

Abstract
We have recently found that celiac disease patient serum-derived autoantibodies targeted against transglutaminase 2 interfere with several steps of angiogenesis, including endothelial sprouting and migration, though the mechanism involved remained to be fully characterized. This study now investigated the processes underlying the antiangiogenic effects exerted by celiac disease patient antibodies on endothelial cells, with particular regard to the adhesion, migration, and polarization signaling pathway. We observed that celiac IgA reduced endothelial cell numbers by affecting adhesion without increasing apoptosis. Endothelial cells in the presence of celiac IgA showed weak attachment, a high susceptibility to detach from fibronectin, and a disorganized extracellular matrix due to a reduction of protein cross-links. Furthermore, celiac patient IgA led to secretion of active transglutaminase 2 from endothelial cells into the culture supernatants. Additionally, cell surface transglutaminase 2 mediated integrin clustering in the presence of celiac IgA was coupled to augmented expression of β1-integrin. We also observed that celiac patient IgA-treated endothelial cells had migratory defects and a less polarized phenotype when compared to control groups, and this was associated with the RhoA signaling pathway. These biological effects mediated by celiac IgA on endothelial cells were partially influenced but not completely abolished by R281, an irreversible extracellular transglutaminase 2 enzymatic activity inhibitor. Taken together, our results imply that celiac patient IgA antibodies disturb the extracellular protein cross-linking function of transglutaminase 2, thus altering cell-extracellular matrix interactions and thereby affecting endothelial cell adhesion, polarization, and motility.
Keywords: Celiac disease antibodies, Endothelial cells, Transglutaminase 2, Integrins, Adhesion, Polarization
Introduction
In the small intestine, an organized array of finger-like villi provides an extensive epithelial surface area for absorptive function. The integrity of the villi relies on an appropriate vascular network providing mechanical support and thus helping to keep the villi tall [1]. In celiac disease, an autoimmune disorder triggered by ingested dietary gluten in susceptible individuals carrying human leukocyte antigen (HLA) DQ2 or DQ8 genotype, the overall architecture of the mucosal vasculature is abnormal [2, 3]. The blood vessel alterations occur in the context of small bowel mucosal villous atrophy, crypt hyperplasia, and inflammation, these constituting typical features of the disease.
Celiac disease is also hallmarked by a strong antibody response targeted against dietary gluten-derived deamidated gliadin peptides and an autoantigen, transglutaminase 2 (TG2) [4–6]. The target of the disease-specific IgA class autoantibodies, TG2, is a highly complex multifunctional protein with both enzymatic and non-enzymatic functions. As an enzyme, TG2 is able to catalyze post-translational modifications of glutamine residues on proteins and peptides through transamidation or deamidation reactions [7]. The TG2-mediated deamidation reaction is of particular importance during the pathogenesis of celiac disease, as distinct gliadin peptides can be deamidated by TG2, resulting in their increased affinity for HLA DQ2 and DQ8, thereby enabling a stronger antigen presentation and inflammatory response [8]. As a transamidase, again, TG2 catalyzes the formation of ε(γ-glutamyl)-lysine crosslinks between various proteins, including fibronectin (FN) and collagen, and this activity plays an important role in the organization of the extracellular matrix [9]. On the other hand, the non-enzymatic functions of TG2 include its role in cell adhesion via action as an FN co-receptor for β integrins [10] and syndecans [11].
The various functions of TG2 are involved in a variety of basic biological processes, including angiogenesis [12, 13]. The modulation of angiogenesis by TG2 is apparently complex, as there are data showing that both increased and decreased TG2 enzymatic activity leads to altered angiogenic response [14–17]. Our group has previously shown that celiac disease autoantibodies targeted against TG2 inhibit angiogenesis [18] and increase vascular permeability [2]. It appears that these outcomes involve increased enzymatic activity of TG2 [2, 19] and the Rho family of small GTPase proteins [2, 20], although the detailed molecular mechanism whereby this occurs is not yet fully understood. This present study sought to provide new insights into the molecular mechanism behind the altered endothelial cell biology and inhibition of angiogenesis mediated by celiac disease patient antibodies.
Materials and methods
Cell culture and reagents
Human umbilical vein endothelial cells (HUVECs) (Lonza Cambrex Bio Science, Walkersville, MD, USA) were maintained in EGM-2 medium (Clonetics, San Diego, CA, USA) with 2 % fetal bovine serum (FBS) (Gibco Invitrogen, Paisley, Scotland) and 15 μg/ml endothelial cell growth supplements (Clonetics). Cells between passages two and six were used in the experiments. In the experiments, the following chemical compounds administered 1 h before the addition of serum IgA were used: a non-reversible and cell-impermeable inhibitor for extracellular TG2 activity, R281 [21], at a final concentration of 200 μM, and a cell-permeable Rho inhibitor C3 transferase (Cytoskeleton, Denver, CO, USA) at 1 μg/ml.
Purification of serum IgA class antibodies
Serum samples from nine biopsy-proven celiac disease patients on a gluten-containing diet and five non-celiac healthy controls were included in the present study. All celiac sera had anti-TG2 antibody titers above 100 U/ml (Celikey; Pharmacia Diagnostic GmbH, Freiburg, Germany) and endomysial antibodies titers 1:≥1,000, while all control sera were negative. Total serum IgA were purified as previously described with anti-Human IgA agarose (Sigma-Aldrich, St. Louis, MO, USA) [18] and diluted in phosphate-buffered saline (PBS) to a final concentration of 100 μg/ml. Purified IgA fractions were used in all experiments at a concentration of 1 μg/ml. The study protocol was approved by the Ethics Committee of Tampere University Hospital, Tampere, Finland. All patients gave written informed consent.
Cell number assay, detachment, and adhesion assay
For the determination of endothelial cell number, 1 × 104 HUVECs were plated on 96-well plates (Nunclon, Thermo Fisher Scientific, Rockford, IL, USA), pre-coated with 5 μg of human plasma FN (Sigma-Aldrich) and grown to confluence. Thereafter, the cells were starved overnight (O/N) in 1 % FBS before a 24-h treatment with IgA. Subsequently, the number of HUVECS was determined by [3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl] tetrazolium bromide CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) (Promega, Madison, WI, USA) following the manufacturer’s instructions. Experiments were performed in quadruplicate and repeated three times. The results are expressed as percentage of HUVECs without any treatment.
In order to study the susceptibility of cells to detach, HUVECs grown and treated as above were trypsinized (Sigma-Aldrich) for 5 min and carefully washed with PBS. The number of adherent cells remaining on the plate was determined by MTS assay as above (Promega). Experiments were performed in quadruplicate and repeated three times. The results are expressed as percentage of HUVECs without any treatment.
Further, to investigate weak versus strong adhesion, confluent HUVEC monolayers grown on six-well plates (Sigma-Aldrich) were starved O/N in 1 % FBS, treated with IgA for 24 h, and detached with 5 mM EDTA in PBS (pH 7.4). Subsequently, 1 × 104 cells in serum-free culture media were allowed to adhere for 1 h on FN-precoated chamber slides (BD-Falcon, Bedford, MA, USA). The cells were carefully washed with PBS (pH 7.4) and then fixed with 4 % paraformaldehyde (4 % PFA). Images of cells from non-overlapping fields were taken at 20× magnification using the Axiovision 3.0 program (Carl Zeiss Vision GmbH, Munich, Germany). Weakly adhering cells (round cells) were counted manually on captured images. The results are given as the percentage of weakly adhering cells among all cells.
Quantification of apoptotic cells
The number of cells undergoing apoptosis was quantified using an Annexin V-FITC kit (Calbiochem, Beckman Coulter, Brea, CA, USA), according to the manufacturer’s instructions. Briefly, 1 × 104 cells in different culture conditions were collected, washed, resuspended in binding buffer, and mixed with Annexin V-FITC and propidium iodine. After 5-min dark incubation at room temperature (RT), cells were analyzed using a flow cytometer (Expo32 ADC, Epics_XL-MCL, Beckman Coulter).
Microtiter plate assay for quantification of surface and matrix expression of proteins
HUVECs were grown to confluence on commercial 96-well plates (Nunclon). After O/N starvation in EGM-2 with 1 % of FBS, they were incubated for 24 h in the presence of purified serum IgA. The cells were then washed with Hanks’ balanced salt solution (HBSS) (Sigma-Aldrich), fixed with 4 % paraformaldehyde (PFA) (Sigma-Aldrich) and blocked with 5 % bovine serum albumin (BSA) (Sigma-Aldrich). The plates were then incubated with primary antibodies against human FN (1:200) (F3648, Sigma-Aldrich), N-ε-(γ-l-glutamyl)-l-lysine isopeptide (1:200) (mab0012, Covalab, Villeurbanne, France), MT1-MMP (1:200) (MAB3328, Millipore, Billerica, MA, USA), β1-integrin (1:200) (MAB1965, Millipore) or vinculin (1:200) (7F9, Santa Cruz Biotechnology, Dallas, TX, USA) for 1 h at 37 °C. After incubation with primary antibodies, the cells were extensively washed and incubated in the presence of secondary horseradish peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulins (1:2,000) (Dako, Glostrup, Denmark) at 37 °C for 1 h. Finally, the peroxidase substrate TMB (Sigma-Aldrich) was used and the enzymatic reaction was stopped with 2.5 M H2SO4. Absorbance was measured at 450 nm by spectrophotometer (Multiskan Ascent, Thermo Labsystems, Santa Clara, CA, USA).
Immunofluorescent stainings
HUVECs were grown to confluence on FN-precoated chamber slides (BD-Falcon) and subjected to different experimental conditions. For visualization of cross-links, the cells were washed with PBS, fixed in 4 % PFA, and then blocked with 1 % BSA. Immunolabeling was carried out using anti-N-ε-(γ-l-glutamyl)-l-lysine isopeptide antibody (1:200) (Covalab), which was detected by Alexa Fluor 488 conjugated secondary anti-mouse antibody (1:3,000) (Invitrogen, Molecular Probes, Leiden, Netherlands). FN staining was performed by incubating live cells grown on chamber slides (BD-Falcon) with anti-human FN (Sigma-Aldrich) diluted 1:200 for 1 h at 37 °C. Thereafter, unbound antibodies were carefully washed with PBS and the cells were fixed with 4 % PFA. Subsequently, non-specific binding was blocked with 1 % BSA and the bound antibodies detected with Alexa Fluor 488 or Alexa Fluor 568 conjugated secondary anti-mouse or anti-rabbit antibodies (1:3000) (Invitrogen). All stainings were visualized by fluorescent microscopy analysis (Olympus Optical Co. Ltd, Tokyo, Japan).
SDS-PAGE and immunoblotting
For Western-blot analysis, confluent HUVECs under different experimental conditions were resuspended in Laemmli loading buffer (63 mM Tris–HCl, 10 % glycerol, 2 % SDS, 0.1 % (w/v) Bromophenol Blue, 2.5–5.0 % beta-mercaptoethanol). Total lysates (10 μl) or cell supernatants were separated by 12 % SDS-PAGE electrophoresis under reducing conditions. After gel electrophoresis and transfer of the proteins to a nitrocellulose membrane (Hybond C-extra, Amersham Biosciences Ltd, Little Chalfont, UK), the nitrocellulose sheets were blocked at RT for 1 h in 5 % non-fat dry milk and incubated O/N at +4 °C using specific primary antibodies. The primary antibodies used here included mouse monoclonal antibodies against TG2, CUB7402 (NeoMarkers, Fremont, CA, USA), β1-integrin (MAB 1965, Chemicon), vinculin (7F9, clone sc-73614, Santa Cruz Biotechnology, Wembley, UK). Monoclonal antibody against γ-tubulin (GTU-88, Sigma-Aldrich) was chosen as the internal loading control. Blots were then washed and immunoreactivity detected by sequential incubation of the membranes with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (Dako) for 1 h at RT. Thereafter, the membranes were analyzed using the chemiluminescence detection system (Amersham, ECL plus Western blotting detection system). Exposed autoradiography films (Kodak, New Haven, CT, USA) were subjected to densitometry investigation using Kodak 1D image analysis software (Kodak). The protein expression was normalized to γ-tubulin and control taken as 100 %. The calculated values are derived from three independent experiments performed in duplicate.
Determination of TG2 activity in culture supernatants
Secreted TG2 enzymatic activity in culture supernatants of endothelial cells was measured by 5-biotinamidopentylamine (5-BP) (Thermo Fisher Scientific) incorporation into FN (Human FN 96-well Multiwell) (BD Biosciences). Supernatants of HUVECs under different experimental conditions were collected after 24 h and subsequently incubated with 40 μM 5-BP for 2 h at 37 °C on FN-precoated plates. After extensive washing with Tris HCl pH 7.4, incorporated 5-BP was detected with a streptavidin biotinylated HRP complex (1:5,000) (Life Science Research Pierce Biotechnology). Lastly, the peroxidase substrate TMB (Sigma-Aldrich) was used and the enzymatic reaction stopped with 2.5 M H2SO4. Absorbance was measured at 405 nM by spectrophotometer (Multiskan Ascent, Thermo Labsystems). In order to verify the presence of TG2 during the procedure, some wells were incubated with CUB7402 (1:200) (NeoMarkers, Fremont, CA, USA) instead of 5-BP. Data are derived from two independent experiments, repeated in quadruplicate.
Integrin clustering assay
Integrin clustering was studied as previously reported [22] with minor modifications. Briefly, 2 × 105 HUVECs cells cultured under different experimental conditions were seeded onto FN-coated (10 μg/ml) chamber slides (BD-Falcon) in serum-free (SE) or in 1 % serum-containing medium (SEM). Cells were left to adhere and grow for 4 h in SE and 24 h in SEM. Live cells were incubated with β1-integrin primary antibody (Chemicon) for 1 h, washed with PBS, fixed with 4 % PFA and permeabilized with 0.25 % Triton X-100. Thereafter, the cells were blocked with 0.1 % BSA in PBS for 25 min, followed by incubation for 1 h with the conjugated secondary antibody Alexa Fluor 568 (Invitrogen). Slides were visualized by fluorescent microscopy analysis (Olympus Optical Co. Ltd).
Golgi reorientation assay
Cell polarity assay was performed as previously described [23], with minor modifications. Briefly, 2 × 105 HUVECs were grown onto FN-coated (10 μg/ml) chamber slides (BD-Falcon) until confluent, starved O/N in culture media 1 % FBS, and subjected to different experimental conditions. After 24 h, endothelial cell monolayers were wounded with a pipette tip, washed with PBS and incubated in serum-free media for 4 h with the monoclonal β-COP antibody (clone M3A5, Sigma-Aldrich). After extensive washings, HUVECs were fixed with 4 % PFA, permeabilized, and then blocked with 0.05 % BSA for 1 h at RT. Golgi apparatus was detected by Alexa Fluor 568 conjugated secondary anti-mouse (1:3,000) (Invitrogen). Actin cytoskeleton changes were visualized using FITC-conjugated phalloidin (Invitrogen). Images were captured at 60× using an inverted microscope (IX51; Olympus Optical Co. Ltd). To quantify endothelial cell polarization, Golgi reorientation at the wounded edges was analyzed. A square was drawn over the nucleus and divided into quadrants. Quadrant A was assigned to the area of the cell between the nucleus and the leading edge. A properly reoriented Golgi was indicated by β-Cop staining entirely within quadrant A. Analyses were performed on 100 cells per experiment to determine the percentage of cells with reoriented Golgi. All stainings were visualized by fluorescent microscopy analysis (Olympus Optical Co. Ltd).
Statistical analysis
Statistical analysis was performed using the non-parametric two-tailed Mann–Whitney U test. Data are presented as means + standard error of means. A p value <0.05 was considered significant.
Results
Celiac disease IgA affects endothelial cell number, adhesion, and detachment
We first addressed the anti-angiogenic properties of celiac disease patient IgA (CD IgA) by investigating their effects on endothelial cell number. We noted that there was a small but statistically significant reduction in the number of HUVECs cultured on FN in the presence of CD IgA for 24 h when compared to cells without treatment or incubated with control IgA (H IgA) (Fig. 1a). As the reduction in cell number might be due to adhesion defects or increased apoptosis, we next investigated these cell parameters in our experimental settings. Interestingly, we observed that in HUVEC cultures supplemented with CD IgA there was a significantly lower number of cells left on the plates after detachment by trypsinization when compared to control groups (Fig. 1b). Additionally, the percentage of round, weakly adherent cells on the FN-coated surface was significantly greater among HUVECs treated with CD IgA than in control cultures (Fig. 1c). Pre-incubation of the cells with the extracellular TG2 enzymatic activity inhibitor R281 was not able to normalize any of the above-mentioned effects (Fig. 1a–c). The numbers of apoptotic cells in control conditions and after treatment with H IgA or CD IgA were comparable (30, 33, and 38 %, respectively).
Fig. 1.

Celiac patient immunoglobulins A (CD IgA) reduces endothelial cell number and affects cell adhesion. a HUVEC cell number in cultures without any supplementation and after 24-h treatment with CD or healthy IgA (H IgA) analyzed by chemical reduction of MTS. b The same experimental setting was used to investigate the number of adherent cells on the plate after 5 min of trypsinization. For a and b, the results were normalized to HUVECs without any treatment and indicated by a dashed line. Data are derived from three independent experiments repeated in quadruplicate. c Quantification of weakly adhering cells to fibronectin-precoated chambers as percentage of total cell number. Before adhesion, the cells were grown for 24 h without any supplementation or with H IgA or CD IgA. Quantification was performed by counting the number of round cells (weak adhesion) in randomly selected microscopic pictures of three independent experiments. In all panels, the bars represent the mean and error bars standard error of mean. A p value <0.05 was considered significant (*p < 0.05, **p < 0.01, and ***p < 0.001) and only statistically significant results are reported. The extracellular transglutaminase 2 activity was inhibited by administering a site-directed, non-permeable inhibitor, R281
The extracellular environment of endothelial cells is altered in the presence of celiac IgA
As we found the attachment of HUVECs to FN to be abnormal in the presence of CD IgA, we next investigated whether the altered expression of FN, a matrix ligand of cell surface TG2 [10], might account for these effects. Interestingly, we found that even though the overall FN expression in total cell lysates was not affected by CD IgA when analyzed by Western blotting (data not shown), there was a slight decrease in its surface expression measured by the microtiter plate assay (Fig. 2a). Moreover, immunofluorescent staining showed differences in the architecture of the extracellular FN network in the presence of CD IgA (Fig. 2b). These effects on FN were not reversed by inhibiting the extracellular TG2 enzymatic activity with R281 (Fig. 2a, b). As FN, among other common extracellular matrix proteins, is polymerized by TG2 cross-linking activity [22, 24, 25], we next determined the relative numbers of stable cross-links in our cell cultures. Using a microtiter plate assay, we observed a statistically significant decrease in N-ε-(γ-l-glutamyl)-l-lysine isopeptide bonds in HUVECs incubated with CD IgA autoantibodies when compared to control groups (Fig. 2c). Similarly, immunostaining of extracellular cross-links supported this finding (Fig. 2d).
Fig. 2.

In endothelial cells, celiac patient immunoglobulin A (CD IgA) affects extracellular fibronectin and cross-links. The relative amount of a fibronectin as well as c the extent of N-ε(γ-l-glutamyl)-l-lysine cross-links were analyzed by a microtiter plate assay in HUVECs with no supplementation or after 24-h treatment in the presence of CD IgA and healthy IgA (H IgA). The results were normalized to HUVECs without any treatment, indicated by the dashed line. Bars represent the mean and error bars indicate standard error of the mean. A p value <0.05 was considered significant (*p < 0.05, **p < 0.01, and ***p < 0.001) and only statistically significant results are reported. Data derived from three independent experiments repeated in quadruplicate are shown. b Representative immunofluorescence stainings of fibronectin in live HUVECs alone or incubated for 24 h with H IgA and CD IgA. Scale bar 100 μM. d Representative immunofluorescence stainings of protein cross-links in control HUVECs with no treatment or incubated for 24 h with H IgA and CD IgA. Cross-links are shown in green and DAPI was used as nuclear counterstain (blue). Scale bar 50 μM. The extracellular TG2 activity was inhibited by administering a site-directed, non-permeable inhibitor, R281
As cell surface TG2 is susceptible to degradation by membrane type 1 matrix metalloproteinase (MT1-MMP) [26, 27], we next studied whether increased MT1-MMP expression could elucidate the adhesion defects observed in endothelial cells grown in the presence of CD IgA. Results obtained by the microtiter plate method showed that in the presence of CD IgA, the expression of MT1-MMP was moderately but significantly increased. This increase could not be prevented by the non-permeable TG2 enzymatic inhibitor, R281 (Fig. 3a). Since increased expression of MT1-MMP is related to extensive proteolysis of cell surface TG2, resulting in the appearance of three major cleavage products of molecular masses of ~75, ~66, and ~56 kDa in culture media [22], we next collected supernatants of HUVECs under different experimental conditions to further investigate this issue by Western blotting. Interestingly, we found two main bands, one of which corresponded to intact TG2 (molecular mass of ~75 kDa) and the other to higher molecular weight TG2 (molecular mass of ~200 kDa) highly expressed in the presence of CD IgA (Fig. 3b). Of note, we also found that TG2 secreted from endothelial cells cultured in the presence of CD IgA was enzymatically active (Fig. 3c). The secretion of TG2 into the supernatants was not prevented by R281, whereas its enzymatic activity was abolished (Fig. 3b, c).
Fig. 3.

Celiac patient immunoglobulins A (CD IgA) increase surface expression of membrane type 1 matrix metalloproteinase (MT1-MMP) and promote transglutaminase 2 (TG2) secretion from endothelial cells. a The relative surface expression of MT1-MMP in endothelial cells without any treatment or in the presence of CD IgA or control non-celiac subject immunoglobulin-A (H IgA) was studied by a microtiter plate method. Bars represent mean MT1-MMP expression as fold of control (dashed line) and error bars indicate standard error of the mean. A p value <0.05 was considered significant (***p < 0.001) and only statistically significant results are reported. Data are derived from three independent experiments repeated in quadruplicate. b A representative Western blot of endothelial cell culture supernatants under different experimental conditions performed with CUB7402 to detect TG2. c The relative activity of secreted TG2 was analyzed by microtiter plate assay. Bars represent mean 5-BP incorporation into fibronectin as fold of control and error bars indicate standard error of the mean. A p value <0.05 was considered significant (*p < 0.05, **p < 0.01) and only statistically significant results are reported. Data are derived from two independent experiments repeated in quadruplicate. R281 indicates a non-permeable, site-directed TG2 enzymatic activity inhibitor
Celiac IgA has an effect on integrin β1 and vinculin expression
Since TG2 has a prominent role in cell adhesion as a β1-integrin co-receptor for FN in a non-enzymatic fashion [10], we next considered β1-integrin expression in HUVECs under different experimental conditions. Immunostaining for β1-integrin revealed that in all groups after 4-h adhesion the cells form similar, punctuate β1-integrin patterns, mainly localized on the perinuclear region in those receiving CD IgA (Fig. 4a). After 24 h, in control cultures β1-integrin was mostly found as punctuate structures along the entire cell body, especially on the periphery of the cells, very likely at focal adhesion sites (Fig. 4b). In contrast, endothelial cells treated with CD IgA were characterized by a prominent β1-immunostaining at the periphery of the cell. In addition, we observed that the contact area of the cells appeared filamentous and lacked a bright β1-integrin signal (Fig. 4b). We also assessed the expression of β1-integrin by Western blotting and by a microtiter plate method after 24-h treatment with serum IgA and found it to be increased in HUVECs cultured in the presence of CD IgA (Fig. 5a–c). Preincubation with the extracellular TG2 enzymatic inhibitor R281 did not affect this increase (Fig. 5c).
Fig. 4.
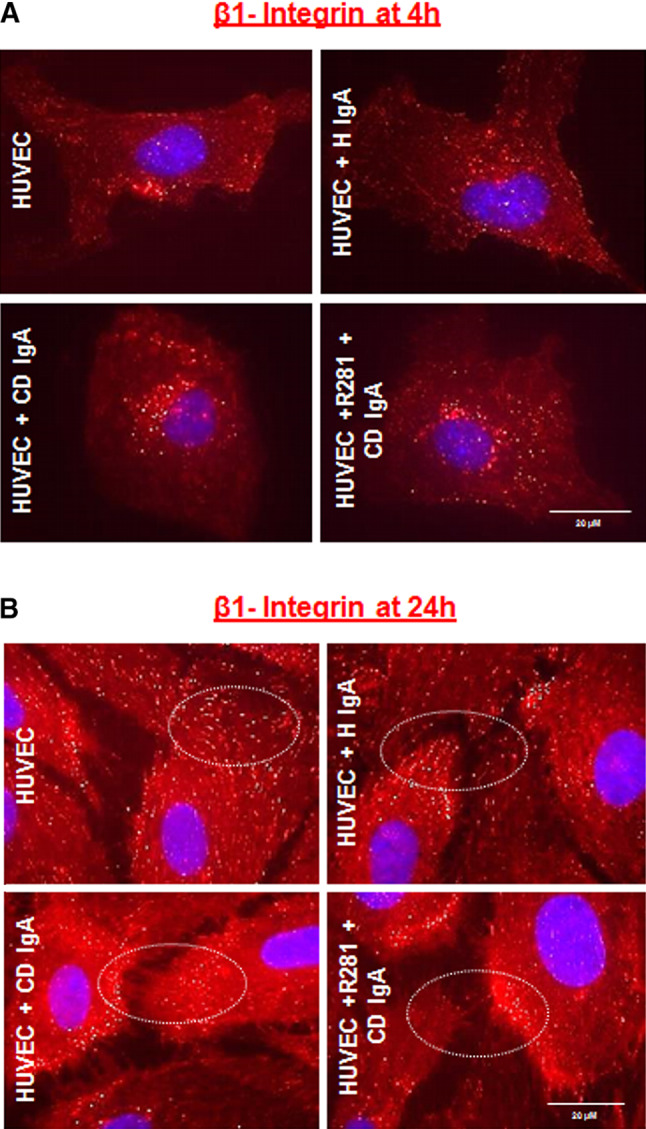
Celiac patient immunoglobulins A (CD IgA) affect β1-integrin clustering in endothelial cells. Representative immunofluorescence stainings of serum-starved endothelial cells (HUVECs) allowed to adhere on fibronectin-precoated chambers for 4 h (in 0 % FBS) (a) and for 24 h (in 1 % FBS) (b) and immunostained with of β1-integrin primary antibody. Differences in β1-integrin (in red) are indicated by white circles and DAPI was used as nuclear counterstaining (blue). Images were selected from three independent experiments. Scale bar 20 μM. The extracellular TG2 activity was inhibited by preincubation of the cells with the non-permeable site-directed inhibitor, R281
Fig. 5.

Celiac patient immunoglobulins A (CD IgA) increase β1-integrin expression in endothelial cells after 24-h treatment. a Quantification of Western-blot analyses of β1-integrin in whole endothelial cell lysates under different experimental conditions. Protein expression was normalized to γ-tubulin and control was taken as 100 %. The dashed line indicates HUVECs without any treatment. Bars represent mean of data derived from four independent experiments repeated in duplicate and error bars indicate standard error of the mean. A p value <0.05 was considered significant (*p < 0.05) and only statistically significant results are reported. b Representative Western blot of β1-integrin in whole endothelial cell lysates under different experimental conditions. c The relative amount of β1-integrin was analyzed by microtiter plate method in unpermeabilized HUVECs alone or incubated with CD IgA and healthy (H IgA) subject’s immunoglobulins. The dashed line indicates HUVECs without any treatment. Bars signify mean and error bars standard error of the mean. A p value <0.05 was considered significant (**p < 0.01, ***p < 0.001) and only statistically significant results are reported. Data are derived from three independent experiments repeated in quadruplicate. The extracellular transglutaminase 2 activity was inhibited by administering a site-directed, non-permeable inhibitor, R281
Adhesion is mediated by the formation of proper focal adhesion sites which include the recruitment of a cytoplasmic protein, vinculin [28] and we thus next investigated its expression. When measured by both the microtiter plate method and Western blotting, we noted a reduction in vinculin expression in cells treated with CD IgA when compared to controls (Fig. 6a–c). As mentioned previously, this decrease was overcome by pre-incubating the cells with R281 (Fig. 6b, c).
Fig. 6.

Celiac patient immunoglobulins A (CD IgA) reduce vinculin protein expression in endothelial cells. a The relative amount of vinculin in HUVECs alone or incubated with CD IgA and healthy (H IgA) subject’s immunoglobulins was studied by microtiter plate assay after 24-h treatment. b Quantification of Western-blot analyses of vinculin in whole endothelial cell lysates under different experimental conditions. Protein expression was normalized to γ-tubulin and control was taken as 100 %. The dashed line indicates HUVECs without any treatment. In a, b, bars represent the mean and error bars indicate standard error of the mean. A p value <0.05 was considered significant (*p < 0.05, **p < 0.01) and only statistically significant results are reported. Data are derived from three independent experiments repeated in quadruplicate. c Representative Western blot of vinculin in whole endothelial cell lysates under different experimental conditions is shown. The extracellular transglutaminase 2 activity was inhibited by administering a site-directed non-permeable inhibitor, R281
Endothelial cell polarization is affected in the presence of celiac disease IgA
Since transient integrin clustering and the establishment of mature focal adhesion sites are necessary for cells to acquire a polarized phenotype [23], we next sought to evaluate this cell parameter by wound healing and Golgi reorientation analysis.
In unwounded endothelial cells monolayers, the Golgi apparatus is randomly oriented [23]. Upon creating a scratch wound edge and allowing for initial cell movement within the denuded space, cells will establish an axis of polarity with lamellipodia and Golgi markers facing the wound edge. Whereas over 90 % of HUVECs incubated with H IgA exhibited Golgi orientation towards the wound edge, only 50 % of HUVECs treated with CD IgA displayed such behavior, with a more disrupted pattern of F-actin and lamellipodia not projected towards the direction of the wound (Fig. 7a–c). Incubation of the cells with R281 prior to CD IgA treatment seemed to ameliorate the defective polarization phenotype even though it did not reverse it totally. In contrast, pre-administration of the C3 transferase inhibitor reversed the polarity defects observed in HUVECs with CD IgA, albeit not to control levels (Fig. 7a–c).
Fig. 7.

Celiac patient immunoglobulins A (CD IgA) inhibit endothelial cell polarization and migration. a Quantification of cell polarization in HUVECs on fibronectin incubated with CD IgA and healthy (H IgA) subject’s immunoglobulins after 24-h treatment by Golgi-reorientation assay. Bars signify the percentage of HUVECs which had reoriented Golgi towards the wound edge and error bars indicate standard error of the mean. A p value <0.05 was considered significant (*p < 0.01, **p < 0.001) and only statistically significant results are reported. More than 500 cells were selected over three independent experiments. b, c Representative immunofluorescence stainings of Golgi by B-COP antibody (red) in HUVECs under different experimental conditions. Actin cytoskeleton was visualized by phalloidin (green) and DAPI (blue) was used as nuclear counterstaining. Images were selected over three independent experiments. The white dashed lines represent the edge of the wound. b Scale bar 100 μM and c scale bar 20 μM. The extracellular TG2 activity was inhibited by administering a site-directed non-permeable inhibitor, R281. Similarly, C3 transferase was administered to inhibit small Rho GTPases in the phenotype
Discussion
The aim of this study was to find new insights into the mechanism of action underlying the antiangiogenic effects exerted by CD IgA in the endothelium, in vitro, without considering the mesenchymal compartment. We observed that the number of endothelial cells was decreased in the presence of CD IgA. This was presumably caused by enhanced susceptibility to detach from FN and an ability to adhere only weakly. Proper cell adhesion relies on the integrity and stabilization of the ECM of the cell [29]. Interestingly, we found that the presence of CD IgA leads to an irregular appearance of the FN network and a reduction in the number of protein cross-links, suggesting that the FN polymerization process catalyzed by extracellular TG2 cross-linking activity is disturbed, this accounting for the abnormal ECM, altered adhesion properties, and reduction in endothelial cell numbers. Of note, inhibition of extracellular TG2 by R281 did not modify the CD IgA-mediated effects. The finding of a decreased ECM cross-links was unexpected, since our previous works [2, 19] have shown that celiac disease autoantibodies increase endothelial TG2 enzymatic activity when measured by incorporation of primary amines. However, this discrepancy might be explained by the fact that transamidation (using a small primary amine substrate) and protein crosslinking by TG2 take place under different conditions. Thus, bearing in mind that the cross-linking activity of TG2 is required for FN assembly and deposition [22, 30], we may speculate that this function of TG2 is inhibited in the presence of CD IgA and leads to decreased expression and abnormal appearance of extracellular FN. A recent work, again, reporting that major auto antigenic epitopes in TG2 recognized by celiac patient autoantibodies are clustered in the N-terminal half of the enzyme [31], in part corresponding to the FN binding site of TG2, allows us to alternatively explain our findings by the inability of anti-human-FN antibody to recognize FN when it is complexed to TG2 and CD IgA. In this regard, our preliminary unpublished data would imply that this indeed might be the case, since in the presence of CD IgA the detection of FN associated with TG2 is reduced.
It has also been reported that treatment of FN-TG2 matrix with TG2 autoantibodies from celiac disease patients reduces cell attachment [32]. Mesin and coworkers have hypothesized that anti-TG2 IgA could displace TG2 from the FN meshwork, and such a mechanism could explain the altered cell adhesion properties [31, 33]. In contrast to this claim, we observed in our in vitro experimental settings that when TG2 is immobilized on FN, it is not released in the presence of CD IgA (unpublished results) nor could any interference be measured between the celiac autoantibodies and FN binding in the presence of Ca2+ [31], which is abundantly available for TG2 in extracellular conditions. On the other hand, we saw augmented secretion of TG2 in endothelial cell culture supernatants treated with CD IgA, independent of the TG2 extracellular activation, since administration of R281 was not capable of preventing this effect. However, we can exclude the involvement of MT1-MMP in this process because only a moderate increase in its expression was observed on the endothelial cell surface of HUVECs-CD IgA which did not promote the proteolytic degradation of TG2. Interestingly, the secreted TG2 was proved to possess transamidase activity. In this regard, a recent work has suggested that the release of TG2 could possibly lead to increased enzymatic activity as the enzyme is brought into solution [31], which would explain our previously reported findings of increased TG2 activity in the presence of celiac IgA. Nevertheless, the increased TG2 amino incorporation activity observed in the presence of celiac patient autoantibodies was measured in an endothelial culture system after supernatants were removed, this pointing towards another mechanism. In addition, externalized TG2 is rapidly incorporated into the ECM [27], as could have occurred in our experimental settings, this explaining the increased TG2 transamidase activity so far observed in the presence of celiac autoantibodies. However, our view is that this might not be the case, since TG2 secretion induced from endothelial cells by CD IgA was not accompanied by a higher expression of extracellular TG2.
Cell-secreted and surface TG2 have different but complementary roles in promoting cell adhesion. At the cell surface, in a non-enzymatic fashion, TG2-FN-mediated integrin clustering potentiates outside-in signaling, while TG2-FN-mediated syndecan-4 binding leads to inside-out integrin signaling to promote cell adhesion and spreading. Functional collaboration between integrin, syndecans, and TG2 is reflected by the alteration of integrin clustering and focal adhesion assembly [34, 35]. We found that in endothelial cells the presence of CD IgA induced the formation of punctate β1-integrin structures after 4 h of adhesion mostly localized around the perinuclear area. After 24 h, a more pronounced alteration in integrin distribution was observed. In fact, cells incubated with CD IgA displayed distinct filaments between the cells coupled to marked β1-integrin immunostaining at the periphery of the cells, in contrast to controls, where more homogeneous punctate arrangements were observed, this probably corresponding to focal adhesion sites. This would suggest that CD IgA induces constitutive clustering and activation of integrins different from the transient integrin clustering required for cell adhesion [34]. This activation was related to increased expression of β1-integrin in the presence of CD IgA.
It has been shown that integrin clustering promotes RhoA activation [34], and this activation is involved in increased vascular permeability in the presence of CD IgA, at least in vitro [2]. Since RhoA affects polarity in migrating cells [23], we next studied directional cell motility in HUVECs under different experimental conditions. We found that endothelial cells when treated with CD IgA lose polarization when compared to control groups. It is noteworthy that the effects exerted by CD IgA were reversed with prior incubation of the cells with C3 transferase inhibitor but not completely by R281, suggesting an important involvement of the RhoA signaling pathway in the polarization defects described above. We however cannot entirely exclude the collaboration of other members of the Ras family, since C3 transferase inhibits the activity of all small GTPases. Moreover, we observed that endothelial cells treated with CD IgA had rounded and less organized focal adhesion sites when compared to controls (unpublished data) together with decreased expression of the late adhesion molecule vinculin. It is of note that R281 was able to ameliorate the expression level of vinculin, suggesting a role for extracellular TG2 enzymatic activity on vinculin expression.
Although a limitation of our study was that we used untreated celiac disease patient’s total serum IgA that likely includes different pools of antibodies in addition to anti-TG2, such as anti-actin, anti-desmin, anti-reticulin autoantibodies [36] as well as anti-deamidated gliadin peptide antibodies, our net results would imply that CD IgA antibodies disturb the extracellular protein cross-linking function of TG2, thus altering endothelial cell–ECM interactions and thereby affecting endothelial cell adhesion, polarization, and motility. These alterations are possibly attributable to altered focal adhesion sites triggered by abnormal ECM organization, the cells seeking to overcome the defect by modulating the expression of β1-integrin, and increased RhoA activity.
Finally, we were intrigued to find that CD IgA promoted the secretion of TG2 into the culture medium from endothelial cells and this secreted TG2 was enzymatically active. Even though the role of TG2 in the extracellular compartment implies its involvement mainly in the stabilization of the ECM and in cell adhesion, we cannot exclude the existence of other undefined functions. In this regard, we might hypothesize that in pathological conditions such as celiac disease, the secretion of TG2 might be related to immune-modulatory functions linked to its active state.
If CD IgA antibodies act on vascular function in vivo similarly to what we have described in the present study in vitro, we could explain the altered small bowel mucosal microvasculature in untreated celiac disease patients [37]. Further studies are thus clearly called for in order to better clarify the mechanism herein proposed, this possibly helping to elucidate the anti-angiogenic effects exerted by CD IgA in the pathogenesis of the disease.
Acknowledgments
We thank Kaisa Teittinen for comments on the manuscript. The Coeliac Disease Study Group was financially supported by the Academy of Finland, the Tampere Graduate Program in Biomedicine and Biotechnology, the Sigrid Juselius Foundation, the Research Fund of the Finnish Coeliac Society, the Competitive State Research Financing of the Expert Responsibility Area of Tampere University Hospital (grant numbers 9P020 and 9P033) and the Pediatric Research Foundation, Elna Kaarina Savolainen’s fund allocated for the development of cancer treatment and the European Commission IAPP grant TRANSCOM (Contract number PIA-GA-2010-251506). Further, the grants OTKA K101788, NK105046 and TÁMOP 4.2.2.11/1/KONV-2012-0023 are also acknowledged.
References
- 1.Martin CA, Perrone EE, Longshore SW, Toste P, Bitter K, Nair R, Guo J, Erwin CR, Warner BW. Intestinal resection induces angiogenesis within adapting intestinal villi. J Pediatr Surg. 2009;44:1077–1082. doi: 10.1016/j.jpedsurg.2009.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Myrsky E, Caja S, Simon-Vecsei Z, Korponay-Szabo IR, Nadalutti C, Collighan R, Mongeot A, Griffin M, Mäki M, Kaukinen K, Lindfors K. Celiac disease IgA modulates vascular permeability in vitro through the activity of transglutaminase 2 and RhoA. Cell Mol Life Sci. 2009;20:3375–3385. doi: 10.1007/s00018-009-0116-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooke WT, Holmes GK. Celiac disease. New York: Churchill Livingstone; 1984. pp. 27–28. [Google Scholar]
- 4.Aleanzi M, Demonte AM, Esper C, Garcilazo S, Waggener M. Celiac disease: antibody recognition against native and selectively deamidated gliadin peptides. Clin Chem. 2001;47:2023–2028. [PubMed] [Google Scholar]
- 5.Sulkanen S, Halttunen T, Laurila K, Kolho KL, Korponay-Szabo IR, Sarnesto A, Savilahti E, Collin P, Mäki M. Tissue transglutaminase autoantibody enzyme-linked immunosorbent assay in detecting celiac disease. Gastroenterology. 1998;115:1322–1328. doi: 10.1016/S0016-5085(98)70008-3. [DOI] [PubMed] [Google Scholar]
- 6.Dieterich W, Laag E, Schöpper H, Volta U, Ferguson A, Gillett H, Riecken EO, Schuppan D. Autoantibodies to tissue transglutaminase as predictors of celiac disease. Gastroenterology. 1998;115:1317–1321. doi: 10.1016/S0016-5085(98)70007-1. [DOI] [PubMed] [Google Scholar]
- 7.Klöck C, Khosla C. Regulation of the activities of the mammalian transglutaminase family of enzymes. Protein Sci. 2012;12:1781–1791. doi: 10.1002/pro.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molberg O, Mcadam SN, Körner R, Quarsten H, Kristiansen C, Madsen L, Fugger L, Scott H, Norén O, Roepstorff P, Lundin KE, Sjöström H, Sollid LM. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4:713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 9.Aeschlimann D, Thomazy V. Protein crosslinking in assembly and remodelling of extracellular matrices: the role of transglutaminases. Connect Tissue Res. 2000;41:1–27. doi: 10.3109/03008200009005638. [DOI] [PubMed] [Google Scholar]
- 10.Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion co receptor for fibronectin. J Cell Biol. 2000;148:825–838. doi: 10.1083/jcb.148.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z, Collighan RJ, Pytel K, Rathbone DL, Li X, Griffin M. Characterization of heparin-binding site of tissue transglutaminase: its importance in cell surface targeting, matrix deposition, and cell signaling. J Biol Chem. 2012;287:13063–13083. doi: 10.1074/jbc.M111.294819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kotsakis P, Griffin M. Tissue transglutaminase in tumour progression: friend or foe? Amino Acids. 2007;33:373–384. doi: 10.1007/s00726-007-0516-1. [DOI] [PubMed] [Google Scholar]
- 13.Nadalutti C, Viiri KM, Kaukinen K, Mäki M, Lindfors K. Extracellular transglutaminase 2 has a role in cell adhesion, whereas intracellular transglutaminase 2 is involved in regulation of endothelial cell proliferation and apoptosis. Cell Prolif. 2011;44:49–58. doi: 10.1111/j.1365-2184.2010.00716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haroon ZA, Hettash JM, Lai TS, Dewhirst MW, Greenberg CS. Tissue transglutaminase is expressed, active and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999;13:1787–1795. doi: 10.1096/fasebj.13.13.1787. [DOI] [PubMed] [Google Scholar]
- 15.Jones RA, Kotsakis P, Johnson TS, Chau DY, Ali S, Melino G, Griffin M. Matrix changes induced by transglutaminase 2 lead to inhibition of angiogenesis and tumor growth. Cell Death Differ. 2006;13:1442–1453. doi: 10.1038/sj.cdd.4401816. [DOI] [PubMed] [Google Scholar]
- 16.Munezane T, Hasegawa T, Suritala, Tanaka A, Okada K, Okita Y. Activation of transglutaminase type 2 for aortic wall protection in a rat abdominal aortic aneurysm formation. J Vasc Surg. 2010;52:967–974. doi: 10.1016/j.jvs.2010.04.049. [DOI] [PubMed] [Google Scholar]
- 17.Jones RA, Wang Z, Dookie S, Griffin M. The role of TG2 in ECV304-related vasculogenic. Amino Acids. 2013;44:89–101. doi: 10.1007/s00726-011-1214-6. [DOI] [PubMed] [Google Scholar]
- 18.Myrsky E, Kaukinen K, Syrjanen M, Korponay-Szabò IR, Maki M, Lindfors K. Coeliac disease-specific autoantibodies targeted against transglutaminase 2 disturb angiogenesis. Clin Exp Immunol. 2008;152:111–119. doi: 10.1111/j.1365-2249.2008.03600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caja S, Myrsky E, Korponay-Szabo IR, Nadalutti C, Sulic AM, Lavric M, Sblattero D, Marzari R, Collighan R, Mongeot A, Griffin M, Mäki M, Kaukinen K, Lindfors K. Inhibition of transglutaminase 2 enzymatic activity ameliorates the anti-angiogenic effects of coeliac disease autoantibodies. Scand J Gastroenterol. 2010;45:421–427. doi: 10.3109/00365520903540822. [DOI] [PubMed] [Google Scholar]
- 20.Martucciello S, Lavric M, Toth B, Korponay-Szabo I, Nadalutti C, Myrsky E, Rauhavirta T, Esposito C, Sulic AM, Sblattero D, Marzari R, Mäki M, Kaukinen K, Lindfors K, Caja S. RhoB is associated with the anti-angiogenic effects of celiac patient transglutaminase 2-targeted autoantibodies. J Mol Med. 2012;90:817–826. doi: 10.1007/s00109-011-0853-0. [DOI] [PubMed] [Google Scholar]
- 21.Griffin M, Mongeot A, Collighan R, Saint RE, Jones RA, Coutts IG, Rathbone DL. Synthesis of potent water-soluble tissue transglutaminase inhibitors. Bioorg Med Chem Lett. 2008;18:5559–5562. doi: 10.1016/j.bmcl.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Forsprecher J, Wang Z, Nelea V, Kaartinen MT. Enhanced osteoblast adhesion on transglutaminase 2-crosslinked fibronectin. Amino Acids. 2009;36:747–753. doi: 10.1007/s00726-008-0125-7. [DOI] [PubMed] [Google Scholar]
- 23.Tomar A, Lim ST, Lim Y, Schlaepfer DD. A FAK-p120RasGAP-p190RhoGAP complex regulates polarity in migrating cells. Cell Sci. 2009;122:3005. doi: 10.1242/jcs.058487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zemskov EA, Janiak A, Hang J, Waghray A, Belkin AM. The role of tissue transglutaminase in cell-matrix interactions. Front Biosci. 2006;11:1057–1076. doi: 10.2741/1863. [DOI] [PubMed] [Google Scholar]
- 25.Scarpellini A, Germack R, Lortat-Jacob H, Muramatsu T, Billett E, Johnson T, Verderio EAM. Heparan sulfate proteoglycans are receptors for the cell-surface trafficking and biological activity of transglutaminase-2. J Biol Chem. 2009;284:18411–18423. doi: 10.1074/jbc.M109.012948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belkin AM, Akimov SS, Zaritskaya LS, Ratnikov BI, Deryugina EI, Strongin AY. Matrix-dependent proteolysis of surface transglutaminase by membrane-type metalloproteinase regulates cancer cell adhesion and locomotion. J Biol Chem. 2001;276:18415–18422. doi: 10.1074/jbc.M010135200. [DOI] [PubMed] [Google Scholar]
- 27.Belkin A. Extracellular TG2: emerging functions and regulation. FEBS J. 2011;278:4704–4716. doi: 10.1111/j.1742-4658.2011.08346.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Demali KA. Vinculin-a dynamic regulator of cell adhesion. Trends Biochem Sci. 2004;11:565–567. doi: 10.1016/j.tibs.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Midwood KS, Williams LV, Schwarzbauer JE. Tissue repair and the dynamics of the extracellular matrix. Int J Biochem Cell Biol. 2004;36:1031–1037. doi: 10.1016/j.biocel.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Al-Jallad HF, Nakano Y, Chen JL, McMillan E, Lefebvre C, Kaartinen MT. Transglutaminase activity regulates osteoblast differentiation and matrix mineralization in MC3T3-E1 osteoblast cultures. Matrix Biol. 2006;25:135–148. doi: 10.1016/j.matbio.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Iversen R, Di Niro R, Stamnaes J, Lundin KE, Wilson PC, Sollid LM. Transglutaminase 2-specific autoantibodies in celiac disease target clustered, N-terminal epitopes not displayed on the surface of cells. J Immunol. 2013;190:5981–5991. doi: 10.4049/jimmunol.1300183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teesalu K, Panarina M, Uibo O, Uibo R, Utt M. Autoantibodies from patients with celiac disease inhibit transglutaminase 2 binding to heparin/heparan sulfate and interfere with intestinal epithelial cell adhesion. Amino Acids. 2012;42:1055–1064. doi: 10.1007/s00726-011-1020-1. [DOI] [PubMed] [Google Scholar]
- 33.Mesin L, Sollid LM, Di Niro R. The intestinal B-cell response in celiac disease. Front Immunol. 2012;3:313. doi: 10.3389/fimmu.2012.00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Janiak A, Zemskov EA, Belkin AM. Cell surface transglutaminase promotes RhoA activation via integrin clustering and suppression of the Src-p190RhoGAP signaling pathway. Mol Biol Cell. 2006;17:1606–1619. doi: 10.1091/mbc.E05-06-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Telci D, Wang Z, Li X, Verderio EA, Humphries MJ, Baccarini M, Basaga H, Griffin M. Fibronectin-tissue transglutaminase matrix rescues RGD-impaired cell adhesion through syndecan-4 and beta1 integrin co-signaling. J Biol Chem. 2008;283:20937–20947. doi: 10.1074/jbc.M801763200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaoul R, Lerner A. Associated autoantibodies in celiac disease. Autoimmun Rev. 2007;8:559–565. doi: 10.1016/j.autrev.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 37.Myrsky E, Syrjänen M, Korponay-Szabo IR, Mäki M, Kaukinen K, Lindfors K. Altered small-bowel mucosal vascular network in untreated coeliac disease. Scand J Gastroenterol. 2009;44:162–167. doi: 10.1080/00365520802400875. [DOI] [PubMed] [Google Scholar]