer specimens [27] and cancer cell lines [28] revealed that the WWOX locus is among the most statistically significant common sites of the whole genome affected by homozygous and hemizygous deletions. More recently, novel somatic mutations in the WWOX sequence, which likely abrogates its protein function, were identified in various tumor types. This was revealed by next generation sequencing and TCGA data analysis (reviewed in [55]). These data further confirm that deregulation of the WWOX gene has an advantage for the cancer cell.
Mouse models of WWOX
The fact that WWOX loss is a common event in human cancer led to the development of mouse models that mimic this loss in order to characterize the in vivo requirement of WWOX for development and tumorigenesis. In 2007, the development of the first Wwox-knockout mouse model was reported [7]. Phenotypic analysis of these mice revealed that WWOX ablation results in growth retardation, metabolic defects, and postnatal lethality within 3–4 weeks of age. Despite this postnatal lethality, juvenile Wwox-knockout mice display focal lesions along the diaphysis of their femurs resembling early osteosarcomas (reviewed in [56]). Since hemizygous deletion of the human WWOX gene is common in human tumors, Wwox-heterozygous (Wwox +/−) mice were also monitored for spontaneous tumor development. It was reported that the incidence of tumor formation in Wwox +/− mice is significantly higher than in wild-type (Wwox +/+) mice [7]. The spontaneous tumors in Wwox +/− mice (B6-129 genetic background) were mainly lung papillary carcinomas [7]. In a subsequent study, it was shown that half of female Wwox +/− on the C3H genetic background develops spontaneous mammary tumors [11]. In many cases, no staining of WWOX was observed in these tumors, suggesting loss of heterozygosity [11]. However, in some other cases WWOX expression was retained [7, 12], implying haploinsufficiency as in the case of other well-known tumors suppressors, e.g., PTEN and p53 [57].
To define the role of WWOX in tumor progression, Wwox +/− and Wwox +/+ mice were treated with chemical carcinogens and the incidence of tumor formation was evaluated. One study used the chemical mutagen ethyl-nitrous urea (ENU) [7] and another study used the established esophageal/forestomach carcinogen N-nitrosomethyl-benzylamine (NMBA) [12]. In both studies, increased tumor incidence and multiplicity in Wwox +/− mice was observed relative to Wwox +/+ mice. These findings provided the first in vivo evidence for the tumor suppressor function of WWOX.
Additional support for the tumor suppressor function of WWOX comes from the work of the laboratory of M. Aldaz. Ludes-Meyers et al. [10] generated a hypomorphic mouse strain that had no detectable WWOX protein in most of the tissues examined. Wwox hypomorphic mice are viable, though they have a significantly shorter lifespan when compared to control wild-type mice. It is important to note that female hypomorphic mice have a higher incidence of spontaneous B cell lymphomas, which is consistent with WWOX functioning as a tumor suppressor.
The early postnatal lethality of conventional Wwox-knockout precluded phenotypic analysis of WWOX ablation in adult tissues. Therefore, conditional knockout (CKO) mouse models allowing tissue-specific ablation of the Wwox alleles were developed. Assessment of these models using a general deleter transgenic mouse strain (EIIA-cre) revealed that WWOX ablation in these CKO mice resembles the phenotypes observed in conventional Wwox-knockout mice [13, 14]. Subsequent studies aimed to specifically delete WWOX in mammary gland epithelia (Wwox MGE−/−). WWOX ablation in these mice was associated with transient defects in mammary ductal growth [58, 59]. Nevertheless, no mammary tumor phenotype was observed in Wwox MGE−/− mice. This phenotype could stem from the fact that the function of WWOX is non-cell autonomous since deletion of Wwox alleles was done using MMTV-cre transgenic mouse, a transgenic line that is mosaically expressed in the luminal cell compartment of the mammary gland. Alternatively, WWOX function could be redundant or compensated by other genes. Another possibility is that WWOX, similar to p53, plays a role in mammary tumor progression. Specific ablation of p53 alleles in mammary gland epithelium, for example, does not lead to mammary tumor formation [60]. However, concurrent deletion of different tumor suppressors, such as BRCA2 or BRCA1, with p53 results in accelerated mammary tumor development [60, 61]. In the same venue, specific deletion of Rb, a well-known tumor suppressor, in mice has also no tumor phenotype in almost all tissues, except in pituitary gland [62]. Whether targeted deletion of Wwox cooperates with loss of other tumor suppressors to accelerate or promote tumorigenesis is yet to be determined.
Emerging functions of the WWOX protein
The localization of the WWOX gene at one of the most active human CFSs has had a major influence on the frequency of its loss or reduction in cancers [2]. Nevertheless, loss of WWOX has also been associated with hypermethylation of its promoter [48] as well as protein degradation [49]. Therefore, it seems highly unlikely that this frequent alteration of WWOX expression does not contribute to a selective advantage for clonal expansion. The WWOX interactome and its ability to associate with multiple protein networks is among the strongest indications that it plays an important role in the neoplastic process. This argues against alteration of WWOX being a passenger event.
The ability of WWOX to interact with a growing list of interesting proteins is mediated mainly through its first WW (WW1) domain [5, 6]. WW domains are among the smallest modular domains that are well known to mediate protein–protein interaction. They are composed of ~35 amino acids that include two signature tryptophan (W) residues (reviewed in [63, 64]). Based on ligand recognition, WW domains of WWOX were reported to interact with PPXY-containing motifs. Several interesting proteins were identified as WWOX partners and are summarized in Table 1. In general, WWOX acts an adaptor protein that regulates localization and transactivation of its partners (reviewed in [4]). For example, WWOX, via its WW1 domain, interacts with the PY-motifs of the C-terminal fragment (CTF) of ErbB4, sequesters it in the cytoplasm or cell membrane preventing it from entering the nucleus, thereby suppressing its transcriptional function [65]. Interestingly, it was found that expression of WWOX and membranous ErbB4 is associated with favorable survival of breast cancer patients, highlighting the clinical significance of WWOX–ErbB4 interaction [66].
Table 1.
WWOX partners and functional outcome
| WWOX interacting partner | Readout | References |
|---|---|---|
| WW-dependent | ||
| TP73 | Promotes p73 transactivation-independent apoptosis | [30] |
| SIMPLE | Unknown | [100] |
| AP2α and γ | Suppresses AP2 transactivation function | [30, 101] |
| ErbB4 intracellular domain (ICD) | Suppresses ErbB4 transactivation function and regulates its localization | [65] |
| c-Jun | Suppresses AP-1 transactivation function | [102] |
| Ezrin | Ezrin mediates the apical membrane localization of WWOX | [103] |
| ACK1 | ACK1 promotes WWOX ubiquitination | [49] |
| RUNX2 | Suppresses Runx2-transactivation | [8] |
| DVL-2 | Inhibits Wnt/β-catenin signaling pathway | [67, 104] |
| ΔNp63α | Suppresses ΔNp63α transactivation function and enhances chemosensitivity | [105] |
| WBP1, 2 | Unknown | [67, 68, 100] |
| HIF1α | Destabilizes HIF1α levels and suppresses its transcriptional activity | [32] |
| ATM | Activates ATM function and enhances proper DDR | [34] |
| Non-WW dependent | ||
| JNK1 | Inhibits JNK1-mediated anti-apoptosis | [74] |
| TP53 | Promotes apoptosis | [106] |
| TAU | Regulates neurodegenerative disorder such as Alzheimer’s disease | [107, 108] |
| MEK1 | Regulates apoptosis (T cell Leukemia) | [107] |
| GSK3β | Promotes neurite outgrowth (neuronal differentiation) | [109] |
| MS study | http://wwox-ms.ekmd.huji.ac.il | [67] |
In a more recent study, mass spectrometry (MS) and phage display experiments were employed to identify putative WWOX-interacting partners [67]. The analysis revealed that WW1 domain of WWOX is indeed the main functional interacting domain. The study revealed several known PY-containing partners of WWOX. This included PPXY containing proteins such as p73 [30], WBP2 [68], and DVL2 [69]. The MS analysis and phage display study also indicated that WW1 domain of WWOX binds LPXY-containing proteins. One such example was the E3 ubiquitin ligase ITCH, which contains two LPXY motifs. Subsequent analysis demonstrated physical and functional interaction between WWOX and ITCH. In fact, it was found that ITCH mediates Lys-63-linked polyubiquitination of WWOX, leading to its nuclear localization and increased cell death [67].
Many of the identified WWOX partners are components of multiprotein complexes involved in molecular processes, including apoptosis, transcription, RNA processing, tight junction, and metabolism [67]. These findings suggest that WWOX acts as an adapter protein and links several individual proteins associated with physiologically important networks. This also sheds light on new emerging roles of the FRAD16 gene product in tumor suppression. Few examples are discussed below.
WWOX modulates function of p53 family proteins
The first WWOX partner to be identified was the p53 homolog, p73 [30]. More recently, our MS analysis also confirmed that WWOX, via its WW1 domain, associates with p73 [67]. TP73 is involved in cell cycle regulation and induction of apoptosis [70, 71]. Like p53, p73 is characterized by the presence of different isoforms of the protein. This is explained by splice variants, and an alternative promoter in the DNA sequence [70, 71]. WWOX binds both p73α and β, but not p73γ, which lacks a PY motif [30]. Upon interacting with WWOX, p73 is sequestered in the cytoplasm. However, an increased rate of apoptosis was observed, suggesting that WWOX might regulate p73 transactivation-independent apoptosis. In a more recent study it was reported that WWOX specifically binds ΔNp63α, but not TAp63 [72]. This protein–protein interaction stabilizes ΔNp63α, through antagonizing the function of the E3 ubiquitin ligase ITCH, inhibits nuclear translocation of ΔNp63α into the nucleus, and suppresses ΔNp63α transactivation function. Additionally, it was found that this functional crosstalk reverses cancer cells’ resistance to cisplatin, mediated by ΔNp63α, and consequently renders these cells more sensitive to undergo apoptosis [72]. Under the same conditions, where WWOX interacts with p73 and ΔNp63α, a direct binding with p53 was not observed. This could be due to a lack of PY motifs in the p53 sequence or that the interaction is indirect or is cell type-specific. Perhaps WWOX is similar to YAP which interacts with p53BP2 [73] that contains a PY motif and hence bridge WWOX association with p53. Nevertheless, the Chang group has shown that the murine WOX1 binds p53 and synergistically enhances p53-mediated apoptosis [3, 74]. Cumulatively, these observations indicate that WWOX partners with the members of the p53 family and regulate cell death.
WWOX modulates TGFβ/SMAD3 signaling in breast cancer
Ferguson et al. [75] have recently shown that WWOX knockdown in normal breast cells results in upregulation of TGFβ/SMAD3 target genes. Using co-immunoprecipitations and GST-pulldowns, it was demonstrated that WWOX directly interacts with SMAD3 and acts as an inhibitor of SMAD3 transcriptional activity by sequestering it in the cytoplasm. This interaction is mediated by a WW1 domain-dependent binding of WWOX with the PY motif of SMAD3. Since TGFβ signaling is aberrant in advanced breast cancers, it is likely that WWOX loss, which is a common event in these cancers, contributes to this deregulation [55]. TGFβ promote tumor invasion and epithelial-to-mesenchymal transition (EMT) [76, 77]. It is therefore of interest to determine whether WWOX modulates EMT of breast cancer cells mediated by TGFβ/SMAD3 signaling and regulates metastasis. Evidence of WWOX anti-metastatic function is emerging; WWOX overexpression has been reported to inhibit migration and invasion of cancer cells [44]. Delineation of WWOX anti-metastatic function remains to be determined.
WWOX modulates HIF1α signaling and affects cell bioenergetics
Cumulative evidence supports a role of WWOX in cellular metabolism. Wwox-deficient mice develop normally, but succumb to lethal hypoglycemia early in life [7, 13, 14], suggesting that WWOX might affect glucose homeostasis. Recent observations in fruit flies have suggested a link between WWOX and mitochondrial metabolic enzymes such as isocitrate dehydrogenase and malate dehydrogenase [33, 78]. In addition, altered levels of dWwox resulted in altered levels of endogenous reactive oxygen species (ROS) [33, 78]. Altogether, these observations led to hypothesize that WWOX might play a key role in cellular metabolism.
In light of these studies, WWOX has been recently identified as a tumor suppressor with emerging roles in regulation of aerobic glycolysis [32]. WWOX controls glycolytic genes’ expression through the regulation of hypoxia-inducible transcription factor 1α (HIF1α) [79]. Specifically, WWOX, via its WW1 domain, physically interacts with HIF1α and functionally modulates its levels and transactivation function. Consistent with this notion, Wwox-deficient cells exhibited increased HIF1α levels and activity and displayed increased levels of HIF1α-target genes and glucose uptake. Remarkably, WWOX deficiency is associated with enhanced glycolysis and diminished mitochondrial respiration, conditions resembling the “Warburg effect” [80]. Genetic and pharmacological inhibition of HIF1α rescued tumorigenic phenotypes of Wwox-deficient cells both in vitro and in vivo [32].
The main physiological mechanism of HIF1α stabilization is in response to low levels of oxygen, by which HIF1α escape the VHL ubiquitination/degradation complex. Oncogenic activation, associated with activation of the RAS-RAF-MAPK, PI3K, PTEN, or AKT pathways can also cause accumulation of HIF1α through unknown mediators. WWOX seems to play a novel role (Fig. 1) by which it destabilizes HIF1α either by direct interaction [32] and/or by affecting ROS cellular levels (unpublished data).
Fig. 1.
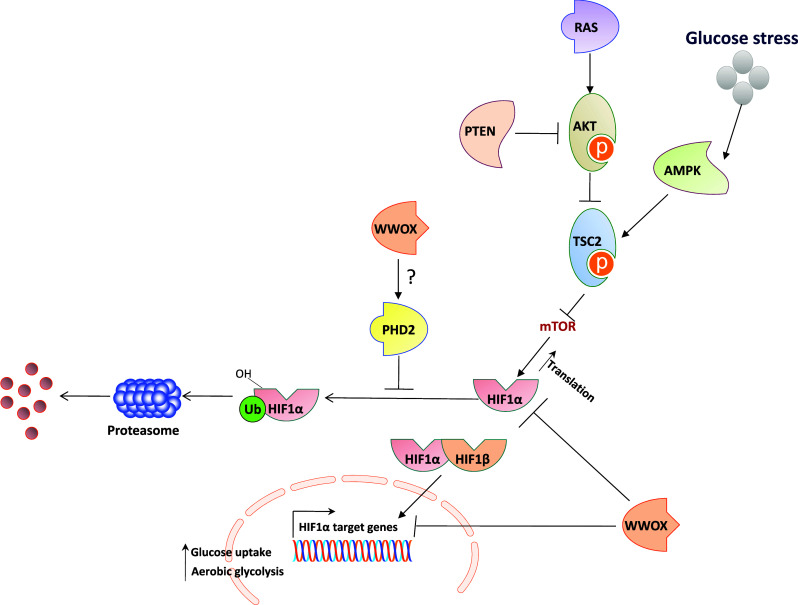
WWOX modulates HIF1α levels and activity. Under hypoxic conditions, HIF1α is stabilized and binds HIF1β to transactivate many target genes, resulting in an increased rate of glycolysis and glucose uptake and inhibiting Krebs cycle. Oncogenic activation of the RAS or AKT pathways can also cause HIF1α accumulation. On one hand, activation of AKT inhibits TSC2 (tuberous sclerosis complex 2), which suppresses the activation of mTOR (mammalian target of rapamycin), resulting in HIF1α protein translation. On the other hand, inactivation of tumor suppressor phosphatase and tensin homolog (PTEN) and Von Hippel-Lindau (VHL) leads to HIF1α accumulation. Recent studies have demonstrated that loss of tumor suppressor WWOX also enhances HIF1α accumulation and its transcriptional function
An emerging hallmark of cancer cells is their adaptation of energy metabolism in order to fuel cell growth and division [81]. These adaptations are directly regulated by many oncogenes and tumor suppressors. They are required to support the energetic and anabolic demands associated with cell growth and proliferation [82]. Intriguingly, it was found that WWOX expression is inversely correlated with levels of HIF1α-target gene, GLUT1 (glucose transporter 1), in breast cancer samples. These findings highlight WWOX as a modulator of breast cancer metabolism [32]. Increased HIF1α levels have been reported in various cancer types [83]. Whether this alteration is also associated with loss of WWOX has yet to be determined. The discovery that WWOX loss activates aerobic glycolysis indicates WWOX’s pleiotropic functions to suppress tumor growth, and opens new venues of cancer metabolic research.
WWOX, DNA damage response, and genomic stability
Genomic instability is a hallmark of almost all cancers and is thought to play an important role in both cancer development and response to therapy [81, 84]. The DNA damage response (DDR) maintains the integrity of the genome in response to DNA damage. DDR is a complex signaling process that either results in cell cycle arrest followed by DNA repair or apoptosis, if the DNA damage is too extensive to be repaired [67, 85, 86]. Key mammalian damage response sensors are ataxia telangiectasia mutated (ATM), ATM and Rad3-related (ATR), and DNA-dependent protein kinases (DNA-PK) [87, 88]. Disruption of the DDR machinery in human cells leads to genomic instability and an increased risk of cancer progression [81, 84].
The fact that WWOX localizes in a CFS has generated a lot of debate on its function as a tumor suppressor. The mechanisms of fragility of CFS, in particular FRA16D, have remained elusive for many years. It is only in recent years that such mechanisms are being molecularly dissected and discovered (see B. Kerem and M. Debatisse chapters in this issue). In the past, this fragility has been attributed to genetic elements, mainly due to formation of secondary structures that halt progression of the replication fork, leading to replication fork collapse and formation of DNA breaks [22]. Recent work, however, has also shed light on the epigenetic mechanisms that contribute to the fragility of FRA16D [21]. Letessier et al. have recently shown that the fragility of FRA16D and FRA3B result from a paucity of replication initiation events ([89] also see chapter by M. Debatisse). These events were shown to be cell-type specific and likely to be CFS-specific as other mechanisms could contribute to their fragility. Together, these findings suggest that a given CFS could be induced in a given type of cancer but not in another. Whether the product of CFS will prove to be functionally relevant in a specific manner, i.e., in one cell type but not in another, is yet to be determined.
Recently, two studies have catalogued the presence of large deletions in a large number of human cancer samples [27] and cell lines [28]. It was concluded that most of these deletions target CFS and large genes [17, 90]. These deletions have been linked to the presence of DNA replication stress. It was in fact suggested that oncogene-induced DNA replication stress preferentially targets CFS due to their sensitivity to DNA damage [18, 19]. CFS were therefore considered as warning sensors since they are the first to be affected upon DNA damage alerting the DDR machinery [20]. Furthermore, emerging evidence has linked products of CFS with genome stability. For example, deficiency of the FHIT gene, encompassing FRA3B, as early as in preneoplastic lesions, induced global genome instability and clonal expansion [91, 92].
Conflicting results were reported on WWOX expression upon exposure to DNA damage. In some reports, WWOX expression was downregulated 24–48 h after UV exposure [46, 93] but was not affected upon ionizing radiation (IR) [93]. In contrast, Lai and colleagues demonstrated that WWOX levels are upregulated upon UV and that it is essential for UVB-induced apoptosis [94]. Consistent with the later, our recent results demonstrate that WWOX expression is increased immediately after IR [34] and UV (unpublished data) exposure. Although WWOX mRNA is upregulated after DSBs, posttranslational modification of the WWOX protein seems to be the predominant cause of WWOX accumulation [34].
If WWOX is induced upon DNA damage, it is plausible to assume that loss of its expression may affect DDR and perhaps genome stability. Indeed, targeted ablation of WWOX, in normal primary cells and cancer cells, was shown to result in delayed activation of DNA damage checkpoint kinase ATM and impaired DNA repair. Furthermore, WWOX knockdown is associated with increased DSBs upon treatment with the radio-mimetic neocarzinostatin (NCS), suggesting that loss of the WWOX fragile gene product renders the genome less stable [34]. Molecular analysis has revealed that WWOX facilitates this function through its functional crosstalk with ATM. Following DNA damage, ATM positively regulates the ligase activity of ITCH [95], which facilitates WWOX ubiquitination at Lys274 [67] and thereby promotes translocation of WWOX into the nucleus [34]. Nuclear WWOX physically interacts with ATM and facilitates ATM monomerization and activation in a positive feed-forward loop manner (Fig. 2). Similar to pharmacological inhibition of ATM, depletion of WWOX or ITCH lead to impaired DDR [34]. These findings argue for a direct function of the gene product of FRA16D, WWOX, in modulating the DNA damage signaling and in maintaining genomic stability.
Fig. 2.

Hypothetical model of WWOX action in DDR. Upon DSBs, ATM is activated and become Ser1981-phosphorylated (pATM). Accordingly, activated ATM monomers phosphorylate numerous substrates including H2A.X, CHK2, p53, and ITCH, which culminates in efficient DDR; i.e., DNA repair or apoptosis. WWOX deficiency leads to an increased number of DSBs upon DNA damage. Following DNA damage, ATM positively enhances ITCH-mediated K63-linked ubiquitination and translocation of WWOX into the nucleus. Nuclear WWOX physically interacts with ATM and mediates ATM monomerization and activation in a positive forward loop manner. When WWOX is lost, ATM function is hampered leading to inefficient DDR
Concluding remarks
Due to the extreme fragility seen at FRA16D and other CFS, it has been debated that alterations at CFS are passenger events in cancer development. It can be argued that this phenomenon is more complicated and that gene products encompassing these sites might have important roles in stress response and in the neoplastic process. Fragility of CFS seems to be highly specific and tissue dependent, and thus alteration of genes within these sites might have selective advantage for tumor growth. In this review, the focus was on WWOX, the product of FRA16D, and its potential driving roles in homeostasis and tumorigenesis was discussed.
Several lines of evidence support WWOX tumor suppressor function. First, WWOX is commonly deleted in numerous types of cancer [35]. Both homozygous and hemizygous deletions were reported but its alteration due to epigenetic and posttranslational mechanisms was also documented. It is therefore possible that one allele of WWOX is lost due to fragility and the other is targeted by other mechanisms fulfilling the Knudson two-hits hypothesis. Second, WWOX replacement in numerous WWOX-negative cancer-derived cell lines caused reduced growth in vitro and tumorigenicity in vivo (reviewed in [6, 29]). Third, analysis of Wwox mutant mice demonstrated that WWOX functions as a bone fide tumor suppressor [7, 11, 12]. Targeted allele deletion of Wwox in a tissue-specific manner is under intensive investigation and should reveal WWOX roles in tumor initiation and progression. Fourth, a number of studies support the hypothesis that loss of WWOX provides a selective advantage in neoplastic transformation. As an example, Ras-mediated transformation of Wwox-deficient cells display increased tumorigenicity when compared to Wwox-sufficient cells [96]. Finally, the WWOX interactome supports a direct role of WWOX as an oncosuppressor. The mechanism of tumor suppression of WWOX involves apoptosis [29, 30], modulation of the extracellular matrix [31], modulation of cell bioenergetics [32, 33], and modulation of the DNA damage response [34]. WWOX appears to interact and regulate the function of different proteins involved in tumor progression. When WWOX is lost, many of these proteins lose their checks, which alters the signaling that feeds into the neoplastic process. Further characterization of WWOX-interacting partners is needed to improve our understanding of the WWOX tumor-suppressor functions and signaling pathways involved. This characterization may also lead to identification of new targets for intervention of tumor development and progression.
The findings regarding WWOX also have broader implications on other CFS products and their role in cancer development. Indeed, gene products of FRA3B (FHIT) (see also K. Huebner’s chapter in this issue), FRA8I (SPIDR) [97, 98] (see also L. Savelyeva chapter in this issue) and FRA15A (RORA) [99] (see also D. Smith chapter in this issue), which are inactivated in multiple tumors, have also been shown to be involved in cellular stress response, DDR, and maintenance of chromosomal integrity. This suggests that their impaired activity may contribute to genomic instability in cancer cells. These observations suggest that some of the products of CFS might play an important role in maintaining genomic stability. Thus, it can be speculated that they function as part of a highly conserved stress response network that is uniquely susceptible to genomic instability in cancer cells. Future work shall further explore and dissect the functions of CFS gene products and their roles in biology and in tumorigenesis.
References
- 1.Bednarek AK, Laflin KJ, Daniel RL, Liao Q, Hawkins KA, Aldaz CM. WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3-24.1, a region frequently affected in breast cancer. Cancer Res. 2000;60(8):2140–2145. [PubMed] [Google Scholar]
- 2.Ried K, Finnis M, Hobson L, Mangelsdorf M, Dayan S, Nancarrow JK, Woollatt E, Kremmidiotis G, Gardner A, Venter D, Baker E, Richards RI. Common chromosomal fragile site FRA16D sequence: identification of the FOR gene spanning FRA16D and homozygous deletions and translocation breakpoints in cancer cells. Hum Mol Genet. 2000;9(11):1651–1663. doi: 10.1093/hmg/9.11.1651. [DOI] [PubMed] [Google Scholar]
- 3.Chang NS, Pratt N, Heath J, Schultz L, Sleve D, Carey GB, Zevotek N. Hyaluronidase induction of a WW domain-containing oxidoreductase that enhances tumor necrosis factor cytotoxicity. J Biol Chem. 2001;276(5):3361–3370. doi: 10.1074/jbc.M007140200. [DOI] [PubMed] [Google Scholar]
- 4.Salah Z, Aqeilan R, Huebner K. WWOX gene and gene product: tumor suppression through specific protein interactions. Future Oncol. 2010;6(2):249–259. doi: 10.2217/fon.09.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aqeilan RI, Croce CM. WWOX in biological control and tumorigenesis. J Cell Physiol. 2007;212(2):307–310. doi: 10.1002/jcp.21099. [DOI] [PubMed] [Google Scholar]
- 6.Del Mare S, Salah Z, Aqeilan RI. WWOX: its genomics, partners, and functions. J Cell Biochem. 2009;108(4):737–745. doi: 10.1002/jcb.22298. [DOI] [PubMed] [Google Scholar]
- 7.Aqeilan RI, Trapasso F, Hussain S, Costinean S, Marshall D, Pekarsky Y, Hagan JP, Zanesi N, Kaou M, Stein GS, Lian JB, Croce CM. Targeted deletion of Wwox reveals a tumor suppressor function. Proc Natl Acad Sci USA. 2007;104(10):3949–3954. doi: 10.1073/pnas.0609783104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aqeilan RI, Hassan MQ, de Bruin A, Hagan JP, Volinia S, Palumbo T, Hussain S, Lee SH, Gaur T, Stein GS, Lian JB, Croce CM. The WWOX tumor suppressor is essential for post-natal survival and normal bone metabolism. J Biol Chem. 2008;283(31):21629–21639. doi: 10.1074/jbc.M800855200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aqeilan RI, Hagan JP, de Bruin A, Rawahneh M, Salah Z, Gaudio E, Siddiqui H, Volinia S, Alder H, Lian JB, Stein GS, Croce CM. Targeted ablation of the WW domain-containing oxidoreductase tumor suppressor leads to impaired steroidogenesis. Endocrinology. 2009;150(3):1530–1535. doi: 10.1210/en.2008-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ludes-Meyers JH, Kil H, Nunez MI, Conti CJ, Parker-Thornburg J, Bedford MT, Aldaz CM. WWOX hypomorphic mice display a higher incidence of B-cell lymphomas and develop testicular atrophy. Genes Chromosomes Cancer. 2007;46(12):1129–1136. doi: 10.1002/gcc.20497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdeen SK, Salah Z, Maly B, Smith Y, Tufail R, Abu-Odeh M, Zanesi N, Croce CM, Nawaz Z, Aqeilan RI. Wwox inactivation enhances mammary tumorigenesis. Oncogene. 2011;30(36):3900–3906. doi: 10.1038/onc.2011.115. [DOI] [PubMed] [Google Scholar]
- 12.Aqeilan RI, Hagan JP, Aqeilan HA, Pichiorri F, Fong LY, Croce CM. Inactivation of the Wwox gene accelerates forestomach tumor progression in vivo. Cancer Res. 2007;67(12):5606–5610. doi: 10.1158/0008-5472.CAN-07-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ludes-Meyers JH, Kil H, Parker-Thornburg J, Kusewitt DF, Bedford MT, Aldaz CM. Generation and characterization of mice carrying a conditional allele of the Wwox tumor suppressor gene. PLoS ONE. 2009;4(11):e7775. doi: 10.1371/journal.pone.0007775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdeen SK, Del Mare S, Hussain S, Abu-Remaileh M, Salah Z, Hagan J, Rawahneh M, Pu XA, Russell S, Stein JL, Stein GS, Lian JB, Aqeilan RI. Conditional inactivation of the mouse Wwox tumor suppressor gene recapitulates the null phenotype. J Cell Physiol. 2013;228(7):1377–1382. doi: 10.1002/jcp.24308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glover TW. Instability at chromosomal fragile sites. Recent Results Cancer Res. 1998;154:185–199. doi: 10.1007/978-3-642-46870-4_11. [DOI] [PubMed] [Google Scholar]
- 16.Glover TW. Common fragile sites. Cancer Lett. 2006;232(1):4–12. doi: 10.1016/j.canlet.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 17.Tsantoulis PK, Kotsinas A, Sfikakis PP, Evangelou K, Sideridou M, Levy B, Mo L, Kittas C, Wu XR, Papavassiliou AG, Gorgoulis VG. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene. 2008;27(23):3256–3264. doi: 10.1038/sj.onc.1210989. [DOI] [PubMed] [Google Scholar]
- 18.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434(7035):864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 19.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr, Kastrinakis NG, Levy B, Kletsas D, Yoneta A, Herlyn M, Kittas C, Halazonetis TD. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434(7035):907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 20.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319(5868):1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 21.Debatisse M, Le Tallec B, Letessier A, Dutrillaux B, Brison O. Common fragile sites: mechanisms of instability revisited. Trends Genet. 2012;28(1):22–32. doi: 10.1016/j.tig.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Ozeri-Galai E, Bester AC, Kerem B. The complex basis underlying common fragile site instability in cancer. Trends Genet. 2012;28(6):295–302. doi: 10.1016/j.tig.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Le Tallec B, Dutrillaux B, Lachages AM, Millot GA, Brison O, Debatisse M. Molecular profiling of common fragile sites in human fibroblasts. Nat Struct Mol Biol. 2011;18(12):1421–1423. doi: 10.1038/nsmb.2155. [DOI] [PubMed] [Google Scholar]
- 24.Le Tallec B, Millot GA, Blin ME, Brison O, Dutrillaux B, Debatisse M. Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 2013;4(3):420–428. doi: 10.1016/j.celrep.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Hosseini SA, Horton S, Saldivar JC, Miuma S, Stampfer MR, Heerema NA, Huebner K. Common chromosome fragile sites in human and murine epithelial cells and FHIT/FRA3B loss-induced global genome instability. Genes Chromosomes Cancer. 2013;52(11):1017–1029. doi: 10.1002/gcc.22097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386(6625):623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 27.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, McHenry KT, Pinchback RM, Ligon AH, Cho YJ, Haery L, Greulich H, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, Buck G, Chen L, Beare D, Latimer C, Widaa S, Hinton J, Fahey C, Fu B, Swamy S, Dalgliesh GL, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463(7283):893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang NS, Hsu LJ, Lin YS, Lai FJ, Sheu HM. WW domain-containing oxidoreductase: a candidate tumor suppressor. Trends Mol Med. 2007;13(1):12–22. doi: 10.1016/j.molmed.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Aqeilan RI, Pekarsky Y, Herrero JJ, Palamarchuk A, Letofsky J, Druck T, Trapasso F, Han SY, Melino G, Huebner K, Croce CM. Functional association between Wwox tumor suppressor protein and p73, a p53 homolog. Proc Natl Acad Sci USA. 2004;101(13):4401–4406. doi: 10.1073/pnas.0400805101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gourley C, Paige AJ, Taylor KJ, Ward C, Kuske B, Zhang J, Sun M, Janczar S, Harrison DJ, Muir M, Smyth JF, Gabra H. WWOX gene expression abolishes ovarian cancer tumorigenicity in vivo and decreases attachment to fibronectin via integrin alpha3. Cancer Res. 2009;69(11):4835–4842. doi: 10.1158/0008-5472.CAN-08-2974. [DOI] [PubMed] [Google Scholar]
- 32.Abu-Remaileh M, Aqeilan RI (2014) Tumor suppressor WWOX regulates glucose metabolism via HIF1alpha modulation. Cell Death Differ [DOI] [PMC free article] [PubMed]
- 33.Dayan S, O’Keefe LV, Choo A, Richards RI. Common chromosomal fragile site FRA16D tumor suppressor WWOX gene expression and metabolic reprogramming in cells. Genes Chromosomes Cancer. 2013;52(9):823–831. doi: 10.1002/gcc.22078. [DOI] [PubMed] [Google Scholar]
- 34.Abu-Odeh M, Salah S, Herbel C, Hofmann TG, Aqeilan RI (2014) WWOX, the common fragile site FRA16D gene product, regulates ATM activation and the DNA damage response. Proc Natl Acad Sci USA (in press) [DOI] [PMC free article] [PubMed]
- 35.Gardenswartz A, Aqeilan RI. WW domain-containing oxidoreductase’s role in myriad cancers: clinical significance and future implications. Exp Biol Med. 2014;239(3):253–263. doi: 10.1177/1535370213519213. [DOI] [PubMed] [Google Scholar]
- 36.Guler G, Uner A, Guler N, Han SY, Iliopoulos D, Hauck WW, McCue P, Huebner K. The fragile genes FHIT and WWOX are inactivated coordinately in invasive breast carcinoma. Cancer. 2004;100(8):1605–1614. doi: 10.1002/cncr.20137. [DOI] [PubMed] [Google Scholar]
- 37.Guler G, Uner A, Guler N, Han SY, Iliopoulos D, McCue P, Huebner K. Concordant loss of fragile gene expression early in breast cancer development. Pathol Int. 2005;55(8):471–478. doi: 10.1111/j.1440-1827.2005.01855.x. [DOI] [PubMed] [Google Scholar]
- 38.Qin HR, Iliopoulos D, Semba S, Fabbri M, Druck T, Volinia S, Croce CM, Morrison CD, Klein RD, Huebner K. A role for the WWOX gene in prostate cancer. Cancer Res. 2006;66(13):6477–6481. doi: 10.1158/0008-5472.CAN-06-0956. [DOI] [PubMed] [Google Scholar]
- 39.Aqeilan RI, Kuroki T, Pekarsky Y, Albagha O, Trapasso F, Baffa R, Huebner K, Edmonds P, Croce CM. Loss of WWOX expression in gastric carcinoma. Clin Cancer Res. 2004;10(9):3053–3058. doi: 10.1158/1078-0432.ccr-03-0594. [DOI] [PubMed] [Google Scholar]
- 40.Donati V, Fontanini G, Dell’Omodarme M, Prati MC, Nuti S, Lucchi M, Mussi A, Fabbri M, Basolo F, Croce CM, Aqeilan RI. WWOX expression in different histological types and subtypes of non-small cell lung cancer. Clinical Cancer Res. 2007;13(3):884–891. doi: 10.1158/1078-0432.CCR-06-2016. [DOI] [PubMed] [Google Scholar]
- 41.Yendamuri S, Kuroki T, Trapasso F, Henry AC, Dumon KR, Huebner K, Williams NN, Kaiser LR, Croce CM. WW domain containing oxidoreductase gene expression is altered in non-small cell lung cancer. Cancer Res. 2003;63(4):878–881. [PubMed] [Google Scholar]
- 42.Kuroki T, Yendamuri S, Trapasso F, Matsuyama A, Aqeilan RI, Alder H, Rattan S, Cesari R, Nolli ML, Williams NN, Mori M, Kanematsu T, Croce CM. The tumor suppressor gene WWOX at FRA16D is involved in pancreatic carcinogenesis. Clin Cancer Res. 2004;10(7):2459–2465. doi: 10.1158/1078-0432.ccr-03-0096. [DOI] [PubMed] [Google Scholar]
- 43.Nakayama S, Semba S, Maeda N, Aqeilan RI, Huebner K, Yokozaki H. Role of the WWOX gene, encompassing fragile region FRA16D, in suppression of pancreatic carcinoma cells. Cancer Sci. 2008;99(7):1370–1376. doi: 10.1111/j.1349-7006.2008.00841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurek KC, Del Mare S, Salah Z, Abdeen S, Sadiq H, Lee SH, Gaudio E, Zanesi N, Jones KB, DeYoung B, Amir G, Gebhardt M, Warman M, Stein GS, Stein JL, Lian JB, et al. Frequent attenuation of the WWOX tumor suppressor in osteosarcoma is associated with increased tumorigenicity and aberrant RUNX2 expression. Cancer Res. 2010;70(13):5577–5586. doi: 10.1158/0008-5472.CAN-09-4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang J, Cogdell D, Yang D, Hu L, Li H, Zheng H, Du X, Pang Y, Trent J, Chen K, Zhang W. Deletion of the WWOX gene and frequent loss of its protein expression in human osteosarcoma. Cancer Lett. 2010;291(1):31–38. doi: 10.1016/j.canlet.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 46.Ishii H, Mimori K, Inageta T, Murakumo Y, Vecchione A, Mori M, Furukawa Y. Components of DNA damage checkpoint pathway regulate UV exposure-dependent alterations of gene expression of FHIT and WWOX at chromosome fragile sites. Mol Cancer Res MCR. 2005;3(3):130–138. doi: 10.1158/1541-7786.MCR-04-0209. [DOI] [PubMed] [Google Scholar]
- 47.Fu J, Qu Z, Yan P, Ishikawa C, Aqeilan RI, Rabson AB, Xiao G. The tumor suppressor gene WWOX links the canonical and noncanonical NF-kappaB pathways in HTLV-I Tax-mediated tumorigenesis. Blood. 2011;117(5):1652–1661. doi: 10.1182/blood-2010-08-303073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iliopoulos D, Guler G, Han SY, Johnston D, Druck T, McCorkell KA, Palazzo J, McCue PA, Baffa R, Huebner K. Fragile genes as biomarkers: epigenetic control of WWOX and FHIT in lung, breast and bladder cancer. Oncogene. 2005;24(9):1625–1633. doi: 10.1038/sj.onc.1208398. [DOI] [PubMed] [Google Scholar]
- 49.Mahajan NP, Whang YE, Mohler JL, Earp HS. Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res. 2005;65(22):10514–10523. doi: 10.1158/0008-5472.CAN-05-1127. [DOI] [PubMed] [Google Scholar]
- 50.Kuroki T, Trapasso F, Shiraishi T, Alder H, Mimori K, Mori M, Croce CM. Genetic alterations of the tumor suppressor gene WWOX in esophageal squamous cell carcinoma. Cancer Res. 2002;62(8):2258–2260. [PubMed] [Google Scholar]
- 51.Driouch K, Prydz H, Monese R, Johansen H, Lidereau R, Frengen E. Alternative transcripts of the candidate tumor suppressor gene, WWOX, are expressed at high levels in human breast tumors. Oncogene. 2002;21(12):1832–1840. doi: 10.1038/sj.onc.1205273. [DOI] [PubMed] [Google Scholar]
- 52.Golan-Gerstl R, Cohen M, Shilo A, Suh SS, Bakacs A, Coppola L, Karni R. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011;71(13):4464–4472. doi: 10.1158/0008-5472.CAN-10-4410. [DOI] [PubMed] [Google Scholar]
- 53.Watanabe A, Hippo Y, Taniguchi H, Iwanari H, Yashiro M, Hirakawa K, Kodama T, Aburatani H. An opposing view on WWOX protein function as a tumor suppressor. Cancer Res. 2003;63(24):8629–8633. [PubMed] [Google Scholar]
- 54.Gao G, Kasperbauer JL, Tombers NM, Wang V, Mayer K, Smith DI. A selected group of large common fragile site genes have decreased expression in oropharyngeal squamous cell carcinomas. Genes Chromosomes Cancer. 2014;53(5):392–401. doi: 10.1002/gcc.22150. [DOI] [PubMed] [Google Scholar]
- 55.Aldaz CM, Ferguson BW, Abba MC (2014) WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochim Biophys Acta [DOI] [PMC free article] [PubMed]
- 56.Del Mare S, Kurek KC, Stein GS, Lian JB, Aqeilan RI. Role of the WWOX tumor suppressor gene in bone homeostasis and the pathogenesis of osteosarcoma. Am J Cancer Res. 2011;1(5):585–594. [PMC free article] [PubMed] [Google Scholar]
- 57.Berger AH, Pandolfi PP. Haplo-insufficiency: a driving force in cancer. J Pathol. 2011;223(2):137–146. doi: 10.1002/path.2800. [DOI] [PubMed] [Google Scholar]
- 58.Abdeen SK, Salah Z, Khawaled S, Aqeilan RI. Characterization of WWOX inactivation in murine mammary gland development. J Cell Physiol. 2013;228(7):1391–1396. doi: 10.1002/jcp.24310. [DOI] [PubMed] [Google Scholar]
- 59.Ferguson BW, Gao X, Kil H, Lee J, Benavides F, Abba MC, Aldaz CM. Conditional Wwox deletion in mouse mammary gland by means of two Cre recombinase approaches. PLoS ONE. 2012;7(5):e36618. doi: 10.1371/journal.pone.0036618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29(4):418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 61.Liu X, Holstege H, van der Gulden H, Treur-Mulder M, Zevenhoven J, Velds A, Kerkhoven RM, van Vliet MH, Wessels LF, Peterse JL, Berns A, Jonkers J. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci USA. 2007;104(29):12111–12116. doi: 10.1073/pnas.0702969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359(6393):295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 63.Salah Z, Alian A, Aqeilan RI. WW domain-containing proteins: retrospectives and the future. Front Biosci. 2012;17:331–348. doi: 10.2741/3930. [DOI] [PubMed] [Google Scholar]
- 64.Sudol M, Recinos CC, Abraczinskas J, Humbert J, Farooq A. WW or WoW: the WW domains in a union of bliss. IUBMB Life. 2005;57(12):773–778. doi: 10.1080/15216540500389039. [DOI] [PubMed] [Google Scholar]
- 65.Aqeilan RI, Donati V, Palamarchuk A, Trapasso F, Kaou M, Pekarsky Y, Sudol M, Croce CM. WW domain-containing proteins, WWOX and YAP, compete for interaction with ErbB-4 and modulate its transcriptional function. Cancer Res. 2005;65(15):6764–6772. doi: 10.1158/0008-5472.CAN-05-1150. [DOI] [PubMed] [Google Scholar]
- 66.Aqeilan RI, Donati V, Gaudio E, Nicoloso MS, Sundvall M, Korhonen A, Lundin J, Isola J, Sudol M, Joensuu H, Croce CM, Elenius K. Association of Wwox with ErbB4 in breast cancer. Cancer Res. 2007;67(19):9330–9336. doi: 10.1158/0008-5472.CAN-07-2147. [DOI] [PubMed] [Google Scholar]
- 67.Abu-Odeh M, Bar-Mag T, Huang H, Kim T, Salah Z, Abdeen SK, Sudol M, Reichmann D, Sidhu S, Kim PM, Aqeilan RI. Characterizing WW domain interactions of tumor suppressor WWOX reveals its association with multiprotein networks. J Biol Chem. 2014;289(13):8865–8880. doi: 10.1074/jbc.M113.506790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McDonald CB, Buffa L, Bar-Mag T, Salah Z, Bhat V, Mikles DC, Deegan BJ, Seldeen KL, Malhotra A, Sudol M, Aqeilan RI, Nawaz Z, Farooq A. Biophysical basis of the binding of WWOX tumor suppressor to WBP1 and WBP2 adaptors. J Mol Biol. 2012;422(1):58–74. doi: 10.1016/j.jmb.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bouteille N, Driouch K, Hage PE, Sin S, Formstecher E, Camonis J, Lidereau R, Lallemand F. Inhibition of the Wnt/beta-catenin pathway by the WWOX tumor suppressor protein. Oncogene. 2009;28:2569–2580. doi: 10.1038/onc.2009.120. [DOI] [PubMed] [Google Scholar]
- 70.Levrero M, De Laurenzi V, Costanzo A, Gong J, Melino G, Wang JY. Structure, function and regulation of p63 and p73. Cell Death Differ. 1999;6(12):1146–1153. doi: 10.1038/sj.cdd.4400624. [DOI] [PubMed] [Google Scholar]
- 71.Melino G, De Laurenzi V, Vousden KH. p73: friend or foe in tumorigenesis. Nat Rev Cancer. 2002;2(8):605–615. doi: 10.1038/nrc861. [DOI] [PubMed] [Google Scholar]
- 72.Salah Z, Bar-mag T, Kohn Y, Pichiorri F, Palumbo T, Melino G, Aqeilan RI. Tumor suppressor WWOX binds to ΔNp63α and sensitizes cancer cells to chemotherapy. Cell Death Dis. 2013;4:e. doi: 10.1038/cddis.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Espanel X, Sudol M. Yes-associated protein and p53-binding protein-2 interact through their WW and SH3 domains. J Biol Chem. 2001;276(17):14514–14523. doi: 10.1074/jbc.M008568200. [DOI] [PubMed] [Google Scholar]
- 74.Chang NS, Doherty J, Ensign A. JNK1 physically interacts with WW domain-containing oxidoreductase (WOX1) and inhibits WOX1-mediated apoptosis. J Biol Chem. 2003;278(11):9195–9202. doi: 10.1074/jbc.M208373200. [DOI] [PubMed] [Google Scholar]
- 75.Ferguson BW, Gao X, Zelazowski MJ, Lee J, Jeter CR, Abba MC, Aldaz CM. The cancer gene WWOX behaves as an inhibitor of SMAD3 transcriptional activity via direct binding. BMC Cancer. 2013;13:593. doi: 10.1186/1471-2407-13-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heldin CH, Vanlandewijck M, Moustakas A. Regulation of EMT by TGFbeta in cancer. FEBS Lett. 2012;586(14):1959–1970. doi: 10.1016/j.febslet.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 77.Moustakas A, Heldin CH. Induction of epithelial-mesenchymal transition by transforming growth factor beta. Semin Cancer Biol. 2012;22(5–6):446–454. doi: 10.1016/j.semcancer.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 78.O’Keefe LV, Colella A, Dayan S, Chen Q, Choo A, Jacob R, Price G, Venter D, Richards RI. Drosophila orthologue of WWOX, the chromosomal fragile site FRA16D tumour suppressor gene, functions in aerobic metabolism and regulates reactive oxygen species. Hum Mol Genet. 2011;20(3):497–509. doi: 10.1093/hmg/ddq495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8(9):705–713. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 80.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 81.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 82.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 83.Semenza GL. HIF-1 and tumor progression: pathophysiology and therapeutics. Trends Mol Med. 2002;8(4 Suppl):S62–67. doi: 10.1016/s1471-4914(02)02317-1. [DOI] [PubMed] [Google Scholar]
- 84.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11(3):220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 85.Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol. 2007;19(2):238–245. doi: 10.1016/j.ceb.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 86.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28(5):739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 87.Shiloh Y. ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev. 2001;11(1):71–77. doi: 10.1016/s0959-437x(00)00159-3. [DOI] [PubMed] [Google Scholar]
- 88.Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434(7033):605–611. doi: 10.1038/nature03442. [DOI] [PubMed] [Google Scholar]
- 89.Letessier A, Millot GA, Koundrioukoff S, Lachages AM, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature. 2011;470(7332):120–123. doi: 10.1038/nature09745. [DOI] [PubMed] [Google Scholar]
- 90.Dereli-Oz A, Versini G, Halazonetis TD. Studies of genomic copy number changes in human cancers reveal signatures of DNA replication stress. Mol Oncol. 2011;5(4):308–314. doi: 10.1016/j.molonc.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Miuma S, Saldivar JC, Karras JR, Waters CE, Paisie CA, Wang Y, Jin V, Sun J, Druck T, Zhang J, Huebner K. Fhit deficiency-induced global genome instability promotes mutation and clonal expansion. PLoS ONE. 2013;8(11):e80730. doi: 10.1371/journal.pone.0080730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Saldivar JC, Miuma S, Bene J, Hosseini SA, Shibata H, Sun J, Wheeler LJ, Mathews CK, Huebner K. Initiation of genome instability and preneoplastic processes through loss of Fhit expression. PLoS Genet. 2012;8(11):e1003077. doi: 10.1371/journal.pgen.1003077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thavathiru E, Ludes-Meyers JH, MacLeod MC, Aldaz CM. Expression of common chromosomal fragile site genes, WWOX/FRA16D and FHIT/FRA3B is downregulated by exposure to environmental carcinogens, UV, and BPDE but not by IR. Mol Carcinog. 2005;44(3):174–182. doi: 10.1002/mc.20122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lai FJ, Cheng CL, Chen ST, Wu CH, Hsu LJ, Lee JY, Chao SC, Sheen MC, Shen CL, Chang NS, Sheu HM. WOX1 is essential for UVB irradiation-induced apoptosis and down-regulated via translational blockade in UVB-induced cutaneous squamous cell carcinoma in vivo. Clin Cancer Res. 2005;11(16):5769–5777. doi: 10.1158/1078-0432.CCR-04-2274. [DOI] [PubMed] [Google Scholar]
- 95.Santini S, Stagni V, Giambruno R, Fianco G, Di Benedetto A, Mottolese M, Pellegrini M, Barila D. ATM kinase activity modulates ITCH E3-ubiquitin ligase activity. Oncogene. 2013;33:1113–1123. doi: 10.1038/onc.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Abu Remaileh M, Aqeilan RI (2014) Tumor suppressor WWOX regulates glucose metabolism via HIF1α modulation. Cell Death Differ (in press) [DOI] [PMC free article] [PubMed]
- 97.Brueckner LM, Hess EM, Schwab M, Savelyeva L. Instability at the FRA8I common fragile site disrupts the genomic integrity of the KIAA0146, CEBPD and PRKDC genes in colorectal cancer. Cancer Lett. 2013;336(1):85–95. doi: 10.1016/j.canlet.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 98.Wan L, Han J, Liu T, Dong S, Xie F, Chen H, Huang J. Scaffolding protein SPIDR/KIAA0146 connects the Bloom syndrome helicase with homologous recombination repair. Proc Natl Acad Sci USA. 2013;110(26):10646–10651. doi: 10.1073/pnas.1220921110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhu Y, McAvoy S, Kuhn R, Smith DI. RORA, a large common fragile site gene, is involved in cellular stress response. Oncogene. 2006;25(20):2901–2908. doi: 10.1038/sj.onc.1209314. [DOI] [PubMed] [Google Scholar]
- 100.Ludes-Meyers JH, Kil H, Bednarek AK, Drake J, Bedford MT, Aldaz CM. WWOX binds the specific proline-rich ligand PPXY: identification of candidate interacting proteins. Oncogene. 2004;23(29):5049–5055. doi: 10.1038/sj.onc.1207680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Aqeilan RI, Palamarchuk A, Weigel RJ, Herrero JJ, Pekarsky Y, Croce CM. Physical and functional interactions between the Wwox tumor suppressor protein and the AP-2gamma transcription factor. Cancer Res. 2004;64(22):8256–8261. doi: 10.1158/0008-5472.CAN-04-2055. [DOI] [PubMed] [Google Scholar]
- 102.Gaudio E, Palamarchuk A, Palumbo T, Trapasso F, Pekarsky Y, Croce CM, Aqeilan RI. Physical association with WWOX suppresses c-Jun transcriptional activity. Cancer Res. 2006;66(24):11585–11589. doi: 10.1158/0008-5472.CAN-06-3376. [DOI] [PubMed] [Google Scholar]
- 103.Jin C, Ge L, Ding X, Chen Y, Zhu H, Ward T, Wu F, Cao X, Wang Q, Yao X. PKA-mediated protein phosphorylation regulates ezrin–WWOX interaction. Biochem Biophys Res Commun. 2006;341(3):784–791. doi: 10.1016/j.bbrc.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 104.Bouteille N, Driouch K, Hage PE, Sin S, Formstecher E, Camonis J, Lidereau R, Lallemand F. Inhibition of the Wnt/beta-catenin pathway by the WWOX tumor suppressor protein. Oncogene. 2009;28(28):2569–2580. doi: 10.1038/onc.2009.120. [DOI] [PubMed] [Google Scholar]
- 105.Salah Z, Bar-mag T, Kohn Y, Pichiorri F, Palumbo T, Melino G, Aqeilan RI. Tumor suppressor WWOX binds to DeltaNp63alpha and sensitizes cancer cells to chemotherapy. Cell Death Dis. 2013;4:e480. doi: 10.1038/cddis.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chang NS, Doherty J, Ensign A, Schultz L, Hsu LJ, Hong Q. WOX1 is essential for tumor necrosis factor-, UV light-, staurosporine-, and p53-mediated cell death, and its tyrosine 33-phosphorylated form binds and stabilizes serine 46-phosphorylated p53. J Biol Chem. 2005;280(52):43100–43108. doi: 10.1074/jbc.M505590200. [DOI] [PubMed] [Google Scholar]
- 107.Lin HP, Chang JY, Lin SR, Lee MH, Huang SS, Hsu LJ, Chang NS. Identification of an in vivo MEK/WOX1 complex as a master switch for apoptosis in T cell leukemia. Genes Cancer. 2011;2(5):550–562. doi: 10.1177/1947601911418498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sze CI, Su M, Pugazhenthi S, Jambal P, Hsu LJ, Heath J, Schultz L, Chang NS. Down-regulation of WW domain-containing oxidoreductase induces Tau phosphorylation in vitro. A potential role in Alzheimer’s disease. J Biol Chem. 2004;279(29):30498–30506. doi: 10.1074/jbc.M401399200. [DOI] [PubMed] [Google Scholar]
- 109.Wang HY, Juo LI, Lin YT, Hsiao M, Lin JT, Tsai CH, Tzeng YH, Chuang YC, Chang NS, Yang CN, Lu PJ. WW domain-containing oxidoreductase promotes neuronal differentiation via negative regulation of glycogen synthase kinase 3beta. Cell Death Differ. 2012;19(6):1049–1059. doi: 10.1038/cdd.2011.188. [DOI] [PMC free article] [PubMed] [Google Scholar]