

Abstract
The neuromuscular junction (NMJ) is the synaptic connection between motor neurons and muscle fibers. It is involved in crucial processes such as body movements and breathing. Its proper development requires the guidance of motor axons toward their specific targets, the development of multi-innervated myofibers, and a selective synapse stabilization. It first consists of the removal of excessive motor axons on myofibers, going from multi-innervation to a single innervation of each myofiber. Whereas guidance cues of motor axons toward their specific muscular targets are well characterized, only few molecular and cellular cues have been reported as clues for selecting and stabilizing specific neuromuscular junctions. We will first provide a brief summary on NMJ development. We will then review molecular cues that are involved in NMJ stabilization, in both pre- and post-synaptic compartments, considering motor neurons and Schwann cells on the one hand, and muscle on the other hand. We will provide links with pathologies and highlight advances that can be brought both by basic research on NMJ development and clinical data resulting from the analyses of neurodegeneration of synaptic connections to obtain a better understanding of this process. The goal of this review is to highlight the findings toward understanding the roles of poly- or single-innervations and the underlying mechanisms of NMJ stabilization.
Keywords: Motor neuron, Axon development, Axon retraction, Stability, Neuromuscular junction, Synapse, Polyinnervation, Agrin
Introduction
The neuromuscular (NM) junction is the synaptic connection between motor neurons and muscle fibers. It is among one of the earliest synapses formed during mammalian development. NM junctions (NMJ) are involved in crucial processes, and their proper functionality governs complex and vital processes such as breathing and body movements. Consequently, improper NMJ formation with abnormal developmental selective synapse stabilization or improper NMJ maintenance later on during life may originate various neurodegenerative diseases.
NMJ remains the best-studied model for understanding the mechanisms involved in synaptogenesis. Its accessibility and size permit analyses of interactions between the nerve and its muscular target. The development of a mature NMJ first requires the guidance of the motor axons toward the specific muscles to be innervated and then the stabilization of the contact. The stabilization of synapses is a key step for both development and function of the nervous system. Several animal models have been used to investigate the cues that are involved in these processes. Neuronal growth cones are located at the tip of the axon and act as chemical sensors of molecular guidance cues present in the environment and targets. Accumulating data have allowed to elucidate mechanisms involved in the guidance of growth cones and motor axons toward their muscle targets in Drosophila, C. elegans, mouse or human. However, only very few reports bring clues explaining the stabilization of NMJ throughout life. The secretion of the Acetylcholine neurotransmitter (Ach)—the only neurotransmitter used in the motor division of the somatic nervous system which behaves as an excitatory neurotransmitter at NMJ in skeletal muscle—is among the main characteristics of the NMJ. Ach activates skeletal muscles and is a major neurotransmitter in the autonomic nervous system. Ach binds to nicotinic acetylcholine receptors (AchR) on skeletal muscle fibers and induces the contraction of skeletal muscle. A third cellular type is present at the NMJ in close proximity to the neuron–muscle synapse. It consists of a class of non-myelinating Schwann cell, called terminal (or peri-synaptic) Schwann cell. Altogether, nerve terminals, muscle fibers and Schwann cells constitute the tripartite cellular synaptic compartment. Several parameters have been considered to analyze the stability of the NMJ: the maintenance of the tripartite cellular synaptic compartment, the number and localization of AchR patches, the number of nerve terminals per fiber according to the developmental stage, Schwann cells capping the nerve terminal, cytoskeleton that coats the in-folded post-synaptic membrane, and the basal lamina that runs through the synaptic cleft—a small space neurons release neurotransmitter molecules into. Here, we will discuss the mechanisms that determine whether a synapse will persist or will be remodeled or eliminated, focusing on key periods that could be considered as “critical periods” (Fig. 1). We will first provide a brief up-to-date summary on NMJ development. Then, we will focus on reviewing most of the molecular cues that are involved in NMJ stabilization, in both pre- and post-synaptic compartments. We will provide links with pathologies and highlight advances that can be brought by both developmental basic data—including molecular candidates involved in pre- and post-synaptic development—and clinical data to get a better understanding of this still poorly understood process. Indeed, we will consider the causal cues that have been hypothesized from the analyses of neurodegeneration of NM synaptic connections following injuries or genetic alterations in cases of amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA) caused by mutations within the survival motor neuron 1 gene. The goal of this review is to highlight the findings that have possible applications for understanding the role of polyinnervation, in particular timely maintenance and retrieval. We will consider the types of muscles and NMJ that are concerned, the physiology and physiopathology of NMJ development, and axon stabilization in peripheral but also central synapses.
Fig. 1.
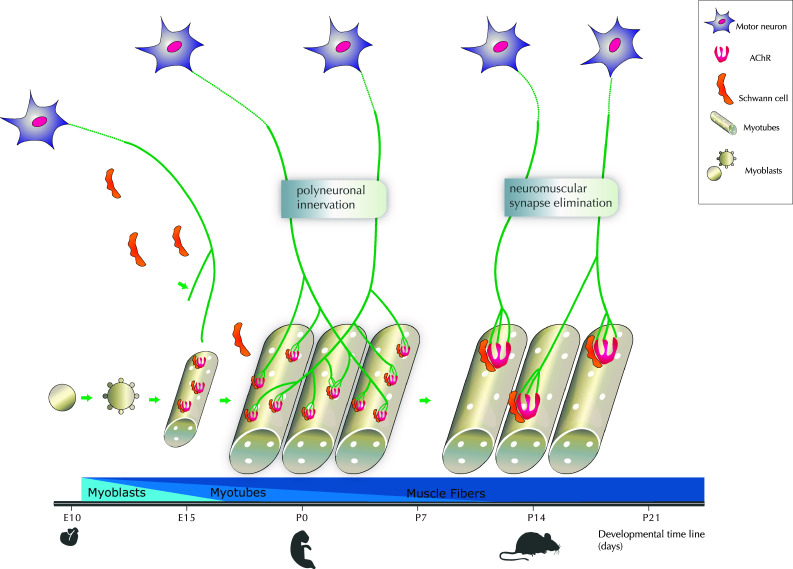
Critical periods of NMJ development with a focus on the elimination of multi-innervation. Motoneurons emit axons toward myotubes from E10 to E15 in mice. In parallel, Schwann cells migrate to reach the NMJ from E16 and cap the terminal. The differentiation of myoblasts into myotubes and muscle fibers is also schematized altogether with the time course. Dispersed acetylcholine receptors (AChRs) are expressed at moderate levels throughout myotubes surface prior to synapse formation, from E15 at the period of polyneuronal innervation, i.e. when each muscle fiber is still innervated by one or several axons of motor neurons (MNs). Postnatally, a critical step with the redrawal of about half multi-innervation occurs between P0 to P7, and the process of synapse elimination is achieved 2 weeks postnatally. AChR clustering occurs in the post-synaptic membrane, altogether with a progressive transition from multiple to single innervation of the NMJ
Development of the NMJ
Motor neurons proliferate in the ventricular zone of the neural tube and become postmitotic between stage 14 and 23, i.e., between E2 and E4.5 in chick embryo [1] and between E9 and E10/E11 in mouse, according to the motor neuronal population. Subsequently, motor neurons differentiate into limb and axial motor neurons. They assume their topographic organization in distinct columns based on the combinatorial expression of homeoproteins [2], in particular unique combinations of LIM-type homeodomain factors [3]. It also allows subtype identity (fast:slow and flexor:extensor) and the formation of proper and characteristic motor circuits whose development is linked to the presence of guidance cues [4]. These multiple classes and subtypes of motor neurons, as fast and slow, alpha (α) and gamma (γ) allow NM-specific contractile and motor functions and physiology ([5 for an extensive review). Alpha motor neurons are the most abundant of these classes driving muscle contraction and they can, in turn, be classified into subtypes according to the contractile properties of the motor units that they form with target muscle fibers: fast-twitch fatigable (FF), fast-twitch fatigue resistant (FR), and slow-twitch fatigue resistant (S) [6]. Gamma-motor neurons innervate intrafusal muscle fibers of the muscle spindle and play complex roles in motor control. We will not insist here on a third not so well-defined population called β-motor neurons [5]. Although the existence of separate programs for the determination of α- and γ-motor neurons identities seems to be a pre-requisite, the identification of early markers is still needed to determine how and when the various populations diverge. Whether genetic cues that will allow this diversity of cell types at early stages of development or differential interactions with the periphery will finally establish a molecular distinction between motor neurons subtypes is still to be determined [7]. Gamma-motor neurons express higher levels of the glial cell line-derived neurotrophic factor (GDNF) receptor subunit GFRα1 than α-motor neurons, and the transcription factor Err3—an orphan nuclear hormone receptor—also becomes restricted to γ- motor neurons during the first two postnatal weeks [7]. Thus, these markers only begin to distinguish γ- from α-motor neurons at postnatal stages, which may suggest a role in the period of polyinnervation retraction and in the selective axon stabilization/retraction that will be detailed below.
Another specificity of motor neurons is their remarkably long axonal length since they can innervate distal muscle targets such as the limbs, thanks to appropriate export of membranes from their cell bodies. Synaptic formation begins as an intrinsic property of axonal terminal to form specific synaptic sites, even in the absence of post-synaptic contacts, through an intrinsic synaptogenic activity. Among the specific features of pre-synaptic differentiation is the formation of active zones (AZ) where a dense network of macromolecules called active zone material (AZM) is attached to the pre-synaptic membranes next to docked vesicles. Later on, retrograde signals, including LRP4 (see below and [8]), will regulate pre-synaptic differentiation at neuromuscular synapses. pre-synaptic active zones are synaptic vesicle release sites that play essential roles in the function and pathology of mammalian NMJs. The molecular mechanisms of active zone organization use pre-synaptic voltage-dependent calcium channels (VDCCs) in NMJs as scaffolding proteins. VDCCs interact extracellularly with the muscle-derived synapse organizer laminin β2, and interact intracellularly with active zone-specific proteins, such as Bassoon, CAST/Erc2/ELKS2alpha, ELKS, Piccolo, and RIMs (for a review, [9]). Muscle innervation by motor neurons leads to a high concentration of acetylcholine receptors (AChRs) in postjunctional membranes of muscle fibers, a complex process that involves AChR aggregation in subsynaptic areas, the dispersion of nonsynaptic AChR-rich sites and local AChR synthesis. The anterograde signals used during development include agrin, a polypeptide used by motor neurons to cluster AChRs and ACh which suppresses AChR subunit gene expression and disassembles AChR clusters in nonsynaptic areas once muscle fibers have been activated. Retrograde neurotrophic information has to be brought from the muscle target transported along the axon to reach the cell bodies, to assert motor neurons survival, which also makes motor neurons great models to study the dialog between synaptic targets and cell bodies to regulate cell survival. A complex interplay exists between axons, Schwann cells and the differentiating muscle fibers, composing a tripartite NM synapse. Schwann cells migrate and contact axon terminals as they branch on young myotubes. Post-synaptic AChR clustering also occurs prenatally. As motor neurons make contact with muscle fibers, the number of motor neurons is greatly reduced. The process of motor neuron elimination takes place between E12 and E14 for phrenic motor neurons—that specifically innervate the diaphragm, the major muscle of respiration—for example, in mice [10]. In most types of motor neurons, up to 50 % of motor neurons are lost by apoptosis at this time, possibly involving motor neurons activity and their functional connections. The neurotrophic theory elaborated by Victor Hamburger and Rita Levi-Montalcini is based on a competition concept between adjacent axons. Some neurons in a population die because trophic molecules are available in only limited amounts by the muscular target during periods of naturally occurring cell death [11]. Indeed, it has become evident that muscles provide signals to regulate differentiation and function of pre-synaptic terminals. In parallel with agrin pathway, the Wnt signaling pathways has been shown to be crucial in mediating nerve–muscle interactions during NMJ formation. Wnt is a family of secreted glycoproteins that have important roles in the development and maturation of the nervous system, including brain patterning, axon guidance and synapse formation. The muscle β-catenin has been shown essential for NMJ development and function, more particularly for pre-synaptic differentiation. The specific suppression of β-catenin in skeletal muscles led to the death of mouse soon after birth, with considerable pre-synaptic defects including the mislocation of primary branches of phrenic nerves and extended secondary branches. Indeed, β-catenin-dependent transcription has been suggested to be necessary for the expression of a necessary retrograde signal protein [12]. β-Catenin may also regulate the expression of synaptic proteins including the AChR [13].
More recent in vivo experiments have studied NM development in mice expressing increased levels of β-catenin in either motor neurons or muscles. β-Catenin overexpression in muscle only, not in motor neurons, increases nerve branching possibly due to an increase in motor neuron numbers but independent of the level on neuromuscular activity. Defasciculation and branching occur prior to the establishment of functional NMJs between phrenic motor neurons and their target diaphragm muscles [14].
During vertebrate NM development, all muscle fibers are transiently innervated by more than one neuron. Later on, during postnatal development, a step of polyneuronal innervation retraction is observed (Fig. 1), leading to motor neuronal mono-innervation. Indeed, although synaptic connections can be stably maintained for prolonged periods, they can be rapidly disassembled during the development and refinement of neural circuitry. This retraction of polyinnervation can be considered as a form of NMJ instability [15]. We will review the mechanisms that are involved in this process.
Activity-dependent competition for synapse elimination
Synapse competition and elimination are a general developmental process both in CNS and PNS which is strongly activity-dependent (Fig. 2). It is well established that the synaptic connections between motor axons and muscle are shaped by activity. Impaired post-synaptic activity at neuromuscular synapses delays the withdrawal of pre-synaptic terminals and synapse elimination [16]. This has been demonstrated extensively for processes occurring late during synaptogenesis in which activity regulates synaptic maturation and refinement [17]. In the absence of activity, NMJs form an aberrant morphology with a reduction of post-synaptic specializations, as demonstrated in rat, Drosophila and mouse [18]. The blockade of neural transmission leads to the loss of synapse elimination, causing aberrant branching of motor axons and multiple innervation of muscle fibers, altogether with modifications of motor neuron survival during normal cell death [19]. Cholinergic transmission is a mediator of the neural control of stability of junctional AChRs in mammals. Accordingly, increasing activity accelerates the transition to mono-innervation. The synchronous activity of motor neurons first favors polyneuronal innervation, whereas asynchronous activity subsequently promotes synapse elimination [20]. Interestingly, the blockade of action potential generation in muscle can inhibit synapse elimination through local signaling [21].
Fig. 2.
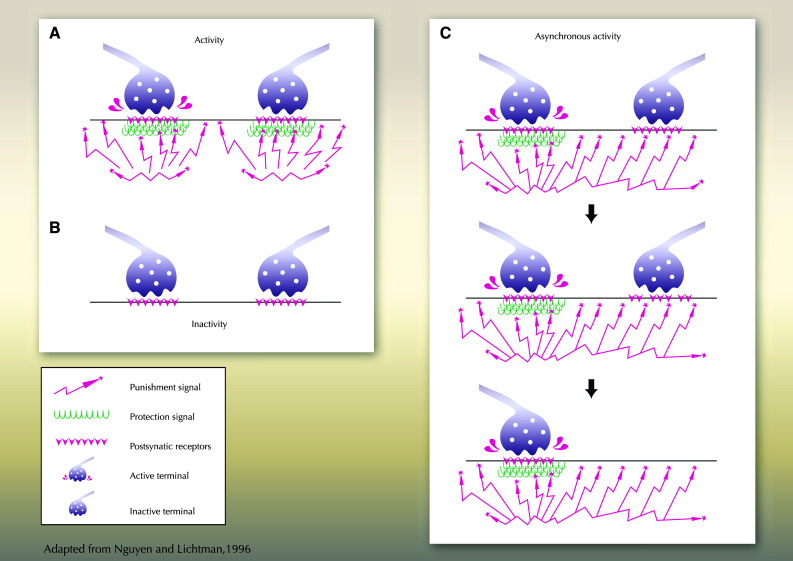
Activity dependent NMJ maintenance/stabilization. a Two motoneurons with synchronous activity are schematized. A similar activity will allow the synthesis of an equivalent amount of punishment (synaptotoxic factor) and reward (synaptotrophic factor) by both MNs, and their survival. b Inactive MNs will not be induced to produce either punishment or reward signal; the absence of competition for survival factor will allow the maintenance of both MNs. c The stabilization of one out of two MNs axons innervating a similar myofiber will be regulated by the activity: the active MN will synthesis both protective (reward) and punishment signals, that will allow its survival, whereas the inactive MN will receive punishment signals from the neighboring active MN only, that will lead to its elimination. Asynchronous AChR activation allows muscle to selectively destabilize synaptic sites to be eliminated
Nevertheless, the precise and successive physiological functions of steps of polyinnervation followed by mono-innervation stabilization—a developmental process that necessarily challenges NMJ activity and function—still remain hypothetical in terms of activity regulation within appropriate physiological ranges. Synaptic activity drives synaptic rearrangement in the vertebrate nervous system, in particular, the competitive process of synapse elimination during early postnatal life. Indeed, more powerful inputs are strongly favored competitors during this process [20], and active synaptic sites can destabilize inactive synapses in their vicinity. Homeostatic stabilization and signaling mechanisms that allow cells to maintain appropriate levels of activity could also control developmental synapse growth and stabilization, or be controlled through retrograde or anterograde processes. Whether multi-innervation removal and mono-innervation at the NMJ are a homeostatic challenge remains to be demonstrated. Anyway, among answers to retrograde signaling that have already been demonstrated to be involved in homeostatic plasticity and compensation at the NMJ, in Drosophila in particular, are the size of readily releasable pool of synaptic vesicles and pre-synaptic calcium influx. It can occur through the post-synaptic inhibition of glutamate receptors, the impairment of muscle excitability, or through the alteration of the rates of innervation received by individual muscles, for Drosophila NMJ in particular as demonstrated through the use of fasciclin mutants (reviewed in [22]). It will be important to demonstrate the cues that are involved in synaptic homeostasis challenges, in addition to modulation of AchR density [23] during normal development and mono-innervation acquisition in mammalian central and peripheral synapses, but also in human health and pathophysiology.
Pre-synaptic components involved in the regulation of NMJ stability
Most of the cues mentioned below are illustrated in Figs. 3 and 4.
Fig. 3.
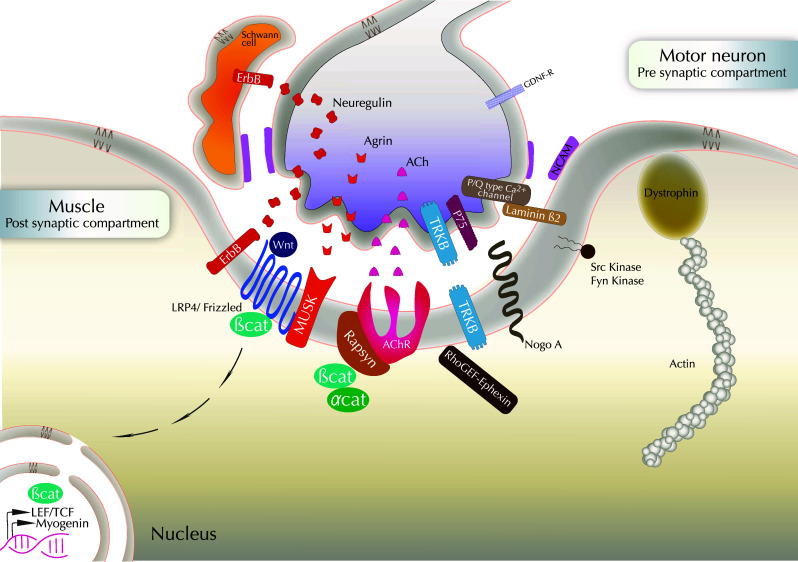
Molecular cues in NMJ formation/stabilization: a cross-talk between pre- and post-synaptic components. The agrin–muscle-specific kinase (MuSK)-rapsyn-AchR pathway is schematized. LRP4 acts as a co-factor for MuSK in agrin signaling. Agrin activates MuSK to cluster AChRs through the cytoplasmic linker protein rapsyn. Neuregulin that binds to ErbB receptors may also induce AChR transcription and agrin would direct AChR clustering. Neuregulin signaling also occurs from the axon to control Schwann cell survival. Schwann cells also belong to the tripartite NMJ with MN and muscle, and are essential for axon maintenance. Homophilic adhesion molecules such as NCAM are expressed on the surface of the three cell types composing the NMJ. Receptors to neurotrophic factors such as TrkB, p75 and GDNF receptors, are expressed at the MN surface. Actin regulators are present in the post-synaptic compartment, in particular Nogo-A, dystrophin and β-catenin. β-Catenin interacts with rapsyn and α-catenin to favor AChR clustering. β-Catenin-dependent transcription is also necessary for NMJ maintenance. Myogenin is involved in AchR expression, stabilization and clustering. Synaptic muscle fiber basal lamina is rich in laminin β2. It binds to and clusters the P/Q-type calcium channels that flank active zones and recruit other pre-synaptic components
Fig. 4.
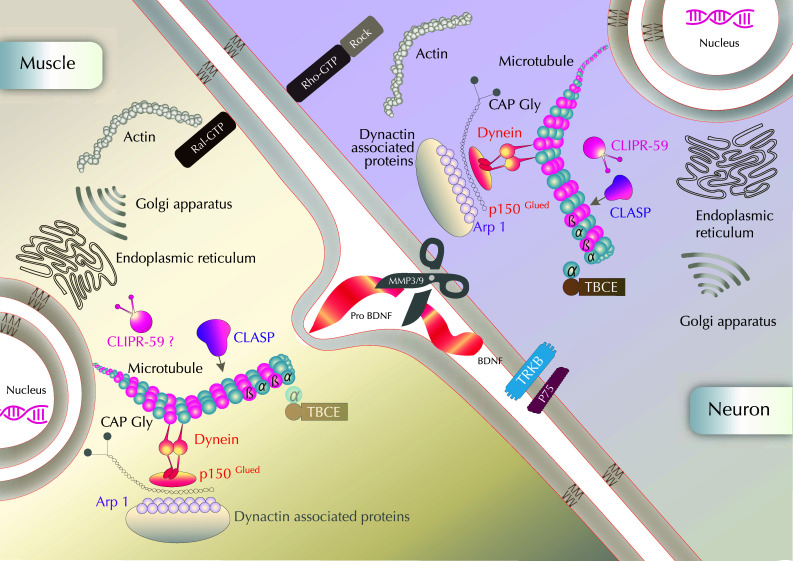
Possible intracellular mechanisms of NMJ stabilization through a focus on actors interacting with cytoskeleton and organelles The dynamics and stability of both actin and microtubules regulate NMJ maintenance. Dynactin complex includes among others Arp1, p150glued and dyneins. Although present in both pre- and post-synaptic compartments, the TBCE protein accumulates at the Golgi apparatus and has been shown to be mainly required for maintenance of microtubules in distal axons so far. CLIPR-59 is located at the trans-Golgi network (TGN) and is proposed to affect protein/membrane trafficking or cytoskeleton remodeling at the NMJ, as well as in the pre-synaptic compartment, although its possible post-synaptic localization and role remain to be further analyzed. Molecular candidates have been proposed to act as synaptotoxic and synaptotrophic cues in NMJ stability: MMP3 and MMP9 located at the active terminals could cleave proBDNF at the active terminal during synaptic competition. The conversion of pro-brain-derived neurotrophic factor (proBDNF) to mature (m)BDNF would be activity-dependent and mediate synaptic competition and cell survival after endocytosis
An extracellular matrix molecule: agrin
Agrin is the key neural factor that controls muscle post-synaptic differentiation. A physiological role of agrin, an essential synaptic organizing protein, is to counteract the destabilizing “antisynaptogenic” effects of the ACh neurotransmitter on nascent post-synaptic sites [24]. Agrin regulates nerve-induced transcriptional activation of several synapse-specific genes. Among them are neuregulins (NRG) that are expressed by motor neurons and activate ErbB receptors in muscle, and AchR [25]. Although initially described as an important inducer of AChR clustering in the post-synaptic membrane, agrin is now defined as a stabilizer of the post-synaptic membrane, rather than an inducer. Thus, the blockade of Neuregulin (NRG)/ErbB signaling also reduces the stability of receptors in agrin-induced AChR clusters in vitro [26]. Mice lacking neuregulin 1 or its receptors Erbb2 or Erbb3 expressed on Schwann cells surface lack Schwann cells [27], and their motoneurons form transient synapses with muscle fibers that fail to be maintained, indicating a crucial role for Schwann cells in NMJ formation and maintenance.
Although secreted by the pre-synaptic compartment, agrin directly modulates the organization of key post-synaptic components involved in NMJ stabilization. Agrin transiently activates the kinase MuSK, but also later on, the Src family kinases (SFKs) required for AChR clusters stabilization [28]. The stabilization of agrin-induced AChR clusters requires Src and Fyn in terms of “adaptor activities”, rather than the kinase activity (Fig. 3). Yes, which also belongs to the Src family kinases, can act with Src downstream ErbB2, and could also be involved in the stabilization process of AChRs clusters [29]. The stabilization of some prepatterned AChR clusters requires the innervation. Indeed, once the muscle has been contacted by the nerve, ACh released by the motor neuron induces a post-synaptic potential which stabilizes previous AChR clusters in the contacted area and prevents AChR clustering in non-contacted areas. Moreover, agrin released by the neuron also stabilizes the AChR clusters and along with neuregulin, strongly increases AChR transcription in subsynaptic nuclei [30].
Agrin mutation can cause congenital myasthenia, with dramatic perturbations of the maintenance of the NMJ [31]. The expression of mutated proteins in muscles destabilizes the wild-type NMJ but not the induction of post-synaptic structures. Some of the post-synaptic congenital myasthenic syndroma (CMS) including fetal akynesia are caused by mutations in agrin, but also in DOK7, GFPT1, musk and Rapsyn, all of them being part of a molecular pathway essential for AChR aggregation and positioning on the post-synaptic membrane that will be detailed below [32].
Adhesion molecules
Several CAMs have been identified at the neuromuscular junction where they regulate synaptic strength by recruiting scaffolding proteins, neurotransmitter receptors and synaptic vesicles in response to the binding of counter-receptors across the synaptic cleft. Among them, cadherins, protocadherins, neuroligins, neurexins, integrins, and immunoglobulin adhesion proteins can be cited. Among cadherins, we will underline the possible role of N-cadherin in the context of NMJ stabilization, but not of axon outgrowth. It accumulates at the neuromuscular junction only a few days after the first synaptic contacts have been established and remains at the adult neuromuscular junction, suggesting a role of this molecule in the stabilization of the mature neuromuscular junction. The presence of N-cadherin has also been described in basal lamina and its association with collagen fibers suggests the release of N-cadherin in the extracellular space [33].
Neurexin is mostly located on the pre-synaptic membrane. It is a synaptic cell adhesion protein critical for synapse formation, maturation and function. Its crucial role has been demonstrated for proper active zone apposition to post-synaptic densities, synaptic growth, and synaptic transmission. In vivo, it acts through the modulation of synaptic architecture and adhesive interactions between pre- and post-synaptic compartments, binding in particular proteins located in the synaptic cleft like Neuroligin.
The trans-synaptic Neurexin–Neuroligin complex can bridge this cleft, providing bidirectional communication across the synaptic cleft. It has recently been proposed in Drosophila that a post-synaptic actin cytoskeleton may function together with the Neurexin–Neuroligin trans-synaptic signaling complex to mediate normal synapse development and pre-synaptic active zone organization [34, 35]. Severe synapse assembly deficits are found in Drosophila melanogaster neurexin (Nrx-1, dnrx) and also neuroligin (Nlg1, dnlg1) mutant [36]. In addition, mutations in these genes in humans have been associated with cognitive disorders such as Autism spectrum disorders, Tourette syndrome and Schizophrenia. Such central pathologies are linked with lower amounts of central synapses, in particular in the dorsolateral prefrontal cortex (DLPC) for Schizophrenia. But it is not known whether this is due to an additional loss of synapses during normal adolescence—linked with reduced abnormal stabilization/increased destabilizing mechanisms—or whether it results from a failure to form a normal complement of synapses during childhood.
However, which adhesion factors establish the essential physical links across synaptic clefts and allow the assembly of synaptic machineries at the contact site in vivo is still unclear, mainly due to the redundancy that may occur among CAMs. Recent studies performed in Drosophila have pointed out the important contribution made by basal membrane-dependent mechanisms in addition to CAM-dependent adhesion [37].
Matrix metalloproteinases
Matrix metalloproteinases are key regulators of the extracellular matrix. Metalloproteases, particularly MMP3 and MMP9 that would be located at the active terminals, have been proposed to cleave proBDNF at the active terminal during synaptic competition in Xenopus NMJ. These proteases are expressed in motor neurons and are highly enriched at the NMJs [38], whereas proBDNF would be mainly secreted by muscle cells. Opposite data concerning the effects of inhibition of MMPs on synapse elimination remain to be reconciled.
Matrix metalloproteinase 3 (MMP3) has also been involved in the regulation of synaptic structure through its ability to cleave agrin and to remove it from the synaptic basal lamina [39]. Antibodies to MMP3 recognize molecules concentrated at the synapses of frog NMJs. NMJs in MMP3 null mutant mice have increased junctional folds and AChR aggregates [40]. The role of MMP3 in NMJ stabilization has not been directly tested, but MMP3 depletion prevents motor endplate degradation following traumatic peripheral nerve injury in KO mice.
It is noteworthy that the changes in synaptic activity will alter the activity of MMP3 at the synapse. Thus, the extracellular matrix is critical to the formation of the synapse, and synaptic activity controls the structure and function of the molecules in the extracellular matrix.
Agrin, microtubule-associated proteins and AChR clusters (Fig. 4)
Cytoplasmic linker associated proteins or CLASPs, are microtubule plus-end tracking proteins. The absence of CLASP2 has been reported to impair the maintenance of the neuromuscular junction with a decreased subsynaptic membrane in muscles, a decrease of synaptic AchRs and of the size of ACh clusters. Thus, the capturing of microtubules at the synaptic membrane through the effect of CLASP2 under the regulation by agrin is strictly required at the NMJ synaptic membrane [41]. Agrin modulates the local capture of dynamic microtubules at agrin-induced acetylcholine receptor clusters through the activation of PI3kinase and GSK3β inactivation, a process largely mediated by CLASP2. PKC activation also accelerates postnatal synapse loss [42] which also occurs in parallel with AChR cluster dispersal post-synaptically.
Dynactin is a multisubunit protein complex composed, among others, of p150Glued doublets that is required for most types of cytoplasmic dynein activity in eukaryotes. In Drosophila, Arp-1 (Actin-related protein-1)/centractin, a subunit of the dynactin complex, has been shown to regulate synapse retraction. Arp-1 dsRNA enhances synapse retraction, and this effect is phenocopied by a mutation in P150/Glued, also a dynactin component. Retraction is associated with a local disruption of the synaptic microtubule cytoskeleton. Altogether, these results suggest that dynactin functions locally within thepre-synaptic arbor to promote synapse stability as demonstrated in Drosophila. Whereas the dynactin complex is found in all tissues including muscle and the central and peripheral nervous systems, Glued is enriched in pre-synaptic NMJ, and only pre-synaptic dynactin function is necessary for synapse stabilization [43].
Another mouse model has provided interesting data about the role of microtubules in axonal degeneration of motor neurons. The tubulin-specific chaperone (TBCE) is a peripheral membrane-associated protein that accumulates at the Golgi apparatus. A function of TBCE is the binding of α-tubulin toward polymerizing microtubules. The mouse model of progressive motor neuronopathy (pmn) is mutated in the TBCE which disappears form the Golgi apparatus of motor neurons, and microtubules are lost in distal axons [44]. The axonal microtubule loss proceeds retrogradely in parallel with the axonal degeneration. Thus axonal tubulin routing from the Golgi apparatus involves tubulin chaperones that are required to allow NMJ stabilization.
Other microtubule-associated proteins and synapse stabilizations
Interestingly, another microtubule-associated protein of the cytoplasmic linker proteins (CLIP) family named CLIP3/CLIPR-59, and mainly localized at the trans-Golgi-network (TGN) has been recently shown to be involved in the stabilization of NMJ perinatally [45]. Indeed, in CLIP3 deficient embryos, animal death occurs perinatally, due to a decreased contraction force of the diaphragm and respiratory failure at birth. Whereas phrenic axon guidance normally occurs until E15, the diaphragm innervation pattern becomes incomplete between E15 and E18.5. Similar defects in axon maintenance have been observed in other muscles, in particular, in hindlimb muscles. The ultrastructural analysis of NMJs revealed that the number of nerve terminals was reduced in the ventral region of the diaphragm in particular, due to a decrease in branching complexity of nerve terminals or in the number of axons at NMJ. A mislocalization of Schwann cells has been reported in CLIP3 KO mice, suggesting premature phagocytosis and the elimination of nerve terminals. The structure and partners of CLIP3, as well as the phenotype of the KO mice suggest that protein/membrane trafficking or cytoskeleton remodeling play a key role for nerve terminal maintenance at the NMJ. Whereas the molecular mechanism of action of CLIP3 has not been elucidated so far, it appears to be necessary to prevent premature motor axon retraction during late embryogenesis. The maintenance of muscle multi-innervation by motor neurons would be strictly required for birth, possibly for the initial breathing and lung expansion and other movements in newborns. Thus, the requirement of the maintenance of multi-innervation perinatally would have a physiological role, and the multi-innervation elimination should not occur prior the postnatal period between P1 and P12. A different hypothesis was proposed from other studies. Synapse elimination is dramatically delayed in a specialized extraocular muscle, the levator palpebrae superioris (LPS). The delayed maturation could have a useful purpose since this specialized eyelid muscle remains immobile during early postnatal development. Thus, the maintenance of the multi-innervation could allow a rest state of the muscle [46]. Further studies will be required to determine whether polyinnervation may allow either a non synchronous but overactivity required at specific steps of development, or allow decreasing and resting activity according to the muscle types and specific NMJ physiology.
Through the analysis of differential defects in various types of motor units in CLIP3 KO, it appears that the selective impairments of synapse stability were possibly linked with specific physiology of motor units. Fast synapsing (FaSyn) and delayed synapsing (DeSyn) muscles have been reported to differ significantly with respect to the initial focal clustering of post-synaptic AChRs, the timing of pre-synaptic maturation, and the maintenance of NMJs in young adult mice [47]. In CLIP3 KO, DeSyn muscles were more affected than FaSyn muscles. In SOD1 mutants, curiously, at the same time, some motor neurons sprout to compensate the degeneration of other motor neurons, indicating distinct sensibilities among the same motor pool. Deciphering the specificity of motor neurons types would possibly allow correlations with distinct stability profiles. During ALS disease, some motor axon branches attempt to compensate for the loss of innervation, resulting in enhanced axonal arbors. An elegant in vivo approach [48] has established that degenerative versus regenerative changes are mainly confined to distinct populations of neurons, but within the same motor pool. Thus, either two types of signals are emitted toward motor neurons with specific characteristics during ALS, or among one motor pool, motor neurons have the ability to answer either by sprouting and by degenerating, or by being hyper-reactive to death signals synthesized in their close vicinity that leads to sprouting cascade.
Post-synaptic components involved in the regulation of NMJ stability
A sequence of required cross-talk between nerve and muscle for their proper maturation has proposed that post-synaptic areas bring the first trigger events. For example, during the development of NMJ (sternomastoid muscle), post-synaptic areas begin to be depleted of AChRs before there is any obvious loss of membrane in the nerve terminal [49]. The matter of stability/stabilization of certain synapses can also be considered as mechanisms involved in the selection of synapses to be eliminated. Nerve terminal withdrawal is accompanied by a loss of acetylcholine receptors (AChRs) at corresponding sites at developing NMJ.
Concomitantly with nerve terminal retraction, a loss of Schwann cell processes occurs in the post-synaptic apparatus. In case of crushes, it has been proposed that changes in Schwann cells occur after alterations in the post-synaptic receptor density. The stabilization of the post-synaptic compartment through a balanced dialog with the pre-synaptic activity may be another way of NMJ stabilization. Among other small GTPases activities that can act on the regulation of NMJ stability could be Ral and the exocyst. It has been reported that Ral mediates activity-dependent growth of post-synaptic membranes [50].
Extracellular matrix and adhesion molecules
Synaptic muscle fiber basal lamina is rich, among other components, in laminin. Laminin β2 mutant mice show vesicles that fail to aggregate near the pre-synaptic membrane and the formation of nerve terminals is severely impaired [51]. Interestingly, Schwann cells and their processes can be abnormally located in the synaptic cleft. This is the case in β2−/− mutants. Synaptic laminins have the ability to inhibit the extension of SC processes. Indeed, the maturation and maintenance of nerve terminals do not require collagen α2, but depend on laminin β2 [9, 52]. Laminin β2 probably binds directly to and clusters the P/Q-type calcium channels that flank active zones, which in turn recruit other pre-synaptic components [53] for a review.
Truncating mutations in the gene encoding the laminin β2 subunit (LAMB2) can cause a severe form of synaptic congenital myasthenic syndroma (CMS) due to various defects in the organization of the NMJ, including the reduction of axon terminal size [54].
At synapses, pre- and post-synaptic cells are in direct contact with each other via cell adhesion molecules (CAMs).
Genetic evidence indicates that cell adhesion molecules of the immunoglobulin superfamily (IgCAMs) are critical for activity-dependent synapse formation at the NMJ in Drosophila and have also been involved in synaptic remodelling during learning in Aplysia. In Drosophila, fasciclin II may play an important role in the maintenance of synapse integrity, particularly in the context of lesion and reinnervation. The role of neural cell adhesion molecule (NCAM), the fascII vertebrate homologue, has been investigated using NCAM−/− mutants. Mice that lack all three major isoforms of neural cell adhesion molecule (NCAM) (180 and 140 kDa transmembrane, and 120 kDa glycosylphosphatidylinositol linked) exhibit major alterations in the maturation of their NMJs [55]. Although functional NMJs form in NCAM-deficient mice, they show multiple alterations in pre-synaptic organization and function. The role of NCAM in the development and maturation of the NMJ was explored by structurally and functionally characterizing NMJs postnatally in NCAM null mutant mice. Both the withdrawal of polyneuronal innervation and the selective accumulation of synaptic vesicle protein in the pre-synaptic terminal were delayed [56].
Whereas many aspects of transmission are normal thanks to a proper assembling of many pre-synaptic and post-synaptic molecules in the absence of NCAM, the latter was indeed required for specific aspects of transmission, including paired-pulse facilitation and reliable transmission with repetitive stimuli, regulating directly or indirectly vesicle mobilization/cycling that are pre-synaptic processes [57].
NCAM null NMJs were unable to maintain effective transmitter output with high-frequency repetitive stimulation, exhibiting both severe initial depression and subsequent cyclical periods of total transmission failures that were of pre-synaptic origin.
In addition to its role in developmental maturation and stabilization of NMJs, the role of NCAM has also been investigated in reinnervation and stabilization of NMJs after nerve injury. Although redundance weakened phenotypes in mouse mutant models, the elimination of polyneuronal innervation was slowed down after nerve crushes [58]. In such models of nerve injury, the absence of NCAM affected the three components of the NMJ, i.e., motor neuron, muscle and Schwann cells, neither prevented nor delayed the recovery of contractile force. Nevertheless, 3 months post lesion (nerve crush), synapses were withdrawn. A loss of fast muscle fibers was also observed, leading to a decrease in contractile force, signs of inappropriate axonal withdrawal and impaired synaptic neurotransmission. Thus, the recovery of contractile force was the same in wild-type and NCAM−/− mice 1 month after nerve injury, but only transiently. NCAM is required to maintain normal synaptic function at reinnervated NMJs, although its loss pre-synaptically or post-synaptically is not sufficient to induce synaptic destabilization, suggesting that NCAM must be absent pre-synaptically and post-synaptically or absent on peri-synaptic terminal Schwann cells to destabilize the synapse after reinnervation [59].
Altogether, these data reveal that NCAM is required both for the normal course of polyneuronal elimination during development, and for maintaining normal muscle function through appropriate stabilization of motor axons after a peripheral nerve injury.
Rapsyn, a 43 kDa cytoplasmic protein, is precisely co-localized with AChRs at the NMJ
It is lost at the same rate as AChRs at junctions undergoing synapse elimination. In MuSK or rapsyn mutant muscle fibers, wild-type nerve terminals underwent continuous remodeling [60]. Normal post-synaptic differentiation appears to be dispensable for initial stages of pre-synaptic differentiation but required for pre-synaptic maturation. When nerves enter transplanted muscles derived from mice lacking muscle-specific receptor tyrosine kinase (MuSK) or rapsyn, wild-type nerve terminals undergo continuous remodeling, suggesting that these muscle components are required to stabilize the immature contacts so that they can mature [60]. Interestingly, biglycans that act as ligands for Musk are proposed to stabilize synapses after P14 once they reach their mature configuration, although they are neither necessary for synapse formation in vivo, nor for the initial AChR clustering in vitro [61]. Thus, NM stabilization would involve specific cues and cellular signaling that are specific of distinct steps of synapses maturation. Among them, LRP4 codes for the post-synaptic low-density lipoprotein receptor-related protein 4. LRP4 is expressed on the surface of the post-synaptic membrane of the NMJ and acts as a receptor for the neurally secreted agrin. Once LRP4 is bound by agrin, MuSK becomes activated. LRP4 acts as a co-factor for MuSK in agrin signaling, interacting with MuSK-like several other proteins in the early stages of synapse development including Dok-7 and Wnt11r [53]. Activated MuSK, together with Dok-7, stimulates rapsyn to concentrate and anchor AChR on the post-synaptic membrane and interacts with other proteins implicated in the assembly and maintenance of the NMJ. Another specific domain of LRP4 functions as an inhibitor of Wnt/beta-catenin signaling. β-Catenin interacts with rapsyn to favor AChR clustering, which also requires interaction with α-catenin. By interacting with rapsyn and α-catenin, β-catenin may link the AChR to the cytoskeleton [13]. Myogenin—a muscle-specific transcription factor—is involved in AchR expression, stabilization and clustering [62]. Some post-synaptic congenital myasthenic syndromes (CMS) are caused by mutations in agrin, musk and Rapsyn, all of them being part of a molecular pathway essential for AChR aggregation and positioning on the post-synaptic membrane [63]. LRP4 has been proposed to be a novel congenital myasthenic syndrome disease gene [64]. In myasthenia gravis (MG), a severely debilitating autoimmune disease that is due to a decrease in the efficiency of synaptic transmission at neuromuscular synapses, antibodies are generated against post-synaptic proteins, including acetylcholine receptors, MuSK, and (Lrp4), which prevent binding between MuSK and Lrp4, and inhibit agrin-stimulated MuSK phosphorylation [65].
Dystroglycan (DGC) is a multi-molecular complex including dystrophin glycoprotein complex altogether with dystrophin, a cytoskeletal protein. The homologue of dystrophin at the NMJ synapse is utrophin. Another cytoplasmic component is α-dystrobrevin. The roles of DGC have been analyzed in KO mouse models. They could be involved in the maintenance of the NMJ, although only analyzed in terms of AChR clusters sizes, which reveals the anchoring of the AChRs in the synaptic membrane as a sign of stabilized mature synapse. But no data have been reported as for axon terminals in these mutants after neurofilament stainings or electron microscopy. It is noteworthy that after denervation, a sequence of molecular loss occurs, syntrophin and dystrophin being lost later than rapsyn and utrophin. In addition, it has been shown that α-dystrobrevin tyrosine phosphorylation is strictly dependent on the functionality of Neuregulin (NRG)/ErbB signaling.
Nogo-A has been first described as an inhibitor of axon growth in the central nervous system. It has been shown that Nogo-A mRNA and protein levels do increase in mSOD1 mouse model as well as in amyotrophic lateral sclerosis (ALS) denervated muscle fiber biopsies. ALS is a fatal paralytic disease that targets motor neurons, leading to motor neurons death and widespread denervation with atrophy of muscle. Clinical observations reported so far can bring interesting cues to get a better understanding of other cellular processes or molecular cues possibly involved in NMJ stabilization. Nogo-A overexpression has also been reported more generally in other muscle pathologies such as peripheral neuropathies [66]. The overexpression of Nogo in patient is limited to oxidative fibers, and the levels of Nogo-A are correlated with the clinical state of the patient [67].
The ectopic expression levels of Nogo-A in the muscles of ALS patients correlate with the severity of clinical symptoms. In wild-type mouse fibers, the overexpression of Nogo-A leads to the shrinkage of the post-synapse and retraction of the pre-synaptic motor ending. Indeed it has been shown that Nogo-A, previously described as acting as an inhibitor of neurite outgrowth, is also able to promote denervation in an ALS model [68]. In ALS, Nogo-A early expression in skeletal muscles can cause the repulsion and the destabilization of the motor nerve terminals with axon elimination and motor neuronal death [68]. The cellular mechanisms may involve the Rho/ROCK pathway, since deleterious effects of Nogo as an axon growth inhibitor are reversed by blocking the Rho/ROCK pathway. Rho/ROCK could directly be involved in axon retraction, and possibly in initial collapse that could precede nerve terminal elimination. Whereas the precise role of RhoGTPase in NMJ stabilization remains to be further analyzed, it has been shown that the RhoGEF-ephexin1 regulates postnatally the stability of AChR clusters in a RhoA-dependent manner, regulating both the structural maturation of the post-synaptic apparatus and the precise neurotransmission of NMJs. Ephexin1 would mediate EphA-dependent dispersal of AChRs by a RhoA-dependent mechanism [69].
Underlying possible mechanisms in the regulation of NMJ stability
The stabilization of axons at the NMJ and the competition at the period of polyneuronal innervation elimination are not linked to apoptosis because they occur postnatally outside the period of motor neuron cell death. Nevertheless, the stabilization of the NMJ involves a synaptic competition that would be mediated by a “punishment” or “elimination” signal produced by the post-synaptic cell, which causes the retraction of some of the terminals, as well as a “protective” or “reward” signal that stabilizes one terminal [70]. A hypothesis to explain synapse elimination has been the active versus inactive synapses involving a competitive process. But no consensus could be reached as for favoring or destabilizing axon maintenance according to activity alone. A common feature of competition at neuromuscular as well as CNS synapses, is that temporally correlated/synchronous activity seems to slow or prevent competition, while uncorrelated/asynchronous activity seems to trigger or enhance competition [71, 72].
Whereas molecular cues involved in this process have remained poorly characterized so far, it has been recently shown that the activity-dependent conversion of pro-brain-derived neurotrophic factor (proBDNF) to mature (m)BDNF mediates synaptic competition [70]. The activity of motor neurons will trigger the proteolytic conversion of proBDNF to mBDNF at nerve terminals whose respective roles are opposite: when two distinct motor neuron axons innervate one myocyte, proBDNF-p75(NTR) signaling promotes the retraction of the less active terminal, whereas mBDNF-tyrosine-related kinase B (TrkB) p75NTR facilitates the stabilization of the more active one. Thus, the activity-dependent conversion of proBDNF to mBDNF may regulate synapse elimination, through the selection of active terminals, both in vivo and in vitro.
In vitro, a recent model consists in proposing a reward signal (mBDNF) which stabilizes the terminal by activating TrkB, whereas proBDNF would act as a default “punishment signal” to actively retract afferent terminals through p75NTR.
The role of other trophic/neurotrophic factors such as bFGF and CNTF has also been investigated. When injected in muscles, they exert powerful and long-lasting effects for the maintenance of polyneuronal innervation [73].
Concluding remarks
Whereas molecular mechanisms that regulate synapse formation have been well documented, little is known about the factors that modulate synaptic stability. Nevertheless, further identifying molecular cues involved in synapse stability would also probably inform on the mechanisms of synapse loss, which is an early and invariant feature of neurodegenerative diseases that can concern central and peripheral synapses. In Alzheimer’s disease (AD), the extent of synapse loss correlates with the severity of the disease. Hence, understanding the molecular mechanisms that underlie synaptic maintenance is crucial to reveal potential targets that will allow the development of therapies to protect synapses. Crossing information from the central nervous system to be applied to peripheral pathologies would be informative. Moreover, the molecular factors that are expressed both during development and adulthood can be of special interest, suggesting their role in synaptic maintenance in the adult. For example, Wnts has been shown to play a central role in the formation and function of neuronal circuits, and could be involved in synapse maintenance in the adult brain [74]. Its role has been studied in satellite cells in muscle, but not in neuromuscular cross-talks and NMJ stability.
It has long been unclear whether disease progression reflects temporally defined selective vulnerabilities and loss of specific synapses or axons, or stochastic loss in progression. Using mouse models, including mSOD1, it has been shown that fast fatigable (FF) motor axons are affected synchronously prior to fast fatigue resistant (FR) motor axons, both at symptom-onset, whereas axons of slow motor neurons are resistant [75].
In human multiple degenerative contexts including ALS, spinal muscular atrophy (SMA), and aging, fast fatigable (FF) motor units degenerate early, whereas motor neurons innervating slow muscles and those involved in eye movement and pelvic sphincter control are strikingly preserved. The diversity of motor neurons in terms of NMJ stabilization could also reveal a great diversity in motor neuron subtypes and help getting a clearer understanding of the cellular and molecular cues involved in normal development and pathological destabilization of NMJ. NMJ dismantlement has been reported to occur generally earlier in multiple degenerative contexts including ALS. A motor unit is defined as a motor neuron (alpha1 or 2) and the muscle fibers it innervates. The twitch speed of a muscle fiber largely depends on the motor neurons that innervate it. Muscle fibers themselves have distinct metabolic capabilities. Slow-twitch fibers rely primarily on oxidative metabolism, whereas Fast-twitch fibers may predominantly perform glycolytic conditions. Some fibers can be both oxidative and glycolytic.
More recently, it has been proposed that the muscle itself would initiate the pathology that then would lead to NMJ destruction, motor neuron degeneration and death. It would be due to an energetic deficit generated by an increase in basal and energetic muscle metabolism, with an increase in the peripheral use of lipids which leads to a reduced adipose tissue accumulation in mSOD1 [76]. The increased muscular metabolism leads to a decrease in fat reserves and a chronic energetic deficit. These results—along with a comparative analysis between the phenotype of mSOD1 mice and ALS patients—suggest new therapeutic strategies including nutritional modifications, for example, hyperlipidic diet [77].
In those diseases, functional changes in axonal transport have been hypothesized and increasing evidence is in favor of a role of ER stress (endoplasmic reticulum stress) in motor neuron degeneration [78]. Among the candidates that are involved in the NMJ stabilization, CLIPR-59 is the first that is located at the trans-Golgi network (TGN) so far. It also represents one of the recent candidates proposed to affect protein/membrane trafficking or cytoskeleton remodeling at the NMJ [45], and to be linked to axonal dieback from the NMJ. Thus, affecting the dynamics of intracellular compartments could itself affect the protein biosynthesis—as it is also the case in pmn model with mutated TBCE—and activate unfolded protein response leading to NMJ destabilization. So far, TBCE has been shown to bind microtubules and to protect against misfolded protein stress in yeast [79]. The extensive characterization of intracellular events involved in axon destabilization will bring cues to get a better understanding of signaling cascades involved in NMJ destabilization, both in normal development and in neurodegenerative disease.
Acknowledgments
We apologize for the authors whose work should have been cited to be fully exhaustive. This work was supported by funds from Institut National de la Santé et de la Recherche and the Fondation pour la Recherche Médicale. We thank Drs Serge Marty, Pascal Maire and Dominique Daegelen for their critical reading of the manuscript, and Annie Goldman for English proof-reading.
Conflict of interest
The authors declare no competing financial interests.
References
- 1.Gould TW, Burek MJ, Sosnowski JM, Prevette D, Oppenheim RW. The spatial-temporal gradient of naturally occurring motoneuron death reflects the time of prior exit from the cell cycle and position within the lateral motor column. Dev Biol. 1999;216:611–621. doi: 10.1006/dbio.1999.9490. [DOI] [PubMed] [Google Scholar]
- 2.Tsuchida T, Ensini M, Morton SB, Baldassare M, Edlund T, Jessell TM, Pfaff SL. Topographic organization of embryonic motor neurons defined by expression of LIM homeobox genes. Cell. 1994;79:957–970. doi: 10.1016/0092-8674(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 3.Sharma K, Leonard AE, Lettieri K, Pfaff SL. Genetic and epigenetic mechanisms contribute to motor neuron pathfinding. Nature. 2000;406:515–519. doi: 10.1038/35020078. [DOI] [PubMed] [Google Scholar]
- 4.Landmesser LT. The acquisition of motoneuron subtype identity and motor circuit formation. Int J Dev Neurosci. 2001;19:175–182. doi: 10.1016/S0736-5748(00)00090-3. [DOI] [PubMed] [Google Scholar]
- 5.Kanning KC, Kaplan A, Henderson CE. Motor neuron diversity in development and disease. Annu Rev Neurosci. 2010;33:409–440. doi: 10.1146/annurev.neuro.051508.135722. [DOI] [PubMed] [Google Scholar]
- 6.Burke RE, Levine DN, Tsairis P, Zajac FE., 3rd Physiological types and histochemical profiles in motor units of the cat gastrocnemius. J Physiol. 1973;234:723–748. doi: 10.1113/jphysiol.1973.sp010369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friese A, Kaltschmidt JA, Ladle DR, Sigrist M, Jessell TM, Arber S. Gamma and alpha motor neurons distinguished by expression of transcription factor Err3. Proc Natl Acad Sci USA. 2009;106:13588–13593. doi: 10.1073/pnas.0906809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yumoto N, Kim N, Burden SJ. Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature. 2012;489:438–442. doi: 10.1038/nature11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishimune H, Valdez G, Jarad G, Moulson CL, Muller U, Miner JH, Sanes JR. Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. J Cell Biol. 2008;182:1201–1215. doi: 10.1083/jcb.200805095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mantilla CB, Sieck GC. Key aspects of phrenic motoneuron and diaphragm muscle development during the perinatal period. J Appl Physiol. 2008;104:1818–1827. doi: 10.1152/japplphysiol.01192.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamburger V. History of the discovery of neuronal death in embryos. J Neurobiol. 1992;23:1116–1123. doi: 10.1002/neu.480230904. [DOI] [PubMed] [Google Scholar]
- 12.Li XM, Dong XP, Luo SW, Zhang B, Lee DH, Ting AK, Neiswender H, Kim CH, Carpenter-Hyland E, Gao TM, et al. Retrograde regulation of motoneuron differentiation by muscle beta-catenin. Nat Neurosci. 2008;11:262–268. doi: 10.1038/nn2053. [DOI] [PubMed] [Google Scholar]
- 13.Zhang B, Luo S, Dong XP, Zhang X, Liu C, Luo Z, Xiong WC, Mei L. Beta-catenin regulates acetylcholine receptor clustering in muscle cells through interaction with rapsyn. J Neurosci. 2007;27:3968–3973. doi: 10.1523/JNEUROSCI.4691-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Lao J, Gao K, Gu Y, Xin Z. Outcome of nerve transfers for traumatic complete brachial plexus avulsion: results of 28 patients by DASH and NRS questionnaires. J Hand Surg Eur. 2012;37:413–421. doi: 10.1177/1753193411425330. [DOI] [PubMed] [Google Scholar]
- 15.Van Essen DC, Gordan H, Soha JM, Fraser SE. Synaptic dynamics at the neuromuscular junction: mechanisms and models. Review. J Neurobiol. 1990;21:223–249. doi: 10.1002/neu.480210115. [DOI] [PubMed] [Google Scholar]
- 16.Bernstein M, Lichtman JW. Axonal atrophy: the retraction reaction. Curr Opin Neurobiol. 1999;9:364–370. doi: 10.1016/S0959-4388(99)80053-1. [DOI] [PubMed] [Google Scholar]
- 17.Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- 18.Duxson MJ. The effect of postsynaptic block on development of the neuromuscular junction in postnatal rats. J Neurocytol. 1982;11:395–408. doi: 10.1007/BF01257985. [DOI] [PubMed] [Google Scholar]
- 19.Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR. Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron. 2002;36:635–648. doi: 10.1016/S0896-6273(02)01020-6. [DOI] [PubMed] [Google Scholar]
- 20.Buffelli M, Busetto G, Cangiano L, Cangiano A. Perinatal switch from synchronous to asynchronous activity of motoneurons: link with synapse elimination. Proc Natl Acad Sci USA. 2002;99:13200–13205. doi: 10.1073/pnas.202471199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Favero M, Massella O, Cangiano A, Buffelli M. On the mechanism of action of muscle fibre activity in synapse competition and elimination at the mammalian neuromuscular junction. Eur J Neurosci. 2009;29:2327–2334. doi: 10.1111/j.1460-9568.2009.06779.x. [DOI] [PubMed] [Google Scholar]
- 22.Frank CA. Homeostatic plasticity at the Drosophila neuromuscular junction. Neuropharmacology. 2014;78:63–74. doi: 10.1016/j.neuropharm.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Pena y Valenzuela I, Pires-Oliveira M, Akaaboune M. PKC and PKA regulate AChR dynamics at the neuromuscular junction of living mice. PLoS One. 2013;8:e81311. doi: 10.1371/journal.pone.0081311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Misgeld T, Kummer TT, Lichtman JW, Sanes JR. Agrin promotes synaptic differentiation by counteracting an inhibitory effect of neurotransmitter. Proc Natl Acad Sci USA. 2005;102:11088–11093. doi: 10.1073/pnas.0504806102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mejat A, Ravel-Chapuis A, Vandromme M, Schaeffer L. Synapse-specific gene expression at the neuromuscular junction. Ann N Y Acad Sci. 2003;998:53–65. doi: 10.1196/annals.1254.008. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt R, Weidner C, Schmelz M. Time course of acetylcholine-induced activation of sympathetic efferents matches axon reflex sweating in humans. J Peripher Nerv Syst. 2011;16:30–36. doi: 10.1111/j.1529-8027.2011.00320.x. [DOI] [PubMed] [Google Scholar]
- 27.Lin W, Sanchez HB, Deerinck T, Morris JK, Ellisman M, Lee KF. Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc Natl Acad Sci USA. 2000;97:1299–1304. doi: 10.1073/pnas.97.3.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mittaud P, Camilleri AA, Willmann R, Erb-Vogtli S, Burden SJ, Fuhrer C. A single pulse of agrin triggers a pathway that acts to cluster acetylcholine receptors. Mol Cell Biol. 2004;24:7841–7854. doi: 10.1128/MCB.24.18.7841-7854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith CL, Mittaud P, Prescott ED, Fuhrer C, Burden SJ. Src, Fyn, and Yes are not required for neuromuscular synapse formation but are necessary for stabilization of agrin-induced clusters of acetylcholine receptors. J Neurosci. 2001;21:3151–3160. doi: 10.1523/JNEUROSCI.21-09-03151.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferraro E, Molinari F, Berghella L. Molecular control of neuromuscular junction development. J Cachexia Sarcopenia Muscle. 2012;3:13–23. doi: 10.1007/s13539-011-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huze C, Bauche S, Richard P, Chevessier F, Goillot E, Gaudon K, Ben Ammar A, Chaboud A, Grosjean I, Lecuyer HA, et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am J Hum Genet. 2009;85:155–167. doi: 10.1016/j.ajhg.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russell JF, Fu YH, Ptacek LJ. Episodic neurologic disorders: syndromes, genes, and mechanisms. Annu Rev Neurosci. 2013;36:25–50. doi: 10.1146/annurev-neuro-062012-170300. [DOI] [PubMed] [Google Scholar]
- 33.Cifuentes-Diaz C, Nicolet M, Goudou D, Rieger F, Mege RM. N-cadherin expression in developing, adult and denervated chicken neuromuscular system: accumulations at both the neuromuscular junction and the node of Ranvier. Development. 1994;120:1–11. doi: 10.1242/dev.120.1.1. [DOI] [PubMed] [Google Scholar]
- 34.Owald D, Khorramshahi O, Gupta VK, Banovic D, Depner H, Fouquet W, Wichmann C, Mertel S, Eimer S, Reynolds E, et al. Cooperation of Syd-1 with Neurexin synchronizes pre- with postsynaptic assembly. Nat Neurosci. 2012;15:1219–1226. doi: 10.1038/nn.3183. [DOI] [PubMed] [Google Scholar]
- 35.Blunk AD, Akbergenova Y, Cho RW, Lee J, Walldorf U, Xu K, Zhong G, Zhuang X, Littleton JT. Postsynaptic actin regulates active zone spacing and glutamate receptor apposition at the Drosophila neuromuscular junction. Mol Cell Neurosci. 2014;61:241–254. doi: 10.1016/j.mcn.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen YC, Lin YQ, Banerjee S, Venken K, Li J, Ismat A, Chen K, Duraine L, Bellen HJ, Bhat MA. Drosophila neuroligin 2 is required presynaptically and postsynaptically for proper synaptic differentiation and synaptic transmission. J Neurosci. 2012;32:16018–16030. doi: 10.1523/JNEUROSCI.1685-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koper A, Schenck A, Prokop A. Analysis of adhesion molecules and basement membrane contributions to synaptic adhesion at the Drosophila embryonic NMJ. PLoS ONE. 2012;7:e36339. doi: 10.1371/journal.pone.0036339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kherif S, Dehaupas M, Lafuma C, Fardeau M, Alameddine HS. Matrix metalloproteinases MMP-2 and MMP-9 in denervated muscle and injured nerve. Neuropathol Appl Neurobiol. 1998;24:309–319. doi: 10.1046/j.1365-2990.1998.00118.x. [DOI] [PubMed] [Google Scholar]
- 39.VanSaun M, Werle MJ. Matrix metalloproteinase-3 removes agrin from synaptic basal lamina. J Neurobiol. 2000;44:369. doi: 10.1002/1097-4695(20000905)44:3<369::AID-NEU7>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 40.Werle MJ. Cell-to-cell signaling at the neuromuscular junction: the dynamic role of the extracellular matrix. Ann N Y Acad Sci. 2008;1132:13–18. doi: 10.1196/annals.1405.035. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt N, Basu S, Sladecek S, Gatti S, van Haren J, Treves S, Pielage J, Galjart N, Brenner HR. Agrin regulates CLASP2-mediated capture of microtubules at the neuromuscular junction synaptic membrane. J Cell Biol. 2012;198:421–437. doi: 10.1083/jcb.201111130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lanuza MA, Santafe MM, Garcia N, Besalduch N, Tomas M, Obis T, Priego M, Nelson PG, Tomas J. Protein kinase C isoforms at the neuromuscular junction: localization and specific roles in neurotransmission and development. J Anat. 2014;224:61–73. doi: 10.1111/joa.12106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eaton BA, Fetter RD, Davis GW. Dynactin is necessary for synapse stabilization. Neuron. 2002;34:729–741. doi: 10.1016/S0896-6273(02)00721-3. [DOI] [PubMed] [Google Scholar]
- 44.Schaefer MK, Schmalbruch H, Buhler E, Lopez C, Martin N, Guenet JL, Haase G. Progressive motor neuronopathy: a critical role of the tubulin chaperone TBCE in axonal tubulin routing from the Golgi apparatus. J Neurosci. 2007;27:8779–8789. doi: 10.1523/JNEUROSCI.1599-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Couesnon A, Offner N, Bernard V, Chaverot N, Backer S, Dimitrov A, Perez F, Molgo J, Bloch-Gallego E. CLIPR-59: a protein essential for neuromuscular junction stability during mouse late embryonic development. Development. 2013;140:1583–1593. doi: 10.1242/dev.087106. [DOI] [PubMed] [Google Scholar]
- 46.Fox MA, Tapia JC, Kasthuri N, Lichtman JW. Delayed synapse elimination in mouse levator palpebrae superioris muscle. J Comp Neurol. 2011;519:2907–2921. doi: 10.1002/cne.22700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pun S, Sigrist M, Santos AF, Ruegg MA, Sanes JR, Jessell TM, Arber S, Caroni P. An intrinsic distinction in neuromuscular junction assembly and maintenance in different skeletal muscles. Neuron. 2002;34:357–370. doi: 10.1016/S0896-6273(02)00670-0. [DOI] [PubMed] [Google Scholar]
- 48.Schaefer AM, Sanes JR, Lichtman JW. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J Comp Neurol. 2005;490:209–219. doi: 10.1002/cne.20620. [DOI] [PubMed] [Google Scholar]
- 49.Balice-Gordon RJ, Lichtman JW. In vivo observations of pre- and postsynaptic changes during the transition from multiple to single innervation at developing neuromuscular junctions. J Neurosci. 1993;13:834–855. doi: 10.1523/JNEUROSCI.13-02-00834.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teodoro RO, Pekkurnaz G, Nasser A, Higashi-Kovtun ME, Balakireva M, McLachlan IG, Camonis J, Schwarz TL. Ral mediates activity-dependent growth of postsynaptic membranes via recruitment of the exocyst. EMBO J. 2013;32:2039–2055. doi: 10.1038/emboj.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP. Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin beta 2. Nature. 1995;374:258–262. doi: 10.1038/374258a0. [DOI] [PubMed] [Google Scholar]
- 52.Fox MA, Sanes JR, Borza DB, Eswarakumar VP, Fassler R, Hudson BG, John SW, Ninomiya Y, Pedchenko V, Pfaff SL, et al. Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell. 2007;129:179–193. doi: 10.1016/j.cell.2007.02.035. [DOI] [PubMed] [Google Scholar]
- 53.Wu H, Xiong WC, Mei L. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. 2010;137:1017–1033. doi: 10.1242/dev.038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maselli RA, Ng JJ, Anderson JA, Cagney O, Arredondo J, Williams C, Wessel HB, Abdel-Hamid H, Wollmann RL. Mutations in LAMB2 causing a severe form of synaptic congenital myasthenic syndrome. J Med Genet. 2009;46:203–208. doi: 10.1136/jmg.2008.063693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Polo-Parada L, Bose CM, Plattner F, Landmesser LT. Distinct roles of different neural cell adhesion molecule (NCAM) isoforms in synaptic maturation revealed by analysis of NCAM 180 kDa isoform-deficient mice. J Neurosci. 2004;24:1852–1864. doi: 10.1523/JNEUROSCI.4406-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rafuse VF, Polo-Parada L, Landmesser LT. Structural and functional alterations of neuromuscular junctions in NCAM-deficient mice. J Neurosci. 2000;20:6529–6539. doi: 10.1523/JNEUROSCI.20-17-06529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Polo-Parada L, Bose CM, Landmesser LT. Alterations in transmission, vesicle dynamics, and transmitter release machinery at NCAM-deficient neuromuscular junctions. Neuron. 2001;32:815–828. doi: 10.1016/S0896-6273(01)00521-9. [DOI] [PubMed] [Google Scholar]
- 58.Sanes JR, Apel ED, Burgess RW, Emerson RB, Feng G, Gautam M, Glass D, Grady RM, Krejci E, Lichtman JW, et al. Development of the neuromuscular junction: genetic analysis in mice. J Physiol Paris. 1998;92:167–172. doi: 10.1016/S0928-4257(98)80004-1. [DOI] [PubMed] [Google Scholar]
- 59.Chipman PH, Franz CK, Nelson A, Schachner M, Rafuse VF. Neural cell adhesion molecule is required for stability of reinnervated neuromuscular junctions. Eur J Neurosci. 2010;31:238–249. doi: 10.1111/j.1460-9568.2009.07049.x. [DOI] [PubMed] [Google Scholar]
- 60.Nguyen QT, Son YJ, Sanes JR, Lichtman JW. Nerve terminals form but fail to mature when postsynaptic differentiation is blocked: in vivo analysis using mammalian nerve-muscle chimeras. J Neurosci. 2000;20:6077–6086. doi: 10.1523/JNEUROSCI.20-16-06077.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Amenta AR, Creely HE, Mercado ML, Hagiwara H, McKechnie BA, Lechner BE, Rossi SG, Wang Q, Owens RT, Marrero E, et al. Biglycan is an extracellular MuSK binding protein important for synapse stability. J Neurosci. 2012;32:2324–2334. doi: 10.1523/JNEUROSCI.4610-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Macpherson PC, Cieslak D, Goldman D. Myogenin-dependent nAChR clustering in aneural myotubes. Mol Cell Neurosci. 2006;31:649–660. doi: 10.1016/j.mcn.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 63.Chevessier F, Faraut B, Ravel-Chapuis A, Richard P, Gaudon K, Bauche S, Prioleau C, Herbst R, Goillot E, Ioos C, et al. Towards the molecular elucidation of congenital myasthenic syndromes: identification of mutations in MuSK. Acta Myol. 2005;24:55–59. [PubMed] [Google Scholar]
- 64.Ohkawara B, Cabrera-Serrano M, Nakata T, Milone M, Asai N, Ito K, Ito M, Masuda A, Ito Y, Engel AG, et al. LRP4 third beta-propeller domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSK signaling in a position-specific manner. Hum Mol Genet. 2014;23:1856–1868. doi: 10.1093/hmg/ddt578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huijbers MG, Zhang W, Klooster R, Niks EH, Friese MB, Straasheijm KR, Thijssen PE, Vrolijk H, Plomp JJ, Vogels P, et al. MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc Natl Acad Sci USA. 2013;110:20783–20788. doi: 10.1073/pnas.1313944110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wojcik S, Engel WK, Askanas V. Increased expression of Noga-A in ALS muscle biopsies is not unique for this disease. Acta Myol. 2006;25:116–118. [PubMed] [Google Scholar]
- 67.Dupuis L, Gonzalez de Aguilar JL, di Scala F, Rene F, de Tapia M, Pradat PF, Lacomblez L, Seihlan D, Prinjha R, Walsh FS, et al. Nogo provides a molecular marker for diagnosis of amyotrophic lateral sclerosis. Neurobiol Dis. 2002;10:358–365. doi: 10.1006/nbdi.2002.0522. [DOI] [PubMed] [Google Scholar]
- 68.Jokic N, Gonzalez de Aguilar JL, Dimou L, Lin S, Fergani A, Ruegg MA, Schwab ME, Dupuis L, Loeffler JP. The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model. EMBO Rep. 2006;7:1162–1167. doi: 10.1038/sj.embor.7400826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shi L, Butt B, Ip FC, Dai Y, Jiang L, Yung WH, Greenberg ME, Fu AK, Ip NY. Ephexin1 is required for structural maturation and neurotransmission at the neuromuscular junction. Neuron. 2010;65:204–216. doi: 10.1016/j.neuron.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Je HS, Yang F, Ji Y, Nagappan G, Hempstead BL, Lu B. Role of pro-brain-derived neurotrophic factor (proBDNF) to mature BDNF conversion in activity-dependent competition at developing neuromuscular synapses. Proc Natl Acad Sci USA. 2012;109:15924–15929. doi: 10.1073/pnas.1207767109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wyatt RM, Balice-Gordon RJ. Activity-dependent elimination of neuromuscular synapses. J Neurocytol. 2003;32:777–794. doi: 10.1023/B:NEUR.0000020623.62043.33. [DOI] [PubMed] [Google Scholar]
- 72.Nguyen QT, Lichtman JW. Mechanism of synapse disassembly at the developing neuromuscular junction. Curr Opin Neurobiol. 1996;6:104–112. doi: 10.1016/S0959-4388(96)80015-8. [DOI] [PubMed] [Google Scholar]
- 73.English AW, Schwartz G. Both basic fibroblast growth factor and ciliary neurotrophic factor promote the retention of polyneuronal innervation of developing skeletal muscle fibers. Dev Biol. 1995;169:57–64. doi: 10.1006/dbio.1995.1126. [DOI] [PubMed] [Google Scholar]
- 74.Purro SA, Galli S, Salinas PC. Dysfunction of Wnt signaling and synaptic disassembly in neurodegenerative diseases. J Mol Cell Biol. 2014;6:75–80. doi: 10.1093/jmcb/mjt049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- 76.Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high-energy diet in a transgenic mouse model. Proc Natl Acad Sci USA. 2004;101:11159–11164. doi: 10.1073/pnas.0402026101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mattson MP, Cutler RG, Camandola S. Energy intake and amyotrophic lateral sclerosis. Neuromol Med. 2007;9:17–20. doi: 10.1385/NMM:9:1:17. [DOI] [PubMed] [Google Scholar]
- 78.Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- 79.Voloshin O, Gocheva Y, Gutnick M, Movshovich N, Bakhrat A, Baranes-Bachar K, Bar-Zvi D, Parvari R, Gheber L, Raveh D. Tubulin chaperone E binds microtubules and proteasomes and protects against misfolded protein stress. Cell Mol Life Sci. 2010;67:2025–2038. doi: 10.1007/s00018-010-0308-8. [DOI] [PMC free article] [PubMed] [Google Scholar]