

Abstract
Gram-negative bacteria can produce specific proteinaceous inhibitors to defend themselves against the lytic action of host lysozymes. So far, four different lysozyme inhibitor families have been identified. Here, we report the crystal structure of the Escherichia coli periplasmic lysozyme inhibitor of g-type lysozyme (PliG-Ec) in complex with Atlantic salmon g-type lysozyme (SalG) at a resolution of 0.95 Å, which is exceptionally high for a complex of two proteins. The structure reveals for the first time the mechanism of g-type lysozyme inhibition by the PliG family. The latter contains two specific conserved regions that are essential for its inhibitory activity. The inhibitory complex formation is based on a double ‘key-lock’ mechanism. The first key-lock element is formed by the insertion of two conserved PliG regions into the active site of the lysozyme. The second element is defined by a distinct pocket of PliG accommodating a lysozyme loop. Computational analysis indicates that this pocket represents a suitable site for small molecule binding, which opens an avenue for the development of novel antibacterial agents that suppress the inhibitory activity of PliG.
Keywords: Lysozyme inhibitor, PliG, Lysozyme, Innate immunity, Inhibitory complex, Crystal structure
Introduction
Lysozyme (EC 3.2.1.17) is an important enzyme of the innate immune system. It makes bacteria sensitive to osmotic lysis by degrading peptidoglycan, a vital component of the cell wall. Specifically, lysozyme catalyzes the hydrolysis of the β1-4 glycosidic bond between N-acetyl muramic acid (NAM) and N-acetyl glucosamine (NAG), which constitute the disaccharide building blocks of peptidoglycan polymers. Based on amino acid sequence similarity, lysozymes in the animal kingdom were classified as c-(conventional or chicken), g-(goose) or i-(invertebrate) type [1]. In their substrate binding cleft, there are six consecutive subsites for binding NAG and/or NAM molecules. These subsites are labelled A–F for c-type and i-type lysozymes, and B–G for g-type lysozymes. In all cases, the bond cleavage occurs between the NAM and NAG molecules occupying subsites D and E [2–4]. C-type and i-type lysozymes cleave the glycosidic bond with net retention of the β-configuration of the anomeric carbon (NAM C1). This reaction proceeds via a two-step mechanism, with the formation and subsequent hydrolysis of a covalent glycosyl–enzyme intermediate [2, 5], whereby a glutamate and an aspartate side chain provide acid/base and nucleophilic assistance, respectively [2, 6]. In contrast to c-type and i-type lysozymes, g-type lysozymes cleave the glycosidic bond in a single step and with inversion of the anomeric carbon configuration from β to α [7]. For goose egg-white lysozyme (GEWL), the archetype g-type lysozyme, the conserved Glu73 is essential for the catalytic activity. It is assumed to act as a general acid, donating a proton to the oxygen in the glycosidic bond [8]. Recently, Helland et al. [9] verified that Asp90 and Asp101 of Atlantic cod lysozyme also participate in catalysis. The authors suggested that these residues ensure the proper positioning of a catalytic water molecule for a nucleophilic attack on the NAM C1 atom. Additional experiments showed that Asp101 is more important for the enzymatic activity than Asp90. In other g-type lysozymes, these two residues are highly conserved; the corresponding residues in Atlantic salmon lysozyme (SalG) are Asp86 and Asp97.
Bacteria can become resistant to lysozyme by making chemical adaptations to their peptidoglycan backbone (reviewed by Davis et al. [10]). Another, more recently discovered, bacterial mechanism conferring lysozyme resistance is the production of proteinaceous lysozyme inhibitors (reviewed by Callewaert et al. [11]). To date, four biochemically distinct lysozyme inhibitor families have been identified. These families are distributed exclusively among Gram-negative bacteria, predominantly the Proteobacteria. In 2001, Monchois et al. [12] described the Ivy (inhibitor of vertebrate lysozyme) family. Ivy proteins are strong inhibitors of c-type lysozymes with some additional weak activity against avian g-type lysozymes [13]. However, they do not inhibit g-type lysozyme from fish or the urochordate Oikopleura dioica [14, 15]. Later, Callewaert et al. [16] identified a family of proteins specifically inhibiting c-type lysozymes, named the MliC/PliC (membrane-associated/periplasmic lysozyme inhibitors of c-type lysozyme). Recently, highly specific inhibitors of both g-type and i-type lysozyme were discovered [17, 18]. They were designated as the PliG (periplasmic lysozyme inhibitor of g-type lysozyme) family and the PliI (periplasmic lysozyme inhibitor of i-type lysozyme) family, respectively.
Since lysozyme inhibitors are safeguards of the bacterial cell wall, they are interesting targets for the development of novel antibacterial agents. To this end, it is essential to understand how these inhibitors interact with their cognate lysozyme. For the Ivy family and the MliC/PliC family, structures of lysozyme–inhibitor complexes have been determined by X-ray crystallography [19, 20]. This structural information has been used in a pilot study to design suppressor molecules that bind to Salmonella enterica serotype Typhimurium PliC (PliC-ST) and prevent its interaction with hen egg-white lysozyme [21]. Also, for the PliI family, the possible mode of its interaction with i-type lysozyme was proposed [22]. Recently, we reported the crystal structures of PliG homologues from Escherichia coli (PliG-Ec, PDB ID 4DY3), S. Typhimurium (PliG-ST, PDB ID 4DY5) and Aeromonas hydrophila (PliG-Ah, PDB ID 4DZG). In addition, we used computational tools and mutagenesis experiments to provide insight into the possible binding mode of these inhibitors to the target lysozyme [23].
Here, we present the crystal structure of PliG-Ec in complex with SalG at the ultrahigh resolution of 0.95 Å. The structure gives a detailed account of the molecular mechanism of lysozyme inhibition by the PliG family. In addition, analysis of the interaction interface reveals a suitable target site for the development of PliG suppressor molecules.
Materials and methods
Expression and purification of the PliG-Ec/SalG complex
PliG-Ec was recombinantly produced in E. coli BL21(DE3) and purified as previously described [23]. To produce recombinant SalG, the pQE-2-SalG expression construct [24] was introduced into E. coli BL21(DE3). A single colony was used to inoculate 1 ml LB containing 100 μg/ml ampicillin. After an 8 h incubation at 37 °C, 500 μl of this pre-culture was used to inoculate 1 L ZYP-5052 auto-induction medium containing 100 μg/ml ampicillin and 0.1 % v/v antifoam SE-15 (Sigma). The culture was grown at 24 °C until reaching OD600nm = 4.0. At that point, the temperature was decreased to 18 °C and the culture was allowed to grow for another 24 h. The cells were pelleted by centrifugation, resuspended in IMAC12.5 buffer (50 mM sodium phosphate, 250 mM NaCl, 12.5 mM imidazole pH 7.5) and lysed using an EmulsiFlex-C5 homogenizer (Avestin) followed by sonication for 4 min with 60 % amplitude and 1 s on/off pulses. The lysate was clarified by centrifugation. The supernatant containing the SalG with attached vector-encoded N-terminal MetLys(His)6-tag was loaded on a 3-ml nickel-chelating column (His60 Ni Superflow™ Resin; Clontech) equilibrated with IMAC12.5. The column was washed with 3 column volumes of IMAC12.5 containing 0.1 % v/v Triton X-100 followed by a further 3 column volumes IMAC12.5 without the detergent. SalG was eluted from the column with 10 column volumes of IMAC 250 (50 mM sodium phosphate, 250 mM NaCl, 250 mM imidazole pH 7.5) while collecting 1 ml fractions. The fractions containing SalG were combined and dialysed overnight at 4 °C against 50 mM HEPES pH7.5 and 10 % w/v glycerol. Next, SalG was loaded on a Hitrap SP HP column (GE Healthcare) equilibrated with buffer IEX1 (50 mM HEPES pH 7.5), and a linear gradient of 0–400 mM NaCl (0–40 % of buffer IEX2 containing 50 mM HEPES pH 7.5 and 1 M NaCl) was applied over 20 column volumes. Fractions containing SalG were pooled and concentrated using an Amicon ultra-centrifugation device with a 3-kDa cutoff (Millipore). Finally, SalG was further purified by size-exclusion chromatography on a Superdex 75 pg 16/60 column (GE Healthcare) equilibrated with SEC buffer (10 mM Tris–HCl, 0.5 mM EDTA and 250 mM KCl at pH 7.5) The final PliG-Ec and SalG samples appeared perfectly pure on SDS-PAGE with Coomassie staining. For the isolation of the PliG-Ec/SalG complex, both proteins were mixed in equimolar amounts and applied on the Superdex 75 pg 16/60 column equilibrated with SEC buffer. Finally, the purified complex was concentrated to 14 mg/ml. The protein concentration was determined by measuring absorbance at 280 nm.
Crystallographic analysis
To screen for crystallization conditions, the commercially available kits, JCSG+ Research and PACT (Qiagen) were used. Hits were further optimized using the hanging-drop method in 24-well XRL plates (Molecular Dimensions) by mixing 1 μl protein solution with 1 μl precipitant solution. After optimization of the initial conditions, PliG-Ec/SalG complex crystals up to 1,000 × 200 × 200 μm in size could be obtained in 1–3 days using 1.8 M tri-ammonium citrate, adjusted to pH 7.0 with HCl, as precipitant. The crystals were flash-cooled in liquid nitrogen in mother liquor and diffraction data were collected at the PROXIMA1 beamline (SOLEIL synchrotron, Gif-sur-Yvette, France). Monochromatic X-rays with λ = 0.8 Å were used to collect 1,400 diffraction images (0.05° rotation/0.25 s exposure) using a Pilatus 6 M detector with shutterless operation. In addition, a highly redundant, long-wavelength dataset (λ = 1.8233 Å) was collected from another crystal to calculate an anomalous difference map [25], revealing bound ions as well as protein sulfur atoms. The data were indexed and integrated using XDS [26].
Following the traditionally used criterion ‹I/σ(I)› > 2, the high resolution limit of the collected data would be about 1.05 Å (‹I/σ(I)› = 2.6, R sym = 72.6 % for the highest resolution bin 1.11–1.05 Å). However, as recently shown by Karplus and Diederichs [27], this would exclude a significant amount of useful data. This is why we used a recent version of XSCALE [26] which implements the statistics suggested in [27], arriving at the high resolution limit of 0.95 Å. Indeed, the highest resolution bin (1.0–0.95 Å) has a Pearson’s intra-dataset correlation coefficient of CC1/2 = 0.235 (Table 1). This corresponds to the estimated correlation between the observed data and the underlying noise-free data CC* = [2CC1/2/(1 + CC1/2)]1/2 = 0.62 in the highest bin, which slightly exceeds the suggested cut-off value of 0.5 [27, 28]. The final scaling was done using SCALA [29].
Table 1.
Crystallographic statistics for the SalG/PliG-Ec complex
| Data collection | ||
| Wavelength (Å) | 0.8 | 1.8233f |
| Resolution (Å) | 43.43–0.95 (1.0–0.95) | 43.65–2.51 (2.65–2.51) |
| Space group | P6 5 | P6 5 |
| Cell parameters (Å) | a = 132.18, c = 42.90 | a = 133.36, c = 43.28 |
| CC 1/2 (%)a,b | 0.998 (0.235) | |
| R sym (%)a,c | 7.0 | 10.4 (20.0) |
| R meas (%)a,d | 8.1 | 10.7 (20.6) |
| Average I/σa(I) | 8.2 | 32.8 (15.9) |
| Completeness (%)a | 98.5 (92.4) | 99.4 (96.1) |
| No. of unique reflectionsa | 265542 | 15254 |
| Redundancya | 3.8 (3) | 38.4 (33.4) |
| Wilson B-factor (Å2) | 6.8 | |
| Mosaicity (°) | 0.043 | |
| Refinement | ||
| Number of protein/solvent atoms | 5092/556 | |
| R work (%) | 13.01 | |
| R free (%) | 13.91 | |
| CC a,ework | 0.973 (0.527) | |
| CC a,efree | 0.973 (0.502) | |
| No. of reflections in the ‘free’ set | 2635 | |
| R.m.s. deviations from ideal values | ||
| Bond lengths (Å)/bond angles (°) | 0.007/1.143 | |
| Average protein/solvent B-factor (Å2) | 12.50/24.2 | |
| Ramachandran plot: favoured/outlier residues (%) | 98.8/0 | |
| Molprobity validation: score/percentile | 0.92/98 |
aNumber in parentheses is for the highest resolution shell
b CC 1/2 = Pearson’s intra-dataset correlation coefficient, as described by Karplus and Diederichs [27]. The values were reported by XDS [26]
c
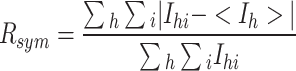 , where I
hi is the ith observation of reflection h and < I
h > is the average intensity of reflection h
, where I
hi is the ith observation of reflection h and < I
h > is the average intensity of reflection h
d
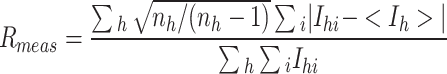 , where n
h is the number of observations of reflection h
, where n
h is the number of observations of reflection h
eCorrelation of experimental intensities with intensities calculated from refined model, as described by Karplus and Diederichs [27]
fLong-wavelength dataset used to calculate the anomalous difference map
The structure was phased by molecular replacement using the structures of SalG (PDB ID 3MGW [30]) and PliG-Ec (PDB ID 4DY3 [23]) as search models in Phaser [31]. Coot [32] and phenix.refine [33] were used in alternating cycles of model building and refinement. Two alternative conformations were observed for the short SalG loop including residue Asp86, correlated with two alternative conformations of PliG-Ec residue Arg115. In addition, a partially occupied chloride ion was identified in a position overlapping with the Asp86 side chain in the inward conformation (see Fig. 4a). Correspondingly, phenix.refine was set to refine occupancies for two groups of atoms: (inward loop conformation + conformation A of Arg115) and (outward loop conformation + conformation B of Arg115 + chloride ion). The quality of the final model was evaluated using MolProbity [34]. All data collection and refinement statistics are shown in Table 1. The PliG-Ec/SalG interface was analyzed using PISA [35]. The crystal structure was submitted to the PDB with accession code 4G9S.
Fig. 4.

Alternative conformations of SalG residue D86 and PliG-Ec residue R115 in the SalG active site. a A 2Fo-Fc electron density map (gray mesh) indicates two possible orientations for both residues, colored in different shades of green and cyan, respectively. An anomalous difference map calculated from the long-wavelength data (yellow mesh) reveals the location of the chloride ion (yellow sphere). b The ‘inward’ conformation of the loop containing D86 (45 % probability) occurs in the absence of the chloride ion, with the D86 residue interacting with Yr47, R115, and R119 of PliG-Ec. The black dashed lines mark hydrogen bonding and ionic interactions. c The ‘outward’ conformation of the loop and the D86 side chain (55 % probability) is stabilized by the chloride ion binding
Small angle X-ray scattering (SAXS) measurements
SAXS data were collected on the European Molecular Biology Laboratory X33 beamline at the DORIS III storage ring (DESY, Hamburg, Germany). SAXS curves were measured over the range of momentum transfer 0.006 < s = 4πsin(θ)/λ < 0.63 Å−1, where 2θ is the total scattering angle and λ = 1.5 Å is the X-ray wavelength. The data for PliG-Ec (1.96, 6.5 and 13.1 mg/ml in 10 mM HEPES, 0.5 mM EDTA and 250 mM KCl at pH 7.5) SalG (0.96, 3.03, 4.97 and 7.82 mg/m in 10 mM Tris–HCl, 0.5 mM EDTA and 250 mM KCl at pH 7.5), the PliG-Ec/SalG complex (1.64, 2.93, 5.37, 9.99 and 13.87 mg/ml in the same buffer as PliG-Ec) and their reference buffers were recorded with eight exposures of 15 s. PRIMUS of the ATSAS program package [36] was used for data processing. To minimize the effect of inter-particle interactions on the scattering intensities, low angle data from low concentration samples were merged with high angle data from the highest concentration samples. The particle distance distribution P(r) was calculated using GNOM [36]. The fits between the experimental scattering curves and the theoretical scattering from atomic models were evaluated using CRYSOL [37].
Results
Structure of the PliG-Ec/SalG complex
SalG and PliG-Ec were recombinantly expressed and purified to homogeneity. After mixing the two proteins in equimolar amounts in 10 mM Tris–HCl buffer, 0.5 mM EDTA and 250 mM KCl (pH 7.5), a stable PliG-Ec/SalG complex was spontaneously formed, as verified using analytical size-exclusion chromatography which produced a single, earlier eluting peak for the complex (Fig. 1a). This observation is in line with the previously established high affinity of PliG-Ec for SalG (K A = 5.81 ± 1.29 × 108 1/M as determined by surface plasmon resonance experiments; [38]). To further characterize the complex formation in solution, we have used SAXS measurements. The scattering curves clearly show that the complex is larger than SalG and PliG-Ec, based on the intensity at zero angle (Fig. 1b). Furthermore, the distance distribution functions calculated from the SAXS curves of the complex and the individual proteins revealed a clear increase in particle size upon complex formation (Fig. 1c).
Fig. 1.

Solution properties of SalG (blue curves), PliG-Ec (red) and their complex (green). a Elution profiles (normalized to the absorbance maximum) of the three samples obtained on an analytical SEC column. b Small-angle X-ray scattering data for the three samples normalized by protein concentration. c Corresponding distance distribution functions. d Fits between the experimental SAXS data (black dots) for the three samples and the theoretical scattering curves (coloured lines) calculated from the corresponding crystal structures (PliG-Ec:PDB ID 4DY3, SalG:PDB ID 3MGW and PliG-Ec/SalG complex: this work). The goodness-of-fit (χ 2) values are reported for each curve. The fits for the three samples were displaced vertically for clarity
Screening and optimization of crystallization conditions yielded high-quality crystals of the PliG-Ec/SalG complex. X-ray diffraction data could be collected up to the ultrahigh resolution of 0.95 Å. The high resolution cut-off was chosen according to the statistical criteria recently proposed in [27]. The asymmetric unit of the crystals reveals a single 1:1 complex of PliG-Ec and SalG (Fig 2a). We found that the complex formation does not involve significant changes in the overall conformation of either protein. Indeed, superimposition of the free PliG-Ec structure (PDB ID 4DY3 [23]) on its equivalent in the complex resulted in the Cα RMSD of 0.57 Å over 107 matched residues (defined as the residues deviating by less than 2 Å upon superposition, as calculated by the MatchMaker tool in Chimera [39]). For the SalG structure (PDB ID 3MGW [30]), the corresponding Cα RMSD was 0.56 Å over 174 matched residues. Furthermore, a theoretical SAXS curve calculated from the crystallographic structure of the complex produced an excellent fit to the experimental SAXS curve, with the goodness-of-fit χ 2 = 0.5 (Fig. 1d). The crystal structure therefore represents the true conformation of the PliG-Ec/SalG complex in solution. The published crystal structures of PliG-Ec and SalG alone also produced excellent fits to the experimental SAXS data (Fig. 1d).
Fig. 2.

Two conserved regions of PliG-Ec interact with the SalG active site. a Stereo view of SalG (green) and PliG-Ec (cyan) in the complex. PliG-Ec residues Y47 in loop 2 of as well as residues R115 and R119 in helix 1 (red labels) interact with E73, D86 and D97 (purple labels) in the active site of SalG. These residues are located in two highly conserved regions (indicated by red boxes in b). Another conserved region of PliG-Ec (marked with orange here and in b) contains the SGxY sequence motif shown to be important for lysozyme inhibition in the PliC/MliC and PliI families. The SalG loop containing residue D86 is shown here in the ‘inward’ conformation (see Fig. 4 for more detail). b Multiple sequence alignment of PliG homologues. Ec, E. coli (NP_287417.1), ST S. Typhimurium (ACY87940.1), Ss Shigella sonnei (YP_310126.1), Et Edwardsiella tarda (YP_003296035.1), P Photobacterium sp. SKA34 (ZP_01159524.1), Va Vibrio angustum (ZP_01234798.1), D Desulfovibrio sp. FW1012B (ZP_06368489.1), Ba B. avium (YP_785764.1), Ah A. hydrophila (YP_854659.1). Red asterisks mark all PliG-Ec residues that are located on the SalG/PliG-Ec interface. The alignment was created with STRAP [48]
Inhibitory mechanism of PliG
The structure of the complex (Fig. 2a) readily reveals that the inhibitory action of PliG is due to the blocking of half of the substrate binding site of the lysozyme. While SalG has only been crystallized in the absence of substrate, six binding subsites, denoted B–G, can be located by superimposing the SalG coordinates from the complex with those of the goose egg-white lysozyme (PDB ID 153L [40]) and Atlantic cod lysozyme (PDB ID 3GXR [9]) structures that have been determined with bound NAG molecules (Fig. 3a). Active SalG processes the polysaccharide substrate by cleaving the glycosidic bond between NAM and NAG moieties located in subsites D and E respectively. As seen from the PliG-Ec/SalG complex structure, once the inhibitor is bound, the subsites B–D are no longer accessible to substrate (Fig. 3b).
Fig. 3.

PliG-Ec blocks half of the substrate binding site on SalG. a Location of the substrate binding subsites in a distinct cleft of the SalG structure shown in surface representation (green). Here, the NAG molecules (orange) were positioned in the SalG structure by superposing the crystal structures of goose egg-white lysozyme including NAG in subsites B–D (PDB ID 153L) and of Atlantic cod lysozyme including NAG in subsites B–C and E–G (PDB ID 3GXR). b The structure of the PliG/SalG complex, with the molecular surfaces of both components shown. Binding of PliG-Ec (cyan) blocks the subsites B–D on SalG
The PliG-Ec/SalG binding interface measures 1,200 Å2, which involves 13.5 and 18 % of the total surface areas of SalG and PliG-Ec, respectively. Detailed analysis of the interface indicates a double ‘key-lock’ type of interaction. The first key-lock element is formed by the insertion of residues in loop 2 and helix 1 (key) into the SalG active site (lock) (Fig. 2a). A multiple sequence alignment of PliG homologues shows that the amino acids in these two regions (residue numbers 46–48 and 115–120, PliG-Ec numbering) are highly conserved (Fig. 2b). Interestingly, the high quality electron density map obtained (Fig. 4a) permits the delineation of two alternative conformations for PliG-Ec Arg115, correlated with two conformations, inward and outward, for the SalG active site loop containing Asp86. The switch between these conformations appears to depend on the presence (55 % probability) of a bound chloride ion most likely originating from the purification buffer. When the chloride atom is absent (Fig 4b), the SalG loop containing Asp86 is folded inwards, allowing PliG-Ec residues Tyr47 (on loop 2), Arg115, and Arg119 (on helix 1) to interact with Asp86 through a hydrogen bond and ionic interactions, respectively, while the Arg115 residue of PliG-Ec makes a salt bridge with the catalytic Glu73 of SalG. However, when the chloride atom is present (Fig. 4c), its negative charge forces Asp86 to fold outwards, away from PliG-Ec Tyr47, Arg115, and Arg119. While in this new conformation, the salt bridge between PliG-Ec Arg115 and SalG Glu73 is retained, the proximal part of the Arg115 side chain is now flipped so that it coordinates the chloride ion together with the main-chain nitrogens of SalG residues 87–90. In addition, Arg119 makes a hydrogen bond with SalG Thr165 in both conformations (Fig. 4b, c).
A pocket on the PliG surface can serve as a target site for the development of suppressor compounds
The second ‘key-lock’ element responsible for the complex formation is the insertion of SalG loop 6 (‘key’) into a distinct pocket (‘lock’) on the PliG-Ec surface (Fig. 5a). The pocket has a considerable volume of 986 Å3. Two adjacent residues, Arg99 and Tyr100 located in SalG loop 6, fit into the PliG-Ec pocket and form hydrophobic interactions and hydrogen bonds with several residues lining it, as described in detail in the legend to Fig. 5b. This strongly suggests that blocking the pocket with a small molecule could be a way to suppress the inhibitory activity of PliG-Ec, as this would render the binding of g-type lysozyme impossible due to steric hindrance. To examine whether this particular PliG-Ec pocket is ‘druggable’, we used fpocket, a computational tool that detects pockets on a protein surface and evaluates their suitability for drug binding based on the hydrophobic density, hydrophobicity, and polarity of the pocket residues [41]. The fpocket analysis, performed on the free PliG-Ec structure (PDB ID 4DY3 [23]), reported a ‘druggability score’ of 0.55 for the discussed PliG-Ec pocket, which is above the suggested threshold of 0.5. We therefore conclude that this pocket indeed represents a promising target site for the development of PliG suppressor compounds.
Fig. 5.

Candidate binding site for PliG suppressor compounds. a In the crystallographic complex, SalG (green) is shown in cartoon representation while PliG-Ec (cyan) is shown in surface representation. Residues R99 and Y100 (shown as yellow sticks) of SalG loop 6 insert into a pocket (gray) on the PliG-Ec surface. The catalytic side chains E73, D86, and D97 of the lysozyme are shown in magenta, while the PliG-Ec residues R115 and R119 are in red (compare with Fig. 2a). b Detailed view of the interactions in the pocket, shown in stereo. Both proteins are shown in cartoon representation with the same colors as in (a). The main-chain carbonyl oxygen of SalG R99 is H-bonded to the side chain of PliG-Ec R110, while the side chain of SalG R99 is H-bonded to the main chain carbonyl atoms of both PliG-Ec Y47 and Q113. Y100 makes hydrophobic interactions with PliG-Ec Y74 and L112 through its ring atoms, while its hydroxyl group makes an H-bond with PliG-Ec D72, and its main chain carbonyl oxygen makes an H-bond with the hydroxyl group of the S83 side chain
Discussion
Here, we report the crystal structure of SalG in complex with PliG-Ec, which is the first to provide insights into the inhibitory mechanism of the PliG family in atomic detail.
The achieved crystallographic resolution of 0.95 Å is exceptionally high for a protein–protein complex. This may be in part explained by the high ordering of the crystal lattice of the PliG-Ec/SalG complex, as manifested by a very low crystal mosaicity value of 0.043°. The crystal lattice contacts are extensive and involve both the inhibitor and the lysozyme. Surprisingly, however, the crystal packing is far from being dense and reveals large solvent channels, with a solvent content of 60 % and a relatively high Matthews coefficient of 3.07 Å3/Da. In fact, about 95 % of all protein crystals diffracting to 0.54–1.2 Å resolution display a denser packing [42]. To the best of our knowledge, only one protein–protein complex available in the PDB diffracted slightly better, namely the complex between bovine trypsin and the Schistocerca gregaria protease inhibitor 1 (PDB ID 2XTT, resolution 0.93 Å [43]). Unlike our PliG-Ec/SalG complex, this trypsin complex displayed the typical dense packing (35 % solvent and a Matthews coefficient of 1.89 Å3/Da). Interestingly, the resolution of the PliG-Ec/SalG complex crystals is much higher than the resolution obtained for its components separately as SalG crystals diffracted to 1.75 Å resolution [30], and PliG-Ec crystals diffracted to 1.25 Å [23]. Moreover, the best resolution reported so far for any g-type lysozyme is only 1.6 Å (goose egg-white lysozyme, PDB ID 153L [40]).
The PliG family shares little overall sequence similarity with the MliC/PliC and the PliI lysozyme inhibitor families, but all three families have a conserved region containing an SGx(x)Y sequence motif [38, 44]. For both the MliC/PliC and the PliI families, the SGx(x)Y motif was shown to be important for the inhibition of their target lysozymes [20, 22]. However, we recently discovered that, in the PliG family, the SGxY motif is located in the beginning of strand β8 (Fig. 2a, b) and is not involved in the g-type lysozyme inhibition [23]. Using a combination of computational analysis and site-directed mutagenesis experiments, we demonstrated that in this case the inhibition critically depends on two different highly conserved regions of PliG-Ec, loop 2 and helix 1. Indeed, a triple PliG-Ec mutant with alanine substitutions for residues Tyr47 (loop 2), Arg115, and Arg119 (helix 1) only retained 8 % of inhibitory activity compared to the wild-type protein [23]. Our crystal structure of the PliG-Ec/SalG complex provides an ultimate support to these observations, revealing that the residues Tyr47, Arg115, and Arg119 directly interact with the active site of g-type lysozyme (Figs. 2, 4). In particular, Arg115 makes a salt bridge with the catalytic Glu73 on SalG, while all three residues make hydrogen bonds and salt bridges with Asp86 of SalG. Moreover, the high resolution of the crystal structure allows us to delineate two possible conformations, inward and outward, for the loop containing SalG Asp86 in the complex. Interestingly, the inward conformation of the loop and Asp86 residue was also seen for the crystal structure of the free SalG (PDB ID 3MGW [30]), while the outward conformation was present in the goose egg-white lysozyme structure (PDB entry 153L [40]). These observations support the view of Helland et al. [9] that the residue Asp86 is flexible and should contribute less than Asp97 to the proper positioning of the nucleophilic water molecule during the catalytic reaction of g-type lysozyme.
Importantly, we found that, apart from the insertion of PliG-Ec loop 2 and helix 1 into the SalG active site, the formation of the inhibitory complex involves the insertion of SalG loop 6 into a distinct, 986 Å3 large pocket on the PliG-Ec surface (Fig. 5). Specifically, the pocket accommodates the bulky side chains of SalG Tyr99 and Arg100. This way, a double ‘key-lock’ type interface is formed with a pronounced shape-complementarity between the lysozyme and the inhibitor. A similar double ‘key-lock’ interface was observed for the complex of hen egg white lysozyme (HEWL, c-type) with P. auruginosa MliC (MliC-Pa) [20]. However, compared to PliG-Ec, MliC-Pa features a smaller (300 Å3) and rather shallow pocket, accommodating only a single, small side chain (Thr47) of HEWL.
Since lysozyme inhibitors impair the bacteriolytic action of lysozyme in the innate immune response, they are interesting targets for the development of novel antibacterial drugs. Recently, Voet et al. [21] provided ‘proof of concept’ thereof by identifying compounds that interfere with the binding of S. typhimurium PliC (PliC-ST) to HEWL, thus suppressing its inhibitory activity. The molecules were selected in silico to occupy the shallow pocket on the PliC-ST surface which accommodates the HEWL loop carrying Thr47. Correspondingly, our crystal structure of the PliG-Ec/SalG complex provides an excellent starting point for the future development of PliG suppressor compounds, as computational analysis suggests that the PliG-Ec pocket accommodating SalG residues Tyr99 and Arg100 is indeed druggable. In silico selection of such compounds can now be based on a pharmacophore model mimicking the interactions of the latter residues with PliG-Ec (Fig 5b). In humans, PliG suppressor compounds could potentially be used for the treatment of eye infections caused by PliG-producing Gram-negative bacteria. Indeed, Huang et al. [45] recently showed that a g-type lysozyme, HLysG2, is produced in the lacrimal gland. Also, in fish and some bird species, g-type lysozyme is a key component of the innate immune system. Therefore, PliG suppressor compounds could also be of use in important industries such as fish and poultry farming. Nowadays, the use of antibiotics in these industries poses a concern for public health since it promotes the emergence of antibiotic-resistant bacteria [46, 47]. Here, the development of PliG suppressor compounds could provide a valuable alternative.
Acknowledgments
Access to the synchrotron beamlines PROXIMA1 at Soleil and X33 at the Deutsches Elektronen-Synchrotron is gratefully acknowledged, with a special ‘thank you’ to Dr. Andrew Thompson for help with ultrahigh-resolution data collection. We also thank Dr. Pavel Afonine for advice on crystallographic refinement. S. Leysen holds a doctoral grant from the K.U. Leuven. L. Vanderkelen holds a doctoral fellowship from the Flemish Institute for the Promotion of Scientific Technological Research (IWT). This work was further financially supported by a Research Grant (G.0363.08) from the Research Foundation-Flanders (F.W.O.-Vlaanderen) and from the KU Leuven Research Fund (research project METH/07/03).
References
- 1.Callewaert L, Michiels CW. Lysozymes in the animal kingdom. J Biosci. 2010;35(1):127–160. doi: 10.1007/s12038-010-0015-5. [DOI] [PubMed] [Google Scholar]
- 2.Goto T, et al. Crystal structure of Tapes japonica Lysozyme with substrate analogue: structural basis of the catalytic mechanism and manifestation of its chitinase activity accompanied by quaternary structural change. J Biol Chem. 2007;282(37):27459–27467. doi: 10.1074/jbc.M704555200. [DOI] [PubMed] [Google Scholar]
- 3.Holler E, Rupley JA, Hess GP. Productive and unproductive lysozyme–chitosaccharide complexes. Equilibrium measurements. Biochemistry. 1975;14(5):1088–1094. doi: 10.1021/bi00676a032. [DOI] [PubMed] [Google Scholar]
- 4.Honda Y, Fukamizo T. Substrate binding subsites of chitinase from barley seeds and lysozyme from goose egg white. Biochim Biophys Acta. 1998;1388(1):53–65. doi: 10.1016/S0167-4838(98)00153-8. [DOI] [PubMed] [Google Scholar]
- 5.Vocadlo DJ, et al. Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature. 2001;412(6849):835–838. doi: 10.1038/35090602. [DOI] [PubMed] [Google Scholar]
- 6.Malcolm BA, et al. Site-directed mutagenesis of the catalytic residues Asp-52 and Glu-35 of chicken egg white lysozyme. Proc Natl Acad Sci USA. 1989;86(1):133–137. doi: 10.1073/pnas.86.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuroki R, Weaver LH, Matthews BW. Structural basis of the conversion of T4 lysozyme into a transglycosidase by reengineering the active site. Proc Natl Acad Sci USA. 1999;96(16):8949–8954. doi: 10.1073/pnas.96.16.8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawamura S, et al. Experimental verification of the crucial roles of Glu73 in the catalytic activity and structural stability of goose type lysozyme. J Biochem. 2006;140(1):75–85. doi: 10.1093/jb/mvj125. [DOI] [PubMed] [Google Scholar]
- 9.Helland R, et al. Crystal structures of g-type lysozyme from Atlantic cod shed new light on substrate binding and the catalytic mechanism. Cell Mol Life Sci. 2009;66(15):2585–2598. doi: 10.1007/s00018-009-0063-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davis KM, Weiser JN. Modifications to the peptidoglycan backbone help bacteria to establish infection. Infect Immun. 2011;79(2):562–570. doi: 10.1128/IAI.00651-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Callewaert L, et al. Guards of the great wall: bacterial lysozyme inhibitors. Trends Microbiol. 2012;20(10):501–510. doi: 10.1016/j.tim.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 12.Monchois V, et al. Escherichia coli ykfE ORFan gene encodes a potent inhibitor of C-type lysozyme . J Biol Chem. 2001;276(21):18437–18441. doi: 10.1074/jbc.M010297200. [DOI] [PubMed] [Google Scholar]
- 13.Callewaert L, et al. Purification of Ivy, a lysozyme inhibitor from Escherichia coli, and characterisation of its specificity for various lysozymes. Enzyme Microb Technol. 2005;37:205–211. doi: 10.1016/j.enzmictec.2005.03.001. [DOI] [Google Scholar]
- 14.Nilsen IW, et al. Urochordates carry multiple genes for goose-type lysozyme and no genes for chicken- or invertebrate-type lysozymes. Cell Mol Life Sci. 2003;60(10):2210–2218. doi: 10.1007/s00018-003-3252-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kyomuhendo P, et al. Structural evidence for lack of inhibition of fish goose-type lysozymes by a bacterial inhibitor of lysozyme. J Mol Model. 2008;14(9):777–788. doi: 10.1007/s00894-008-0317-9. [DOI] [PubMed] [Google Scholar]
- 16.Callewaert L, et al. A new family of lysozyme inhibitors contributing to lysozyme tolerance in gram-negative bacteria. PLoS Pathog. 2008;4(3):e1000019. doi: 10.1371/journal.ppat.1000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Herreweghe JM, et al. Lysozyme inhibitor conferring bacterial tolerance to invertebrate type lysozyme. Cell Mol Life Sci. 2010;67:1177–1188. doi: 10.1007/s00018-009-0241-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanderkelen L, et al. Identification of a bacterial inhibitor against g-type lysozyme. Cell Mol Life Sci CMLS. 2011;68(6):1053–1064. doi: 10.1007/s00018-010-0507-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abergel C, et al. Structure and evolution of the Ivy protein family, unexpected lysozyme inhibitors in Gram-negative bacteria. Proc Natl Acad Sci USA. 2007;104(15):6394–6399. doi: 10.1073/pnas.0611019104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yum S, et al. Structural basis for the recognition of lysozyme by MliC, a periplasmic lysozyme inhibitor in Gram-negative bacteria. Biochem Biophys Res Commun. 2009;378(2):244–248. doi: 10.1016/j.bbrc.2008.11.039. [DOI] [PubMed] [Google Scholar]
- 21.Voet A, et al. Structure based discovery of small molecule suppressors targeting bacterial lysozyme inhibitors. Biochem Biophys Res Commun. 2011;405(4):527–532. doi: 10.1016/j.bbrc.2011.01.053. [DOI] [PubMed] [Google Scholar]
- 22.Leysen S, et al. Molecular basis of bacterial defense against host lysozymes: X-ray structures of periplasmic lysozyme inhibitors PliI and PliC. J Mol Biol. 2011;405(5):1233–1245. doi: 10.1016/j.jmb.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 23.Leysen S, et al. Structural characterization of the PliG lysozyme inhibitor family. J Struct Biol. 2012;180(1):235–242. doi: 10.1016/j.jsb.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 24.Kyomuhendo P, Myrnes B, Nilsen IW. A cold-active salmon goose-type lysozyme with high heat tolerance. Cell Mol Life Sci. 2007;64(21):2841–2847. doi: 10.1007/s00018-007-7372-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mueller-Dieckmann C, et al. On the routine use of soft X-rays in macromolecular crystallography. Part IV. Efficient determination of anomalous substructures in biomacromolecules using longer X-ray wavelengths. Acta Crystallogr D Biol Crystallogr. 2007;63(Pt 3):366–380. doi: 10.1107/S0907444906055624. [DOI] [PubMed] [Google Scholar]
- 26.Kabsch W (2010) XDS. Acta Crystallogr D Biol Crystallogr 66(Pt 2): 125–32 [DOI] [PMC free article] [PubMed]
- 27.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336(6084):1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333(4):721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 29.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 1):72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 30.Kyomuhendo P, et al. Thermodynamics and structure of a salmon cold active goose-type lysozyme. Comp Biochem Physiol B Biochem Mol Biol. 2010;156(4):254–263. doi: 10.1016/j.cbpb.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 31.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallographica D, Biol Crystallogr 60(Pt 12 Pt 1):2126–32 [DOI] [PubMed]
- 33.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis IW, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35(Web Server issue):W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 36.Petoukhov MV, et al. ATSAS 2.1—towards automated and web-supported small-angle scattering data analysis. J Appl Crystallogr. 2007;40(s1):s223–s228. [Google Scholar]
- 37.Svergun D, Barberato C, Koch MHJ. CRYSOL—a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J Appl Crystallogr. 1995;28(6):768–773. doi: 10.1107/S0021889895007047. [DOI] [Google Scholar]
- 38.Vanderkelen L, et al. Identification of a bacterial inhibitor against g-type lysozyme. Cell Mol Life Sci. 2011;68(6):1053–1064. doi: 10.1007/s00018-010-0507-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 40.Weaver LH, Grutter MG, Matthews BW. The refined structures of goose lysozyme and its complex with a bound trisaccharide show that the “goose-type” lysozymes lack a catalytic aspartate residue. J Mol Biol. 1995;245(1):54–68. doi: 10.1016/S0022-2836(95)80038-7. [DOI] [PubMed] [Google Scholar]
- 41.Schmidtke P, Barril X. Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J Med Chem. 2010;53(15):5858–5867. doi: 10.1021/jm100574m. [DOI] [PubMed] [Google Scholar]
- 42.Kantardjieff KA, Rupp B. Matthews coefficient probabilities: improved estimates for unit cell contents of proteins, DNA, and protein-nucleic acid complex crystals. Protein Sci. 2003;12(9):1865–1871. doi: 10.1110/ps.0350503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wahlgren WY, et al. The catalytic aspartate is protonated in the Michaelis complex formed between trypsin and an in vitro evolved substrate-like inhibitor: a refined mechanism of serine protease action. J Biol Chem. 2011;286(5):3587–3596. doi: 10.1074/jbc.M110.161604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Herreweghe JM, et al. Lysozyme inhibitor conferring bacterial tolerance to invertebrate type lysozyme. Cell Mol Life Sci. 2010;67(7):1177–1188. doi: 10.1007/s00018-009-0241-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang P, et al. Characterization and expression of HLysG2, a basic goose-type lysozyme from the human eye and testis. Mol Immunol. 2011;48(4):524–531. doi: 10.1016/j.molimm.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 46.Angulo FJ, Nargund VN, Chiller TC. Evidence of an association between use of anti-microbial agents in food animals and anti-microbial resistance among bacteria isolated from humans and the human health consequences of such resistance. J Vet Med B Infect Dis Vet Public Health. 2004;51(8–9):374–379. doi: 10.1111/j.1439-0450.2004.00789.x. [DOI] [PubMed] [Google Scholar]
- 47.Cabello FC. Heavy use of prophylactic antibiotics in aquaculture: a growing problem for human and animal health and for the environment. Environ Microbiol. 2006;8(7):1137–1144. doi: 10.1111/j.1462-2920.2006.01054.x. [DOI] [PubMed] [Google Scholar]
- 48.Gille C, Frommel C. STRAP: editor for STRuctural alignments of proteins. Bioinformatics. 2001;17(4):377–378. doi: 10.1093/bioinformatics/17.4.377. [DOI] [PubMed] [Google Scholar]