

Abstract
Pentameric ligand-gated ion channel (pLGIC) receptors exhibit desensitization, the progressive reduction in ionic flux in the prolonged presence of agonist. Despite its pathophysiological importance and the fact that it was first described over half a century ago, surprisingly little is known about the structural basis of desensitization in this receptor family. Here, we explain how desensitization is defined using functional criteria. We then review recent progress into reconciling the structural and functional basis of this phenomenon. The extracellular–transmembrane domain interface is a key locus. Activation is well known to involve conformational changes at this interface, and several lines of evidence suggest that desensitization involves a distinct conformational change here that is incompatible with activation. However, major questions remain unresolved, including the structural basis of the desensitization-induced agonist affinity increase and the mechanism of pore closure during desensitization.
Keywords: Single channel kinetics, Ligand-gated ion channel, Acetylcholine, Glycine, GABA, 5-HT, Electrophysiology
Introduction
The nicotinic acetylcholine receptor (nAChR) cation channel, which mediates neuromuscular transmission and excitatory synaptic neurotransmission, has been the standard model ligand-gated receptor since it was first characterized by Bernard Katz and colleagues in the 1950s. In 1957, Del Castillo and Katz [1] first proposed the two-state model of receptor activation, which essentially posited structurally distinct steps for ligand binding and channel opening. This was closely followed by a study in which Katz and Thesleff [2] attempted to describe the process of desensitization as observed at the frog endplate in response to acetylcholine application. This extended the previous model by proposing that the nAChR could exist in the resting shut, liganded open, and desensitized shut states. Acetylcholine binding to a resting shut channel initially induces the nAChR to open, but prolonged acetylcholine exposure induces the desensitized shut state. Then, following dissociation of acetylcholine, the channel returns from the desensitized state directly to the resting shut state. Using this simple model as a starting point, researchers over the past 50 years have generated more and more complex and precise kinetic models to describe nAChR function [2–5]. Conventionally, the resting shut, open, and desensitized states have been defined on the basis of unique pairings of two salient properties of the channel: their affinity for the activating ligand and their ion-conducting status. Thus, resting shut channels have a low ligand affinity and are non-conducting, open channels have a high ligand affinity and conduct ions, whereas desensitized channels also have a high ligand affinity, but are non-conducting. High to saturating concentrations of ligand increase the likelihood of the channels entering desensitized states over that of liganded shut states. However, channels can nevertheless desensitize in the presence of low ligand concentrations, and even in the absence of ligand (see Fig. 4), albeit with a lower occupancy.
Fig. 4.

A general reaction mechanism describing any ligand-gated ion channel that accommodates the binding of two ligand molecules. Un-, mono- and di-liganded shut (S), open (O), and desensitized (D) configurations are shown. The forward and backward arrows between connected states are associated with corresponding rate constants, which quantify the transition frequencies to and from the states. Only the diliganded opening (β), shutting (α), entry into desensitized (δ), and ligand dissociation (k−1) rate constants are shown
Although members of the extensive pLGIC receptor family are involved in diverse physiological processes, they are best known for mediating most of the fast neurotransmission in the brain. It is often difficult to demonstrate a physiological role for pLGIC desensitization at a specific synapse because the net decay rate of a synaptic current is due to a combination of interlinked factors including the transmitter unbinding rate, the channel shutting rate, the transmitter clearance rate, the intrinsic receptor desensitization rate, and dendritic filtering. However, computational modeling has suggested important roles for desensitization in controlling the magnitude, decay rate, and frequency of synaptic currents elicited by fast repetitive stimulation [6]. Experimentally, it has proved more straightforward to demonstrate a causative relationship between a pathological process and a change in the propensity of a pLGIC receptor to be desensitized. For example, it has been shown that concentrations of nicotine in the brains of cigarette smokers are sufficient to desensitize presynaptic nAChRs on GABAergic inputs onto dopaminergic neurons in the ventral tegmental area, thus reducing GABAergic inhibitory drive and producing a shift towards increased activation of the dopamine reward system [7]. Several neurological disorders are also caused by hereditary mutations that change the ensemble current (i.e., the net current of many channels) desensitization rate of pLGIC receptors. For example, autosomal dominant nocturnal frontal lobe epilepsy is caused by the neuronal nAChR α4-S248F mutation, which yields acetylcholine-evoked currents with amplitudes comparable to those of control responses but with a higher sensitivity and faster desensitization to the endogenous neurotransmitter [8]. Similarly, hereditary mutations to muscle nAChR genes may cause congenital myasthenic syndromes by altering the desensitization rate of motor endplate nAChRs [9]. Finally, hereditary mutations that selectively alter the desensitization rates of GlyRs and GABAARs, respectively, are thought to cause human startle disease [10] and childhood absence epilepsy with febrile seizures [11].
Recent crystal structures of pLGIC receptor isoforms have provided useful insights into the structures of the shut and open states [12–15]. Although several apparently incompatible models have been proposed to account for the shut-to-open structural transition [16], it is reasonable to conclude that we are moving rapidly to the point where the common elements of the pLGIC receptor close–open transition can be discerned from those elements that are specific to particular pLGIC receptor subtypes. Unfortunately, despite the physiological and pathological importance of desensitization in pLGICs [17], almost nothing is known about the conformational changes that mediate this process. However, some important new insights into the mechanisms of desensitization have recently emerged, and this review will attempt to integrate these findings into a coherent model of pLGIC structure and function.
pLGIC receptor structure
pLGICs display a broad range of activation, desensitization, and deactivation kinetics that parallels the size and functionally diversity of the family. This functional variety renders pLGICs an ideal family for studying the general principles of the structure–function relationship of channel desensitization, both on an ensemble current and single channel level. pLGIC receptors conduct either anions or cations and are activated by relatively small ligands. In addition to the nAChR, prominent eukaryotic members of this family include the anion selective GABA type-A receptor (GABAAR), glycine receptor (GlyR), and glutamate receptor (GluClR), and the cation selective serotonin type-3 receptor (5HT3R) and zinc receptor. All pLGICs comprise pentameric arrangements of identical or similar subunits, and have representatives in both eukaryotes and prokaryotes [18]. The five subunits come together to form a central ion permeation pathway, which incorporates a physical barrier (gate) to ion flux [19, 20] located within the narrow transmembrane portion of the pathway (Fig 1a). Each subunit can be divided into three domains, each of which act as semi-independent modular units in the complete pentamer [21, 22] (Fig. 1b). The extracellular domain, which is typically ~220 amino acids long, contains an α-helix near its amino terminal end followed by a series of 10 β-strands. The β-strands are organized into two β-sheets that together form a twisted β-sandwich structure with two hydrophobic cores (Fig. 1b). Ligand binding pockets are formed at the subunit interfaces, by three domains (binding loops A–C) from the ‘+’ side of the interface and three domains (binding ‘loops’ D–F) from the ‘−’ side of the interface. These ligand binding pockets are situated about 5 nm above the gate. The transmembrane domain consists of 20 (4 per subunit) α-helical segments, referred to as M1–M4. These form concentric rings around the central ion pore, which is directly lined by the five M2s. A segment of variable length connecting M3 and M4 (M3–M4 linker) forms the bulk of the intracellular domain. The M3–M4 linker can be as short as three amino acids, as found in some prokaryotic pLGICs [18, 22], to over 180 in some Metazoan subunits, such as in the human α4 subunit of the GABAAR [23].
Fig. 1.

a A top view schematic of a pLGIC showing its five-fold symmetry and the central ion permeation pathway. The five M2 α-helical segments, located within transmembrane domain, line a pore. Each subunit is given a different colour. b A side view schematic of a pLGIC showing two of the three functional domains, the extracellular and transmembrane (demarcated by black horizontal bars). The intracellular domain is not shown. The images were made using the crystal structure of the C. elegans α GluClR 3RIF [5]
Channel activation involves long-range conformational rearrangements that are initiated by the ligand binding reaction [24, 25]. This local change in conformation then propagates in a sequence of sub-domain movements throughout the receptor as a ‘wave’ [25, 26], with the ultimate functional outcome being the disruption of the permeation gate to produce an open, ion-conducting channel. In the continuous presence of high ligand concentrations the channel adopts desensitized configurations that are refractory to further activation. Several domains located at the boundary of extracellular and transmembrane domains communicate conformational changes across this boundary. This network of interacting domains include the C-terminus of β-strand 10 (or the ‘pre-M1’ domain which links the extracellular domain to the M1 transmembrane helix), β1–β2 loop (loop 2), β8–β9 loop (loop 9), β6–β7 loop (loop 7) and the linker connecting M2 to M3 (M2–M3 linker) (Fig. 2) [27, 28]. The interface between transmembrane and intracellular domains has also been shown to transmit structural rearrangements that give rise to functional states, mainly via the M1–M2 linker [10, 29, 30] and the proximal portion of the M3–M4 linker [31–33].
Fig. 2.

a Schematic of a single subunit indicating the extracellular-transmembrane domain and transmembrane-intracellular domain interfaces (arrows). The intracellular domain has been omitted. b A higher magnification view of a single subunit showing the elements of the contributing to the interface between extracellular and transmembrane domains. The elements are; loop 9 (green), loop 2 (light blue), loop 7 (dark blue), pre-M1 (purple) and N terminus of M1 (olive). Also shown are the other transmembrane α helices, including the pore-lining M2 (orange). The images were made using the crystal structure of the C. elegans α GluClR 3RIF [5]
Ensemble and single channel desensitization
Desensitization in pLGICs is manifest in ensemble currents as a decline in the initial peak amplitude, reaching a plateau (steady-state) in the persistent presence of agonist. This phase of the current generally proceeds with a slower time-course than the initial activation phase [2] and represents an equilibrium between channels that enter into desensitized states and those that exit desensitized states and reactivate (Fig. 3a). Ensemble currents represent the time and ligand concentration dependent average behaviour of the entire channel population and are fitted to the sum of exponential terms of the form [34]:
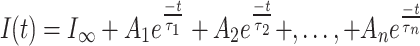 |
1 |
where I(t) is the current at time t, I ∞ is the steady state current, A n is the amplitude of the nth component, τn is the decay time constant of the nth component, and (n + 1) is the number of functional states in the underlying mechanism. Typically, one or two components are required to fit the desensitization phase of the ensemble current [2, 35–38]. Time constants for desensitization determined in this way range between <1 ms, as in the homomeric α7 nAChRs [36], to intermediate values (~100 ms), such as in α1β2γ2 GABAARs [35] (Fig. 3a; Table 1) to channels that show little current decay, such as homomeric GABAARs formed by ρ1 subunits [39, 40] or GlyRs formed by α1 subunits [41]. Perturbations, such as may be caused by particular ligands, mutations,or post-translational mechanisms, are inferred to affect channel desensitization on the basis of altered time constants (or relative amplitudes) of the fitted components. This method of analysis, although widely used, is phenomenological and provides little insight into what might be happening structurally to the channels. The time constants have no physical correlates and do not correspond to any functional states, at least not unless specifically demonstrated to do so within the context of a mechanism [35–38] (see, e.g., Fig. 4).
Fig. 3.
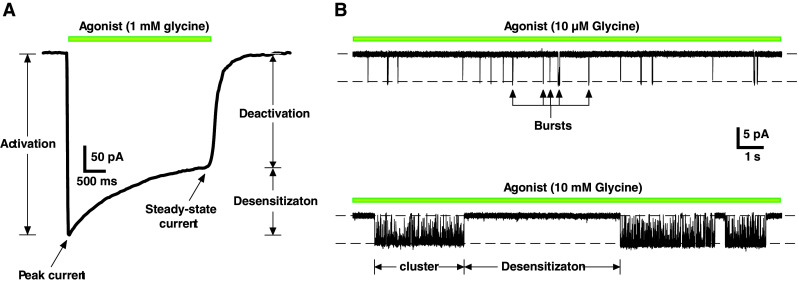
a Whole-cell current recorded from an HEK293 cell expressing homomeric α1 GlyRs showing the phases of an ensemble current in response to a saturating concentration of glycine. b Single channel currents recorded from outside-out patches expressing α1 GlyRs. The records show short bursts of activity at low ligand concentrations (top), which group into clusters at saturating ligand concentrations. The clusters are separated by non-conducting periods where the channel adopts desensitized states (bottom)
Table 1.
Single channel and ensemble channel parameters
| Channela | Single channel parameters | |||
|---|---|---|---|---|
| Opening (β; s−1) | Shutting (α; s−1) | Cluster duration (ms) | Burst duration (ms) | |
| α GlyR | 2–4 × 104 [91, 92] | 0.8–1 × 103 [91] | 1.1 × 103 [92] | 2–7 [91] |
| αβ GlyR | 1.3 × 105 [43, 53] | 7 × 103 [43, 53] | 1.0 × 103 [43] | 6 [43] |
| αβγ GABAAR | 2–3 × 103 [52, 93] | 300–900 [52] | 100–500 [52, 93] | 5–50 [52, 94] |
| Muscle nAChR | 8–10 × 104 [95, 96] | 2–3 × 103 [53] | 50, 250–500 [42, 95] | 2–4 [95] |
| α7 nAChR | – | 700 [36] | 0.35–0.60 [36, 97] | – |
| 5HT3R | – | – | 1.2 × 103 [36] | 700 [36] |
| Channela | Ensemble channel parametersb | |||
|---|---|---|---|---|
| Activationc (s−1) | Desensitizationd (entry; ms) | Desensitizatione (exit; ms) | Deactivationf (ms) | |
| α1 GlyR | 2–4 × 103 [41, 98] | 30, 300, 1.5 × 103 [98, 99] | 0.5–2.0 × 103 [99] | 6–9, 20–50 [41, 99] |
| α1β GlyR | 3 × 103 [99] | 10, 100, 0.5 × 103 [99] | 04–1.4 × 103 [99] | 7, 30 [99] |
| αβγ GABAAR | 1.1–6.0 × 103 [35, 94, 100–103] | 10–50, 100–400, 0.8–10 × 103 [35, 94, 103–106] | 60–500, 13–D25 × 103 [103, 104, 106] | 6–30, 100–300 [35, 94, 100, 104, 106] |
| Muscle nAChR | 6–8 × 104 [96] | 15, 50–70, 1 × 103 [2, 45, 95] | 3–270 × 103 [2, 45] | 1–3 [95] |
| α7 nAChR | 7 × 103 [36] | 0.40 [36] | 1.1 × 103 [36] | – |
| 5HT3AR | 100–400 [38, 75, 77] | 0.8–3 × 103 [36, 38, 75, 77] | 5 × 103 [38] | 770–3 × 103 [38, 77] |
aAll parameters are for wild-type channels, estimated using physiological ligand
bFrom excised macropatches or small cells
cFrom 10–90 % rise-times in response to 1–5 ms pulses of ligand
dRefer to references for relative amplitudes of components
eUsing paired-pulse protocol
fParameters depend on subunit composition
On the level of a single ion channel, the durations of shut and open intervals within a record are also described by exponential distributions, and can be written as [34]:
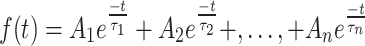 |
2 |
Desensitizing ligand concentrations elicit a distinctive pattern of single channel activity. Fig. 3b shows a comparison between the effects of a low (~EC10) and a high (saturating) glycine concentration at recombinantly expressed homomeric α1 GlyRs. The low concentration induces relatively brief conducting episodes known as bursts (Fig. 3b, top). At saturating concentrations, these bursts coalesce into well-defined clusters that are separated by non-conducting periods, where the channel is fully liganded and adopts desensitized configurations (Fig. 3b, bottom). Cluster lengths vary from <1 ms, as in the homomeric α7 nAChRs, which are so brief they are referred to as ‘bursts’ [36], to ~500 ms in the muscle nAChR [42] to ≥1 s for GlyRs [43, 44] (Table 1; see also Fig. 3b). Shut intervals that correspond to desensitized nAChRs have been estimated to extend up to 5 min [45]. The mean cluster length at high agonist concentrations has been determined to approximate the ensemble current desensitization time constants [36, 46]. This is a reflection of the relative slowness of entry into a desensitized state (e.g., δ in Fig. 4) relative to the channel shutting rate (α), so that the fully liganded channel has the opportunity to oscillate between open and shut configurations for extended periods before entering desensitized states [47]. Similarly, theoretical and experimental studies suggest that the lengths of bursts, principally the slowest component induced by low ligand concentrations, determine the deactivation phase of ensemble currents [5, 34, 37, 48]. Burst kinetics demonstrate that the opening rate constant (β) is greater than the rate constant for ligand dissociation (k−1), so here the channel oscillates between open and shut states until the ligand dissociates to terminate the burst [34, 46].
As ensemble currents, including those observed at synapses, represent the net behavior of many channels, factors that affect the proportions of functional states in a population at a given time will in turn shape the ensemble current. These factors include different activating and modulating ligands and their concentrations, voltage [49], and mutations (both naturally occurring and synthetic), and are best quantified within the framework of a mechanism.
Functional mechanisms of desensitization
Functional mechanisms for channel behavior are generally derived by postulating plausible reaction schemes and fitting them to experimental data by statistical methods [34, 50]. An example of such a mechanism is provided in Fig. 4. These mechanisms include functional states and the rate constants that govern the transitions between connected states, which are calculated from the distributions of conducting and non-conducting periods in single channel records. The mechanisms are fitted to clearly defined bursts and clusters of activity that are excised from the records (Fig. 3b). This process of data selection offers a reasonable guarantee that the data represent the activity of a single ion channel, thus eliminating the need to correct for multiple channels of unknown number. The minimum number of states in the underlying mechanism can be inferred from the corresponding number of components in the conducting and non-conducting distributions obtained from isolated burst or clusters of activity [34, 51]. Some of the shut components in clusters induced at concentrations that increase the occupancy of desensitized states may represent relatively short-lived desensitized configurations. Most often, however, because the precise number of channels in a given patch is not known, the long quiescent periods corresponding to salient desensitized states cannot be interpreted and are usually deliberately overlooked. This is because one cannot be certain if consecutive clusters, and the non-conducting period between them, are due to the same ion channel [34], even when precautions are taken to exclude segments of single channel record that contain unambiguous evidence of multiple channel activity (such as channel stacking). For this reason, the majority of published mechanisms omit treatment of desensitization altogether (see, e.g., [52, 53]).
Ad hoc methods of deriving mechanisms that include desensitized states were first postulated for the amphibian nAChR [2], and have since been utilized successfully to describe the desensitization of other pLGICs [35–38, 42]. A reductionist approach is taken with a comprehensive mechanism, such as that shown in Fig. 4, by ‘pruning’ it or identifying dominant pathways leading to desensitized states, often with the aid of ensemble current data [35–37, 42], or in some cases exclusively using ensemble currents [38, 54]. For instance, using single channel recordings, it was determined that heteromeric nAChRs enter into desensitized states (A2D; Fig. 4) faster when the channels are fully liganded and open (A2O) than shut (A2S), establishing the dominant pathway for entry into desensitization as A2S → A2O → A2D, with a desensitization rate constant for ACh of ~4 s−1. This result strongly suggests that desensitization is dependent on channel activation and the status of the gate rather than ligand occupancy, an inference supported by the use of different ligands [42]. A study that examined the burst characteristics of the same channel in conjunction with macropatch (ensemble) data has also concluded that, although wild-type channels desensitize too slowly for desensitized states to have a significant effect on the shape of the synaptic current, mutations that increase the β/α ratio (Fig. 4), including those that might cause disease, or decrease the ligand unbinding rate constant from the A2S state, can affect the time-course of nAChR current deactivation [37]. These mutations essentially increase the probability of the channels entering desensitized configurations during the deactivation phase of synaptic currents, and this suggests that the contribution of desensitized channels to ensemble deactivation is not only affected by the rate constants connected to desensitized states but also states that are predominant pathways for burst termination, such as A2S.
An extreme case where entry into desensitized states is rapid and the rate constants leading away from desensitized states are small is in the homomeric α7 AChRs. In this channel, entry into desensitization is likely the main pathway to burst termination, making it the main determinant of current decay. Moreover, the slow recovery from desensitization sets the refractory period for reactivation by successive pulses of ligand at synapses containing this channel [36]. Studies of desensitization in α1β2γ2 GABAARs, using exposure to 3H-ligand after the ligand was washed off the activated channels provide evidence that in this channel ligand unbinding from desensitized states occurs faster than the rate constants exiting desensitized states (resensitization). These data demonstrate that α1β2γ2 GABAARs can remain in desensitized configurations after the ligand has dissociated from the channel and re-bind ligand (radiolabeled) with a higher affinity than it does at non-desensitized channels [54].
As the determination of rate constants leading away from desensitized states derived from the durations of long non-conducting periods in single channel records is confounded by the problem of channel number, rapid ligand perfusion onto macropatches can provide estimates of rates of recovery from desensitized configurations. The technique involves a ligand application protocol consisting of a sequence of paired ligand pulses (applications), separated by a variable interpulse interval. The first (conditioning) pulse is of sufficiently long duration (say, 1 s) to induce desensitization whereas the second (test) pulse is briefer (say, 50 ms). Sufficient time is allowed between application pairs for the channels to recover. The peak amplitude of the test pulse relative to that of the conditioning pulse is a measure of the number of activatable channels that have exited desensitized states (resensitized) in the intervening period. Plots of relative current peak between response pairs as a function of interpulse interval produces a pattern of data that is well fit by mutliexponential functions, revealing a multistep recovery process from desensitization [36–38]. A multicomponent desensitization process is also evinced by the observation that longer conditioning pulse exposures result in longer time-courses of recovery from desensitization. This suggests multiple desensitized states in the mechanism, some of which having slow recovery rate constants [45].
There are other benefits in using both equilibrium (single channel) and non-equilibrium (ensemble) data to investigate desensitization within the context of a mechanism. Distinguishing mechanisms, especially ones that contain the same number of shut, open, and desensitized states cannot usually be performed on the basis of single channel kinetic analysis alone. Similar mechanisms can produce the same statistical outcome in terms of predicting lifetimes of conducting and non-conducting states in a record [55]. This limitation necessitates testing similar mechanisms under different experimental conditions and evaluating them on the basis of their accuracy in describing and simulating all the data. On inspection of any mechanism, such as in Fig. 4, it becomes apparent that the initial conditions prior to channel activation between the steady state and non-equilibrium circumstances differ. Steady state activations, especially in high ligand concentrations, are much more likely to begin from a partially or full liganded channel, whereas channels in ensemble currents activate from unliganded shut states in response to a step increase in the ligand from zero to some pre-defined concentration. Hence, ensemble currents have an average longer latency prior to entering conducting configurations, as most channels begin further up-stream in the mechanism when first exposed to a ligand.
Ultimately, quantitative functional mechanisms of channel desensitization must be related to channel structure. The multiple desensitized, shut, and open states in a reaction mechanism must have physical correlates that occur in a reaction sequence that coincides with the mechanism. A significant step towards understanding the sequence of structural rearrangements adopted by a channel in response to ligand binding is the technique of Φ-value analysis [56, 57]. However, this method has yet to be applied to desensitization, mainly owing to the abovementioned difficulty of counting channel numbers in single channel records. Moreover, Φ-value analysis data cannot yet be overlayed onto a complex functional mechanisms, and seems to be most applicable to mechanisms whose states are connected in linear sequence [56]. Another limitation to investigating discrete desensitized states is the paucity of information about protein structures that mediate desensitization per se. Structural elements of pLGICs that affect shut to open isomerisations are far better characterized [16, 25, 26, 58, 59] than those specifically affecting desensitization. Although mutations to many structural segments of pLGICs have been identified that do alter ensemble desensitization, it remains to be established if any of these specifically affect the entry and exit rate constants associated to desensitized states in a mechanism. They may, instead, alter the lifetimes of open states by altering rate constants connected to them, or affecting the lifetimes of connected shut states along the pathway to channel opening [16].
Structural mechanisms of desensitization
Evidence for a separate desensitization gate in the pore
It is first necessary to consider whether the structure of the desensitized state is different from that of the resting shut state. Evidence for desensitized states being distinct from liganded shut states has been furnished by single channel electrophysiological studies. Furthermore, electron microscopic images of the muscle nAChR have revealed structural differences between shut (unliganded), open, and desensitized configurations [60–62]. These images reveal global differences in protein structure including evidence for a structural realignment at the transmembrane domains between open and shut states. A systematic mutagenesis study involving functional nAChRs has provided more direct evidence that the pore-lining M2 domains differ in structure between shut, open, and desensitized states [19]. Notably, this study provided evidence for two permeation gates, one corresponding to a shut state and the other to a desensitized state. Both gates are situated within the M2 domains and there is overlap between them, but nevertheless are sufficiently different to suggest that channel desensitization is accompanied by unique structural rearrangements in this part of the receptor. Single channel kinetic analysis of nAChRs also supports the notion of a distinct desensitized gate. Calculated desensitization rate constants were not affected by ligands with different activation efficacies (β/α ratios), neither did mutations near the binding sites for ACh that altered the ligand binding reaction or the β/α ratio [42]. Blocking the pore of these channels also had no affect on the desensitization rate constant. These results, in conjunction with the determination that the channels desensitize much faster from A2O than A2S and recover faster when ligand unbinds, was reconciled with a two-gate structural model consisting of an activation gate and a desensitized gate [42] (Fig. 5). A study that examined the pore-blocking effects on single channel nAChR currents also drew similar conclusions regarding the two-gate model of activation and desensitization. Single channel activations exhibited two characteristics that changed as a function of increasing the concentration of the blocking agent. First, the open intervals within discrete activations lengthened as a result of a slowing of the shutting rate constant (α) for channel activation. Second, the entry rates into desensitized states were unaffected when the channel was blocked. These results are consistent with the pore blocker affecting the activation gate, and implies a second, structurally distinct gate that was unaffected by the pore blocker, which mediates densensitizaton [63].
Fig. 5.

A cartoon schematic illustrating the salient features of channel activation and desensitization and the two-gate mechanism. The unliganded shut (S) channel binds two molecules of ligand (orange), which initially induce a structural reorientation of the extracellular domain (dark grey) producing a liganded shut state (A 2 S). This is followed by a structural change in the transmembrane domain (red), which disrupts the activation gate to produce an open channel (A 2 O). The continued presence of ligand elicits a further structural change in the extracellular domain (light brown) and transmembrane domain (dark brown) that occludes the pore with a second desensitization gate
A structurally separate desensitization gate has also been posited for the α1β2γ2 GABAAR. Under circumstances where channels activated with a low open probability, such as low efficacy, non-desensitizing activators or mutant channels, the current–voltage (I–V) relationship rectified outwardly (greater relative current at positive potentials). Conversely, channel activation that produces high channel open probability, such as high (desensitizing) ligand concentrations or activation enhancing modulators, resulted in an attenuation of net outward current to linearize the I–V relationship. The other notable observation was that ensemble currents showed enhanced desensitization at positive potentials (outward current). These data were reconciled by postulating two, structurally separate gates: one that acted to increase open probability at positive potentials (activation gate), producing the outward rectification, and another that antagonized the first gate to diminish current at positive potentials and linearize the I–V (desensitization gate) [49]. Another recent study on the same GABAARs showed that access of pore-blocking agents increased in the presence of activating concentrations of GABA, but decreased in the presence of desensitization concentrations, consistent with open and desensitized states being structurally different at the level of the channel pore. This was supported by the observation that an M2–M3 linker mutant that reduced spontaneous activation (reduced β/α) also reduced access to pore blockers, and, as the mutant also reduced desensitization, high concentrations of GABA were now less effective at inhibiting blocker access compared to wild-type channels [64].
Desensitization-specific conformational changes elsewhere in the receptor
The structural basis of pLGIC receptor desensitization has been probed using the voltage clamp fluorometry (VCF) technique in the α1 homomeric GlyR [30]. This technique takes advantage of the fact that changes in the quantum efficiency of small molecule fluorophores may occur in response to changes in their immediate chemical microenvironment. VCF involves introducing a cysteine into a receptor domain of interest and covalently tagging it with a sulfhydryl-labeled fluorophore, commonly a rhodamine derivative. VCF is unique in its ability to report conformational rearrangements in real time at defined locations on the receptor surface [65]. The α1 GlyR VCF study compared the time-course of agonist-induced fluorescent changes at numerous extracellular and transmembrane sites in the absence of fast desensitization with those observed at the same labeled sites after an intracellular mutation had been introduced to dramatically enhance the desensitization rate. This enabled the authors to distinguish those conformational changes that remain unchanged throughout the period of agonist-binding from those that tracked the desensitization rate. Although no evidence was found for conformational changes associated with desensitization at any of the labeled sites near the glycine-binding site, the authors did identify desensitization-specific conformational changes in loop 2, the pre-M1 segment, and the M1 domain (Fig. 2) of the α1 GlyR. Figure 6a shows examples of current and fluorescence responses recorded simultaneously from slow- and fast-desensitizing GlyRs, each of which incorporates a rhodamine label in loop 2 (at residue A52C). Because fluorescence decay parallels the current decay in the fast-desensitizing receptor (Fig. 6b), it was concluded that the fluorophore is detecting a local conformational change associated with desensitization. Note also that, in the prolonged presence of glycine, the fluorescence response of the fast-desensitizing receptor stabilized at a level higher than the resting shut state level, indicating that the conformation of the desensitized state is distinct from that of the resting shut state. Labeled sites in both the pre-M1 domain and the M1 domain also reported conformational changes associated with desensitization. Together, the results imply that desensitization involves a specific reorganization of molecular interactions at the extracellular-transmembrane domain interface.
Fig. 6.

Combined current and fluorescence recording of rhodamine-labeled A52C and A52C–A248L mutant GlyRs reveals a conformational change specifically associated with receptor desensitization. The A248L mutation in the M1–M2 loop induces fast desensitization. a Examples of current (black) and fluorescence (red) responses induced by a saturating glycine concentration in both GlyRs. Note the biphasic fluorescence response and the glycine-induced plateau in the double mutant GlyR. b Averaged time constants for current and fluorescence decay recorded from double mutant GlyRs plotted as a function of glycine concentration. There was no significant difference at any concentration, confirming that the fluorescence response reported a conformational change associated with desensitization. Figure modified from [30]
Although VCF studies on other pLGIC receptor family members have yet to systematically characterize the location of labeled sites that report desensitization-specific conformational changes, the limited information available suggests the above model may also apply to other members of this receptor family. For example, fluorophores attached to residues in or near GABAAR agonist-binding sites reveal no measureable fluorescence change during desensitization of GABA-gated currents [66, 67]. However, a fluorophore attached to an M2–M3 loop residue in the muscle nAChR exhibited an agonist-induced fluorescence change that correlated with the current desensitization rate [68]. These data are consistent with the above model whereby desensitization of pLGIC receptor family members involves a specific conformational change at the extracellular––transmembrane domain interface.
A common feature of liganded open and desensitized configurations is their relatively high ligand affinity. Differences in ligand affinity between open and desensitized channels tend to be in the range of only a few-fold, such as in α1β2γ2 GABAARs [54] and nAChRs [59, 69], although more significant changes to affinity have been reported for this channel in a mutation to the ACh binding site [70]. Coupled with mutations to ligand binding pockets that show no appreciable affect on desensitization kinetics [54], and mutations that exhibit desensitization in the absence of ligand [59], it would be reasonable to infer that the structural differences at the binding pockets between open and desensitized states are subtle. A recent study on the α7 AChR provides evidence that unique arrangements of hydrogen bonding between activating ligand and binding pocket correspond to activated and desensitized states of the channel [71]. In contrast, desensitization can be profoundly influenced by mutating parts of the channel downstream of the pockets [36, 72]. The VCF data on the α1 GlyRs [30] revealed that the molecular processes of channel activation and desensitization recruit the same structural elements, but in different ways. Chimeric studies of the fast desensitizing α7 nAChR and the slow desensitizing 5HT3AR have clearly demonstrated that the M2–M3 linker, loop 7, loop 9, and the N-terminal end of the M1 domain are all involved in modulating the entry and exit rates of channel desensitization. By systematically swapping these segments between the two parent channels, it was deduced that loop 7 and loop 9 make independent contributions to the desensitization kinetics, as does the M1 domain, mainly by regulating single channel cluster durations and open dwell times [36]. Another study supported and further developed the notion that the molecular coupling compatibility between segments at the extracellular and transmembrane interface mediates channel desensitization in the α7 nAChR [72]. The investigators here found that mutations in the M2 domain that increased spontaneous channel activity and leftward shifted the ensemble agonist concentration–response relationship—evidence that the efficacy of activation (β/α) had increased—was tightly correlated to a marked decrease in ensemble desensitization. Furthermore, when these gating enhancer mutations were combined with others that disrupted the normal interactions between the M2––M3 linker, loop 2, and loop 7, which resulted in ablation of channel function, the impairment was counteracted and accompanied by an increase in desensitization. These data led the investigators to conclude that desensitization was a function of the relative strength of interactions at the extracellular-transmembrane interface (Fig. 2) and the energetics of disrupting the gate [72]. These intriguing interpretations clearly do not require two separate gates or any structural differences that would constitute distinct transduction pathways for activation and desensitization.
Structural elements that are common to activation and ensemble desensitization have been reported for many of the best-characterized pLGICs. The mutational effects on desensitization are dependent on both the nature of the residue and the pLGIC under study. M2 mutations have been shown to affect desensitization in α7 nAChRs [72, 73], α4β2 nAChRs [73, 74], 5HT3ARs [75], and ρ1 GABAARs [76]. Of course, deriving the inference that M2 mutations actually affect desensitization from ensemble current data comes with the caveat that the mutation may have stabilized the open state(s) by, say, reducing β, and may not have affected desensitization at all [16]. M2–M3 linker mutations that influence desensitization have been reported in GABAARs [64] and 5HT3ARs [77]. Mutations to the pre-M1 and M1 domains have been shown to modulate desensitization kinetics in α7 nAChRs [36], α1 GlyRs [30, 78], α1β2γ2 GABAARs [79, 80], and 5HT3ARs [36, 81]. At the transmembrane––intracellular domain interface,desensitization can be affected to mutations in the M1–M2 linker in the α1 GlyR [10, 29, 82] and the M3–M4 linker in heteromeric nAChRs [33], α1β2γ2 GABAARs [32], α3 GlyRs [83], and 5HT3ARs [31, 84]. These studies (and many others not mentioned here) demonstrate that the pathways for activation and desensitization are intimately linked, and emphasize the need to combine fluorometric techniques, such as VCF, with studies based on ensemble currents and single channel measurements in conjunction with channel state mechanisms to aid in disentangling the two processes at a molecular level.
X-ray crystallography has the potential to provide us with structural models of functional channel states. Soaking the crystals in ligands for state-selective durations [85] or using state-favoring ligands [86] or mutations [87] is the main method used for isolating a particular state for crystallographic studies. However, functional studies are exposing the present-day limitations of crystallography in obtaining images of functionally salient states. For instance, channels crystallized in the presence and absence of modulators that induce non-conducting states exhibit no difference in the structure of the pore [88]. Similarly, channels that were supposedly crystallized in open states [12, 15] are more likely to represent desensitized channels [85, 89], as revealed by functional experiments. Other functional studies demonstrate that, in the presence of ligand, the channels desensitize, whereas in its absence the channels are shut, but here too the crystal images reveal little difference in structure [14, 90]. Even more remarkably, crystal structures of channels bearing mutations that favor the open state are similar to those in the absence of ligand, where the channel is much more likely to adopt a shut configuration [87]. Clearly, the full prospects of crystallography are not yet upon us, so it would be hasty indeed to apply crystallography in the absence of the functionally verified states.
Concluding remarks
Desensitization is mediated by a global conformational change involving the extracellular, transmembrane, and intracellular domains. The key issue is to determine which structural elements of that global conformational change produce the increase in channel affinity and which produce the channel closure that is observed in the continued presence of agonist. If the structural basis of these mechanisms can be resolved, then we are well on the way towards assigning structural configurations to the functional states as identified by kinetic analysis of single channel and ensemble currents.
The affinity increase that accompanies desensitization must be due to a conformational change in the agonist binding site. To date, such conformational changes have proved difficult to detect, and only one study [71] has proposed a change in a molecular interaction between an agonist and its binding site that might mediate this affinity increase. The extent to which this result is applicable to other desensitizing pLGIC receptor subtypes is yet to be investigated.
It is now well established that channel activation is mediated by a specific reorganization of molecular interactions at the extracellular–transmembrane domain interface. Several recent functional studies, described in detail above, have demonstrated that activation and desensitization correlate with distinct sets of conformational changes at this interface. We therefore propose that desensitization is mediated by a distinct set of conformational changes that is incompatible with activation, and thereby closes the channel. However, the question of whether the desensitized shut state and the resting shut state are structurally identical is yet to be unequivocally resolved.
Acknowledgments
Research in the authors’ laboratory is funded by the Australian Research Council and the National Health and Medical Research Council.
References
- 1.Del Castillo J, Katz B. Interaction at end-plate receptors between different choline derivatives. Proc R Soc Lond B. 1957;146:369–381. doi: 10.1098/rspb.1957.0018. [DOI] [PubMed] [Google Scholar]
- 2.Katz B, Thesleff S. A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol. 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moffatt L, Hume RI. Responses of rat P2X2 receptors to ultrashort pulses of ATP provide insights into ATP binding and channel gating. J Gen Physiol. 2007;130:183–201. doi: 10.1085/jgp.200709779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silberberg SD, et al. Ivermectin Interaction with transmembrane helices reveals widespread rearrangements during opening of P2X receptor channels. Neuron. 2007;54:263–274. doi: 10.1016/j.neuron.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 5.Wyllie DJ, et al. Single-channel activations and concentration jumps: comparison of recombinant NR1a/NR2A and NR1a/NR2D NMDA receptors. J Physiol. 1998;510(Pt 1):1–18. doi: 10.1111/j.1469-7793.1998.001bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones MV, Westbrook GL. The impact of receptor desensitization on fast synaptic transmission. Trends Neurosci. 1996;19:96–101. doi: 10.1016/S0166-2236(96)80037-3. [DOI] [PubMed] [Google Scholar]
- 7.Mansvelder HD, et al. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–919. doi: 10.1016/S0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 8.Bertrand D, et al. How mutations in the nAChRs can cause ADNFLE epilepsy. Epilepsia. 2002;43(Suppl 5):112–122. doi: 10.1046/j.1528-1157.43.s.5.16.x. [DOI] [PubMed] [Google Scholar]
- 9.Sine SM, et al. Naturally occurring mutations at the acetylcholine receptor binding site independently alter ACh binding and channel gating. J Gen Physiol. 2002;120:483–496. doi: 10.1085/jgp.20028568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saul B, et al. Novel GLRA1 missense mutation (P250T) in dominant hyperekplexia defines an intracellular determinant of glycine receptor channel gating. J Neurosci. 1999;19:869–877. doi: 10.1523/JNEUROSCI.19-03-00869.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bowser DN, et al. Altered kinetics and benzodiazepine sensitivity of a GABAA receptor subunit mutation [gamma 2(R43Q)] found in human epilepsy. Proc Natl Acad Sci USA. 2002;99:15170–15175. doi: 10.1073/pnas.212320199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bocquet N, et al. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- 13.Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hilf RJ, Dutzler R. X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature. 2008;452:375–379. doi: 10.1038/nature06717. [DOI] [PubMed] [Google Scholar]
- 15.Hilf RJ, Dutzler R. Structure of a potentially open state of a proton-activated pentameric ligand-gated ion channel. Nature. 2009;457:115–118. doi: 10.1038/nature07461. [DOI] [PubMed] [Google Scholar]
- 16.Auerbach A. The gating isomerization of neuromuscular acetylcholine receptors. J Physiol. 2010;588:573–586. doi: 10.1113/jphysiol.2009.182774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giniatullin R, et al. Desensitization of nicotinic ACh receptors: shaping cholinergic signaling. Trends Neurosci. 2005;28:371–378. doi: 10.1016/j.tins.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 18.Tasneem A, et al. Identification of the prokaryotic ligand-gated ion channels and their implications for the mechanisms and origins of animal Cys-loop ion channels. Genome Biol. 2005;6:R4. doi: 10.1186/gb-2004-6-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson G, Karlin A. Acetylcholine receptor channel structure in the resting, open, and desensitized states probed with the substituted-cysteine-accessibility method. Proc Natl Acad Sci USA. 2001;98:1241–1248. doi: 10.1073/pnas.98.3.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyazawa A, et al. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- 21.Jansen M, et al. Modular design of Cys-loop ligand-gated ion channels: functional 5-HT3 and GABA rho1 receptors lacking the large cytoplasmic M3M4 loop. J Gen Physiol. 2008;131:137–146. doi: 10.1085/jgp.200709896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goyal R, et al. Engineering a prokaryotic Cys-loop receptor with a third functional domain. J Biol Chem. 2011;286:34635–34642. doi: 10.1074/jbc.M111.269647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang W, et al. Cloning and characterization of the human GABAA receptor alpha 4 subunit: identification of a unique diazepam-insensitive binding site. Eur J Pharmacol. 1995;291:319–325. doi: 10.1016/0922-4106(95)90072-1. [DOI] [PubMed] [Google Scholar]
- 24.Purohit P, Auerbach A. Energetics of gating at the apo-acetylcholine receptor transmitter binding site. J Gen Physiol. 2010;135:321–331. doi: 10.1085/jgp.200910384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purohit P, et al. A stepwise mechanism for acetylcholine receptor channel gating. Nature. 2007;446:930–933. doi: 10.1038/nature05721. [DOI] [PubMed] [Google Scholar]
- 26.Grosman C, et al. Mapping the conformational wave of acetylcholine receptor channel gating. Nature. 2000;403:773–776. doi: 10.1038/35001586. [DOI] [PubMed] [Google Scholar]
- 27.Miller PS, Smart TG. Binding, activation and modulation of Cys-loop receptors. Trends Pharmacol Sci. 2010;31:161–174. doi: 10.1016/j.tips.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Xiu X, et al. A unified view of the role of electrostatic interactions in modulating the gating of Cys loop receptors. J Biol Chem. 2005;280:41655–41666. doi: 10.1074/jbc.M508635200. [DOI] [PubMed] [Google Scholar]
- 29.Lynch JW, et al. Identification of intracellular and extracellular domains mediating signal transduction in the inhibitory glycine receptor chloride channel. EMBO J. 1997;16:110–120. doi: 10.1093/emboj/16.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Q, Lynch JW. Activation and desensitization induce distinct conformational changes at the extracellular-transmembrane domain interface of the glycine receptor. J Biol Chem. 2011;286:38814–38824. doi: 10.1074/jbc.M111.273631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu XQ, et al. An interaction involving an arginine residue in the cytoplasmic domain of the 5-HT3A receptor contributes to receptor desensitization mechanism. J Biol Chem. 2006;281:21781–21788. doi: 10.1074/jbc.M600676200. [DOI] [PubMed] [Google Scholar]
- 32.O’Toole KK, Jenkins A. Discrete M3–M4 intracellular loop subdomains control specific aspects of gamma-aminobutyric acid type A receptor function. J Biol Chem. 2011;286:37990–37999. doi: 10.1074/jbc.M111.258012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen XM, et al. Subunit-specific contribution to agonist binding and channel gating revealed by inherited mutation in muscle acetylcholine receptor M3–M4 linker. Brain. 2005;128:345–355. doi: 10.1093/brain/awh364. [DOI] [PubMed] [Google Scholar]
- 34.Colquhoun DH, Hawkes AG (1995) The principles of the stochastic interpretation of ion-channel mechanisms. In: Single-channel recordings. Plenum, New York
- 35.Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 36.Bouzat C, et al. The interface between extracellular and transmembrane domains of homomeric Cys-loop receptors governs open-channel lifetime and rate of desensitization. J Neurosci. 2008;28:7808–7819. doi: 10.1523/JNEUROSCI.0448-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elenes S, et al. Desensitization contributes to the synaptic response of gain-of-function mutants of the muscle nicotinic receptor. J Gen Physiol. 2006;128:615–627. doi: 10.1085/jgp.200609570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Solt K, et al. Differential effects of serotonin and dopamine on human 5-HT3A receptor kinetics: interpretation within an allosteric kinetic model. J Neurosci. 2007;27:13151–13160. doi: 10.1523/JNEUROSCI.3772-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amin J, Weiss DS. Homomeric rho 1 GABA channels: activation properties and domains. Recept Channels. 1994;2:227–236. [PubMed] [Google Scholar]
- 40.Yang J, et al. Kinetic properties of GABA rho1 homomeric receptors expressed in HEK293 cells. Biophys J. 2006;91:2155–2162. doi: 10.1529/biophysj.106.085431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis TM, et al. Kinetic determinants of agonist action at the recombinant human glycine receptor. J Physiol. 2003;549:361–374. doi: 10.1113/jphysiol.2002.037796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Auerbach A, Akk G. Desensitization of mouse nicotinic acetylcholine receptor channels: a two-gate mechanism. J Gen Physiol. 1998;112:181–197. doi: 10.1085/jgp.112.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burzomato V, et al. Single-channel behavior of heteromeric alpha1beta glycine receptors: an attempt to detect a conformational change before the channel opens. J Neurosci. 2004;24:10924–10940. doi: 10.1523/JNEUROSCI.3424-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krashia P, et al. The long activations of alpha2 glycine channels can be described by a mechanism with reaction intermediates (“flip”) J Gen Physiol. 2011;137:197–216. doi: 10.1085/jgp.201010521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elenes S, Auerbach A. Desensitization of diliganded mouse muscle nicotinic acetylcholine receptor channels. J Physiol. 2002;541:367–383. doi: 10.1113/jphysiol.2001.016022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edmonds B, et al. Mechanisms of activation of muscle nicotinic acetylcholine receptors and the time course of endplate currents. Annu Rev Physiol. 1995;57:469–493. doi: 10.1146/annurev.ph.57.030195.002345. [DOI] [PubMed] [Google Scholar]
- 47.Sakmann B, et al. Single acetylcholine-activated channels show burst-kinetics in presence of desensitizing concentrations of agonist. Nature. 1980;286:71–73. doi: 10.1038/286071a0. [DOI] [PubMed] [Google Scholar]
- 48.Colquhoun D, Hawkes AG, Mersushkin A, Edmonds B. Properties of single ion channel currents elicitied by a pulse of agonist concentration or voltage. Philos Trans R Soc Lond A. 1997;355:1743–1786. doi: 10.1098/rsta.1997.0090. [DOI] [Google Scholar]
- 49.O’Toole KK, Jenkins A. The apparent voltage dependence of GABAA receptor activation and modulation is inversely related to channel open probability. Mol Pharmacol. 2012;81:189–197. doi: 10.1124/mol.111.074476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Milescu LS, et al. Maximum likelihood estimation of ion channel kinetics from macroscopic currents. Biophys J. 2005;88:2494–2515. doi: 10.1529/biophysj.104.053256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shelley C, Magleby KL. Linking exponential components to kinetic states in Markov models for single-channel gating. J Gen Physiol. 2008;132:295–312. doi: 10.1085/jgp.200810008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keramidas A, Harrison NL. The activation mechanism of alpha1beta2gamma2S and alpha3beta3gamma2S GABAA receptors. J Gen Physiol. 2010;135:59–75. doi: 10.1085/jgp.200910317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lape R, et al. On the nature of partial agonism in the nicotinic receptor superfamily. Nature. 2008;454:722–727. doi: 10.1038/nature07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang Y, et al. Desensitization mechanism of GABA receptors revealed by single oocyte binding and receptor function. J Neurosci. 2002;22:7982–7990. doi: 10.1523/JNEUROSCI.22-18-07982.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kienker P. Equivalence of aggregated Markov models of ion-channel gating. Proc R Soc Lond B. 1989;236:269–309. doi: 10.1098/rspb.1989.0024. [DOI] [PubMed] [Google Scholar]
- 56.Zhou Y, et al. Phi-value analysis of a linear, sequential reaction mechanism: theory and application to ion channel gating. Biophys J. 2005;89:3680–3685. doi: 10.1529/biophysj.105.067215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Auerbach A. How to turn the reaction coordinate into time. J Gen Physiol. 2007;130:543–546. doi: 10.1085/jgp.200709898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lape R, et al. The alpha1K276E Startle disease mutation reveals multiple intermediate states in the gating of glycine receptors. J Neurosci. 2012;32:1336–1352. doi: 10.1523/JNEUROSCI.4346-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Purohit P, Auerbach A. Unliganded gating of acetylcholine receptor channels. Proc Natl Acad Sci USA. 2009;106:115–120. doi: 10.1073/pnas.0809272106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Unwin N, et al. Arrangement of the acetylcholine receptor subunits in the resting and desensitized states, determined by cryoelectron microscopy of crystallized Torpedo postsynaptic membranes. J Cell Biol. 1988;107:1123–1138. doi: 10.1083/jcb.107.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 62.Unwin N. Acetylcholine receptor channel imaged in the open state. Nature. 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- 63.Purohit Y, Grosman C. Block of muscle nicotinic receptors by choline suggests that the activation and desensitization gates act as distinct molecular entities. J gen physiol. 2006;127:703–717. doi: 10.1085/jgp.200509437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Othman NA, et al. Influences on blockade by t-butylbicyclo-phosphoro-thionate of GABA(A) receptor spontaneous gating, agonist activation and desensitization. J Physiol. 2012;590:163–178. doi: 10.1113/jphysiol.2011.213249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pless SA, Lynch JW. Illuminating the structure and function of Cys-loop receptors. Clin Exp Pharmacol Physiol. 2008;35:1137–1142. doi: 10.1111/j.1440-1681.2008.04954.x. [DOI] [PubMed] [Google Scholar]
- 66.Akk G, et al. Pharmacology of structural changes at the GABA(A) receptor transmitter binding site. Br J Pharmacol. 2011;162:840–850. doi: 10.1111/j.1476-5381.2010.01083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Muroi Y, et al. Local and global ligand-induced changes in the structure of the GABA(A) receptor. Biochemistry. 2006;45:7013–7022. doi: 10.1021/bi060222v. [DOI] [PubMed] [Google Scholar]
- 68.Dahan DS, et al. A fluorophore attached to nicotinic acetylcholine receptor beta M2 detects productive binding of agonist to the alpha delta site. Proc Natl Acad Sci USA. 2004;101:10195–10200. doi: 10.1073/pnas.0301885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grosman C, Auerbach A. The dissociation of acetylcholine from open nicotinic receptor channels. Proc Natl Acad Sci USA. 2001;98:14102–14107. doi: 10.1073/pnas.251402498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sine SM, et al. Mutation of the acetylcholine receptor alpha subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron. 1995;15:229–239. doi: 10.1016/0896-6273(95)90080-2. [DOI] [PubMed] [Google Scholar]
- 71.Wang J, et al. Potential state-selective hydrogen bond formation can modulate the activation and desensitization of the alpha7 nicotinic acetylcholine receptor. J Biol Chem. 2012;287(26):21957–21969. doi: 10.1074/jbc.M112.339796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang J, et al. Desensitization of alpha7 nicotinic receptor is governed by coupling strength relative to gate tightness. J Biol Chem. 2011;286:25331–25340. doi: 10.1074/jbc.M111.221754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Revah F, et al. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature. 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- 74.Matsushima N, et al. Mutation (Ser284Leu) of neuronal nicotinic acetylcholine receptor alpha 4 subunit associated with frontal lobe epilepsy causes faster desensitization of the rat receptor expressed in oocytes. Epilepsy Res. 2002;48:181–186. doi: 10.1016/S0920-1211(01)00336-9. [DOI] [PubMed] [Google Scholar]
- 75.Gunthorpe MJ, et al. The 4′lysine in the putative channel lining domain affects desensitization but not the single-channel conductance of recombinant homomeric 5-HT3A receptors. J Physiol. 2000;522(Pt 2):187–198. doi: 10.1111/j.1469-7793.2000.00187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martinez-Torres A, Miledi R. A single amino acid change within the ion-channel domain of the gamma-aminobutyric acid rho1 receptor accelerates desensitization and increases taurine agonism. Arch Med Res. 2004;35:194–198. doi: 10.1016/j.arcmed.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 77.Hu XQ, Lovinger DM. Role of aspartate 298 in mouse 5-HT3A receptor gating and modulation by extracellular Ca2+ . J Physiol. 2005;568:381–396. doi: 10.1113/jphysiol.2005.092866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Castaldo P, et al. A novel hyperekplexia-causing mutation in the pre-transmembrane segment 1 of the human glycine receptor alpha1 subunit reduces membrane expression and impairs gating by agonists. J Biol Chem. 2004;279:25598–25604. doi: 10.1074/jbc.M311021200. [DOI] [PubMed] [Google Scholar]
- 79.Bianchi MT, et al. Structural determinants of fast desensitization and desensitization-deactivation coupling in GABAa receptors. J Neurosci. 2001;21:1127–1136. doi: 10.1523/JNEUROSCI.21-04-01127.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Engblom AC, et al. Point mutation in the first transmembrane region of the beta 2 subunit of the gamma–aminobutyric acid type A receptor alters desensitization kinetics of gamma–aminobutyric acid- and anesthetic-induced channel gating. J Biol Chem. 2002;277:17438–17447. doi: 10.1074/jbc.M111215200. [DOI] [PubMed] [Google Scholar]
- 81.Lobitz N, et al. A single amino-acid in the TM1 domain is an important determinant of the desensitization kinetics of recombinant human and guinea pig alpha-homomeric 5-hydroxytryptamine type 3 receptors. Mol Pharmacol. 2001;59:844–851. doi: 10.1124/mol.59.4.844. [DOI] [PubMed] [Google Scholar]
- 82.Breitinger HG, et al. Molecular dynamics simulation links conformation of a pore-flanking region to hyperekplexia-related dysfunction of the inhibitory glycine receptor. Chem Biol. 2004;11:1339–1350. doi: 10.1016/j.chembiol.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 83.Breitinger HG, et al. Hydroxylated residues influence desensitization behaviour of recombinant alpha3 glycine receptor channels. J Neurochem. 2002;83:30–36. doi: 10.1046/j.1471-4159.2002.01109.x. [DOI] [PubMed] [Google Scholar]
- 84.McKinnon NK, et al. Length and amino acid sequence of peptides substituted for the 5-HT3A receptor M3M4 loop may affect channel expression and desensitization. PLoS One. 2012;7:e35563. doi: 10.1371/journal.pone.0035563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gonzalez-Gutierrez G, Grosman C. Bridging the gap between structural models of nicotinic receptor superfamily ion channels and their corresponding functional states. J Mol Biol. 2010;403:693–705. doi: 10.1016/j.jmb.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pan J, et al. Structure of the pentameric ligand-gated ion channel ELIC cocrystallized with its competitive antagonist acetylcholine. Nat Commun. 2012;3:714. doi: 10.1038/ncomms1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gonzalez-Gutierrez G, et al. Mutations that stabilize the open state of the Erwinia chrisanthemi ligand-gated ion channel fail to change the conformation of the pore domain in crystals. Proc Natl Acad Sci USA. 2012;109:6331–6336. doi: 10.1073/pnas.1119268109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nury H, et al. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature. 2011;469:428–431. doi: 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- 89.Parikh RB, et al. Structure of the M2 transmembrane segment of GLIC, a prokaryotic Cys loop receptor homologue from Gloeobacter violaceus, probed by substituted cysteine accessibility. J Biol Chem. 2011;286:14098–14109. doi: 10.1074/jbc.M111.221895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zimmermann I, Dutzler R. Ligand activation of the prokaryotic pentameric ligand-gated ion channel ELIC. PLoS Biol. 2011;9:e1001101. doi: 10.1371/journal.pbio.1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Beato M, et al. Openings of the rat recombinant alpha 1 homomeric glycine receptor as a function of the number of agonist molecules bound. J Gen Physiol. 2002;119:443–466. doi: 10.1085/jgp.20028530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Beato M, et al. The activation mechanism of alpha1 homomeric glycine receptors. J Neurosci. 2004;24:895–906. doi: 10.1523/JNEUROSCI.4420-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lema GM, Auerbach A. Modes and models of GABA(A) receptor gating. J Physiol. 2006;572:183–200. doi: 10.1113/jphysiol.2005.099093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Haas KF, Macdonald RL. GABAA receptor subunit gamma2 and delta subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol. 1999;514(Pt 1):27–45. doi: 10.1111/j.1469-7793.1999.027af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jahn K, et al. Deactivation and desensitization of mouse embryonic- and adult-type nicotinic receptor channel currents. Neurosci Lett. 2001;307:89–92. doi: 10.1016/S0304-3940(01)01929-2. [DOI] [PubMed] [Google Scholar]
- 96.Maconochie DJ, Steinbach JH. The channel opening rate of adult- and fetal-type mouse muscle nicotinic receptors activated by acetylcholine. J Physiol. 1998;506(Pt 1):53–72. doi: 10.1111/j.1469-7793.1998.053bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fucile S, et al. The single-channel properties of human acetylcholine alpha 7 receptors are altered by fusing alpha 7 to the green fluorescent protein. Proc Natl Acad Sci USA. 2002;99:3956–3961. doi: 10.1073/pnas.052699599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Grewer C. Investigation of the alpha(1)-glycine receptor channel-opening kinetics in the submillisecond time domain. Biophys J. 1999;77:727–738. doi: 10.1016/S0006-3495(99)76927-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mohammadi B, et al. Kinetic analysis of recombinant mammalian alpha(1) and alpha(1)beta glycine receptor channels. Eur Biophys J. 2003;32:529–536. doi: 10.1007/s00249-003-0286-y. [DOI] [PubMed] [Google Scholar]
- 100.Keramidas A, et al. The pre-M1 segment of the alpha1 subunit is a transduction element in the activation of the GABAA receptor. J Physiol. 2006;575:11–22. doi: 10.1113/jphysiol.2005.102756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Maconochie DJ, et al. How quickly can GABAA receptors open? Neuron. 1994;12:61–71. doi: 10.1016/0896-6273(94)90152-X. [DOI] [PubMed] [Google Scholar]
- 102.Burkat PM, et al. Dominant gating governing transient GABA(A) receptor activity: a first latency and Po/o analysis. J Neurosci. 2001;21:7026–7036. doi: 10.1523/JNEUROSCI.21-18-07026.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Scheller M, Forman SA. Coupled and uncoupled gating and desensitization effects by pore domain mutations in GABA(A) receptors. J Neurosci. 2002;22:8411–8421. doi: 10.1523/JNEUROSCI.22-19-08411.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Boileau AJ, et al. Effects of gamma2S subunit incorporation on GABAA receptor macroscopic kinetics. Neuropharmacology. 2003;44:1003–1012. doi: 10.1016/S0028-3908(03)00114-X. [DOI] [PubMed] [Google Scholar]
- 105.Bianchi MT, Macdonald RL. Slow phases of GABA(A) receptor desensitization: structural determinants and possible relevance for synaptic function. J Physiol. 2002;544:3–18. doi: 10.1113/jphysiol.2002.020255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Krampfl K, et al. Molecular analysis of the A322D mutation in the GABA receptor alpha-subunit causing juvenile myoclonic epilepsy. Eur J Neurosci. 2005;22:10–20. doi: 10.1111/j.1460-9568.2005.04168.x. [DOI] [PubMed] [Google Scholar]