

Abstract
Affinity proteins have advanced the field of targeted therapeutics due to their generally higher specificity compared to small molecular compounds. However, side effects caused by on-target binding in healthy tissues are still an issue. Here, we design and investigate a prodrug strategy for improving tissue specificity of Affibody molecules in future in vivo studies. The prodrug Affibody (pro-Affibody) against the HER2 receptor was constructed by fusing a HER2-specific Affibody (ZHER2) to an anti-idiotypic Affibody (anti-ZHER2). The linker was engineered to comprise a substrate peptide for the cancer-associated matrix metalloprotease 1 (MMP-1). The hypothesis was that the binding surface of ZHER2 would thereby be blocked from interacting with HER2 until the substrate peptide was specifically hydrolyzed by MMP-1. Binding should thereby only occur where MMP-1 is overexpressed, potentially decreasing on-target toxicities in normal tissues. The pro-Affibody was engineered to find a suitable linker and substrate peptide, and the different constructs were evaluated with a new bacterial display assay. HER2-binding of the pro-Affibody was efficiently masked and proteolytic activation of the best variant yielded over 1,000-fold increase in apparent binding affinity. Biosensor analysis revealed that blocking of the pro-Affibody primarily affected the association phase. In a cell-binding assay, the activated pro-Affibody targeted native HER2 on cancer cells as opposed to the non-activated pro-Affibody. We believe this prodrug approach with proteolytic activation is promising for improving tissue specificity in future in vivo targeting applications and can hopefully be extended to other Affibody molecules and similar affinity proteins as well.
Keywords: HER2, Bacterial display, Combinatorial protein engineering, Staphylococcal display, Directed evolution, Cell surface display, Flow cytometry, MMP-1
Introduction
Monoclonal antibodies (mAbs) have become a well-established class of drugs for targeted therapeutics in a range of human diseases, such as cancer [1, 2]. More recently, non-immunoglobulin based protein scaffolds have been reported as promising alternatives to traditional mAbs [3]. Common for all these affinity proteins is that they can be engineered to very high affinity and specificity [4]. However, although affinity proteins generally demonstrate a considerably higher specificity for their molecular targets than small molecular compounds, on-target related side effects are still an issue. Indeed, most drug targets are also present at various degrees in normal tissues resulting in toxicities or even making the target non-druggable [5–7].
Prodrugs that are inactive in normal tissue and selectively activated by the often dysregulated microenvironment in diseased tissue is an attractive approach to circumvent side effects mediated by target binding in healthy tissue [8]. Proteases are often locally and specifically upregulated in cancer [9]. An example is the matrix metalloprotease family (MMPs), comprising zinc-dependent endopeptidases that are capable of degrading the extracellular matrix (ECM) [10]. Several members of the MMP family are highly expressed in tumors and their upregulation is usually associated with poor disease outcome. Matrix metalloprotease 1 (MMP-1) is for example often upregulated in breast cancer and facilitates the invasiveness of breast cancer cells. Upregulation of MMP-1 also correlates with overexpression of human epidermal growth factor receptor 2 (HER2) in breast cancer cells [11–16]. The tissue specificity of an affinity protein could potentially be improved by exploiting the disease-specific upregulation of such proteases, thereby reducing side effects caused by target binding in normal tissue. An engineered “pro-drug” variant of Cetuximab that was masked by a protease-sensitive peptide has been investigated in preclinical studies, demonstrating similar efficacy as Cetuximab and reduced toxicity [17]. Similarly, a VCAM-1-specific mAb for targeting of atherosclerotic plaque was linked to a paratope-blocking peptide and shown to be specifically activated by disease-associated proteases [18]. The binding activity of a TNF prodrug and a CD95L prodrug were also shown to be restored upon processing by tumor-associated proteases [19, 20].
Affibody molecules are very small (58 aa) alpha-helical non-immunoglobulin based affinity proteins [21]. Properties such as small size, efficient production in prokaryotic hosts (or by chemical peptide synthesis), cysteine-free sequence, high solubility and flexible pharmacokinetics make Affibody molecules promising alternatives for a wide variety of applications, including: targeted drug delivery, cytokine blocking in inflammatory diseases and molecular imaging. Affibody molecules have been generated to a large number of different disease-related targets, and are often demonstrating monovalent affinity in the low nanomolar to picomolar range as well as high specificity similar to what is normally achieved with mAbs [22, 23]. Several Affibody molecules are currently in preclinical and clinical development. A HER2-specific Affibody molecule (ZHER2) with 22 pM monovalent affinity for the receptor has previously been developed and investigated for molecular imaging and targeted delivery of payloads [21]. The binder is currently investigated in the clinic as a HER2-targeted PET imaging agent (clinicaltrials.gov).
Here, we design and evaluate a new prodrug strategy for potentially improving tissue specificity of Affibody molecules in future in vivo studies. The small size combined with an independent and fast folding of Affibody molecules facilitates construction of various multimer formats. Our approach was to design such dimeric constructs based on two Affibody molecules in which one is masking the activity of the other. So-called anti-idiotypic Affibody molecules have been generated to a number of binders and are demonstrating specific and reversible binding to the other Affibody molecule [21]. We used an anti-idiotypic Affibody directed against ZHER2 for engineering of a “prodrug-like” Affibody molecule (pro-Affibody). The new pro-Affibody is composed of: (1) a targeting domain, (2) a blocking domain, (3) a linker and (4) a protease substrate sequence within the linker (Fig. 1). Here, we used the high-affinity HER2-specific Affibody molecule (ZHER2) [23] as targeting domain, which was genetically fused by a linker to an anti-idiotypic Affibody molecule (anti-ZHER2) [24] that specifically binds ZHER2 and hopefully blocks its binding surface from interacting with HER2 (Fig. 1). We decided to use a small protein-domain with a stable three-dimensional structure as blocker rather than a linear peptide to minimize potential unspecific proteolysis in serum. The linker contained a substrate for MMP-1. MMP-1 upregulation is associated with high HER2 expression in breast cancer cells [16]. The new pro-Affibody approach is based on the hypothesis that as long as the linker is intact, the local apparent concentration of the blocking domain will be high and the equilibrium will be shifted towards the blocked state even though the affinity between the two moieties is moderate (K D ~ 270 nM). Upon proteolysis of the linker, the blocking domain is no longer connected after dissociation from ZHER2, leading to a shift in equilibrium towards the free state (Fig. 1). In principle, the non-activated pro-Affibody should be blocked from interacting with HER2 in normal tissue and when activated by MMP-1 in diseased tissue, the binding capacity of ZHER2 should be restored. Binding should thereby preferentially occur in diseased tissue where MMP-1 is overexpressed, thus potentially decreasing on-target toxicities in normal tissues (Fig. 1). For investigation of the new pro-Affibody concept, we used an in-house developed bacterial display technology in combination with flow-cytometric analysis. The analysis included different linkers as well as various MMP-1 substrate sequences and the best-performing variant demonstrated over 1,000-fold difference in affinity between non-activated and activated pro-Affibody. Soluble pro-Affibodies were further characterized using SPR-based biosensor technology as well as in cancer cell assays.
Fig. 1.

Schematic picture describing the pro-Affibody concept. a The pro-Affibody is constructed by fusing a target binding Affibody molecule (green) to an anti-idiotypic Affibody molecule (purple), intended to mask the antigen-binding surface from interacting with its target, via a protease-cleavable linker. The masking prevents target binding until selective proteolysis releases the masking domain and restores binding capacity of the Affibody molecule. b Covalent linkage of the masking domain to the target-binding domain results in a large increase in local concentration, which shifts the equilibrium towards the bound (blocked) state. After proteolysis of the substrate peptide within the linker, the blocking domain can diffuse upon dissociation, which leads to a shift in equilibrium towards the free (active) state. c The target-binding domain within the pro-Affibody will only be able to bind to its target when the pro-Affibody is activated by proteases that are upregulated in diseased tissue, potentially reducing side effects caused by on-target, but off-tissue binding
Materials and methods
Labeling of HER2
Biotinylation of recombinant human HER2/Fc chimera (Sino Biological Inc., Beijing, China) was performed using biotin-XX sulfo succinimidyl ester, sodium salt (Invitrogen, Carlsbad, CA, USA) in NaHCO3 (0.1 M, pH 8.5) with a 50-fold molar excess of biotin and incubated at room temperature for 1 h. The reaction was stopped by adding Glycine (0.1 M) followed by buffer exchange to PBS using a PD MiniTrap G-25 column (GE Healthcare, Uppsala, Sweden). The protein concentration was determined by spectrophotometry at 280 nm.
Subcloning into the staphylococcal display vector
The gene encoding for the HER2-binding Affibody molecule (ZHER2:342) [23] C-terminally fused to the anti-idiotypic Affibody molecule (anti-ZHER2 and previously denoted ZE01) [24] with a connecting dummy sequence was ordered from Genscript (Piscataway, NJ, USA). The gene was digested and ligated into the staphylococcal display vector pSCZ1 [25, 26]. The dummy was replaced by three different linkers, each constructed by two hybridized oligonucleotides that were ligated into the vector. The linkers consisted of the MMP-1 substrate peptide GPQAIAGQ flanked by (GGGGS)X repeats, where X represents 1, 2 or 3. To generate a pro-Affibody with an irrelevant fusion partner, an Affibody molecule with no affinity towards ZHER2:342 (ZHER3) [27] was amplified by PCR and ligated into the vector and replacing anti-ZHER2. A different substrate peptide was constructed by introducing a mutation in the P1 position of the substrate peptide, generating GPQGIAGQ. The mutation was introduced by the QuikChange site-directed mutagenesis method (Agilent Technologies, Inc., Santa Clara, CA, USA). All the resulting plasmids were transformed to E. coli TOP10 (Invitrogen, Carlsbad, CA, USA) followed by plasmid preparation and transformation into electrocompetent S. carnosus TM300 [28] according to previously described protocol [29].
Analysis of activation by MMP-1 with staphylococcal surface display
Staphylococcal cells displaying the different pro-Affibody variants were inoculated into 10 ml tryptic soy broth supplemented with yeast extract (TSB+Y; Merck, Darmstadt, Germany) supplemented with 10 μg/ml chloramphenicol and grown overnight at 37 °C and 150 rpm. About 107 bacterial cells from respectively ON cultures were washed twice with 800 μl 1× PBSP (phosphate-buffered saline supplemented with 0.1 % Pluronic® F108 NF Surfactant, pH 7.4; BASF Corporation, Mount Olive, NJ, USA) and pelleted by centrifugation (3,500g, 6 min, 4 °C). Bacteria were resuspended either in assay buffer (50 mM Tris, 10 mM CaCl2, 150 mM NaCl, 0.05 % Brij35, pH 7.5) supplemented with 25 nM MMP-1 (Enzo Life Sciences, Farmingdale, NY, USA) or in assay buffer only and incubated for 2 h at 37 °C. Bacteria were washed three times with 800 μl 1× PBSP and incubated with 100 μl of the indicated concentration of biotinylated HER2 at room temperature for 45 min with gentle mixing. Bacteria were washed twice with 800 μl ice-cold 1XPBSP and incubated on ice with 2 μg/ml streptavidin, R-phycoerythrin conjugate (SAPE) and 225 nM Alexa Fluor 647-human serum albumin (HSA) conjugate for 15 min. After two washes as above, the bacterial cells were resuspended in 250 μl ice-cold 1× PBSP and analyzed using a Gallios™ flow cytometer (Beckman Coulter, CA, USA).
Expression of soluble pro-Affibody molecules
The genes encoding the different pro-Affibody variants were cloned into pET26b-(+) (Novagen, Madison, WI, USA) generating pro-Affibody constructs with a C-terminal His6-tag. The resulting plasmids were purified and transformed into E. coli BL21* cells (Life Technologies, Carlsbad, CA, USA). Overnight cultures were used to inoculate 100 ml TSB+Y supplemented with 50 μg/ml kanamycin. The bacterial cells were grown at 37 °C until OD600 ~1 when protein expression was induced by adding isopropyl-beta-d-thiogalactopyranoside (IPTG) to a final concentration of 1 mM followed by culturing at 25 °C ON. Cells were harvested by centrifugation and resuspended in wash buffer (500 mM NaCl, 50 mM Na2HPO4, pH 8) followed by sonication, centrifugation and filtration. The proteins were purified under native conditions on TALON® Metal Affinity Resin (Clontech Laboratories, Inc., Mountain View, CA, USA). The column was washed with wash buffer and proteins were eluted with wash buffer supplemented with 500 mM imidazole. Finally, buffer was exchanged to PBS with PD-10 columns (GE Healthcare, Uppsala, Sweden).
MMP-1 treatment of soluble pro-Affibody molecules
2 μg of the different pro-Affibody variants were incubated in 20 μl assay buffer (50 mM HEPES, 10 mM CaCl2, 20 μM ZnCl2, 0.05 % Brij35, pH 7.5) with 25 nM MMP-1 or assay buffer only for 2 h at 37 °C. The reaction was stopped by adding EDTA to a final concentration of 10 mM.
Characterization of pro-Affibody molecules by surface plasmon resonance
HER2 binding was measured for pro-Affibody molecules using surface plasmon resonance (Biacore 3000, GE Healthcare). Approximately 1,800 RU of HER2/Fc was immobilized by amine coupling on CM-5 sensor chip according to the manufacturer’s recommendations. One surface on the chip was activated and deactivated and used as reference cell during injections. A modified HBS-EP buffer (50 mM HEPES, 150 mM NaCl, 10 mM EDTA, 0.05 % Brij35, pH 7.5) was used as running buffer. For surface plasmon resonance measurements, 150 mM NaCl was added to MMP-1 treated pro-Affibody molecules from above to reduce buffer effects during measurements. Both protease-treated and untreated pro-Affibody variants were injected at 25 μl/min for 5 min, followed by dissociation for 5 min. The surfaces were regenerated with two injections of 25 mM HCl. Responses from the reference surface were subtracted as well as the response from a buffer sample or a sample with MMP-1 for non-activated and activated pro-Affibody, respectively.
Cell binding
The HER2 high-expressing cell line SKOV3 was cultured as recommended by the supplier (ATCC, Manassas, VA). Trypsin-treated cells, 500,000 in each tube, were washed with PBS + 0.1 % BSA (Saveen & Werner, Limhamn, Sweden). The cells were incubated with 25 nM of pro-Affibody variants previously treated with or without MMP-1 in PBS + 0.1 % BSA for 1 h at room temperature. The cells were washed with PBS + 0.1 % BSA and a goat anti-Affibody antibody was added to a final concentration of 4 μg/ml. The cells were incubated for 50 min on ice and washed with PBS + 0.1 % BSA followed by incubation with Alexa Fluor® 647-labeled donkey anti-goat antibody (1 μg/ml) on ice. After wash in PBS + 0.1 % BSA, the cells were analyzed using a Gallios™ flow cytometer (Beckman Coulter, CA).
Results
Analysis of pro-Affibody with staphylococcal surface display
To improve tissue specificity in future in vivo targeting studies, we designed a pro-drug variant of an Affibody molecule that would be activated by specific proteolytic hydrolysis. For investigation of the pro-Affibody concept, we employed an in-house developed bacterial display technology [25, 26, 30] together with flow cytometry. The bacterial display technology is based on recombinant expression and display of proteins on the surface of Gram-positive Staphylococcus carnosus (Fig. 2a, b). An advantage with bacterial display for analysis is that it obviates the purification step of recombinant proteins, thereby potentially increasing throughput of the assay. Recombinant pro-Affibodies were subcloned to a staphylococcal display vector in fusion to a reporter tag for simultaneous monitoring of surface expression level (Fig. 2a). The pro-Affibodies were enzymatically anchored in the C-termini to the peptidoglycan in the staphylococcal cell wall. The bacterial display assay for analysis of pro-Affibodies included: (a) expression and display of recombinant pro-Affibodies on bacteria, (b) incubation of bacteria with protease (MMP-1), (c) incubation with labeled HER2, (d) flow-cytometric analysis (Fig. 2c). The multivalent display on bacteria should make the assay quantitative and fluorescence intensity would hopefully correlate with proteolytic cleavage.
Fig. 2.
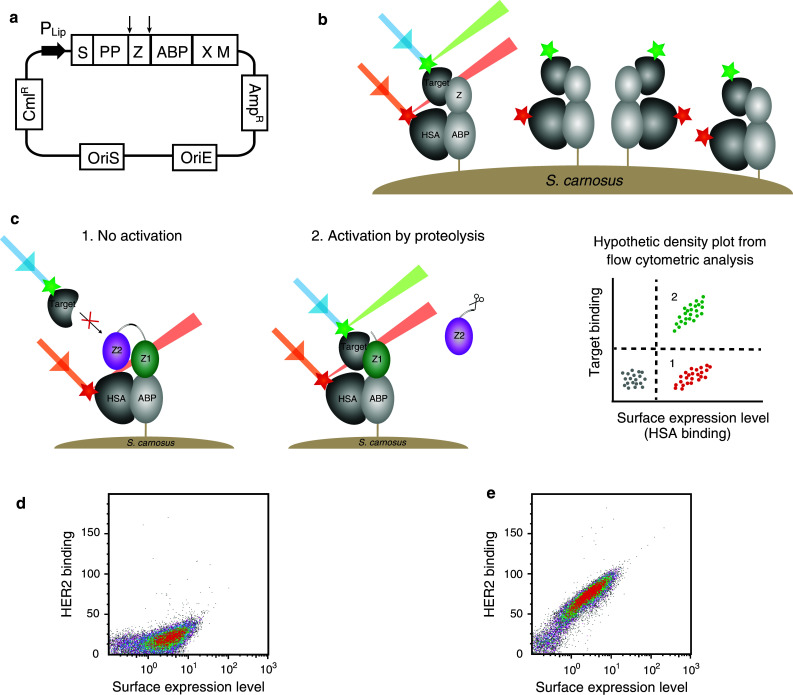
Staphylococcal surface display for characterization of the pro-Affibody. a Schematic illustration of the display vector with relevant features indicated. P Lip S. hyicus promoter region; S secretion signal, PP propeptide, Z Affibody molecule, ABP albumin binding protein, used as surface expression reporter tag, XM region for cell wall anchoring. b Schematic picture showing the staphylococcal surface display technology when displaying a monomeric Affibody molecule. Bacterial cells are incubated with fluorescently labeled target and HSA, labeled with a different fluorophore for monitoring of surface expression levels, and then analyzed with flow cytometry. c Schematic picture showing the staphylococcal surface display of a pro-Affibody and hypothetic dot plot from flow cytometry. Bacteria displaying the non-activated pro-Affibody exhibit the signal for surface expression only (1), whereas bacteria displaying pro-Affibody that has been activated by recombinant protease exhibit signals for surface expression as well as target binding (2). d Representative density plot of the non-activated pro-Affibody displayed on S. carnosus, incubated with 20 nM HER2 and analyzed in the flow cytometer, showing the HER2-binding signal on the y-axis and the surface expression level on the x-axis. e Same as in d, but for the pro-Affibody that has been activated with MMP-1. Experiments were performed in duplicates on different days using freshly prepared samples and reagents
The anti-idiotypic Affibody (anti-ZHER2) was generated in a previous study [24], but it has never been demonstrated whether it blocks the interaction between ZHER2 and the receptor. Hence, to investigate potential blocking and to assess the prodrug concept, the recombinant staphylococci displaying the HER2-specific pro-Affibody were analyzed by flow cytometry. Bacteria were incubated with labeled HER2 receptor to analyze if the anti-idiotypic domain was able to mask the binding site and inhibit the interaction. For comparison, one sample was treated with protease (MMP-1) prior to incubation with labeled HER2 and analysis. The results demonstrated that the masking domain inhibited binding of ZHER2 to the HER2 receptor as intended (Fig. 2d). Moreover, analysis of the proteolytically-treated bacteria revealed a substantial shift in fluorescence, demonstrating that the pro-Affibody was activated by MMP-1 and regained its binding activity (Fig. 2e).
To verify that masking was a result from specific interaction between the two domains and not due to unspecific sterical hindrance, the HER2-binding Affibody molecule (ZHER2) was fused to a control Affibody molecule (ZHER3) with no measurable affinity towards ZHER2. This negative control pro-Affibody was subcloned to the staphylococcal vector, expressed on bacteria and recombinant bacteria were analyzed in the flow cytometer. The assay showed that fusing the negative-control blocking domain to ZHER2 had no effect on HER2-binding and no significant shift was observed after proteolytic digestion (Fig. 3c). The results thus verified that the observed masking of the pro-Affibody was due to specific binding of the anti-idiotypic domain to the HER2-binding paratope of ZHER2.
Fig. 3.

Engineering and evaluation of bacteria-displayed pro-Affibody molecules. a Representative results from flow-cytometric analysis of efficiency of masking and proteolytic activation of different linkers connecting the two domains of the pro-Affibody. The protease substrate is flanked by one (green), two (red) or three (blue) G4S repeats. Light colors represent the non-activated and dark colors the activated pro-Affibody. b Representative results from flow-cytometric analysis of two different substrate peptides, GPQAIAGQ (green) and GPQGIAGQ (gray). Pro-Affibody molecules containing the substrates were tested for their ability to be activated by MMP-1. c Representative results from flow-cytometric analysis of negative control pro-Affibody (ZHER2 fused to ZHER3). The HER2-binding Affibody molecule was fused to a non-binding Affibody molecule (ZHER3) as the “masking” domain, which did not mask before proteolysis (purple) and was not affected by proteolysis (black). Shown in green is the non-activated pro-Affibody with the masking domain (anti-ZHER2). d Binding to HER2 of activated pro-Affibody (green) and non-activated pro-Affibody (red). HER2 concentration was titrated from 0.1 to 1,000 nM. Dots represent mean values (±SD) of MFI measured by flow cytometry from two independent experiments. Solid line represents fitted data by non-linear regression to a 1:1 binding model. All experiments were performed at least in duplicates on different days using freshly prepared samples and reagents
Analysis of linker lengths
Next, we assessed the influence of linker length on masking efficiency and activation by proteolysis. Three distinct pro-Affibodies containing different linker lengths were therefore subcloned to the staphylococcal display vector and compared using the bacterial display assay. The linkers consisted of the MMP-1 protease substrate (PS) flanked by one, two or three G4S repeats. The flow-cytometric analysis demonstrated that all three variants with different linker lengths exhibited efficient masking before proteolysis as well as HER2 binding after proteolysis (Fig. 3a). Interestingly, the construct with the shortest linker (G4S-PS-G4S) showed the highest fluorescence signal after activation by proteolysis (Fig. 3a). Based on this result and that a smaller protein with a shorter linker might be more favorable in terms of tissue penetration, serum stability and developability, the pro-Affibody with the shortest linker was chosen for further experiments.
Analysis of protease substrates
In a previous study, a panel of different substrate peptides was analyzed in terms of proteolytic hydrolysis by MMP-1 [31]. According to their published ranking, we used one of the most optimal and specific substrates (GPQA#IAGQ; # indicates the bond that is cleaved) in our pro-Affibody. To verify that activation of the pro-Affibody correlated with the previously published data, an alternate substrate sequence that was reported less efficient was engineered into the linker. The pro-Affibody with the alternate substrate (GPQG#IAGQ) was subcloned into the staphylococcal vector and displayed on bacteria. The analysis demonstrated that the substitution in the P1 position of the substrate peptide decreased the proteolytic activation of the pro-Affibody and the HER2-binding signal intensity was around 3-fold lower than for the original substrate peptide (Fig. 3b). The results hence indicated that ranking of protease substrates conducted with other published methods might be predictive for the efficiency of proteolytic activation of the pro-Affibody.
Quantification of masking and activation
The experiments above demonstrated that the pro-Affibody was efficiently masked from binding to HER2 and that specific proteolysis by MMP-1 activated the targeting domain.
To quantify the masking and activation we sought to determine the affinity of non-activated and activated pro-Affibody for HER2 using the bacterial display assay [32]. HER2-binding was measured in the flow cytometer before and after proteolysis at varying HER2 concentrations. Protease-activated pro-Affibody demonstrated an apparent affinity in the single-digit nanomolar range, while non-activated pro-Affibody was efficiently masked even at a HER2 concentration of 1 μM. Extrapolation of the titration suggested that there was at least a 1,000-fold increase in apparent binding affinity after exposure to MMP-1 (Fig. 3d).
Subcloning, production and purification of soluble pro-Affibodies
The bacterial display assay was an efficient method for analysis of pro-Affibodies and the results demonstrated that the prodrug approach was promising. To verify the results obtained with surface-displayed pro-Affibodies, we subcloned the optimal variant to an expression vector for production of soluble recombinant protein. The protein was expressed in fusion to a His-tag in E. coli and purified using IMAC. SDS-PAGE of the soluble pro-Affibody demonstrated high purity with no detectable contaminants and the correct size (Fig. 4a). The pro-Affibody was thereafter treated with MMP-1 and analyzed by SDS-PAGE, revealing two bands of expected sizes, demonstrating specific hydrolysis of the substrate peptide within the soluble recombinant protein (Fig. 4a).
Fig. 4.

Characterization of soluble non-activated and protease activated pro-Affibody. a Representative SDS-PAGE of non-activated (1) and activated pro-Affibody (2). M represents molecular weight marker with indicated sizes in kDa. Protein production, purification, enzymatic hydrolysis and SDS-PAGE analysis was performed in duplicates on different days. b Representative sensorgram from Biacore experiments with HER2 immobilized on the chip surface. The response from 100 nM of the activated (green) and non-activated pro-Affibody (red) was compared to the HER2-binding Affibody molecule (ZHER2) (blue) and the two non-fused monomeric domains of the pro-Affibody molecule mixed in an equimolar ratio (black). c Representative sensorgram from Biacore experiments with HER2 immobilized on the chip surface. The response from 100, 300 and 900 nM of the activated (green) and non-activated pro-Affibody (red). All biosensor experiments were performed at least in duplicates using freshly prepared samples and reagents
Characterization of the pro-Affibody with Surface plasmon resonance
To investigate the masking and activation of the pro-Affibody in more detail, we used an SPR-based biosensor assay. Protease-activated and non-activated pro-Affibody molecules were injected over a sensor chip with HER2 immobilized on the surface (Fig. 4b). The original monomeric ZHER2 was also included in the experiment for comparison. The activated pro-Affibody showed a high response and a slow dissociation after the injection, while the non-activated pro-Affibody resulted in a very low signal despite a relatively high injected concentration (100 nM) (Fig. 4b). When comparing the activated pro-Affibody to the original HER2 binder (ZHER2), the sensorgrams were almost identical (Fig. 4b). However, the activated pro-Affibody had a somewhat decreased apparent on-rate. This is likely due to the presence of the released blocking domain in the solution, which slightly reduced the active concentration of ZHER2. In a control experiment, we therefore injected a mix of the two monomeric moieties (ZHER2 and anti-ZHER2) in equimolar concentrations, and the obtained sensorgram was essentially identical to that obtained for the activated pro-Affibody, confirming the hypothesis (Fig. 4b).
To be able to study any potential weak interaction of non-activated pro-Affibody with the receptor, the concentration was increased up to 900 nM. Both activated and non-activated pro-Affibody molecules were injected over the sensor chip with HER2 immobilized on the surface (Fig. 4c). The activated pro-Affibody was saturated already at 100 nM and resulted in similar responses at 300 and 900 nM. The non-activated pro-Affibody demonstrated binding at the higher concentrations, although much weaker compared to the activated variant. Please note that the non-activated pro-Affibody has roughly twice the molecular weight compared to the activated variant, resulting in a higher response per bound molecule in the assay (i.e., double theoretical R max). Importantly, the results showed that it was mainly the association that was dramatically reduced by the masking of the pro-Affibody, whereas the off-rate was basically unaffected as in theory (Fig. 4c).
Analysis of cancer cell targeting of pro-Affibody
The results from the bacterial display assays and the SPR experiments demonstrated that the pro-Affibody was able to bind to the extracellular domain of recombinantly produced HER2. For future tumor-targeting studies in vivo, we also wanted to verify binding to native HER2 on cancer cells. Flow cytometry was therefore employed to assess the ability of activated and non-activated pro-Affibody to bind to HER2-overexpressing SKOV3 cells (Fig. 5a). The pro-Affibody was treated with MMP-1 or buffer prior to incubation with the cells and the samples were thereafter analyzed in the flow cytometer. The results from the experiment showed a large increase in fluorescence intensity for cells incubated with the activated pro-Affibody compared to cells incubated with the non-activated pro-Affibody, confirming that both the masking and the activation worked as intended and that the pro-Affibody was also able to target native HER2 on cancer cells (Fig. 5b).
Fig. 5.
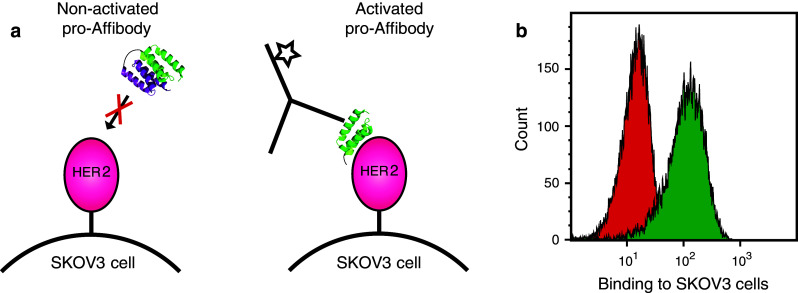
Flow cytometric analysis of cancer cell binding of protease activated pro-Affibody. a The pro-Affibody was treated with MMP1 (activated) or buffer only (non-activated) before HER2-overexpressing SKOV3 cells were labeled with the non-activated or activated pro-Affibody followed by a fluorescently labeled anti-Affibody antibody. b Representative histograms from flow-cytometric analysis of binding to SKOV3 cells of activated (green) and non-activated (red) pro-Affibody. The cancer cell assay was performed in duplicate using freshly prepared samples and reagents
Discussion
Like most affinity proteins (e.g., mAbs), Affibody molecules typically demonstrate very high specificity for their antigens and potential off-target related adverse effects are generally much milder than for small molecular compounds. However, despite their high specificity, affinity proteins can still cause on-target related toxicity considering that many biomarkers are present at low or moderate levels also in healthy tissue. A general strategy often exploited for small molecular drugs is development of prodrugs, which are systemically inert and selectively activated at the site of disease [8]. The maximum tolerated dose is thus often higher for a successful prodrug, which results in a wider therapeutic window. Hopefully, this leads in turn to more effective treatments and that a broader range of targets become druggable.
In this work, we have investigated a new strategy of a prodrug concept for Affibody molecules to potentially improve their tissue specificity for future in vivo studies. We utilized that proteases are often locally upregulated in diverse pathologies [9] and therefore have been used to activate prodrugs [17–20]. The design of the new pro-Affibody was based on a HER2-targeting Affibody molecule (ZHER2) in fusion to an anti-idiotypic Affibody molecule (anti-ZHER2) that specifically binds to the HER2-binding domain. The anti-ZHER2 was originally developed as an affinity ligand for efficient purification of ZHER2 [24]. Here we explored if the anti-idiotypic domain could also mask the HER2-binding activity of ZHER2. By incorporation of a substrate peptide for the cancer-associated protease MMP-1 in the connecting linker, we envisioned that the masking domain might be released upon specific proteolysis, resulting in an Affibody that is inert in circulation and selectively activated in the tumor. This approach would be especially important for targeted payload delivery where even a very low dose might cause toxicity to normal cells.
The data from the analysis of the new pro-Affibody revealed that fusion of the anti-idiotypic domain to the HER2-specific binder indeed resulted in inhibition of the receptor binding activity, thereby also suggesting that the binding sites are at least partially overlapping. We could also show that the prodrug strategy worked as intended, and proteolytic activation of the pro-Affibody increased the apparent binding affinity over 1,000-fold. The theory is that covalent linkage of the masking domain drives the equilibrium towards the blocked state due to the high local concentration of the masking domain. Therefore, a masking domain with a much weaker affinity than that between the target and the targeting domain can be used. Ideally, the masking domain should have a fast off-rate to enable rapid dissociation followed by diffusion upon proteolysis, thereby shifting the equilibrium towards the free and active state. In our pro-Affibody, we used a high affinity (22 pM) HER2-targeting Affibody molecule and a blocking domain with relatively fast off-rate (0.058 s−1). In theory, masking should mainly influence the association of the binding reaction by principally lowering the active concentration of ZHER2. We investigated the kinetics of the interaction with HER2 using SPR and demonstrated that the main effect of masking was indeed a dramatically slower association, while the dissociation was similar as for the activated pro-Affibody. A control pro-Affibody that was composed of the HER2-binder in fusion with a non-binding Affibody was still able to target the receptor, verifying that masking by the anti-idiotypic domain was specific. Moreover, mutations in the substrate peptide resulted in a substantial decrease in activation efficiency, showing that proteolysis was specific to the substrate site. We also evaluated various linkers that differed in length. All tested linkers were suitable for blocking and interestingly; the shortest linker was most efficiently hydrolyzed. If speculating, this could potentially be due to that it is more constrained and therefore will fit better into the substrate-binding pocket of the protease. It should be noted that even though we have engineered a dimeric construct, the overall molecular weight of the pro-Affibody is still very low and the best-performing variant with the shortest linker has a size of merely 16 kDa (i.e., smaller than even one scFv). Importantly, activated pro-Affibody could efficiently target native HER2 on cancer cells in an in vitro assay, whereas the non-activated pro-Affibody remained inert, indicating a potential for the HER2-specific pro-Affibody in future tumor-targeting studies in vivo.
In summary, the study has demonstrated that the new pro-Affibody design is functioning as intended. The anti-idiotypic domain masks the HER2-binding site on the targeting domain and efficiently inhibits the interaction. Proteolytic treatment results in specific cleavage of the substrate peptide followed by dissociation of the blocking domain and activation of the pro-Affibody. Here we used MMP-1 for activation, but which protease and peptide substrate will be optimal for activation in different diseases as well as the stability of the substrate peptide towards endogenous proteases will require extensive investigations. It has, however, been demonstrated that a wide range of proteases are implicated in various disorders and available for such efforts, which increases the applicability of the strategy. We believe that the results obtained here are likely to hold true also for other proteases. Here we have demonstrated the prodrug concept for ZHER2, however, given the straightforward generation of anti-idiotypic Affibody molecules, we believe that the concept could be extended to other Affibody molecules to improve their tissue specificity. The modified bacterial display assay that was developed here greatly facilitated the work in this study and we anticipate that it will be a powerful tool also in the future for development of new pro-Affibody molecules.
Finally, the prodrug approach using anti-idiotypic blocking domains is probably not only efficient for Affibody molecules and might be extended to other small affinity proteins (e.g., Darpins, Knottins, Anticalins, etc.). Moreover, the described bacterial-based assay may become a valuable method for such efforts.
Acknowledgments
The authors wish to thank Stig Linder (Department of Oncology-Pathology, Karolinska Institute, Sweden) and Stefan Ståhl (School of Biotechnology, KTH, Sweden) for scientific advice, Frida Kalm for help with cloning and Hanna Lindberg for help will cell culturing. The Swedish Research council (Vetenskapsrådet, VR) is acknowledged for funding.
Conflict of interest
JL is a member of the Technical Advisory Board at Affibody AB. Other authors declare no financial or commercial conflict of interest.
References
- 1.Strebhardt K, Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat Rev Cancer. 2008;8(6):473–480. doi: 10.1038/nrc2394. [DOI] [PubMed] [Google Scholar]
- 2.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10(5):317–327. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Löfblom J, Frejd FY, Ståhl S. Non-immunoglobulin based protein scaffolds. Curr Opin Biotechnol. 2011 doi: 10.1016/j.copbio.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 4.Ståhl S, Kronqvist N, Jonsson A, Löfblom J. Affinity proteins and their generation. J Chem Technol Biotechnol. 2013;88(1):25–38. doi: 10.1002/jctb.3929. [DOI] [Google Scholar]
- 5.Kamba T, McDonald DM. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br J Cancer. 2007;96(12):1788–1795. doi: 10.1038/sj.bjc.6603813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez EA. Cardiac toxicity of ErbB2-targeted therapies: what do we know? Clin Breast Cancer. 2008;8(Suppl 3):S114–S120. doi: 10.3816/CBC.2008.s.007. [DOI] [PubMed] [Google Scholar]
- 7.Segaert S, Van Cutsem E. Clinical signs, pathophysiology and management of skin toxicity during therapy with epidermal growth factor receptor inhibitors. Ann Oncol Off J Euro Soc Med Oncol/ESMO. 2005;16(9):1425–1433. doi: 10.1093/annonc/mdi279. [DOI] [PubMed] [Google Scholar]
- 8.Huttunen KM, Raunio H, Rautio J. Prodrugs-from serendipity to rational design. Pharmacol Rev. 2011;63(3):750–771. doi: 10.1124/pr.110.003459. [DOI] [PubMed] [Google Scholar]
- 9.Overall CM, Dean RA. Degradomics: systems biology of the protease web. Pleiotropic roles of MMPs in cancer. Cancer Metastasis Rev. 2006;25(1):69–75. doi: 10.1007/s10555-006-7890-0. [DOI] [PubMed] [Google Scholar]
- 10.Duffy MJ, Maguire TM, Hill A, McDermott E, O’Higgins N. Metalloproteinases: role in breast carcinogenesis, invasion and metastasis. Breast Cancer Res BCR. 2000;2(4):252–257. doi: 10.1186/bcr65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boström P, Söderström M, Vahlberg T, Söderström K-O, Roberts PJ, Carpén O, Hirsimäki P. MMP-1 expression has an independent prognostic value in breast cancer. BMC Cancer. 2011;11:348. doi: 10.1186/1471-2407-11-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eck SM, Hoopes PJ, Petrella BL, Coon CI, Brinckerhoff CE. Matrix metalloproteinase-1 promotes breast cancer angiogenesis and osteolysis in a novel in vivo model. Breast Cancer Res Treat. 2009;116(1):79–90. doi: 10.1007/s10549-008-0085-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Köhrmann A, Kammerer U, Kapp M, Dietl J, Anacker J. Expression of matrix metalloproteinases (MMPs) in primary human breast cancer and breast cancer cell lines: new findings and review of the literature. BMC Cancer. 2009;9:188. doi: 10.1186/1471-2407-9-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mannello F. What does matrix metalloproteinase-1 expression in patients with breast cancer really tell us? BMC Med. 2011;9:95. doi: 10.1186/1741-7015-9-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGowan PM, Duffy MJ. Matrix metalloproteinase expression and outcome in patients with breast cancer: analysis of a published database. Ann Oncol Off J Eur Soc Med Oncol/ESMO. 2008;19(9):1566–1572. doi: 10.1093/annonc/mdn180. [DOI] [PubMed] [Google Scholar]
- 16.Park YH, Jung HH, Ahn JS, Im Y-H. Ets-1 upregulates HER2-induced MMP-1 expression in breast cancer cells. Biochem Biophys Res Commun. 2008;377(2):389–394. doi: 10.1016/j.bbrc.2008.09.135. [DOI] [PubMed] [Google Scholar]
- 17.Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EEM, Liang TW, Wong C, Bessette PH, Kamath K, Moore SJ, Sagert JG, Hostetter, Han F, Gee J, Flandez J, Markham K, Nguyen M, Krimm M, Wong KR, Liu S, Daugherty PS, West JW, Lowman HB. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci Trans Med. 2013;5(207):207ra144. doi: 10.1126/scitranslmed.3006682. [DOI] [PubMed] [Google Scholar]
- 18.Erster O, Thomas JM, Hamzah J, Jabaiah AM, Getz JA, Schoep TD, Hall SS, Ruoslahti E, Daugherty PS. Site-specific targeting of antibody activity in vivo mediated by disease-associated proteases. J Control Release Off J Control Release Soc. 2012;161(3):804–812. doi: 10.1016/j.jconrel.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerspach J, Müller D, Münkel S, Selchow O, Nemeth J, Noack M, Petrul H, Menrad A, Wajant H, Pfizenmaier K. Restoration of membrane TNF-like activity by cell surface targeting and matrix metalloproteinase-mediated processing of a TNF prodrug. Cell Death Differ. 2006;13(2):273–284. doi: 10.1038/sj.cdd.4401735. [DOI] [PubMed] [Google Scholar]
- 20.Watermann I, Gerspach J, Lehne M, Seufert J, Schneider B, Pfizenmaier K, Wajant H. Activation of CD95L fusion protein prodrugs by tumor-associated proteases. Cell Death Differ. 2007;14(4):765–774. doi: 10.1038/sj.cdd.4402051. [DOI] [PubMed] [Google Scholar]
- 21.Löfblom J, Feldwisch J, Tolmachev V, Carlsson J, Ståhl S, Frejd FY. Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010;584(12):2670–2680. doi: 10.1016/j.febslet.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 22.Malm M, Kronqvist N, Lindberg H, Gudmundsdotter L, Bass T, Frejd FY, Höidén-Guthenberg I, Varasteh Z, Orlova A, Tolmachev V, Ståhl S, Löfblom J. Inhibiting HER3-mediated tumor cell growth with affibody molecules engineered to low picomolar affinity by position-directed error-prone PCR-like diversification. PLoS One. 2013;8(5):e62791. doi: 10.1371/journal.pone.0062791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orlova A, Magnusson M, Eriksson TLJ, Nilsson M, Larsson B, Höidén-Guthenberg I, Widström C, Carlsson J, Tolmachev V, Ståhl S, Nilsson FY. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 2006;66(8):4339–4348. doi: 10.1158/0008-5472.CAN-05-3521. [DOI] [PubMed] [Google Scholar]
- 24.Wållberg H, Löfdahl P-K, Tschapalda K, Uhlén M, Tolmachev V, Nygren P-K, Ståhl S. Affinity recovery of eight HER2-binding affibody variants using an anti-idiotypic affibody molecule as capture ligand. Protein Expr Purif. 2011;76(1):127–135. doi: 10.1016/j.pep.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 25.Kronqvist N, Löfblom J, Jonsson A, Wernérus H, Ståhl S. A novel affinity protein selection system based on staphylococcal cell surface display and flow cytometry. Protein Eng Des Sel PEDS. 2008;21(4):247–255. doi: 10.1093/protein/gzm090. [DOI] [PubMed] [Google Scholar]
- 26.Kronqvist N, Malm M, Rockberg J, Hjelm B, Uhlén M, Ståhl S, Löfblom J. Staphylococcal surface display in combinatorial protein engineering and epitope mapping of antibodies. Recent Pat Biotechnol. 2010;4(3):171–182. doi: 10.2174/187220810793611536. [DOI] [PubMed] [Google Scholar]
- 27.Kronqvist N, Malm M, Göstring L, Gunneriusson E, Nilsson M, Höidén Guthenberg I, Gedda L, Frejd FY, Ståhl S, Löfblom J. Combining phage and staphylococcal surface display for generation of ErbB3-specific Affibody molecules. Protein Eng Des Sel PEDS. 2011;24(4):385–396. doi: 10.1093/protein/gzq118. [DOI] [PubMed] [Google Scholar]
- 28.Götz F. Staphylococcus carnosus: a new host organism for gene cloning and protein production. Soc Appl Bacteriol Symp Ser. 1990;19:49S–53S. doi: 10.1111/j.1365-2672.1990.tb01797.x. [DOI] [PubMed] [Google Scholar]
- 29.Löfblom J, Kronqvist N, Uhlén M, Ståhl S, Wernérus H. Optimization of electroporation-mediated transformation: Staphylococcus carnosus as model organism. J Appl Microbiol. 2007;102(3):736–747. doi: 10.1111/j.1365-2672.2006.03127.x. [DOI] [PubMed] [Google Scholar]
- 30.Löfblom J. Bacterial display in combinatorial protein engineering. Biotechnol J. 2011 doi: 10.1002/biot.201100129. [DOI] [PubMed] [Google Scholar]
- 31.Clendeninn NJ, Appelt K, editors. Matrix metalloproteinase inhibitors in cancer therapy. New York: Humana Press; 2001. [Google Scholar]
- 32.Löfblom J, Sandberg J, Wernérus H, Ståhl S. Evaluation of staphylococcal cell surface display and flow cytometry for postselectional characterization of affinity proteins in combinatorial protein engineering applications. Appl Environ Microbiol. 2007;73(21):6714–6721. doi: 10.1128/AEM.01432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]